 Mengrou Geng1,2
Mengrou Geng1,2 Xin Jiang
Xin Jiang Ying Xin
Ying Xin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 08 February 2024
Sec. Cardiovascular Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1324393
This article is part of the Research TopicCardiovascular Diseases Related to Diabetes and Obesity, volume IVView all 11 articles
Diabetes is a metabolic disease characterized by hyperglycemia, which induces the production of AGEs, ROS, inflammatory cytokines, and growth factors, leading to the formation of vascular dysfunction and target organ damage, promoting the development of diabetic complications. Diabetic nephropathy, retinopathy, and cardiomyopathy are common complications of diabetes, which are major contributors to disability and death in people with diabetes. Long non-coding RNAs affect gene transcription, mRNA stability, and translation efficiency to influence gene expression for a variety of biological functions. Over the past decade, it has been demonstrated that dysregulated long non-coding RNAs are extensively engaged in the pathogenesis of many diseases, including diabetic complications. Thus, this review discusses the regulations of long non-coding RNAs on the primary pathogenesis of diabetic complications (oxidative stress, inflammation, fibrosis, and microvascular dysfunction), and some of these long non-coding RNAs may function as potential biomarkers or therapeutic targets for diabetic complications.
Diabetes mellitus (DM) has become a worldwide epidemic, already affecting one-sixteenth of the global population in 2021, and the prevalence continues to rise annually, with the number of people with the disease expected to reach 783.2 million worldwide by 2045, posing an increasingly serious threat to humanity (1). Type 1 diabetes mellitus (T1DM), an autoimmune disease marked by complete insulin insufficiency as a result of autoimmune β-cell destruction, accounts for approximately 5–10% of all cases of diabetes (2). Furthermore, more than 90% of diabetic individuals have type 2 diabetes mellitus (T2DM), which is characterized by insulin resistance and relative insulin deficiency (2). Therefore, the relative or absolute deficiency of insulin in diabetes induces hyperglycemia and various metabolic signaling disorders that target organs throughout the body and ultimately lead to diabetic complications. Diabetic nephropathy (DN) is one of the most common microvascular complications in diabetic patients and today accounts for almost 40% of all end-stage renal disease (ESRD) (3). The prevalence of diabetic retinopathy (DR) can reach 34.1% and is the leading cause of blindness in adults (4). Diabetic cardiomyopathy (DCM) is difficult to diagnose, has an insidious onset, and is a major cause of death in diabetic patients (5). The burden of the disease and high cost are driven by the presence of chronic diabetic complications, and patients with complications would increase health expenditure by 3.36 times higher compared to those without complications(6).
Recent theories on the development of diabetic complications state that multiple cellular pathways are activated by hyperglycemia and dyslipidemia, including activation in polyol pathway flux, intracellular formation of advanced glycation end products (AGEs), expression of the receptor for AGEs and its activating ligands, activation of protein kinase C (PKC) and hexosamine pathway. The activation of these pathways results in production of reactive oxygen species (ROS) (e.g., superoxide anion) and epigenetic changes (DNA methylation, histone modifications, and the expression of non-coding RNAs), which produce growth factors and proinflammatory cytokines that motivate oxidative stress, fibrosis, inflammation, and vascular dysfunction. This leads to pathogenetic alterations and adversely affects endothelial cells, vascular smooth muscle cells (VSMCs), monocytes, and key targets such as retinal cells, cardiomyocytes, and renal cells leading to diabetic complications (7). Although these are common mechanisms in most vascular complications of diabetes, the pathological process and symptoms of the disease can change depending on the target cells and organs. For instance, transforming growth factor β (TGF-β) signaling is activated in a variety of cells in the diabetic kidney and is involved in increased synthesis and deposition of extracellular matrix, ultimately leading to glomerulosclerosis and tubulointerstitial fibrosis. Microvascular dysfunction is a characteristic feature in diabetic retinopathy, with increased microvascular permeability and vascular exudation in the early stages, as well as late neovascular capillary formation. Diabetic cardiomyopathy is seen with inadequate microvascular blood flow and reduced myocardial perfusion, leading to focal necrosis and scar formation, which in turn leads to changes in cardiac structure and function (Figure 1). Available anti-diabetic drugs commonly used on the market, such as metformin and sulfonylureas, are presently efficient in regulating hyperglycemia, but they cannot completely prevent the occurrence and progression of its complications. Sometimes these drugs have adverse effects such as liver, heart, and kidney toxicity, hypoglycemia, and gastrointestinal reactions. Consequently, it is essential to understand the underlying molecular mechanisms to develop more effective treatments.

Figure 1 The pathogenesis of diabetic complications and the role of lncRNAs. Diabetes and its attendant metabolic disorders can activate multiple signaling pathways that promote ROS (e.g., superoxide anion) production and dysregulated expression of lncRNAs. These events can lead to the development of pivotal pathological events that consequently have an impact on the progression of DN, DCM, and DR. Abbreviations: HG, high glucose; HF, high fat; AGEs, advanced glycation end products; PKC, protein kinase C; ROS, reactive oxygen species; LncRNA, long non-coding RNA; MiRNA, microRNA; CircRNA, circular RNA; Histone PTMs, histones post-translational modifications; DNAMe, DNA methylation; DN, diabetic nephropathy; DR, diabetic retinopathy; DCM, diabetic cardiomyopathy.
Long non-coding RNAs (lncRNAs) are non-coding RNAs with over 200 nucleotides, which have gained increasing attention from researchers because of their tissue-specific expression patterns and rich regulatory mechanisms. Currently, lncRNAs have been found to regulate gene expression at the transcriptional and post-transcriptional levels. At the transcriptional level, lncRNAs primarily participate in chromatin modification and remodeling, leading to the expression or repression of a large number of genes. Post-transcriptional regulation involves mRNA splicing, translation, and stability. LncRNAs also can regulate protein stability by involving post-translational modifications associated with protein degradation. LncRNAs are increasingly recognized as epigenetic regulators to participate in the development of diabetes and diabetic complications (Figure 1) (8). Because of the different onset of T1DM and T2DM, lncRNA can play roles on different targets. In the chronic autoimmune disease of T1DM, it has been recently revealed that viral infections are involved in the attack of pancreatic islet β-cells by immune cells. Evidences showed that lncRNAs Lnc13 and antiviral response gene inducer (ARGI) are upregulated in viral infection and activate the proinflammatory chemokine secretion and antiviral responses (9, 10). In T2DM, lncRNAs are mainly responsible for the insulin resistance. For example, Guo et al. discovered LncRNA Reg1cp was mainly expressed in the islet and its mutation was a risk factor for T2DM. Mutant Reg1cp increased insulin resistance via inhibiting polypyrimidine tract binding protein 1 (PTBP1) phosphorylation and the PTBP1-AdipoR1 pathway (11). However, lncRNAs also have significant effects on the progression of DM and diabetic complications through regulating the mainly pathogenic progress of oxidative stress, inflammation, cell death, fibrosis, and vascular proliferation. For instance, metastasis associated lung adenocarcinoma transcript 1 (MALAT1) was reported to interact with nuclear factor erythroid 2-related factor 2 (Nrf2) as a negative regulator. MALAT1 ablation activates Nrf2-regulated antioxidant genes expression and reduces ROS accumulation and oxidative stress, resulting in lower inflammation, sensitivity to insulin signaling and improved β-cell function (12). In this paper, biomedical articles published on lncRNAs and diabetic complications in the Pubmed database was searched and the action of lncRNAs in the pathogenesis of DN, DCM, and DR will be reviewed, and this information provides a theoretical basis for the potential use of lncRNAs as therapeutic targets for complications.
DN is one of the primary microvascular complications of diabetes mellitus. Pathologically, DN is characterized by the enlargement of the glomerular mesangial expansion and accumulation of extracellular matrix (ECM) proteins. This leads to glomerulosclerosis and fibrosis in the tubulointerstitial region. In addition, there is damage to the capillary endothelium and the glomerular filtration membrane due to the death of podocytes. All of these factors contribute to kidney dysfunction, which manifests itself early in the form of microproteinuria, reduced glomerular filtration rate, and eventually progresses to end-stage renal disease (ESRD) (13). The current standard treatment for DN involves the use of RAS inhibitors and hypoglycemic agents to manage blood pressure and glucose levels. Unfortunately, these conventional therapies are ineffective in preventing the progression of the disease to ESRD. Many promising novel medications for the treatment of DN have also encountered setbacks in phase 3 clinical trials due to issues such as toxicity. Therefore, there is a growing interest in the development of biomarkers that can predict the early stages of the disease in order to adopt preventative therapy (14). Certain lncRNAs are abnormally expressed in patients with DN and are considered potential biomarkers for its diagnosis. Further research has shown that these lncRNAs have an impact on renal fibrosis and damage to podocytes in diabetic nephropathy, which ultimately affects kidney function (Table 1).
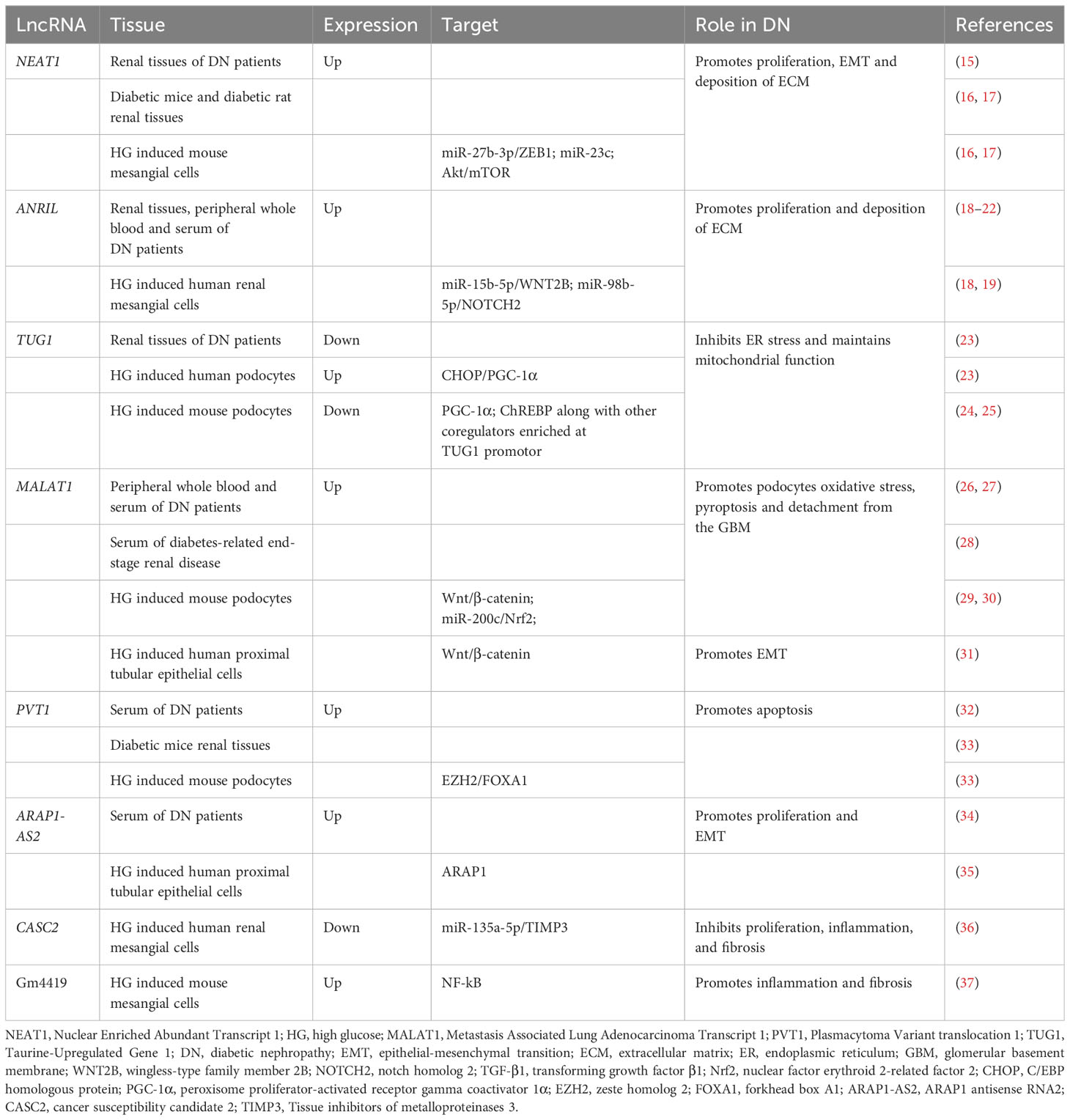
Table 1 The roles of lncRNAs in DN.
