 Peng Pi
Peng Pi Liqing Zeng
Liqing Zeng Zhipeng Zeng1
Zhipeng Zeng1 Yan Wang
Yan Wang- 1School of Sports Medicine and Rehabilitation, Beijing Sport University, Beijing, China
- 2School of Physical Education, Qiqihar University, Heilongjiang, Qiqihar, China
- 3College of Physical Education and Health, East China Normal University, Shanghai, China
Osteoarthritis (OA) is a common degenerative joint disease that can affect almost any joint, mainly resulting in joint dysfunction and pain. Worldwide, OA affects more than 240 million people and is one of the leading causes of activity limitation in adults. However, the pathogenesis of OA remains elusive, resulting in the lack of well-established clinical treatment strategies. Recently, energy metabolism alterations have provided new insights into the pathogenesis of OA. Accumulating evidence indicates that glucose metabolism plays a key role in maintaining cartilage homeostasis. Disorders of glucose metabolism can lead to chondrocyte hypertrophy and extracellular matrix degradation, and promote the occurrence and development of OA. This article systematically summarizes the regulatory effects of different enzymes and factors related to glucose metabolism in OA, as well as the mechanism and potential of various substances in the treatment of OA by affecting glucose metabolism. This provides a theoretical basis for a better understanding of the mechanism of OA progression and the development of optimal prevention and treatment strategies.
1 Introduction
Articular cartilage (AC) is a complex connective tissue, which is responsible for bearing, shock absorption and lubrication of joints of the whole body (1). Chondrocyte is the main cell type in normal human cartilage, which is differentiated from mesenchymal cells and accounts for 1-5% of cartilage tissue (2). Chondrocytes are surrounded by the pericellular matrix (PCM), which is considered a buffer of physical forces between chondrocytes and the extracellular matrix (ECM) (3). When chondrocytes are subjected to abnormal mechanical stimulation, such as overloading or joint injury, their metabolic balance is altered, leading to matrix loss and tissue degeneration, which can lead to osteoarthritis (OA) (4, 5). OA is one of the most common and diverse degenerative diseases with complex pathogenesis that affects all joint tissues, characterized by cartilage degeneration, osteophyte formation, meniscal degeneration, subchondral bone remodeling, joint inflammation and fibrosis of the infrapatellar fat pad (6–12). The main clinical manifestations of OA patients are AC degeneration, subchondral bone sclerosis, synovitis, osteophyte formation, joint pain and disability (13). Globally, OA affects 10% - 12% of the adult population and this number is expected to increase rapidly over the next two decades (6). Although many risk factors are known to contribute to OA, including aging, obesity, genetics, joint biomechanics, and previous joint damage, the pathogenesis of OA remains unclear (14, 15). Due to the difficulty of cartilage recovery after damage, at present, there is no other more effective treatment except the use of analgesic drugs and the selection of surgical joint replacement as the final treatment option for OA (16, 17). Considering the huge population affected by OA, it not only affects the quality of life of patients, but also imposes a huge economic burden on society (7). It is crucial to investigate the pathogenesis of OA and explore new therapeutic approaches.
Energy metabolism is an important regulator of cellular function (18). Under adverse conditions, most mammalian cells undergo an energy metabolic transition (i.e., switching from a static regulatory state to an active metabolic state) to maintain energy homeostasis and promote cell survival (19–21). It has been shown that energy metabolism is critical for cartilage homeostasis and is drastically altered in chondrocytes of OA cartilage (16). In addition, recent studies have shown that chondrocytes can undergo metabolic alterations in response to different stimuli (18, 22). Normally, chondrocytes in vivo tend to produce ATP through glycolysis rather than oxidative phosphorylation (OXPHOS) due to the relatively hypoxic environment and maintain a stable ECM by balancing anabolic and catabolic processes (16). However, the TCA cycle and electron transport chain (ETC) efficiently produce ATP in the presence of oxygen through OXPHOS (23). Furthermore, stressed cells, such as immune cells and cancer cells, are capable of generating ATP by aerobic glycolysis when oxygen is available (18). Such cellular metabolic alterations are not only important for the maintenance of energy balance, but probably also critical for alterations in cellular function and signaling (18). Hence, regulation of metabolism may play a vital role in the treatment of OA.
As the main content of energy metabolism, lipid metabolism, amino acid (AA) metabolism and glucose metabolism play a crucial role in the pathogenesis of OA (21). The pathogenesis of OA involves a variety of lipid metabolic changes in cartilage, subchondral bone, and periosteum, which interact with inflammatory mediators to affect the development of lesions (24). Furthermore, some adipokines can directly regulate inflammation and promote the development of OA, thereby affecting joint health (25). Since a variety of AA have been found to be abnormally expressed in chondrocytes, it has also been proposed that the establishment and development of OA are related to the alterations in AA metabolism and profiles, including glutamate- and arginine-family AA as well as their related metabolites (26). Additionally, increasing evidence has shown that glucose metabolism plays a key role in maintaining cartilage homeostasis. Disorders of glucose metabolism can lead to chondrocyte hypertrophy and ECM degradation, contributing to the development of OA (27). It is suggested that targeting enzymes and factors affecting glucose metabolism may be a major breakthrough in the treatment of OA.
At present, many reviews have summarized the role of lipid metabolism and AA metabolism in the pathogenesis of OA in detail. However, no article has specifically targeted a comprehensive summary of the association between glucose metabolism and OA. This narrative review focused on the research literature related to glucose metabolism and OA, aiming to summarize the potential mechanisms of glucose metabolism in the regulation of OA, and provide a reference for further research on the role of glucose metabolism in the treatment of OA and other related diseases. We used “osteoarthritis,” “glucose,” “metabolism,” “mechanism” and “therapeutic” as keywords. Then we searched PubMed, Embase and Web of science for methodological papers on articles from inception to September 2023, especially recently 5 years. Inclusion criteria included: (a) research on OA and its related diseases, (b) the experimental subjects were humans or animals, (c) targeting proteins or factors related to glucose metabolism. Exclusion criteria included: (a) article was not written in English, (b) the full-text was not available or published, (c) conference papers, (d) reviews or meta-analysis. A total of 950 relevant articles were retrieved, and 81 were finally selected for this review.
2 An overview of glucose metabolism
Glucose plays a crucial role in cartilage development, stability, repair, and remodeling (19). Like most cells, chondrocytes use glucose metabolism to produce ATP as their primary energy source, and at the same time, also require glucose as a structural precursor for glycosaminoglycan synthesis (28). There are three major pathways involved in glucose metabolism: glycolysis, pentose phosphate pathway, and tricarboxylic acid cycle (28–30). Among them, glycolysis is tightly regulated as it is required to provide energy for cell survival and specific cellular functions such as inflammation (31). Dysregulation or inhibition of any pathway can lead to lactic acidosis and/or energy deficit, ultimately resulting in abnormal cellular processes that contribute to the formation of several chronic diseases (31). Notably, abnormal glucose metabolism dysregulation has been demonstrated in human cancers as well as abnormal vascular smooth muscle cells, endothelial cells, and human rheumatoid arthritis (RA) chondrocytes, suggesting that glucose metabolism disorders are associated with a variety of diseases (32).
OA is now considered to be a metabolism-related disease, and its establishment and development are closely related to inflammation and metabolism (33). In OA, low-grade inflammation leads to chondrocyte hypoxia, which changes energy metabolism from a resting state to an active state (34). At this time, as the primary energy source for chondrocytes, glycolysis increases to meet the energy demand of chondrocytes (35). Specifically, unlike many tissues, AC has no blood, nerves, or lymphatic vessels, and the synovial fluid in the joint is its source of nutrients (36). Therefore, oxygen and glucose are more difficult to obtain compared to plasma. Its relatively hypoxic environment results in lower energy production by OXPHOS (27). Since glycolysis is a process of rapid ATP generation, chondrocytes are extremely dependent on glycolysis for energy production. However, excessive enhancement of glycolysis also leads to decreased energy acquisition, which inhibits cell proliferation and differentiation. Disturbance of glycolytic metabolism can lead to chondrocyte hypertrophy and ECM degradation (27, 37). Accumulating evidence indicated that glucose metabolism disorders are the cause of OA. Among them, different enzymes and factors play an important role in the process of glucose metabolism, and may also be involved in the pathogenesis of OA. Targeting glucose metabolism is expected to make a major breakthrough in the pathogenesis and treatment of OA. Therefore, in the following few sections, we summarized the potential targets such as related enzymes and factors involved in glucose metabolism in OA as well as therapeutic substances, respectively.
3 Role of glucose metabolism mediated by different enzymes in OA
3.1 Role of GLUT1-mediated glucose metabolism in OA
Since glucose is a large molecule, its diffusion across a membrane is difficult. Hence, in most cell types, glucose is transported across the plasma membrane through facilitated diffusion along a concentration gradient and does not require energy (38, 39). Glucose metabolism in normal AC has been demonstrated to respond to anabolic and catabolic signals by regulating the expression of multiple facilitative glucose transporters of the Glut family, such as glucose transporter type 1 (GLUT1). GLUT1 is up-regulated under hypoxic and glucose-deprived conditions to enhance glucose uptake and decreased under high-glucose conditions to balance glucose levels in chondrocytes (40). Earlier studies in vitro showed that the loss of GLUT1 gene leads to AC cytopenia and proteoglycan loss in mice, indicating that GLUT1-mediated glucose metabolism is required for chondrocyte growth (41). Recent studies have found in mice that genetic defects in chondrocyte GLUT1 during development cause chondrodysplasia, which may result in structural and mechanical abnormalities of AC and lead to OA in adulthood (41–43). In addition, Li et al. (44) revealed that GLUT1 programming changes play an important role in increased AC matrix degradation and susceptibility to OA in female prenatal caffeine exposure (PCE) offspring rats. At the same time, they also established a PCE-induced intrauterine growth retardation (IUGR) model in rats to elucidate the role of altered glucose transport function mediated by altered GC- insulin-like growth factor-1 (IGF1)-GLUT1 axis programming in the development of fetal-originated OA and its epigenetic mechanism, providing a theoretical basis for the early prevention and treatment of OA. Furthermore, using genetic and metabolic pathways, Wang et al. (28) found in mice that GLUT1-mediated glucose metabolism is required for normal growth plate and showed that GLUT1 loss-of-function (LOF) causes GP cartilage and AC abnormalities. The latest evidence found in mouse suggests that forced expression of GLUT1 in AC is able to prevent OA progression. Notably, in vitro studies of human OA chondrocytes, the sustained elevation of GLUT1 expression can increase glucose uptake and produce excess advanced glycation end products (AGEs), which degrade cartilage (45). Moreover, Pfander et al. (46) found in human OA AC that the amount of GLUT1 increased with the severity of OA. A study of human OA chondrocytes reported that although OA chondrocytes were able to adapt to glucose deprivation, high glucose exposure failed to down-regulate GLUT-1 expression. This may be a crucial pathogenic mechanism by which diseases characterized by hyperglycemia promote degenerative changes in chondrocytes, thereby contributing to the progression of OA (47).
