 Tibor Szénási
Tibor Szénási Gábor Turu
Gábor Turu László Hunyady
László Hunyady- 1Institute of Enzymology, Research Center for Natural Sciences, Centre of Excellence of the Hungarian Academy of Sciences, Budapest, Hungary
- 2Department of Physiology, Faculty of Medicine, Semmelweis University, Budapest, Hungary
β-arrestins, which have multiple cellular functions, were initially described as proteins that desensitize rhodopsin and other G protein-coupled receptors. The cytoskeletal system plays a role in various cellular processes, including intracellular transport, cell division, organization of organelles, and cell cycle. The interactome of β-arrestins includes the major proteins of the three main cytoskeletal systems: tubulins for microtubules, actins for the actin filaments, and vimentin for intermediate filaments. β-arrestins bind to microtubules and regulate their activity by recruiting signaling proteins and interacting with assembly proteins that regulate the actin cytoskeleton and the intermediate filaments. Altered regulation of the cytoskeletal system plays an essential role in the development of Alzheimer’s, Parkinson’s and other neurodegenerative diseases. Thus, β-arrestins, which interact with the cytoskeleton, were implicated in the pathogenesis progression of these diseases and are potential targets for the treatment of neurodegenerative disorders in the future.
1 Introduction
The diverse functions of the cytoskeletal system include cell protection, cytokinesis, cell motility, intracellular transport, cell division, and organization of organelles within the cell. In eukaryotes, three types of cytoskeleton exist: microtubules, actin microfilaments, and intermediate filaments. The clinical importance of these systems is indicated by the findings that their dysfunction may initiate neurodegenerative or cancerous processes (1).
Arrestins were first discovered as proteins responsible for quenching the light-induced photoreceptor response in the retina (2). Activated rhodopsin is phosphorylated by rhodopsin-kinase on serine/threonine (S/T) amino acids at its C-terminus. This visual arrestin recognizes and binds to the phosphorylated rhodopsin and uncouples it from its heterotrimeric G-protein partner, transducin. Later, it was discovered that β2 adrenergic receptor (β2AR) shares the seven-transmembrane topology with the rhodopsin protein. It has also been recognized that β2AR undergoes similar phosphorylation as rhodopsin, and the kinase responsible for this modification was called β2-adrenergic receptor kinase (βARK1), which is now named G protein-coupled receptor kinase 2 (GRK2). Two different visual receptor kinases (GRKs) and arrestins (arrestin1 and 4) were identified, together with five additional non-visual GRKs and two arrestins (arrestin2 and 3, also called β-arrestin1 and β-arrestin2) (3). Non-visual arrestins are expressed in all tissues. They bind to activated and phosphorylated GPCRs and uncouple them from their cognate heterotrimeric G-proteins. In the case of many GPCRs, this process is followed by β-arrestin-mediated transportation to the clathrin-coated pits and internalization of the receptor (4). β-arrestins are localized to the cytoplasm and, in the case of the β-arrestin1, also to the nucleus, which localization is quickly changed upon stimulation. First, β-arrestins translocate to the cell membrane and bind to the receptors, and then, they follow the receptors to clathrin-coated pits or even to intracellular vesicles (see below). β-arrestins form two interactions with GPCRs: they can bind to the phosphorylated C-terminal part of the receptors, and/or their finger loop domain is inserted into the transducer-binding region of the receptor, forming the so-called “core-interaction” (3, 5–8).
Based on the strength of the binding between β-arrestins and GPCRs, two groups of receptors can be distinguished: class A receptors bind to β-arrestins with weaker affinity, and the interaction is restricted to the vicinity of the cell membrane, while class B receptors bind β-arrestins with high affinity and remain bound to them even in the endosomes (9). The interaction stability is determined by the C-terminal interaction, exchanging the C-termini between receptors can switch class A receptors to class B types, and vice versa (9). The interaction stability is determined by interactions between positively charged amino acids in the β-arrestin and the phosphorylated serine-threonine amino acids on the receptor C-terminus, or in the case of some receptors, on the third intracellular loop (10–15). β-arrestins are critical regulators of multiple cellular processes and signaling pathways, such as scaffolding, receptor desensitization, GPCR endocytosis and trafficking, transcriptional regulation, cell growth and survival, cytoskeletal rearrangement and cell migration, and other specialized functions (16–19). β-arrestins exist in distinct conformational states: free, receptor-bound, microtubule-associated (20), and associated with other partner proteins. The interactome of β-arrestin proteins includes the prominent component proteins for the three major type elements of the cytoskeleton: actins for the actin filaments, tubulins for the microtubules, and vimentin for the intermediate filaments (16). These cytoskeleton-associated proteins, including multiple isoforms of actins, are abundant in cells, and increasing evidence indicates that the interaction between these proteins and β-arrestins may have functional consequences.
2 Interactions of β-arrestin proteins and the cytoskeleton
2.1 Interactions between β-arrestin proteins and the microtubular system
The microtubule network, an integral component of the eukaryotic cytoskeleton, plays a critical role in cell division, differentiation, maintenance of cell shape and polarization, motility and intracellular transport, and formation of pathological inclusion bodies (21, 22) (Figures 1, 2). Microtubules (MT) are composed of the highly conserved dimers of α- and β-tubulin and form the microtubule network (22). Dynamic instability is characteristic of microtubules and is central to their functions by allowing their rapid spatial and temporal reorganization and differentiation in response to environmental factors or signals (23).
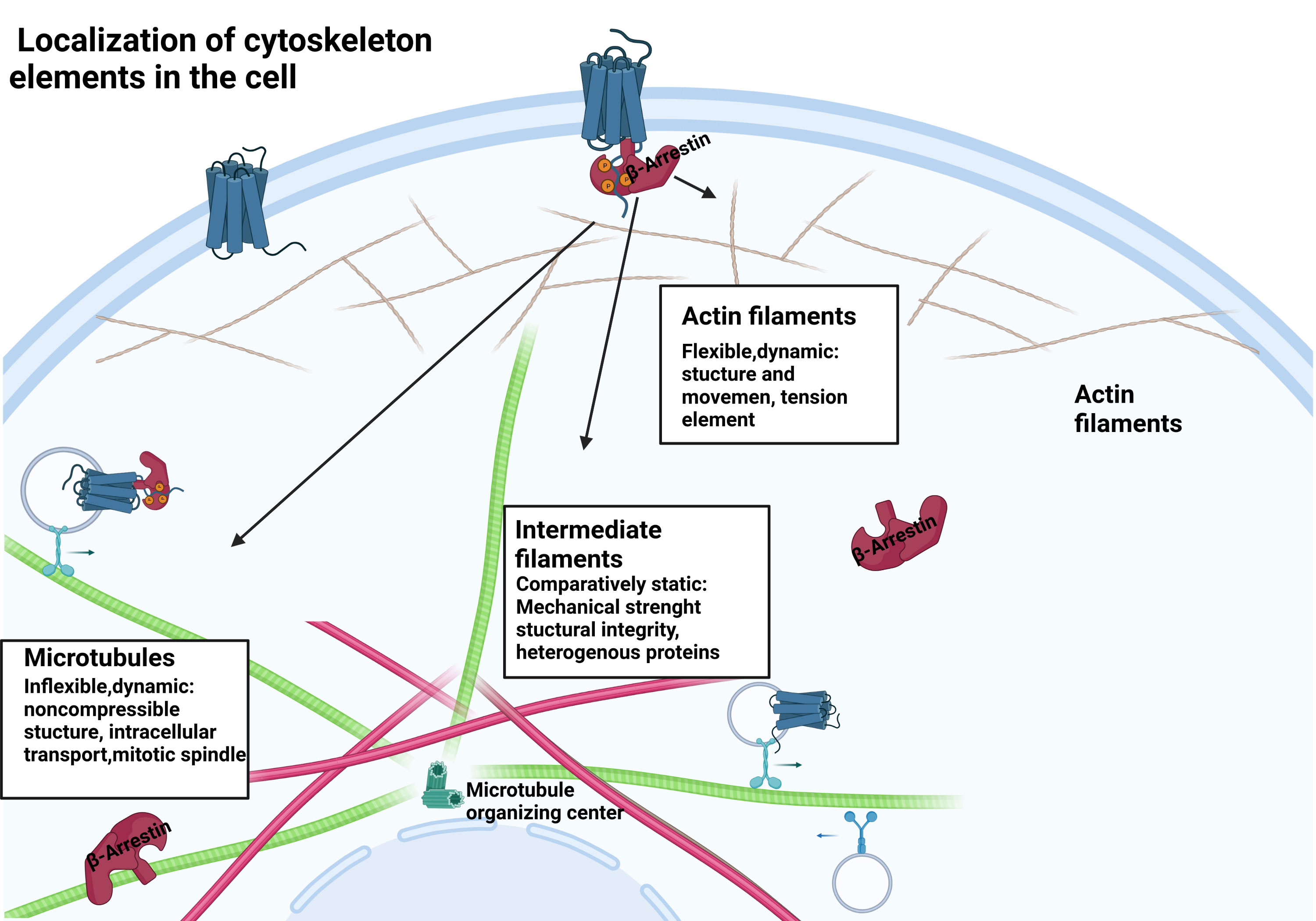
Figure 1 β-arrestin protein location within the cell. β-arrestins exist in distinct conformational states: free, receptor-bound, and microtubule-associated.