Renal fibrosis has been recognized as one of the most crucial processes for the development of DN and is significantly associated with DN prognosis. Anti-fibrotic treatment significantly improves renal function. Renal fibrosis is manifested as excessive deposition of the ECM. It is widely accepted that myofibroblasts play a major role in the synthesis and secretion of ECM under pathological conditions (38). Mesangial cells and renal tubular epithelial cells are considered to be an important precursor cell type of myofibroblasts in DN, transformed into myofibroblasts by epithelial mesenchymal transition (EMT) in response to high sugar stimulation (38, 39). Lately, there is evidence pointing to the involvement of lncRNAs.
Nuclear Enriched Abundant Transcript 1(NEAT1) has been reported to dysregulate in DN (15). Previously, it was shown that AKT/mTOR is a key signaling pathway initiated by the kidney in response to high glucose contributing to glomerular hypertrophy (40). NEAT1 upregulation has positive effects on mesangial cell growth and secretion of ECM by increasing AKT and mTOR phosphorylation levels (16). Moreover, there is evidence that NEAT1 takes part in renal fibrosis by advancing the EMT process. Zinc finger E-box binding homeobox 1(ZEB1), a key molecule in EMT initiation and activation, is upregulated by NEAT1 by sponging miR-27b-3p. NEAT1 deficiency significantly reduces the secretion of EMT proteins (E-calmodulin, N-calmodulin) from mesangial cells (17). It also activates bovine serum albumin (BSA)-mediated EMT and fibrosis in HK-2 cells via the ERK1/2 pathway. Silencing of NEAT1 reversed renal tubular epithelial cells migration and the expression of mesenchymal markers such as α-SMA and inhibited the transformation of renal tubular epithelial cells into myofibroblasts. And NEAT1 is the most significantly repressed lncRNA in kidney tissue of Klotho (an antiaging protein) overexpressing diabetic mice. These results imply that targeted NEAT1 implicates the protective effect of Klotho on renal tubular epithelial cell fibrosis and EMT (41). Furthermore, ARAP1 antisense RNA2 (ARAP1-AS2) leads to cytoskeletal rearrangement by interacting with ARAP1, and MALAT1 activates the Wnt/β-catenin pathway to promote the transformation of renal tubular epithelial cells into myofibroblasts (31, 35).
LncRNAs engage in renal fibrosis by promoting ECM secretion. Antisense Non-coding RNA in the INK4 Locus (ANRIL), also known as cell Cycle protein-Dependent Kinase Inhibitor 2B Antisense RNA1 (CDNK2B-AS1), is discovered to be elevated in the renal tissues of people with diabetic nephropathy and has been linked to the development of DN via a variety of pathways (18–22). ANRIL knockout has a protective on diabetic mouse kidneys, revealing a reduction in urine output and albumin creatinine levels, as well as decreased mesangial matrix depositions and fibronectin levels (42). The underlying mechanism displayed that CDKN2B-AS1 interference reverses the ECM accumulation and mesangial cell growth by regulating the miR-15b-5p/Wingless-Type family member 2B (WNT2B) axis (18). Notch homolog 2 (NOTCH2) is one of the important receptors in the NOTCH pathway, which also mediates renal fibrosis. Xiao et al, display that NOTCH2 acts as a target of ANRIL facilitates apoptosis and fibrosis of high glucose-treated HK-2 cells, and is overturned by miR-98-5p overexpression (21).
Moreover, LncRNA cancer susceptibility candidate 2 (CASC2) is reported to exert a protective role in DN by modulating the inflammation. Tissue inhibitors of metalloproteinases 3 (TIMP3) is identified as endogenous specific inhibitors of matrix metalloproteinases in the kidney. CASC2 functions as competing endogenous RNA (ceRNA) to upregulate TIMP3 expression by sponging of miR-135a-5p and alleviates inflammatory response and fibrosis of mesangial cells (36). Gm4419 was highly expressed in renal tissues of DN mice and formed positive feedback with p50, the subunit of NF-kB. The pro-inflammatory and fibrosis biomarkers were upregulated in mesangial cells when Gm4419 was overexpressed (37).
Podocytes are highly specialized terminally differentiated cells that, together with the glomerular basement membrane (GBM) and endothelial cells constitute the glomerular filtration barrier, which leads to a significant correlation between podocyte damage and the severity of proteinuria. LncRNAs are also discovered to be a partial participant in the podocyte damage in the development of DN.
LncRNA Taurine-Upregulated Gene 1 (TUG1) is poorly expressed in the renal tissues of people with DN (23). Recent studies have identified the precise regulation of TUG1 by high-glucose (HG) environments and the downstream regulatory mechanisms of TUG1 that link cellular metabolic states to cellular life activities. The study conducted by Long et al, found that HG enhances the transportation of the transcription factor ChREBP and other coregulators, such as MAX dimerization protein (MLX), MAX dimerization protein 1 (MXD1), and histone deacetylase 1 (HDAC1) to the nucleus. These co-regulators are particularly abundant in the TUG1 promoter and suppress TUG1 expression (24). TUG1 exhibits an evident negative effect on the expressions of markers of endoplasmic reticulum stress (ERS) in the cultured podocytes treated with HG, such as eukaryotic translation initiation factor 2α (eIF2), glucose-regulated protein (GRP78), and C/EBP homologous protein (CHOP). TUG1 significantly enhances peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α) expression by deregulating the inhibitory effect of CHOP on PGC-1α. PGC-1α is a transcriptional activator that is significantly associated with mitochondrial morphology and dynamics and plays a protective role in kidney injury. Thus, TUG1 overexpression rescues HG-induced podocyte loss and reduced number the of podocytes (43). Further investigation of the renoprotective mechanism of TUG1/PGC1 signaling reveals that PGC1 is necessary for TUG1 maintenance of the mitochondrial biogenesis, dynamics, redox, and bioenergetics of podocytes, which is partly mediated by negatively regulating the transcription of arginase 2 (AGR2)(25).
MALAT1 expression is considerably higher in DN patients than in T2DM patients, and it can be utilized to identify DN in conjunction with other biomarkers (ACR, creatinine, and 1-MG) (26, 28). Besides this, MALAT1 correlates directly with biomarkers of podocyte damage (synaptopodin, podocalyxin), and exerts negative effects upon the podocytes (27). Further research demonstrates that MALAT1 may play a role in the detachment of podocytes from GBM. P-cadherin is a key component of the slit diaphragm and was found to be associated with podocyte adhesion(44). MALAT1 is upregulated in the nucleus of high glucose-treated podocytes and is involved in variable splicing of β-catenin. MALAT1 reduction increases P-cadherin levels and reduces podocyte damage (29). Moreover, the knockdown of MALAT1 protects MPC-5 cells from HG-induced pyroptosis and oxidative stress through upregulation of the Nrf2 expression (30).
Plasmacytoma Variant translocation 1 (PVT1) is the first lncRNA suspects to be involved in kidney diseases, and two studies in 2007 reported the role of PVT1 in mediating susceptibility to ESRD caused by type 2 and type 1 diabetes, providing a rationale for PVT1 as a candidate gene for ESRD (45, 46). There is strong evidence from subsequent studies that PVT1 is essential in renal parenchymal cell injury and increases in the serum of patients with diabetic nephropathy (32, 47). Recently, it has been shown that PVT1 localizes to the nucleus of podocytes and silences forkhead box A1(FOXA1) expression by recruiting zeste homolog 2 (EZH2) to the FOXA1 promoter region. FOXA1 is a transcription factor that has been identified to regulate apoptosis through inducing the expression of Bcl-2. PVT1 silencing or overexpression of FOXA1 attenuates podocyte apoptosis in vitro and in vivo (33).
The above lncRNAs summarized in Table 1 are dysregulated in DN and have been found to play a role in DN by regulating multiple pathological processes such as mesangial cell proliferation, ECM deposition, podocyte detachment, and apoptosis, and may be useful as new promising therapeutic target.
DCM is one of the most serious diabetic complications and was first identified in 1972. Rubler et al, reveal that this disease occurs in diabetic patients who develop heart failure in the absence of coronary artery disease, hypertension, and valvular heart disease (48). In the early stage, it is characterized by hyperglycemia caused by insulin resistance and increased free fatty acid levels, and only diastolic dysfunction has not yet appeared in the structural and morphological changes of cardiomyocytes. Later, with metabolic disorders and long-term neurohumoral abnormalities, myocardial cell death increases and interstitial fibrosis impairs systolic and diastolic function. Further decline in cardiac compliance in late DCM increases the prevalence of heart failure (49). LncRNAs have been found to regulate different forms of death (apoptosis, pyroptosis, and autophagy) as well as fibrosis in DCM (Figure 2).