Overall, the GLUT1 transport function is essential for glucose uptake in cartilage. Targeting GLUT1 to regulate glucose metabolism may alter chondrocyte maturation processes, including cell proliferation, hypertrophy and cartilage matrix production, suggesting that GLUT1-mediated glucose metabolism plays a key role in cartilage ossification and development. Therefore, GLUT1 may be a potential target for the treatment of OA. However, the exact mechanisms by which glucose metabolism maintains AC health may be multifaceted and warrant further investigation.
3.2 Role of HK2-mediated glucose metabolism in OA
Hexokinases (HKs) catalyze the first step of glucose metabolism, that is, the conversion of glucose to glucose-6-phosphate (G6P) (48). Hexokinase 2 (HK2), an isoform of HK, has a high affinity for glucose compared with other enzymes. Notably, to maintain a concentration gradient, HK2 initiates major glucose utilization pathways such as glycolysis, pentose phosphate pathway, and OXPHOS while promoting glucose entry into the cell (29, 49). Recently, Bustamante et al. (50) explored the role of HK2 in bone and cartilage injury, and found that overexpression of HK2 can lead to increased RNA expression levels of proinflammatory cytokines such as interleukin (IL)-6 and IL-8 in human OA fibroblast-like synoviocytes (FLS, a heterogenous cell population of the synovium that produces certain components of the synovial fluid and AC), suggesting a potential therapeutic effect of HK2 on OA and may be safer than overall glycolysis inhibition (51). Additionally, Wu et al. (31) investigated the energy metabolism of human primary chondrocytes in OA, as well as the role of glycolysis on AC under inflammatory conditions and found that gene expression of HK2 was increased in severe OA cartilage. Although no significant changes in HK2 were found at the overall protein level, an increase in G6P was found at the metabolomic level of cartilage collected from severely injured region, indicating an elevated level of HK2 activity in OA.
In general, HK2 plays an important role in cartilage glycolysis by regulating cellular metabolism, which greatly expands our understanding of glucose metabolism in the pathogenesis of OA. New treatments for OA or related alternative outcomes may be developed in the future, especially small-molecule drugs that target HK2 downstream proteins or kinases.
3.3 Role of LDHA-mediated glucose metabolism in OA
Lactate dehydrogenase A (LDHA), the reduced form of nicotinamide-adenine dinucleotide (NADH), is one of the key enzymes in aerobic glycolysis, which is mainly used to catalyze the last step of glycolysis, i.e. the conversion of pyruvate and finally into lactate via the alternative oxidation of the cofactor, from reduced form of NADH to oxidized form (NAD+) (32, 52). In the normoxic environment, glucose is catalyzed to produce pyruvate, which then enters the mitochondria and undergoes OXPHOS through the TCA cycle. Conversely, under a hypoxic state, high-throughput glycolysis occurs primarily in the cytoplasm via LDHA (32). Previous studies have reported that LDHA is highly expressed in aerobic glycolytic cancer lineages, suggesting that the up-regulation of LDHA can be induced in a hypoxic state (53). In addition, the aberrant expression of LDHA may be an important initiator of RA joints (54, 55). A recent study explored the effect of glycolytic inhibition on hyaluronic acid synthetase 2 (HAS2) synthesis and inflammation development in synovial fibroblasts (SFs) from TMJOA patients and found that the use of a specific inhibitor of LDHA inhibited lactate secretion and promoted HAS2 and hyaluronic acid (HA) synthesis in temporomandibular joint OA (TMJOA) SFs (32). HA is an important matrix component in cartilage. High molecular weight HA can participate in the anti-inflammatory process by forming a protective coating of chondrocytes (56). Interestingly, recent in vivo results in a rat OA model showed that the combination of HA and lactose-modified chitosan (Chitlac; CTL) inhibited cartilage degradation more than HA alone (57). This provides a new therapeutic approach for OA. Furthermore, Arra et al. (18) have shown in studies of mouse and human OA chondrocytes that a metabolic shift in cellular metabolism toward glycolysis and LDHA reprogramming may occur in chondrocytes under inflammatory conditions. Moreover, they also found a pro-reactive oxygen species (ROS) formation function of LDHA in chondrocytes under inflammatory states (18). ROS are considered to be one of the potential factors leading to OA disease due to their ability to modify proteins and lipids, damage DNA and other cellular adverse reactions (58–61). Although ROS play an important signaling function under normal physiological conditions, elevated ROS as a mediator of disease progression in OA is considered to carry significant pathological risks (62). Besides, it has been specifically reported that upregulation of pyruvate kinase M2 (PKM2) and LDHA expression was found in OA chondrocytes compared to healthy controls, indicating an increased activity of glycolysis (18, 63). This suggests a shift from anti-inflammatory dependent OXPHOS and TCA cycle to pro-inflammatory dependent glycolytic energy in OA chondrocytes, identifying LDHA as a potential target for OA therapy.
In summary, LDHA is a key glycolytic enzyme that promotes the conversion of pyruvate to lactate and ROS production in chondrocytes under inflammatory conditions. Since there are many pathways for ROS formation, further studies are needed to prove its source. Previous studies have shown that LDHA plays an important role in the development of RA as well as TMJOA, suggesting the potential of LDHA in the treatment of OA.
3.4 Role of PKM2-mediated glucose metabolism in OA
PKM2 is one of the major rate-limiting enzymes in glycolysis and plays an important role in the regulation of glycolysis by catalyzing phosphoenolpyruvate (PEP) to pyruvate and ATP (63). Yang et al. (63) investigated the involvement of PKM2 in the pathogenesis of OA by exploring its role in glycolysis and collagen matrix production in OA chondrocytes. PKM2 expression has been reported to be upregulated in isolated OA chondrocytes compared to healthy control chondrocytes. In addition, glucose consumption and lactate production were significantly inhibited in the PKM2 knockout group, while GLUT1, LDHA and hypoxia-inducible factor 1α (HIF-1α) were decreased, suggesting that PKM2 knockout inhibited glycolysis in OA chondrocytes. Furthermore, the hydrolysis of cartilage ECM proteins is the main pathogenesis of OA, and PKM2 gene knockout reduces the production of extracellular collagen matrix, which in turn leads to the degeneration of AC in OA. Interestingly, when PKM2 expression is upregulated in OA chondrocytes, lactate accumulates under the catalysis of LDHA, forming an acidic environment that inhibits chondrocyte matrix synthesis and possibly promotes OA cartilage degeneration (64, 65).
In conclusion, PKM2 expression and activity were increased in OA chondrocytes, while PKM2 knockdown inhibited chondrocyte glycolysis and OA cartilage proliferation, suggesting that PKM2 can regulate glycolysis and ECM production, which provides new insights into PKM2 as a potential target for the treatment of OA.
3.5 Role of PFKFB3-mediated glucose metabolism in OA
Fructose 2, 6-diphosphate (F2,6BP) is one of the important regulators of glycolysis and an allosteric activator of phosphofructokinase-1 (PFK-1). F2,6BP is mainly synthesized and degraded by 6-phosphofructose-2-kinase/fructose 2, 6-bisphosphatase (PFKFB1/2/3/4). Among them, PFKFB3 has a high kinase/phosphatase activity, therefore, it is the most widely expressed in the human tissues, and is also the main studied isomerase (66). At present, it has been confirmed that PFKFB3 can affect the physiological activities of various tissues. Interestingly, PFKFB3 expression can regulate glycolysis in tumor cells and cancer cells (67, 68). In addition, inhibition of PFKFB3 was found to be associated with decreased insulin-stimulated glucose uptake, GLUT4 translocation, AKT signaling, and glycolytic flux in cancer cells and adipocytes (69). Based on this, PFKFB3 may play a role in OA cartilage and its dysfunction may participate in the pathogenesis of OA. Data from Qu et al. showed that PFKFB3 expression was decreased in OA cartilage tissue as well as tumor necrosis factor (TNF)-α- or IL-1β-regulated chondrocytes, accompanied by decreased glucose utilization, ATP production, and lactate production, suggesting that PFKFB3 is impaired in OA and contributes to glycolysis disorders (70). Similarly, inhibition of PFKFB3 in FLS cells from synovium of patients with inflammatory arthritis correlated with decreased glucose uptake and lactate production (54, 71). More importantly, PFKFB3 was reported to enhance the cell viability of chondrocytes through the PI3K/AKT/C/EBP homologous protein (CHOP) signaling pathway (70). It is also capable of reducing caspase 3 activation and promoting aggrecan and type II collagen expression in OA cartilage explants and chondrocytes, possessing potential to be a target for OA prevention and treatment.
