 Shengju Wang
Shengju Wang Shuai Qin1
Shuai Qin1 Qiu Chen
Qiu Chen- 1Department of Nephrology, The First Affiliated Hospital of Guizhou University of Traditional Chinese Medicine, Guiyang, Guizhou, China
- 2Department of Endocrinology, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan, China
- 3Diabetes Department, Jiaxing Hospital of Traditional Chinese Medicine, Jiaxing, Zhejiang, China
Diabetic nephropathy (DN) is one of the most serious chronic microvascular abnormalities of diabetes mellitus and the major cause of uremia. Accumulating evidence has confirmed that fibrosis is a significant pathological feature that contributes to the development of chronic kidney disease in DN. However, the exact mechanism of renal fibrosis in DN is still unclear, which greatly hinders the treatment of DN. Chinese herbal medicine (CHM) has shown efficacy and safety in ameliorating inflammation and albuminuria in diabetic patients. In this review, we outline the underlying mechanisms of renal fibrosis in DN, including oxidative stress (OS) generation and OS-elicited ASK1-p38/JNK activation. Also, we briefly summarize the current status of CHM treating DN by improving renal fibrosis. The treatment of DN by inhibiting ASK1 activation to alleviate renal fibrosis in DN with CHM will promote the discovery of novel therapeutic targets for DN and provide a beneficial therapeutic method for DN.
Introduction
Approximately 50% of diabetes patients develop diabetic nephropathy (DN) (1). DN is one of the major microvascular diseases of diabetes mellitus and is currently the most common cause of chronic renal failure among diabetic patients (2–5). DN has significant long-term impacts on the morbidity and mortality of diabetes patients. The distinctive features of DN are glomerular basement membrane thickening, matrix accumulation in both mesangium and tubulointerstitium, and podocyte dysfunction or depletion, leading to the occurrence of renal function decline (6–9).
Fibrosis is a process marked by excessive extracellular matrix (ECM) deposits that contribute to functional parenchyma damage by fibrotic tissue (10). The role of fibrotic alterations in DN development has been identified as a critical in favoring chronic renal failure in diabetes mellitus (11, 12). Studies have confirmed that the grade of the fibrotic changes within the renal cortical interstitium is positively correlated with serum creatinine levels in adults with DN (13–15). Renal fibrosis is characterized by certain pathological hallmarks encompassing myofibroblast activation, ECM, and renal inflammation. The kidney gradually becomes incapable of repairing itself because of progressing tissue lesions and inflammation, eventually resulting in renal fibrosis (16–18). However, renal fibrosis is a multifactorial and dynamic process that induces many cellular changes in response to injurious triggers. Studies have recognized many factors involved in the development of fibrosis of DN, including oxidative stress (OS), transforming growth factor (TGF)-β, and inflammatory cytokines (such as IL-1/6 and TNF-α) (19, 20).
OS and renal fibrosis
OS is always followed by an increase in the production of reactive oxygen species (ROS). A continuous hyperglycemic state contributes to the occurrence of OS when the local antioxidant functionality loses its ability to quench the overproduction of ROS (21, 22). Sufficient evidence has confirmed that OS has a significant impact on the pathogenesis of multiple diabetic complications, such as DN (23). OS is increased in diabetic renal tissue due to elevated inflammation, hypoxia, overproduction of NADPH oxidases, exhaustion of antioxidant proteins, and mitochondrial dysfunction (24). OS is one of the principal factors that hasten glomerulosclerosis and tubulointerstitial fibrosis, which is the common process contributing to uremia in DN (25–27). OS-activated redox-sensitive signals facilitate parenchymal cell dysfunction through apoptosis and necrosis and boost unchaining of inflammatory mediators (e.g., IL1/6 and INF-α). Inflammation and apoptosis significantly contribute to the activation and accumulation of myofibroblasts, and the overproduction of ECM cells leading to fibrosis in DN (28).
Furthermore, sustained hyperglycemia and OS promote the excessive production of advanced glycation end products (AGEs) (29). When the AGEs that are primarily cleared by the kidneys are elevated (30), they can induce the glomerular cells to release transforming growth factor-β1 (TGF-β1), causing glomerular sclerosis and interstitial tubular damage in response to the elevated production of ECM (31, 32). The augmentation of ECM results in renal fibrosis mainly produced by the recruitment of mesangial cells, promoting ECM accumulation, thickening of the glomerular and tubular membranes, and disability of the podocytes, finally causing cell death (33, 34).
Apoptosis in renal fibrosis
Apoptosis is characterized by a decrease in volume, cell surface blebbing, chromatin shrinkage, internucleosomal fragmentation of DNA, and production of apoptosis bodies; all these processes are indispensable in the maintenance of normal tissue homeostasis (35). However, excessive apoptosis or its dysfunction causes various pathological fibrosis processes (36). It has been shown that a persistent hyperglycemic state elicits apoptosis and leads to the decline of renal function in DN (37). A hyperglycemic environment amplifies apoptosis in multiple cell types within DN, encompassing the proximal tubule epithelial, endothelial, and interstitial cells (38, 39). Renal interstitial fibrosis is the ultimate common pathological consequence of chronic renal disease (40, 41), marked by tubular atrophy and ECM deposition (42). Apoptosis of the tubular epithelial cells is one of the hallmarks of tubular atrophy and interstitial fibrosis (43, 44). Apoptosis has been found in renal tubular cells from patients with DN, while the blockade of renal tubular cell apoptosis retards fibrosis (45–47).
OS and ROS are identified as apoptosis stimulators and regulators (48, 49). Exogenous ROS causes apoptosis in many types of cells (50); however, OS and ROS alone are incapable of inducing apoptosis, thus requiring the involvement of other cell death signaling pathways.
ASK1 and p38/JNK
p38/JNK and renal fibrosis
Renal biopsy analysis from patients has displayed overexpression of p38 in many glomerular injuries and DN (51, 52), especially within proliferative forms of glomerulonephritis (51). Overproduction of p38 promotes the accumulation of numerous proinflammatory and profibrotic cytokines, such as TNFα, TGF-β1, and monocyte chemoattractant protein-1 (MCP-1). In humans and rodents with DN, p38 is overexpressed in a wide range of renal cells, including podocytes, endothelial cells, tubular cells, mesangial cells, macrophages, and myofibroblasts, and is associated with disease development (51–53). These studies indicate that p38 signaling is a great contributor to the development of kidney inflammation and fibrosis.
There is accumulating evidence confirming the notion that c-Jun N-terminal kinase (JNK) serves as a powerful proapoptotic mechanism in stressed cells (54). It has been found that overexpression of JNK is capable of driving the production of apoptogenic mediators such as cytochrome c (55), as well as proinflammatory and profibrotic mediators such as IL-6 and TNF-β1 (56, 57), which contribute to fibrotic renal disease (58). Overactivation of JNK in renal cells can favor fibroblast and collagen production. Simultaneously, JNK signaling can hasten profibrotic TGF-β1 signaling, playing an essential role in the progression of fibrotic kidney disease (59). JNK expression activated by mediators such as TNF-α, IL-1/6, and angiotensin II promotes TGF-β1 generation via stimulation of the activator protein-1 (AP-1) (60, 61). Additionally, TGF-β1 positively promotes mitogen-activated protein kinase (MAPK) activation through ASK1 (62, 63). The JNK signal transduction pathway also promotes the tubular generation of thrombospondin-1, leading to positive feedback that stimulates the production of TGF-β1 (61). Hence, overexpression of JNK in renal cells, especially in tubular epithelial cells, causes ongoing renal injuries with renal cell necrosis followed by apoptosis leading to tubulointerstitial lesions via the enhancement of inflammation and fibrosis (64–67).
