
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 03 January 2024
Sec. Translational and Clinical Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1320516
 Luis Martínez-Heredia1
Luis Martínez-Heredia1 Manuel Muñoz-Torres1,2,3,4*
Manuel Muñoz-Torres1,2,3,4* Raquel Sanabria-de la Torre1,5
Raquel Sanabria-de la Torre1,5 Ángela Jiménez-Ortas1,6
Ángela Jiménez-Ortas1,6 Francisco Andújar-Vera7,8,9Trinidad González-Cejudo1,10
Francisco Andújar-Vera7,8,9Trinidad González-Cejudo1,10 Victoria Contreras-Bolívar2
Victoria Contreras-Bolívar2 Sheila González-Salvatierra1,2,3José María Gómez-Vida11
Sheila González-Salvatierra1,2,3José María Gómez-Vida11 Cristina García-Fontana1,2,4*
Cristina García-Fontana1,2,4* Beatriz García-Fontana1,2,4,12
Beatriz García-Fontana1,2,4,12Introduction: Hypophosphatasia (HPP) is an inborn metabolic error caused by mutations in the ALPL gene encoding tissue non-specific alkaline phosphatase (TNSALP) and leading to decreased alkaline phosphatase (ALP) activity. Although the main characteristic of this disease is bone involvement, it presents a great genetic and clinical variability, which makes it a systemic disease.
Methods: Patients were recruited based on biochemical assessments. Diagnosis was made by measuring serum ALP and pyridoxal 5-phosphate levels and finally by Sanger sequencing of the ALPL gene from peripheral blood mononuclear cells. Characterization of the new variants was performed by transfection of the variants into HEK293T cells, where ALP activity and cellular localization were measured by flow cytometry. The dominant negative effect was analyzed by co-transfection of each variant with the wild-type gene, measuring ALP activity and analyzing cellular localization by flow cytometry.
Results: Two previously undescribed variants were found in the ALPL gene: leucine 6 to serine missense mutation (c.17T>C, L6S) affecting the signal peptide and threonine 167 deletion (c.498_500delCAC, T167del) affecting the vicinity of the active site. These mutations lead mainly to non-pathognomonic symptoms of HPP. Structural prediction and modeling tools indicated the affected residues as critical residues with important roles in protein structure and function. In vitro results demonstrated low TNSALP activity and a dominant negative effect in both mutations. The results of the characterization of these variants suggest that the pleiotropic role of TNSALP could be involved in the systemic effects observed in these patients highlighting digestive and autoimmune disorders associated with TNSALP dysfunction.
Conclusions: The two new mutations have been classified as pathogenic. At the clinical level, this study suggests that both mutations not only lead to pathognomonic symptoms of the disease, but may also play a role at the systemic level.
Hypophosphatasia (HPP) is a rare genetic and, in some cases, a lethal disease characterized mainly by bone and tooth mineralization defects (1). This disease is caused by one or more loss-of-function mutations in the ALPL gene encoding tissue non-specific alkaline phosphatase protein (TNSALP) (2). The prevalence of this disease is difficult to estimate due to the lack of knowledge of this disorder leading to a high rate of underdiagnosis. In 2011, Mornet et al. reported a mild HPP prevalence of 1/6,370 for Europe (3). However, our research group, in 2019, indicated that the prevalence of mild HPP in Spain may be higher than previously thought (1/3,100) (4). In 2021, Mornet et al. reevaluated the prevalence of mild HPP in Europe (1/2,430), which aligned with the prevalence we observed in Spain (5). Nevertheless, our most recent study, published in 2023, has revealed a new estimate for mild HPP prevalence in Spain, nearly double the earlier figure (1/1,692 vs. 1/3,100) (6). During the last 20 years, the perception of this disease has evolved significantly from a rare, recessive, bone disease to a systemic disease with higher incidence than previously reported and dominant inheritance in the mild forms (7).
TNSALP is a homodimer ectoenzyme (8) belonging to the alkaline phosphatase (ALP) family (EC 3.1.3.1). There are four other tissue-specific phosphatases in this family: intestinal (IALP), placental (PALP) and germ cell (GCALP) alkaline phosphatases, which are encoded by the ALPI, ALPP and ALPP2 genes respectively. These enzymes hydrolyze monoester bonds from their substrates to yield inorganic phosphate (Pi). TNSALP acts on inorganic pyrophosphate (PPi) to yield Pi, one of the substrates to produce hydroxyapatite crystals. Pyridoxal-5-phosphate (PLP), also known as vitamin B6, is another major substrate of TNSALP and a precursor of some neurotransmitters. This molecule is hydrolyzed to pyridoxal (PL) to cross the blood-brain barrier where it will be reconverted to PLP (2, 9) (Figure 1A). Other TNSALP substrates described include lipopolysaccharide (LPS) (10), adenosine triphosphate (ATP) (11) and phosphorylated osteopontin (12).