3.6 Role of AMPK-mediated glucose metabolism in OA
AMP-activated protein kinase (AMPK) is a major regulator of energy metabolism and balance, possessing the potential to be a therapeutic target for metabolic disorders and aging-related diseases, including OA (7, 72, 73). A reduction in AMPK activity was found in both human and mouse knee OA cartilage as assessed by phosphorylation of specific threonines in AMPKα1, the AMPK-activated α subunit, suggesting that AMPK activity in chondrocytes plays an important role in maintaining joint function stability and the development of OA (74). Indeed, Zhou et al. (75) found in a mouse model that using cartilage-specific, tamoxifen-inducible AMPKα knockout mice exhibited accelerated severity of surgically induced OA compared to wild-type mice, and at 12 months, male mice developed severe spontaneous aging-related OA lesions. These results demonstrate that chondrocyte-specific AMPKα depletion in adulthood leads to accelerated OA progression (29, 74). Notably, the activation of AMPK is partially achieved by maintaining glycolysis. Specifically, OA-associated degradation features in AC are a downstream consequence of altered metabolism of resident chondrocytes. These OA features include enhanced production of extracellular proteases such as MMPs. Ohashi et al. (76) found that chondrocytes were more dependent on glycolysis as a source of cellular ATP after il-1β treatment, while only a small fraction was derived from OXPHOS. Blocking IL-1β-enhanced MMP13 levels using galactose substitution (a known activator of OXPHOS and inhibitor of glycolysis) in OA chondrocytes led to a reverse increase in phospho-AMPK-related biomarkers, confirming AMPK as a downstream regulator during OA glycolysis.
Current studies have confirmed the important role of AMPK and aerobic glycolysis in the pathogenesis of OA and provided clues for further exploration.
All in all, the role of glucose metabolism mediated by different enzymes in OA is shown in Table 1 and Figure 1.
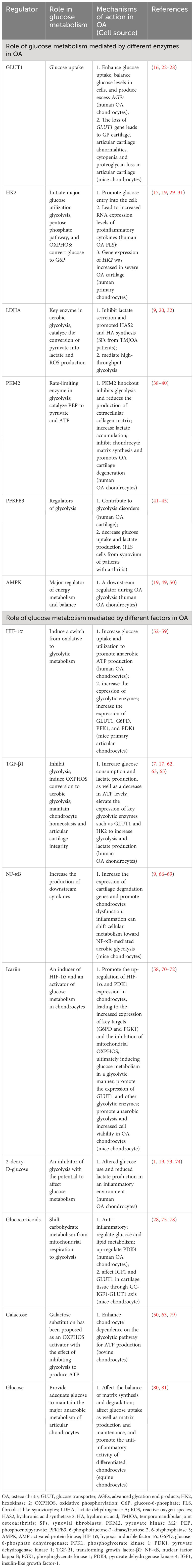
Table 1 The role of targeted regulation of glucose metabolism in OA.
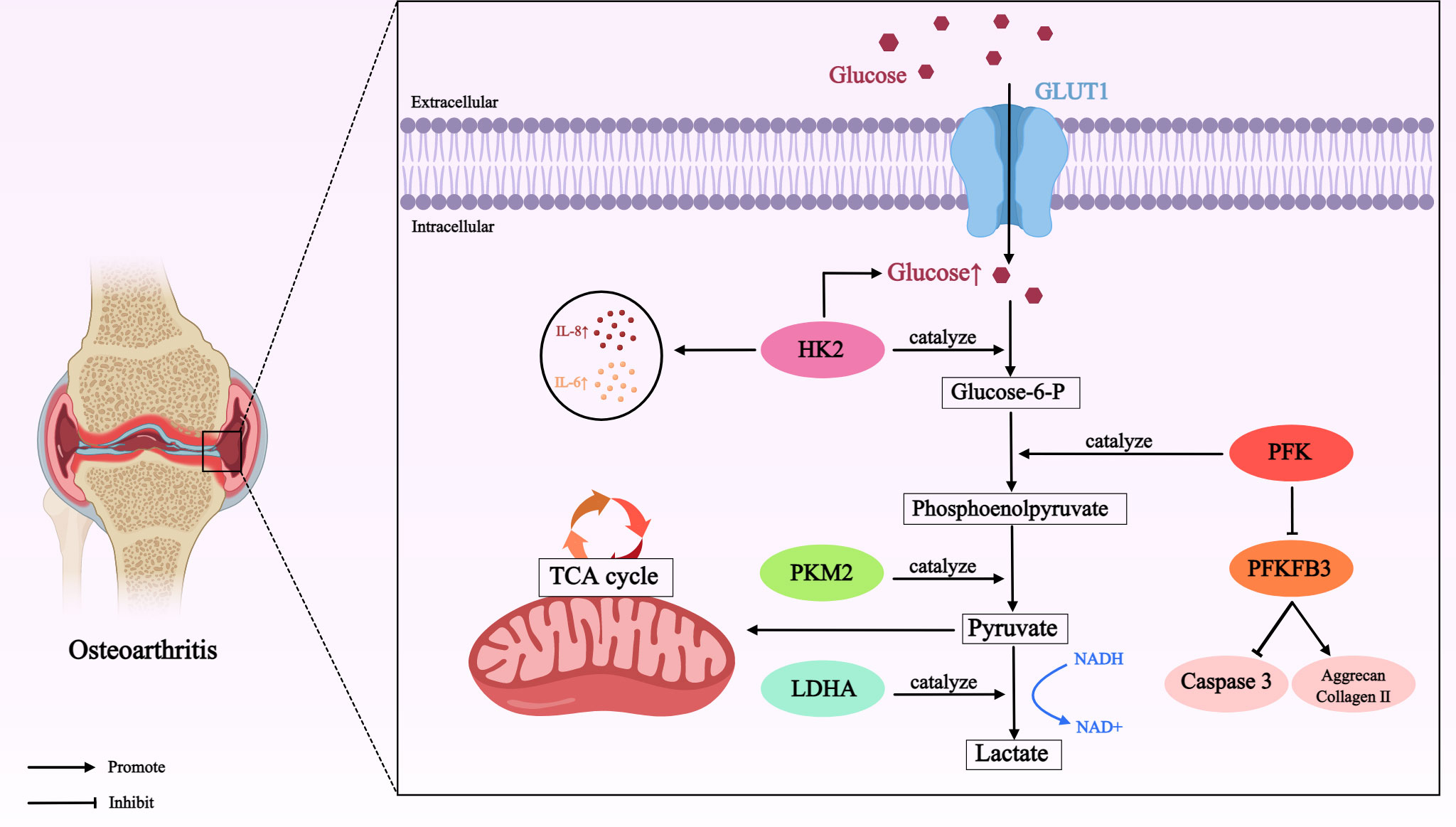
Figure 1 Role of glucose metabolism mediated by different enzymes in OA. In OA cartilage, glucose is transported by GLUT1 (a passive transporter that relies on the concentration gradient as the driving force for the movement of glucose molecules across the membrane) into chondrocytes for metabolism. Many enzymes involved in glucose metabolism, such as HK2, PKM2, LDHA, PFK, PFKFB3, initiate the major pathways of glucose utilization (including glycolysis) in this process, thereby directly or indirectly affecting the development of OA. Glucose transporter type 1 (GLUT1); hexokinase 2 (HK2); interleukin-8 (IL-8); interleukin-6 (IL-6); phosphofructokinase (PFK); 6-phosphofructose-2-kinase/fructose 2, 6-bisphosphatase 3 (PFKFB3); pyruvate kinase M2 (PKM2); lactate dehydrogenase A (LDHA); nicotinamide-adenine dinucleotide (NADH).
4 Role of glucose metabolism mediated by different factors in OA
4.1 Role of HIF-1α-mediated glucose metabolism in OA
HIF-1 is a highly conserved transcription factor and a major regulator of cellular adaptation to hypoxic stimulation (77). The activity of HIF-1 is primarily dependent on its alpha subunit (HIF-1α) (78). Because cartilage is relatively thick, chondrocytes at the surface of the joint are exposed to only about 6% to 10% of oxygen, while the deepest chondrocytes have access to even less oxygen in the cartilage (79). In hypoxic cells, HIF-1α can induce a switch from oxidative to glycolytic metabolism. Moreover, it is of critical importance in maintaining chondrocytes activity and integrity, cartilage energy production, and regulating chondrogenesis, energy metabolism, and matrix synthesis (80, 81). Studies have shown that the treatment of OA can significantly down-regulate the expression of HIF-1α gene, suggesting that HIF-1α plays an important role in the pathological mechanism of OA (82). Yudoh et al. (83) revealed that OA chondrocytes lacking HIF-1α fail to produce energy and maintain ECM growth under both normoxic and hypoxic conditions, suggesting that chondrocytes may regulate ATP levels and ECM production during OA through adaptive functions of the HIF-1α gene. Metabolically, activated OA chondrocytes rely on HIF-1 to increase glucose uptake and utilization to promote anaerobic ATP production, thereby compensating for the accelerated energy expenditure during OA (84). HIF-1α also increases the expression of glycolytic enzymes, such as HIF-1α augments the expression of phosphoglycerate kinase, which converts 1, 3-diphosphoglycerate to 3-phosphoglycerate during glycolysis (85). Besides, under the influence of the complex of HIF-1α, HIF-1β and HREs, the expression of GLUT1, G6P dehydro-genase (G6PD), phosphoglycerate kinase 1 (PGK1) and pyruvate dehydrogenase kinase 1 (PDK1) is increased, which leads to promoting glucose transfer and anaerobic glycolysis (79, 86). Furthermore, vascular endothelial growth factor (VEGF) and erythropoietin (EPO) have been reported to increase oxygen and nutrient delivery, as well as metabolic adaptations, thereby preventing chondrocyte death in the growth plate. Under hypoxia, HIF-1α promotes glycolysis by inducing the expression of both (87). Therefore, further studies on the role of HIF-1α in OA and its potential mechanism may provide a new therapeutic target for the treatment of OA.