ASK1
Numerous publications indicate that novel apoptotic pathways play a vital role in the pathogenesis and progression of DN. Apoptosis signal-regulating kinase (ASK) 1 is an upstream kinase in the MAPK pathway. The MAPK pathway promotes OS-elicited renal apoptosis, kidney inflammation, and fibrosis, favoring DN development (68–70). Increased expression levels of ASK1 have been identified in kidney biopsy tissues from adults with DN (71). ASK1 stimulates the activation of p38 and JNK in response to the pathological OS, modulating different stress-induced responses in development and cellular function (72). The downstream MAPKs, p38, and JNK are significantly elevated in a range of renal tissues such as the glomerulus, vasculature, and renal tubule interstitium, thereby inducing apoptosis, inflammation, fibrosis, and renal damage in humans (73–75).
ASK1-p38/JNK pathway
Studies have shown that the deposition of glomerular ECMs is elevated as long as the MAPK signaling pathways are activated in the diabetic state. ROS quickly stimulates p38 and JNK through distinct signaling pathways, such as ASK1. Additionally, inhibitors of p38/JNK by either genetic or pharmacological methods enhance the inhibition of apoptosis activated by either ASK1 or ROS. Experiments have shown that in ASK1−/− cells, p38/JNK is not stimulated in response to ROS. Decreased expression levels of p38/JNK are critical underlying mechanisms of resistance to ROS-induced apoptosis (76). Activation of a preponderant negative mutant of p38/JNK also improved resistance to ROS-elicited apoptotic cell death (77). Hence, inhibition of the ASK1-p38/JNK pathway can be an important approach that protects against OS-induced apoptosis, which is a central mechanism of renal inflammation and fibrosis in DN (78–80).
Therapeutic strategies for renal fibrosis
Antioxidants
It is evident that oxidative injury induces apoptosis, necrosis, and cellular dysfunction in many pathologies (81, 82), implying that antioxidants may be an effective treatment for many diseases (82, 83). Although many ROS inhibitors have been recommended to treat DN, they fail to scavenge ROS within DN due to non-specificity, impotence, and insufficient pharmaceutical activity (84, 85). Moreover, the actions of the antioxidants may be based on interference within ROS-induced pathways (84, 85). Hence, effective antioxidant therapies should be focused on an understood specific OS/ROS-related target or pathway that contributes to the pathological process of a disease in a defined organ or tissue. Specific targets have been proposed that will benefit the pathological progression of oxidative damage while avoiding direct targeting of non-specific OS or ROS. Therefore, the ASK1-p38/JNK axis is a specific mechanism driving the progression of myofibroblast generation and renal inflammation and fibrosis in DN, making it a beneficial target for treating DN.
p38 or JNK inhibitor
Selective inhibition of p38 is efficacious in rodents with renal damage (51, 86, 87). However, these outcomes have not been replicated in clinical practice due to the impotence and side effects on other organs and tissues (88). Hence, the application prospect of p38 inhibition in a clinical setting is still uncertain. Clinical trials have demonstrated that JNK inhibitors alleviate fibrosis in lung and liver disease (89, 90). Studies have also shown that JNK inhibitors can block the progression of fibrotic renal changes in a rodent model (91, 92). However, many questions exist, primarily about the side effects. Studies have shown that although JNK inhibition in rodents can significantly protect the kidneys from inflammation and fibrosis damage, it can also aggravate albuminuria while alleviating renal inflammation (93–96). Moreover, the relative action of JNK1 and JNK2 in different types of renal damage is still unestablished. Furthermore, whether the combined JNK1 and JNK2 inhibitions will impact normal health is still unknown. Also, the application of JNK inhibition is not well understood, considering the actions of JNK expression in a wide range of cells following renal damage. p38 and JNK inhibitors have exhibited both helpful and detrimental actions within disease states (91); thus, there may be limitations to inhibiting all p38 and JNK targets as a disease state therapeutic. Therefore, the upstream kinases of the p38/JNK pathway could be an alternative target for fibrotic renal disease in DN.
ASK1 inhibition
ASK1 is a critical mediator of apoptosis, inflammation, and renal fibrosis and is an upstream kinase in the p38 and JNK pathways (95). ASK1 inhibition in clinical trials has already shown promising antifibrotic efficacies in non-alcoholic steatohepatitis (NASH) (97). Therefore, it will be beneficial to clarify whether the actions are translated into fibrosis within DN. Inhibition of ASK1 can halt inflammation, myofibroblast production, and collagen accumulation in rodents with fibrotic kidney disease (98). Data demonstrate that ASK1−/− mice develop normally. However, they have delayed pathological organ remolding, fibrotic changes in multiple disease states, reduced apoptosis-induced cell loss, and ROS generation (99–101), implicating that therapeutic targeting of ASK1 can be a viable way to inhibit the progression of oxidative damage, organ remolding, and fibrotic changing (102).
Chinese herbal medicine
Although current drugs such as angiotensin-converting enzyme inhibitor (ACEI) and angiotensin receptor blocker (ARB) are highly recommended for the treatment of DN because they can protect the renal function of people with DN via blood glucose and pressure control, they are a single-target and insufficient to delay DN progress to diabetic end-stage kidney disease (103). Therefore, discovering a beneficial agent for DN is a priority within modern medical research. Traditional Chinese medicine (TCM) is part of a therapeutic strategy widely used in treating diabetes and its complications in China for centuries due to having less toxic and adverse reactions (104–106). Herbs are the primary approach for medical treatment in TCM. Chinese herbal medicine (CHM) has shown efficacy in alleviating OS, inflammation, and albuminuria in diabetic patients (107). TCM has been increasingly used to prevent renal damage due to its clinical efficacy and safety (108). Considering the different mechanisms involved in the pathogenesis of renal fibrosis, CHM displays multiangled and multichanneled properties that are being applied in clinical practice to fight against renal fibrosis (109) (Table 1).
