 Gan Wang
Gan Wang Tian-Yi Ma1
Tian-Yi Ma1 Kang Huang
Kang Huang Shi-Juan Lu
Shi-Juan Lu Jian-Jun Li
Jian-Jun Li- 1Department of Cardiology, Haikou Affiliated Hospital of Central South University Xiangya School of Medicine, Haikou, Hainan, China
- 2State Key Laboratory of Cardiovascular Diseases, Fu Wai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Diabetic cardiomyopathy (DCM), one of the common complications of diabetes, presents as a specific cardiomyopathy with anomalies in the structure and function of the heart. With the increasing prevalence of diabetes, DCM has a high morbidity and mortality worldwide. Recent studies have found that pyroptosis, as a programmed cell death accompanied by an inflammatory response, exacerbates the growth and genesis of DCM. These studies provide a theoretical basis for exploring the potential treatment of DCM. Therefore, this review aims to summarise the possible mechanisms by which pyroptosis promotes the development of DCM as well as the relevant studies targeting pyroptosis for the possible treatment of DCM, focusing on the molecular mechanisms of NLRP3 inflammasome-mediated pyroptosis, different cellular pyroptosis pathways associated with DCM, the effects of pyroptosis occurring in different cells on DCM, and the relevant drugs targeting NLRP3 inflammasome/pyroptosis for the treatment of DCM. This review might provide a fresh perspective and foundation for the development of therapeutic agents for DCM.
1 Introduction
Diabetes mellitus (DM) remains a crucial public health concern. Diabetes and its complications have brought a huge medical burden to people all over the world (1, 2). There is an undeniable connection between diabetes and cardiovascular disease. Some studies have confirmed that DM can increase the likelihood of heart failure, irrespective of usual heart failure risk factors including hypertension and coronary heart disease (2). Cardiovascular complications resulting from DM are among the primary factors that contribute to mortality in patients with diabetes (1). There are currently two main types of diabetes: type 1 diabetes(T1DM), which is mainly characterized by insulin deficiency, and type 2 diabetes(T2DM), which is mainly characterized by insulin resistance (3, 4). These two main types of DM can cause microvascular and macrovascular damage, leading to a variety of diabetes-related complications such as diabetic nephropathy, diabetic cardiomyopathy (DCM), diabetic retinopathy, and so on (5).
DCM is a distinctive cardiomyopathy defined by abnormal cardiac structure and function independent of other cardiac risk factors like coronary heart disease and hypertension (6). The pathophysiological mechanisms of DCM may be partially different in different types of DM, but less is known about the differences in the pathogenesis of DCM between different types of DM. In an analysis of a multiethnic sample, Eguchi et al. found that T2DM was positively associated with increased left ventricular mass, independent of factors such as obesity and race (7). However, few studies have shown that T1DM can promote the increase of left ventricular mass, which may be related to the younger age and insulin therapy in patients with type 1 diabetes (8). Unlike T1DM, T2DM is mainly caused by hyperglycaemia due to insulin resistance and hyperinsulinemia (3, 4). Insulin signaling in cells mainly involves two related pathways: the insulin receptor substrate 1 (IRS-1) pathway and the mitogen-activated protein kinase (MAPK) pathway (9). These two pathways crosstalk to form a complex and balanced insulin signaling system. However, insulin resistance breaks this balance, making the MAPK pathway dominate, and cell growth and metabolism become imbalanced, eventually leading to cardiac fibrosis and diastolic dysfunction (6, 9).
The development of DCM can be roughly divided into two stages: the early stage is marked by left ventricular hypertrophy and diastolic dysfunction, while heart failure with systolic dysfunction characterizes the late stage (10). Various metabolic disturbances, such as hyperglycaemia and hyperinsulinaemia, are present in DM. These disturbances have been found to cause cardiac hypertrophy and diastolic dysfunction, which are the primary manifestations in most DCM patients. As a result, DCM mainly leads to heart failure with preserved ejection fraction (HFpEF) (11–13). The occurrence and development of DCM is the result of a variety of factors. Previous reviews have summarized the possible mechanisms of DCM including insulin resistance, cardiac inflammation, advanced glycation end products (AGEs), and angiotensin II (Ang II) (14, 15). Nevertheless, the precise pathogenesis of DCM remains unclear.
Cardiomyocyte death is one of the key links in the development of DCM. In the last few years, the role of various programmed cell death in DCM has received extensive attention. There are also many ways of cell death in DCM, including cell apoptosis, autophagy, cell necrosis, and ferroptosis (16, 17). Pyroptosis is a recently identified type of programmed cell death that comes with an inflammatory response. In the process of DCM, metabolic disorders such as glucose and lipids are one of the triggers of myocardial injury. Inflammation plays an essential role in it (18, 19). At the same time, many scholars believe that inflammation plays a different role in different types of heart failure. Heart failure with reduced ejection fraction (HFrEF) is mainly related to myocardial ischemia and myocarditis, while HFpEF shows a greater association with inflammation (20). Clinically, the majority of patients with DCM have predominantly HFpEF, and pyroptosis accompanied by inflammation may be an important pathogenic factor of DM and DCM. Therefore, it is imperative to investigate the related mechanisms and therapeutic interventions.
2 Conception of pyroptosis
Pyroptosis is one of programmed cell death (21). The term ‘ pyroptosis ‘ was first coined by Cookson and Brennan in 2001. The word consists of ‘ pyro ‘ and ‘ ptosis ‘. Pyro ‘ means fire, which means inflammation accompanied by pyroptosis, while ‘ ptosis ‘ means fall, consistent with other programmed cell death (22). Pyroptosis is mainly triggered by inflammasomes and executed by the caspase and Gasdermin families (23, 24). Furthermore, pyroptosis can manifest in diverse cell types, including the digestive system, urinary system, central nervous system, reproductive system, and cardiovascular system (25). Pyroptosis promotes the development of multiple chronic diseases (26).
3 Molecular mechanism of pyroptosis
The Gasdermin family and the Caspase family play an important role in pyroptosis. The Gasdermin family is predominantly found in the gastrointestinal tract and skin (27, 28). The Gasdermin family mainly consists of six proteins: GASDMA, GASDMB, GASDMC, GASDMD, GASDME, and DFNB59 (28). In addition to DFNB59, the family members exhibit two conserved domains: the N-terminal pore-forming domain and the C-terminal repression domain (28). The cleavage and release of the N domains can lead to the creation of oligomeric pores, which have a diameter of approximately 10-20 nm (29). Pyroptosis is also regulated by caspases, an evolutionarily conserved family of cysteine proteases (30). Caspases hydrolyze a variety of cellular protein substrates to coordinate cell apoptosis, and caspases are also the key players in pyroptosis (31). The molecular mechanism of pyroptosis is mainly divided into three pathways (Figure 1): canonical pathway, non-canonical pathway, and Caspase-3/-8-mediated pathway (32). The canonical pathway is mainly mediated by Caspase-1, while the non-canonical pathway is independent of Caspase-1, mainly mediated by Caspase-4/-5/-11 (Caspase-4/-5 in humans, Caspase-11 in mice) (28, 32).
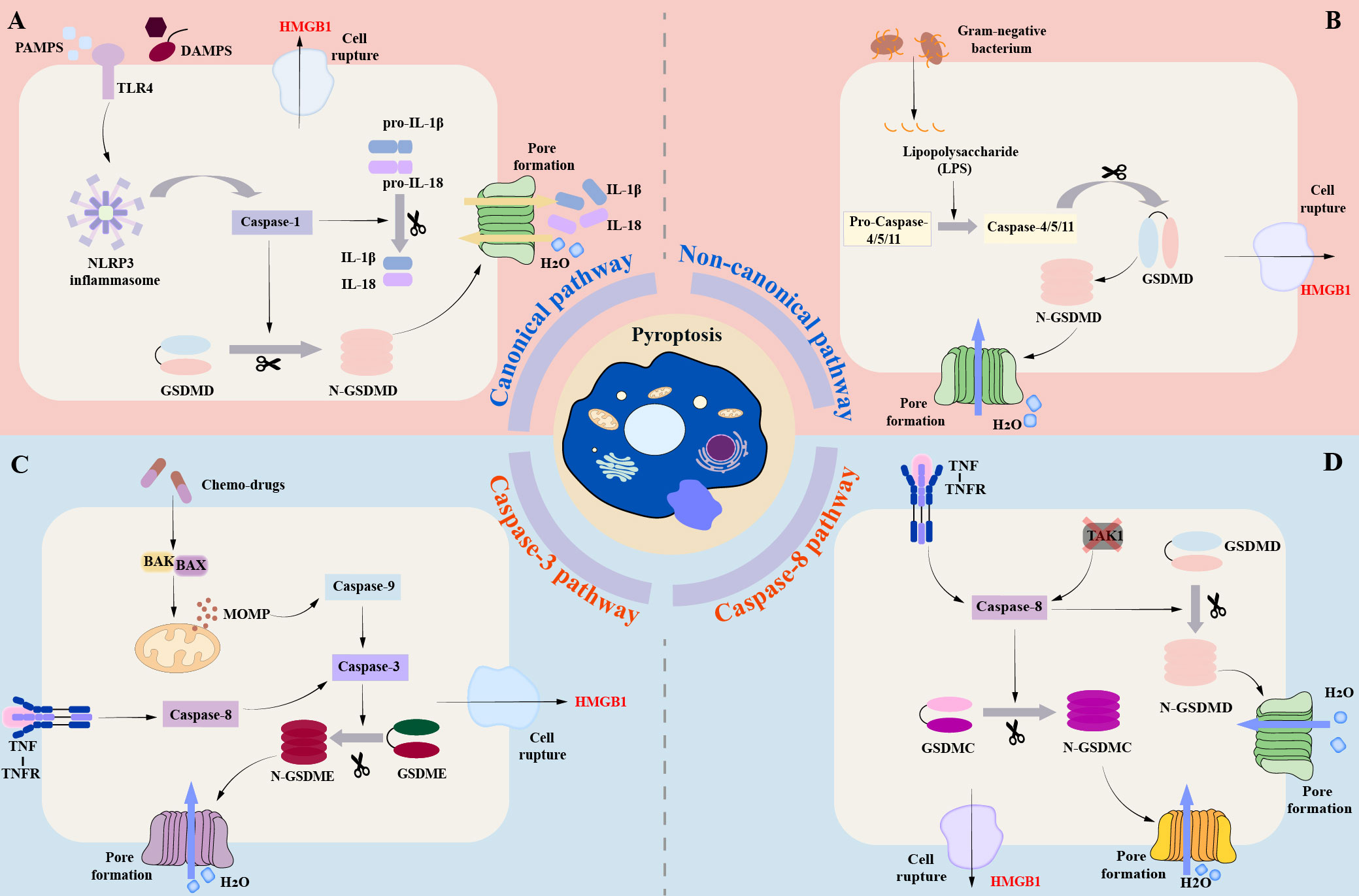
Figure 1 Brief molecular mechanism of Pyroptosis. (A) (canonical pathway): In the canonical pathway, when the host cell receptor recognizes various stimuli, it mainly includes pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), which can promote the activation of downstream pro-Caspase-1 into mature Csapase-1, and promote the assembly of inflammasome. Mature Caspase-1 can cleave GSDMD to form N-GASDMD. Subsequently, the N-terminal poreforming domain of GSDMD can non-selectively penetrate the cell membrane to form membrane pores, which further leads to cell swelling, cracking, and death. (B) (Non-canonical pathway): In the non-canonical pathway, LPS can directly bind to the CARD domain of Caspase-4 / 5 / 11 to achieve activation. The activated Caspase-4 / 5 / 11 can also cleave GSDMD to form N-GSDMD, thus promoting the occurrence of pyroptosis. (C) (Caspase-3-mediated pathway): Unlike the canonical and non-canonical pathways, activated Caspase-3 mainly mediates the formation of membrane pores by cutting GSDME and promoting the N-GSDME domain to the cell membrane, leading to the occurrence of pyroptosis. (D) (Caspase-8-mediated pathway): Under the stimulation of TNF-α, Caspase-8 can also specifically cleave GSDMC to produce N-GSDMC to induce pyroptosis.