4.2 Role of TGF-β1-mediated glucose metabolism in OA
Transforming growth factor (TGF)-β1 is a multifunctional cell regulatory factor, mainly involved in the process of growth and development, proliferation and differentiation, ECM synthesis and immune response (88, 89). Recent studies have indicated that TGF-β1 can inhibit glycolysis by inducing natural regulatory T cells and down-regulating the expression of critical enzymes of glycolysis such as GLUT1 and HK2 (29, 90). TGF-β1 stimulation can promote the glycolytic process in dermal fibroblasts, while inhibition of glycolysis is able to impede TGF-β1-induced profibrosis, suggesting that TGF-β1 plays a dominant role in inducing OXPHOS conversion to aerobic glycolysis (91). Additionally, accumulating evidence supports that the maintenance of AC morphology and the intracellular environment of chondrocytes relies on the regulatory function of TGF-β1 (16, 21). Wang et al. (16) found an increase in glucose consumption and lactate production, as well as a decrease in ATP levels, in OA chondrocytes from humans treated with TGF-β1. Moreover, TGF-β1 stimulation significantly elevated the expression of key glycolytic enzymes such as GLUT 1 and HK2 to increase glycolysis and lactate production. Interestingly, HK1 was also found to be upregulated under TGF-β1 treatment (16). Likewise, Ni et al. detected that TGF-β1 increased HK1 and HK2 expression and simultaneously upregulated HK activity in c-Kit cell lines, suggesting that TGF-β1 has the potential to directly regulate HK and providing important clues for TGF-β1-mediated OA pathogenesis (92). However, the current therapeutic potential of TGF-β1 in OA is still controversial, as it plays both protective and detrimental roles in OA. Therefore, the molecular mechanisms of glucose metabolism in OA targeting TGF-β1 deserve further attention to provide insights into the regulation of energy metabolism and lead to exploring new therapeutic approaches for effective management of OA by modulating cellular metabolism.
4.3 Role of NF-κB -mediated glucose metabolism in OA
Nuclear factor kappa B (NF-κB) is a transcription factor that widely presents in a variety of cell lines and participates in various cellular inflammatory responses (93). Mechanistically, in chondrocytes, NF-κB activation may play an important role in the progression of OA by increasing the expression of cartilage degradation genes and promoting chondrocytes dysfunction (18). An earlier study found that NF-κB activation is involved in regulating cartilage differentiation (94). In addition, Sang et al. (95) found that glycolysis and microenvironment remodeling could be promoted by activating the Ca2+/NF-κB axis to increase the production of downstream cytokines, suggesting that the role played by NF-κB signaling is critical in the regulation of glycolytic activity. In addition, Wang et al. (96) reported a positive correlation between HK2 expression and inflammatory cell infiltration and cartilage destruction in arthritic rats treated with glycolysis inhibitors, implying that HK2 glycolysis inhibitors are closely related to NF-κB activation and inhibition. Furthermore, Arra et al. (18) showed that inflammation can shift cellular metabolism toward aerobic glycolysis mediated by NF-κB, further confirming the strong links between inflammatory and metabolic responses in OA chondrocytes. The above evidence clearly establishes that NF-κB activation can mediate glycolytic process to drive OA pathological changes, but further studies are needed to reveal the precise association and underlying mechanism.
In conclusion, the role of glucose metabolism mediated by different factors in OA is presented in Table 1 and Figure 2.
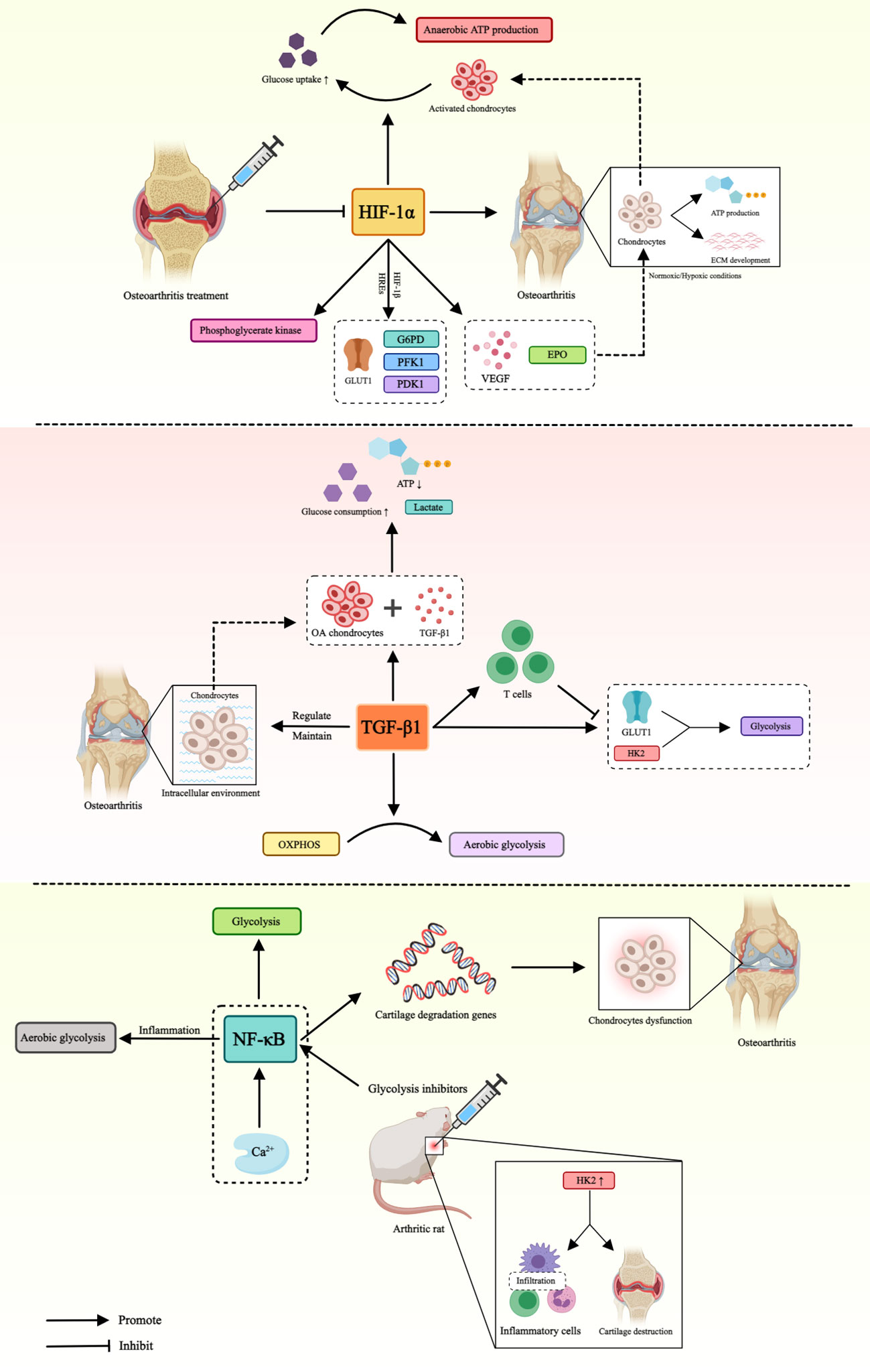
Figure 2 Role of glucose metabolism mediated by different factors in OA. This figure illustrates that HIF-1α, TGF-β and NF-κB affect the pathogenesis of OA by mediating different pathways of glucose metabolism. Hypoxia-inducible factor-1α (HIF-1α), glucose transporter type 1 (GLUT1), hypoxia-inducible factor-β1 (HIF-β1), hypoxia-responsive elements (HREs), glucose-6-phosphate dehydro-genase (G6PD), phosphoglycerate kinase 1 (PFK1), pyruvate dehydrogenase kinase 1 (PDK1), vascular endothelial-derived growth factor (VEGF), erythropoietin (EPO), transforming growth factor-β1 (TGF-β), hexokinase 2 (HK2), oxidative phosphorylation (OXPHOS), nuclear factor kappa B (NF-κB).
5 Potential therapeutic substances for OA that affect glucose metabolism
Targeting glucose metabolism switches, especially glucose transporters and regulatory factors involved in mediating glucose metabolism, is challenging but has potential therapeutic implications in OA. The following substances (summarized in Table 1) play an important role in inhibiting OA development by affecting glucose metabolism.
5.1 Icariin
Icariin (ICA), a flavonoid compound extracted from Herba Epimedii (HEP), is known to be an inducer of HIF-1α and an activator of glucose metabolism in chondrocytes (86, 97). As reported by Wang et al. (86) in chondrocyte experiments in mice, ICA treatment upregulated the expression of SOX9, a key ECM regulator that contributes to the chondrocyte phenotype, suggesting that ICA can promote chondrocyte viability and formation. Specifically, as a glucose carrier and a downstream target of HIF-1α, GLUT1 is activated after ICA treatment. Similarly, ICA treatment also enhanced the expression of PGK1, which is both a critical enzyme in anaerobic glycolysis and a downstream target of HIF-1α (86, 98). PDK1 is capable of limiting oxygen consumption by preventing TCA cycle, thereby promoting anaerobic glycolysis. Additionally, ICA has been shown to up-regulate the expression of G6PD, which is a rate-limiting enzyme in the pentose phosphate pathway and an important regulator of energy expenditure and glucose metabolism. Mechanistically, ICA treatment promoted the up-regulation of HIF-1α and PDK1 expression in chondrocytes, leading to the increased expression of key targets (G6PD and PGK1) and the inhibition of mitochondrial OXPHOS, ultimately inducing glucose metabolism in a glycolytic manner (86). To sum up, ICA treatment promoted the expression of GLUT1 and other glycolytic enzymes, which contributed to anaerobic glycolysis and increased cell viability in OA chondrocytes (99). Therefore, ICA is expected to represent a novel treatment for OA.
5.2 2-deoxy-D-glucose
2-deoxy-D-glucose (2-DG) is widely known as an inhibitor of glycolysis with the potential to affect glucose metabolism (100). Ying et al. (6) demonstrated that glucose metabolism is an inflammatory target for OA progression in vivo by intraperitoneal injection of 2-DG. The results suggest that 2-DG attenuates the development of OA in mice, and the underlying mechanism may be related to the inhibition of subchondral bone sclerosis and cartilage degeneration. Meanwhile, they also found that 2-DG had a protective effect on destabilization of the medial meniscus surgery (DMM)-induced OA mice by inhibiting glycolysis. Previous studies have recommended an effective dose of 2-DG to sufficiently inhibit glycolytic activity in animals, but it remains important to optimize this dose to prevent cartilage damage in OA (101). In addition, Wu et al. (31) found in primary chondrocytes collected from patients undergoing total knee arthroplasty surgery that altered glucose use and reduced lactate production in an inflammatory environment have been proposed as potential mechanisms by which 2-DG inhibits the expression of cartilage catabolic genes by suppressing glycolysis. Terabe et al. (102) also demonstrated that 2DG is a potent blocker of the glycolytic pathway in chondrocytes in IL1β-induced human OA chondrocytes and bovine chondrocytes. Overall, the glycolytic inhibitor 2-DG provides an experimental therapeutic option for OA, but further studies are still needed to observe the potential effects of 2-DG on chondrogenesis and overall anabolic activity.