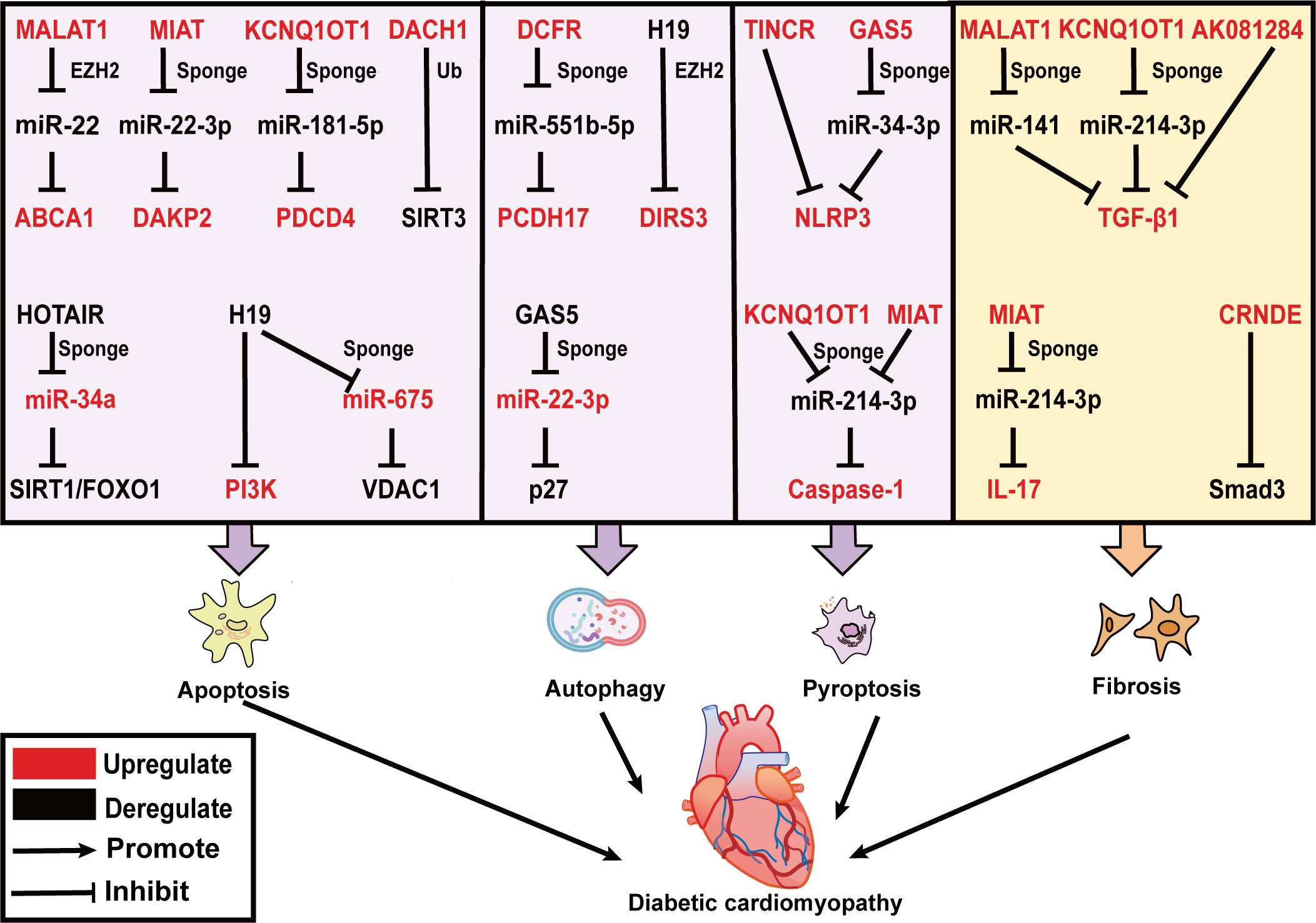
Figure 2 LncRNAs effect on DCM by regulating cardiac apoptosis, autophagy, pyroptosis and fibrosis. LncRNAs are mainly involved in three modes of death including apoptosis, autophagy, and pyroptosis in DCM. MALAT1, MIAT, KCNQ1OT1, and DACH1 promote cardiomyocyte apoptosis, while HOTAIR and H19 prevent cardiomyocyte apoptosis. DCRF and H19 inhibit, and GAS5 promotes cardiomyocyte autophagy. TINCR, GAS5, KCNQ1OT1, and MIAT trigger cardiomyocyte pyroptosis. In addition, KCNQ1OT1, MALAT1, AK081284, and MIAT induce TGF-β1 secretion and cardiac fibrosis. CRNDE inhibits Smad3 phosphorylation and suppresses cardiac fibrosis. The upregulated lncRNAs, miRNAs and target genes in DCM are represented in red color; while those downregulated are represented in black color. Abbreviations: MALAT1, Metastasis Associated Lung Adenocarcinoma Transcript 1; MIAT, Myocardial Infarction Associated Transcript; KCNQ1OT1, KCNQ1 Opposite Strand/Antisense Transcript 1; DACH1, Dachshund Family Transcription Factor 1; GAS5, Growth Stabilization Specific Transcript; TINCR, Terminal Differentiation-induced NcRNA; CRNDE, Colorectal Neoplasia Differentially Expressed; Ub, Ubiquitination; ABCA1, ATP-binding cassette transporter A1; DAPK2, death-associated protein kinase 2; PDCD4, programmed cell death protein 4; SIRT3, sirtuin 3; EZH2, zeste homolog 2; NLRP3, NOD-like receptor family pyrin domain containing 3; TGF-β, transforming growth factor β; VDAC1, voltage dependent anion channel 1; IL-17, interleukins-17.
Increased cardiac apoptosis has been indicated as a leading cause of a major risk factor for the development of DCM (50), as supported by the evidence that cardiomyocyte apoptosis is 85 times more prevalent in the biopsied cardiac tissue of DCM patients than in control non-diabetic hearts. Some lncRNAs have been reported to be upregulated in DCM mice and promote apoptosis in cardiomyocytes. MALAT1 knockdown can restore cardiac function and suppress cardiomyocyte apoptosis by inhibiting ATP-binding cassette transporter A1 (ABCA1) expression and raising miR-22 expression. In this study, MALAT1 can interact with EZH2 and recruit it to the miR-22 promoter region, where it might epigenetically suppress miR-22 transcription in cardiomyocytes (51). MiR-22-3p is directly targeted with Myocardial Infarction Associated Transcript (MIAT) in an AGO2-dependent manner. MIAT increases the death-associated protein kinase 2 (DAPK2) levels via sponging miR-22-3p, promoting apoptosis in cardiomyocytes in diabetic rats (52). Recently programmed cell death protein 4 (PDCD4) is considered to be involved in the progression of diabetic cardiomyocytes (53), which serves as a tumor suppressor in prior studies (54). KCNQ1 Opposite Strand/Antisense Transcript 1 (KCNQ1OT1) can serve as a ceRNA for miR-181a-5p to regulate the expression of PDCD4, which contributes to the inflammatory response and apoptosis in human cardiomyocytes under HG conditions (55).
Mitochondrial dysfunction and ROS are of great interest to trigger apoptosis, and it is recognized that lncRNAs are engaged in this process. SIRT3 can enhance the capacity of mitochondria to eliminate overproduction of ROS by deacetylating and activating superoxide dismutase (SOD). SIRT3 belongs to the sirtuin (SIRT) family, which is a primary mitochondrial deacetylase. In neonatal mouse ventricular cardiomyocytes (NMVCs) exposed to HG conditions, lncRNA Dachshund Family Transcription Factor 1 (DACH1) overexpression notably increases ROS accumulation and apoptosis by promoting SIRT3 ubiquitination (56). In contrast, HOX Transcript Antisense Intergenic RNA (HOTAIR) alleviates oxidative stress and myocardial death of DCM via sponging miR-34a and activating the SIRT1/FOXO1 pathway (57), is specifically downregulated in DCM patients and serves as a promising biomarker for DCM (58). Furthermore, ROS accumulate in the endoplasmic reticulum (ER), increasing the number of misfolded proteins and finally causing ERS. H19 plays a protective role in the progression of DCM. It restores left ventricular dysfunction in the heart of STZ‐induced diabetic mice, as well as under HG culture, suppresses ERS-elicited myocardial apoptosis by activating PI3K in HL-1 cells (59). Additionally, voltage dependent anion channel 1 (VDAC1) plays a crucial role in mitochondria-mediated apoptosis. H19-derived miR-675 (60), through downregulation of its target VDAC1, represses hyperglycemia-mediated oxidative stress and apoptosis in cardiomyocytes (61).
Autophagy is a ubiquitous process, that is responsible for eliminating harmful protein aggregates, intracellular pathogens, and superfluous proteins by the lysosomes (62). Autophagy has been controversial in the sense of being beneficial or disadvantageous to the heart. In general, appropriate levels of autophagy protect cardiomyocytes from apoptosis, while its excessive activation leads to autophagic cell death (50). This accounts for the fact that the effect of lncRNA-regulated autophagy on cardiac function is also two-sided. Growth Stabilization Specific Transcript (GAS5) promotes autophagy to ameliorate cardiomyocyte hypertrophy, myocardial fiber breakage, and mitigated synthesis of collagen. Mechanistically, GAS5 positively regulates p27 gene by binding with miR-221-3p and raising the levels of p62 and LC3B II, reversing the inhibition of autophagy in HG-processed H9c2 cells (63, 64). Conversely, DCRF, a newly discovered lncRNA, is boosted in the myocardium of STZ‐induced diabetic mice (65). It is mainly expressed in cardiomyocyte cytoplasm and is directly targeted at miR-551b-5p. Protocadherin 17 (PCDH17), which belongs to the protocadherin gene family, has been evidenced to be linked with the activation of autophagy in cancer cells (66, 67). DCRF can enhance PCDH17 expression by sponging miR-551b-5p, thus promoting autophagy in cardiomyocytes of STZ‐induced diabetic rats. Reduced expression of DCFR alleviates myocardial fibrosis and restores cardiac function. Likewise, H19 overexpression inhibited autophagy to improve cardiac function in T1DM rats. H19 can interact with enhancer of EZH2 to exert effects on DIRAS family GTPase 3 (DIRAS3) transcription in cardiomyocytes, which results in epigenetically suppressing DIRAS3 and activating mTOR signaling to inhibit autophagy (68).
Although both pyroptosis and apoptosis are forms of programmed death, pyroptosis leads to the breakdown of the plasma membrane and rapid release of large amounts of inflammatory contents into the extracellular compartment to induce inflammation, whereas apoptosis is immunologically silent and the contents of the dying cell are contained within apoptotic bodies (69). The canonical pathway of pyroptosis is through the activation of caspase1 by NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, which converts interleukins 1beta and 18 (IL-1β and IL-18) precursor into mature forms while cleaving gasdermin D (GSDMD), forming pores in the plasma membrane, and resulting in cell swelling and lysis (70).