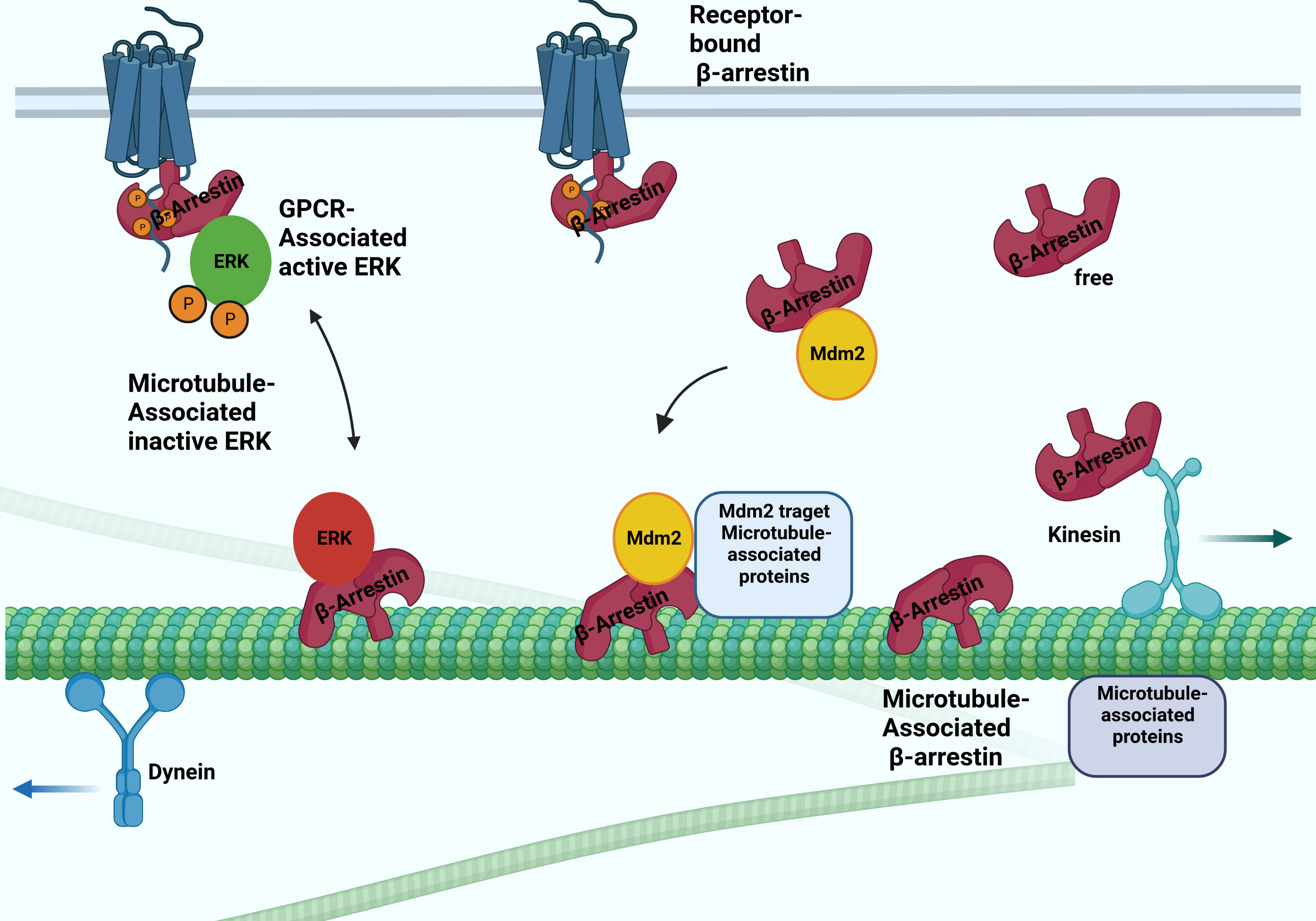
Figure 2 Possible interactions between arrestins and microtubular system. The β-arrestin proteins binding to microtubules involve the conformation change. They bind to the microtubule system and regulate their activity by recruiting ERK and Mdm2. The β-arrestin2 interacts with the kinesin-like protein KIF3A motor protein.
Interaction with the microtubular system was first demonstrated in vivo in the case of arrestin1, a visual arrestin, in rod photoreceptors, and it has been reported that it determines the differential subcellular localization of its splice variants (24). The interaction of rod arrestin with the microtubule system is increased in the dark. The light-dependent but energy-independent translocation of rod arrestin is involved in light and dark adaptation (25). Interestingly, visual arrestin N- and C-domains or truncated arrestin(1-378) bind microtubules substantially better than the full-length protein. The MT-bound arrestin’s conformation is different from the free and receptor-bound conformations. The exchange of two elements on the concave sides of the N- and C- terminal domains reverses both the receptor specificity and the relative ability of rod-arrestin and β-arrestin1 to bind microtubules, indicating that the receptor- and MT-binding sites overlap (26). Thus, β-arrestin proteins cannot interact with microtubules and receptors simultaneously. It was also shown that MT-bound arrestin recruits extracellular signal-regulated kinase 2 (ERK)1/2 proteins and mouse double minute 2 (MDM2), an E3 ubiquitin ligase, and regulates their activity: the translocation ERK to MTs by β-arrestin1 reduces the level of ERK activation (27). Although β-arrestin1 and rod-arrestin bind both polymerized and unpolymerized tubulin, they do not affect microtubule polymerization. These reports described a new non-receptor binding partner of arrestin proteins and showed that the interaction between arrestins and the microtubule system localizes specific signaling molecules to the cytoskeleton. β-arrestin-dependent recruitment of the protein kinase ERK silences ERK, whereas its recruitment to the ubiquitin E3 ligase Mdm2 directs Mdm2 to its cytoskeletal substrates (Figure 2). α-methylserotonin, an agonist of 5HT2A/C receptors, caused a β-arrestin-dependent stimulation of ERK1/2 phosphorylation in the dendrites of cortical neurons. The siRNA-mediated depletion of one of the β-arrestins suppressed this process. The depletion also counteracted the serotonin 5HT-1A receptor-mediated disruption of microtubule stability, which is required for normal N-methyl-D-aspartate (NMDA) receptor transport (28). On the other hand, targeting Mdm2 to microtubules by β-arrestin redirects Mdm2 activity toward MT-associated proteins, significantly increasing partner ubiquitination (27).