5.3 Glucocorticoids
Glucocorticoids (GCs) are steroid hormones produced endogenously by the adrenal cortex. In target cells, they bind to glucocorticoid receptors (GR) to form complexes and then migrate into the nucleus (103, 104). Clinically, intra-articular injection of GCs is widely used to relieve various types of inflammation and pain (103, 105). There are two potential mechanisms underlying the anti-inflammatory effects: 1) enhancing the expression of anti-inflammatory genes through the binding of GR-steroid complexes to glucocorticoid response elements (GREs); 2) inhibiting the activity of inflammatory transcription factors such as NF-κB and activator protein 1 (AP-1) (103). More importantly, GCs are also recommended for the treatment of knee OA. In OA chondrocytes, in addition to their anti-inflammatory effects, GCs also partially mediates their effects on OA cartilage by regulating glucose and lipid metabolism. Interestingly, pyruvate dehydrogenase kinase 4 (PDK4) was one of the most significantly up-regulated genes by dexamethasone (a glucocorticoid). Since this gene is able to inhibit pyruvate dehydrogenase activity and prevent the progression of pyruvate produced by glycolysis to OXPHOS, the role of GCs may be to shift carbohydrate metabolism from mitochondrial respiration to glycolysis (106). Furthermore, Li et al. (44) reported that GCs could affect IGF1 and GLUT1 in cartilage tissue through the GC-IGF1-GLUT1 axis. In detail, IGF1 can promote the up-regulation of GLUT1 expression, leading to the accumulation of AGEs in chondrocytes, thereby promoting the inflammation and matrix degradation of AC, and ultimately inducing OA. Therefore, future studies to further elucidate the role of GCs in glucose metabolism associated with OA pathophysiology may be an interesting avenue to investigate.
5.4 Galactose
Galactose substitution has been proposed as an OXPHOS activator with the effect of inhibiting glycolysis to produce ATP (91). In addition, galactose substitution is a common method to study the downstream effects of metabolic function in cancer or normally proliferating cells. Typically, G6P converted from galactose is involved in the glycolytic pathway, and the conversion rate of galactose to G6P is slower than that of glucose (107). Thus, pyruvate produced through the glycolytic metabolism of galactose produces less ATP, forcing the cell to increase its dependence on ATP produced by OXPHOS. Ohashi et al. (76) observed in bovine and human primary articular chondrocytes a strong dependence of chondrocytes on ATP produced by the glycolytic pathway in glucose-rich medium, which became more prominent upon stimulation with IL-1β and, in turn, echoed the absence of ATP produced by OXPHOS. In contrast, chondrocytes in galactose-substituted medium showed a significant reduction in glycolysis and an increase in ATP produced by mitochondrial activity due to the enhanced overall mitochondrial function, suggesting that repairing mitochondrial dysfunction may be one of the effective strategies for the treatment of OA by targeting catabolism. Similarly, Lane et al. (108) found significant reductions in lactate production and maximal lactate dehydrogenase activity in healthy bovine primary chondrocytes with increasing time of galactose culture, suggesting inhibition of chondrocyte glycolysis and decreased dependence of cellular ATP production on the glycolytic pathway, confirming the ability of galactose culture to alter chondrocyte metabolism.
5.5 Glucose
Glucose plays an essential role in the maintenance of chondrocyte metabolism, and adequate glucose supply is required to maintain the major anaerobic metabolism of articular chondrocytes. In addition, glucose is also a key precursor for the synthesis of ECM macromolecules by these cells (109). Therefore, inflammatory diseases such as OA may have a higher glucose requirement, but the demand for chondrocytes is limited due to the avascular nature of cartilage tissue. It should be emphasized that limited glucose supply carries the risk of impairing cellular function and may lead to an imbalance in matrix synthesis and degradation, resulting in OA (56). Of interest, Sopasakis et al. (56) investigated the pivotal role of glucose as a fuel source for matrix generation and cartilage repair in conditions of OA using differentiated equine chondrocytes pellets in a 3D model in vitro. The results showed that in the presence of inflammatory conditions associated with OA, heightened glucose levels not only facilitate glucose uptake, HA synthesis and enhance matrix production and maintenance, but also promote the anti-inflammatory activity of differentiated chondrocytes. This indicates that sufficient glucose supply to chondrocytes promotes cartilage repair by interrupting the harmful inflammatory cycle and inducing the synthesis of HA, suggesting that intra-articular injection of glucose is beneficial to the early treatment of OA, and its combination with other therapies is expected to improve the therapeutic effect by regulating chondrocytes metabolism and inducing anti-inflammatory processes. Notably, a large number of studies have shown that both diabetes mellitus and type 2 diabetes mellitus are significantly associated with OA, and diabetic patients have a higher risk of OA, suggesting that hyperglycemia may induce or aggravate OA (110, 111). Li et al. (112) demonstrated in high glucose-stimulated rat FLS in the in vitro cocultivation system that hyperglycemia leads to the accumulation of AGEs in FLS through the HIF-1α-GLUT1 pathway, thereby increasing the release of FLS inflammatory factors, which in turn induces chondrocyte degradation and promotes OA development. Of note, after being activated by AGEs, the receptor of AGEs (RAGE) can trigger downstream inflammatory signals to participate in a variety of degenerative diseases. Therefore, RAGE inhibitors are expected to block the proinflammatory effects of high glucose.
Prolotherapy is a safe nonsurgical regenerative injection technique that stimulates the production of growth factors and cytokines, and promotes growth of normal cell and tissue by injecting small amounts of a stimulating solution (including hypertonic dextrose, morrhuate sodium, dextrose/phenol/glycerin solution, or platelet-rich plasma) into painful and degenerative joints, ligaments, and adjacent joint spaces, leading to the regeneration of previously damaged ligaments, tendons, and other intra-articular structures (113). A previous systematic review suggests that dextrose prolotherapy may benefit patients with knee OA by promoting tissue repair and improving range of motion and joint stability. OA patients treated with dextrose showed significant improvement compared to those receiving saline injection and normal treatment (114). In addition, another systematic review including 10 articles indicated that dextrose prolotherapy has been shown to have better therapeutic effects on pain in patients with OA compared to exercise, and that it has similar effects to platelet-rich plasma and steroid injections (115). However, although the latest systematic review showed that dextrose prolotherapy is beneficial in reducing pain and improving function in patients with knee OA, there is a high risk of bias and heterogeneity in the results (116, 117). While individuals may experience short-term relief, questions remain about the long-term durability of the treatment. This uncertainty can make it challenging to assess the suitability of treatment for sustained management of knee OA. Additionally, the dextrose concentration/volume, interval and duration of treatment, as well as injection site and technique may differ. The absence of standardized treatment protocols for dextrose prolotherapy contributes to variability in how the procedure is performed. Therefore, despite the promising results, there is still a need for larger clinical trials with a standardized treatment regimen and long-term findings to show improvement in the quality of life consistently.
6 Conclusions
In conclusion, improving the glucose metabolism is conducive to treating OA, mainly through regulating the glycolysis. Due to the hypoxic environment, chondrocytes are highly dependent on glycolysis, which is the main source of energy for chondrocytes. Thus, in OA chondrocytes, the regulation of glucose transport and glycolytic pathways is thought to influence the pathogenesis of the disease, and the process involves the responses of multiple enzymes. Among them, GLUT1, HK2, LDHA, PKM2, PFKFB3, and AMPK are closely related to cell growth, proliferation, metabolism, and apoptosis, and play a vital role in the development of OA mainly by affecting the glycolytic process. In addition, many studies have reported key factors in the process of glucose metabolism, suggesting their involvement in the pathogenesis of OA. This review emphasizes the potential mechanisms of HIF-1α, TGF-β1, NF-κB and their regulation pathway in energy metabolism and progression of OA. They are also important targets for the regulation of glycolysis. More importantly, ICA, 2-DG, GC, galactose and glucose have been shown to inhibit OA by regulating glucose metabolism, which provides new insights into the treatment of OA. However, different administration methods, concentrations and dosages still need to be further explored to ensure the best results.
7 Future perspectives
Energy metabolism is central to the maintenance of cartilage function. Accumulating studies are interested in the role of glucose metabolism in the pathogenesis and therapeutic mechanisms of OA. In an adverse microenvironment, energy conversion to glycolysis-dominated glucose metabolism is critical for immune response and inflammatory pathway activation during OA progression. From a translation perspective, the pathogenesis of OA is considered to be related to differences in glucose utilization between normal and diseased chondrocytes (16, 31). Targeting glucose transporters and rate-limiting enzymes during glucose metabolism may be an important strategy for the treatment of OA. However, future studies are needed to further confirm the comprehensive details of the specific regulatory mechanisms and signaling pathways. In addition, studies on the key factors affecting glucose metabolism such as HIF-1α, TGF-β1, NF-κB, and their mediated signaling pathways are expected to further reveal the potential mechanisms regulating energy metabolism in OA, which will contribute to developing small molecule drugs targeting the downstream proteins or kinases. Furthermore, metabolomics techniques have suggested some potential metabolic pathway alterations in OA. Future research of OA should focus on specific phenotypes and related molecular pathways. The disease-related alterations of specific components and regulatory molecules in glucose metabolism pathways such as glycolysis, TCA cycle, and their interactions in OA progression warrant further investigation. From a therapeutic perspective, it will also be critical to explore the potential of glucose metabolism shifts in current effective OA therapies, such as physical exercise and weight loss, and the response to experimental drugs that affect cellular glucose metabolism. Similarly, immunometabolism should also be used as a key means of understanding OA pathophysiology in the future.
Author contributions
PP: Writing – original draft, Writing – review & editing. LZ: Writing – original draft, Writing – review & editing. ZZ: Writing – review & editing. KZ: Writing – review & editing. BH: Writing – review & editing. XB: Writing – review & editing. YW: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the fundamental research funds for the central universities (The laboratory of exercises rehabilitation science).