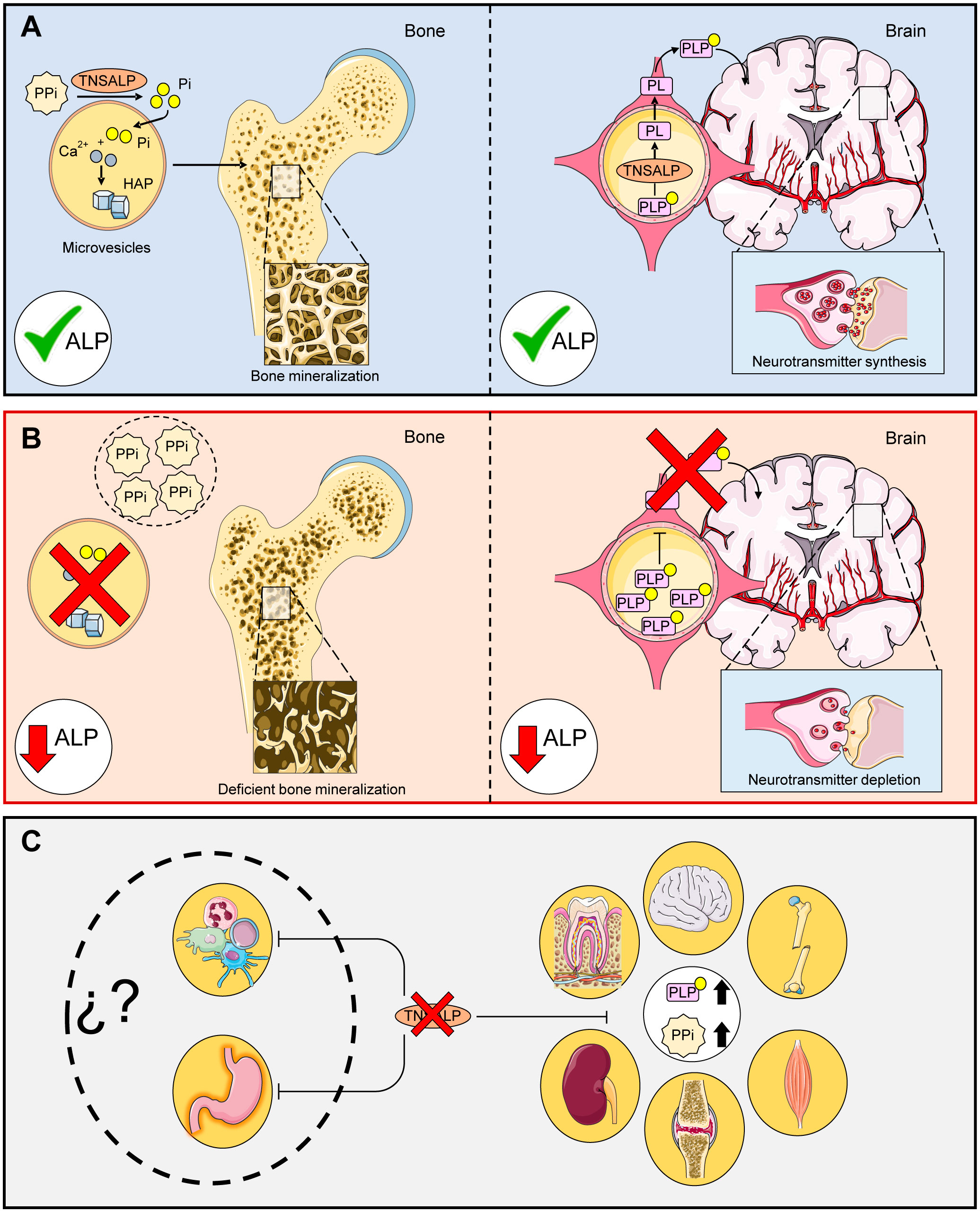
Figure 1 Main functions of TNSALP in the body. (A) shows the correct function of TNASLP: PPi is hydrolyzed to Pi for subsequent conversion to hidroxyhapatite crystals (HAP) in microvesicles via the action of TNSALP. PLP is hydrolyzed to PL by TNSALP to cross the blood-brain barrier, where it is subsequently reconstituted into PLP and leads to the formation of neurotransmitters. (B) shows the molecular effects related to defective TNSALP: Loss of function in TNSALP produces an accumulation of its substrates. PPi inhibits bone mineralization while PLP cannot cross the blood-brain barrier, decreasing the production of neurotransmitters. (C) shows clinical manifestations related to defective TNSALP: The accumulation of PPi alters calcium/phosphate homeostasis, causing bone, tooth, renal and joint damage, while increased levels of PLP lead to a decrease in B6 supply at the neurological level, which can lead to seizures, muscle and respiratory problems. However, the autoimmune and digestive implications have not been explored to date.