It has been first reported that rod arrestin can form oligomers at physiological concentrations, and monomeric visual arrestin binds to rhodopsin, whereas microtubules can bind both arrestin1 monomers and tetramers (29). β-arrestin1 exhibited dimerization throughout crystallization (30), indicating that it may oligomerize (31). Oligomerization of β-arrestin1 is facilitated in the presence of inositol-hexakisphosphate (IP6) (32, 33). The purpose of β-arrestin1 oligomerization remains poorly understood. Besides thecytoplasmic and plasma membrane (in activated cells) localization of β-arrestin1, its presence in the nucleus was also reported (34). β-arrestin2 does not localize to the nucleus since the C-terminus of β-arrestin2, in contrast to that of β-arrestin1, has a nuclear export signal (35, 36). However, hetero-oligomerization with β-arrestin2 dramatically changed the subcellular distribution of β-arrestin1, which became predominantly cytosolic (37). It has also been reported that although monomeric β-arrestin1 can enter the nucleus, its homo-oligomers are mostly cytosolic (32), suggesting that homo- and hetero-oligomerization of β-arrestin1 cause similar changes in its subcellular localization. A proteomics-based screening study on the arrestin interactome revealed that over three hundred distinct proteins coprecipitated with β-arrestin1 or β-arrestin2 under different conditions (16). This study confirmed the interaction of β-arrestin proteins with tubulin and has identified new interactions with resident and accessory cytoskeletal proteins. Interestingly, β-arrestin1 binds kinesin family member 3A (KIF3A) and filamin-A, whereas β-arrestin2 binds cofilin-1 (actin assembly protein) (16) (Figures 2, 3) and localizes in primary cilia (38, 39). Primary cilia are antenna-like structures on the surface of most vertebrate cells, which regulate essential signaling pathways during development and play crucial roles in tissue homeostasis (40). Lack of β-arrestin2 resulted in ciliogenesis defects and uncontrolled proliferation in β-arrestin2 KO mouse embryonic fibroblast (MEF) cells (39). β-arrestin2 interacts with 14-3-3 proteins and kinesin KIF3A (39), and β-arrestin proteins are required for the formation of a “translocation complex”, including KIF3A and Smo, that promote activation of downstream transcriptional targets in mammalian cells and activity-dependent localization of Smo to primary cilia (38). Microtubule-independent localization of β-arrestin2 to the proximal region of centrioles has been reported (38). Endogenous β-arrestin2 is localized at the centrosome during the entire cell cycle, and β-arrestin2 is co-localized with the γ-tubulin and acetyl-tubulin staining (41).
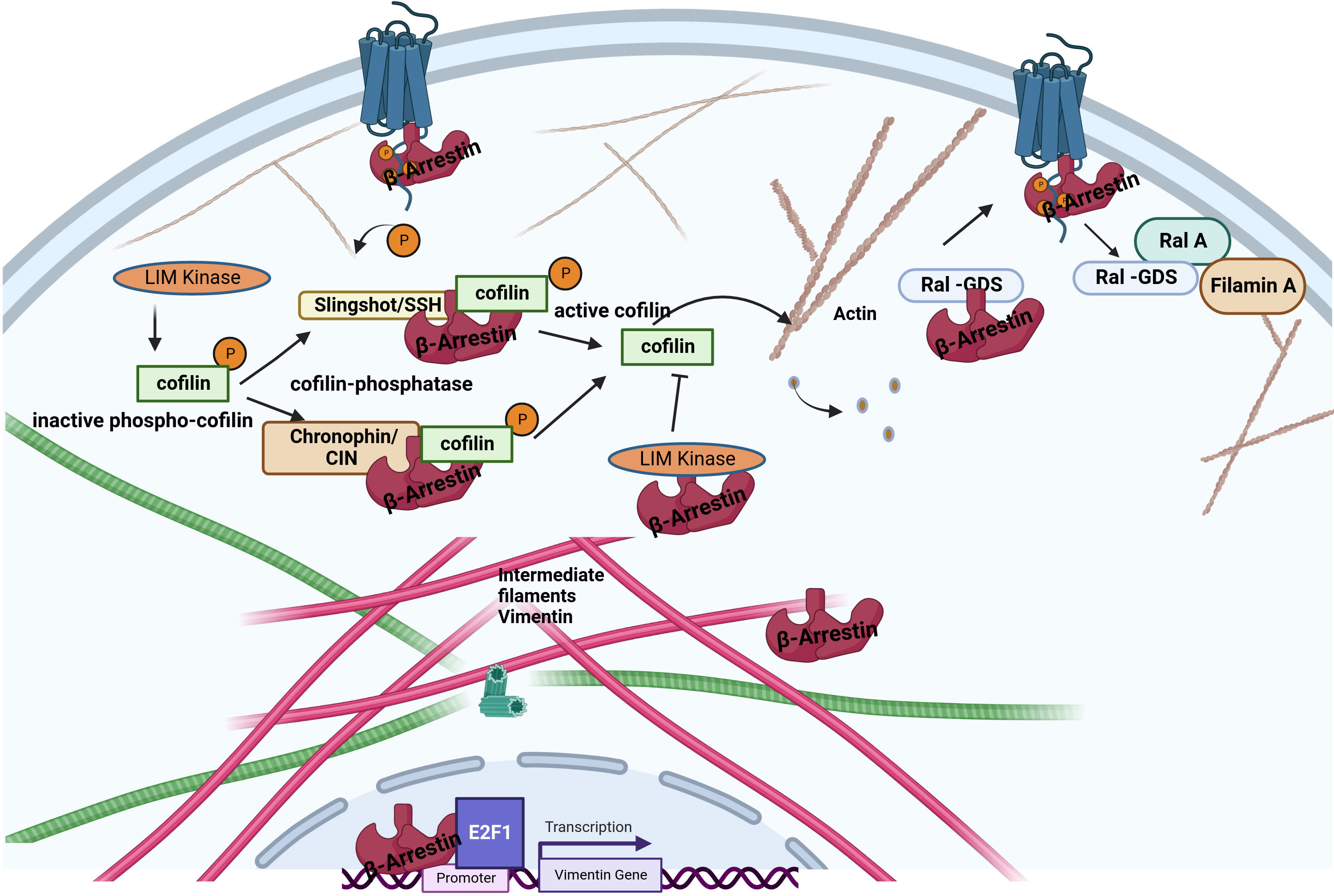
Figure 3 Interaction between β-arrestin proteins and other cytoskeletal elements. β-arrestins directly bind actin filaments and vimentin. Increased cofilin activity results from the scaffolding of cofilin by the upstream cofilin phosphatases chronophin and slingshot. The increased cofilin activity is also related to LIMK inhibition by arrestins. Older filaments are cut by cofilin, producing new filament seeds. β-arrestin2 binds to the guanine exchange factor for GTPase, Ral1(Ral-GDS). After receptor activation, RAL-GDS is released, which activates Ras-related protein Ral-A (RalA). RalA interacts with Filamin-A to promote actin rearrangement. β-arrestin1 can enhance vimentin gene expression in association with the transcription factor E2F1.
2.2 β-arrestin interactions with actin
The actin cytoskeleton is responsible for mediating various cellular processes, including cell structure determination, organelle transport, cell migration, axonal growth, and endocytosis or phagocytosis. Reorganization of the cytoskeleton is a crucial process during alterations of cell shape, signal transduction, movement, and many other dynamic cellular processes. Mitogen-activated protein (MAP) kinase, c-Jun N-terminal kinase, and p38 kinases play critical roles in regulating dynamic changes of the cytoskeleton because several extracellular signals that promote cell shape change converge on MAP kinases, which phosphorylate downstream targets involved in the regulation of the actin cytoskeleton (42). During cell migration, the actin filaments are rapidly reorganized by the formation of new barbed ends and by cutting and severing existing filaments. This reorganization occurs partly through the binding of various actin cross-linking and assembly proteins (43). Bhattacharya et al. have identified by co-immunoprecipitation and yeast two-hybrid screening that Ral-GDS, a stimulator of Ral-GDP dissociation, is a β-arrestin-binding protein in human neutrophil leukocytes (44). These authors proved that chemoattractant receptor-induced regulation of β-arrestin-Ral-GDS protein complexes is necessary for Ral activation, leading to cytoskeleton reorganization. After the recruitment of β-arrestins to the activated GPCR, Ral-GDS dissociates from the β-arrestin and becomes activated (45). Ral-GDS stimulates RalA GTPase at the leading edge of the actin cytoskeleton where the β-arrestin-ERK scaffolds are located. After GPCR activation, β-arrestins also induce stress fiber formation and reorganization of the actin cytoskeleton by interacting with RhoGEFs or RhoGAPs (46). Stress fiber formation and actin structure polymerization are required for cell movement, adhesion, and chemotaxis (47). After disruption of the actin cytoskeleton dynamics by toxins β-arrestin2, but not β-arrestin1, could rescue agonist-induced endocytosis of the β isoform of the thromboxane A2 receptor (TPβ). The use of β-arrestin2 mutants showed that this was an AP2- and a clathrin-dependent mechanism (48).