Acknowledgments
We would like to thank the fundamental research funds for the central universities (The laboratory of exercises rehabilitation science) for supporting our research. Illustrations were created by image.medpeer.cn.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Patel JM, Wise BC, Bonnevie ED, Mauck RL. Systematic review and guide to mechanical testing for articular cartilage issue engineering. Tissue Eng Part C Methods. (2019) 25:593–608. doi: 10.1089/ten.TEC.2019.0116
2. Pettenuzzo S, Arduino A, Belluzzi E, Pozzuoli A, Fontanella CG, Ruggieri P, et al. Biomechanics of chondrocytes and chondrons in healthy conditions and osteoarthritis: A review of the mechanical characterisations at the microscale. Biomedicines. (2023) 11:1942. doi: 10.3390/biomedicines11071942
3. Guilak F, Alexopoulos LG, Upton ML, Youn I, Choi JB, Cao L, et al. The pericellular matrix as a transducer of biomechanical and biochemical signals n articular cartilage. Ann N Y Acad Sci. (2006) 1068:498–512. doi: 10.1196/annals.1346.011
4. Griffin TM, Guilak F. The role of mechanical loading in the onset and progression of osteoarthritis. Exerc Sport Sci Rev. (2005) 33:195–200. doi: 10.1097/00003677-200510000-00008
5. Grodzinsky AJ, Levenston ME, Jin M, Frank EH. Cartilage tissue remodeling in response to mechanical forces. Annu Rev BioMed Eng. (2000) . 2:691–713. doi: 10.1146/annurev.bioeng.2.1.691
6. Ying J, Wang P, Shi Z, Xu J, Ge Q, Sun Q, et al. Inflammation-mediated aberrant glucose metabolism in subchondral bone induces osteoarthritis. Stem Cells. (2023) 41:482–92. doi: 10.1093/stmcls/sxad012
7. Lin Z, Miao J, Zhang T, He M, Zhou X, Zhang H, et al. d-Mannose suppresses osteoarthritis development in vivo and delays IL-1beta-induced degeneration in vitro by enhancing autophagy activated via the AMPK pathway. BioMed Pharmacother. (2021) 135:111199. doi: 10.1016/j.biopha.2020.111199
8. Yao Q, Wu X, Tao C, Gong W, Chen M, Qu M, et al. Osteoarthritis: pathogenic signaling pathways and therapeutic targets. Signal Transduct Target Ther. (2023) 8:56. doi: 10.1038/s41392-023-01330-w
9. Fu W, Chen S, Yang R, Li C, Gao H, Li J, et al. Cellular features of localized microenvironments in human meniscal degeneration: a single-cell transcriptomic study. Elife. (2022) 11:e79585. doi: 10.7554/eLife.79585
10. Molnar V, Matisic V, Kodvanj I, Bjelica R, Jelec Z, Hudetz D, et al. Cytokines and chemokines involved in osteoarthritis pathogenesis. Int J Mol Sci. (2021) 22:9208. doi: 10.3390/ijms22179208
11. Hunter DJ, Bierma-Zeinstra S. Osteoarthritis. Lancet. (2019) 393:1745–59. doi: 10.1016/S0140-6736(19)30417-9
12. Dai B, Zhu Y, Li X, Liang Z, Xu S, Zhang S, et al. Blockage of osteopontin-integrin beta3 signaling in infrapatellar fat pad attenuates osteoarthritis in Mice. Adv Sci (Weinh). (2023) 10:e2300897. doi: 10.1002/advs.202300897
13. Felson DT. Clinical practice. Osteoarthritis of the knee. N Engl J Med. (2006) 354:841–8. doi: 10.1056/NEJMcp051726
14. Blüher M. Metabolically healthy obesity. Endocr Rev. (2020) 41:bnaa004. doi: 10.1210/endrev/bnaa004
15. Palazzo C, Nguyen C, Lefevre-Colau MM, Rannou F, Poiraudeau S. Risk factors and burden of osteoarthritis. Ann Phys Rehabil Med. (2016) 59:134–8. doi: 10.1016/j.rehab.2016.01.006
16. Wang C, Silverman RM, Shen J, O'Keefe RJ. Distinct metabolic programs induced by TGF-beta1 and BMP2 in human articular chondrocytes with osteoarthritis. J Orthop Translat. (2018) 12:66–73. doi: 10.1016/j.jot.2017.12.004
17. Katz JN, Arant KR, Loeser RF. Diagnosis and treatment of hip and knee osteoarthritis: A review. JAMA. (2021) 325:568–78. doi: 10.1001/jama.2020.22171
18. Arra M, Swarnkar G, Ke K, Otero JE, Ying J, Duan X, et al. LDHA-mediated ROS generation in chondrocytes is a potential therapeutic target for osteoarthritis. Nat Commun. (2020) 11:3427. doi: 10.1038/s41467-020-17242-0
19. Kong P, Chen R, Zou FQ, Wang Y, Liu MC, Wang WG. HIF-1alpha repairs degenerative chondrocyte glycolytic metabolism by the transcriptional regulation of Runx2. Eur Rev Med Pharmacol Sci. (2021) 25:1206–14. doi: 10.26355/eurrev_202102_24823
20. Rabinowitz JD, Enerbäck S. Lactate: the ugly duckling of energy metabolism. Nat Metab. (2020) 2:566–71. doi: 10.1038/s42255-020-0243-4
21. Mobasheri A, Rayman MP, Gualillo O, Sellam J, van der Kraan P, Fearon U. The role of metabolism in the pathogenesis of osteoarthritis. Nat Rev Rheumatol. (2017) 13:302–11. doi: 10.1038/nrrheum.2017.50
22. Lu S, Wang Y. Nonmetabolic functions of metabolic enzymes in cancer development. Cancer Commun (Lond). (2018) 38:63. doi: 10.1186/s40880-018-0336-6
23. Vander HMG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
24. Su Z, Zong Z, Deng J, Huang J, Liu G, Wei B, et al. Lipid metabolism in cartilage development, degeneration, and regeneration. Nutrients. (2022) 14:3984. doi: 10.3390/nu14193984
25. Wang T, He C. Pro-inflammatory cytokines: The link between obesity and osteoarthritis. Cytokine Growth Factor Rev. (2018) 44:38–50. doi: 10.1016/j.cytogfr.2018.10.002
26. Li Y, Xiao W, Luo W, Zeng C, Deng Z, Ren W, et al. Alterations of amino acid metabolism in osteoarthritis: its implications for nutrition and health. Amino Acids. (2016) 48:907–14. doi: 10.1007/s00726-015-2168-x
27. Tan C, Li L, Han J, Xu K, Liu X. A new strategy for osteoarthritis therapy: Inhibition of glycolysis. Front Pharmacol. (2022) 13:1057229. doi: 10.3389/fphar.2022.1057229
28. Wang C, Ying J, Niu X, Li X, Patti GJ, Shen J, et al. Deletion of Glut1 in early postnatal cartilage reprograms chondrocytes toward enhanced glutamine oxidation. Bone Res. (2021) 9:38. doi: 10.1038/s41413-021-00153-1
29. Bao C, Zhu S, Song K, He C. HK2: a potential regulator of osteoarthritis via glycolytic and non-glycolytic pathways. Cell Commun Signal. (2022) 20:132. doi: 10.1186/s12964-022-00943-y
30. Jiang D, Guo J, Liu Y, Li W, Lu D. Glycolysis: an emerging regulator of osteoarthritis. Front Immunol. (2023) 14:1327852. doi: 10.3389/fimmu.2023.1327852
31. Wu X, Liyanage C, Plan M, Stark T, McCubbin T, Barrero RA, et al. Dysregulated energy metabolism impairs chondrocyte function in osteoarthritis. Osteoarthritis Cartilage. (2023) 31:613–26. doi: 10.1016/j.joca.2022.11.004
32. Li HM, Guo HL, Xu C, Liu L, Hu SY, Hu ZH, et al. Inhibition of glycolysis by targeting lactate dehydrogenase A facilitates hyaluronan synthase 2 synthesis in synovial fibroblasts of temporomandibular joint osteoarthritis. Bone. (2020) 141:115584. doi: 10.1016/j.bone.2020.115584
33. Berenbaum F, Griffin TM, Liu-Bryan R. Review: Metabolic regulation of inflammation in osteoarthritis. Arthritis Rheu-matol. (2017) 69:9–21. doi: 10.1002/art.39842
34. Kim H, Kang D, Cho Y, Kim JH. Epigenetic regulation of chondrocyte catabolism and anabolism in osteoarthritis. Mol Cells. (2015) 38:677–84. doi: 10.14348/molcells.2015.0200
35. Li X, Sun J, Xu Q, Duan W, Yang L, Wu X, et al. Oxymatrine inhibits colorectal cancer metastasis via attenuating PKM2-Mediated aerobic glycolysis. Cancer Manag Res. (2020) 12:9503–13. doi: 10.2147/CMAR.S267686
36. Mobasheri A, Bondy CA, Moley K, Mendes AF, Rosa SC, Richardson SM, et al. Facilitative glucose transporters in articular chondrocytes. Expression, distribution and functional regulation of GLUT isoforms by hypoxia, hypoxia mimetics, growth factors and pro-inflammatory cytokines. Adv Anat Embryol Cell Biol. (2008) 200:1–84.