In the canonical pathway, pattern-recognition receptors (PRRs) of the host cell are identified by recognizing a variety of stimuli, including two main categories, pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) (32, 33). Toll-like receptor 4 (TLR4), which is part of the pattern recognition receptor, is able to interact with PAMPs and DAMPs to initiate the NF-κB pathway (34). NF-κB binds to the inhibitory protein IκB and remains inactive in the cytoplasm (35). However, IKKβ phosphorylation can ubiquitinate IκBα, resulting in its dissociation from NF-κB (36, 37). Activated NF-κB is able to relocate to the nucleus, which ultimately initiates the transcription of genes associated with inflammation (38). In DCM, elevated glucose levels stimulate advanced glycation end product (AGE) synthesis, which binds with receptors for AGEs (RAGE) causing activation of the NF-κB pathway through phosphorylation of IKKβ (39). Inflammasomes are macromolecular protein complexes that play an important role in the immune system. The NLRP3 inflammasome is the most extensively studied, consisting of pyrin domain-containing NOD-like receptor protein 3 (NLRP3), CARD-containing apoptosis-associated speck-like protein (ASC), and effector protein Caspase-1 (40, 41). Typical activation of the NLRP3 inflammasome consists of two stages: initiation and subsequent activation. Initiation is mainly achieved via the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway (34). Studies have confirmed that the activation of NLRP3 inflammasome is a key step in pyroptosis, which also plays an essential role in promoting the occurrence and development of diabetic complications such as DCM (42, 43). The activated NLRP3 inflammasome can cleave pro-Caspase to generate active Caspase-1. Activated Caspase-1 can promote the maturation of IL-1β and IL-18, in addition, it can also cleave GSDMD to form C-GSDMD and N-GSDMD. Subsequently, its N-terminal pore-forming domain oligomerizes in the membrane and binds to phosphatidic acid (PA) and phosphatidylserine (PS) to promote the formation of cell membrane pores (44). The GSDMD-mediated membrane pore is a non-selective channel that promotes the release of inflammatory substances such as IL-1β and IL-18 in cells (44–46). At the same time, extracellular fluid can enter the cell to cause cell swelling, so that the cells form bubble-like protrusions (pyroptotic bodies), eventually leading to cell membrane rupture, and finally HMGB1 and ATP flow out of the cell, causing pyroptosis (44–46).
In the non-canonical pathway mediated by Caspase-4/-5/-11, LPS can directly interact with the CARD domain of Caspase-4/-5/-11, promote the cleavage of GSDMD by active Caspase, and trigger pyroptosis (27, 32, 33, 47). Activated Caspase-4/-5/-11 could not cleave pro-IL-1β and pro-IL-18, but activated caspase-11 could cleave pannexin-1 and induce ATP release (27, 32, 33, 47). ATP binds to the P2X7 receptor and mediates potassium ion release. The efflux of ATP and potassium ions can promote the activation of NLRP3 inflammasome (41, 48). The activated NLRP3 inflammasome can promote the maturation of IL-1β and IL-18 and release them into the extracellular space through the membrane pores formed by GSDMD (27, 33, 45, 47).
Caspase-3 and Caspase-8 were previously thought to be apoptosis-related caspases, which did not stimulate GSDM to induce pyroptosis. In recent years, researchers have found that Caspase-3 and Caspase-8 can also mediate pyroptosis (32, 49, 50). Caspase-3 can induce pyroptosis by cleaving GSDME (51). Caspase-3 is mainly initiated by various chemotherapeutic drugs (52, 53). In the process of chemotherapy, chemotherapeutic drugs can initiate BAK, BAX activation, and oligomerization of the mitochondrial outer membrane, resulting in mitochondrial outer membrane permeability (MOMP) (52, 53). Subsequently, Caspase-9 and downstream Caspase-3 were activated to mediate the cleavage of GSDME, resulting in C-GSDME and N-GSDME.N-GSDME was ectopic to the plasma membrane and promoting the formation of membrane pores (54). Under the stimulation of TNF-α, Caspase-8 can specifically cleave GSDMC to produce N-GSDMC, and then induce pyroptosis (32, 49, 55). In addition, inhibition of TGF-β-activated kinase-1 (TAK1) can cause Caspase-8-dependent GSDMD cleavage during Yersinia infection, which in turn leads to pyroptosis (56). Interestingly, death receptor signaling-induced Caspase-8 can also activate Caspase-3 (57). In addition, recent studies have confirmed that GSDME and Caspase-3 can be cleaved by granzyme B at the same site (58). Granzymes can directly cleave GSDME and induce pyroptosis. At the same time, Liu et al.also found that CAR-T cells can release granzyme to activate Caspase-3 in target cells, causing extensive pyroptosis (59). Therefore, some scholars have also referred to this pathway as the granzyme-mediated pyroptosis pathway (32).
4 The differences between pyroptosis and apoptosis
Pyroptosis and apoptosis are both programmed cell death, and there are some similarities between them. For example, both of them rely on Caspases, which damage DNA and cell membrane blebbing (29, 32, 60). However, Regardless of morphology or mechanism, pyroptosis is different from apoptosis (Table 1). Compared with apoptosis, pyroptosis is significantly different in that pyroptosis can promote the occurrence of inflammation, which can lead to cell swelling and incomplete cell membranes (29, 32). In addition, although both of them can cause DNA damage, unlike apoptosis, the nucleus remains intact during pyroptosis, and the nucleus is broken when apoptosis occurs (61, 62). The plasma membrane undergoes diverse morphological alterations during these two processes. During apoptosis, apoptotic bodies can form, while pyroptotic bodies may develop during pyroptosis (29). Apoptotic bodies are subcellular structures that consist of intracellular material, including DNA, organelles, and nuclear debris, and their formation is dependent on membrane blebbing (63, 64). Pyroptotic bodies are a new cellular morphological structure discovered by observing cells undergoing pyroptosis through Time-lapse electron microscopy (29). The nature of pyroptotic bodies remains unclear, but it is interesting to note that pyroptotic bodies share a resemblance to apoptotic bodies, with diameters of about 1-5 μm (45). Furthermore, pyroptosis undermines the integrity of the plasma membrane. Cell death was previously usually classified as apoptosis and necrosis (65, 66). Apoptosis is an active, programmed, non-inflammatory form of cell death. Whereas necrosis is a passive, accidental, non-programmed, with inflammatory response (65, 66). Subsequently, the researchers uncovered another form of cell death that has the characteristics of necrosis but can be highly regulated, called regulated necrosis (67, 68). Both necroptosis, also referred to as programmed necrosis, and pyroptosis are forms of regulated necrosis, characterized by the disruption of cytoplasmic membrane integrity and cellular content leakage (67, 69). There are numerous similarities between necroptosis and pyroptosis, both of which have the potential to initiate inflammation (70). Furthermore, studies conducted on animals have indicated that necroptosis is significant in the occurrence of myocardial infarction, renal ischemia, and ischemia-reperfusion in stroke (71–73). RAW-asc cells have the ability to undergo necroptosis and pyroptosis when exposed to various stimuli. Through electron microscopy observation, Chen et al. noted that necroptotic cells become rounded and eventually rupture explosively, while pyroptosis produces flat pyroptotic bodies before cell rupture occurs. This research also affirms that pyroptosis, in contrast to MLKL channel-mediated necroptosis, requires the creation of non-selective pores by GSDMD-N to enable cell lysis (45).
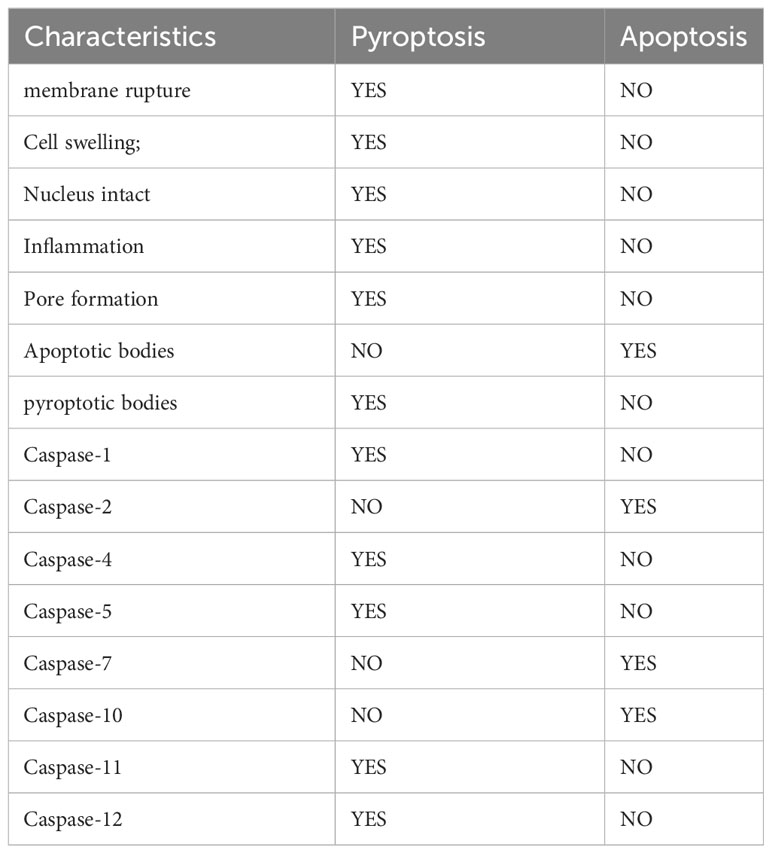
Table 1 The differences between pyroptosis and apoptosis.
The caspase family functions critically in pyroptosis as well as apoptosis. Caspases, when activated, can lead to programmed cell death. Additionally, they can also determine the type of cell death that takes place (30, 31, 74, 75). According to their functions, mammalian Caspase can be classified into two types: Apoptotic caspases and Inflammatory caspases (75). Apoptotic caspases encompass Caspase-2, -7, and -10, while inflammatory caspases mainly include Caspase-1, -4, -5, -11and-12 (30, 75, 76). Although the Caspases involved in apoptosis and pyroptosis are not all the same, Caspase-3,-6,-8,-9 have now been shown to play a role in both apoptosis and pyroptosis (32). Inflammatory caspases can accomplish pyroptosis by cleaving different gasdermin. In contrast to inflammatory caspases, apoptotic caspases primarily execute a type of programmed cell death termed apoptosis, which is immunologically silent (77). Depending on their role in the process of cell death, caspases can be classified as either initiation proteins, including caspase-2, -8, -9, and -10, or effector proteases, namely caspase-3, -6 and -7 (77). Notably, most of the caspases that induce pyroptosis can also induce apoptosis in the absence of the corresponding GSDM proteins, implying that cleavage of GSDM by inflammatory caspases can convert apoptosis into pyroptosis (78). Meanwhile, specific cleavage of GSDM by inflammatory caspases is necessary for pyroptosis (74).
5 Mechanisms of pyroptosis-triggered DCM
The development of DCM is a complex process driven by multiple factors. Chronic inflammation is one of the key drivers of DCM development. Chronic inflammation can lead to cellular death by triggering the activation of inflammatory vesicles such as NLRP3 (79, 80). Pyroptosis can promote the development of DCM through multiple pathways (Figure 2), and both myocardial cells and non-myocardial cells can undergo pyroptosis to accelerate the development of DCM (Figure 3) (25). Understanding the effects of the different pathways of pyroptosis and the cell types that undergo cellular pyroptosis on DCM may provide possible therapeutic targets for DCM.
