
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 01 March 2024
Sec. Adrenal Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1336306
This article is part of the Research TopicAdvances in Diagnostics and Management of Adrenal TumorsView all 24 articles
 Marta Araujo-Castro1,2*†
Marta Araujo-Castro1,2*† Paola Parra3Patricia Martín Rojas-Marcos3Miguel Paja Fano4,5Marga González Boillos6
Paola Parra3Patricia Martín Rojas-Marcos3Miguel Paja Fano4,5Marga González Boillos6 Eider Pascual-Corrales1,2
Eider Pascual-Corrales1,2 Ana María García Cano7
Ana María García Cano7 Jorge Gabriel Ruiz-Sanchez8Almudena Vicente Delgado9Emilia Gómez Hoyos10Rui Ferreira11Iñigo García Sanz12Mònica Recasens Sala13Rebeca Barahona San Millan13María José Picón César14,15Patricia Díaz Guardiola16
Jorge Gabriel Ruiz-Sanchez8Almudena Vicente Delgado9Emilia Gómez Hoyos10Rui Ferreira11Iñigo García Sanz12Mònica Recasens Sala13Rebeca Barahona San Millan13María José Picón César14,15Patricia Díaz Guardiola16 Carolina M. Perdomo17
Carolina M. Perdomo17 Laura Manjón-Miguélez18,19Rogelio García Centeno20
Laura Manjón-Miguélez18,19Rogelio García Centeno20 Ángel Rebollo Román21Paola Gracia Gimeno22
Ángel Rebollo Román21Paola Gracia Gimeno22 Cristina Robles Lázaro23Manuel Morales-Ruiz24
Cristina Robles Lázaro23Manuel Morales-Ruiz24 María Calatayud25Simone Andree Furio Collao25Diego Meneses6Miguel Sampedro Nuñez26Verónica Escudero Quesada27Elena Mena Ribas28Alicia Sanmartín Sánchez28Cesar Gonzalvo Diaz29
María Calatayud25Simone Andree Furio Collao25Diego Meneses6Miguel Sampedro Nuñez26Verónica Escudero Quesada27Elena Mena Ribas28Alicia Sanmartín Sánchez28Cesar Gonzalvo Diaz29 Cristina Lamas29María del Castillo Tous30Joaquín Serrano Gotarredona31Theodora Michalopoulou Alevras32Eva María Moya Mateo33
Cristina Lamas29María del Castillo Tous30Joaquín Serrano Gotarredona31Theodora Michalopoulou Alevras32Eva María Moya Mateo33 Felicia A. Hanzu34
Felicia A. Hanzu34Purpose: To compare the clinical and hormonal characteristics of patients with familial hyperaldosteronism (FH) and sporadic primary aldosteronism (PA).
Methods: A systematic review of the literature was performed for the identification of FH patients. The SPAIN-ALDO registry cohort of patients with no suspicion of FH was chosen as the comparator group (sporadic group).
Results: A total of 360 FH (246 FH type I, 73 type II, 29 type III, and 12 type IV) cases and 830 sporadic PA patients were included. Patients with FH-I were younger than sporadic cases, and women were more commonly affected (P = 0.003). In addition, the plasma aldosterone concentration (PAC) was lower, plasma renin activity (PRA) higher, and hypokalemia (P < 0.001) less frequent than in sporadic cases. Except for a younger age (P < 0.001) and higher diastolic blood pressure (P = 0.006), the clinical and hormonal profiles of FH-II and sporadic cases were similar. FH-III had a distinct phenotype, with higher PAC and higher frequency of hypokalemia (P < 0.001), and presented 45 years before sporadic cases. Nevertheless, the clinical and hormonal phenotypes of FH-IV and sporadic cases were similar, with the former being younger and having lower serum potassium levels.
Conclusion: In addition to being younger and having a family history of PA, FH-I and III share other typical characteristics. In this regard, FH-I is characterized by a low prevalence of hypokalemia and FH-III by a severe aldosterone excess causing hypokalemia in more than 85% of patients. The clinical and hormonal phenotype of type II and IV is similar to the sporadic cases.
Primary aldosteronism (PA) is the most common cause of secondary hypertension, accounting for 10% of hypertensive patients in the general setting and of 20% in patients with refractory hypertension (1). Approximately 95% of these cases are sporadic, with the remaining 5% caused by a pathogenic variant in genes that result in an increase in the transcription and expression of CYP11B2, which is responsible for aldosterone synthesis (familial hyperaldosteronism (FH) types II, III, and IV) or a fusion of the CYP11B2 and CYP11B1 genes, which is responsible for FH-I or glucocorticoid-remediable PA (GRA) (2).
Sutherland et al. described the familial occurrence of PA for the first time in 1966 (3). Lifton et al. (4) demonstrated the genetic basis of this familial form: GRA caused by a hybrid gene composed of ACTH-regulated 11b-hydroxylase (CYP11B1) regulatory sequences and aldosterone synthase (CYP11B2) coding sequences. Following that, a number of FH type I cases have been described in the literature (5–29). Gordon et al. (30) described the first case of FH type II several years after the discovery of FH-I. Since its first description, more than 80 cases have been reported in the literature (6, 31–41). More recently, two additional kinds of genetic PA have been identified: type III caused by a pathogenic mutation in KCNJ5 (42) and type IV caused by a CACNA1H pathogenic variant (43).
However, few studies have directly compared the clinical and hormonal aspects of the familial and sporadic PA cases (11, 33), despite the well-known description of the familial cases. Michael Stowasser (33) was the first to compare 88 consecutive PA patients and 13 FH type II patients. There were no differences in age at presentation, sex incidence, and biochemical parameters between both groups. Another series comparing family members with positive (n = 21) and negative (n = 18) study for FH-I found that body mass index (BMI) was lower and plasma aldosterone concentration (PAC) higher in FH-I; however, the control group did not have PA in this study (26). This study design was comparable with the Litchfield study (25). A subsequent study matched 79 GRA-positive patients and 114 GRA-negative unilateral PA patients by age, gender, and BMI. They found that being younger and having a lower PAC were associated with a higher probability of FH-I (11). Nevertheless, the majority of these studies contained a small number of patients and only evaluated the differences between sporadic PA cases and FH type I and type II cases.
Considering this background, the aim of our study was to identify the clinical and hormonal features linked to FH by comparing a large cohort of FH patients to a group of sporadic PA cases. Based on that information, we will aim to determine which PA patients should be genetically tested.