Double β-arrestin1 and 2 double knockout MEFs have been used to study β-arrestin function for over two decades (49). These cells have an unusual size and shape and are dramatically different from wild-type MEF cells. The actin cytoskeletal system of β-arrestin KO MEF cells, plated on fibronectin, is also different from that of the wild-type cells, and the size of these cells is much larger. However, retroviral expression of β-arrestin1 or 2 rescued the morphological phenotype of the double β-arrestin1 and 2 KO MEF cells (50).
2.3 β-arrestin interactions with intermediate filaments
The intermediate filaments include vimentin, a structural protein found in mesenchymal cells (51). A vimentin monomer has non-helical N-terminal and C-terminal domains and a central α-helical domain (52). The interaction between β-arrestins and vimentin was first reported by Xiao et al. based on co-immunoprecipitation and mass spectroscopy data (16). Pillai et al. have demonstrated that β-arrestin1, but not β-arrestin2, is required for nicotine-induced epithelial-mesenchymal transition and metastasis formation. The increased levels of β-arrestin1, vimentin, and fibronectin in tumors from smokers and nonsmokers suggest that these molecules contribute to the growth and progression of non–small cell lung cancer cells. At the same time, the induction of these genes may be an essential mechanism by which nicotine exerts its tumor-promoting functions. Interestingly, β-arrestin1 was required to induce mesenchymal promoters following stimulation by the nicotinic acetylcholine receptor but played no role in the TGFβ-mediated induction of the vimentin and fibronectin genes. The β-arrestin1 may be co-operated with the E2F1 transcription factor in the transcriptional regulation of these genes (53).
2.4 Roles of β-arrestin proteins in the regulation of cell migration and chemotaxis
Filamin-A, an actin cross-linking protein, can serve as a scaffold for several binding partners and is a crucial regulator of cell migration. Similar to β-arrestins, this protein can localize various kinases at the leading edge and interacts with β-arrestins downstream of the angiotensin receptor to control chemotaxis, indicating that the interaction between these two proteins may reach different levels of actin dynamics during cancer cell motility (54, 55). Downregulation of β-arrestin causes decreased GPCR internalization and chemotaxis (56).
Cofilin is one of the essential proteins which participate in the dynamic reorganization of the cytoskeleton (57). The binding of cofilin to actin filaments causes its cleavage, and the number of ends increases, either shortening or lengthening the filament, depending on the physiological situation. Cofilin is a target of the opposing actions of LIM kinase, which inactivate it by phosphorylation at Ser3, and specific phosphatases, such as slingshot (SSH) and chronophine (CIN), which activate it, enabling the fine-tuning of the regulation of the actin cytoskeleton dynamics (Figure 3) (57, 58). β-arrestins are tight regulators of cofilin phosphorylation in response to upstream GPCR stimuli by interacting with LIM kinase and Ser/Thr phosphatases, including slingshot and chronophine, to control the direction and force of membrane expansion required for cell movement (19, 59). The degree of control depends on the GPCR and the cellular context. During chemotaxis, chemotactic signals, such as those from PAR-2 stimulation, promote the local formation of free actin barbed ends, membrane protrusions, and leading-edge formation by interacting with cofilin and phosphatases or directly binding to LIM kinase to antagonize its ability to phosphorylate and inactivate cofilin (54, 60). β-arrestin2 has also been shown to negatively regulate the migration of dendritic cells and play a role in inflammatory diseases (61).
3 The role of β-arrestin proteins in cytoskeleton-associated neurodegenerative disease
Neurodegenerative diseases cause progressive neuronal dysfunction, toxicities, and death (62). The most common of these are Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), and Huntington’s disease (HD). GPCRs and cytoskeletal proteins play important roles in these disorders and can also serve as therapeutic targets (1, 62). Abnormal aggregates of cytoskeletal proteins are pathologic signatures of neurodegenerative diseases. These aggregated proteins of the neuronal cytoskeleton characteristically include aggregates of the neuronal intermediate filaments and inclusions containing the microtubule-associated protein (MAP) tau protein (1). The pathological features of AD have three main parts, 1) extracellular senile plaques due to the deposition of beta-amyloid peptide (Aβ), 2) intracellular neurofibrillary tangles due to tau phosphorylation, and 3) neuronal cell death (63, 64). Aβ, a 4 kDa fragment of the amyloid precursor protein (APP), is widely produced by brain neurons and astrocytes to a lesser extent. Two sequential proteolytic cleavages of APP by β-secretase at the ectodomain and γ-secretase at the intramembrane site generate Aβ (65). MAP proteins, such as tau, regulate the extent and rate of microtubule assembly and play important roles in morphogenetic processes, such as axonal outgrowth (66). In AD, tau is phosphorylated at sites that are normally not phosphorylated in adulthood or is significantly phosphorylated at sites that are normally phosphorylated in adult tau (67). Arrestin proteins regulate these and other proteins implicated in the development of neurodegenerative diseases.
Changes in β-arrestin and GRK expression were first reported in Parkinson’s disease with dementia (68). Amyloid-β peptide (Aβ) can bind to the β2-adrenergic receptor and cause allosteric receptor activation leading to cAMP- and arrestin-mediated signaling (69, 70). Aβ treatment can activate the β2-adrenergic receptor-arrestin-MAPK pathway in prefrontal cortex primer neurons. It was reported that MEK, an upstream regulator of ERK1/2 MAP kinases, promotes the phosphorylation of tau, and β-arrestin2 is required for the Aβ-induced ERK1/2 phosphorylation (71). These data suggest that Aβ-induced MEK phosphorylation simultaneously leads to β-arrestin2-dependent ERK1/2 activation and arrestin-independent tau phosphorylation (71).
The γ-secretase complex plays a pivotal role in the production of the Aβ. Undertanding the processes involved in its regulation might be essential to developing new therapies (72). The γ-secretase complex is composed of four integral membrane proteins: the catalytic component presenilin 1(Ps1) or 2 (Ps2) and the essential cofactors nicastrin (Nct), anterior pharynx defective 1 (Aph1) and presenilin enhancer 2 (Pen 2) (73). It has been proposed that β-arrestin1 plays a role in AD pathogenesis through the regulation of this complex since depletion of β-arrestin1 expression by specific siRNA reduced γ-secretase activity and Aβ40 and Aβ42 production in neurons. Moreover, when APP/PS1 mice, a model of the AD (74), were crossed with β-arrestins1−/− mice, offspring had lower amounts of Aβ deposits compared to β-arrestins1+/+ mice and ameliorated memory deficits (75). In other experiments, Thathiah et al. isolated embryonic neuronal cultures from wild-type and β-arrestin proteins knock-out mice. The amounts of endogenous Aβ40 and Aβ42 were substantially reduced in β-arrestins2−/− but not β-arrestins1−/− neurons, suggesting that β-arrestin2 is involved endogenously in the modulation of neuronal Aβ production (76). Although the role of the β-arrestin1 in these two experiments is somewhat controversial, the different timescale of the siRNA vs. knockout models might explain these differences. Nevertheless, the levels of both β-arrestin proteins were increased in the brains of patients with AD (75, 77) (Figure 4A). Pontrello et al. have demonstrated that deletion of β-arrestin2 can protect against Aβ-induced loss of dendritic spines in hippocampal neurons, further supporting the role of the β-arrestin2 in the pathogenesis of AD (78). Overexpression of both β-arrestins can cause an increase in Aβ peptide generation (75, 76). β-arrestins interact with the Aph1a subunit of the γ-secretase complex but not with Nct, Ps1, and Pen2 subunits offering a mechanism through which the effects of the arrestin could be explained (75, 76).