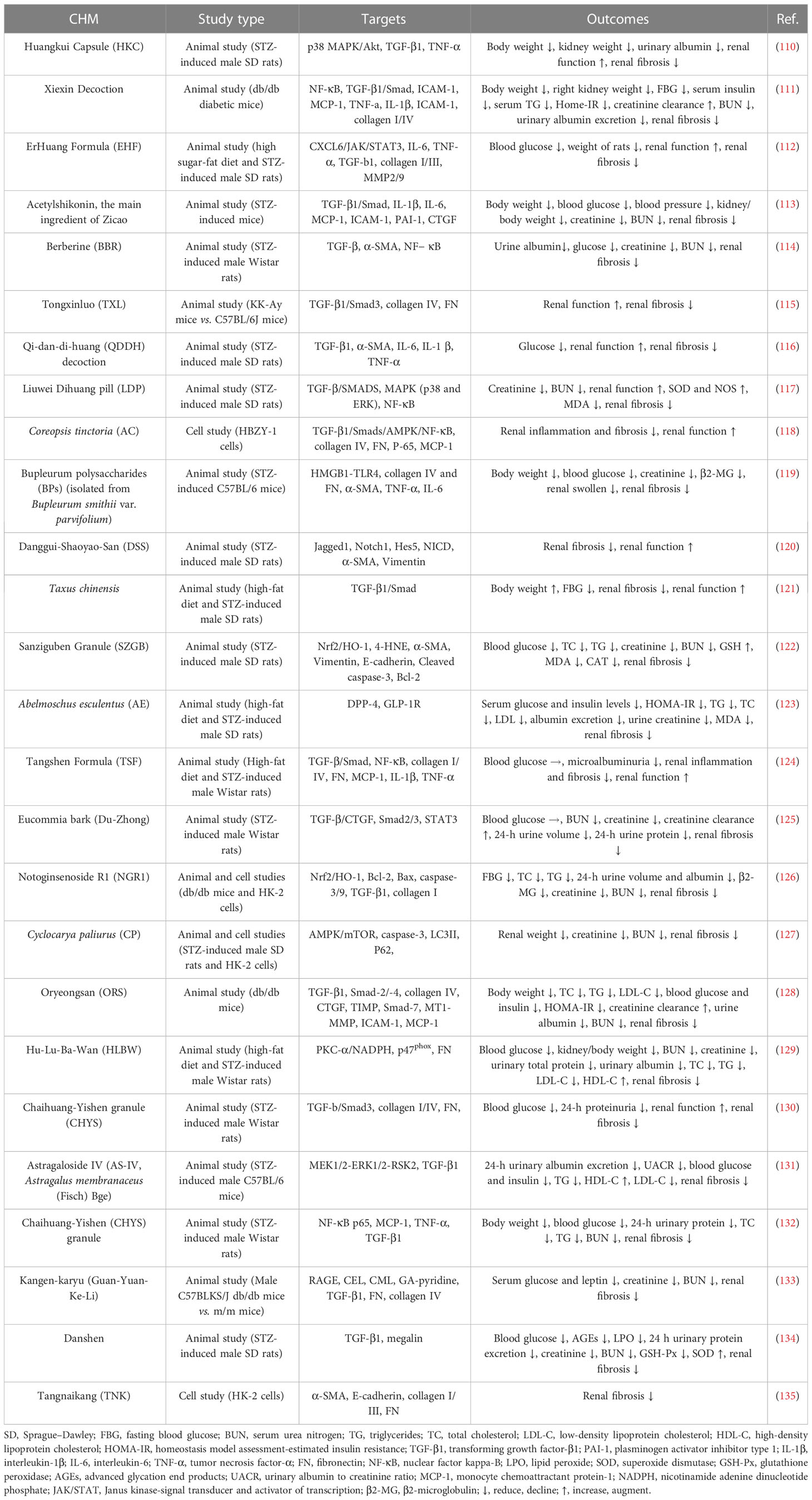
Table 1 Studies on the regulatory mechanism of CHM in the treatment of renal fibrosis in DN.
Conclusions and perspectives
In conclusion, modern treatments against blood glucose and pressure control can delay but inadequately block the development of multiple renal damages in DN. Hence, developing new therapeutic strategies that target the specific mechanisms of renal damage is becoming urgent. Mechanisms associated with stress-related inflammation and fibrosis are critical for kidney damage in DN and are not targeted by current small-molecule therapeutics.
The underlying pathological mechanisms of fibrosis in DN are closely associated with stress-elicited activation of the p38 and JNK pathways in various types of renal cells (91). Therapeutic inhibitors target these two protein kinases and block inflammation and fibrosis progression in animal models of renal disease (91); however, the toxic side effects such as liver toxicity, aggravation of albuminuria, and dizziness should not be ignored (88, 95, 96). Therefore, targeting the upstream signaling would be an optimal option. ASK1 is an upstream kinase of p38 and JNK and is critical for mediating numerous disease states such as apoptosis, inflammation, and renal fibrosis (95). More importantly, activation of ASK1 only happens in pathological conditions. Therefore, ASK1 can be seen as a novel target to halt the pathological progress of p38 and JNK activation in OS-related fibrotic renal change of DN.
CHM has demonstrated anti-inflammation and antifibrotic efficacy in several studies in rodents with renal fibrosis. We will carry out studies using CHM as an ASK1 inhibitor in an animal model to retard the progression of renal fibrosis in DN. When available, the data from the established DN rodents will be critical to further clarify the beneficial therapeutic properties of CHM as a potential ASK1 inhibitor, making it an ideal candidate therapy for treating patients with DN (Figure 1).
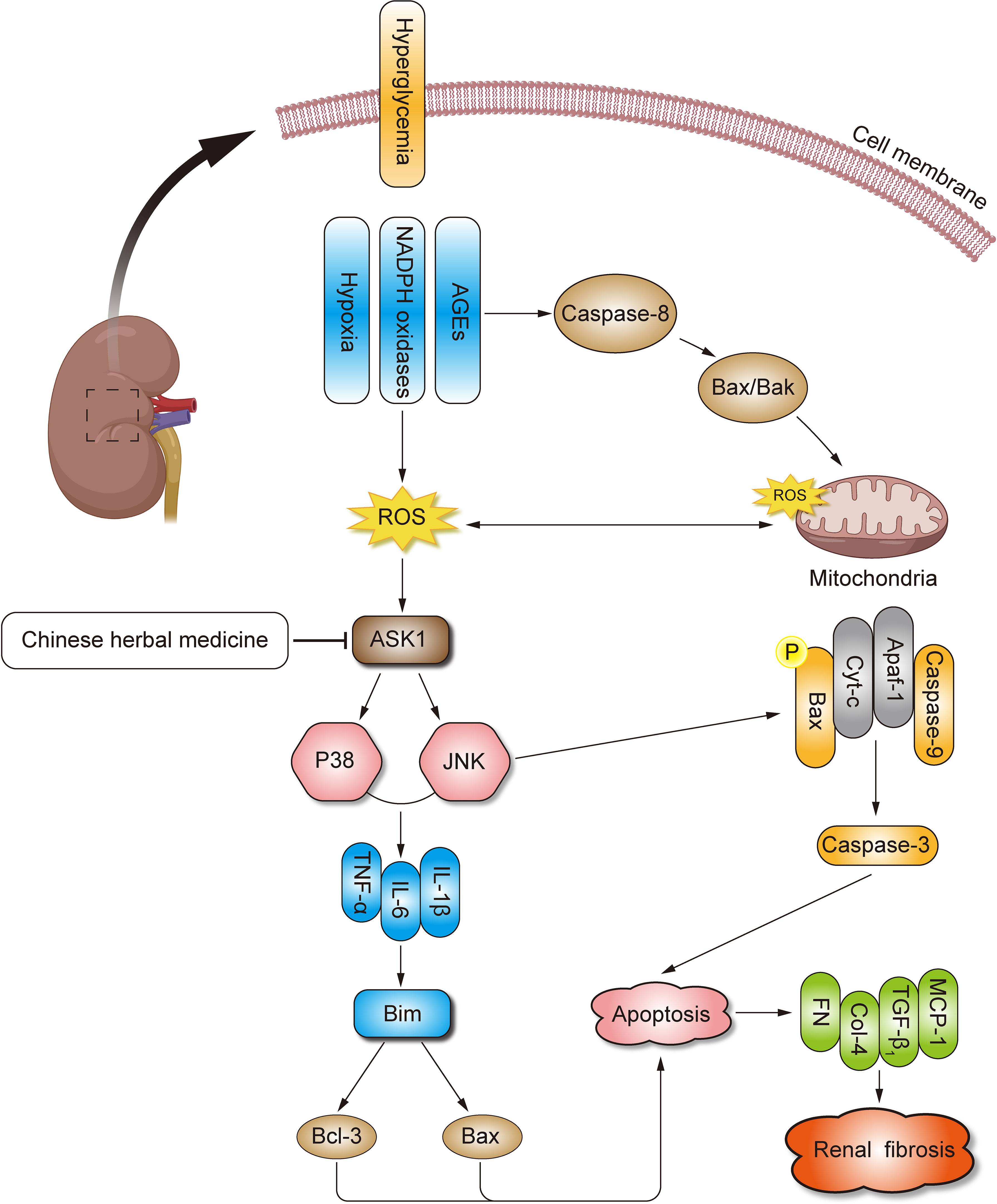
Figure 1 Mechanisms of CHM for DN. DN, diabetic nephropathy; CHM, Chinese herbal medicine; OS, oxidative stress; ECM, extracellular matrix; TGF-β, transforming growth factor-β; ROS, reactive oxygen species; AGEs, advanced glycation end products; MCP-1, monocyte chemoattractant protein-1; JNK, c-Jun N terminal kinase; AP-1, activator protein-1; ASK1, apoptosis signal-regulating kinase 1; MAPK, mitogen-activated protein kinase; NASH, non-alcoholic steatohepatitis; STZ, streptozotocin.