The control group of sporadic cases was extracted from the Spanish Primary Aldosteronism (SPAIN-ALDO) Registry of the Spanish Endocrinology and Nutrition Society (SEEN). As previously described (44), this is a multicenter collaborative study involving patients with a diagnosis of PA who were followed up in 35 Spanish tertiary hospitals between January 2018 and July 2023. At the time of data analysis (11/08/2023), the registry contained 855 patients with PA.
We excluded the following patients from the sporadic group: (i) those patients with a positive genetic study (n = 1); (ii) those with familial history of PA and no available negative genetic study for FH (n = 13); (iii) patients who underwent genetic testing due to high suspicion and whose results were pending (n = 7); and (iv) patients diagnosed of PA before the age of 30 and no available negative genetic study for FH (n = 4). Thus, 830 cases were included. Of these 830 patients, only 18 underwent genetic study for FH, with negative results (Figure 1).
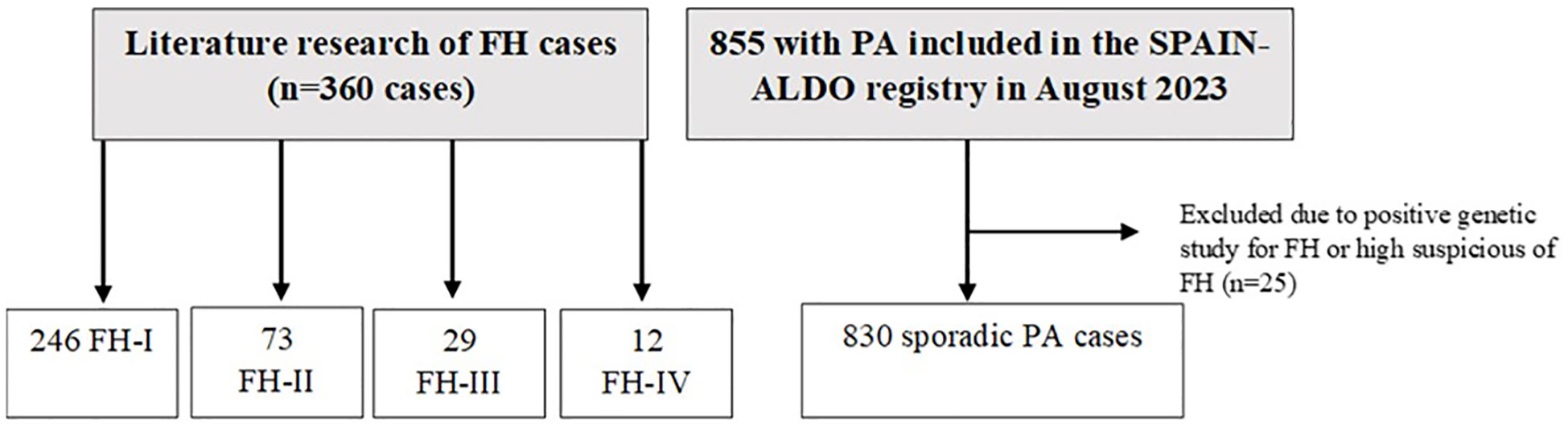
Figure 1 Study population. PA, primary aldosteronism; FH, familial hyperaldosteronism.
For the diagnosis of FH, we followed the following criteria: Diagnosis of FH-I was performed by long-PCR amplification of the hybrid gene (CYP11B1/CYP11B2); diagnosis of FH-II was made when a mutation in CLCN2 was detected or when there was a family history of PA in at least two family members; diagnosis of FH-III when a pathogenic variant in KCNJ5 was demonstrated and FH-IV if a pathogenic variant in CACNA1H was detected.
PA diagnosis was made following the recommendations of the clinical international guidelines for PA (45, 46). Blood samples for plasma renin activity (PRA) and/or concentration (PRC) and PAC were collected from all patients. As we have previously described (47), those patients (n = 340) who did not meet the criteria of overt PA (hypokalemia, PAC >18 ng/dL and pathological aldosterone-to-renin ratio) underwent at least one of the following confirmatory tests: oral sodium loading, saline infusion test, captopril challenge test, and/or fludrocortisone suppression test.
The findings of the adrenal venous sampling (AVS) and/or the outcomes after adrenalectomy were used to differentiate between unilateral and bilateral PA. A total of 326 out of 830 patients underwent AVS, with 190 being successful. Unilateral disease was assumed if the lateralization index of the aldosterone to cortisol ratio was ≥4.0 on the dominant vs. non-dominant side during ACTH stimulation or at least two times higher under unstimulated conditions (1, 6). In patients without successful AVS, unilateral disease was assumed if a complete biochemical cure after surgery was obtained (n = 186). The PASO classification criteria were used to define biochemical and clinical cure for PA after adrenalectomy (48).
The FH cases were identified using the SANRA scale (49). The search strategy was conducted in PubMed without a date filter until 12/08/2023. The following keywords were used in the search: familial primary aldosteronism [TI]: 136 results; familial hyperaldosteronism [TI]: 61 results; hereditary aldosteronism [title]: 18 results; inherited primary aldosteronism [TI]: 16 results; dexamethasone-suppressible aldosteronism: 2 results; and glucocorticoid-remediable aldosteronism [TI]: 49 results. Potentially relevant articles were retrieved after reading the title, abstract, or whole article, and we discarded repeated articles. Only articles written in English were considered. The articles found through these searches as well as the pertinent references listed in those papers were reviewed. After that, 53 original articles were included: 25 articles about FH-I (5–29), 12 about FH-II (6, 31–41), 13 about FAH-III (32, 42, 50–60), and 3 about FAH-IV (43, 61, 62) (Figure 2).