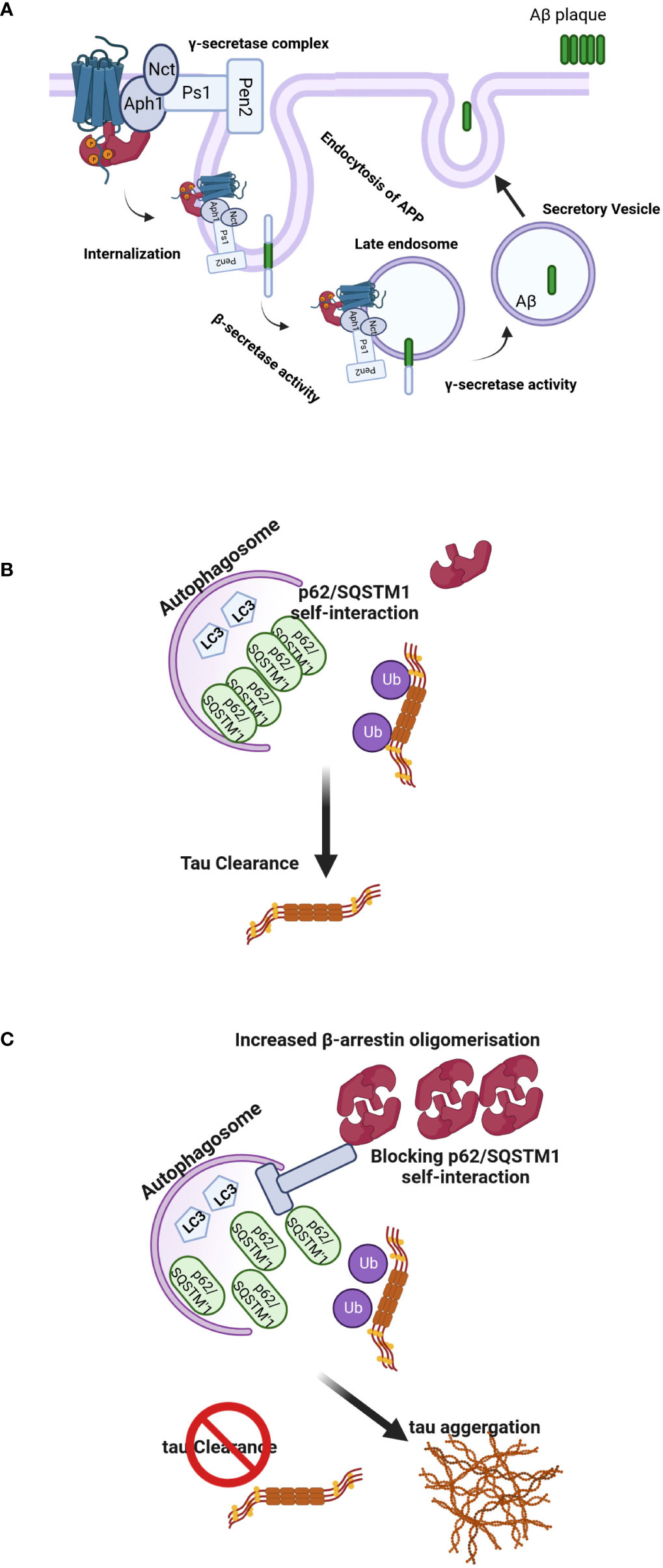
Figure 4 (A) Regulation of Aβ-related pathology by β-arrestin2. Activation of GPCRs by their ligands increases the affinity for β-arrestin2. Recruitment of β-arrestin2 to GPCRs ends up interacting with the γ-secretase complex via the Aph1 subunit. β-arrestin2 mediates the internalization of GPCRs and localizes γ-secretase to late endosomes, where the acidic environment often increases its activation. APP is cleaved by the β-secretase, and C-terminal APP fragments are produced, the direct substrates of the γ-secretase. These fragments are often subsequently cleaved by the γ-secretase to provide Aβ and APP intracellular domains. The produced Aβ is often discharged into intercellular space via secretory vesicles, resulting in extracellular amyloid plaque formation. (B, C) Model of β-arrestins-promoted tauopathy in AD and FTD. In healthy brains, monomeric β-arrestins regulate GPCR trafficking, and there is no excess of oligomeric β-arrestins, and thus misfolded tau is efficiently ubiquitylated and targeted for autophagy clearance. However, in FTD form-tau brains, β-arrestins oligomers are increased, inhibiting p62/SQSTM1-mediated autophagy, leading to failure of misfolded/aggregated tau to be efficiently cleared.
The other mechanism by which β-arrestins might regulate the amount of Aβ and tau is the degradation of these proteins, further complicating the picture. Both Aβ and tau proteins can be degraded by the ubiquitin-proteasome system (UPS) and autophagy-lysosome system (ALS), a process called clearance of Aβ and tau (64). The upregulation of β-arrestin1 and β-arrestin2 was an early event after Aβ25-35 exposure, which was accompanied by induction of autophagy. Downregulation of β-arrestin1 resulted in decreased autophagic flux and enhancement of Aβ25-35 induced cell death, whereas depletion of β-arrestin2 reversed to some extent Aβ25-35 cytotoxicity. Liu et al. demonstrated that β-arrestin2, but not β-arrestin1, was critical for autophagy activation and preferentially regulated the nicotinic acetylcholine receptor α7 subunit expression in the membrane, which is responsible for the neuroprotective effect of nicotine. Suppression of β-arrestin2 increased the plasma membrane expression of this receptor, attenuating Aβ25-35 toxicity (79).
Interestingly, oligomerization of β-arrestin2 is required for tau stability. Overexpression of β-arrestin2 in tau-expressing HeLa cells inhibited the bafilomycin A-induced increase in LC3-positive puncta, suggesting that β-arrestin2 inhibits autophagy at or before LC3. The β-arrestin2 oligomers increase tau levels by blocking the self-interaction of p62, which is the initial step essential for autophagy flux. These data have demonstrated that the oligomerized form of β-arrestin2 reduces the elimination of tau protein by interfering with p62-mediated autophagy (Figures 4B, C). The p62/SQSTM1 interacts with polyubiquitinated tau through its ubiquitin-associated domain and serves a unique role in the regulation of tau proteasomal degradation (80). Thathiah et al. have proposed that the development of small molecule β-arrestin2 oligomerization inhibitors may have therapeutic relevance for intervention in frontotemporal dementia (FTD) form-tau to enhance the elimination of the tau protein without exerting significant side effects via GPCR signaling pathways. Agents that promote the degradation of misfolded aggregated proteins, such as tau, are attractive therapeutic targets for neurodegenerative diseases (81). Cargo-bound p62/SQSTM1 promotes autophagosome maturation by converting LC3 to its lipidated active form, LC3.II (82). This data is in agreement with the inhibitory effect of β-arrestin1 on the formation of LC3 puncta and the reduction in p62-LC3 colocalization. These data are consistent with the observed role of β-arrestin1 in impeding p62/SQSTM1 flux and impairing destruction of misfolded tau (83) and underline the potential of β-arrestin proteins as drug targets for the therapy of neurodegenerative diseases. FTD includes a spectrum of clinical syndromes associated with various neurodegenerative diseases. In patients with FTD, the primarily affected regions are the frontal and temporal lobes (84). Therefore, FTD is also regarded as frontotemporal lobar degeneration. β-arrestin2 protein and mRNA were significantly increased in the tau-FTD brain samples compared to the controls and in the brain samples of the P301S transgenic mice compared to the brains of the non-transgenic mice (85). A recent report has shown that β-arrestin1 levels are increased in the brains of FTD patients, and β-arrestins are essential for the β2 adrenergic receptor and mGluR2 glutamate receptor-mediated increase in pathogenic tau (83). Increased β-arrestin1 also causes the accumulation of pathogenic tau, whereas its reduction alleviates tau-induced pathology and rescues the impaired synaptic plasticity and cognitive abilities in PS19 mice. Biochemical and cellular studies show that these effects of β-arrestin1 were mediated by the destabilization of microtubules and impairment of p62/SQSTM1 autophagy flux due to the interference with p62/SQSTM1 body formation, which promotes pathogenic tau accumulation.
Other β-arrestin partner proteins have been also implicated in the pathogenesis of neurodegenerative diseases, such as ERK, Filamin-A, and cofilin (86, 87). For example, the activated form of cofilin, a partner of both β-arrestin proteins, exacerbates tau pathology by interfering with tau-mediated microtubule dynamics (88) and may contribute to the cytoskeletal pathogenesis in Alzheimer’s disease (87).
In Parkinson’s disease, β-arrestin proteins play a role in the microglia-mediated inflammation and the pathogenesis of this disease (89). The E3 ubiquitin protein ligase Parkin directly interacts with both β-arrestins, and Parkin promotes Mdm2 binding to β-arrestin proteins (90). Interestingly, certain anti-Parkinson’s disease drugs (e.g. levodopa and piribedil) induced β-arrestin2 and dopamine D2 receptor-mediated Aβ elevation in Alzheimer’s disease model cells (91). On the other hand, in macaque and mouse models of Parkinson’s disease, β-arrestin2 overexpression has been shown to ameliorate dyskinesia symptoms, which develop in patients during levodopa treatment (92).