According to the present research, by regulating NLRP3 and caspase-1, lncRNAs have a significant impact on the emergence of DCM. Cardiac dysfunction in diabetic rats is significantly reversed by MCC950 (NLRP3 inhibitor). Similarly, HG treated neonatal rat ventricular myocytes and H9c2 cells exhibit characteristic pyroptosis promoted by elevating Terminal Differentiation-induced NcRNA (TINCR). RNA pull-down assays reveal that NLRP3 mRNA is prominently enriched by TINCR, and TINCR knockdown accelerates NLRP3 mRNA degradation in cardiomyocytes to inhibit pyroptosis (71). GAS5, acting as a ceRNA and being downregulated in DCM mice, forms a feedback loop with the NLRP3 negative regulator AHR and miR-34-3p to alleviate pyroptosis in HL-1 cells (72). The expression of lncRNA KCNQ1OT1 is found to rise in HG-induced cardiac fibroblasts and diabetic mice. The binding of KCNQ1OT1 with its target of miR-214-3p disrupts the interaction of miR-214-3p with caspase-1, leading to the initiation of primary mouse cardiac fibroblast pyroptosis (73). Similarly, bioinformatic prediction analysis indicates that miR-214-3p potentially contains both MIAT and caspase-1-binding sites. Silencing MIAT by a small interfering RNA suppresses the expression of caspase-1, IL-1β, IL-18, and GSDMD, and ameliorates cardiac pyroptosis in C57BL/6 mice (74).
Myocardial cell death stimulates inflammation and subsequent myofibroblasts activation, leading to the formation of reparative fibrosis. Fibrosis is one of the key factors in the development of DCM, leading to ventricular remodeling, contractile failure, and diastolic dysfunction. Cardiac fibroblasts (CFs) converted to myofibroblasts (MFs), which display boost levels of collagens and alpha‐SMA (a marker of CFs activation into MFs), are required for cardiac fibrosis (75). TGF-β1/Smads signaling pathway plays a crucial role in the transformation of CFs into MFs, and it significantly promotes myocardial fibrosis. A growing body of evidence suggests that lncRNAs take part in the dysregulation of the TGF-β1/Smads signaling pathway in DCM. LncRNA MALAT1 directly increases the TGF-β1 expression in HG-treated CFs by acting as a miR-141 sponge. Ablation of MALAT1 alleviates cardiac interstitial fibrosis and enhances cardiac contractility in diabetic mice (76). Interleukins-17 (IL-17) protein expression is upregulated in HG-treated fibroblasts. IL-17 ultimately promotes fibroblast proliferation and secretion of TGF-β1 and α-SMA through increased expression of lncRNA AK081284, which promotes fibrosis (77). MIAT as an upstream molecule of IL-17, is responsible for increasing IL-17 production by sponging miR-214-3p in cardiomyocytes (78). KCNQ1OT1 also targets miR-214-3p and attenuates the inhibition of TGF-β1/Smads pathway activation by miR-214-3p (73). Zheng et al, demonstrate that Smad3‐Colorectal Neoplasia Differentially Expressed (CRNDE) negative feedback loop exerts in mouse neonatal CFs. LncRNA CRNDE can compete with TGF-β1 to bind Smad3 through rSBEs, thereby preventing TGF-β-mediated phosphorylation of smad3. Smad3, in turn, activates CRNDE transcription. Accordingly, silencing CRNDE elevates CFs collagen deposition and aggravates left ventricular ejection fraction (79).
Overall, above mentioned lncRNAs are involved in regulating different cell death pathways and fibrosis in DCM, which is summarized in Figure 2. Some lncRNA can even directly link cell death to fibrosis, for illustration, MIAT and KCNQ1OT1 can both bind miR-214-3p through the ceRNA mechanism and promote pyroptosis and fibrosis in DCM (73, 78). These important lncRNAs have the potential to become new targets for the treatment of DCM in the future.
DR is a frequent consequence of diabetes, both type 1 and type 2. The severity of DR is influenced by age and the progression of the disease (80). Prolonged oxidative stress, release of pro-inflammatory factors and vascular endothelial growth factor (VEGF) induced by DM damage neurovascular and endothelial cells. This results in increasing vascular permeability, angiogenesis, and impairment of the blood-retinal barrier (BRB). DR has historically been divided into two types: non-proliferative diabetic retinopathy (NPDR) and proliferative diabetic retinopathy (PDR) (81). Microaneurysms and blood vessel leakage are features of NPDR in the early stages, which are followed by swelling and blood vessel obstruction in the later phases. PDR involves the growth of new blood vessels behind the retina and vitreous. VEGF is a target for treatment, and it can lead to regression of vascular lesions and improvement in the severity of DR. However, VEGF treatment requires frequent administration and is most effective in advanced disease stages. This means that new therapeutic targets other than VEGF need to be found (82). The retina is a neural tissue and neurodegeneration has been demonstrated to occur earlier than vascular abnormalities both in animal models and DR patients(83). Researchers have discovered that lncRNAs play a critical role in the development of retinal neurodegeneration and vascular dysfunction. They also have the potential to be innovative treatments.
Müller cells are the major glial cells in the retina, spanning the entire retina and mediating neuronal and vascular interactions, thus dominating the retina (83). Recent studies have demonstrated that Müller cells are crucial for the development of DR and that may be connected to the proinflammatory cytokines released from them (84). Zhang et al, suggest that C-myc impacts the release of proinflammatory cytokine by mediating MIAT/thioredoxin-interacting protein (TXNIP) pathway. C-myc binds to the MIAT promoter and up-regulates it expression which is markedly promoted by HG stimulation. Furthermore, MIAT binding to TXNIP protein restrains TXNIP ubiquitination degradation. Previous studies have suggested that TXINP leads IL-1β maturation and inflammation during DR development. As a result, MIAT silence diminishes the effects of HG on the release of IL-1β, tumor necrosis factor-alpha (TNF-α), and interleukins-6 (IL-6) from Müller cells, and C-myc over-expression abrogates the impact (85). LncRNA OGRU is a newly identified transcript that is found to be markedly up-regulated in serum samples of diabetic patients with DR and plays a strong role in regulating inflammation and oxidative stress. OGRU silencing restores Nrf2 protein levels and inhibits nuclear factor kappa-beta (NF-kB) activation in DR rat retinal tissue. OGRU over-expression and miR-320 knockdown can increase ROS production by restraining Nrf2 activation and are reversed through decreasing ubiquitin-specific protease14 (USP14) expression. USP14 deletion also greatly limits the function of IkBa ubiquitination to accelerate NF-kB activation. To further explore the potential of OGRU as a therapeutic target, intraocular injection of OGRU shRNA in diabetic rats is found to inhibit OGRU expression in animals and improve neuronal survival and glial activation. OGRU is also involved in angiogenesis and vascular leakage in DR progression, marked by the release of VEGF and TGF-β1 from Müller cells (86).
MALAT1 has a protective effect on DR. MALAT1 knockdown inhibits Müller cell viability in vitro and in vivo. Interestingly, the rate of retinal ganglion cells (RGCs) apoptosis is significantly decreased when co-cultured with Müller cell by the releasing of neuroprotective factors, glial cell-derived neurotrophic factor (GDNF), brain-derived neurotrophic factor (BDNF), neurotrophin nerve growth factor (NGF) and neurotrophin-4 (NT-4), and this protective effect is weakened by MALAT1 silencing (87). Aquaporin-4 (AQP4) is the major water channel protein of the central system and is involved in water crossing the blood-brain barrier (88). AQP4 Antisense RNA 1 (AQP4-AS1) is transcribed from the antisense strand of the AQP4 gene, and is positively regulated in the aqueous humor of diabetic patients. AQP4-AS1 negatively regulates AQP4 mRNA in glucose-induced human Müller cells and diabetic retinas. Under the context of high glucose, AQP4-AS1 silencing reverses human Müller cells apoptosis, RGC cell damage as well as the proliferation and migration of endothelial cells co-cultured with Müller cells. Intravitreal injection of AQP4-AS1 shRNA in diabetic mice silences its expression, improves retinal dysfunction, and attenuates vascular leakage (89). This suggests that Müller cells play an important role in DR neurovascular crosstalk and dysregulation, and then lncRNAs, which have an important regulatory role in it, are an option for therapeutic targets (Figure 3).
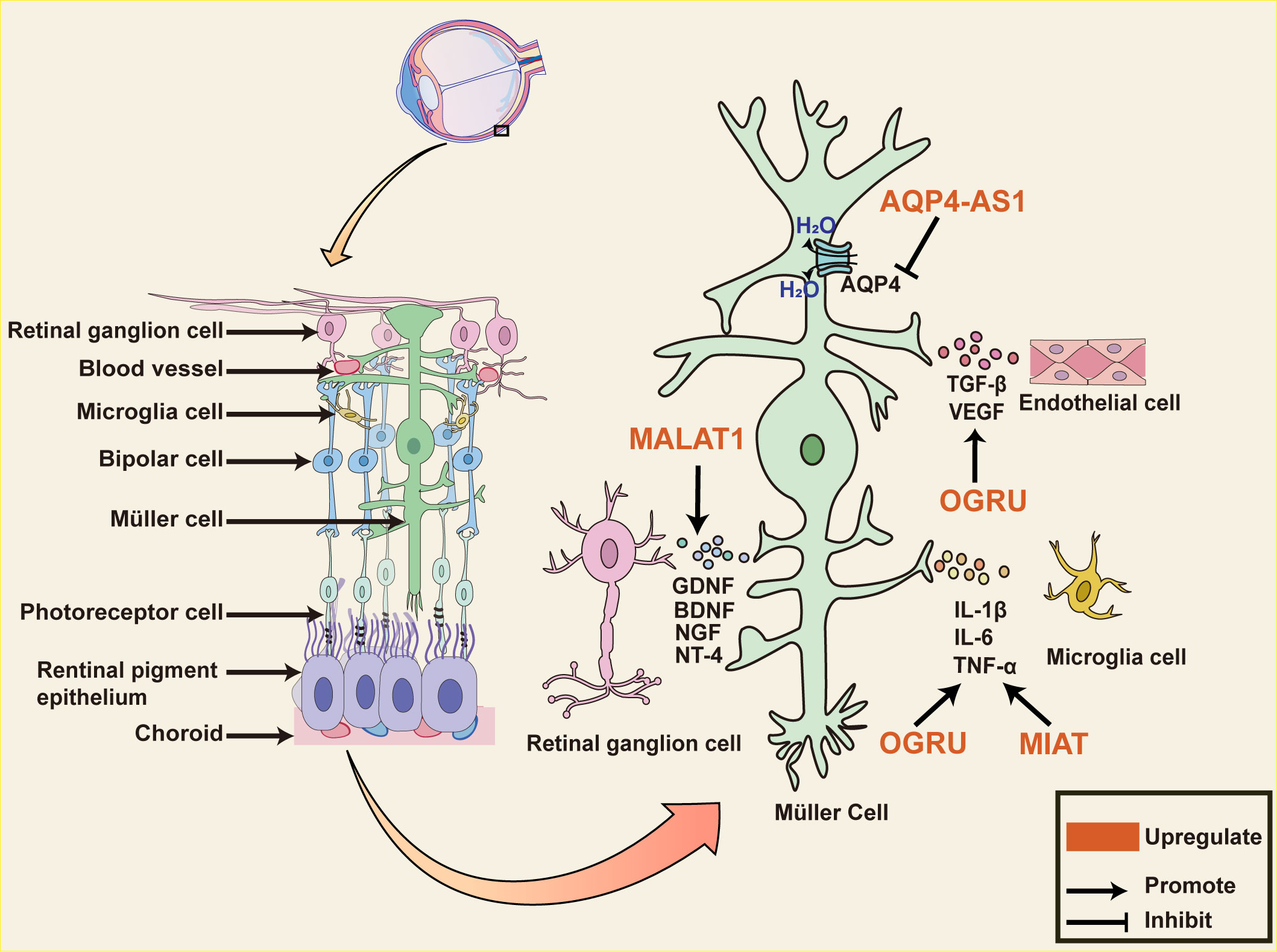
Figure 3 The role of lncRNAs in diabetic retinal neurodegeneration. The diagram shows Müller cells span the entire retina and interact with almost all cells within the retina. Dysregulated lncRNA in Müller cells impacts various pathophysiological events in diabetic retinopathy. MIAT and OGRU promote the release of pro-inflammatory mediators such as TNF-α, IL-1β, IL-17, and IL-6 from Müller cells. OGRU also promotes the release of VEGF and TGF-β1, which are involved in angiogenesis and vascular leakage during DR progression. MALAT1 upregulates the expression of neurotrophic factors, including GDNF, NT-4, BDNF, and NGF in the retina of optic nerve transection rat, decreasing the number of apoptotic RGCs. AQP4 is the major water channel protein of the central system and is negatively regulated through AQP4-AS1 in glucose-induced human Müller cells. AQP4-AS1 silencing reverses Müller cells and RGC cell apoptosis, endothelial cell proliferation, and migration, improving retinal functions. Abbreviations: MIAT, Myocardial Infarction Associated Transcript; MALAT1, Metastasis Associated Lung Adenocarcinoma Transcript 1; AQP4-AS1, AQP4 Antisense RNA 1; DR, diabetic retinopathy; RGCs, retinal ganglion cells; GDNF, Glial cell-derived neurotrophic factor; BDNF, brain-derived neurotrophic factor; NGF, neurotrophin nerve growth factor; NT-4, neurotrophin-4; VEGF, vascular endothelial growth factor; TGF-β, transforming growth factor β; IL-6, interleukins-6; TNF-α, tumor necrosis factor alpha; IL-1β, interleukins 1beta.