Author contributions
SW and SQ contributed to the conceptualization, writing and editing, and original draft preparation. BC contributed by analyzing the data and editing the table and figure. JZ and QC contributed through supervision, validation, reviewing, and funding. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Medical Service and Guarantee Capacity Improvement Subsidy Funds (Major and Difficult Diseases, grant number CYW2019079, to QC) and the National Natural Science Foundation of China (81460728 to JZ).
Acknowledgments
The authors thank AiMi Academic Services (www.aimieditor.com) for the English language editing and review services.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tuttle KR, Bakris GL, Bilous RW, Chiang JL, de Boer IH, Goldstein-Fuchs J, et al. Diabetic kidney disease: a report from an ADA consensus conference. Am J Kidney Dis (2014) 64(4):510–33. doi: 10.1053/j.ajkd.2014.08.001
2. Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, Johansen K, et al. 'United States renal data system 2011 annual data report: atlas of chronic kidney disease & end-stage renal disease in the United States. Am J Kidney Dis (2012) 59(1 Suppl 1):A7, e1–420. doi: 10.1053/j.ajkd.2011.11.015
3. Collins AJ, Foley RN, Chavers B, Gilbertson D, Herzog C, lshani A, et al. US Renal data system 2013 annual data report. Am J Kidney Dis (2014) 63(1 Suppl):A7. doi: 10.1053/j.ajkd.2013.11.001
4. Remuzzi G, Macia M, Ruggenenti P. Prevention and treatment of diabetic renal disease in type 2 diabetes: the BENEDICT study. J Am Soc Nephrol JASN (2006) 17(4 Suppl 2):S90–97. doi: 10.1681/ASN.2005121324
5. Zoja C, Zanchi C, Benigni A. Key pathways in renal disease progression of experimental diabetes. Nephrology dialysis Transplant (2015) 30 Suppl 4:iv54–59. doi: 10.1093/ndt/gfv036
6. Chen HY, Zhong X, Huang XR, Meng XM, You Y, Chung AC, et al. MicroRNA-29b inhibits diabetic nephropathy in db/db mice. Mol Ther (2014) 22(4):842–53. doi: 10.1038/mt.2013.235
7. Chen HY, Huang XR, Wang W, Li JH, Heuchel RL, Chung AC, et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes (2011) 60(2):590–601. doi: 10.2337/db10-0403
8. Valk EJ, Bruijn JA, Bajema IM. Diabetic nephropathy in humans: pathologic diversity. Curr Opin Nephrol hypertension (2011) 20(3):285–9. doi: 10.1097/MNH.0b013e328345bc1c
9. Haneda M, Utsunomiya K, Koya D, Babazono T, Mariya T, Makino H, et al. A new classification of diabetic nephropathy 2014: a report from joint committee on diabetic nephropathy. Clin Exp Nephrol (2015) 19(1):1–5. doi: 10.1007/s10157-014-1057-z
10. Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Kbnigshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol (2019) 15(12):705–30. doi: 10.1038/s41584-019-0322-7
11. Zeng LF, Xiao Y, Sun L. A glimpse of the mechanisms related to renal fibrosis in diabetic nephropathy. Adv Exp Med Biol (2019) 1165:49–79. doi: 10.1007/978-981-13-8871-2_4
12. Eddy AA. Molecular basis of renal fibrosis. Pediatr Nephrol (Berlin Germany) (2000) 15(3-4):290–301. doi: 10.1007/s004670000461
13. Nguyen TQ, Tarnow L, Andersen S, Hovind P, Parving HH, Goldschmeding R, et al. Urinary connective tissue growth factor excretion correlates with clinical markers of renal disease in a large population of type 1 diabetic patients with diabetic nephropathy. Diabetes Care (2006) 29(1):83–8. doi: 10.2337/diacare.29.01.06.dc05-1670
14. Yonemoto S, Machiguchi T, Nomura K, Minakata T, Nanno M, Yoshida H. Correlations of tissue macrophages and cytoskeletal protein expression with renal fibrosis in patients with diabetes mellitus. Clin Exp Nephrol (2006) 10(3):186–92. doi: 10.1007/s10157-006-0426-7
15. Chowdhury P, Sacks SH, Sheerin NS. Toll-like receptors TLR2 and TLR4 initiate the innate immune response of the renal tubular epithelium to bacterial products. Clin Exp Immunol (2006) 145(2):346–56. doi: 10.1111/j.1365-2249.2006.03116.x
16. Rice EK, Nikolic-Paterson DJ, David JR, Bucala R, Metz CN, Atkins RC, et al. Macrophage accumulation and renal fibrosis are independent of macrophage migration inhibitory factor in mouse obstructive nephropathy. Nephrol (Carlton Vic) (2004) 9(5):278–87. doi: 10.1111/j.1440-1797.2004.00319.x
17. Zheng S, Powell DW, Zheng F, Kantharidis P, Gnudi L. Diabetic nephropathy: proteinuria, inflammation, and fibrosis. J Diabetes Res (2016) 2016:5241549. doi: 10.1155/2016/5241549
18. Duffield JS. Cellular and molecular mechanisms in kidney fibrosis. J Clin Invest (2014) 124(6):2299–306. doi: 10.1172/JCI72267
19. Kanwar YS, Wada J, Sun L, Xie P, Wallner EI, Chen S, et al. Diabetic nephropathy: mechanisms of renal disease progression. Exp Biol Med (Maywood NJ) (2008) 233(1):4–11. doi: 10.3181/0705-MR-134
20. Ha H, Lee HB. Reactive oxygen species amplify glucose signalling in renal cells cultured under high glucose and in diabetic kidney. Nephrol (Carlton Vic) (2005) 10 Suppl:S7–10. doi: 10.1111/j.1440-1797.2005.00448.x
21. Sedeek M, Nasrallah R, Touyz RM, Hebert RL. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol JASN (2013) 24(10):1512–8. doi: 10.1681/ASN.2012111112
22. Babizhayev MA, Strokov IA, Nosikov VV, Savel'yeva EL, Sitnikov VF, Yegorov YE, et al. The role of oxidative stress in diabetic neuropathy: generation of free radical species in the glycation reaction and gene polymorphisms encoding antioxidant enzymes to genetic susceptibility to diabetic neuropathy in population of type I diabetic patients. Cell Biochem biophysics (2015) 71(3):1425–43. doi: 10.1007/s12013-014-0365-y
23. Luc K, Schramm-Luc A, Guzik TJ, Mikolajczyk TP. Oxidative stress and inflammatory markers in prediabetes and diabetes. J Physiol Pharmacol (2019) 70(6). doi: 10.26402/jpp.2019.6.01
24. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. New Engl J Med (2001) 345(12):861–9. doi: 10.1056/NEJMoa011161
25. Su H, Wan C, Song A, Qiu Y, Xiong W, Zhang C. Oxidative stress and renal fibrosis: mechanisms and therapies. Adv Exp Med Biol (2019) 1165:585–604. doi: 10.1007/978-981-13-8871-2_29
26. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest (2014) 124(6):2333–40. doi: 10.1172/JCI72271
27. Ponchiardi C, Mauer M, Najafian B. Temporal profile of diabetic nephropathy pathologic changes. Curr Diabetes Rep (2013) 13(4):592–9. doi: 10.1007/s11892-013-0395-7