TNSALP is involved in numerous pleiotropic processes (13, 14) and is expressed in most tissues and organs according to the Human Protein Atlas (Web References), particularly in bone, kidney and liver. The most frequent clinical manifestation of HPP is high rates of bone fragility and fractures as well as neurological disorders. This is due to the accumulation of their substrates that inhibit bone mineralization and lead to a decrease in neurotransmitter precursors (Figure 1B). In addition, some forms of rickets and osteomalacia have been linked to deficiencies in this enzyme (15, 16). However, these clinical features do not apply to all HPP patients due to the vast variety of mutations as well as the different types of inheritance. The clinical features of this disease range from completely asymptomatic patients to complete absence of bone mineralization and fetal death (17). The disease conditions can affect different organs and systems including skeletal (17, 18), muscular (19), dental (20), neurological (21), respiratory (17), renal (22) and articular (23) due to accumulation of their substrates (Figure 1C).
There are more than 500 described variants in the ALPL gene according to the Leiden Open Variation Database (LOVD) website, 628 nonsynonymous variants in The Genome Aggregation Database (gnomAD) website and 438 nucleotide variants and 14 structural variants in ALPL mutation database (Web References) which strongly contributes to the wide phenotypic heterogeneity of this disease. In addition, many of these variants have a dominant negative effect (DNE), which further exacerbates this phenotypic variability. DNE is defined as the decrease below 50% of TNSALP activity when the wild-type (WT) allele and the mutated allele are co-expressed at the same levels (24, 25). Due to these factors, is very difficult to establish a relationship between genotype and phenotype in HPP patients. In this context, del Angel et al. (2020), using structural model predictions in silico algorithms, observed associations between residual enzymatic between residual enzyme activity and variant origin, variant type, affected protein domain, and HPP subtype (26). However, although there appears to be a relationship between low in vitro TNSALP levels and disease severity, there are other variables that influence the development of clinical manifestations in affected patients.
In this study, two previously undescribed mutations are presented in two patients recently diagnosed with childhood-onset HPP, with no familial relationship and with completely different clinical features. The aim is to characterize each of the new mutations at the genetic, structural and functional levels to establish a relationship with the clinical features. Clinical characteristics and biochemical parameters have been useful in establishing a correct diagnosis of the rest of the relatives. In this context, it is worth emphasizing the importance of establishing a geno-phenotypic relationship for each newly identified mutation to provide more information and better patient management.
Two fifteen-year-old male patients were evaluated in the Endocrinology Unit of the University Hospital Clínico San Cecilio of Granada following the algorithm developed by García-Fontana et al. (4). Neither of the two patients took vitamin B6 supplements. Patients with secondary causes of hypophosphatasemia such as malnutrition, magnesium and zinc deficiencies, haemochromatosis or certain therapies, were excluded (2, 27). Two venous blood samples were taken from each patient at the Clinical Analysis Unit of the University Hospital Clínico San Cecilio; one was for ALP and PLP determinations, and the other one was used for ALPL gene sequencing. Written informed consent was obtained from their legal guardians and an individualized and personal interview was conducted on potentially related HPP symptoms. This study was approved by the ethics committee of Granada following the principles of the World Medical Association Declaration of Helsinki (Project ID: 0777-M1-20. Research Ethics Committee of Granada Center [(CEI-Granada) on 8 May 2019].
ALP activity was measured bichromatically at 410/480 nm by conversion of p-nitrophenyl phosphate to p-nitrophenol in the presence of magnesium, zinc and 2-amino-2-methyl-1-propanol as phosphate acceptor at pH 10.4 from blood samples on AU5800 analyzers (Beckman Coulter) according to the method recommended by the International Federation of Clinical Chemistry. ALP determinations were performed in the Clinical Analysis Laboratory of University Hospital Clínico San Cecilio. The reference values for fifteen-year-old males were 75-312 IU/L following the values indicated in the CALIPER study adjusted by age and sex (28).
Plasma PLP levels were measured by high-pressure liquid chromatography (HPLC) at the Clinical Unit of the University Hospital Niño Jesús (Madrid). Chromatographic determination was determined using an isocratic HPLC system. For PLP detection, an emission laser at 320 nm and a fluorescence detector was used. Reference values (3.6-18 ng/mL) were established by University Hospital Niño Jesús and for fifteen-year-old males.
The DNA used for sequencing was collected from peripheral blood lymphocytes and the polymerase chain reaction (PCR) of the ALPL gene was performed following the method described by Riancho et al. (29). The PCR product underwent Sanger sequencing and the canonical sequence (Gene ID: 249, RefSeq: NM_000478.6 (ALPL Transcript variant 1), UniProt: P05186-1) was used as a reference. The alternative ALPL transcripts and their corresponding protein sequences has been included in Supplementary Tables S1, S2. Finally, a copy variant number study was performed by multiple ligation probe amplification (MLPA) (MRCHolland) and the results were analyzed using the SeqPilot program (JSI Medical System). The Biomedical Diagnostic Center of the Clinic Hospital of Barcelona provided the sequencing results.