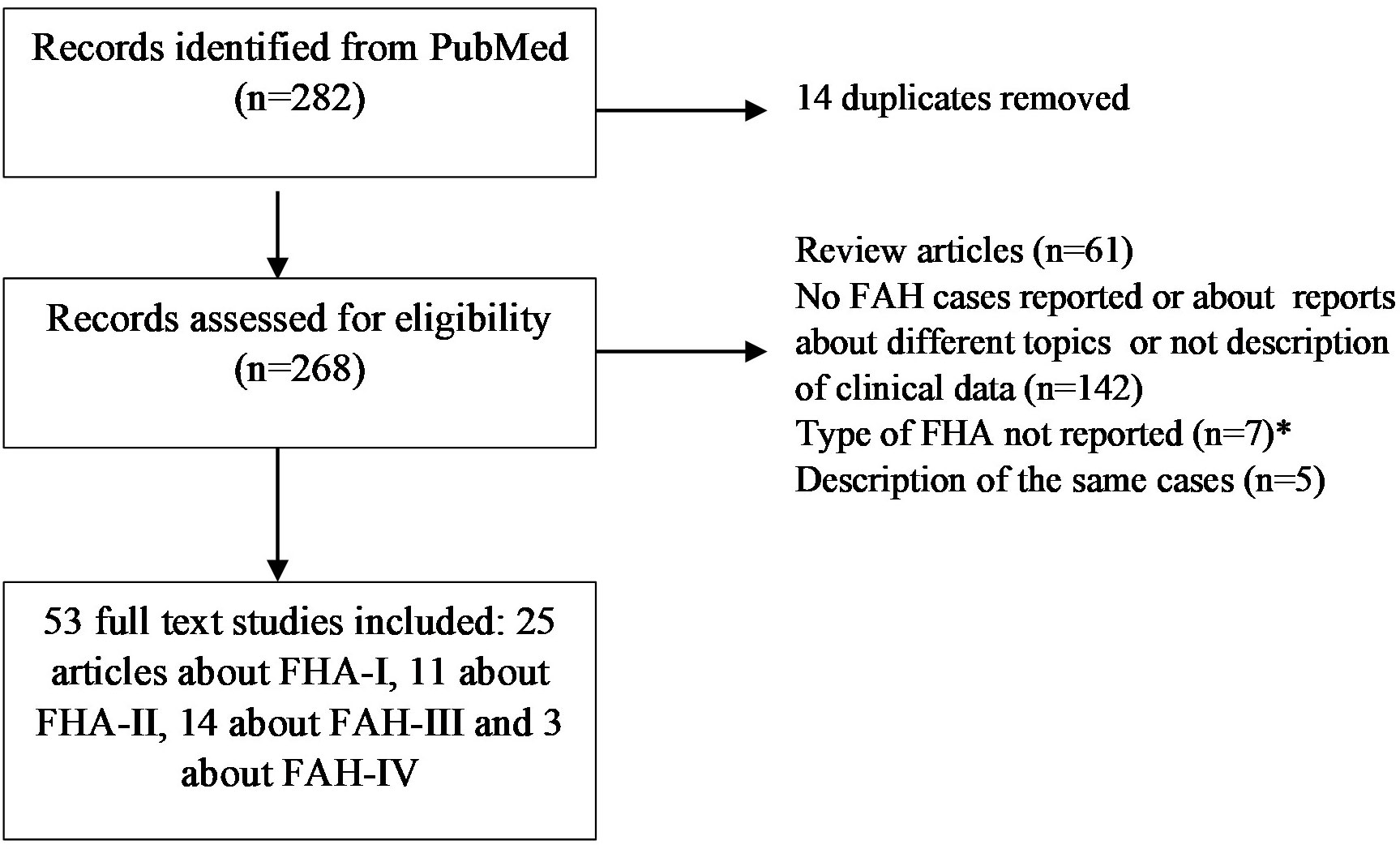
Figure 2 Flow-chart for identification of FAH cases. FH, familial hyperaldosteronism.
All statistical analyses were conducted with STATA.15. Shapiro–Wilk’s test was used to assess the normality of continuous variables. All data are expressed as the mean and standard deviation for normally distributed variables and the median (and range) for non-normally distributed variables. Student’s t-test was used to compare quantitative variables and the X2 test for qualitative variables between two groups. In all cases, a two-tailed P value < 0.05 was considered statistically significant.
A total of 246 patients with confirmed FH-I were compared with 830 cases of sporadic PA. Patients with FH-I were found to be younger than sporadic cases, and women were more commonly affected than men. On the other hand, sporadic cases had higher PAC and lower PRA than FH-I. Besides, hypokalemia was uncommon (12%) in FH-I patients, but it reached a prevalence of 60% in sporadic cases. 40.3% of the FH-I patients (n = 77/129) were normotensive. No other differences were detected between the two groups (Table 1).
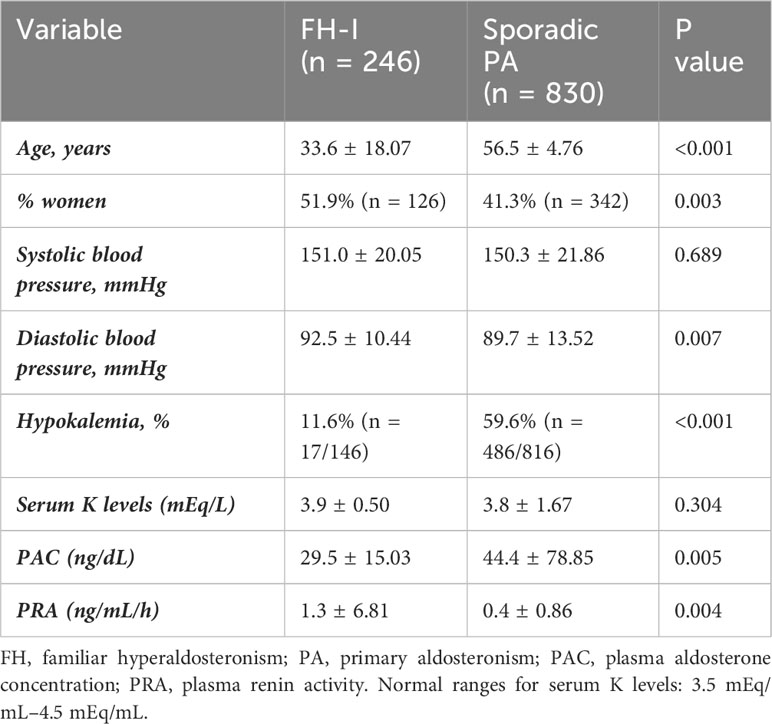
Table 1 Differences between FH-I and sporadic PA cases.
We compared 73 FH-II patients to 830 sporadic PA patients. The clinical and hormonal profiles were similar, except for a younger age and higher diastolic blood pressure in the group of FH-II (Table 2).