4 Concluding remarks and future perspectives
The interactions between β-arrestin proteins and the cytoskeleton, and its assembly proteins that determine the dynamics of the skeleton, are critical mechanisms for several physiological and pathological processes. The β-arrestinome (93), the interactome of β-arrestin proteins, contains more than 400 cytosolic and nuclear protein partners, suggesting various functions for these proteins. Interactions between β-arrestin proteins and the cytoskeletal system play essential roles in neurodegenerative diseases, especially Alzheimer’s. In addition, due to their role in cytoskeletal rearrangement and movement, β-arrestin proteins may also be relevant targets for cancer therapy. Experimental approaches to inhibit arrestin function by cell-specific genetic ablation or downregulation by siRNA have been pursued, including aptamers, the oligonucleotides with structures that allow selective binding to the surface of pathological target proteins and inhibit protein-protein interactions. Arrestin2-specific nucleotide aptamers have been developed, and Kotula et al. have reported that β-arrestin2-specific aptamers not only interfere with β-arrestin-dependent signaling but also inhibit the malignant progression in leukemic cell models and samples from human patients (94). Therefore, there is a need for novel approaches that exploit the therapeutic potential of β-arrestin2 without interfering with their physiological roles. Instead of small molecules, targeting β-arrestin oligomers or cell- specific targeting of these proteins may lead to novel therapeutic approaches to treat neurodegenerative and other diseases.
Author contributions
All listed authors have made a substantial, direct, and intellectual contribution to the work and have approved it for publication.
Funding
This work was supported by the Hungarian National Research, Development and Innovation Office Grants OTKA [K-139231, FK-138862 and PD-124061], the János Bolyai Research Scholarship and János Bolyai Research Scholarship Plus of the Hungarian Academy of Sciences BO/00807/21 and ÚNKP-21-5-SE-29.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cairns NJ, Lee VMY, Trojanowski JQ. The cytoskeleton in neurodegenerative diseases. J Pathol (2004) 204(4):438–49. doi: 10.1002/path.1650
2. Pfister C, Chabre M, Plouet J, Tuyen VV, De Kozak Y, Faure JP, et al. Retinal s antigen identified as the 48K protein regulating light-dependent phosphodiesterase in rods. Science (1985) 228:891–3. doi: 10.1126/science.2988124
3. Caron MG, Barak LS. A brief history of the β-arrestins. Methods Mol Biol (2019) 1957:3–8. doi: 10.1007/978-1-4939-9158-7_1
4. Jiang H, Galtes D, Wang J, Rockman HA. G Protein-coupled receptor signaling: transducers and effectors. Am J Physiol Cell Physiol (2022) 323(3):C731–48. doi: 10.1152/ajpcell.00210.2022
5. Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature (2014) 512(7513):218–22. doi: 10.1038/nature13430
6. Wisler JW, Rockman HA, Lefkowitz RJ. Biased G protein-coupled receptor signaling: Changing the paradigm of drug discovery. Circulation (2018) 137(22):2315–7. doi: 10.1161/CIRCULATIONAHA.117.028194
7. Turu G, Balla A, Hunyady L. The role of β-arrestin proteins in organization of signaling and regulation of the AT1 angiotensin receptor. Front Endocrinol (2019) 10:519. doi: 10.3389/fendo.2019.00519
8. Seyedabadi M, Gharghabi M, Gurevich EV, Gurevich VV. Receptor-arrestin interactions: The GPCR perspective. Biomolecules (2021) 11(2). doi: 10.3390/biom11020218
9. Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem (2000) 275(22):17201–10. doi: 10.1074/jbc.M910348199
10. Shenoy SK, Lefkowitz RJ. Receptor-specific ubiquitination of β-arrestin directs assembly and targeting of seven-transmembrane receptor signalosomes *. J Biol Chem (2005) 280(15):15315–24. doi: 10.1074/jbc.M412418200
11. Shukla AK, Manglik A, Kruse AC, Xiao K, Reis RI, Tseng WC, et al. Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature (2013) 497(7447):137–41. doi: 10.1038/nature12120
12. Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell (2017) 170(3):457–69.e13. doi: 10.1016/j.cell.2017.07.002
13. Tóth AD, Prokop S, Gyombolai P, Várnai P, Balla A, Gurevich VV, et al. Heterologous phosphorylation-induced formation of a stability lock permits regulation of inactive receptors by β-arrestins. J Biol Chem (2018) 293(3):876–92. doi: 10.1074/jbc.M117.813139
14. Yang Z, Yang F, Zhang D, Liu Z, Lin A, Liu C, et al. Phosphorylation of G protein-coupled receptors: From the barcode hypothesis to the flute model. Mol Pharmacol (2017) 92(3):201–10. doi: 10.1124/mol.116.107839
15. Maharana J, Sarma P, Yadav MK, Saha S, Singh V, Saha S, et al. Structural snapshots uncover a lock-and-key type conserved activation mechanism of β-arrestins by GPCRs. bioRxiv (2022) 2022.10.10.511556. doi: 10.1101/2022.10.10.511556
16. Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci U S A (2007) 104(29):12011–6. doi: 10.1073/pnas.0704849104
17. Lefkowitz RJ. Arrestins come of age: a personal historical perspective. Prog Mol Biol Transl Sci (2013) 118:3–18. doi: 10.1016/B978-0-12-394440-5.00001-2
18. Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol (2018) 25(1):4–12. doi: 10.1038/s41594-017-0011-7
19. Ahn S, Shenoy SK, Luttrell LM, Lefkowitz RJ. SnapShot: β-arrestin functions. Cell (2020) 182(5):1362–2. doi: 10.1016/j.cell.2020.07.034
20. Gurevich VV, Gurevich EV, Uversky VN. Arrestins: structural disorder creates rich functionality. Protein Cell (2018) 9(12):986–1003. doi: 10.1007/s13238-017-0501-8
21. Conde C, Cáceres A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci (2009) 10(5):319–32. doi: 10.1038/nrn2631
22. de Forges H, Bouissou A, Perez F. Interplay between microtubule dynamics and intracellular organization. Int J Biochem Cell Biol (2012) 44(2):266–74. doi: 10.1016/j.biocel.2011.11.009
23. Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature (1984) 312(5991):237–42. doi: 10.1038/312237a0
24. Nair KS, Hanson SM, Kennedy MJ, Hurley JB, Gurevich VV, Slepak VZ. Direct binding of visual arrestin to microtubules determines the differential subcellular localization of its splice variants in rod photoreceptors. J Biol Chem (2004) 279(39):41240–8. doi: 10.1074/jbc.M406768200
25. Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, et al. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron (2005) 46(4):555–67. doi: 10.1016/j.neuron.2005.03.023
26. Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem (2006) 281(14):9765–72. doi: 10.1074/jbc.M510738200
27. Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song X, et al. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol (2007) 368:375–87. doi: 10.1016/j.jmb.2007.02.053
28. Yuen EY, Jiang Q, Chen P, Feng J, Yan Z. Activation of 5-HT2A/C receptors counteracts 5-HT1A regulation of n-methyl-D-aspartate receptor channels in pyramidal neurons of prefrontal cortex. J Biol Chem (2008) 283(25):17194–204. doi: 10.1074/jbc.M801713200
29. Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, et al. Structure and function of the visual arrestin oligomer. EMBO J (2007) 26(6):1726–36. doi: 10.1038/sj.emboj.7601614
30. Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 a: possible mechanism of receptor binding and membrane translocation. Structure (2001) 9(9):869–80. doi: 10.1016/s0969-2126(01)00644-x
31. Gurevich VV, Gurevich EV. Solo vs. chorus: Monomers and oligomers of arrestin proteins. Int J Mol Sci (2022) 23(13). doi: 10.3390/ijms23137253
32. Milano SK, Kim YM, Stefano FP, Benovic JL, Brenner C. Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J Biol Chem (2006) 281(14):9812–23. doi: 10.1074/jbc.M512703200
33. Hanson SM, Vishnivetskiy SA, Hubbell WL, Gurevich VV. Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry (2008) 47(3):1070–5. doi: 10.1021/bi7021359
34. Hoeppner CZ. Identification of a nuclear localization sequence in beta-Arrestin1:Implications in NF- kB activation (2012). Chicago: University of Illinois. Available at: https://indigo.uic.edu/articles/thesis/Identification_of_a_Nuclear_Localization_Sequence_in_Beta-Arrestin1_Implications_in_NF-_kB_Activation/10949873 (Accessed cited 2022 Sep 23).