After confirmation of the presence of mutations in the ALPL gene in the two patients, available relatives were recruited to perform ALP blood measurements, interviews about their clinical history, and ALPL gene sequencing. According to de IFCC, the reference values for adult males were 43-115 IU/L while for females were 33-98 IU/L.
MutPred, PROVEAN and Mutation Taster algorithms were used to predict the consequences of protein mutations. Combined annotation-dependent depletion (CADD) was used to rank mutations according to impact and was compared to the mutation significance cut-off (MSC) obtained for CADD scores (30). Both mutations have already been introduced into the VarSome database with references NM_000478:c.17T>C for the L6S variant and NM_001369805.2:c.498_500del for the T167del variant.
To determine the degree of conservation of the amino acids affected by the new variants in the TNSALP protein of Homo sapiens (P05186-1), a multiple sequence analysis (MSA) was performed. Twenty species encompassing different types of vertebrates including different classes such as reptiles, amphibians, birds, fish and mammals belonging to different orders were chosen for the analysis. Among the different orders of mammals, rodents, artiodactyls, perissodactyls, carnivores and primates of the genera macaca, pongo, pan and gorilla were chosen (Supplementary Table S3). Multiple sequence alignment was performed using the ClustalW tool of the Unipro UGENE V.45.0 software (31).
For 3D modelling, the complete atomic model was predicted using AlphaFold2_advanced (32). The models with the highest scores in the local distance difference test (pLDDT) were chosen. Finally, the visualization and preparation of the figures were performed using Chimera X software (33).
Human embryonic kidney cells 293T (HEK293T) were used. Briefly, cells were cultured at 37°C and 5% CO2 with Dulbecco’s Modified Eagle Medium (DMEM) High Glucose (pH 7.2) (Biowest) supplemented with 10% fetal bovine serum (Capricorn scientific), 5% Ham’s F12 Nutrient Medium (Biowest) and 1% of 100X Antibiotic-Antimycotic (Biowest).
The vectors used were constructed by modifying the pcDNA3.1 plasmid. The ALPL gene with the study variants L6S (pcDNA3.1:ALPL c.17T>C) and T167del (pcDNA3.1:ALPL c.498_500delCAC) was inserted in this plasmid. The pcDNA3.1 plasmid with the WT ALPL gene insertion (pcDNA3.1: ALPL) was used as a positive control to functionally characterize the identified variants. The empty vector (EV) without insertion (pcDNA3.1) was used as a negative control to monitor the basal expression of the ALPL gene at the cellular level. The different variants of the ALPL gene were inserted between the HindIII and BamHI restriction sequences belonging to the multi-cloning site. All vectors were supplied by GenScript.
Before HEK293T cells transfection, 150000 cells were grown per well in 24-well plates. After 24 hours, transient transfection was performed by adding to each well 50 µL of serum-free DMEM containing 1.5 µL of LipoD293 DNA in vitro transfection reagent (SignaGen Laboratories) and 500 ng of the corresponding plasmid. Co-transfections were carried out by mixing 250 ng of each variant with 250 ng of WT obtaining 500 ng of total reaction. The cells were incubated for 18 hours with the mixture and after that time, 2 mL of DMEM supplemented with serum was added. Finally, the cells were incubated for 24 hours and harvested for the different assays. The term homozygous will refer to cells transfected with a single plasmid while the term heterozygous will refer to cells co-transfected unless otherwise indicated.
RT-qPCR was performed to determine the exogenous levels of ALPL gene expression by each construction. Firstly, RNA was collected from each transfected culture using the RNeasy® Mini Kit (Qiagen) and treated with DNase (Qiagen). For cDNA synthesis, 600 ng of template RNA and the iScript cDNA synthesis kit (BioRad) were used following the manufacturer’s protocol. For quantitative PCR, PowerUP SYBR Green Master Mix (Thermo Fisher Scientific) was used in the CFX96 real-time thermal cycler (BioRad). Gene expression was normalized using the ribosomal protein L13 (RPL13) used as a constitutive gene. The set of primers used for the determination of ALPL expression is listed in Supplementary Table S4. Finally, the results were analyzed by using the ΔΔCt method.
Cell viability was determined by using FITC Annexin V Apoptosis Detection Kit I (BD Biosciences) following the manufacturer’s protocol. Cells that were negative for both propidium iodide and Annexin V expression were considered viable cells, while cells that were positive for Annexin V, individually or together with propidium iodide, were considered apoptotic cells.
Antigenic density was performed following the protocol developed by Lopez-Perez et al. (34). Briefly, the cells were washed with PBS and incubated with 2 µL of BV421 Mouse Anti-Human Alkaline Phosphatase (BD Biosciences) for 20 minutes. Then, the cells were fixed with 4% formaldehyde for 20 minutes. Finally, the cells were washed twice with PBS and resuspended in 100 µL. To standardize the expression of TNSALP variants on cell membranes, 5 µL of CountBright™ Absolute Count Beads (Invitrogen) were resuspended in 100 µL of PBS. All results were obtained with the BD FACSAria III Cell Sorter flow cytometer (BD Biosciences) on a logarithmic scale. Antigenic density was calculated as the ratio of the median intensities of the TNSALP-positive cells versus the median intensity obtained by the CountBrigh Absolute Count Beads.