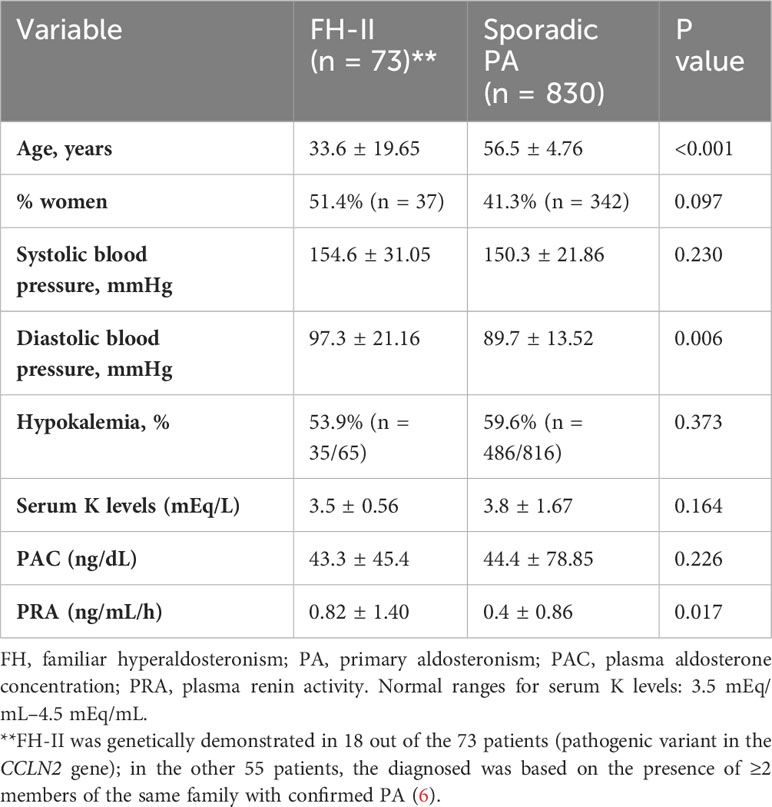
Table 2 Differences between FH-II and sporadic PA cases.
The 29 cases of FH type III and the 12 cases of FH-IV were compared with the sporadic cases. FH-III showed a distinct phenotype, with higher PAC, lower PRA, lower serum potassium levels, and a younger age at PA diagnosis, presenting 45 years earlier than the sporadic cases (Table 3). Due to problems in achieving proper blood pressure control, 17 of the 29 patients with FH-III underwent bilateral adrenalectomy.
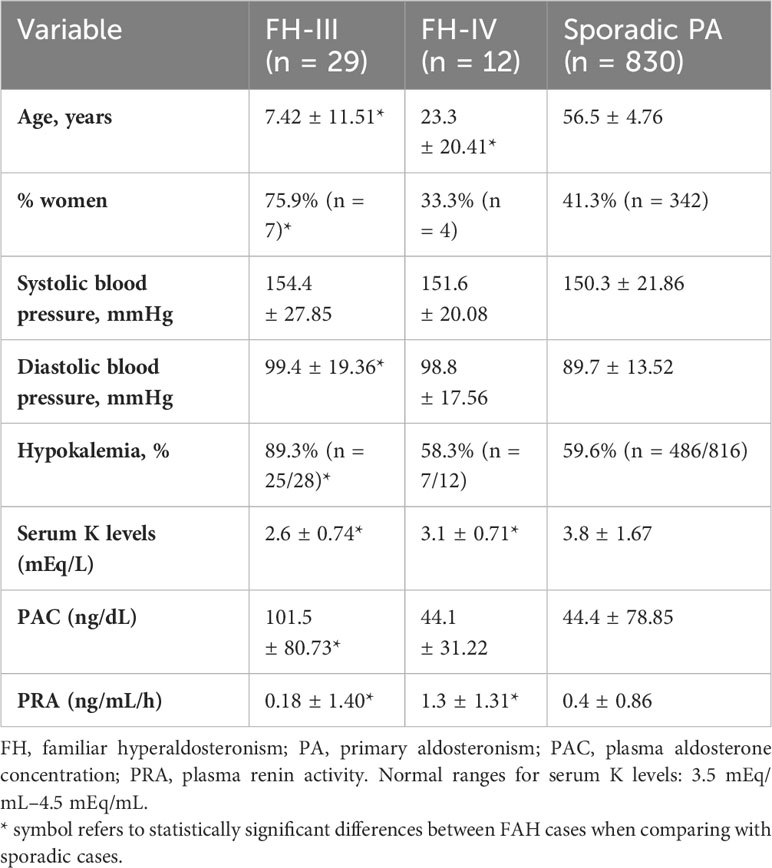
Table 3 Differences between FH-III and IV and sporadic PA cases.
The clinical and hormonal phenotypes of FH type IV and sporadic cases were similar, except for lower age and serum potassium levels and higher PRA at presentation in the former (Table 3).
The most consistent finding in FH patients was the younger age of the four types when compared with sporadic PA cases. Because FH is transmitted by autosomal dominant inheritance, the history of other members of the family with PA is another key indicator to infer hereditary PA (63). In fact, the current general recommendation for screening hereditary PA is for patients with early onset and a positive family history of PA (64).
When we compared FH type I and sporadic cases, we found that, in addition to being younger, FH-I had more severe hypertension than in sporadic PA patients. In this regard, it has been previously reported that severe hypertension in infancy and early adulthood is the most typical presentation of GRA (5). Nevertheless, some cases are just moderately hypertensive, if at all (65). According to this, we found that 40% of the FH-I patients had normal blood pressure levels and were classed as normotensive. However, even in normotensive FH-I patients, the aldosterone excess is associated with increased left ventricular wall thickness and reduced diastolic function when compared with normotensive subjects without GRA (23). Another notable feature of FH-I cases in our study was the low prevalence of hypokalemia, which occurred in less than 12% of the cases. Accordingly, a prevalence of 13% was described in the PATOGEN study (6) and in the Aglony et al. series (5). The real cause of hypokalemia in individuals with PA is due to the fact that the CYP11B1/CYP11B2 hybrid gene is unknown; however, it may be connected to the fact that aldosterone secretion is regulated by ACTH instead of angiotensin II. Thus, the mineralocorticoid effect may be expected to be reduced. This hypothesis is supported by the observation that PRA was less suppressed in FH-I than in sporadic cases. Considering our results, it is important to take into account that FH-I may be hypokalemic or normokalemic. Thus, the presence of normal serum potassium levels does not discount genetic testing particularly in families where the penetrance of hypertension is inevitably variable.
In relation to FH type II, we found that the clinical and hormonal profile was comparable with sporadic cases, apart from a younger age and slightly higher diastolic blood pressure in the group of FH-II. Type II FH is caused by a pathogenic mutation in the CLCN2 gene, leading to elevated intracellular Ca2+ concentration, which triggers depolarization and aldosterone secretion (43). The only previous study that compared sporadic and FH-II cases described that both groups had similar potassium levels and blood pressure levels. In addition, since the prevalence of aldosterone producing adenomas was not significantly different in FH-II and sporadic PA patients, the former group may have had radiological features similar to sporadic cases (6). Considering that the clinical picture does not allow to differentiate it from sporadic cases and taking into account that FH-II is the most common form of hereditary PA (6), the most recent Endocrinology PA guidelines recommend screening for familial FH type II in all hypertensive patients who have relatives with FH (66). However, we are aware that some patients classified as FH-II may be sporadic cases coexisting within the same family since until the discovery of the underlying genetic cause of FH-II, all cases with two or more positive cases of primary aldosteronism in the same family were classified as FH type II.