Normal connections between endothelium in the retinal microvascular system are essential for maintaining vascular function. Vascular endothelial (VE)-calmodulin is a key molecule that mediates interendothelial cell junctions (90). LncRNAs modify VE-calmodulin production through a rich mechanism. Highly HOTAIR expression has been shown in DR patients in several studies and has been indicated as an important epigenetic mediator in vascular dysfunction (91). HOTAIR acts as a scaffold for lysine demethylase 1A (LSD1) and represses VE-cadherin transcription by decreasing H3K4me3 levels on its promoter (92). MALAT1 and VE-cadherin are up-regulated while miR-125b is down-regulated in human retina microvascular endothelial cells (hRMECs) treated with HG. MALAT1 can competitively bind to miR-125b against VE-cadherin at the site of the 3’-untranslated region (3’-UTR), leading to the up-regulation of VE-cadherin(93). Protein kinase C β (PRKCB), a serine-threonine kinase, ubiquitinates VE-calmodulin leading to increased endothelial permeability in retinal vasculature (94). Vascular endothelial‐associated lncRNA‐2 (VEAL2) was identified as a novel lncRNA expressed in human umbilical vein endothelial cells (HUVECs). It compete with DAG for binding to the C1 structural domain of PRKCB2 leading to its activation. PRKCB2 translocation to the cell membrane is inhibited by VEAL2 overexpression and is observed mainly in the cytoplasm, thereby partially reversing endothelial permeability (95).
VEGF has been used as an anti-vascular proliferation target and applied in the clinical treatment of DR, but the efficacy has not been satisfactory. VEGF-A mRNA and protein levels are changed under the regulation of HOTAIR. HOTAIR not only alters VEGF-A epigenetic activation but also forms a complex with LSD1 to increase hypoxia inducible factor 1 subunit alpha (HIF-1α) production and promote HIF-1α-mediated transcriptional activation of VEGF-A (92). MALAT1 can likewise directly promote the expression of HIF-1α and VEGF-A through sponging miRNA. According to recent research, HIF1A antisense RNA 2 (HIF1A-AS2), the antisense transcript of HIF-1, is strongly and positively linked with HIF-1α and VEGF and is higher in peripheral blood in NPDR patients as well as in those with proliferative diabetic retinopathy (PDR) (96). Further explore the therapeutic potential of lncRNA-targeted VEGF against microvascular proliferation in vivo. In fibrovascular membranes (FVMs) of PDR patients, the co-expression of lncRNA Testis Development Related Gene 1 (TDRG1) and VEGF around the vessels is observed with immunofluorescence staining. Knockdown of TDRG1 notably represses the HG-induced VEGF expression, resulting in levels close to normal. TDRG1 silencing rescues hyperglycemia-induced HREC dysfunction, including reducing cell proliferation ability, improving HREC leakage, inhibiting cell migration, and maintaining the tube network formation (97). More and more lncRNAs have been shown to modulate VEGF (Table 2.). Among them MALAT1, Urothelial Carcinoma-Associated 1 (UCA1), TUG1, and linc00174 control VEGF production by regulating various miRNAs via the ceRNA pathway, which suggests that these lncRNAs could serve as potential targets for treating vascular proliferative imbalance in DR patients(98–101).
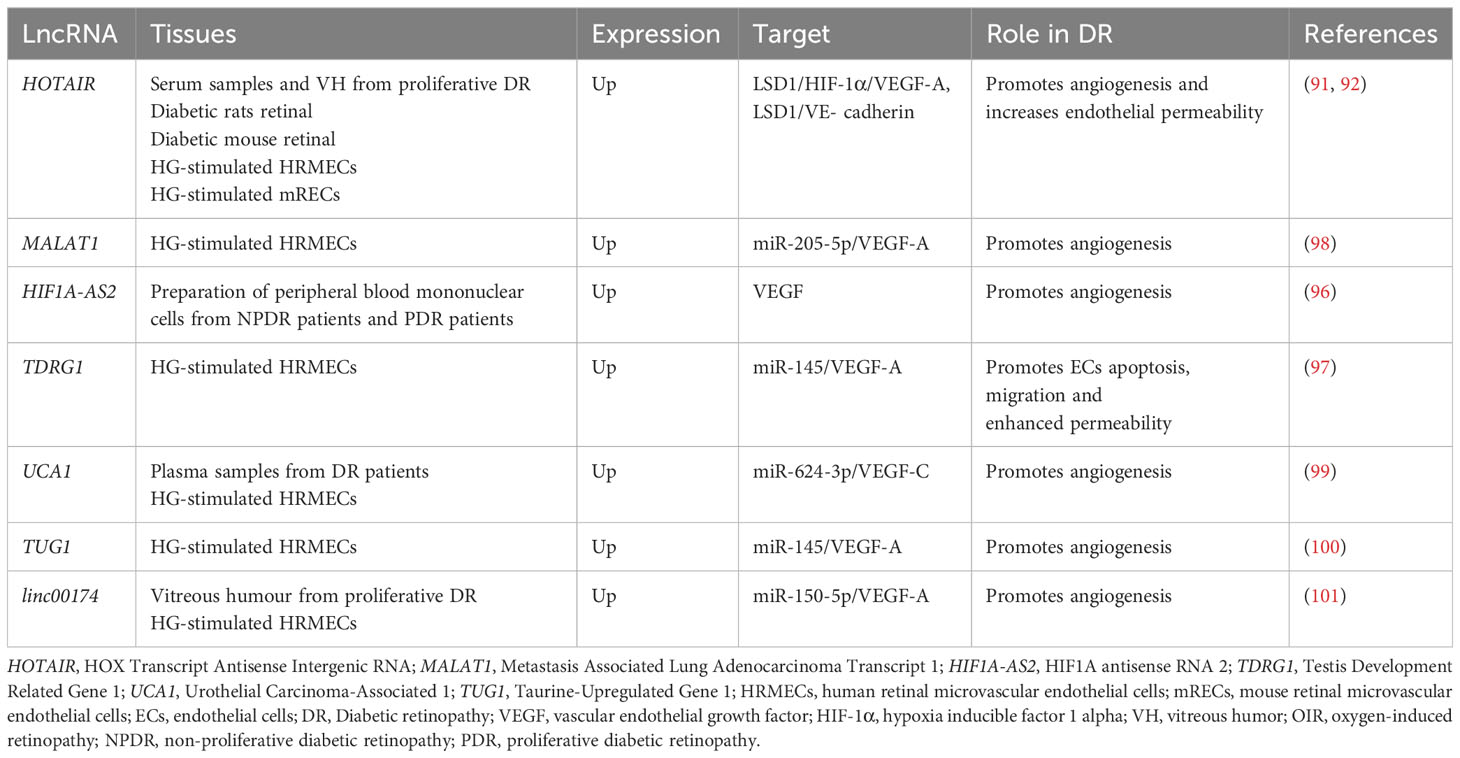
Table 2 LncRNA regulation on VEGF is involved in the progression of DR.
LncRNAs are stable in a variety of body fluids, such as blood, plasma, serum, and urine, which can be used as a novel non-invasive biomarker for diabetic complications. Some lncRNAs have a diagnostic role in diabetic complications. ANRIL and MALAT1 are upregulated in patients with DN, as the biomarker for the diagnosis of diabetic kidney disease (22, 26). TINCR and HOTAIR are downregulated in serum and myocardial biopsies of patients with DCM and can be used to effectively distinguish patients with DCM from healthy controls (58;102). LncRNAs are also known to act as prognostic molecules in DR. According to the receiver operating characteristic (ROC) curve, MALAT1 and HOTAIR can be used as promising new biomarkers for predicting the severity of DR. Comparing NPDR with PDR patients, upregulation of serum HOTAIR and MALAT1 was detected in PDR (103). Distinct lncRNA phenotype combinations may be able to discriminate DR patient sub-groups (NPDR and PDR). In the NPDR group, the most prevalent phenotype is MIAT/WISPER/ZFAS1/H19, while the prevalent lncRNA phenotypes in the PDR group is HOTAIR/ANRIL/HULC/H19. LncRNA variants may predict treatment outcomes. Following anti-VEGF therapy, DR patients with the TUG1 A or MIAT T/C exhibit worse therapeutic efficacy (104). Unfortunately, further validation in an expanded population is necessary due to the limited sample size included in this study.
Interestingly, lncRNAs are better suited as ideal candidates for therapeutic intervention because not encoding proteins. TUG1 overexpression maintains mitochondrial morphology and dynamics in podocytes, silencing KCNQ1OT1 alleviates myocardial dysfunction and attenuates myocardial fibrosis, targeting AQP4-AS1 for the treatment of diabetic retinal neurovascular dysfunction, which is demonstrated in animal models (73, 89, 105). Nevertheless, for possible reasons such as off-target effects, adverse effects on cells other than those targeted, and lack of suitable delivery vehicles, the lack of lncRNA-based therapeutic approaches in human trials.