TNSALP activity was measured at a wavelength of 450 nm by spectrophotometry (Dynex Technologies) using the Alkaline Phosphatase Detection Kit (Abnova) from cell extracts according to the manufacturer’s recommended protocol.
Each experiment was performed in triplicate. Saphiro-Wilk test was used to test the normal distribution of data. To evaluate the differences between groups, the one-way ANOVA test was used followed by Tukey HSD. P-values below 0.05 were considered significant. All tests were conducted with GraphPad Prism 9.5.1.
Table 1 shows the results of the biochemical analyses of each patient. The blood ALP activity in Patient 1 (P1) and 2 (P2) had persistently low ALP activity (73 IU/L and 45 IU/L respectively) while the PLP concentration were 6.5 and 2.5 times higher (118 µg/L and 45.5 µg/L respectively) than the normal values. Regarding clinical manifestations, P1 showed symptoms related to digestive, neurological, endocrine and muscular systems, gastroesophageal reflux disease (GERD) and dyspepsia, pes valgus deformity, vitamin D deficiency and high blood pressure (HBP) while P2 presented clinical manifestations related to autoimmune diseases such as Crohn’s disease and inverse psoriasis in addition to vitamin D deficiency. P2 was treated with calciferol and immunosuppressants. Interestingly, none of the patients presented pathognomonic symptoms of HPP. After two years of follow-up, both patients maintained persistently low levels of ALP activity (P1: 42 IU/L; P2 38 IU/L).

Table 1 Anthropometric and clinical parameters of HPP patients.
P1 presented a missense mutation in heterozygosis in the second exon of the ALPL gene (c.17T>C) changing a leucine for serine in position 6 of the protein sequence (L6S). P2 was found to be heterozygous for a three-nucleotide in-frame deletion (c.498_500del) that resulted in the loss of the threonine residue at position 167 (T167del) in the TNSALP sequence. Figure 2A shows a schematic context of the newly identified variants at genomic, transcriptional and protein levels. Both mutations have not been previously described in the scientific literature as they have not been found in any database such as genomeAD LOVD or Clinical relevant Variation (ClinVar) (Web References).
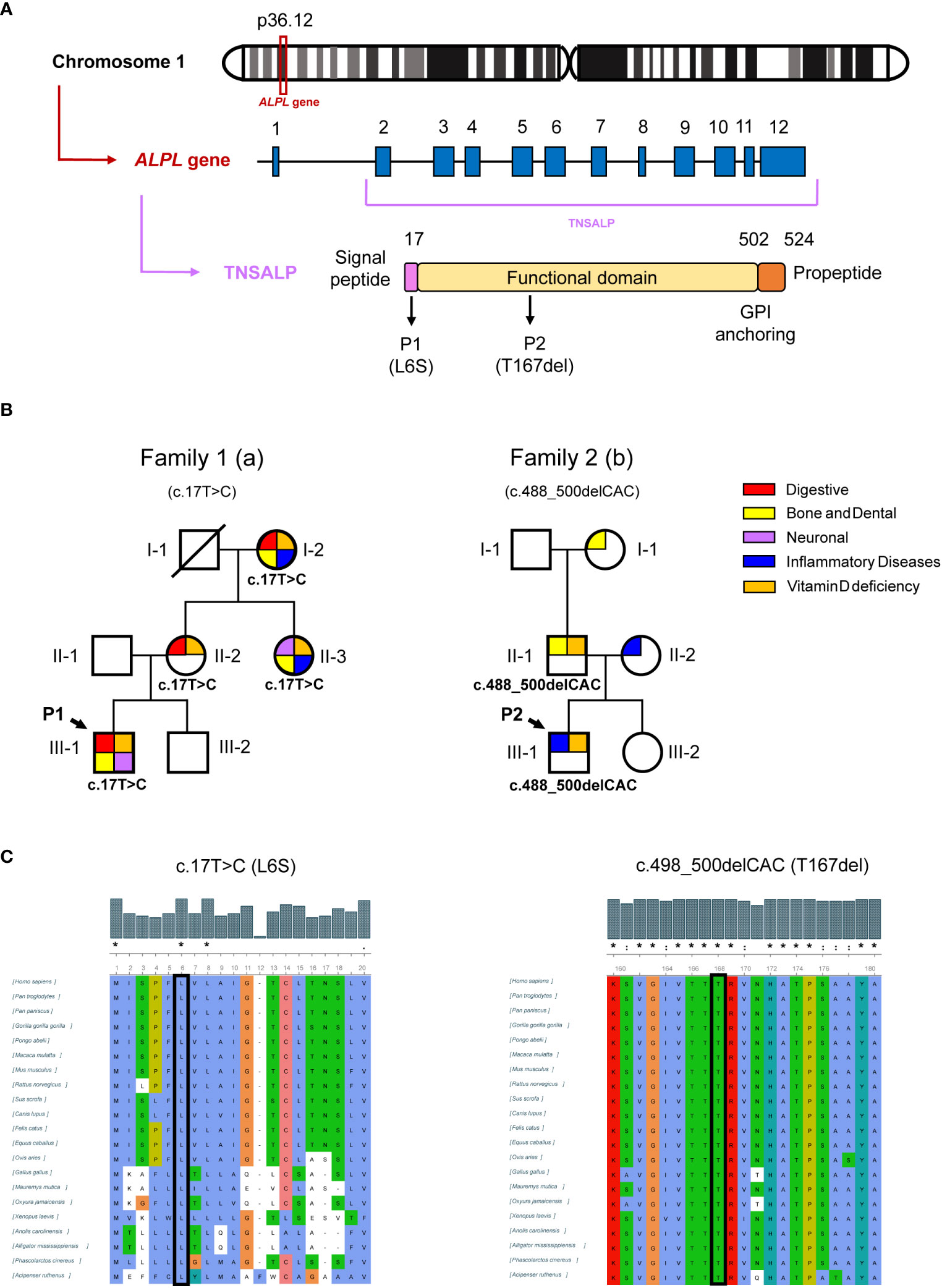