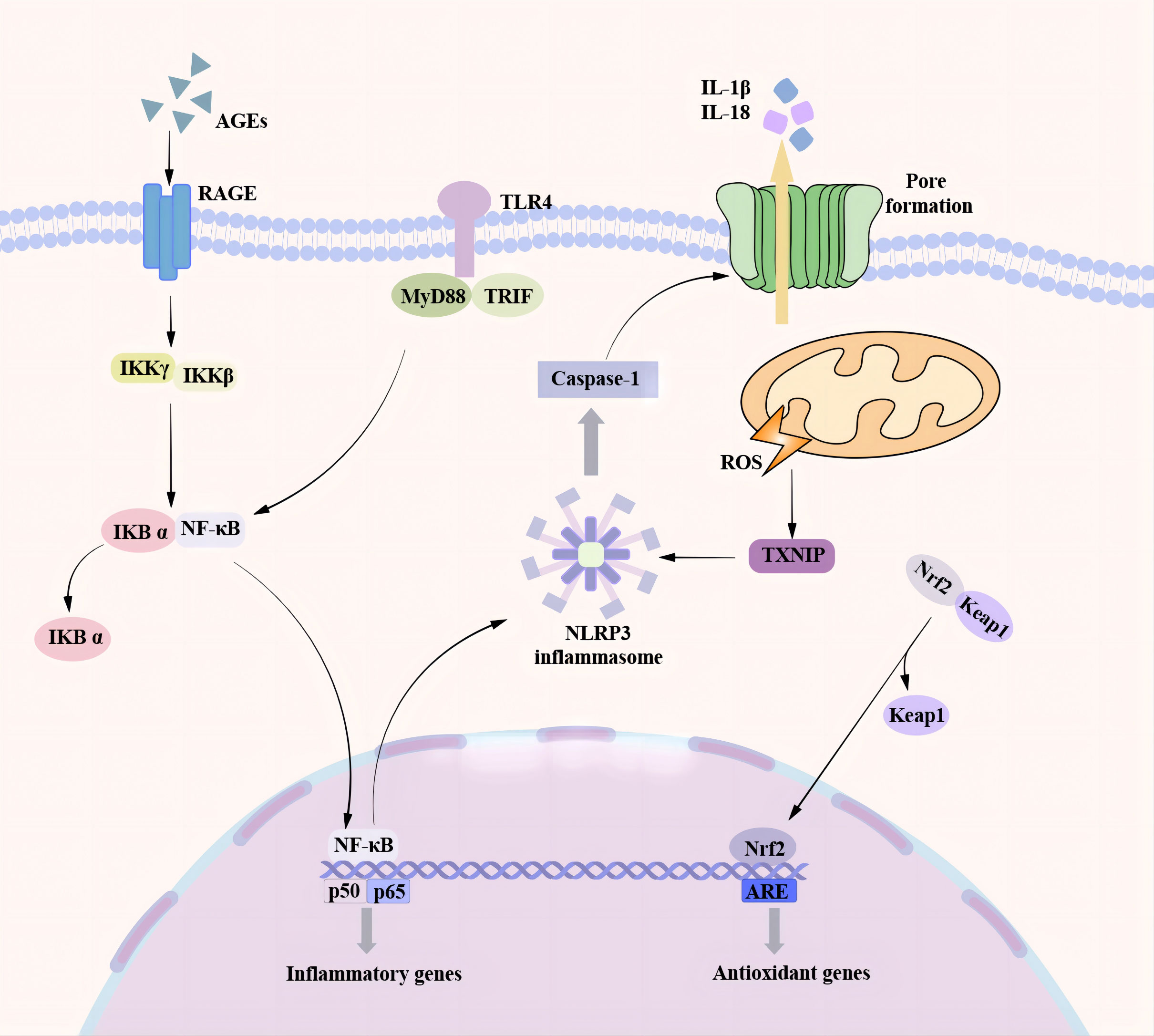
Figure 2 Pyroptosis pathway associated with DCM. There are many pathways involved in the process of pyroptosis promoting the occurrence and development of DCM. In the NF-κB / NLRP3 signaling pathway, the TLR4 receptor senses various stimulating factors, separates the NF-κB in the cytoplasm from the inhibitory protein IκB, and transfers to the nucleus, resulting in an increase in the expression of NLRP3 and the activation of NLRP3 inflammasome. A high glucose environment can induce the production of a large number of ROS. TXNIP is a ROS-dependent NLRP3 inflammasome activation regulator. A large amount of ROS can promote the binding of TXNIP to NLRP3 and trigger the activation of NLRP3 inflammasome. As a transcription factor, Nrf2 can regulate cardiac homeostasis by controlling various antioxidant genes to inhibit oxidative stress. Nrf2 is transferred to the nucleus under the action of ROS and oxidative stress and binds to the promoter region of the antioxidant response element (ARE) to promote the production of antioxidant enzymes and protect cardiomyocytes. At the same time, Nrf2 can also inhibit NF-κB to reduce the formation of NLRP3 inflammasome and inhibit pyroptosis.
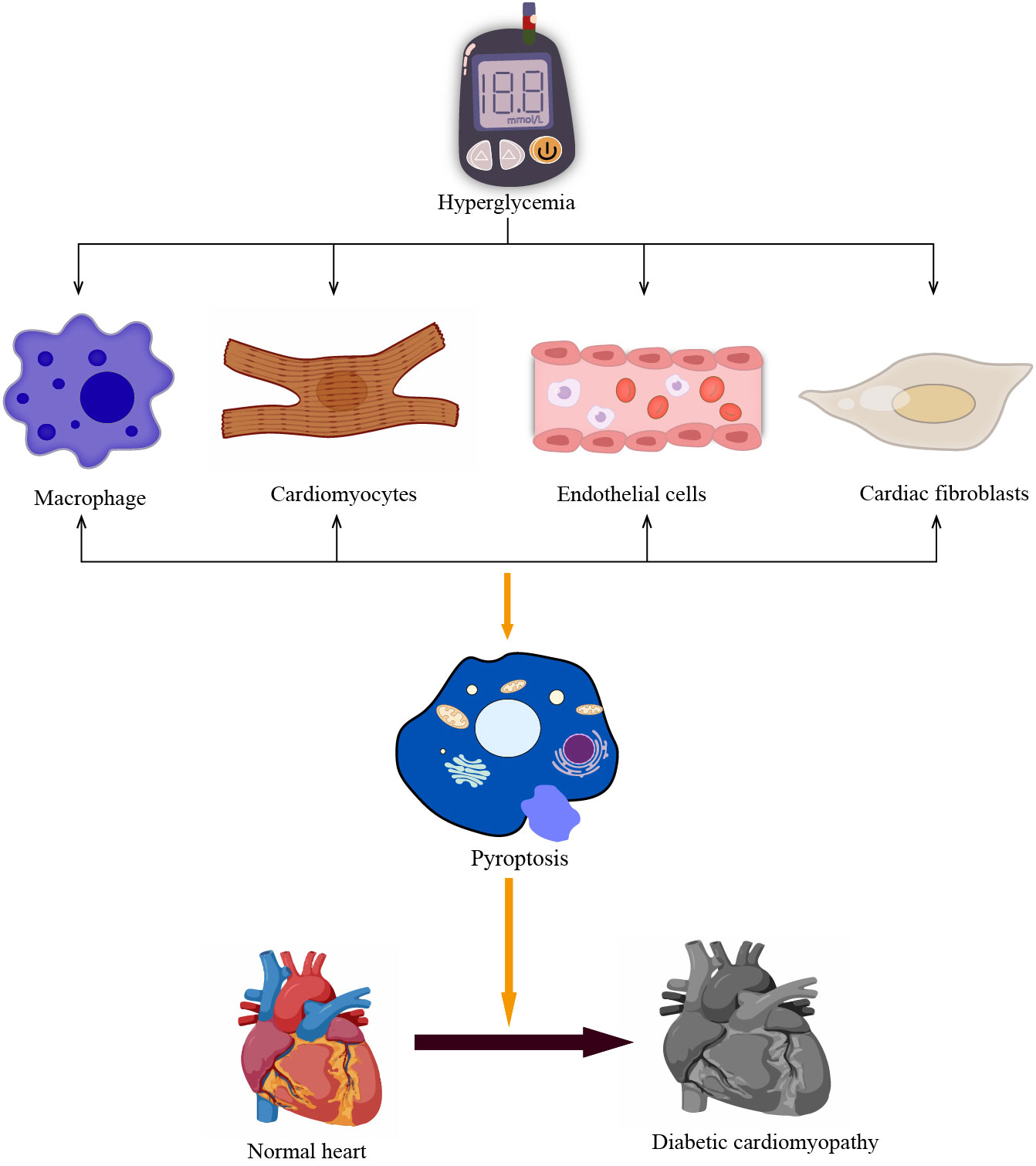
Figure 3 Pyroptosis in different cells promotes the development of DCM. The prolonged hyperglycemia causes pyroptosis of non-myocardial cells such as cardiomyocytes and macrophages, vascular endothelial cells, and cardiac fibroblasts, promoting poor cardiac remodeling and accelerating the development of DCM.
5.1 NLRP3 regulatory pathways associated with DCM
5.1.1 NF-κB/NLRP3 inflammasome pathway
The NF-κB pathway promotes the activation of NLRP3 inflammasome, which are involved in multiple chronic inflammatory diseases such as Ulcerative Colitis, lupus nephritis, rheumatoid arthritis, and so on (81–83). In recent years, NF-κB-related signaling pathways have been recognized as crucial in the pathophysiology of DCM. Luo et al. observed severe metabolic disturbances in diabetic rats and increased expression of NLRP3, ASC, caspase-1, and IL-1β in a rat model of type 2 diabetes induced by a high-fat (HF) diet and low-dose streptozotocin (STZ). Furthermore, it was shown that NF-κB is involved in the activation of NLRP3 inflammasome in high glucose-induced H9C2 cells (84). Another study demonstrated that the use of tilianin and syringin in this model not only ameliorated metabolic disorders and improved cardiac function in diabetic rats but also significantly increased the expression of TLR4, NF-Kb, NLRP3, IL-1β (85).In the STZ-induced C57BL/6 mouse model, it was observed that DCM was prevented in STZ-induced diabetic mice by inhibiting NF-κB and NLRP3 (86). High glucose (HG) enhances the expression of IL-1β, NLRP3, caspase-1, and ASC along with TLR4 protein levels in HUVECs. These effects were reversed upon TLR4 silencing (87). Besides, the study revealed that in H9C2 cells, high glucose led to an increased expression of NLRP3 inflammatory vesicles, TLR4, and NF-κB. However, the utilization of exogenous H2S effectively inhibited the activation of the TLR4/NF-Kb/NLRP3 pathway (88). All the experiments mentioned indicate the crucial involvement of TLR4/NF-κB/NLRP3 inflammasome pathway in the onset and progression of DCM. Targeting a specific link in this pathway can offer a new therapeutic approach for DCM.