The regulatory role of lncRNAs in diabetic complications offers the possibility of finding new therapeutic targets. Interfering with the expression or function of lncRNAs, which involved in the diabetes-induced oxidative stress, apoptosis, and inflammation has the potential to improve the pathological process of diabetic complications. As mentioned above, MALAT1 is strongly associated with the progression of DN, DR and DCM, as well as it is up-regulated in peripheral blood mononuclear cells (PBMCs) from type 2 diabetes patients. (106), suggesting that MALAT1 is a crucial target molecule and biomarker for diabetic complications. Therefore, MALAT1 should be investigated more deeply as an essential therapeutic target in future studies. Moreover, TUG1 may be used as a therapeutic target for DN, KCNQ1OT1 is specific for the interference of DCM, while AQP4-AS1 for DR.
Current lncRNA targeting methods include the use of small interfering RNAs (siRNA), antisense oligonucleotides (ASOs), and the CRISPR/Cas9 system, which are delivered in vivo via a variety of vectors including viral vectors, liposomes, and exosomes. However, given the safety and delivery difficulties, CRISPR/Cas9 systems and viral vectors are more limited to basic research, and other approaches targeting lncRNAs also face a few concerns. The most significant issue is that the function and potential downstreams of the lncRNAs chosen to be targeted are still well understudied, and inadequate elucidation of their roles in vivo. Therefore, the use of lncRNA-targeted drugs in the clinic may face unintended consequences. In future, a better understanding of the mechanisms of lncRNA will pave the way for early diagnosis and the design of better treatments to reduce the morbidity and mortality of diabetic complications.
MG: Investigation, Software, Writing – original draft. WL: Investigation, Software, Writing – original draft. JL: Resources, Writing – original draft. GY: Resources, Writing – original draft, Software. YT: Resources, Writing – original draft. XJ: Conceptualization, Writing – review & editing. YX: Conceptualization, Funding acquisition, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Natural Science Foundation of China (No. 82170369), the Jilin Provincial Science and Technology Foundation (Nos.20210509003RQ and 20230402002GH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Eizirik DL, Pasquali L, Cnop M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol (2020) 16(7):349–62. doi: 10.1038/s41574-020-0355-7
3. Dagar N, Das P, Bisht P, Taraphdar AK, Velayutham R, Arumugam S. Diabetic nephropathy: A twisted thread to unravel. Life Sci (2021) 278:119635. doi: 10.1016/j.lfs.2021.119635
4. Lin YK, Gao B, Liu L, Ang L, Mizokami-Stout K, Pop-Busui R, et al. The Prevalence of Diabetic Microvascular Complications in China and the USA. Curr Diabetes Rep (2021) 21(6):16. doi: 10.1007/s11892-021-01387-3
5. Gulsin GS, Athithan L, Mccann GP. Diabetic cardiomyopathy: prevalence, determinants and potential treatments. Ther Adv Endocrinol Metab (2019) 10:2042018819834869. doi: 10.1177/2042018819834869
6. Williams R, Van Gaal L, Lucioni C. Assessing the impact of complications on the costs of Type II diabetes. Diabetologia (2002). doi: 10.1007/s00125-002-0859-9
7. Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia (2015) 58(3):443–55. doi: 10.1007/s00125-014-3462-y
8. Bridges MC, Daulagala AC, Kourtidis A. LNCcation: lncRNA localization and function. J Cell Biol (2021) 220(2):e202009045. doi: 10.1083/jcb.202009045
9. Gonzalez-Moro I, Olazagoitia-Garmendia A, Colli ML, Cobo-Vuilleumier N, Postler TS. Marselli L, et al. The T1D-associated lncRNA Lnc13 modulates human pancreatic β cell inflammation by allele-specific stabilization of STAT1 mRNA. Proc Natl Acad Sci U.S.A. (2020) 117(16):9022–31. doi: 10.1073/pnas.1914353117
10. González-Moro I, Garcia-Etxebarria K, Mendoza LM, Fernández-Jiménez N, Mentxaka J, Olazagoitia-Garmendia A, et al. LncRNA ARGI Contributes to Virus-Induced Pancreatic β Cell Inflammation Through Transcriptional Activation of IFN-Stimulated Genes. Adv Sci (Weinh) (2023) 10(25):e2300063. doi: 10.1002/advs.202300063
11. Guo WH, Guo Q, Liu YL, Yan DD, Jin L, Zhang R, et al. Mutated lncRNA increase the risk of type 2 diabetes by promoting β cell dysfunction and insulin resistance. Cell Death Dis (2022) 13(10):904. doi: 10.1038/s41419-022-05348-w
12. Chen J, Ke S, Zhong L, Wu J, Tseng A, Morpurgo B, et al. Long noncoding RNA MALAT1 regulates generation of reactive oxygen species and the insulin responses in male mice. Biochem Pharmacol (2018) 152:94–103. doi: 10.1016/j.bcp.2018.03.019
13. Persson F, Rossing P. Diagnosis of diabetic kidney disease: state of the art and future perspective. Kidney Int Suppl (2011) 2018) 8(1):2–7. doi: 10.1016/j.kisu.2017.10.003
14. Cherney DZI, Bakris GL. Novel therapies for diabetic kidney disease. Kidney Int Suppl (2011) 2018) 8(1):18–25. doi: 10.1016/j.kisu.2017.10.005
15. Liao L, Chen J, Zhang C, Guo Y, Liu W, Liu W, et al. LncRNA NEAT1 Promotes High Glucose-Induced Mesangial Cell Hypertrophy by Targeting miR-222-3p/CDKN1B Axis. Front Mol Biosci (2020) 7:627827. doi: 10.3389/fmolb.2020.627827
16. Huang S, Xu Y, Ge X, Xu B, Peng W, Jiang X, et al. Long noncoding RNA NEAT1 accelerates the proliferation and fibrosis in diabetic nephropathy through activating Akt/mTOR signaling pathway. J Cell Physiol (2019) 234(7):11200–7. doi: 10.1002/jcp.27770
17. Wang X, Xu Y, Zhu YC, Wang YK, Li J, Li XY, et al. LncRNA NEAT1 promotes extracellular matrix accumulation and epithelial-to-mesenchymal transition by targeting miR-27b-3p and ZEB1 in diabetic nephropathy. J Cell Physiol (2019) 234(8):12926–33. doi: 10.1002/jcp.27959
18. Chang J, Yu Y, Fang Z, He H, Wang D, Teng J, et al. Long non-coding RNA CDKN2B-AS1 regulates high glucose-induced human mesangial cell injury via regulating the miR-15b-5p/WNT2B axis. Diabetol Metab Syndr (2020) 12(1):109. doi: 10.1186/s13098-020-00618-z
19. Li Y, Zheng LL, Huang DG, Cao H, Gao YH, Fan ZC. LNCRNA CDKN2B-AS1 regulates mesangial cell proliferation and extracellular matrix accumulation via miR-424-5p/HMGA2 axis. BioMed Pharmacother (2020) 121:109622. doi: 10.1016/j.biopha.2019.109622
20. Wang J, Zhao SM. LncRNA-antisense non-coding RNA in the INK4 locus promotes pyroptosis via miR-497/thioredoxin-interacting protein axis in diabetic nephropathy. Life Sci (2021) 264:118728. doi: 10.1016/j.lfs.2020.118728
21. Xiao M, Bai S, Chen J, Li Y, Zhang S, Hu Z. CDKN2B-AS1 participates in high glucose-induced apoptosis and fibrosis via NOTCH2 through functioning as a miR-98-5p decoy in human podocytes and renal tubular cells. Diabetol Metab Syndr (2021) 13(1):107. doi: 10.1186/s13098-021-00725-5
22. Zhu Y, Dai L, Yu X, Chen X, Li Z, Sun Y, et al. Circulating expression and clinical significance of LncRNA ANRIL in diabetic kidney disease. Mol Biol Rep (2022) 49(11):10521–9. doi: 10.1007/s11033-022-07843-x
23. Shen H, Ming Y, Xu C, Xu Y, Zhao S, Zhang Q. Deregulation of long noncoding RNA (TUG1) contributes to excessive podocytes apoptosis by activating endoplasmic reticulum stress in the development of diabetic nephropathy. J Cell Physiol (2019). doi: 10.1002/jcp.28153
24. Long J, Galvan DL, Mise K, Kanwar YS, Li L, Poungavrin N, et al. Role for carbohydrate response element-binding protein (ChREBP) in high glucose-mediated repression of long noncoding RNA Tug1. J Biol Chem (2020) 295(47):15840–52. doi: 10.1074/jbc.RA120.013228
25. Li L, Long J, Mise K, Galvan DL, Overbeek PA, Tan L, et al. PGC1α is required for the renoprotective effect of lncRNA Tug1 in vivo and links Tug1 with urea cycle metabolites. Cell Rep (2021) 36(6):109510. doi: 10.1016/j.celrep.2021.109510
26. Zhou LJ, Yang DW, Ou LN, Guo XR, Wu BL. Circulating Expression Level of LncRNA Malat1 in Diabetic Kidney Disease Patients and Its Clinical Significance. J Diabetes Res (2020) 2020:4729019. doi: 10.1155/2020/4729019
27. Petrica L, Hogea E, Gadalean F, Vlad A, Vlad M, Dumitrascu V, et al. Long noncoding RNAs may impact podocytes and proximal tubule function through modulating miRNAs expression in Early Diabetic Kidney Disease of Type 2 Diabetes Mellitus patients. Int J Med Sci (2021) 18(10):2093–101. doi: 10.7150/ijms.56551
28. Fawzy MS, Abu Alsel BT, Al Ageeli E, Al-Qahtani SA, Abdel-Daim MM, Toraih EA. Long non-coding RNA MALAT1 and microRNA-499a expression profiles in diabetic ESRD patients undergoing dialysis: a preliminary cross-sectional analysis. Arch Physiol Biochem (2020) 126(2):172–82. doi: 10.1080/13813455.2018.1499119
29. Hu M, Wang R, Li X, Fan M, Lin J, Zhen J, et al. LncRNA MALAT1 is dysregulated in diabetic nephropathy and involved in high glucose-induced podocyte injury via its interplay with β-catenin. J Cell Mol Med (2017) 21(11):2732–47. doi: 10.1111/jcmm.13189