28. Chow BSM, Allen TJ. Mouse models for studying diabetic nephropathy. Curr Protoc Mouse Biol (2015) 5(2):85–94. doi: 10.1002/9780470942390.mo140192
29. Monnier VM, Mustata GT, Biemel KL, Reihl O, Lederer MO, Zhenyu D, et al. Cross-linking of the extracellular matrix by the maillard reaction in aging and diabetes: an update on "a puzzle nearing resolution". Ann N Y Acad Sci (2005) 1043:533–44. doi: 10.1196/annals.1333.061
30. Sterenczak KA, Willenbrock S, Barann M, Klemke M, Soller JT, Eberle N, et al. Cloning, characterisation, and comparative quantitative expression analyses of receptor for advanced glycation end products (RAGE) transcript forms. Gene (2009) 434(1-2):35–42. doi: 10.1016/j.gene.2008.10.027
31. Forbes JM, Thallas V, Thomas MC, Founds HW, Burns WC, Jerums G, et al. The breakdown of preexisting advanced glycation end products is associated with reduced renal fibrosis in experimental diabetes. FASEB J (2003) 17(12):1762–4. doi: 10.1096/fj.02-1102fje
32. Zhou G, Li C, Cai L. Advanced glycation end-products induce connective tissue growth factor-mediated renal fibrosis predominantly through transforming growth factor beta-independent pathway. Am J Pathol (2004) 165(6):2033–43. doi: 10.1016/S0002-9440(10)63254-3
33. Manda G, Checherita AI, Comanescu MV, Hinescu ME. Redox signaling in diabetic nephropathy: hypertrophy versus death choices in mesangial cells and podocytes. Mediators Inflammation (2015) 2015:604208. doi: 10.1155/2015/604208
34. Lee HB, Yu MR, Yang Y, Jiang Z, Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J Am Soc Nephrol JASN (2003) 14(8 Suppl 3):S241–245. doi: 10.1097/01.ASN.0000077410.66390.0F
35. Verleih M, Borchel A, Rebl A, Brenmoehl J, Kuhn C, Goldammer T. A molecular survey of programmed cell death in rainbow trout: structural and functional specifications of apoptotic key molecules. Comp Biochem Physiol Part B Biochem Mol Biol (2019) 230:57–69. doi: 10.1016/j.cbpb.2019.01.012
36. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology (2014) 147(4):765–783.e764. doi: 10.1053/j.gastro.2014.07.018
37. Adeghate E. Molecular and cellular basis of the aetiology and management of diabetic cardiomyopathy: a short review. Mol Cell Biochem (2004) 261(1-2):187–91. doi: 10.1023/B:MCBI.0000028755.86521.11
38. Allen DA, Harwood S, Varagunam M, Raftery MJ, Yaqoob MM. High glucose-induced oxidative stress causes apoptosis in proximal tubular epithelial cells and is mediated by multiple caspases. FASEB J (2003) 17(8):908–10. doi: 10.1096/fj.02-0130fje
39. Dröge W. Free radicals in the physiological control of cell function. Physiol Rev (2002) 82(1):47–95. doi: 10.1152/physrev.00018.2001
40. Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol JASN (2010) 21(2):212–22. doi: 10.1681/ASN.2008121226
41. Humphreys BD. Mechanisms of renal fibrosis. Annu Rev Physiol (2018) 80:309–26. doi: 10.1146/annurev-physiol-022516-034227
42. Bohle A, Christ H, Grund KE, Mackensen S. The role of the interstitium of the renal cortex in renal disease. Contributions to Nephrol (1979) 16:109–14. doi: 10.1159/000402883
43. Johnson A, DiPietro LA. Apoptosis and angiogenesis: an evolving mechanism for fibrosis. FASEB J (2013) 27(10):3893–901. doi: 10.1096/fj.12-214189
44. Docherty NG, O'Sullivan OE, Healy DA, Fitzpatrick JM, Watson RW. Evidence that inhibition of tubular cell apoptosis protects against renal damage and development of fibrosis following ureteric obstruction. Am J Physiol Renal Physiol (2006) 290(1):F4–13. doi: 10.1152/ajprenal.00045.2005
45. Ren Y, Du C, Yan L, Wei J, Wu H, Shi Y, et al. CTGF siRNA ameliorates tubular cell apoptosis and tubulointerstitial fibrosis in obstructed mouse kidneys in a Sirt1-independent manner. Drug design Dev Ther (2015) 9:4155–71. doi: 10.2147/DDDT.S86748
46. Li H, Xu Y, Zhang Q, Xu H, Xu Y, Ling K. Microvesicles containing miR-34a induce apoptosis of proximal tubular epithelial cells and participate in renal interstitial fibrosis. Exp Ther Med (2019) 17(3):2310–6. doi: 10.3892/etm.2019.7197
47. Huang JS, Chuang LY, Guh JY, Huang YJ, Hsu MS. Antioxidants attenuate high glucose-induced hypertrophic growth in renal tubular epithelial cells. Am J Physiol Renal Physiol (2007) 293(4):F1072–1082. doi: 10.1152/ajprenal.00020.2007
48. Carmody RJ, Cotter TG. Signalling apoptosis: a radical approach. Redox Rep Commun Free Radical Res (2001) 6(2):77–90. doi: 10.1179/135100001101536085
49. Chandra J, Samali A, Orrenius S. Triggering and modulation of apoptosis by oxidative stress. Free Radical Biol Med (2000) 29(3-4):323–33. doi: 10.1016/S0891-5849(00)00302-6
50. Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxid Redox Signal (2002) 4(3):405–14. doi: 10.1089/15230860260196209
51. Stambe C, Nikolic-Paterson DJ, Hill PA, Dowling J, Atkins R. p38 mitogen-activated protein kinase activation and cell localization in human glomerulonephritis: correlation with renal injury. J Am Soc Nephrol JASN (2004) 15(2):326–36. doi: 10.1097/01.ASN.0000108520.63445.E0
52. Adhikary L, Chow F, Nikolic-Paterson DJ, Stambe C, Dowling J, Atkins RC, et al. Abnormal p38 mitogen-activated protein kinase signalling in human and experimental diabetic nephropathy. Diabetologia (2004) 47(7):1210–22. doi: 10.1007/s00125-004-1437-0
53. Stambe C, Atkins RC, Hill PA, Nikolic- Paterson DJ. Activation and cellular localization of the p38 and JNK MAPK pathways in rat crescentic glomerulonephritis. Kidney Int (2003) 64(6):2121–32. doi: 10.1046/j.1523-1755.2003.00324.x
54. Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front bioscience J virtual library (2003) 8:d369–391. doi: 10.2741/999
55. Aoki H, Kang PM, Hampe J, Yoshimura K, Noma T, Matsuzaki M, et al. Direct activation of mitochondrial apoptosis machinery by c-jun n-terminal kinase in adult cardiac myocytes. J Biol Chem (2002) 277(12):10244–50. doi: 10.1074/jbc.M112355200
56. Chow FY, Nikolic-Paterson DJ, Ozols E, Atkins RC, Rollin BJ, Tesch GH. Monocyte chemoattractant protein-1 promotes the development of diabetic renal injury in streptozotocin-treated mice. Kidney Int (2006) 69(1):73–80. doi: 10.1038/sj.ki.5000014
57. de Borst MH, Prakash J, Sandovici M, Klok PA, Hamming I, Kok RJ, et al. C-jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J Pharmacol Exp Ther (2009) 331(3):896–905. doi: 10.1124/jpet.109.154179