5.1.2 Reactive oxygen species/thioredoxin interacting protein/NLRP3 inflammasome pathway
ROS is a by-product produced by specific enzymes during mitochondrial respiration or metabolism (89). ROS has two sides, it has a beneficial effect while also causing damage to the body (89). Long-term adverse environments such as hyperglycaemia, hyperlipidemia, and chronic inflammation can lead to the up-regulation of ROS-producing enzyme activities, resulting in a large amount of ROS and inducing oxidative stress (89, 90). It has been shown that HG causes a significant increase in ROS production in H9C2 cardiomyocytes, leading to the activation of NLRP3 inflammatory complexes via cytochrome C-dependent pathways (91). TXNIP serves as a regulator of NLRP3 inflammasome activation dependent on ROS. Researchers have shown that ROS promotes the binding of TXNIP to NLRP3, leading to the activation of the NLRP3 inflammasome (92–94). Suppression of ROS significantly diminished the expression of TXNIP, NLRP3 inflammatory vesicles, and IL-1β in H9C2 cells induced with high glucose. Meanwhile, suppression of TXNIP expression through the use of TXNIP-siRNA plasmid decreased downstream caspase-1 and IL-1β activation. Moreover, Silencing NLRP3 attenuated the expression of caspase-1 and IL-1β, while alleviating left ventricular dysfunction and reversing myocardial remodeling in diabetic rats on the HF diet and in low-dose STZ-induced diabetic rats (95). These indicate that ROS and the resulting oxidative stress can cause negative changes in the structure of the heart and hasten the progression of DCM (89, 93). Furthermore, in a rat INS-1 pancreatic β-cell-related study, Liu et al. found that HG significantly increased the expression of TXNIP, NLRP3, and caspase-1 in the cells and that TXNIP showed a dose-dependent relationship with glucose concentration and that TXNIP expression increased with increasing glucose concentration (96). These all suggest that the ROS/TXNIP/NLRP3 inflammasome pathway plays an important role in DM and may accelerate the progression of DCM.
5.1.3 Nrf2-related pathways
Nuclear factor-erythroid-2-related factor 2 (Nrf2) is a transcription factor that is expressed in a variety of tissues and organs. Studies have demonstrated that the Nrf2 signaling pathway is closely related to various cardiac diseases, which can regulate cardiac homeostasis by inhibiting oxidative stress by controlling various antioxidant genes (97). Under physiological conditions, Nrf2 binds to and is present in the cytoplasm with its repressor Kelch-like epichlorohydrin-associated protein 1 (Keap1) (97, 98). However, large amounts of ROS and oxidative stress dissociate Nrf2 from Keap1, causing Nrf2 to transfer to the nucleus to bind to the promoter region of the antioxidant response element (ARE), which activates antioxidant genes and promotes antioxidant enzyme production (97, 98). These antioxidant enzymes have anti-inflammatory and antioxidant effects, protecting cardiomyocytes from myocardial damage caused by diabetes and high glucose oxidation (98). A study showed that Nrf2 upregulated the antioxidant protein HO-1 expression in STZ-induced SD rats and HG-cultured H9C2 cells, suppressed cardiomyocyte pyroptosis and impeded the progression of DCM (99). In contrast, blocking the Nrf2 pathway with ML385 did not decrease the expression of proteins associated with cellular pyroptosis (NLRP3, caspase-1, etc.). Furthermore, this study discovered that Nrf2 activation impedes NF-κB, leading to a decrease in the production of NLRP3 inflammasomes and the inhibition of pyroptosis, ultimately leading to cardioprotection (99). Li et al. observed similar results in C57BL/6 mice, where the use of Luteolin increased Nrf2 expression, attenuated cardiac oxidative stress, and provided cardioprotection (100). Another experiment using the Nrf2 knockout vector showed that cardiomyocytes were highly susceptible to high glucose-induced injury. The expression of NLRP3, caspase-1, IL-18, and IL-1β was also significantly increased. Meanwhile, DM rats treated with ML385 to suppress Nrf2 showed more severe myocardial fibrosis and cardiac enlargement (101). All of these studies provide valuable evidence that the Nrf2-related pathway may serve as a future target for treating DCM.
5.2 Effect of cardiomyocyte pyroptosis on DCM
Pyroptosis can occur in various cells of the heart, including cardiomyocytes, macrophages, fibroblasts, and endothelial cells. Cardiomyocytes are the main cells that make up the heart. Cardiomyocytes enable the heart to perform systolic and diastolic functions and help the heart pump blood throughout the body (102). The death of cardiomyocytes, whether acute or chronic, can cause irreversible damage to the heart. The activation of NLRP3 inflammasome plays an important role in the process of cardiomyocyte pyroptosis promoting cardiac dysfunction (43, 103, 104). When myocardial ischemia occurs, the potassium efflux in cardiomyocytes increases, thereby up-regulating the expression of NIMA-associated kinase 7 (NEK7) and promoting the activation of NLRP3 inflammasome (105–108). The activation of NLRP3 inflammasome in cardiomyocytes under pressure overload mainly depends on the regulation of NF-κB by calcium/calmodulin-dependent kinase II (CaMKII) (109–111). Hyperglycaemia promotes high ROS production, which binds TXNIP to NLRP3, thereby promoting the activation of NLRP3 inflammasome (43). Moreover, a high glucose environment can promote the pyroptosis of various cells in the heart, thereby accelerating the development of DCM (43). The death of cardiomyocytes plays an important role in the progression of DCM (80). New evidence also confirms that cardiomyocyte pyroptosis induced by NLRP3 inflammasome is a critical step in the progression of DCM (43, 112).
An experiment demonstrated that modulation of the miR-34b-3p/AHR axis in high glucose-induced HL-1 cells inhibited the activation of NLRP3 inflammasome and attenuated NLRP3-mediated pyroptosis in HL-1 cells. Furthermore, the hindrance of pyroptosis resulted in enhanced cardiac function and a reduction in cardiac hypertrophy among DCM mice (113). The expression of the NLRP3 inflammasome has been found to increase when PNRCMs are exposed to HG. This leads to pyroptosis in these cells (114). Likewise, within cardiomyocytes, such as those induced by HGin AC-16 and H9C2 cell lines, as well as primary cardiomyocytes separated from neonatal C57BL/6 mice, there was an increase in the expression of specific focal death-associated proteins, including caspase-1, NLRP3, and other proteins (115, 116). The conducted in vivo and in vitro experiments exhibit that suppressing cardiomyocyte pyroptosis ameliorates cardiac function and adverse remodeling besides delaying DCM progression. Furthermore, these outcomes highlight pyroptosis as a plausible therapeutic target for DCM.
5.3 Impact of non-cardiomyocyte pyroptosis on DCM
5.3.1 Macrophages
Macrophage pyroptosis also plays an important role in complications caused by hyperglycaemia (117). Macrophages constitute a vital aspect of the immune system, performing functions such as identification, phagocytosis, secretion, regulation of immunity, and maintenance of body homeostasis (118, 119). A study found that in diabetic mouse models, persistent hyperglycaemia can lead to macrophage dysfunction in which activation of NLRP3 inflammasome plays an important role. Partial macrophage dysfunction induced by hyperglycaemia can be restored by inhibiting the activation of cathepsin B and NLRP3 inflammasome (117). Simultaneously, researchers discovered that the activation of NLRP3 inflammasome in M1 macrophages can exacerbate cardiac dysfunction and encourage myocardial morphological changes in a post-stroke diabetic mouse model. The implementation of CY-09 to inhibit NLRP3 inflammasome has been shown to ameliorate cardiac function in diabetic mice (119). These findings suggest that the activation of NLRP3 inflammasome and pyroptosis in macrophages may contribute to the development of DCM.
5.3.2 Fibroblasts
DCM may manifest cardiac interstitial fibrosis in addition to cardiomyocyte hypertrophy. Cardiac fibroblasts are effector cells in the process of myocardial fibrosis (120). When the heart suffers various injuries, cardiac fibroblasts can repair the heart by promoting the formation of collagen and extracellular matrix. However, when the damage is excessive, this repair matrix causes cardiac fibrosis, resulting in a decrease in cardiac compliance. In the diabetic state, metabolites such as glycosylation end products (AGEs) promote fibroblast differentiation into myofibroblasts (120, 121). Pyroptosis also plays an important role in the process of cardiac fibrosis. Ren et al. discovered that HG caused an increase in caspase-1, IL-1β mRNA, and protein expression in fibroblasts, as well as a similar trend in GSDMD protein expression. However, the administration of ranolazine led to a reduction in cardiac fibrosis in diabetic rats by inhibiting fibroblast pyroptosis and decreasing collagen deposition through the regulation of miR-135b (122). Another study also indicated that the expression of NLRP3, IL-1β, and GSDMD-N increased in fibroblasts following inducement by high glucose. Conversely, the inhibition of inflammation and pyroptosis in fibroblasts improved cardiac function and reduced fibrosis in diabetic C57BL/6 mice (123). These studies all confirm that fibroblasts play an important role in the progression of DCM and that modulation of fibroblast pyroptosis may alleviate cardiac fibrosis and delay the onset of DCM.
5.3.3 Vascular endothelial cells
Vascular endothelial cell dysfunction is also the key pathological basis for the occurrence and development of DCM (124). Vascular endothelial cells form a semi-permeable barrier between circulating blood and the extravascular matrix, and this endothelial barrier regulates cellular connectivity to maintain homeostasis in the body (125). During the development of DCM, activated NLRP3 inflammasome induces cellular pyroptosis releasing large quantities of pro-inflammatory factors IL-1β and IL-18. These pro-inflammatory factors subsequently bind to cell-surface receptors and enhance the expression of adhesion and chemokine in the endothelium, while increasing leukocyte adhesion and extravasation. This ultimately leads to the disruption of intercellular junctions, endothelial barrier dysfunction, and increased vascular permeability, which also facilitates the infiltration of pro-inflammatory cells and pro-inflammatory factors and accelerates adverse cardiac remodeling (124, 125). Increased NLRP3, caspase-1, and other cell death-related proteins were observed in HUVEC cells exposed to HG (126). A further study has verified that NLRP3 inflammasome-induced pyroptosis, can harm vascular endothelial cells and reduce the density of cardiac microvessels, resulting in negative cardiac restructuring (127).
6 Potential therapies of pyroptosis/NLRP3 in DCM
The pathogenesis of DCM is very complex. Although its mechanism has been continuously improved for decades, there is still no effective treatment for DCM, which also makes the morbidity and mortality of DCM still high. As many researchers have found that pyroptosis plays an important role in DCM, targeted inhibition of pyroptosis and NLRP3 inflammasome may provide a good direction for the treatment of DCM. At present, these substances mainly include hypoglycemic drugs, phytochemicals, NLRP3 inflammasome, pyroptosis inhibitors, non-coding RNA, and so on (Table 2).
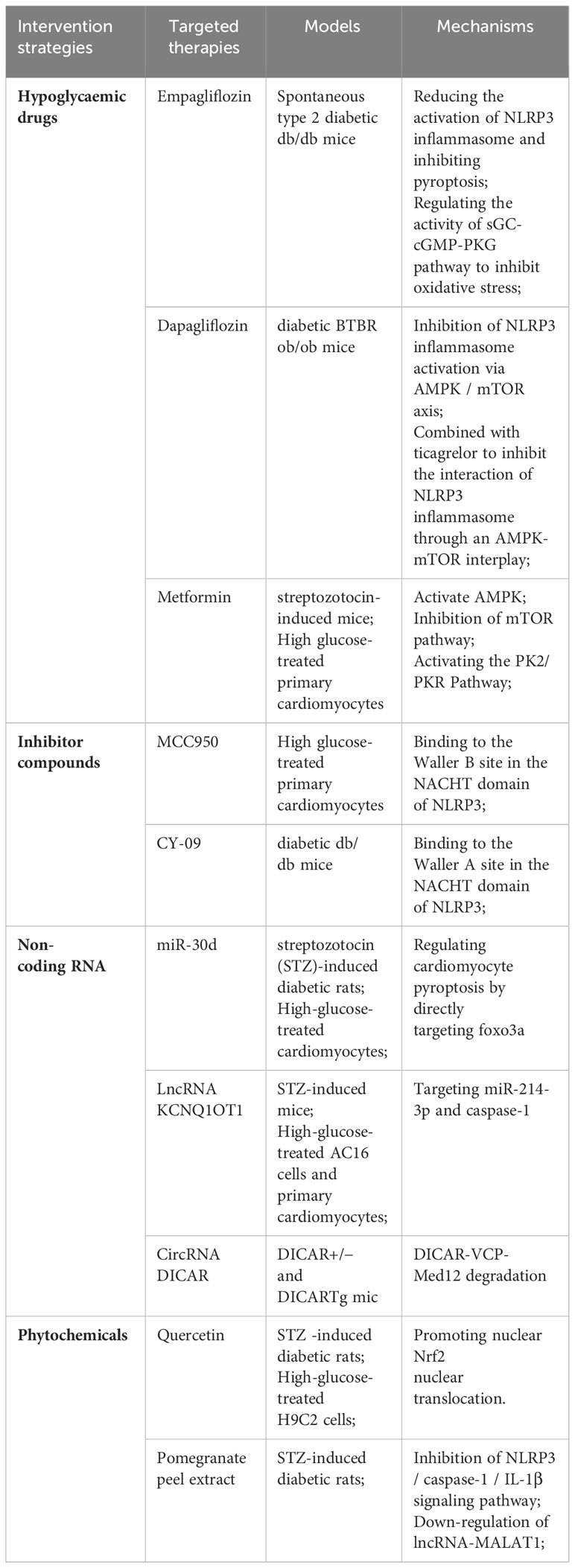
Table 2 Therapies and mechanisms targeting NLRP3 inflammasome/pyroptosis.