35. Scott MGH, Pierotti V, Storez H, Lindberg E, Thuret A, Muntaner O, et al. Cooperative regulation of extracellular signal-regulated kinase activation and cell shape change by filamin a and beta-arrestins. Mol Cell Biol (2006) 26(9):3432–45. doi: 10.1128/MCB.26.9.3432-3445.2006
36. Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem (2006) 281(30):21491–9. doi: 10.1074/jbc.M603659200
37. Storez H, Scott MGH, Issafras H, Burtey A, Benmerah A, Muntaner O, et al. Homo- and hetero-oligomerization of beta-arrestins in living cells. J Biol Chem (2005) 280(48):40210–5. doi: 10.1074/jbc.M508001200
38. Kovacs JJ, Whalen EJ, Liu R, Xiao K, Kim J, Chen M, et al. β-Arrestin–mediated localization of smoothened to the primary cilium. Science (2008) 320(5884):1777–81. doi: 10.1126/science.1157983
39. Molla-Herman A, Boularan C, Ghossoub R, Scott MGH, Burtey A, Zarka M, et al. Targeting of β-Arrestin2 to the centrosome and primary cilium: Role in cell proliferation control. PloS One (2008) 3:e3728. doi: 10.1371/journal.pone.0003728
40. Ishikawa H, Marshall WF. Ciliogenesis: building the cell’s antenna. Nat Rev Mol Cell Biol (2011) 12(4):222–34. doi: 10.1038/nrm3085
41. Molla-Herman A, Davis KM, Mykytyn K, Benmerah A. Monitoring β-arrestin 2 targeting to the centrosome, basal body, and primary cilium by fluorescence microscopy [Internet]. Beta-Arrestins (2019), 271–89. doi: 10.1007/978-1-4939-9158-7_17
42. Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci (2004) 117(Pt 20):4619–28. doi: 10.1242/jcs.01481
43. Blanchoin L, Boujemaa-Paterski R, Sykes C, Plastino J. Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev (2014) 94(1):235–63. doi: 10.1152/physrev.00018.2013
44. Bhattacharya M, Anborgh PH, Babwah AV, Dale LB, Dobransky T, Benovic JL, et al. β-arrestins regulate a Ral-GDS–ral effector pathway that mediates cytoskeletal reorganization. Nat Cell Biol (2002) 4:547–55. doi: 10.1038/ncb821
45. Bhattacharya M, Anborgh PH, Babwah AV, Dale LB, Dobransky T, Benovic JL, et al. Beta-arrestins regulate a ral-GDS ral effector pathway that mediates cytoskeletal reorganization. Nat Cell Biol (2002) 4(8):547–55. doi: 10.1038/ncb821
46. Barnes WG, Reiter E, Violin JD, Ren XR, Milligan G, Lefkowitz RJ. β-arrestin 1 and Gαq/11 coordinately activate RhoA and stress fiber formation following receptor stimulation. J Biol Chem (2005) 280:8041–50. doi: 10.1074/jbc.m412924200
47. Meyer G, Feldman EL. Signaling mechanisms that regulate actin-based motility processes in the nervous system. J Neurochem (2002). doi: 10.1046/j.1471-4159.2002.01185.x
48. Laroche G, Rochdi MD, Laporte SA, Parent JL. Involvement of actin in agonist-induced endocytosis of the G protein-coupled receptor for thromboxane A2. J Biol Chem (2005) 280:23215–24. doi: 10.1074/jbc.m414071200
49. Kohout TA. Beta -arrestin 1 and 2 differentially regulate heptahelical receptor signaling and trafficking. Proc Natl Acad Sci (2001) 98:1601–6. doi: 10.1073/pnas.041608198
50. Cleghorn WM, Bulus N, Kook S, Gurevich VV, Zent R, Gurevich EV. Non-visual arrestins regulate the focal adhesion formation via small GTPases RhoA and Rac1 independently of GPCRs. Cell Signal (2018) 42:259–69. doi: 10.1016/j.cellsig.2017.11.003
51. Herrmann H, Aebi U. Intermediate filaments and their associates: multi-talented structural elements specifying cytoarchitecture and cytodynamics. Curr Opin Cell Biol (2000) 12(1):79–90. doi: 10.1016/S0955-0674(99)00060-5
52. Fuchs E, Weber K. Intermediate filaments: structure, dynamics, function, and disease. Annu Rev Biochem (1994) 63:345–82. doi: 10.1146/annurev.bi.63.070194.002021
53. Pillai S, Trevino J, Rawal B, Singh S, Kovacs M, Li X, et al. β-Arrestin-1 mediates nicotine-induced metastasis through E2F1 target genes that modulate epithelial–mesenchymal transition. Cancer Res (2015) 75:1009–20. doi: 10.1158/0008-5472.can-14-0681
54. Rosanò L, Bagnato A. New insights into the regulation of the actin cytoskeleton dynamics by GPCR/β-arrestin in cancer invasion and metastasis. Int Rev Cell Mol Biol (2019) 346:129–55. doi: 10.1016/bs.ircmb.2019.03.002
55. Scott MGH, Le Rouzic E, Périanin A, Pierotti V, Enslen H, Benichou S, et al. Differential nucleocytoplasmic shuttling of β-arrestins: Characterization of a leucine-rich nuclear export signal in β-arrestin2. Boll Soc Ital Biol Sper (2002) 277(40):P37693–701. doi: 10.1074/jbc.M207552200
56. Hunton DL, Barnes WG, Kim J, Ren XR, Violin JD, Reiter E, et al. Beta-arrestin 2-dependent angiotensin II type 1A receptor-mediated pathway of chemotaxis. Mol Pharmacol (2005) 67(4):1229–36. doi: 10.1124/mol.104.006270
57. Bravo-Cordero JJ, Magalhaes MAO, Eddy RJ, Hodgson L, Condeelis J. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol (2013) 14(7):405–15. doi: 10.1038/nrm3609
58. DeFea KA. Arrestins in actin reorganization and cell migration. Prog Mol Biol Transl Sci (2013) 118:205–22. doi: 10.1016/B978-0-12-394440-5.00008-5
59. McGovern KW, DeFea KA. Molecular mechanisms underlying beta-arrestin-dependent chemotaxis and actin-cytoskeletal reorganization. Handb Exp Pharmacol (2014) 219:341–59. doi: 10.1007/978-3-642-41199-1_17
60. Zoudilova M, Ge L, Kumar P, Bokoch GM, DeFea KA. Beta-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J Biol Chem (2007) 21(28):20634–46. doi: 10.1096/fasebj.21.6.a993-d
61. Cai Y, Yang C, Yu X, Qian J, Dai M, Wang Y, et al. Deficiency of β-arrestin 2 in dendritic cells contributes to autoimmune diseases. J Immunol (2019) 202:407–20. doi: 10.4049/jimmunol.1800261
62. Azam S, Haque ME, Jakaria M, Jo SH, Kim IS, Choi DK. G-Protein-Coupled receptors in CNS: A potential therapeutic target for intervention in neurodegenerative disorders and associated cognitive deficits. Cells (2020) 9(2). doi: 10.3390/cells9020506
63. Du X, Wang X, Geng M. Alzheimer’s disease hypothesis and related therapies. Transl Neurodegener (2018) 7(2). doi: 10.1186/s40035-018-0107-y
64. Xin SH, Tan L, Cao X, Yu JT, Tan L. Clearance of amyloid beta and tau in alzheimer’s disease: from mechanisms to therapy. Neurotox Res (2018) 34(3):733–48. doi: 10.1007/s12640-018-9895-1
65. Hampel H, Hardy J, Blennow K, Chen C, Perry G, Kim SH, et al. The amyloid-β pathway in alzheimer’s disease. Mol Psychiatry (2021) 26(10):5481–503. doi: 10.1038/s41380-021-01249-0
66. Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell (1992) 3(10):1141–54. doi: 10.1091/mbc.3.10.1141
67. Lee G, Leugers CJ. Tau and tauopathies. Prog Mol Biol Transl Sci (2012) 107:263–93. doi: 10.1016/B978-0-12-385883-2.00004-7