37. Kudelko M, Chan CW, Sharma R, Yao Q, Lau E, Chu IK, et al. Label-free quantitative proteomics reveals survival mechanisms developed by hypertrophic chondrocytes under ER stress. J Proteome Res. (2016) 15:86–99. doi: 10.1021/acs.jproteome.5b00537
38. Lee WC, Guntur AR, Long F, Rosen CJ. Energy metabolism of the osteoblast: Implications for osteoporosis. Endocr Rev. (2017) 38:255–66. doi: 10.1210/er.2017-00064
39. Lizak B, Szarka A, Kim Y, Choi KS, Nemeth CE, Marcolongo P, et al. Glucose transport and transporters in the endomembranes. Int J Mol Sci. (2019) 20:5898. doi: 10.3390/ijms20235898
40. Li K, Ji X, Seeley R, Lee WC, Shi Y, Song F, et al. Impaired glucose metabolism underlies articular cartilage degeneration in osteoarthritis. FASEB J. (2022) 36:e22377. doi: 10.1096/fj.202200485R
41. Lee SY, Abel ED, Long F. Glucose metabolism induced by Bmp signaling is essential for murine skeletal development. Nat Commun. (2018) 9:4831. doi: 10.1038/s41467-018-07316-5
42. Aigner T, Richter W. OA in 2011: Age-related OA–a concept emerging from infancy? Nat Rev Rheumatol. (2012) 8:70–2. doi: 10.1038/nrrheum.2011.206
43. Pitsillides AA, Beier F. Cartilage biology in osteoarthritis–lessons from developmental biology. Nat Rev Rheumatol. (2011) 7:654–63. doi: 10.1038/nrrheum.2011.129
44. Qing XL, Lin LW, Yi ZW, Liang L, Hui H, Liao BC. Programming changes in GLUT1 mediated the accumulation of AGEs and matrix degradation in the articular cartilage of female adult rats after prenatal caffeine exposure. Pharmacol Res. (2020) 151:104555. doi: 10.1016/j.phrs.2019.104555
45. Rasheed Z, Akhtar N, Haqqi TM. Advanced glycation end products induce the expression of interleukin-6 and inter-leukin-8 by receptor for advanced glycation end product-mediated activation of mitogen-activated protein kinases and nuclear factor-kappaB in human osteoarthritis chondrocytes. Rheumatol (Oxford). (2011) 50:838–51. doi: 10.1093/rheumatology/keq380
46. Pfander D, Cramer T, Swoboda B. Hypoxia and HIF-1alpha in osteoarthritis. Int Orthop. (2005) 29:6–9. doi: 10.1007/s00264-004-0618-2
47. Rosa SC, Goncalves J, Judas F, Mobasheri A, Lopes C, Mendes AF. Impaired glucose transporter-1 degradation and increased glucose transport and oxidative stress in response to high glucose in chondrocytes from osteoarthritic versus normal human cartilage. Arthritis Res Ther. (2009) 11:R80. doi: 10.1186/ar2713
48. Fang J, Luo S, Lu Z. HK2: Gatekeeping microglial activity by tuning glucose metabolism and mitochondrial functions. Mol Cell. (2023) 83:829–31. doi: 10.1016/j.molcel.2023.02.022
49. Lee HJ, Li CF, Ruan D, He J, Montal ED, Lorenz S, et al. Non-proteolytic ubiquitination of Hexokinase 2 by HectH9 controls tumor metabolism and cancer stem cell expansion. Nat Commun. (2019) 10:2625. doi: 10.1038/s41467-019-10374-y
50. Bustamante MF, Oliveira PG, Garcia-Carbonell R, Croft AP, Smith JM, Serrano RL, et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann Rheum Dis. (2018) 77:1636–43. doi: 10.1136/annrheumdis-2018-213103
51. Maglaviceanu A, Wu B, Kapoor M. Fibroblast-like synoviocytes: Role in synovial fibrosis associated with osteoarthritis. Wound Repair Regener. (2021) 29:642–9. doi: 10.1111/wrr.12939
52. Sharma D, Singh M, Rani R. Role of LDH in tumor glycolysis: Regulation of LDHA by small molecules for cancer therapeutics. Semin Cancer Biol. (2022) 87:184–95. doi: 10.1016/j.semcancer.2022.11.007
53. Pucino V, Bombardieri M, Pitzalis C, Mauro C. Lactate at the crossroads of metabolism, inflammation, and autoimmunity. Eur J Immunol. (2017) 47:14–21. doi: 10.1002/eji.201646477
54. Biniecka M, Canavan M, McGarry T, Gao W, McCormick J, Cregan S, et al. Dysregulated bioenergetics: a key regulator of joint inflammation. Ann Rheum Dis. (2016) 75:2192–200. doi: 10.1136/annrheumdis-2015-208476
55. Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical role of glucose metabolism in rheumatoid arthritis fibroblast-like synoviocytes. Arthritis Rheumatol. (2016) 68:1614–26. doi: 10.1002/art.39608
56. Rotter SV, Wickelgren R, Sukonina V, Brantsing C, Svala E, Hansson E, et al. Elevated glucose levels preserve glucose uptake, hyaluronan production, and low glutamate release following interleukin-1β stimulation of differentiated chondrocytes. Cartilage. (2019) 10:491–503. doi: 10.1177/1947603518770256
57. Tarricone E, Elia R, Mattiuzzo E, Faggian A, Pozzuoli A, Ruggieri P, et al. The viability and anti-inflammatory effects of hyaluronic acid-chitlac-tracimolone acetonide- beta-cyclodextrin complex on human chondrocytes. Cartilage. (2021) 13:920S–4S. doi: 10.1177/1947603520908658
58. Davies CM, Guilak F, Weinberg JB, Fermor B. Reactive nitrogen and oxygen species in interleukin-1-mediated DNA damage associated with osteoarthritis. Osteoarthritis Cartilage. (2008) 16:624–30. doi: 10.1016/j.joca.2007.09.012
59. Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med. (2019) 132:73–82. doi: 10.1016/j.freeradbiomed.2018.08.038
60. Drevet S, Gavazzi G, Grange L, Dupuy C, Lardy B. Reactive oxygen species and NADPH oxidase 4 involvement in osteoarthritis. Exp Gerontol. (2018) 111:107–17. doi: 10.1016/j.exger.2018.07.007
61. Altindag O, Erel O, Aksoy N, Selek S, Celik H, Karaoglanoglu M. Increased oxidative stress and its relation with collagen metabolism in knee osteoarthritis. Rheumatol Int. (2007) 27:339–44. doi: 10.1007/s00296-006-0247-8
62. Lepetsos P, Papavassiliou KA, Papavassiliou AG. Redox and NF-kappaB signaling in osteoarthritis. Free Radic Biol Med. (2019) 132:90–100. doi: 10.1016/j.freeradbiomed.2018.09.025
63. Yang X, Chen W, Zhao X, Chen L, Li W, Ran J, et al. Pyruvate kinase M2 modulates the glycolysis of chondrocyte and extracellular matrix in osteoarthritis. DNA Cell Biol. (2018) 37:271–7. doi: 10.1089/dna.2017.4048
64. High RA, Ji Y, Ma YJ, Tang Q, Murphy ME, Du J, et al. In vivo assessment of extracellular pH of joint tissues using acidoCESTUTE MRI. Quant Imaging Med Surg. (2019) 9:1664–73. doi: 10.21037/qims.2019.08.11
65. Wilkins RJ, Hall AC. Control of matrix synthesis in isolated bovine chondrocytes by extracellular and intracellular pH. J Cell Physiol. (1995) 164:474–81. doi: 10.1002/jcp.1041640305
66. Eelen G, Cruys B, Welti J, de Bock K, Carmeliet P. Control of vessel sprouting by genetic and metabolic determinants. Trends Endocrinol Metab. (2013) 24:589–96. doi: 10.1016/j.tem.2013.08.006
67. Liu J, Liu ZX, Wu QN, Lu YX, Wong CW, Miao L, et al. Long noncoding RNA AGPG regulates PFKFB3-mediated tumor glycolytic reprogramming. Nat Commun. (2020) 11:1507. doi: 10.1038/s41467-020-15112-3
68. Shi L, Pan H, Liu Z, Xie J, Han W. Roles of PFKFB3 in cancer. Signal Transduct Target Ther. (2017) 2:17044. doi: 10.1038/sigtrans.2017.44
69. Trefely S, Khoo PS, Krycer JR, Chaudhuri R, Fazakerley DJ, Parker BL, et al. Kinome screen identifies PFKFB3 and glucose metabolism as important regulators of the insulin/insulin-like growth factor (IGF)-1 signaling pathway. J Biol Chem. (2015) 290:25834–46. doi: 10.1074/jbc.M115.658815
70. Qu J, Lu D, Guo H, Miao W, Wu G, Zhou M. PFKFB3 modulates glycolytic metabolism and alleviates endoplasmic reticulum stress in human osteoarthritis cartilage. Clin Exp Pharmacol Physiol. (2016) 43:312–8. doi: 10.1111/1440-1681.12537
71. Fearon U, Hanlon MM, Wade SM, Fletcher JM. Altered metabolic pathways regulate synovial inflammation in rheumatoid arthritis. Clin Exp Immunol. (2019) 197:170–80. doi: 10.1111/cei.13228
72. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
73. Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab. (2018) 27:299–313. doi: 10.1016/j.cmet.2017.10.009
74. Li J, Zhang B, Liu WX, Lu K, Pan H, Wang T, et al. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann Rheum Dis. (2020) 79:635–45. doi: 10.1136/annrheumdis-2019-216713
75. Zhou S, Lu W, Chen L, Ge Q, Chen D, Xu Z, et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep. (2017) 7:43245. doi: 10.1038/srep43245
76. Ohashi Y, Takahashi N, Terabe K, Tsuchiya S, Kojima T, Knudson CB, et al. Metabolic reprogramming in chondrocytes to promote mitochondrial respiration reduces downstream features of osteoarthritis. Sci Rep. (2021) 11:15131. doi: 10.1038/s41598-021-94611-9
77. Cowman SJ, Koh MY. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer. (2022) 8:28–42. doi: 10.1016/j.trecan.2021.10.004
78. Xu D, Li C. Regulation of the SIAH2-HIF-1 axis by protein kinases and its implication in cancer therapy. Front Cell Dev Biol. (2021) 9:646687. doi: 10.3389/fcell.2021.646687
79. Zeng CY, Wang XF, Hua FZ. HIF-1alpha in osteoarthritis: from pathogenesis to therapeutic implications. Front Pharmacol. (2022) 13:927126. doi: 10.3389/fphar.2022.927126