Type III FH is a severe form of PA characterized by extensive adrenocortical hyperplasia and hybrid steroid synthesis (42). The genetic defect is located in the KCNJ5 gene. Women are disproportionately afflicted, accounting for more than 75% of all cases described in the literature. A higher prevalence of women has also been described in those sporadic PA patients who harbor a somatic pathogenic variant in KCNJ5 (67). As we found, FH-III is frequently detected at a very young age and PAC reached double values than in sporadic cases. In addition, serum potassium levels are typically low, with a prevalence of hypokalemia nearing 90%. In fact, over 60% of these patients required a bilateral adrenalectomy to achieve adequate blood pressure control. Nevertheless, the severity of the PA varies depending on the KCNJ5 pathogenic variant; for example, p.T158A, p.I157S, p.E145Q, and p.G151R are associated with early-onset severe PA (68) whereas p.G151E and p.Y152C present with mild PA (50, 59).
Only few cases of FH-IV have been reported in the literature (43). FH-IV is caused by germline defects in CACNA1H. Type IV, like FH-III, manifests at a very young age but later than the FH III cases. When compared with sporadic cases, serum potassium levels are also lower, but the degree of decline seems to be less severe than in FH type III. Patients with a family history of FH and early diagnosis of PA should be screened for familial FH type IV. Thus, the indications are identical to those for FH-III (45). In fact, if there is a suspicion of genetic PA, it is recommended that all genes be tested.
We are aware that our study has some limitations. The main limitation is that the definition of sporadic in the control group of sporadic patients was based on the epidemiological and clinical characteristics since the majority of patients in the SPAIN-ALDO did not undergo genetic testing. Nevertheless, cases with any suspicion of familial origin were excluded. Second, we have included all the cases reported in the literature following our strategy research that have available clinical and hormonal data, including patients from all over the world. Thus, we know that ethnic differences may also play a role in the many clinical and hormonal phenotypes detected. Thus, specific studies comparing sporadic and familial cases of the same ethnicity should be conducted. However, despite these limitations, this is the largest study comparing all the patients with FH of different types as well as sporadic PA cases.
In addition to being younger and having a family history of PA, FH-I and III share other typical characteristics. In this regard, FH-I is characterized by a low prevalence of hypokalemia and FH-III by a severe aldosterone excess causing hypokalemia in more than 85% of patients. The clinical and hormonal phenotype of types II and IV is similar to the sporadic cases. However, all genes should be tested if there is a possibility of genetic PA.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by CEIm Hospital Ramón y Cajal, Madrid. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because Retrospective nature of the study.
MA: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. PP: Writing – review & editing. PR: Writing – review & editing. MF: Writing – review & editing. MB: Writing – review & editing. EP: Writing – review & editing. AC: Writing – review & editing. JR: Writing – review & editing. AD: Writing – review & editing. EH: Writing – review & editing. RF: Writing – review & editing. IS: Writing – review & editing. MS: Writing – review & editing. RM: Writing – review & editing. MC: Writing – review & editing. PG: Writing – review & editing. CP: Writing – review & editing. LM: Writing – review & editing. RC: Writing – review & editing. ÁR: Writing – review & editing. PG: Writing – review & editing. CL: Writing – review & editing. MM: Writing – review & editing. MC: Writing – review & editing. SC: Writing – review & editing. DM: Writing – review & editing. MN: Writing – review & editing. VQ: Writing – review & editing. ER: Writing – review & editing. AS: Writing – review & editing. CD: Writing – review & editing. CO: Writing – review & editing. MT: Writing – review & editing. JG: Writing – review & editing. TM: Writing – review & editing. EM: Writing – review & editing. FH: Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pilz S, Grubler MR, Theiler-Schwetz V, Malle O, Trummer C. The unrecognized prevalence of primary aldosteronism. Ann Intern Med. (2020) 173:682. doi: 10.7326/L20-1094
2. Araujo-Castro M, Martín Rojas-Marcos P, Parra Ramírez P. Familial forms and molecular profile of primary hyperaldosteronism. Hipertens y Riesgo Vasc. (2022) 39:167–73. doi: 10.1016/J.HIPERT.2022.05.007
3. Sutherland DJ, Ruse JL, Laidlaw JC. Hypertension, increased aldosterone secretion and low plasma renin activity relieved by dexamethasone. Can Med Assoc J. (1966) 95:1109–19.
4. Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, et al. A chimaeric llβ-hydroxylase/aldosterone synthase gene causes glucocorticoid-remediable aldosteronism and human hypertension. Nature. (1992) 355:262–5. doi: 10.1038/355262a0
5. Aglony M, Martínez-Aguayo A, Carvajal CA, Campino C, García H, Bancalari R, et al. Frequency of familial hyperaldosteronism type 1 in a hypertensive pediatric population: clinical and biochemical presentation. Hypertens (Dallas Tex 1979). (2011) 57:1117–21. doi: 10.1161/HYPERTENSIONAHA.110.168740
6. Mulatero P, Tizzani D, Viola A, Bertello C, Monticone S, Mengozzi G, et al. Prevalence and characteristics of familial hyperaldosteronism: The PATOGEN study (Primary aldosteronism in TOrino-GENetic forms). Hypertension. (2011) 58:797–803. doi: 10.1161/HYPERTENSIONAHA.111.175083
7. Sanga V, Lenzini L, Seccia TM, Rossi GP. Familial hyperaldosteronism type 1 and pregnancy: successful treatment with low dose dexamethasone. Blood Press. (2021) 30:133–7. doi: 10.1080/08037051.2020.1863771
8. Carvajal CA, Campino C, Martinez-Aguayo A, Tichauer JE, Bancalari R, Valdivia C, et al. A new presentation of the chimeric CYP11B1/CYP11B2 gene with low prevalence of primary aldosteronism and atypical gene segregation pattern. Hypertens (Dallas Tex 1979). (2012) 59:85–91. doi: 10.1161/HYPERTENSIONAHA.111.180513
9. Shahrrava A, Moinuddin S, Boddu P, Shah R. A case of glucocorticoid remediable aldosteronism and thoracoabdominal aneurysms. Case Rep Endocrinol. (2016) 2016:1–4. doi: 10.1155/2016/2017571
10. Methe H, Pehlivanli S. Glucocorticoid-remediable aldosteronism in a young adult with a family history of Conn’s syndrome. Clin Case Rep. (2018) 6:416–9. doi: 10.1002/CCR3.1377
11. Cheng CY, Liao HW, Peng KY, Chen TH, Lin YH, Chueh JS, et al. Characteristics and outcomes in primary aldosteronism patients harboring glucocorticoid-remediable aldosteronism. Biomedicines. (2021) 9. doi: 10.3390/BIOMEDICINES9121816
12. Carvajal CA, Stehr CB, González PA, Riquelme EM, Montero T, Santos MJ, et al. A de novo unequal cross-over mutation between CYP11B1 and CYP11B2 genes causes familial hyperaldosteronism type I. J Endocrinol Invest. (2011) 34:140–4. doi: 10.1007/BF03347044
13. Kamrath C, Maser-Gluth C, Haag C, Schulze E. Diagnosis of glucocorticoid-remediable aldosteronism in hypertensive children. Horm Res Paediatr. (2011) 76:93–8. doi: 10.1159/000326524
14. Yokota K, Ogura T, Kishida M, Suzuki J, Otsuka F, Mimura Y, et al. Japanese family with glucocorticoid-remediable aldosteronism diagnosed by long-polymerase chain reaction. Hypertens Res. (2001) 24:589–94. doi: 10.1291/HYPRES.24.589
15. Dluhy RG, Anderson B, Harlin B, Ingelfinger J, Lifton R. Glucocorticoid-remediable aldosteronism is associated with severe hypertension in early childhood. J Pediatr. (2001) 138:715–20. doi: 10.1067/MPD.2001.112648
16. Campino C, Trejo P, Carvajal CA, Vecchiola A, Valdivia C, Fuentes CA, et al. Pregnancy normalized familial hyperaldosteronism type I: a novel role for progesterone? J Hum Hypertens. (2015) 29:138–9. doi: 10.1038/JHH.2014.49
17. Lee IS, Kim SY, Jang HW, Kim MK, Lee JH, Lee YH, et al. Genetic analyses of the chimeric CYP11B1/CYP11B2 gene in a Korean family with glucocorticoid-remediable aldosteronism. J Korean Med Sci. (2010) 25:1379–83. doi: 10.3346/JKMS.2010.25.9.1379
18. Gill JR, Bartter FC. Overproduction of sodium-retaining steroids by the zona glomerulosa is adrenocorticotropin-dependent and mediates hypertension in dexamethasone-suppressible aldosteronism. J Clin Endocrinol Metab. (1981) 53:331–7. doi: 10.1210/JCEM-53-2-331
19. Fallo F, Sonino N, Armanini D, Luzzi T, Pedini F, Pasini C, et al. A new family with dexamethasone-suppressible hyperaldosteronism: aldosterone unresponsiveness to angiotensin II. Clin Endocrinol (Oxf). (1985) 22:777–85. doi: 10.1111/j.1365-2265.1985.tb00168.x
20. Seeman T, Widimský J, Hampf M, Bernhardt R. Abolished nocturnal blood pressure fall in a boy with glucocorticoid-remediable aldosteronism. J Hum Hypertens. (1999) 13:823–8. doi: 10.1038/SJ.JHH.1000918
21. Liu X, Jin L, Zhang H, Ma W, Song L, Zhou X, et al. A Chinese pedigree with glucocorticoid remediable aldosteronism. Hypertens Res. (2021) 44:1428–33. doi: 10.1038/S41440-021-00685-3
22. Fallo F, Pilon C, Williams TA, Sonino N, Morra Di Cella S, Veglio F, et al. Coexistence of different phenotypes in a family with glucocorticoid-remediable aldosteronism. J Hum Hypertens. (2004) 18:47–51. doi: 10.1038/SJ.JHH.1001636
23. Stowasser M, Sharman J, Leano R, Gordon RD, Ward G, Cowley D, et al. Evidence for abnormal left ventricular structure and function in normotensive individuals with familial hyperaldosteronism type I. J Clin Endocrinol Metab. (2005) 90:5070–6. doi: 10.1210/jc.2005-0681
24. Stowasser M, Huggard PR, Rossetti TR, Bachmann AW, Gordon RD. Biochemical evidence of aldosterone overproduction and abnormal regulation in normotensive individuals with familial hyperaldosteronism type I. J Clin Endocrinol Metab. (1999) 84:4031–6. doi: 10.1210/JCEM.84.11.6159
25. Litchfield WR, New MI, Coolidge C, Lifton RP, Dluhy RG. Evaluation of the dexamethasone suppression test for the diagnosis of glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. (1997) 82:3570–3. doi: 10.1210/JCEM.82.11.4381
26. Mulatero P, Di Cella SM, Williams TA, Milan A, Mengozzi G, Chiandussi L, et al. Glucocorticoid remediable aldosteronism: low morbidity and mortality in a four-generation italian pedigree. J Clin Endocrinol Metab. (2002) 87:3187–91. doi: 10.1210/JCEM.87.7.8647
27. Al Romhain B, Young AMH, Battacharya JJ, Suttner N. Intracranial aneurysm in a patient with glucocorticoid-remediable aldosteronism. Br J Neurosurg. (2015) 29:715–7. doi: 10.3109/02688697.2015.1023775