58. Grynberg K, Ma FY, Nikolic-Paterson DJ. The JNK signaling pathway in renal fibrosis. Front Physiol (2017) 8:829. doi: 10.3389/fphys.2017.00829
59. Stewart AG, Thomas B, Koff J. TGF-β: master regulator of inflammation and fibrosis. Respirology (Carlton Vic) (2018) 23(12):1096–7. doi: 10.1111/resp.13415
60. Lee KY, Ito K, Hayashi R, Jazrawi EP, Barnes PJ, Adcock IM. NF-kappaB and activator protein 1 response elements and the role of histone modifications in IL-1beta-induced TGF-beta1 gene transcription. J Immunol (Baltimore Md 1950) (2006) 176(1):603–15. doi: 10.4049/jimmunol.176.1.603
61. Naito T, Masaki T, Nikolic-Paterson DJ, Tanji C, Yorioka N, Kohno N. Angiotensin II induces thrombospondin-1 production in human mesangial cells via p38 MAPK and JNK: a mechanism for activation of latent TGF-beta1. Am J Physiol Renal Physiol (2004) 286(2):F278–287. doi: 10.1152/ajprenal.00139.2003
62. Seong HA, Manoharan R, Ha H. Coordinate activation of redox-dependent ASK1/TGF-β signaling by a multiprotein complex (MPK38, ASK1, SMADs, ZPR9, and TRX) improves glucose and lipid metabolism in mice. Antioxid Redox Signal (2016) 24(8):434–52. doi: 10.1089/ars.2015.6325
63. Tran P, Ho SM, Kim BG, Vuong TA, Leem YE, Bae GU, et al. TGF-β-activated kinase 1 (TAK1) and apoptosis signal-regulating kinase 1 (ASK1) interact with the promyogenic receptor cdo to promote myogenic differentiation via activation of p38MAPK pathway. J Biol Chem (2012) 287(15):11602–15. doi: 10.1074/jbc.M112.351601
64. Khalil N, Xu YD, O'Connor R, Duronio V. Proliferation of pulmonary interstitial fibroblasts is mediated by transforming growth factor-beta1-induced release of extracellular fibroblast growth factor-2 and phosphorylation of p38 MAPK and JNK. J Biol Chem (2005) 280(52):43000–9. doi: 10.1074/jbc.M510441200
65. Huang LL, Nikolic-Paterson DJ, Ma FY, Tesch GH. Aldosterone induces kidney fibroblast proliferation via activation of growth factor receptors and PI3K/MAPK signalling. Nephron Exp Nephrol (2012) 120(4):e115–122. doi: 10.1159/000339500
66. Panzhinskiy E, Zawada WM, Stenmark KR, Das M. Hypoxia induces unique proliferative response in adventitial fibroblasts by activating PDGFβ receptor-JNK1 signalling. Cardiovasc Res (2012) 95(3):356–65. doi: 10.1093/cvr/cvs194
67. Ren S, Johnson BG, Kida Y, Ip C, Davidson KC, Lin SL, et al. LRP-6 is a coreceptor for multiple fibrogenic signaling pathways in pericytes and myofibroblasts that are inhibited by DKK-1. Proc Natl Acad Sci U.S.A. (2013) 110(4):1440–5. doi: 10.1073/pnas.1211179110
68. Mueller PW, Rogus JJ, Cleary PA, Zhao Y, Smiles AM, Steffes MW, et al. Genetics of kidneys in diabetes (GoKinD) study: a genetics collection available for identifying genetic susceptibility factors for diabetic nephropathy in type 1 diabetes. J Am Soc Nephrol JASN (2006) 17(7):1782–90. doi: 10.1681/ASN.2005080822
69. Dluhy RG, McMahon GT. Intensive glycemic control in the ACCORD and ADVANCE trials. New Engl J Med (2008) 358(24):2630–3. doi: 10.1056/NEJMe0804182
70. Tesch GH, Ma FY, Han Y, Liles JT, Breckenridge DG, Nikolic- Paterson DJ. ASK1 inhibitor halts progression of diabetic nephropathy in Nos3-deficient mice. Diabetes (2015) 64(11):3903–13. doi: 10.2337/db15-0384
71. Liles JT, Corkey BK, Notte GT, Budas GR, Lansdon EB, Hinojosa-Kirschenbaum F, et al. ASK1 contributes to fibrosis and dysfunction in models of kidney disease. J Clin Invest (2018) 128(10):4485–500. doi: 10.1172/JCI99768
72. Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (2012) 337(6099):1190–5. doi: 10.1126/science.1222794
73. Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature (2011) 473(7345):43–9. doi: 10.1038/nature09906
74. Li Q, Seo JH, Stranger B, McKenna A, Pe'er I, Laframboise T, et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell (2013) 152(3):633–41. doi: 10.1016/j.cell.2012.12.034
75. Nathan DM, Bayless M, Cleary P, Genuth S, Gubitosi Klug - R, Lachin JM, et al. Diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: advances and contributions. Diabetes (2013) 62(12):3976–86. doi: 10.2337/db13-1093
76. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, et al. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep (2001) 2(3):222–8. doi: 10.1093/embo-reports/kve046
77. Li Y, Arita Y, Koo HC, Davis JM, Kazzaz JA. Inhibition of c-jun n-terminal kinase pathway improves cell viability in response to oxidant injury. Am J Respir Cell Mol Biol (2003) 29(6):779–83. doi: 10.1165/rcmb.2003-0087RC
78. Kim YS, Jhon DY, Lee KY. Involvement of ROS and JNK1 in selenite-induced apoptosis in Chang liver cells. Exp Mol Med (2004) 36(2):157–64. doi: 10.1038/emm.2004.22
79. Yoon SO, Park SJ, Yoon SY, Yun CH, Chung AS. Sustained production of H(2)O(2) activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-kappa b pathway. J Biol Chem (2002) 277(33):30271–82. doi: 10.1074/jbc.M202647200
80. Yoshizumi M, Kogame T, Suzaki Y, Fujita Y, Kyaw M, Kirima K, et al. Ebselen attenuates oxidative stress-induced apoptosis via the inhibition of the c-jun n-terminal kinase and activator protein-1 signalling pathway in PC12 cells. Br J Pharmacol (2002) 136(7):1023–32. doi: 10.1038/sj.bjp.0704808
81. Halliwell B. Oxidative stress and neurodegeneration: where are we now? J neurochemistry (2006) 97(6):1634–58. doi: 10.1111/j.1471-4159.2006.03907.x
82. Gur S, Abdel-Mageed AB, Sikka SC, Hellstrom WJG. Advances in stem cell therapy for erectile dysfunction. Expert Opin Biol Ther (2018) 18(11):1137–50. doi: 10.1080/14712598.2018.1534955
83. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell (2005) 120(4):483–95. doi: 10.1016/j.cell.2005.02.001
84. Schmidt HH, Stocker R, Vollbracht C, Paulsen G, Riley D, Daiber A, et al. Antioxidants in translational medicine. Antioxid Redox Signal (2015) 23(14):1130–43. doi: 10.1089/ars.2015.6393
85. Murphy MP. Antioxidants as therapies: can we improve on nature? Free Radical Biol Med (2014) 66:20–3. doi: 10.1016/j.freeradbiomed.2013.04.010
86. Jung DS, Li JJ, Kwak SJ, Lee SH, Park J, Song YS, et al. FR167653 inhibits fibronectin expression and apoptosis in diabetic glomeruli and in high-glucose-stimulated mesangial cells. Am J Physiol Renal Physiol (2008) 295(2):F595–604. doi: 10.1152/ajprenal.00624.2007