6.1 Hypoglycaemic drugs
Sodium-glucose co-transporter 2 inhibitor (SGLT2i) is one of the commonly used hypoglycemic drugs. SGLT2i plays a hypoglycemic role mainly by increasing urinary glucose excretion. In addition to lowering blood glucose, SGLT2i also has an extraordinary cardioprotective effect, which can reduce cardiovascular mortality and heart failure hospitalization rates in patients with type 2 diabetes (128). SGLT2i plays a role in delaying the progression of DCM by regulating metabolism, improving mitochondrial function, inhibiting oxidative stress, and reducing programmed cell death (129).
Empagliflozin is one of the typical SGLT2i, which has been widely used in patients with diabetes (130). A study showed that empagliflozin reduced the activation of NLRP3 inflammasome and attenuated pyroptosis caused by NLRP3 inflammasome in diabetic mice. In addition, this study also found that empagliflozin can inhibit oxidative stress by affecting the activity of the sGC-cGMP-PKG pathway, thus exerting cardioprotective effects (130). Dapagliflozin can inhibit SGLT2 to reduce NLRP3 inflammasome activation and delay the progression of DCM in type 2 diabetic mice. At the same time, it has a better inhibitory effect when combined with DPP-4 inhibitor Saxagliptin (131). It has also been found that ticagrelor, a P2Y12 receptor antagonist, acts synergistically with dagliflozin to slow the progression of DCM by attenuating the activation of NLRP3 inflammatory vesicles via the AMPK/mTOR axis (132).
Metformin is a kind of biguanide drug derived from herbaceous plants. It is the first-line treatment of diabetes. It can protect the heart in a variety of ways (133). Metformin was found to improve DCM by down-regulating the expression of NLRP3, Caspase-1, and IL-1β through AMP-activated protein kinase (AMPK) in cardiomyocytes treated with high glucose and diabetic mouse models (134). Studies have also shown that metformin can reduce the adverse effects of hyperglycaemia on streptozotocin-induced diabetic mice through the PK2/PKR pathway (135). Metformin can also produce incredible results when co-administered with other drugs. Ye et al. found that metformin significantly suppressed oxidative stress and inflammation when co-administered with the lipid-lowering drug atorvastatin, providing better protection against DCM (131). Metformin combined with hydrogen, Cocoa-Carob Blend, and Dendrobium Mixture can also show more effective cardioprotective effects (136–138).
In addition, certain hpoglycaemic drugs have demonstrated the ability to impede cellular pyroptosis in non-diabetic conditions. This implies that glucose-lowering drugs may potentially enhance DCM by inhibiting cellular pyroptosis. Zhao et al. induced HUVEC cells with oxidized LDL and observed that SGLT2i decreased cellular pyroptosis while enhancing endothelial cell dysfunction (139). In the case of ischemia/reperfusion injury, metformin inhibits NLRP3 inflammasome activation and exerts cardioprotective effects through the AMPK pathway (140).
6.2 Inhibiting compounds
NLRP3 inflammasome plays an important role in pyroptosis promoting the occurrence and development of DCM (42, 43). Inhibition of NLRP3 inflammasome can greatly inhibit pyroptosis and delay the progression of DCM. MCC950 is a small molecule that can effectively inhibit NLRP3 inflammasome (141). MCC950 binds to the Waller B site in the NACHT domain of the NLRP3 inflammasome, thereby blocking ATP hydrolysis and inhibiting NLRP3 inflammasome formation (142, 143). In high glucose-induced cardiomyocytes, the use of MCC950 can inhibit the expression of NLRP3, down-regulate the expression of pyroptosis-related proteins, and alleviate high glucose-induced LDH leakage (114). In a study in streptozotocin-induced diabetic mice, MCC950 was also found to delay the development of various complications associated with diabetes, including diabetic retinopathy, diabetes-associated atherosclerosis, and diabetic encephalopathy, by inhibiting the NLRP3 inflammasome and its downstream inflammatory response (144–146). The effect of MCC950 on DCM is still insufficient, but the above experimental results are undoubtedly encouraging. The exact effect of MCC950 on DCM and its related mechanisms still needs to be improved by researchers.
CY-09 is a compound that can effectively inhibit NLRP3 by directly binding to the ATP-binding motif of the NACHT domain of NLRP3 and inhibiting NLRP3 ATPase activity, thereby inhibiting the assembly and activation of the NLRP3 inflammasome (147). Jiang et al.also studied the mechanism of CY-09 inhibiting NLRP3 ATPase activity. The NLRP3 NACHT domain contains two sequences that are important for ATPase activity. The Walker A motif is an important motif for ATP binding, while the Walker B motif is essential for ATPase activity (148). Unlike MCC950, CY-09 binds to the Waller A site in NLRP3, thereby blocking the binding of NLRP3 to ATP, thereby blocking ATPase activity (147). CY-09 significantly ameliorates metabolic disorders in a diabetic mouse model (147), and can also alleviate inflammation, oxidative stress, and fibrosis in diabetic mice by selectively inhibiting NLRP3 inflammasome, thereby improving renal damage in diabetic nephropathy (149). Furthermore, CY-09 also alleviated insulin resistance and hepatocyte steatosis in diabetic mice (150). In terms of cardiac protection, CY-09 can inhibit the activation of NLRP3 inflammasome in M1 polarized macrophages and improve cardiac dysfunction after ischemic stroke (119). The above experiments show that CY-09 may also be used to delay DCM, but there are few studies on the relationship between CY-09 and DCM.
6.3 Non-coding RNA
In the process of pyroptosis accelerating the occurrence and development of DCM, non-coding RNA also plays an important regulatory role. Non-coding RNA (ncRNA) includes many types, including microRNA (miRNA), long non-coding RNA (lncRNA), and circular RNA (circRNA) (151). Among them, miRNA is the most studied type of ncRNA (151). MiRNA is a class of endogenous small non-coding RNA molecules with a length of about 20 nucleotides. It can play a role in biological processes such as cell proliferation, differentiation, and apoptosis by inhibiting or activating gene expression (152). MiRNAs can be stably expressed in different body fluids, and the level of this miRNA in the blood can also change at different stages of the development of DCM (152). Therefore, miRNA may be a potential biomarker for DCM (152, 153). MiR-21, miR-30d, miR-223, and other miRNAs were found to be up-regulated in DCM, whereas miR-1, miR-9, miR-150, and other miRNAs were found to be down-regulated in diabetic conditions (153, 154). Recent studies have found that some miRNAs can slow down the development of DCM by regulating pyroptosis. In vivo and in vitro experiments confirmed that the expression of miRNA-30d in the diabetic group was significantly increased, and its increased expression up-regulated the expression of pro-inflammatory factors such as Caspase-1 and IL-1βand directly inhibited the expression of foxo3a and its downstream proteins, which promoted the pyroptosis of cardiomyocytes in DCM. However, these effects were reversed after the knockdown of miR-30d (155). Xu et al. also found that miR-223 is highly expressed in H9C2 cardiomyocytes induced by high glucose, and the use of miR-223 inhibitors can attenuate the activation of NLRP3 inflammasome and alleviate myocardial fibrosis, thereby delaying the development of DCM and protecting the heart (154). In the diabetic mouse model induced by high glucose and high fat, Deng et al.also found that miR-223 can regulate the expression of NLRP3 to reduce damage to endothelial cells (156). Meanwhile, it also shows that in the development of DCM, in addition to the myocardial cells themselves, endothelial cell death also plays an important role in promoting it.
In recent years, lncRNAs and circRNAs have also been found to play a regulatory role in DCM. LncRNAs refer to transcription RNA molecules with a length of more than 200 nucleotides, but they do not have protein-coding ability (157). LncRNA KCNQ1OT1 expression is increased in diabetic patients, high glucose-induced cardiomyocytes, and diabetic mouse models. Silencing KCNQ1OT1 inhibits pyroptosis by targeting miR-214-3p and Caspase-1, and also ameliorates abnormal cytoskeleton structure and calcium overload, improving cardiac structure and function (116). CircRNAs are joint regulators in various diseases. CircRNA DICAR has been shown to alleviate DCM, and knockout of DICAR can enhance pyroptosis in DCM (158).
6.4 Phytochemicals
In recent years, some substances extracted from herbs have received extensive attention in academia due to their anti-inflammatory effects. Flavonoids are the most abundant phytochemicals in plants, which can alleviate DCM by reducing myocardial oxidative stress and inflammation, so they have also been widely studied (159). Quercetin is a natural flavonoid. Zhang et al.found that quercetin increased the expression of antioxidant proteins such as HO-1 through the Nrf2 pathway and inhibited myocardial pyroptosis (99). Pomegranate peel extract was found to improve cardiac hypertrophy and myocardial fibrosis in diabetic rat models. The study also found that pomegranate peel extract may protect DCM by inhibiting the NLRP3/caspase-1/IL-1β signaling pathway and down-regulating the expression of lncRNA-MALAT1 (160).
Colchicine is a tricyclic alkaloid that is mainly used to treat inflammatory diseases such as gout. In cardiovascular diseases, colchicine is used in acute and recurrent pericarditis, coronary syndrome, atrial fibrillation, and heart failure (161). Colchicine can inhibit the activation of NLRP3 inflammasome by inhibiting the P2X7 receptor and blocking potassium efflux. Moreover, colchicine can also inhibit microtubule synthesis, promote microtubule degradation, and inhibit the assembly of NLRP3 inflammasome (162). Given the important role of colchicine in inhibiting the activation of NLRP3 inflammasome, it may have the potential to treat DCM. However, there are few studies on the role of colchicine in DCM.
7 Conclusion
DCM has been recognized for decades, and a large number of studies have been carried out to explore its potential pathophysiological mechanisms. However, the incidence of DCM is still increasing with the increasing prevalence of diabetes. The pathogenesis of DCM has been refined through continuous exploration by researchers, and several new drugs have been shown to have beneficial effects in vivo and in vitro models. Pyroptosis, as a programmed cell death with inflammation, plays an important role in the occurrence and development of DCM. Understanding the different pyroptosis pathways associated with DCM and the effects of pyroptosis of different cells on DCM will help us to find new therapeutic targets. Some researchers have also found that the intervention of pyroptosis by hypoglycemic drugs and inhibitors targeting NLRP3 inflammasome can delay the progression of DCM. However, the exact mechanism between DCM and pyroptosis has not yet been clarified, and it still needs further exploration by researchers.