30. Zuo Y, Chen L, He X, Ye Z, Li L, Liu Z, et al. Atorvastatin Regulates MALAT1/miR-200c/NRF2 Activity to Protect Against Podocyte Pyroptosis Induced by High Glucose. Diabetes Metab Syndr Obes (2021) 14:1631–45. doi: 10.2147/dmso.S298950
31. Zhang J, Jiang T, Liang X, Shu S, Xiang X, Zhang W, et al. lncRNA MALAT1 mediated high glucose-induced HK-2 cell epithelial-to-mesenchymal transition and injury. J Physiol Biochem (2019) 75(4):443–52. doi: 10.1007/s13105-019-00688-2
32. Zhong W, Zeng J, Xue J, Du A, Xu Y. Knockdown of lncRNA PVT1 alleviates high glucose-induced proliferation and fibrosis in human mesangial cells by miR-23b-3p/WT1 axis. Diabetol Metab Syndr (2020) 12:33. doi: 10.1186/s13098-020-00539-x
33. Liu DW, Zhang JH, Liu FX, Wang XT, Pan SK, Jiang DK, et al. Silencing of long noncoding RNA PVT1 inhibits podocyte damage and apoptosis in diabetic nephropathy by upregulating FOXA1. . Exp Mol Med (2019) 51(8):1–15. doi: 10.1038/s12276-019-0259-6
34. Yang Y, Lv X, Fan Q, Wang X, Xu L, Lu X, et al. Analysis of circulating lncRNA expression profiles in patients with diabetes mellitus and diabetic nephropathy: Differential expression profile of circulating lncRNA. Clin Nephrol (2019) 92(1):25–35. doi: 10.5414/cn109525
35. Li L, Xu L, Wen S, Yang Y, Li X, Fan Q. The effect of lncRNA-ARAP1-AS2/ARAP1 on high glucose-induced cytoskeleton rearrangement and epithelial-mesenchymal transition in human renal tubular epithelial cells. J Cell Physiol (2020) 235(7-8):5787–95. doi: 10.1002/jcp.29512
36. Zhu D, Wu X, Xue Q. Long non-coding RNA CASC2 restrains high glucose-induced proliferation, inflammation and fibrosis in human glomerular mesangial cells through mediating miR-135a-5p/TIMP3 axis and JNK signaling. Diabetol Metab Syndr (2021) 13(1):89. doi: 10.1186/s13098-021-00709-5
37. Yi H, Peng R, Zhang LY, Sun Y, Peng HM, Liu HD, et al. LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3 inflammasome-mediated inflammation in diabetic nephropathy. Cell Death Dis (2017) 8(2):e2583. doi: 10.1038/cddis.2016.451
38. Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int (2015) 87(2):297–307. doi: 10.1038/ki.2014.287
39. Gao J, Wang W, Wang F, Guo C. LncRNA-NR_033515 promotes proliferation, fibrogenesis and epithelial-to-mesenchymal transition by targeting miR-743b-5p in diabetic nephropathy. BioMed Pharmacother (2018) 106:543–52. doi: 10.1016/j.biopha.2018.06.104
40. Nagai K, Matsubara T, Mima A, Sumi E, Kanamori H, Iehara N, et al. Gas6 induces Akt/mTOR-mediated mesangial hypertrophy in diabetic nephropathy. Kidney Int (2005) 68(2):552–61. doi: 10.1111/j.1523-1755.2005.00433.x
41. Yang YL, Xue M, Jia YJ, Hu F, Zheng ZJ, Wang L, et al. Long noncoding RNA NEAT1 is involved in the protective effect of Klotho on renal tubular epithelial cells in diabetic kidney disease through the ERK1/2 signaling pathway. Exp Mol Med (2020) 52(2):266–80. doi: 10.1038/s12276-020-0381-5
42. Thomas AA, Feng B, Chakrabarti S. ANRIL regulates production of extracellular matrix proteins and vasoactive factors in diabetic complications. Am J Physiol Endocrinol Metab (2018) 314(3):E191–e200. doi: 10.1152/ajpendo.00268.2017
43. Shen H, Ming Y, Xu C, Xu Y, Zhao S, Zhang Q. Deregulation of long noncoding RNA (TUG1) contributes to excessive podocytes apoptosis by activating endoplasmic reticulum stress in the development of diabetic nephropathy. J Cell Physiol (2019) 234(9):15123–33. doi: 10.1002/jcp.28153
44. Reiser J, Kriz W, Kretzler M, Mundel P. The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol (2000) 11(1):1–8. doi: 10.1681/asn.V1111
45. Millis MP, Bowen D, Kingsley C, Watanabe RM, Wolford JK. Variants in the plasmacytoma variant translocation gene (PVT1) are associated with end-stage renal disease attributed to type 1 diabetes. Diabetes (2007) 56(12):3027–32. doi: 10.2337/db07-0675
46. Hanson RL, Craig DW, Millis MP, Yeatts KA, Kobes S, Pearson JV, et al. Identification of PVT1 as a candidate gene for end-stage renal disease in type 2 diabetes using a pooling-based genome-wide single nucleotide polymorphism association study. Diabetes (2007) 56(4):975–83. doi: 10.2337/db06-1072
47. Yu D, Yang X, Zhu Y, Xu F, Zhang H, Qiu Z. Knockdown of plasmacytoma variant translocation 1 (PVT1) inhibits high glucose-induced proliferation and renal fibrosis in HRMCs by regulating miR-23b-3p/early growth response factor 1 (EGR1). Endocr J (2021) 68(5):519–29. doi: 10.1507/endocrj.EJ20-0642
48. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol (1972) 30(6):595–602. doi: 10.1016/0002-9149(72)90595-4
49. Jia G, Hill MA, Sowers JR. Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res (2018) 122(4):624–38. doi: 10.1161/circresaha.117.311586
50. Wei J, Zhao Y, Liang H, Du W, Wang L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm Sin B (2022) 12(1):1–17. doi: 10.1016/j.apsb.2021.08.026
51. Wang C, Liu G, Yang H, Guo S, Wang H, Dong Z, et al. MALAT1-mediated recruitment of the histone methyltransferase EZH2 to the microRNA-22 promoter leads to cardiomyocyte apoptosis in diabetic cardiomyopathy. Sci Total Environ (2021) 766:142191. doi: 10.1016/j.scitotenv.2020.142191
52. Zhou X, Zhang W, Jin M, Chen J, Xu W, Kong X. lncRNA MIAT functions as a competing endogenous RNA to upregulate DAPK2 by sponging miR-22-3p in diabetic cardiomyopathy. Cell Death Dis (2017) 8(7):e2929. doi: 10.1038/cddis.2017.321
53. Zhang J, Zhang M, Yang Z, Huang S, Wu X, Cao L, et al. PDCD4 deficiency ameliorates left ventricular remodeling and insulin resistance in a rat model of type 2 diabetic cardiomyopathy. BMJ Open Diabetes Res Care (2020) 8(1):e001081. doi: 10.1136/bmjdrc-2019-001081
54. Lu K, Chen Q, Li M, He L, Riaz F, Zhang T, et al. Programmed cell death factor 4 (PDCD4), a novel therapy target for metabolic diseases besides cancer. Free Radic Biol Med (2020) 159:150–63. doi: 10.1016/j.freeradbiomed.2020.06.016
55. Zhao SF, Ye YX, Xu JD, He Y, Zhang DW, Xia ZY, et al. Long non-coding RNA KCNQ1OT1 increases the expression of PDCD4 by targeting miR-181a-5p, contributing to cardiomyocyte apoptosis in diabetic cardiomyopathy. Acta Diabetol (2021) 58(9):1251–67. doi: 10.1007/s00592-021-01713-x
56. Zhang Q, Li D, Dong X, Zhang X, Liu J, Peng L, et al. LncDACH1 promotes mitochondrial oxidative stress of cardiomyocytes by interacting with sirtuin3 and aggravates diabetic cardiomyopathy. . Sci China Life Sci (2022) 65(6):1198–212. doi: 10.1007/s11427-021-1982-8
57. Gao L, Wang X, Guo S, Xiao L, Liang C, Wang Z, et al. LncRNA HOTAIR functions as a competing endogenous RNA to upregulate SIRT1 by sponging miR-34a in diabetic cardiomyopathy. J Cell Physiol (2019) 234(4):4944–58. doi: 10.1002/jcp.27296
58. Qi K, Zhong J. LncRNA HOTAIR improves diabetic cardiomyopathy by increasing viability of cardiomyocytes through activation of the PI3K/Akt pathway. Exp Ther Med (2018) 16(6):4817–23. doi: 10.3892/etm.2018.6755
59. Wang S, Duan J, Liao J, Wang Y, Xiao X, Li L, et al. LncRNA H19 inhibits ER stress induced apoptosis and improves diabetic cardiomyopathy by regulating PI3K/AKT/mTOR axis. Aging (2022) 14(16):6809–28. doi: 10.18632/aging.204256
60. Cai X, Cullen BR. The imprinted H19 noncoding RNA is a primary microRNA precursor. RNA (2007) 13(3):313–6. doi: 10.1261/rna.351707
61. Li X, Wang H, Yao B, Xu W, Chen J, Zhou X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci Rep (2016) 6:36340. doi: 10.1038/srep36340
62. Ghosh R, Pattison JS. Macroautophagy and Chaperone-Mediated Autophagy in Heart Failure: The Known and the Unknown. Oxid Med Cell Longev (2018) 2018:8602041. doi: 10.1155/2018/8602041
63. Wu QQ, Liu C, Cai Z, Xie Q, Hu T, Duan M, et al. High-mobility group AT-hook 1 promotes cardiac dysfunction in diabetic cardiomyopathy via autophagy inhibition. Cell Death Dis (2020) 11(3):160. doi: 10.1038/s41419-020-2316-4
64. Chen D, Zhang M. GAS5 regulates diabetic cardiomyopathy via miR−221−3p/p27 axis−associated autophagy. Mol Med Rep (2021) 23(2):135. doi: 10.3892/mmr.2020.11774
65. Feng Y, Xu W, Zhang W, Wang W, Liu T, Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics (2019) 9(15):4558–66. doi: 10.7150/thno.31052
66. Hu X, Sui X, Li L, Huang X, Rong R, Su X, et al. Protocadherin 17 acts as a tumour suppressor inducing tumour cell apoptosis and autophagy, and is frequently methylated in gastric and colorectal cancers. J Pathol (2013) 229(1):62–73. doi: 10.1002/path.4093
67. Wu JC, Wang FZ, Tsai ML, Lo CY, Badmaev V, Ho CT, et al. Se-Allylselenocysteine induces autophagy by modulating the AMPK/mTOR signaling pathway and epigenetic regulation of PCDH17 in human colorectal adenocarcinoma cells. Mol Nutr Food Res (2015) 59(12):2511–22. doi: 10.1002/mnfr.201500373
68. Zhuo C, Jiang R, Lin X, Shao M. LncRNA H19 inhibits autophagy by epigenetically silencing of DIRAS3 in diabetic cardiomyopathy. Oncotarget (2017) 8(1):1429–37. doi: 10.18632/oncotarget.13637
69. Ketelut-Carneiro N, Fitzgerald KA. Apoptosis, Pyroptosis, and Necroptosis-Oh My! The Many Ways a Cell Can Die. J Mol Biol (2022) 434(4):167378. doi: 10.1016/j.jmb.2021.167378