Figure 2 Genetic results of patients and relatives. (A) shows schematic representation of the location of the ALPL gene on chromosome 1, the ALPL gene with twelve exons and the TNSALP precursor protein with its N-terminal signal peptide, the functional domain, the C-terminal glycosylphosphatidylinositol (GPI) anchor binding site and the propeptide sequence. (B) shows genogram of the two families affected by HPP. The pathologies that affect each individual are summarized by categories. (C) shows the representation of the conservation of the human TNSALP mutated residues after MSA by ClustalW. The symbols (*,:.) represent identical, conserved and semi-conserved substitutions respectively while the absence of a symbol represents a lack of amino acid characteristic conservation. The whole alignment is shown in Supplementary Material.
The results of the pathogenicity predictions are shown in Table 2. P1 showed a disparity of results regarding the prognosis of the severity of the disease caused by the mutation. On the contrary, the P2 mutation was classified as pathogenic in all the programs used.
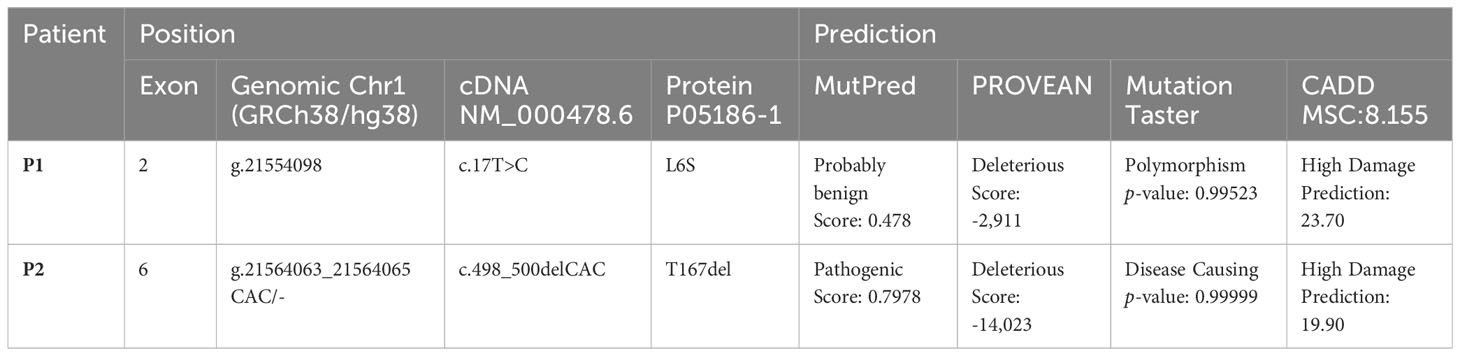
Table 2 Features of new mutations found in patients 1 and 2.
After identifying mutations in P1 and P2, serum ALP levels of close relatives were studied and those who presented persistently low levels underwent genetic study to investigate the origin of these mutations while those who presented serum ALP values within the normal range were discarded from the study following the clinical algorithm criteria described by our group (4). Table 3 shows the results for relatives of both patients affected by the new variants. In Family 1, I-2a, II-2a, and II-3a had the same mutation as P1, with consistently low levels of ALP activity (18, 20, and 25 IU/L, respectively). I-2a was treated with cholecalciferol and presented digestive disorders such as chronic diarrhea and diverticulitis. This patient also presented inflammatory diseases such as arteritis or polymyalgia rheumatica with current treatment. Regarding pathognomonic symptoms of HPP, this patient lost several teeth at an early age. II-2a presented with chronic gastritis, parathyroidism and a giant cell tumor affecting the little finger. Both I-2a and II-2a have elevated levels of interleukin 6 (IL6), indicating a pro-inflammatory state. II-3a had fibromyalgia, tooth infections, psoriatic arthritis, and other skeletal abnormalities detailed in Table 3. In Family 2, the P2 mutation came from II-1b, who also had low ALP activity (below 43 IU/L), tooth loss, and high blood pressure. It appears this variant was inherited from I-2b, who had a similar tooth loss condition associated with odontohypophosphatasia. All carriers of the new variants in the ALPL gene from both families presented vitamin D deficiency (<30 ng/mL). Figure 2B illustrates these findings in a family tree with clinical and sequencing results.