Author contributions
GW: Writing – original draft, Writing – review & editing. TM: Writing – review & editing. KH: Writing – review & editing. JZ: Writing – review & editing. SL: Writing – review & editing. JL: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Glovaci D, Fan W, Wong ND. Epidemiology of diabetes mellitus and cardiovascular disease. Curr Cardiol Rep (2019) 21:21. doi: 10.1007/s11886-019-1107-y
2. Murtaza G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D G, Virk HUH, Khalid M, Lavie CJ, Ventura H, Mukherjee D, et al. Diabetic cardiomyopathy - A comprehensive updated review. Prog Cardiovasc Dis (2019) 62:315–26. doi: 10.1016/j.pcad.2019.03.003
3. Type 1 diabetes mellitus. Nature Reviews Disease Primers. Available at: https://www.nature.com/articles/nrdp201716 (Accessed November 9, 2023).
4. Molecular and metabolic mechanisms of insulin resistance and β-cell failure in type 2 diabetes . Nature Reviews Molecular Cell Biology. Available at: https://www.nature.com/articles/nrm2327 (Accessed November 9, 2023).
5. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev (2013) 93:137–88. doi: 10.1152/physrev.00045.2011
6. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. SpringerLink. Available at: https://link.springer.com/article (Accessed August 25, 2023).
7. Eguchi K, Boden-Albala B, Jin Z, Rundek T, Sacco RL, Homma S, et al. Association between diabetes mellitus and left ventricular hypertrophy in a multiethnic population. Am J Cardiol (2008) 101:1787–91. doi: 10.1016/j.amjcard.2008.02.082
8. Hölscher ME, Bode C, Bugger H. Diabetic cardiomyopathy: does the type of diabetes matter? Int J Mol Sci (2016) 17:2136. doi: 10.3390/ijms17122136
9. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
10. Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, Martino FD, et al. Diabetic cardiomyopathy: definition, diagnosis, and therapeutic implications. Heart Failure Clinics (2019) 15:341–7. doi: 10.1016/j.hfc.2019.02.003
11. Oktay AA, Paul TK, Koch CA, Lavie CJ. Diabetes, cardiomyopathy, and heart failure, in: Endotext (2023). MDText.com, Inc. Available at: https://www.ncbi.nlm.nih.gov/sites/books/NBK560257/ (Accessed November 6, 2023).
12. Seferović PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J (2015) 36:1718–27. doi: 10.1093/eurheartj/ehv134
13. Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart Fail Rev (2013) 18:149–66. doi: 10.1007/s10741-012-9313-3
14. Diabetic Cardiomyopathy: From Mechanism to Management in a Nutshell . Ingenta Connect. Available at: https://www.ingentaconnect.com/content/ben/emiddt/2021/00000021/00000002/art00009 (Accessed August 25, 2023).
15. Liu Q, Wang S, Cai L, Miura T. Diabetic cardiomyopathy and its mechanisms: Role of oxidative stress and damage. J Diabetes Invest 5(6):623–34. doi: 10.1111/jdi.12250
16. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol 11:42. doi: 10.3389/fphar.2020.00042
17. Wei J, Zhao Y, Liang H, Du W, Wang L. Preliminary evidence for the presence of multiple forms of cell death in diabetes cardiomyopathy. Acta Pharm Sin B (2022) 12:1–17. doi: 10.1016/j.apsb.2021.08.026
18. Elia E, Ministrini S, Carbone F, Montecucco F. Diabetic cardiomyopathy and inflammation: development of hostile microenvironment resulting in cardiac damage. Minerva Cardiol Angiol (2022) 70:357–69. doi: 10.23736/s2724-5683.20.05454-7
19. Ramesh P, Yeo JL, Brady EM, McCann GP. Role of inflammation in diabetic cardiomyopathy. Therapeutic Advances in Endocrinology and Metabolism (2022) 13:20420188221083530. doi: 10.1177/20420188221083530
20. Mesquita T, Lin YN, Ibrahim A. Chronic low-grade inflammation in heart failure with preserved ejection fraction. Aging Cell (2021) 20(9):e13453. doi: 10.1111/acel.13453
21. Wang Y, Kanneganti T-D. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J (2021) 19:4641–57. doi: 10.1016/j.csbj.2021.07.038
22. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol (2001) 9:113–4. doi: 10.1016/S0966-842X(00)01936-3
23. Zhaolin Z, Guohua L, Shiyuan W, Zuo W. Role of pyroptosis in cardiovascular disease. Cell Proliferation (2019) 52:e12563. doi: 10.1111/cpr.12563
24. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
25. Wei Y, Yang L, Pandeya A, Cui J, Zhang Y, Li Z. Pyroptosis-induced inflammation and tissue damage. J Mol Biol (2022) 434:167301. doi: 10.1016/j.jmb.2021.167301
26. Wu Y, Zhang J, Yu S, Li Y, Zhu J, Zhang K, et al. Cell pyroptosis in health and inflammatory diseases. Cell Death Discovery (2022) 8:1–8. doi: 10.1038/s41420-022-00998-3
27. Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol (2017) 27:673–84. doi: 10.1016/j.tcb.2017.05.005
28. Hsu S-K, Li C-Y, Lin I-L, Syue W-J, Chen Y-F, Cheng K-C, et al. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics (2021) 11:8813–35. doi: 10.7150/thno.62521
29. Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res (2018) 28:9–21. doi: 10.1038/cr.2017.133
30. Kesavardhana S, Malireddi RKS, Kanneganti T-D. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol (2020) 38:567–95. doi: 10.1146/annurev-immunol-073119-095439
31. Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell (2009) 138:838–54. doi: 10.1016/j.cell.2009.08.021
32. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Sig Transduct Target Ther (2021) 6:1–21. doi: 10.1038/s41392-021-00507-5
33. Burdette BE, Esparza AN, Zhu H, Wang S. Gasdermin D in pyroptosis. Acta Pharm Sin B (2021) 11:2768–82. doi: 10.1016/j.apsb.2021.02.006
34. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18:2114–27. doi: 10.1038/s41423-021-00740-6
35. Barnes PJ. Nuclear factor-κB. Int J Biochem Cell Biol (1997) 29:867–70. doi: 10.1016/S1357-2725(96)00159-8
36. Karin M, Ben-Neriah Y. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol (2000) 18:621–63. doi: 10.1146/annurev.immunol.18.1.621
37. Chen Z. Ubiquitination in signaling to and activation of IKK. Immunol Rev (2012) 246(1):95–106. doi: 10.1111/j.1600-065X.2012.01108.x
38. Bhatt D, Ghosh S. Regulation of the NF-κB-mediated transcription of inflammatory genes. Front Immunol (2014) 5:71. doi: 10.3389/fimmu.2014.00071
39. Peng M-L, Fu Y, Wu C-W, Zhang Y, Ren H, Zhou S-S. Signaling pathways related to oxidative stress in diabetic cardiomyopathy. Front Endocrinol (Lausanne) (2022) 13:907757. doi: 10.3389/fendo.2022.907757
40. Sharma BR, Kanneganti T-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol (2021) 22:550–9. doi: 10.1038/s41590-021-00886-5
41. Jin C, Flavell RA. Molecular mechanism of NLRP3 inflammasome activation. J Clin Immunol (2010) 30:628–31. doi: 10.1007/s10875-010-9440-3
42. Yu Z-W, Zhang J, Li X, Wang Y, Fu Y-H, Gao X-Y. A new research hot spot: The role of NLRP3 inflammasome activation, a key step in pyroptosis, in diabetes and diabetic complications. Life Sci (2020) 240:117138. doi: 10.1016/j.lfs.2019.117138
43. Zhang L, Ai C, Bai M, Niu J, Zhang Z. NLRP3 inflammasome/pyroptosis: A key driving force in diabetic cardiomyopathy. Int J Mol Sci (2022) 23:10632. doi: 10.3390/ijms231810632
44. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
45. Chen X, He W, Hu L, Li J, Fang Y, Wang X, et al. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res (2016) 26:1007–20. doi: 10.1038/cr.2016.100
46. Rühl S, Broz P. Regulation of lytic and non-lytic functions of gasdermin pores. J Mol Biol (2022) 434:167246. doi: 10.1016/j.jmb.2021.167246
47. Gong W, Shi Y, Ren J. Research progresses of molecular mechanism of pyroptosis and its related diseases. Immunobiology (2020) 225:151884. doi: 10.1016/j.imbio.2019.11.019
48. Pelegrin P. P2X7 receptor and the NLRP3 inflammasome: Partners in crime. Biochem Pharmacol (2021) 187:114385. doi: 10.1016/j.bcp.2020.114385
49. Jiang M, Qi L, Li L, Wu Y, Song D, Li Y. Caspase-8: A key protein of cross-talk signal way in “PANoptosis” in cancer. Int J Cancer (2021) 149:1408–20. doi: 10.1002/ijc.33698
50. Ketelut-Carneiro N, Fitzgerald KA. Apoptosis, pyroptosis, and necroptosis-oh my! The many ways a cell can die. J Mol Biol (2022) 434:167378. doi: 10.1016/j.jmb.2021.167378
51. Jiang M, Qi L, Li L, Li Y. The caspase-3/GSDME signal pathway as a switch between apoptosis and pyroptosis in cancer. Cell Death Discovery (2020) 6:1–11. doi: 10.1038/s41420-020-00349-0
52. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature (2017) 547:99–103. doi: 10.1038/nature22393
53. Hu L, Chen M, Chen X, Zhao C, Fang Z, Wang H, et al. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis (2020) 11:1–17. doi: 10.1038/s41419-020-2476-2
54. Wang Y-Y, Liu X-L, Zhao R. Induction of pyroptosis and its implications in cancer management. Front Oncol (2019) 9:971. doi: 10.3389/fonc.2019.00971
55. Hou J, Wang S, Miao R, Zhang X, Hung M-C. Detection of gasdermin C-mediated cancer cell pyroptosis. In: Fink SL, editor. Pyroptosis: Methods and Protocols. New York: NY: Springer US (2023). p. 135–46. doi: 10.1007/978-1-0716-3040-2_11
56. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science (2018) 362:1064–9. doi: 10.1126/science.aau2818
57. Zhang J, Zhou B, Sun R, Ai Y, Cheng K, Li F, et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res (2021) 31:980–97. doi: 10.1038/s41422-021-00506-9
58. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579:415–20. doi: 10.1038/s41586-020-2071-9
59. Liu Y, Fang Y, Chen X, Wang Z, Liang X, Zhang T, et al. Gasdermin E-mediated target cell pyroptosis by CAR T cells triggers cytokine release syndrome. Sci Immunol (2020) 5:eaax7969. doi: 10.1126/sciimmunol.aax7969
60. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18(5):1106–21. doi: 10.1038/s41423-020-00630-3
61. Miao EA, Rajan JV, Aderem A. Caspase-1-induced pyroptotic cell death. Immunol Rev (2011) 243(1):206–14. doi: 10.1111/j.1600-065X.2011.01044.x
62. Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infection Immun (2005) 73(4):1907–16. doi: 10.1128/iai.73.4.1907-1916.2005
63. Battistelli M, Falcieri E. Apoptotic bodies: particular extracellular vesicles involved in intercellular communication. Biology (2020) 9:21. doi: 10.3390/biology9010021