28. Vonend O, Altenhenne C, Büchner NJ, Dekomien G, Maser-Gluth C, Weiner SM, et al. A German family with glucocorticoid-remediable aldosteronism. Nephrol Dial Transplant. (2007) 22:1123–30. doi: 10.1093/NDT/GFL706
29. Lin YF, Peng KY, Chang CH, Hu YH, Wu VC, Chueh JS, et al. Adrenalectomy completely cured hypertension in patients with familial hyperaldosteronism type I who had somatic KCNJ5 mutation. J Clin Endocrinol Metab. (2019) 104:5462–6. doi: 10.1210/JC.2019-00689
30. Gordon RD, Stowasser M, Tunny TJ, Klemm SA, Finn WL, Krek AL. Clinical and pathological diversity of primary aldosteronism, including a new familial variety. Clin Exp Pharmacol Physiol. (1991) 18:283–6. doi: 10.1111/j.1440-1681.1991.tb01446.x
31. Lafferty AR, Torpy DJ, Stowasser M, Taymans SE, Lin JP, Huggard P, et al. A novel genetic locus for low renin hypertension: familial hyperaldosteronism type II maps to chromosome 7 (7p22). J Med Genet. (2000) 37:831–5. doi: 10.1136/JMG.37.11.831
32. Fernandes-Rosa FL, Daniil G, Orozco IJ, Göppner C, El Zein R, Jain V, et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat Genet. (2018) 50:355–61. doi: 10.1038/S41588-018-0053-8
33. Stowasser M, Gordon RD, Tunny TJ, Klemm SA, Finn WL, Krek AL. Familial hyperaldosteronism type II: five families with a new variety of primary aldosteronism. Clin Exp Pharmacol Physiol. (1992) 19:319–22. doi: 10.1111/j.1440-1681.1992.tb00462.x
34. Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, Nelson-Williams C, et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat Genet. (2018) 50:349–54. doi: 10.1038/s41588-018-0048-5
35. Sukor N, Mulatero P, Gordon RD, So A, Duffy D, Bertello C, et al. Further evidence for linkage of familial hyperaldosteronism type II at chromosome 7p22 in Italian as well as Australian and South American families. J Hypertens. (2008) 26:1577–82. doi: 10.1097/HJH.0B013E3283028352
36. Malagon-Rogers M. Non-glucocorticoid-remediable aldosteronism in an infant with low-renin hypertension. Pediatr Nephrol. (2004) 19:235–6. doi: 10.1007/S00467-003-1339-2
37. Torpy DJ, Gordon RD, Lin JP, Huggard PR, Taymans SE, Stowasser M, et al. Familial hyperaldosteronism type II: description of a large kindred and exclusion of the aldosterone synthase (CYP11B2) gene. J Clin Endocrinol Metab. (1998) 83:3214–8. doi: 10.1210/JCEM.83.9.5086
38. Pallauf A, Schirpenbach C, Zwermann O, Fischer E, Morak M, Holinski-Feder E, et al. The prevalence of familial hyperaldosteronism in apparently sporadic primary aldosteronism in Germany: a single center experience. Horm Metab Res. (2012) 44:215–20. doi: 10.1055/S-0031-1299730
39. Elphinstone MS, Gordon RD, So A, Jeske YWA, Stratakis CA, Stowasser M. Genomic structure of the human gene for protein kinase A regulatory subunit R1-beta (PRKAR1B) on 7p22: no evidence for mutations in familial hyperaldosteronism type II in a large affected kindred. Clin Endocrinol (Oxf). (2004) 61:716–23. doi: 10.1111/j.1365-2265.2004.02155.x
40. Ise T, Shimoda A, Takakuwa H, Kato T, Izumiya Y, Shimizu K, et al. A chimeric CYP11B1/CYP11B2 gene in glucocorticoid-insuppressible familial hyperaldosteronism. Clin Endocrinol (Oxf). (2001) 55:131–4. doi: 10.1046/j.1365-2265.2001.01192.x
41. Somekh NN, Finkielstein D. A mother/daughter case of familial hyperaldosteronism. Clin Cardiol. (2010) 33:E68–9. doi: 10.1002/CLC.20654
42. Geller DS, Zhang J, Wisgerhof MV, Shackleton C, Kashgarian M, Lifton RP. A novel form of human mendelian hypertension featuring nonglucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab. (2008) 93:3117–23. doi: 10.1210/JC.2008-0594
43. Scholl UI, Stölting G, Nelson-Williams C, Vichot AA, Choi M, Loring E, et al. Recurrent gain of function mutation in calcium channel CACNA1H causes early-onset hypertension with primary aldosteronism. Elife. (2015) 4:e06315. doi: 10.7554/eLife.06315
44. Ruiz-Sánchez JG, Paja-Fano M, González Boillos M, Pla Peris B, Pascual-Corrales E, García Cano AM, et al. Impact of obesity on clinical characteristics of Primary Aldosteronism patients at diagnosis and post-surgical response. J Clin Endocrinol Metab. (2023) 109(1):e379–88. doi: 10.1210/CLINEM/DGAD400
45. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2016) 101:1889–916. doi: 10.1210/jc.2015-4061
46. Monticone S, Sconfienza E, D’Ascenzo F, Buffolo F, Satoh F, Sechi LA, et al. Renal damage in primary aldosteronism: A systematic review and meta-analysis. J Hypertens. (2020) 38(1):3–12. doi: 10.1097/HJH.0000000000002216
47. Parra Ramírez P, Martín Rojas-Marcos P, Paja Fano M, González Boillos M, Peris BP, Pascual-Corrales E, et al. Is adrenal venous sampling always necessary to differentiate between unilateral and bilateral primary aldosteronism? Lesson from the SPAIN-ALDO register. Endocrine. (2023). doi: 10.1007/s12020-023-03609-y
48. Williams TA, Lenders JWM, Mulatero P, Burrello J, Rottenkolber M, Adolf C, et al. Outcomes after adrenalectomy for unilateral primary aldosteronism: an international consensus on outcome measures and analysis of remission rates in an international cohort. Lancet Diabetes Endocrinol. (2017) 5:689–99. doi: 10.1016/S2213-8587(17)30135-3
49. Baethge C, Goldbeck-Wood S, Mertens S. SANRA-a scale for the quality assessment of narrative review articles. Res Integr Peer Rev. (2019) 4. doi: 10.1186/S41073-019-0064-8
50. Mulatero P, Tauber P, Zennaro MC, Monticone S, Lang K, Beuschlein F, et al. KCNJ5 mutations in European families with nonglucocorticoid remediable familial hyperaldosteronism. Hypertension. (2012) 59:235–40. doi: 10.1161/HYPERTENSIONAHA.111.183996
51. Takizawa N, Tanaka S, Nishimoto K, Sugiura Y, Suematsu M, Ohe C, et al. Familial hyperaldosteronism type 3 with a rapidly growing adrenal tumor: an in situ aldosterone imaging study. Curr Issues Mol Biol. (2021) 44:128–38. doi: 10.3390/CIMB44010010