70. Fang Y, Tian S, Pan Y, Li W, Wang Q, Tang Y, et al. Pyroptosis: A new frontier in cancer. BioMed Pharmacother (2020) 121:109595. doi: 10.1016/j.biopha.2019.109595
71. Meng L, Lin H, Huang X, Weng J, Peng F, Wu S. METTL14 suppresses pyroptosis and diabetic cardiomyopathy by downregulating TINCR lncRNA. Cell Death Dis (2022) 13(1):38. doi: 10.1038/s41419-021-04484-z
72. Xu Y, Fang H, Xu Q, Xu C, Yang L, Huang C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle (2020) 19(22):3054–65. doi: 10.1080/15384101.2020.1831245
73. Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis (2018) 9(10):1000. doi: 10.1038/s41419-018-1029-4
74. Xiao W, Zheng D, Chen X, Yu B, Deng K, Ma J, et al. Long non-coding RNA MIAT is involved in the regulation of pyroptosis in diabetic cardiomyopathy via targeting miR-214-3p. iScience (2021) 24(12):103518. doi: 10.1016/j.isci.2021.103518
75. Liu M, López De Juan Abad B, Cheng K. Cardiac fibrosis: Myofibroblast-mediated pathological regulation and drug delivery strategies. Adv Drug Delivery Rev (2021) 173:504–19. doi: 10.1016/j.addr.2021.03.021
76. Che H, Wang Y, Li H, Li Y, Sahil A, Lv J, et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-β1/Smads signaling in diabetic cardiomyopathy. FASEB J (2020) 34(4):5282–98. doi: 10.1096/fj.201902692R
77. Zhang Y, Zhang YY, Li TT, Wang J, Jiang Y, Zhao Y, et al. Ablation of interleukin-17 alleviated cardiac interstitial fibrosis and improved cardiac function via inhibiting long non-coding RNA-AK081284 in diabetic mice. J Mol Cell Cardiol (2018) 115:64–72. doi: 10.1016/j.yjmcc.2018.01.001
78. Qi Y, Wu H, Mai C, Lin H, Shen J, Zhang X, et al. LncRNA-MIAT-Mediated miR-214-3p Silencing Is Responsible for IL-17 Production and Cardiac Fibrosis in Diabetic Cardiomyopathy. Front Cell Dev Biol (2020) 8:243. doi: 10.3389/fcell.2020.00243
79. Zheng D, Zhang Y, Hu Y, Guan J, Xu L, Xiao W, et al. Long noncoding RNA Crnde attenuates cardiac fibrosis via Smad3-Crnde negative feedback in diabetic cardiomyopathy. FEBS J (2019) 286(9):1645–55. doi: 10.1111/febs.14780
80. Tan TE, Wong TY. Diabetic retinopathy: Looking forward to 2030. Front Endocrinol (Lausanne) (2022) 13:1077669. doi: 10.3389/fendo.2022.1077669
81. Yau JW, Rogers SL, Kawasaki R, Lamoureux EL, Kowalski JW, Bek T, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care (2012) 35(3):556–64. doi: 10.2337/dc11-1909
82. Couturier A, Rey PA, Erginay A, Lavia C, Bonnin S, Dupas B, et al. Widefield OCT-Angiography and Fluorescein Angiography Assessments of Nonperfusion in Diabetic Retinopathy and Edema Treated with Anti-Vascular Endothelial Growth Factor. Ophthalmology (2019) 126(12):1685–94. doi: 10.1016/j.ophtha.2019.06.022
83. Carpi-Santos R, De Melo Reis RA, Gomes FCA, Calaza KC. Contribution of Müller Cells in the Diabetic Retinopathy Development: Focus on Oxidative Stress and Inflammation. Antioxidants (Basel) (2022) 11(4):617. doi: 10.3390/antiox11040617
84. Yang S, Qi S, Wang C. The role of retinal Müller cells in diabetic retinopathy and related therapeutic advances. Front Cell Dev Biol (2022) 10:1047487. doi: 10.3389/fcell.2022.1047487
85. Zhang J, Chen C, Wu L, Wang Q, Chen J, Zhang S, et al. C-myc contributes to the release of Müller cells-derived proinflammatory cytokines by regulating lncRNA MIAT/XNIP pathway. Int J Biochem Cell Biol (2019) 114:105574. doi: 10.1016/j.biocel.2019.105574
86. Fu S, Zheng Y, Sun Y, Lai M, Qiu J, Gui F, et al. Suppressing long noncoding RNA OGRU ameliorates diabetic retinopathy by inhibition of oxidative stress and inflammation via miR-320/USP14 axis. Free Radic Biol Med (2021) 169:361–81. doi: 10.1016/j.freeradbiomed.2021.03.016
87. Yao J, Wang XQ, Li YJ, Shan K, Yang H, Wang YN, et al. Long non-coding RNA MALAT1 regulates retinal neurodegeneration through CREB signaling. EMBO Mol Med (2016) 8(4):346–62. doi: 10.15252/emmm.201505725
88. Verkman AS, Anderson MO, Papadopoulos MC. Aquaporins: important but elusive drug targets. Nat Rev Drug Discov (2014) 13(4):259–77. doi: 10.1038/nrd4226
89. Li X, Zhu J, Zhong Y, Liu C, Yao M, Sun Y, et al. Targeting long noncoding RNA-AQP4-AS1 for the treatment of retinal neurovascular dysfunction in diabetes mellitus. EBioMedicine (2022) 77:103857. doi: 10.1016/j.ebiom.2022.103857
90. Navaratna D, Mcguire PG, Menicucci G, Das A. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes (2007) 56(9):2380–7. doi: 10.2337/db06-1694
91. Biswas S, Feng B, Chen S, Liu J, Aref-Eshghi E, Gonder J, et al. The Long Non-Coding RNA HOTAIR Is a Critical Epigenetic Mediator of Angiogenesis in Diabetic Retinopathy. Invest Ophthalmol Vis Sci (2021) 62(3):20. doi: 10.1167/iovs.62.3.20
92. Zhao D, Zhao Y, Wang J, Wu L, Liu Y, Zhao S, et al. Long noncoding RNA Hotair facilitates retinal endothelial cell dysfunction in diabetic retinopathy. Clin Sci (Lond) (2020) 134(17):2419–34. doi: 10.1042/cs20200694
93. Liu P, Jia SB, Shi JM, Li WJ, Tang LS, Zhu XH, et al. LncRNA-MALAT1 promotes neovascularization in diabetic retinopathy through regulating miR-125b/VE-cadherin axis. Biosci Rep (2019) 39(5):BSR20181469. doi: 10.1042/bsr20181469
94. Haidari M, Zhang W, Willerson JT, Dixon RA. Disruption of endothelial adherens junctions by high glucose is mediated by protein kinase C-β-dependent vascular endothelial cadherin tyrosine phosphorylation. Cardiovasc Diabetol (2014) 13:105. doi: 10.1186/1475-2840-13-105
95. Sehgal P, Mathew S, Sivadas A, Ray A, Tanwar J, Vishwakarma S, et al. LncRNA VEAL2 regulates PRKCB2 to modulate endothelial permeability in diabetic retinopathy. EMBO J (2021) 40(15):e107134. doi: 10.15252/embj.2020107134
96. Atef MM, Shafik NM, Hafez YM, Watany MM, Selim A, Shafik HM, et al. The evolving role of long noncoding RNA HIF1A-AS2 in diabetic retinopathy: a cross-link axis between hypoxia, oxidative stress and angiogenesis via MAPK/VEGF-dependent pathway. Redox Rep (2022) 27(1):70–8. doi: 10.1080/13510002.2022.2050086
97. Gong Q, Dong W, Fan Y, Chen F, Bian X, Xu X, et al. LncRNA TDRG1-Mediated Overexpression of VEGF Aggravated Retinal Microvascular Endothelial Cell Dysfunction in Diabetic Retinopathy. Front Pharmacol (2019) 10:1703. doi: 10.3389/fphar.2019.01703
98. Tan A, Li T, Ruan L, Yang J, Luo Y, Li L, et al. Knockdown of Malat1 alleviates high-glucose-induced angiogenesis through regulating miR-205-5p/VEGF-A axis. Exp Eye Res (2021) 207:108585. doi: 10.1016/j.exer.2021.108585
99. Yan H, Yao P, Hu K, Li X, Li H. Long non-coding ribonucleic acid urothelial carcinoma-associated 1 promotes high glucose-induced human retinal endothelial cells angiogenesis through regulating micro-ribonucleic acid-624-3p/vascular endothelial growth factor C. J Diabetes Investig (2021) 12(11):1948–57. doi: 10.1111/jdi.13617
100. Shi Q, Tang J, Wang M, Xu L, Shi L. Knockdown of Long Non-coding RNA TUG1 Suppresses Migration and Tube Formation in High Glucose-Stimulated Human Retinal Microvascular Endothelial Cells by Sponging miRNA-145. Mol Biotechnol (2022) 64(2):171–7. doi: 10.1007/s12033-021-00398-5
101. Wang JJ, Wu KF, Wang DD. A novel regulatory network of linc00174/miR-150-5p/VEGFA modulates pathological angiogenesis in diabetic retinopathy. Can J Physiol Pharmacol (2021) 99(11):1175–83. doi: 10.1139/cjpp-2021-0036
102. Chen Y, Tan S, Liu M, Li J. LncRNA TINCR is downregulated in diabetic cardiomyopathy and relates to cardiomyocyte apoptosis. Scand Cardiovasc J (2018) 52(6):335–9. doi: 10.1080/14017431.2018.1546896
103. Shaker OG, Abdelaleem OO, Mahmoud RH, Abdelghaffar NK, Ahmed TI, Said OM, et al. Diagnostic and prognostic role of serum miR-20b, miR-17-3p, HOTAIR, and MALAT1 in diabetic retinopathy. IUBMB Life (2019) 71(3):310–20. doi: 10.1002/iub.1970
104. Mohammad HMF, Abdelghany AA, Al Ageeli E, Kattan SW, Hassan R, Toraih EA, et al. Long Non-Coding RNAs Gene Variants as Molecular Markers for Diabetic Retinopathy Risk and Response to Anti-VEGF Therapy. Pharmgenomics Pers Med (2021) 14:997–1014. doi: 10.2147/pgpm.S322463
105. Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest (2016) 126(11):4205–18. doi: 10.1172/jci87927
Keywords: long non-coding RNAs, diabetic nephropathy, diabetic retinopathy, diabetic cardiomyopathy, microRNA
Citation: Geng M, Liu W, Li J, Yang G, Tian Y, Jiang X and Xin Y (2024) LncRNA as a regulator in the development of diabetic complications. Front. Endocrinol. 15:1324393. doi: 10.3389/fendo.2024.1324393
Received: 19 October 2023; Accepted: 16 January 2024;
Published: 08 February 2024.
Edited by:
Chika Ifeanyi Chukwuma, Central University of Technology, South AfricaReviewed by:
Olakunle Sanni, Stellenbosch University, South AfricaCopyright © 2024 Geng, Liu, Li, Yang, Tian, Jiang and Xin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Jiang, amlhbmd4QGpsdS5lZHUuY24=; Ying Xin, eGlueUBqbHUuZWR1LmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.