64. Santavanond JP, Rutter SF, Atkin-Smith GK, Poon IKH. Apoptotic bodies: mechanism of formation, isolation and functional relevance. Subcell Biochem (2021) 97:61–88. doi: 10.1007/978-3-030-67171-6_4
65. Kanduc D, Mittelman A, Serpico R, Sinigaglia E, Sinha AA, Natale C, et al. Cell death: Apoptosis versus necrosis (Review). Int J Oncol (2002) 21:165–70. doi: 10.3892/ijo.21.1.165
66. D’Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int (2019) 43:582–92. doi: 10.1002/cbin.11137
67. Tonnus W, Meyer C, Paliege A, Belavgeni A, von Mässenhausen A, Bornstein SR, et al. The pathological features of regulated necrosis. J Pathol (2019) 247:697–707. doi: 10.1002/path.5248
68. Laster SM, Wood JG, Gooding LR. Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J Immunol (1988) 141:2629–34. doi: 10.4049/jimmunol.141.8.2629
69. Tsuchiya K. Inflammasome-associated cell death: Pyroptosis, apoptosis, and physiological implications. Microbiol Immunol (2020) 64:252–69. doi: 10.1111/1348-0421.12771
70. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ (2019) 26:99–114. doi: 10.1038/s41418-018-0212-6
71. Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller J-O, et al. Two independent pathways of regulated necrosis mediate ischemia–reperfusion injury. Proc Natl Acad Sci U.S.A. (2013) 110:12024–9. doi: 10.1073/pnas.1305538110
72. Luedde M, Lutz M, Carter N, Sosna J, Jacoby C, Vucur M, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res (2014) 103:206–16. doi: 10.1093/cvr/cvu146
73. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1:112–9. doi: 10.1038/nchembio711
74. Shi J, Zhao Y, Wang K J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526(7575):660–5. doi: 10.1038/nature15514
75. Van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity (2019) 50:1352–64. doi: 10.1016/j.immuni.2019.05.020
76. Ross C, Chan AH, von Pein JB, Maddugoda MP, Boucher D, Schroder K. Inflammatory caspases: toward a unified model for caspase activation by inflammasomes. Annu Rev Immunol (2022) 40:249–69. doi: 10.1146/annurev-immunol-101220-030653
77. Man SM, Kanneganti T-D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat Rev Immunol (2016) 16:7–21. doi: 10.1038/nri.2015.7
78. Tsuchiya K. Switching from apoptosis to pyroptosis: gasdermin-elicited inflammation and antitumor immunity. Int J Mol Sci (2021) 22:426. doi: 10.3390/ijms22010426
79. Cai Z, Yuan S, Luan X, Feng J, Deng L, Zuo Y, et al. Pyroptosis-related inflammasome pathway: A new therapeutic target for diabetic cardiomyopathy. Front Pharmacol (2022) 13:842313. doi: 10.3389/fphar.2022.842313
80. Zeng C, Wang R, Tan H. Role of pyroptosis in cardiovascular diseases and its therapeutic implications. Int J Biol Sci (2019) 15:1345–57. doi: 10.7150/ijbs.33568
81. Jing M, Yang J, Zhang L, Liu J, Xu S, Wang M, et al. Celastrol inhibits rheumatoid arthritis through the ROS-NF-κB-NLRP3 inflammasome axis. Int Immunopharmacol (2021) 98:107879. doi: 10.1016/j.intimp.2021.107879
82. Zhao J, Zhang H, Huang Y, Wang H, Wang S, Zhao C, et al. Bay11-7082 attenuates murine lupus nephritis via inhibiting NLRP3 inflammasome and NF-κB activation. Int Immunopharmacol (2013) 17:116–22. doi: 10.1016/j.intimp.2013.05.027
83. Zeng J, Zhang D, Wan X, Bai Y, Yuan C, Wang T, et al. Chlorogenic Acid Suppresses miR-155 and Ameliorates Ulcerative Colitis through the NF-κB/NLRP3 Inflammasome Pathway. Mol Nutr Food Res (2020) 64:2000452. doi: 10.1002/mnfr.202000452
84. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PloS One (2014) 9:e104771. doi: 10.1371/journal.pone.0104771
85. Yao J, Li Y, Jin Y, Chen Y, Tian L, He W. Synergistic cardioptotection by tilianin and syringin in diabetic cardiomyopathy involves interaction of TLR4/NF-κB/NLRP3 and PGC1a/SIRT3 pathways. Int Immunopharmacol (2021) 96:107728. doi: 10.1016/j.intimp.2021.107728
86. Xu L, Chen R, Zhang X, Zhu Y, Ma X, Sun G, et al. Scutellarin protects against diabetic cardiomyopathy via inhibiting oxidative stress and inflammatory response in mice. Ann Palliat Med (2021) 10:2481–93. doi: 10.21037/apm-19-516
87. Leng B, Zhang Y, Liu X, et al. Astragaloside IV suppresses high glucose-induced NLRP3 inflammasome activation by inhibiting TLR4/NF-κB and caSR. Mediators Inflamm (2019) 2019:1082497. doi: 10.1155/2019/1082497
88. Huang Z, Zhuang X, Xie C, Hu X, Dong X, Guo Y, et al. Exogenous hydrogen sulfide attenuates high glucose-induced cardiotoxicity by inhibiting NLRP3 inflammasome activation by suppressing TLR4/NF-κB pathway in H9c2 cells. Cell Physiol Biochem (2016) 40:1578–90. doi: 10.1159/000453208
89. García N, Zazueta C, Aguilera-Aguirre L. Oxidative stress and inflammation in cardiovascular disease. Oxid Med Cell Longev (2017) 2017:5853238. doi: 10.1155/2017/5853238
90. Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med (2020) 7:12. doi: 10.3389/fcvm.2020.00012
91. Zhang H, Chen X, Zong B, Yuan H, Wang Z, Wei Y, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med (2018) 22:4437–48. doi: 10.1111/jcmm.13743
92. Yoshihara E. TXNIP/TBP-2: A master regulator for glucose homeostasis. Antioxidants (Basel) (2020) 9:765. doi: 10.3390/antiox9080765
93. Dominic A, Le N-T, Takahashi M. Loop between NLRP3 inflammasome and reactive oxygen species. Antioxid Redox Signal (2022) 36:784–96. doi: 10.1089/ars.2020.8257
94. Han Y, Xu X, Tang C, Gao P, Chen X, Xiong X, et al. Reactive oxygen species promote tubular injury in diabetic nephropathy: The role of the mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol (2018) 16:32–46. doi: 10.1016/j.redox.2018.02.013
95. Luo B, Li B, Wang W, Liu X, Liu X, Xia Y, et al. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc Drugs Ther (2014) 28:33–43. doi: 10.1007/s10557-013-6498-1
96. Liu S, Tang G, Duan F, Zeng C, Gong J, Chen Y, et al. MiR-17-5p inhibits TXNIP/NLRP3 inflammasome pathway and suppresses pancreatic β-cell pyroptosis in diabetic mice. Front Cardiovasc Med (2021) 8:768029. doi: 10.3389/fcvm.2021.768029
97. Wu X, Huang L, Liu J. Relationship between oxidative stress and nuclear factor-erythroid-2-related factor 2 signaling in diabetic cardiomyopathy (Review). Exp Ther Med (2021) 22:678. doi: 10.3892/etm.2021.10110
98. Tang Z, Wang P, Dong C, Zhang J, Wang X, Pei H. Oxidative stress signaling mediated pathogenesis of diabetic cardiomyopathy. Oxid Med Cell Longev (2022) 2022:5913374. doi: 10.1155/2022/5913374
99. Wei Z, Jing Z, Pinfang K, Chao S, Shaohuan Q. Quercetin inhibits pyroptosis in diabetic cardiomyopathy through the nrf2 pathway. J Diabetes Res (2022) 2022:9723632. doi: 10.1155/2022/9723632
100. Li L, Luo W, Qian Y, Zhu W, Qian J, Li J, et al. Luteolin protects against diabetic cardiomyopathy by inhibiting NF-κB-mediated inflammation and activating the Nrf2-mediated antioxidant responses. Phytomedicine (2019) 59:152774. doi: 10.1016/j.phymed.2018.11.034
101. Wei Z, Pinfang K, Jing Z, Zhuoya Y, Shaohuan Q, Chao S. Curcumin improves diabetic cardiomyopathy by inhibiting pyroptosis through AKT/nrf2/ARE pathway. Mediators Inflammation (2023) 2023:e3906043. doi: 10.1155/2023/3906043
102. Karbassi E, Fenix A, Marciano S, Muraoka N, Nakamura K, Yang X, et al. Cardiomyocyte maturation: advances in knowledge and implications for regenerative medicine. Nat Rev Cardiol (2020) 17(6):341–59. doi: 10.1038/s41569-019-0331-x
103. Wang Y, Liu X, Shi H, Yu Y, Yu Y, Li M, et al. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin Trans Med (2020) 10:91–106. doi: 10.1002/ctm2.13
104. Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol (2020) 34:101523. doi: 10.1016/j.redox.2020.101523
105. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature (2019) 570:338–43. doi: 10.1038/s41586-019-1295-z
106. Shen S, Wang Z, Sun H, Ma L. Role of NLRP3 inflammasome in myocardial ischemia-reperfusion injury and ventricular remodeling. Med Sci Monitor: Int Med J Exp Clin Res (2022) 28:e934255. doi: 10.12659/MSM.934255
107. Zhou L, Yang S, Zou X. Farrerol alleviates myocardial ischemia/reperfusion injury by targeting macrophages and NLRP3. Front Pharmacol (2022) 13:879232. doi: 10.3389/fphar.2022.879232
108. Li Y, Sun X, Liu X, Li J, Li X, Wang G, et al. P2X7R-NEK7-NLRP3 inflammasome activation: A novel therapeutic pathway of qishen granule in the treatment of acute myocardial ischemia. JIR (2022) 15:5309–26. doi: 10.2147/JIR.S373962
109. Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J. CaMKIIδ-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight (2018) 3:e97054. doi: 10.1172/jci.insight.97054
110. Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by ca2+/calmodulin-dependent protein kinase II δ Signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation (2018) 138:2530–44. doi: 10.1161/CIRCULATIONAHA.118.034621
111. Suetomi T, Miyamoto S, Brown JH. Inflammation in nonischemic heart disease: initiation by cardiomyocyte CaMKII and NLRP3 inflammasome signaling. Am J Physiol Heart Circ Physiol (2019) 317(5):H877–90. doi: 10.1152/ajpheart.00223.2019
112. Ding K, Song C, Hu H, Yin K, Huang H, Tang H. The role of NLRP3 inflammasome in diabetic cardiomyopathy and its therapeutic implications. Oxid Med Cell Longev (2022) 2022:3790721. doi: 10.1155/2022/3790721
113. Xu Y, Fang H, Xu Q, Xu C, Yang L, Huang C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle (2020) 19:3054–65. doi: 10.1080/15384101.2020.1831245
114. Gao G, Fu L, Xu Y, Tao L, Guo T, Fang G, et al. Cyclovirobuxine D Ameliorates Experimental Diabetic Cardiomyopathy by Inhibiting Cardiomyocyte Pyroptosis via NLRP3 in vivo and in vitro. Front Pharmacol (2022) 13:906548. doi: 10.3389/fphar.2022.906548
115. Xie Y, Huang Y, Ling X, Qin H, Wang M, Luo B. Chemerin/CMKLR1 axis promotes inflammation and pyroptosis by activating NLRP3 inflammasome in diabetic cardiomyopathy rat. Front Physiol (2020) 11:381. doi: 10.3389/fphys.2020.00381
116. Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X, et al. LncRNA KCNQ1OT1 mediates pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem (2018) 50:1230–44. doi: 10.1159/000494576