52. Tamura A, Nishimoto K, Seki T, Matsuzawa Y, Saito J, Omura M, et al. Somatic KCNJ5 mutation occurring early in adrenal development may cause a novel form of juvenile primary aldosteronism. Mol Cell Endocrinol. (2017) 441:134–9. doi: 10.1016/J.MCE.2016.07.031
53. Adachi M, Muroya K, Asakura Y, Sugiyama K, Homma K, Hasegawa T. Discordant genotype-phenotype correlation in familial hyperaldosteronism type III with KCNJ5 gene mutation: a patient report and review of the literature. Horm Res Paediatr. (2014) 82:138–42. doi: 10.1159/000358197
54. Greco RG, Carroll JE, Morris DJ, Grekin RJ, Melby JC. Familial hyperaldosteronism, not suppressed by dexamethasone. J Clin Endocrinol Metab. (1982) 55:1013–6. doi: 10.1210/JCEM-55-5-1013
55. Gomez-Sanchez CE, Qi X, Gomez-Sanchez EP, Sasano H, Bohlen MO, Wisgerhof M. Disordered zonal and cellular CYP11B2 enzyme expression in familial hyperaldosteronism type 3. Mol Cell Endocrinol. (2017) 439:74–80. doi: 10.1016/J.MCE.2016.10.025
56. Tong A, Liu G, Wang F, Jiang J, Yan Z, Zhang D, et al. A novel phenotype of familial hyperaldosteronism type III: concurrence of aldosteronism and Cushing’s syndrome. J Clin Endocrinol Metab. (2016) 101:4290–7. doi: 10.1210/JC.2016-1504
57. Pons Fernández N, Moreno F, Morata J, Moriano A, León S, De Mingo C, et al. Familial hyperaldosteronism type III a novel case and review of literature. Rev Endocr Metab Disord. (2019) 20:27–36. doi: 10.1007/S11154-018-9481-0
58. Charmandari E, Sertedaki A, Kino T, Merakou C, Hoffman DA, Hatch MM, et al. A novel point mutation in the KCNJ5 gene causing primary hyperaldosteronism and early-onset autosomal dominant hypertension. J Clin Endocrinol Metab. (2012) 97. doi: 10.1210/JC.2012-1334
59. Monticone S, Hattangady NG, Penton D, Isales CM, Edwards MA, Williams TA, et al. a Novel Y152C KCNJ5 mutation responsible for familial hyperaldosteronism type III. J Clin Endocrinol Metab. (2013) 98:E1861–5. doi: 10.1210/JC.2013-2428
60. Maria AG, Suzuki M, Berthon A, Kamilaris C, Demidowich A, Lack J, et al. Mosaicism for KCNJ5 causing early-onset primary aldosteronism due to bilateral adrenocortical hyperplasia. Am J Hypertens. (2020) 33:124–30. doi: 10.1093/AJH/HPZ172
61. Wulczyn K, Perez-Reyes E, Nussbaum RL, Park M. Primary aldosteronism associated with a germline variant in CACNA1H. BMJ Case Rep. (2019) 12:e229031. doi: 10.1136/BCR-2018-229031
62. Daniil G, Fernandes-Rosa FL, Chemin J, Blesneac I, Beltrand J, Polak M, et al. CACNA1H mutations are associated with different forms of primary aldosteronism. EBioMedicine. (2016) 13:225–36. doi: 10.1016/J.EBIOM.2016.10.002
63. Karwacka I, Obołończyk Ł, Kaniuka-Jakubowska S, Bohdan M, Sworczak K. Progress on genetic basis of primary aldosteronism. Biomedicines. (2021) 9:1708. doi: 10.3390/BIOMEDICINES9111708
64. Mulatero P, Monticone S, Deinum J, Amar L, Prejbisz A, Zennaro MC, et al. Genetics, prevalence, screening and confirmation of primary aldosteronism: a position statement and consensus of the Working Group on Endocrine Hypertension of The European Society of Hypertension. J Hypertens. (2020) 38:1919–28. doi: 10.1097/HJH.0000000000002510
65. Stowasser M, Bachmann AW, Huggard PR, Rossetti TR, Gordon RD. Severity of hypertension in familial hyperaldosteronism type I: relationship to gender and degree of biochemical disturbance. J Clin Endocrinol Metab. (2000) 85:2160–6. doi: 10.1210/JCEM.85.6.6651
66. Funder JW. Primary aldosteronism: Mutations, mechanisms, prevalence, and public health. Hypertension. (2019) 74:458–66. doi: 10.1161/HYPERTENSIONAHA.119.12935
67. Pitsava G, Faucz FR, Stratakis CA, Hannah-Shmouni F. Update on the genetics of primary aldosteronism and aldosterone-producing adenomas. Curr Cardiol Rep. (2022) 24:1189–95. doi: 10.1007/s11886-022-01735-z.
Keywords: primary aldosteronism, familial hyperaldosteronism, genetic study, pathogenic variant, plasma aldosterone concentration
Citation: Araujo-Castro M, Parra P, Martín Rojas-Marcos P, Paja Fano M, González Boillos M, Pascual-Corrales E, García Cano AM, Ruiz-Sanchez JG, Vicente Delgado A, Gómez Hoyos E, Ferreira R, García Sanz I, Recasens Sala M, Barahona San Millan R, Picón César MJ, Díaz Guardiola P, Perdomo CM, Manjón-Miguélez L, García Centeno R, Rebollo Román Á, Gracia Gimeno P, Robles Lázaro C, Morales-Ruiz M, Calatayud M, Furio Collao SA, Meneses D, Sampedro Nuñez M, Escudero Quesada V, Mena Ribas E, Sanmartín Sánchez A, Gonzalvo Diaz C, Lamas C, del Castillo Tous M, Serrano Gotarredona J, Michalopoulou Alevras T, Moya Mateo EM and Hanzu FA (2024) Differences in the clinical and hormonal presentation of patients with familial and sporadic primary aldosteronism. Front. Endocrinol. 15:1336306. doi: 10.3389/fendo.2024.1336306
Received: 10 November 2023; Accepted: 06 February 2024;
Published: 01 March 2024.
Edited by:
Roberta Giordano, University of Turin, ItalyReviewed by:
Sergei Tevosian, University of Florida, United StatesCopyright © 2024 Araujo-Castro, Parra, Martín Rojas-Marcos, Paja Fano, González Boillos, Pascual-Corrales, García Cano, Ruiz-Sanchez, Vicente Delgado, Gómez Hoyos, Ferreira, García Sanz, Recasens Sala, Barahona San Millan, Picón César, Díaz Guardiola, Perdomo, Manjón-Miguélez, García Centeno, Rebollo Román, Gracia Gimeno, Robles Lázaro, Morales-Ruiz, Calatayud, Furio Collao, Meneses, Sampedro Nuñez, Escudero Quesada, Mena Ribas, Sanmartín Sánchez, Gonzalvo Diaz, Lamas, del Castillo Tous, Serrano Gotarredona, Michalopoulou Alevras, Moya Mateo and Hanzu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marta Araujo-Castro, bWFydGEuYXJhdWpvQHNhbHVkLm1hZHJpZC5vcmc=
†ORCID: Marta Araujo-Castro, orcid.org/0000-0002-0519-0072
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.