87. Wada T, Furuichi K, Sakai N, Hisada Y, Kobayashi K, Mukaida N, et al. Involvement of p38 mitogen-activated protein kinase followed by chemokine expression in crescentic glomerulonephritis. Am J Kidney Dis (2001) 38(6):1169–77. doi: 10.1053/ajkd.2001.29206
88. Hammaker D, Firestein GS. Go upstream, young man": lessons learned from the p38 saga. Ann rheumatic Dis (2010) 69 Suppl 1(Suppl 1):i77–82. doi: 10.1136/ard.2009.119479
89. van der Velden JL, Ye Y, Nolin JD, Hoffman SM, Chapman DG, Lahue KG, et al. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin Trans Med (2016) 5(1):36. doi: 10.1186/s40169-016-0117-2
90. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Kappe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med (2014) 6(8):1062–74. doi: 10.15252/emmm.201403856
91. Ma FY, Liu J, Nikolic-Paterson DJ. The role of stress-activated protein kinase signaling in renal pathophysiology. Braz J Med Biol Res (2009) 42(1):29–37. doi: 10.1590/s0100-879x2008005000049
92. Ma FY, Flanc RS, Tesch GH, Han Y, Atkins RC, Bennett BL, et al. A pathogenic role for c-jun amino-terminal kinase signaling in renal fibrosis and tubular cell apoptosis. J Am Soc Nephrol JASN (2007) 18(2):472–84. doi: 10.1681/ASN.2006060604
93. Flanc RS, Ma FY, Tesch GH, Han Y, Atkins RC, Bennett BL, et al. A pathogenic role for JNK signaling in experimental anti-GBM glomerulonephritis. Kidney Int (2007) 72(6):698–708. doi: 10.1038/sj.ki.5002404
94. Ma FY, Flanc RS, Tesch GH, Bennett BL, Friedman GC, Nikolic- Paterson DJ. Blockade of the c-jun amino terminal kinase prevents crescent formation and halts established anti-GBM glomerulonephritis in the rat. Lab investigation; J Tech Methods Pathol (2009) 89(4):470–84. doi: 10.1038/labinvest.2009.2
95. Ijaz A, Tejada T, Catanuto P, Xia X, Elliot SJ, Lenz O, et al. Inhibition of c-jun n-terminal kinase improves insulin sensitivity but worsens albuminuria in experimental diabetes. Kidney Int (2009) 75(4):381–8. doi: 10.1038/ki.2008.559
96. Lim AK, Ma FY, Nikolic-Paterson DJ, Ozols E, Young MJ, Bennett BL, et al. Evaluation of JNK blockade as an early intervention treatment for type 1 diabetic nephropathy in hypertensive rats. Am J Nephrol (2011) 34(4):337–46. doi: 10.1159/000331058
97. Challa TD, Wueest S, Lucchini FC, Dedual M, Modica S, Borsigova M, et al. Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol Med (2019) 11(10):e10124. doi: 10.15252/emmm.201810124
98. Ma FY, Tesch GH, Nikolic-Paterson DJ. ASK1/p38 signaling in renal tubular epithelial cells promotes renal fibrosis in the mouse obstructed kidney. Am J Physiol Renal Physiol (2014) 307(11):F1263–1273. doi: 10.1152/ajprenal.00211.2014
99. Tesch GH, Ma FY, Nikolic-Paterson DJ. Targeting apoptosis signal-regulating kinase 1 in acute and chronic kidney disease. Anatomical Rec (Hoboken NJ 2007) (2020) 303(10):2553–60. doi: 10.1002/ar.24373
100. Yamaguchi O, Higuchi Y, Hirotani S, Kashiwase K, Nakayama H, Hikoso S, et al. Targeted deletion of apoptosis signal-regulating kinase 1 attenuates left ventricular remodeling. Proc Natl Acad Sci U.S.A. (2003) 100(26):15883–8. doi: 10.1073/pnas.2136717100
101. Tasaki T, Yamada S, Guo X, Tanimoto A, Wang KY, Nabeshima A, et al. Apoptosis signal-regulating kinase 1 deficiency attenuates vascular injury-induced neointimal hyperplasia by suppressing apoptosis in smooth muscle cells. Am J Pathol (2013) 182(2):597–609. doi: 10.1016/j.ajpath.2012.10.008
102. Yamashita T, Yamamoto E, Kataoka K, Nakamura T, Matsuba S, Tokutomi Y, et al. Apoptosis signal-regulating kinase-1 is involved in vascular endothelial and cardiac remodeling caused by nitric oxide deficiency. Hypertension (Dallas Tex 1979) (2007) 50(3):519–24. doi: 10.1161/HYPERTENSIONAHA.107.092049
103. Kanasaki K, Taduri G, Koya D. Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol (2013) 4:7. doi: 10.3389/fendo.2013.00007
104. Tong XL, Dong L, Chen L, Zhen Z. Treatment of diabetes using traditional Chinese medicine: past, present and future. Am J Chin Med (2012) 40(5):877–86. doi: 10.1142/S0192415X12500656
105. Shi X, Lu XG, Zhan LB, Qi X, Liang LN, Hu SY, et al. The effects of the Chinese medicine ZiBu PiYin recipe on the hippocampus in a rat model of diabetes-associated cognitive decline: a proteomic analysis. Diabetologia (2011) 54(7):1888–99. doi: 10.1007/s00125-011-2147-z
106. Zhao HL, Sui Y, Qiao CF, Yip KY, Leung RK, Tsui SK, et al. Sustained antidiabetic effects of a berberine-containing Chinese herbal medicine through regulation of hepatic gene expression. Diabetes (2012) 61(4):933–43. doi: 10.2337/db11-1164
107. Xiao Y, Liu Y, Yu K, Zhou L, Bi J, Cheng J, et al. The effect of chinese herbal medicine on albuminuria levels in patients with diabetic nephropathy: a systematic review and meta-analysis. Evidence-Based complementary Altern Med eCAM (2013) 2013:937549. doi: 10.1155/2013/937549
108. Jiang M, Yang J, Zhang C, Liu B, Chan K, Cao H, et al. Clinical studies with traditional Chinese medicine in the past decade and future research and development. Planta Med (2010) 76(17):2048–64. doi: 10.1055/s-0030-1250456
109. Shen YL, Wang SJ, Rahman K, Zhang LJ, Zhang H. Chinese Herbal formulas and renal fibrosis: an overview. Curr Pharm design (2018) 24(24):2774–81. doi: 10.2174/1381612824666180829103355
110. Han W, Ma Q, Liu Y, Wu W, Tu Y, Huang L, et al. Huangkui capsule alleviates renal tubular epithelial-mesenchymal transition in diabetic nephropathy via inhibiting NLRP3 inflammasome activation and TLR4/NF-κB signaling. Phytomedicine Int J phytotherapy phytopharmacology (2019) 57:203–14. doi: 10.1016/j.phymed.2018.12.021
111. Wu JS, Shi R, Lu X, Ma YM, Cheng NN. Combination of active components of xiexin decoction ameliorates renal fibrosis through the inhibition of NF-κB and TGF-β1/Smad pathways in db/db diabetic mice. PloS One (2015) 10(3):e0122661. doi: 10.1371/journal.pone.0122661
112. Shen YL, Jiang YP, Li XQ, Wang SJ, Ma MH, Zhang CY, et al. ErHuang formula improves renal fibrosis in diabetic nephropathy rats by inhibiting CXCL6/JAK/STAT3 signaling pathway. Front Pharmacol (2019) 10:1596. doi: 10.3389/fphar.2019.01596
113. Li Z, Hong Z, Peng Z, Zhao Y, Shao R. Acetylshikonin from zicao ameliorates renal dysfunction and fibrosis in diabetic mice by inhibiting TGF-β1/Smad pathway. Hum Cell (2018) 31(3):199–209. doi: 10.1007/s13577-017-0192-8