117. Lian D, Zhu L, Yu Y, Zhang X, Lin Y, Liu J, et al. Kakonein restores hyperglycemia-induced macrophage digestion dysfunction through regulation of cathepsin B-dependent NLRP3 inflammasome activation. J Leukoc Biol (2022) 112:143–55. doi: 10.1002/JLB.3MA0821-418R
118. Mosser DM, Hamidzadeh K, Goncalves R. Macrophages and the maintenance of homeostasis. Cell Mol Immunol (2021) 18:579–87. doi: 10.1038/s41423-020-00541-3
119. Lin H-B, Wei G-S, Li F-X, Guo W-J, Hong P, Weng Y-Q, et al. Macrophage-NLRP3 inflammasome activation exacerbates cardiac dysfunction after ischemic stroke in a mouse model of diabetes. Neurosci Bull (2020) 36:1035–45. doi: 10.1007/s12264-020-00544-0
120. Levick SP, Widiapradja A. The diabetic cardiac fibroblast: mechanisms underlying phenotype and function. Int J Mol Sci (2020) 21:970. doi: 10.3390/ijms21030970
121. Zhao J, Randive R, Stewart JA. Molecular mechanisms of AGE/RAGE-mediated fibrosis in the diabetic heart. World J Diabetes (2014) 5:860–7. doi: 10.4239/wjd.v5.i6.860
122. Ren L, Chen X, Nie B, Qu H, Ju J, Bai Y. Ranolazine Inhibits Pyroptosis via Regulation of miR-135b in the Treatment of Diabetic Cardiac Fibrosis. Front Mol Biosci (2022) 9:806966. doi: 10.3389/fmolb.2022.806966
123. Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis (2018) 9:1–13. doi: 10.1038/s41419-018-1029-4
124. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin (2019) 40:1–8. doi: 10.1038/s41401-018-0042-6
125. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis (2020) 11:776. doi: 10.1038/s41419-020-02985-x
126. Shah W, Zhao Q, Wang S, Zhang M, Ma H, Guan Y, et al. Polydatin improves vascular endothelial function by maintaining mitochondrial homeostasis under high glucose conditions. Sci Rep (2023) 13:16550. doi: 10.1038/s41598-023-43786-4
127. Feng Y-H, Li L-F, Zhang Q, Zhang J-H, Huang Y, Lv Y-L, et al. Microtubule associated protein 4 (MAP4) phosphorylation reduces cardiac microvascular density through NLRP3-related pyroptosis. Cell Death Discovery (2021) 7:213. doi: 10.1038/s41420-021-00606-w
128. Nikolic M, Zivkovic V, Jovic JJ, Sretenovic J, Davidovic G, Simovic S, et al. SGLT2 inhibitors: a focus on cardiac benefits and potential mechanisms. Heart Fail Rev (2022) 27:935–49. doi: 10.1007/s10741-021-10079-9
129. Huang K, Luo X, Liao B, Li G, Feng J. Insights into SGLT2 inhibitor treatment of diabetic cardiomyopathy: focus on the mechanisms. Cardiovasc Diabetol (2023) 22:86. doi: 10.1186/s12933-023-01816-5
130. Xue M, Li T, Wang Y, Chang Y, Cheng Y, Lu Y, et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin Sci (Lond) (2019) 133:1705–20. doi: 10.1042/CS20190585
131. Ye Y, Bajaj M, Yang H-C, Perez-Polo JR, Birnbaum Y. SGLT-2 inhibition with dapagliflozin reduces the activation of the nlrp3/ASC inflammasome and attenuates the development of diabetic cardiomyopathy in mice with type 2 diabetes. Further augmentation of the effects with saxagliptin, a DPP4 inhibitor. Cardiovasc Drugs Ther (2017) 31:119–32. doi: 10.1007/s10557-017-6725-2
132. Chen H, Tran D, Yang H-C, Nylander S, Birnbaum Y, Ye Y. Dapagliflozin and ticagrelor have additive effects on the attenuation of the activation of the NLRP3 inflammasome and the progression of diabetic cardiomyopathy: an AMPK-mTOR interplay. Cardiovasc Drugs Ther (2020) 34:443–61. doi: 10.1007/s10557-020-06978-y
133. Foretz M, Guigas B, Bertrand L, Pollak M, Viollet B. Metformin: from mechanisms of action to therapies. Cell Metab (2014) 20:953–66. doi: 10.1016/j.cmet.2014.09.018
134. Yang F, Qin Y, Wang Y, Meng S, Xian H, Che H, et al. Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int J Biol Sci (2019) 15:1010–9. doi: 10.7150/ijbs.29680
135. Yang Z, Wang M, Zhang Y, Cai F, Jiang B, Zha W, et al. Metformin ameliorates diabetic cardiomyopathy by activating the PK2/PKR pathway. Front Physiol (2020) 11:425. doi: 10.3389/fphys.2020.00425
136. Zou R, Nie C, Pan S, Wang B, Hong X, Xi S, et al. Co-administration of hydrogen and metformin exerts cardioprotective effects by inhibiting pyroptosis and fibrosis in diabetic cardiomyopathy. Free Radic Biol Med (2022) 183:35–50. doi: 10.1016/j.freeradbiomed.2022.03.010
137. García-Díez E, López-Oliva ME, Caro-Vadillo A, Pérez-Vizcaíno F, Pérez-Jiménez J, Ramos S, et al. Supplementation with a cocoa-carob blend, alone or in combination with metformin, attenuates diabetic cardiomyopathy, cardiac oxidative stress and inflammation in zucker diabetic rats. Antioxidants (Basel) (2022) 11:432. doi: 10.3390/antiox11020432
138. Qin W, Zhao X, Tai J, Qin G, Yu S. Combination of dendrobium mixture and metformin curbs the development and progression of diabetic cardiomyopathy by targeting the lncRNA NEAT1. Clinics (Sao Paulo) (2021) 76:e2669. doi: 10.6061/clinics/2021/e2669
139. Quanwei Z, Hui L, Danan L, Caiwei G, Long C. Dapagliflozin attenuates endothelial cell pyroptosis and dysfunction induced by oxidized low-density lipoprotein. Chin J Tissue Eng Res (2024) 28:80. doi: 10.12307/2023.773
140. Zhang J, Huang L, Shi X, Yang L, Hua F, Ma J, et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging (Albany NY) (2020) 12:24270–87. doi: 10.18632/aging.202143
141. Wu D, Chen Y, Sun Y, Gao Q, Li H, Yang Z, et al. Target of MCC950 in inhibition of NLRP3 inflammasome activation: a literature review. Inflammation (2020) 43:17–23. doi: 10.1007/s10753-019-01098-8
142. Bakhshi S, Shamsi S. MCC950 in the treatment of NLRP3-mediated inflammatory diseases: Latest evidence and therapeutic outcomes. Int Immunopharmacol (2022) 106:108595. doi: 10.1016/j.intimp.2022.108595
143. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
144. Ge K, Wang Y, Li P, Li M, Zhang W, Dan H, et al. Down-expression of the NLRP3 inflammasome delays the progression of diabetic retinopathy. Microvasc Res (2022) 139:104265. doi: 10.1016/j.mvr.2021.104265
145. Sharma A, Choi JSY, Stefanovic N, Al-Sharea A, Simpson DS, Mukhamedova N, et al. Specific NLRP3 inhibition protects against diabetes-associated atherosclerosis. Diabetes (2021) 70:772–87. doi: 10.2337/db20-0357
146. Hong P, Gu R-N, Li F-X, Xiong X-X, Liang W-B, You Z-J, et al. NLRP3 inflammasome as a potential treatment in ischemic stroke concomitant with diabetes. J Neuroinflamm (2019) 16:121. doi: 10.1186/s12974-019-1498-0
147. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214:3219–38. doi: 10.1084/jem.20171419
148. MacDonald JA, Wijekoon CP, Liao K-C, Muruve DA. Biochemical and structural aspects of the ATP-binding domain in inflammasome-forming human NLRP proteins. IUBMB Life (2013) 65:851–62. doi: 10.1002/iub.1210
149. Yang M, Zhao L. The selective NLRP3-inflammasome inhibitor CY-09 ameliorates kidney injury in diabetic nephropathy by inhibiting NLRP3- inflammasome activation. Curr Med Chem (2023) 30:3261–70. doi: 10.2174/0929867329666220922104654
150. Sun K, Wang J, Lan Z, Li L, Wang Y, Li A, et al. Sleeve gastroplasty combined with the NLRP3 inflammasome inhibitor CY-09 reduces body weight, improves insulin resistance and alleviates hepatic steatosis in mouse model. Obes Surg (2020) 30:3435–43. doi: 10.1007/s11695-020-04571-8
151. Zhang W, Xu W, Feng Y, Zhou X. Non-coding RNA involvement in the pathogenesis of diabetic cardiomyopathy. J Cell Mol Med (2019) 23:5859–67. doi: 10.1111/jcmm.14510
152. Jakubik D, Fitas A, Eyileten C, Jarosz-Popek J, Nowak A, Czajka P, et al. MicroRNAs and long non-coding RNAs in the pathophysiological processes of diabetic cardiomyopathy: emerging biomarkers and potential therapeutics. Cardiovasc Diabetol (2021) 20:55. doi: 10.1186/s12933-021-01245-2
153. Guo R, Nair S. Role of microRNA in diabetic cardiomyopathy: From mechanism to intervention. Biochim Biophys Acta Mol Basis Dis (2017) 1863:2070–7. doi: 10.1016/j.bbadis.2017.03.013
154. Xu D, Zhang X, Chen X, Yang S, Chen H. Inhibition of miR-223 attenuates the NLRP3 inflammasome activation, fibrosis, and apoptosis in diabetic cardiomyopathy. Life Sci (2020) 256:117980. doi: 10.1016/j.lfs.2020.117980
155. Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis (2014) 5:e1479. doi: 10.1038/cddis.2014.430
156. Deng B, Hu Y, Sheng X, Zeng H, Huo Y. miR-223-3p reduces high glucose and high fat-induced endothelial cell injury in diabetic mice by regulating NLRP3 expression. Exp Ther Med (2020) 20:1514–20. doi: 10.3892/etm.2020.8864
157. Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nat Rev Genet (2009) 10:155–9. doi: 10.1038/nrg2521
158. Yuan Q, Sun Y, Yang F, Yan D, Shen M, Jin Z, et al. CircRNA DICAR as a novel endogenous regulator for diabetic cardiomyopathy and diabetic pyroptosis of cardiomyocytes. Signal Transduct Target Ther (2023) 8:99. doi: 10.1038/s41392-022-01306-2
159. Jubaidi FF, Zainalabidin S, Taib IS, Hamid ZA, Budin SB. The potential role of flavonoids in ameliorating diabetic cardiomyopathy via alleviation of cardiac oxidative stress, inflammation and apoptosis. Int J Mol Sci (2021) 22:5094. doi: 10.3390/ijms22105094
160. Abo-Saif MA, Ragab AE, Ibrahim AO, Abdelzaher OF, Mehanyd ABM, Saber-Ayad M, et al. Pomegranate peel extract protects against the development of diabetic cardiomyopathy in rats by inhibiting pyroptosis and downregulating LncRNA-MALAT1. Front Pharmacol (2023) 14:1166653. doi: 10.3389/fphar.2023.1166653
161. Imazio M, Nidorf M. Colchicine and the heart. Eur Heart J (2021) 42:2745–60. doi: 10.1093/eurheartj/ehab221
Keywords: pyroptosis, diabetic cardiomyopathy, NLRP3 inflammasome, inflammation, mechanism
Citation: Wang G, Ma T-Y, Huang K, Zhong J-H, Lu S-J and Li J-J (2024) Role of pyroptosis in diabetic cardiomyopathy: an updated review. Front. Endocrinol. 14:1322907. doi: 10.3389/fendo.2023.1322907
Received: 17 October 2023; Accepted: 06 December 2023;
Published: 05 January 2024.
Edited by:
Ramoji Kosuru, Versiti Blood Research Institute, United StatesReviewed by:
Lorenzo Da Dalt, University of Milan, ItalyIgnacio Norambuena-Soto, University of Chile, Chile
Copyright © 2024 Wang, Ma, Huang, Zhong, Lu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiang-Hua Zhong, emhvbmczODgyQDE2My5jb20=; Shi-Juan Lu, MTE1NzQxNjY3NkBxcS5jb20=; Jian-Jun Li, bGlqaWFuanVuOTM4QDEyNi5jb20=