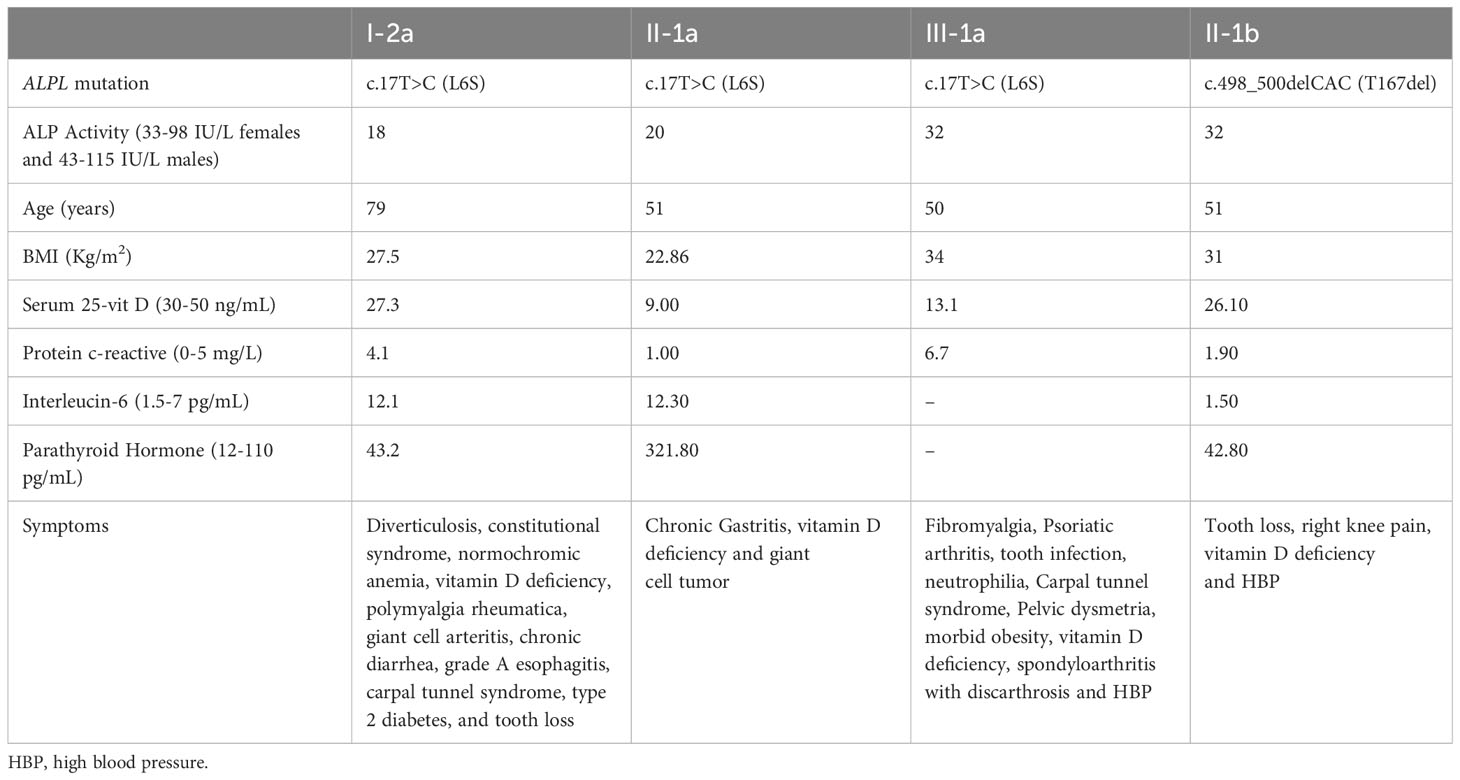
Table 3 Clinical results of relatives affected with HPP.
To know the degree of conservation of the mutated amino acids, MSA was performed using TNSALP sequences. Twenty animal TNSALP sequences were chosen for MSA (Supplementary Figure S1).
The results obtained demonstrated that the beginning of the sequence is quite poorly conserved except for three amino acids, M1, L6 and L8, which match all the sequences analyzed. Regarding the mutation of P2, the T167 matches throughout all aligned sequences (Figure 2C).