114. Li Z, Zhang W. Protective effect of berberine on renal fibrosis caused by diabetic nephropathy. Mol Med Rep (2017) 16(2):1055–62. doi: 10.3892/mmr.2017.6707
115. Wu XM, Gao YB, Xu LP, Zou DW, Zhu ZY, Wang XL, et al. Tongxinluo inhibits renal fibrosis in diabetic nephropathy: involvement of the suppression of intercellular transfer of TGF-[Formula: see text]1-containing exosomes from GECs to GMCs. Am J Chin Med (2017) 45(5):1075–92. doi: 10.1142/S0192415X17500586
116. Ma F, Li L, Wang Q, Chen Z, You Y, Gao P, et al. Qi-dan-di-huang decoction alleviates diabetic nephropathy by inhibiting the NF-kappaB pathway. Front bioscience (Landmark edition) (2019) 24:1477–86. doi: 10.2741/4792
117. Xu ZJ, Shu S, Li ZJ, Liu YM, Zhang RY, Zhang Y. Liuwei dihuang pill treats diabetic nephropathy in rats by inhibiting of TGF-β/SMADS, MAPK, and NF-kB and upregulating expression of cytoglobin in renal tissues. Medicine (2017) 96(3):e5879. doi: 10.1097/MD.0000000000005879
118. Yao L, Li J, Li L, Li X, Zhang R, Zhang Y, et al. Coreopsis tinctoria nutt ameliorates high glucose-induced renal fibrosis and inflammation via the TGF-β1/SMADS/AMPK/NF-κB pathways. BMC complementary Altern Med (2019) 19(1):14. doi: 10.1186/s12906-018-2410-7
119. Liu ZZ, Weng HB, Zhang LJ, Pan LY, Sun W, Chen HX, et al. Bupleurum polysaccharides ameliorated renal injury in diabetic mice associated with suppression of HMGB1-TLR4 signaling. Chin J Natural Medicines (2019) 17(9):641–9. doi: 10.1016/S1875-5364(19)30078-0
120. Xiaobing L, Chunling N, Wenyu C, Yan C, Zhenzhen L. Effect of danggui-Shaoyao-San-containing serum on the renal tubular epithelial-mesenchymal transition of diabetic nephropathy. Curr Pharm Biotechnol (2020) 21(12):1204–12. doi: 10.2174/1389201021666200416094318
121. Weng HB, Han WK, Xiong YW, Jin ZH, Lan Z, Liu C, et al. Taxus chinensis ameliorates diabetic nephropathy through down-regulating TGF-β1/Smad pathway. Chin J Natural Medicines (2018) 16(2):90–6. doi: 10.1016/S1875-5364(18)30034-7
122. Zhang C, Li Q, Lai S, Yang L, Shi G, Wang Q, et al. Attenuation of diabetic nephropathy by sanziguben granule inhibiting EMT through Nrf2-mediated anti-oxidative effects in streptozotocin (STZ)-induced diabetic rats. J ethnopharmacology (2017) 205:207–16. doi: 10.1016/j.jep.2017.05.009
123. Peng CH, Lin HC, Lin CL, Wang CJ, Huang CN. Abelmoschus esculentus subfractions improved nephropathy with regulating dipeptidyl peptidase-4 and type 1 glucagon-like peptide receptor in type 2 diabetic rats. J Food Drug Anal (2019) 27(1):135–44. doi: 10.1016/j.jfda.2018.07.004
124. Zhao T, Sun S, Zhang H, Huang X, Yan M, Dong X, et al. Therapeutic effects of tangshen formula on diabetic nephropathy in rats. PloS One (2016) 11(1):e0147693. doi: 10.1371/journal.pone.0147693
125. Niu HS, Liu IM, Niu CS, Ku PM, Hsu CT, Cheng JT. Eucommia bark (Du-zhong) improves diabetic nephropathy without altering blood glucose in type 1-like diabetic rats. Drug design Dev Ther (2016) 10:971–8. doi: 10.2147/DDDT.S98558
126. Zhang B, Zhang X, Zhang C, Shen Q, Sun G, Sun X. Notoginsenoside R1 protects db/db mice against diabetic nephropathy via upregulation of Nrf2-mediated HO-1 expression. Molecules (Basel Switzerland) (2019) 24(2). doi: 10.3390/molecules24020247
127. Zhang XX, Jiang CH, Liu Y, Lou DX, Huang YP, Gao M, et al. Cyclocarya paliurus triterpenic acids fraction attenuates kidney injury via AMPK-mTOR-regulated autophagy pathway in diabetic rats. Phytomedicine (2019) 64:153060. doi: 10.1016/j.phymed.2019.153060
128. Yoon JJ, Lee YJ, Kang DG, Lee HS. Protective role of oryeongsan against renal inflammation and glomerulosclerosis in db/db mice. Am J Chin Med (2014) 42(6):1431–52. doi: 10.1142/S0192415X14500906
129. Zhou L, Dong H, Huang Y, Xu L, Zou X, Wang K, et al. Hu-Lu-Ba-Wan attenuates diabetic nephropathy in type 2 diabetic rats through PKC- α /NADPH oxidase signaling pathway. Evidence-Based complementary Altern Med eCAM (2013) 2013:504642. doi: 10.1155/2013/504642
130. Zhao TT, Zhang HJ, Lu XG, Huang XR, Zhang WK, Wang H, et al. Chaihuang-yishen granule inhibits diabetic kidney disease in rats through blocking TGF-β/Smad3 signaling. PloS One (2014) 9(3):e90807. doi: 10.1371/journal.pone.0090807
131. Song G, Han P, Sun H, Shao M, Yu X, Wang W, et al. Astragaloside IV ameliorates early diabetic nephropathy by inhibition of MEK1/2-ERK1/2-RSK2 signaling in streptozotocin-induced diabetic mice. J Int Med Res (2018) 46(7):2883–97. doi: 10.1177/0300060518778711
132. Zhang H, Zhao T, Gong Y, Dong X, Zhang W, Sun S, et al. Attenuation of diabetic nephropathy by chaihuang-yishen granule through anti-inflammatory mechanism in streptozotocin-induced rat model of diabetics. J ethnopharmacology (2014) 151(1):556–64. doi: 10.1016/j.jep.2013.11.020
133. Okamoto T, Park CH, Noh JS, Toriizuka K, Sei Y, Park JC, et al. Hepato-/reno-protective activity of Chinese prescription kangen-karyu through inhibition of AGE formation and fibrosis-related protein expression in type 2 diabetes. J Pharm Pharmacol (2011) 63(7):952–9. doi: 10.1111/j.2042-7158.2011.01299.x
134. Yin D, Yin J, Yang Y, Chen S, Gao X. Renoprotection of danshen injection on streptozotocin-induced diabetic rats, associated with tubular function and structure. J ethnopharmacology (2014) 151(1):667–74. doi: 10.1016/j.jep.2013.11.025
Keywords: diabetic nephropathy, renal fibrosis, Chinese herbal medicine, oxidative stress, ASK1
Citation: Wang S, Qin S, Cai B, Zhan J and Chen Q (2023) Promising therapeutic mechanism for Chinese herbal medicine in ameliorating renal fibrosis in diabetic nephropathy. Front. Endocrinol. 14:932649. doi: 10.3389/fendo.2023.932649
Received: 30 April 2022; Accepted: 22 June 2023;
Published: 14 July 2023.
Edited by:
In-Kyu Lee, Kyungpook National University, Republic of KoreaReviewed by:
Shangang Zhao, University of Texas Southwestern Medical Center, United StatesTomas Obsil, Charles University, Czechia
Copyright © 2023 Wang, Qin, Cai, Zhan and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jihong Zhan, emhhbmppaG9uZzAyNUBnenkuZWR1LmNu; Qiu Chen, Y2hlbnFpdTEwMDVAY2R1dGNtLmVkdS5jbg==
†These authors have contributed equally to this work