68. Bychkov ER, Gurevich VV, Joyce JN, Benovic JL, Gurevich EV. Arrestins and two receptor kinases are upregulated in parkinson’s disease with dementia. Neurobiol Aging (2008) 29(3):379–96. doi: 10.1016/j.neurobiolaging.2006.10.012
69. Wang D, Govindaiah G, Liu R, De Arcangelis V, Cox CL, Xiang YK. Binding of amyloid β peptide to β 2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J (2010) 24:3511–21. doi: 10.1096/fj.10-156661
70. Wang D, Yuen EY, Zhou Y, Yan Z, Xiang YK. Amyloid beta peptide-(1-42) induces internalization and degradation of beta2 adrenergic receptors in prefrontal cortical neurons. J Biol Chem (2011) 286(36):31852–63. doi: 10.1074/jbc.M111.244335
71. Wang D, Fu Q, Zhou Y, Xu B, Shi Q, Igwe B, et al. β2 adrenergic receptor, protein kinase a (PKA) and c-jun n-terminal kinase (JNK) signaling pathways mediate tau pathology in Alzheimer disease models. J Biol Chem (2013) 288(15):10298–307. doi: 10.1074/jbc.M112.415141
72. Zhang X, Li Y, Xu H, Zhang YW. The γ-secretase complex: from structure to function. Front Cell Neurosci (2014) 8:427. doi: 10.3389/fncel.2014.00427
73. De Strooper B. Aph-1, pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron (2003) 38(1):9–12. doi: 10.1016/S0896-6273(03)00205-8
74. Serneels L, Van Biervliet J, Craessaerts K, Dejaegere T, Horré K, Van Houtvin T, et al. Gamma-secretase heterogeneity in the Aph1 subunit: relevance for alzheimer’s disease. Science (2009) 324(5927):639–42. doi: 10.1126/science.1171176
75. Liu X, Zhao X, Zeng X, Bossers K, Swaab DF, Zhao J, et al. β-arrestin1 regulates γ-secretase complex assembly and modulates amyloid-β pathology. Cell Res (2013) 23(3):351–65. doi: 10.1038/cr.2012.167
76. Thathiah A, Horré K, Snellinx A, Vandewyer E, Huang Y, Ciesielska M, et al. β-arrestin 2 regulates aβ generation and γ-secretase activity in alzheimer’s disease. Nat Med (2013) 19:43–9. doi: 10.1038/nm.3023
77. Bossers K, Wirz KTS, Meerhoff GF, Essing AHW, van Dongen JW, Houba P, et al. Concerted changes in transcripts in the prefrontal cortex precede neuropathology in alzheimer’s disease. Brain (2010) 133(Pt 12):3699–723. doi: 10.1093/brain/awq258
78. Pontrello CG, Sun MY, Lin A, Fiacco TA, DeFea KA, Ethell IM. Cofilin under control of β-arrestin-2 in NMDA-dependent dendritic spine plasticity, long-term depression (LTD), and learning. Proc Natl Acad Sci U S A (2012) 109(7):E442–51. doi: 10.1073/pnas.1118803109
79. Liu YQ, Jia MQ, Xie ZH, Liu XF, Hui-Yang, Zheng XL, et al. Arrestins contribute to amyloid beta-induced cell death via modulation of autophagy and the α7nAch receptor in SH-SY5Y cells. Sci Rep (2017) 7(1):3446. doi: 10.1038/s41598-017-01798-x
80. Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem (2005) 94(1):192–203. doi: 10.1111/j.1471-4159.2005.03181.x
81. Thathiah A. β-Arrestin2 arrests the clearance of tau in FTLD. Proc Natl Acad Sci U S A (2020) 117(13):6968–70. doi: 10.1073/pnas.2001455117
82. Cha-Molstad H, Yu JE, Feng Z, Lee SH, Kim JG, Yang P, et al. p62/SQSTM1/Sequestosome-1 is an n-recognin of the n-end rule pathway which modulates autophagosome biogenesis. Nat Commun (2017) 8(1):102. doi: 10.1038/s41467-017-00085-7
83. Woo JA, Yan Y, Kee TR, Cazzaro S, McGill Percy KC, Wang X, et al. β-arrestin1 promotes tauopathy by transducing GPCR signaling, disrupting microtubules and autophagy. Life Sci Alliance (2022) 5(3). doi: 10.26508/lsa.202101183
84. Jicha GA, Nelson PT. Management of frontotemporal dementia: targeting symptom management in such a heterogeneous disease requires a wide range of therapeutic options. Neurodegener Dis Manage (2011) 1(2):141–56. doi: 10.2217/nmt.11.9
85. Woo JAA, Liu T, Fang CC, Castaño MA, Kee T, Yrigoin K, et al. β-Arrestin2 oligomers impair the clearance of pathological tau and increase tau aggregates. Proc Natl Acad Sci U S A (2020) 117(9):5006–15. doi: 10.1073/pnas.1917194117
86. Feuillette S, Deramecourt V, Laquerriere A, Duyckaerts C, Delisle MB, Maurage CA, et al. Filamin-a and myosin VI colocalize with fibrillary tau protein in alzheimer’s disease and FTDP-17 brains. Brain Res (2010) 1345:182–9. doi: 10.1016/j.brainres.2010.05.007
87. Kang DE, Woo JA. Cofilin, a master node regulating cytoskeletal pathogenesis in alzheimer’s disease. J Alzheimers Dis (2019) 72(s1):S131–44. doi: 10.3233/JAD-190585
88. Woo JAA, Liu T, Fang CC, Cazzaro S, Kee T, LePochat P, et al. Activated cofilin exacerbates tau pathology by impairing tau-mediated microtubule dynamics. Commun Biol (2019) 2:112. doi: 10.1038/s42003-019-0359-9
89. Fang Y, Jiang Q, Li S, Zhu H, Xu R, Song N, et al. Opposing functions of β-arrestin 1 and 2 in parkinson’s disease via microglia inflammation and Nprl3. Cell Death Differ (2021) 28(6):1822–36. doi: 10.1038/s41418-020-00704-9
90. Ahmed MR, Zhan X, Song X, Kook S, Gurevich VV, Gurevich EV. Ubiquitin ligase parkin promotes Mdm2–arrestin interaction but inhibits arrestin ubiquitination. Biochemistry (2011) 50(18):3749–63. doi: 10.1021/bi200175q
91. Lu J, Li X, Wang Q, Pei G. Dopamine D2 receptor and β-arrestin 2 mediate amyloid-β elevation induced by anti-parkinson’s disease drugs, levodopa and piribedil, in neuronal cells. PloS One (2017) 12(3):e0173240. doi: 10.1371/journal.pone.0173240
92. Urs NM, Bido S, Peterson SM, Daigle TL, Bass CE, Gainetdinov RR, et al. Targeting β-arrestin2 in the treatment of l-DOPA–induced dyskinesia in parkinson’s disease. Proc Natl Acad Sci (2015) 112:E2517–26. doi: 10.1073/pnas.1502740112
93. Crépieux P, Poupon A, Langonné-Gallay N, Reiter E, Delgado J, Schaefer MH, et al. A comprehensive view of the β-arrestinome. Front Endocrinol (2017) 8:32. doi: 10.3389/fendo.2017.00032
Keywords: arrestin, microtubule, Alzheimer’s disease, actin, cytoskeleton, neurodegeneration
Citation: Szénási T, Turu G and Hunyady L (2023) Interactions between β-arrestin proteins and the cytoskeletal system, and their relevance to neurodegenerative disorders. Front. Endocrinol. 14:957981. doi: 10.3389/fendo.2023.957981
Received: 31 May 2022; Accepted: 04 January 2023;
Published: 09 February 2023.
Edited by:
Katarina Nikolic, University of Belgrade, SerbiaReviewed by:
Jung Alexa Woo, Case Western Reserve University, United StatesVsevolod V. Gurevich, Vanderbilt University, United States
Sadashiva Karnik, Case Western Reserve University, United States
Copyright © 2023 Szénási, Turu and Hunyady. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: László Hunyady, aHVueWFkeS5sYXN6bG9AdHRrLmh1
†These authors have contributed equally to this work