80. Schipani E, Ryan HE, Didrickson S, Kobayashi T, Knight M, Johnson RS. Hypoxia in cartilage: HIF-1alpha is essential for chondrocyte growth arrest and survival. Genes Dev. (2001) 15:2865–76. doi: 10.1101/gad.934301
81. Stegen S, Laperre K, Eelen G, Rinaldi G, Fraisl P, Torrekens S, et al. HIF-1α metabolically controls collagen synthesis and modification in chondrocytes. Nature. (2019) 565:511–5. doi: 10.1038/s41586-019-0874-3
82. Malkov MI, Lee CT, Taylor CT. Regulation of the hypoxia-inducible factor (HIF) by pro-inflammatory cytokines. Cells. (2021) 10:2340. doi: 10.3390/cells10092340
83. Yudoh K, Nakamura H, Masuko-Hongo K, Kato T, Nishioka K. Catabolic stress induces expression of hypoxia-inducible factor (HIF)-1 alpha in articular chondrocytes: involvement of HIF-1 alpha in the pathogenesis of osteoarthritis. Arthritis Res Ther. (2005) 7:R904–14. doi: 10.1186/ar1765
84. Pfander D, Swoboda B, Cramer T. The role of HIF-1alpha in maintaining cartilage homeostasis and during the pathogenesis of osteoarthritis. Arthritis Res Ther. (2006) 8:104. doi: 10.1186/ar1894
85. Semenza GL, Roth PH, Fang HM, Wang GL. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J Biol Chem. (1994) 269:23757–63. doi: 10.1016/S0021-9258(17)31580-6
86. Wang P, Xiong X, Zhang J, Qin S, Wang W, Liu Z. Icariin increases chondrocyte vitality by promoting hypoxia-inducible factor-1alpha expression and anaerobic glycolysis. Knee. (2020) 27:18–25. doi: 10.1016/j.knee.2019.09.012
87. Stegen S, Carmeliet G. Hypoxia, hypoxia-inducible transcription factors and oxygen-sensing prolyl hydroxylases in bone development and homeostasis. Curr Opin Nephrol Hypertens. (2019) 28:328–35. doi: 10.1097/MNH.0000000000000508
88. Wang J, Xiang H, Lu Y, Wu T. Role and clinical significance of TGF−β1 and TGF−βR1 in Malignant tumors (Review). Int J Mol Med. (2021) 47:. doi: 10.3892/ijmm.2021.4888
89. Moreau JM, Velegraki M, Bolyard C, Rosenblum MD, Li Z. Transforming growth factor-β1 in regulatory T cell biology. Sci Immunol. (2022) 7:eabi4613. doi: 10.1126/sciimmunol.abi4613
90. Jeong JH, Jang HJ, Kwak S, Sung GJ, Park SH, Song JH, et al. Novel TGF-beta1 inhibitor antagonizes TGF-beta1-induced epithelial-mesenchymal transition in human A549 lung cancer cells. J Cell Biochem. (2019) 120:977–87. doi: 10.1002/jcb.27460
91. Henderson J, Duffy L, Stratton R, Ford D, O'Reilly S. Metabolic reprogramming of glycolysis and glutamine metabolism are key events in myofibroblast transition in systemic sclerosis pathogenesis. J Cell Mol Med. (2020) 24:14026–38. doi: 10.1111/jcmm.16013
92. Ni Z, Deng J, Potter CMF, Nowak WN, Gu W, Zhang Z, et al. Recipient c-Kit lineage cells repopulate smooth muscle cells of transplant arteriosclerosis in mouse models. Circ Res. (2019) 125:223–41. doi: 10.1161/CIRCRESAHA.119.314855
93. Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. (2020) 5:209. doi: 10.1038/s41392-020-00312-6
94. Caron MM, Emans PJ, Surtel DA, Cremers A, Voncken JW, Welting TJ, et al. Activation of NF-κB/p65 facilitates early chondrogenic differentiation during endochondral ossification. PloS One. (2012) 7:e33467. doi: 10.1371/journal.pone.0033467
95. Sang LJ, Ju HQ, Liu GP, Tian T, Ma GL, Lu YX, et al. LncRNA CamK-A regulates Ca(2+)-signaling-mediated tumor microenvironment remodeling. Mol Cell. (2018) 72:71–83.e7. doi: 10.1016/j.molcel.2018.08.014
96. Wang Y, Xian H, Qi J, Wei F, Cheng X, Li S, et al. Inhibition of glycolysis ameliorate arthritis in adjuvant arthritis rats by inhibiting synoviocyte activation through AMPK/NF-кB pathway. Inflammation Res. (2020) 69:569–78. doi: 10.1007/s00011-020-01332-2
97. Wang G, Li X, Li N, Wang X, He S, Li W, et al. Icariin alleviates uveitis by targeting peroxiredoxin 3 to modulate retinal microglia M1/M2 phenotypic polarization. Redox Biol. (2022) 52:102297. doi: 10.1016/j.redox.2022.102297
98. Chen J, Zhang M, Liu Y, Zhao S, Wang Y, Wang M, et al. Histone lactylation driven by mROS-mediated glycolytic shift promotes hypoxic pulmonary hypertension. J Mol Cell Biol. (2023) 14:mjac073. doi: 10.1093/jmcb/mjac073
99. Zheng L, Zhang Z, Sheng P, Mobasheri A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res Rev. (2021) 66:101249. doi: 10.1016/j.arr.2020.101249
100. Raman APS, Kumari K, Jain P, Vishvakarma VK, Kumar A, Kaushik N, et al. In silico evaluation of binding of 2-deoxy-D-glucose with mpro of nCoV to combat COVID-19. Pharmaceutics. (2022) 14;135. doi: 10.3390/pharmaceutics14010135
101. Taubmann J, Krishnacoumar B, Bohm C, Faas M, Muller DIH, Adam S, et al. Metabolic reprogramming of osteoclasts represents a therapeutic target during the treatment of osteoporosis. Sci Rep. (2020) 10:21020. doi: 10.1038/s41598-020-77892-4
102. Terabe K, Ohashi Y, Tsuchiya S, Ishizuka S, Knudson CB, Knudson W. Chondroprotective effects of 4-methylumbelliferone and hyaluronan synthase-2 overexpression involve changes in chondrocyte energy metabolism. J Biol Chem. (2019) 294:17799–817. doi: 10.1074/jbc.RA119.009556
103. Pemmari A, Leppanen T, Hamalainen M, Moilanen T, Vuolteenaho K, Moilanen E. Widespread regulation of gene expression by glucocorticoids in chondrocytes from patients with osteoarthritis as determined by RNA-Seq. Arthritis Res Ther. (2020) 22:271. doi: 10.1186/s13075-020-02289-7
104. Hardy RS, Raza K, Cooper MS. Therapeutic glucocorticoids: mechanisms of actions in rheumatic diseases. Nat Rev Rheumatol. (2020) 16:133–44. doi: 10.1038/s41584-020-0371-y
105. Bannuru RR, Osani MC, Vaysbrot EE, Arden NK, Bennell K, Bierma-Zeinstra SMA, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthritis Cartilage. (2019) 27:1578–89. doi: 10.1016/j.joca.2019.06.011
106. Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. (2001) 358:69–77. doi: 10.1042/0264-6021:3580069
107. Aguer C, Gambarotta D, Mailloux RJ, Moffat C, Dent R, McPherson R, et al. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PloS One. (2011) 6:e28536. doi: 10.1371/journal.pone.0028536
108. Lane RS, Fu Y, Matsuzaki S, Kinter M, Humphries KM, Griffin TM. Mitochondrial respiration and redox coupling in articular chondrocytes. Arthritis Res Ther. (2015) 17:54. doi: 10.1186/s13075-015-0566-9
109. Mobasheri A. Glucose: an energy currency and structural precursor in articular cartilage and bone with emerging roles as an extracellular signaling molecule and metabolic regulator. Front Endocrinol (Lausanne). (2012) 3:153. doi: 10.3389/fendo.2012.00153
110. Eitner A, Culvenor AG, Wirth W, Schaible HG, Eckstein F. Impact of diabetes mellitus on knee osteoarthritis pain and physical and mental status: Data from the osteoarthritis initiative. Arthritis Care Res (Hoboken). (2021) 73:540–8. doi: 10.1002/acr.24173
111. Veronese N, Cooper C, Reginster JY, Hochberg M, Branco J, Bruyere O, et al. Type 2 diabetes mellitus and osteoarthritis. Semin Arthritis Rheum. (2019) 49:9–19. doi: 10.1016/j.semarthrit.2019.01.005
112. Li Q, Wen Y, Wang L, Chen B, Chen J, Wang H, et al. Hyperglycemia-induced accumulation of advanced glycosylation end products in fibroblast-like synoviocytes promotes knee osteoarthritis. Exp Mol Med. (2021) 53:1735–47. doi: 10.1038/s12276-021-00697-6
113. Goswami A. Prolotherapy. J Pain Palliat Care Pharmacother. (2012) 26:376–8. doi: 10.3109/15360288.2012.734900
114. Hauser RA, Lackner JB, Steilen-Matias D, Harris DK. A systematic review of dextrose prolotherapy for chronic musculoskeletal pain. Clin Med Insights Arthritis Musculoskelet Disord. (2016) 9:139–59. doi: 10.4137/CMAMD.S39160
115. Bae G, Kim S, Lee S, Lee WY, Lim Y. Prolotherapy for the patients with chronic musculoskeletal pain: systematic review and meta-analysis. Anesth Pain Med (Seoul). (2021) 16:81–95. doi: 10.17085/apm.20078
116. Wee TC, Neo EJR, Tan YL. Dextrose prolotherapy in knee osteoarthritis: A systematic review and meta-analysis. J Clin Orthop Trauma. (2021) 19:108–17. doi: 10.1016/j.jcot.2021.05.015
Keywords: osteoarthritis, glucose metabolism, cartilage, glycolysis, therapeutic
Citation: Pi P, Zeng L, Zeng Z, Zong K, Han B, Bai X and Wang Y (2024) The role of targeting glucose metabolism in chondrocytes in the pathogenesis and therapeutic mechanisms of osteoarthritis: a narrative review. Front. Endocrinol. 15:1319827. doi: 10.3389/fendo.2024.1319827
Received: 17 October 2023; Accepted: 19 February 2024;
Published: 06 March 2024.
Edited by:
Xiaonan Liu, Johns Hopkins Medicine, United StatesCopyright © 2024 Pi, Zeng, Zeng, Zong, Han, Bai and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Wang, d3l3ZWl3ZWlAMTI2LmNvbQ==
†These authors have contributed equally to this work