After simulation in AlphaFold2_advanced, the highest-ranked predictions based on the pLDDT score were chosen for WT (Figure 3A) and both new variants.
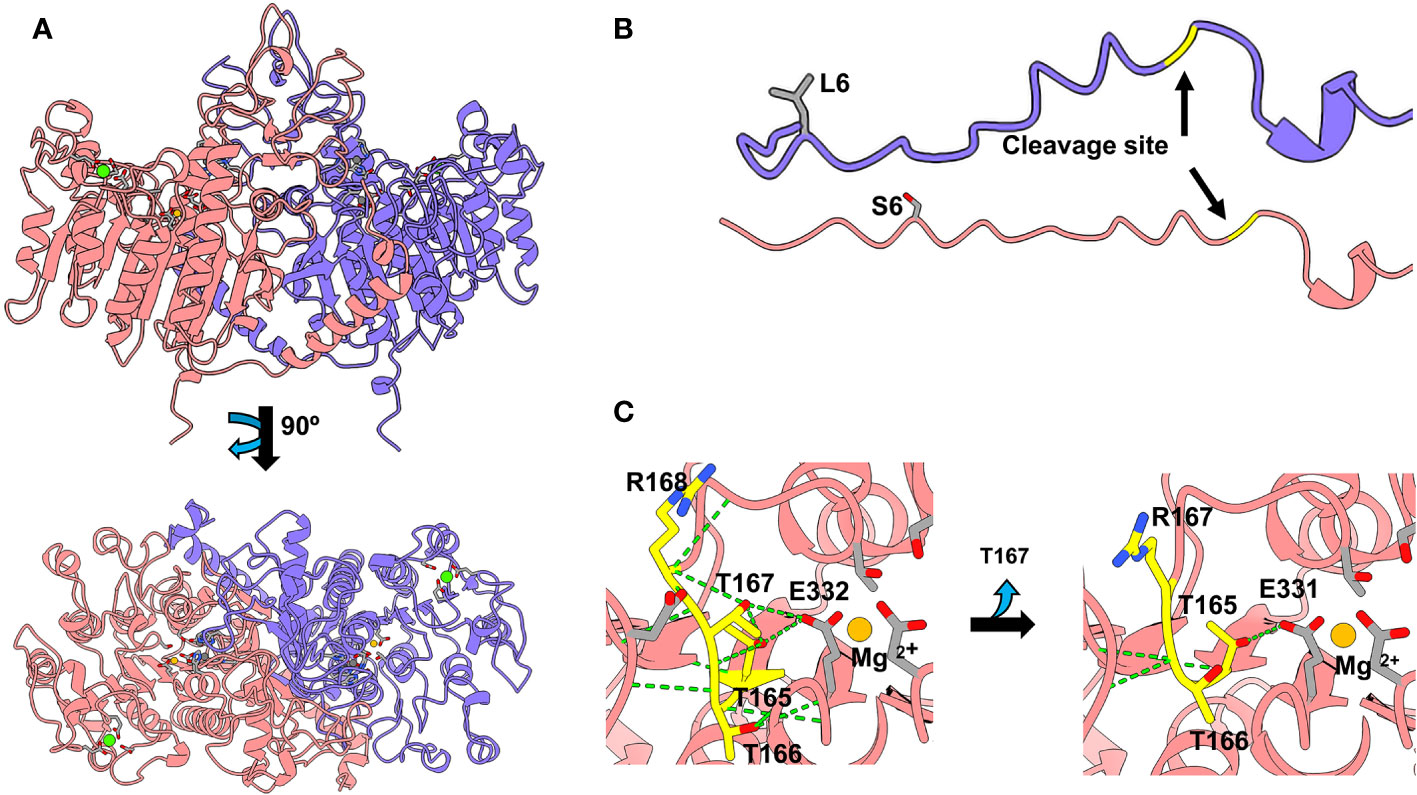
Figure 3 TNSALP 3D modelling based on the algorithm developed in AlphaFold2_advanced and visualized in Chimera X (Web References). (A) shows the visualization of TNSALP WT top and bottom structure. (B) shows the representation of the WT TNSALP and variant L6S signal peptide. (C) shows the representation of the structural characteristics of the T167del mutation.
L6S affected the signal peptide, while the catalytic core was not affected (Figure 3B). In contrast, T167del produces the shortening of a beta-loop in the vicinity of the active site of the protein. This loop is built by three consecutive threonines and one arginine (165TTTR168). T165 and T167 establish H-bonds with E332 which interacts directly with the Mg2+ of the active center. T166 generates hydrogen bonds with G334 and R335, forming an alpha helix above the catalytic site (Figure 3C). The shortening of the loop due to the deletion of T167 causes the loss of most of the hydrogen bonds, including those that affect the catalytic site.
Results of ALPL gene expression by RT-qPCR showed that cells transfected with the mutated vectors and with the WT vector had a 45-55 fold change compared to EV (p<0.001). There was no significant difference between the mutations and WT (Figure 4A).
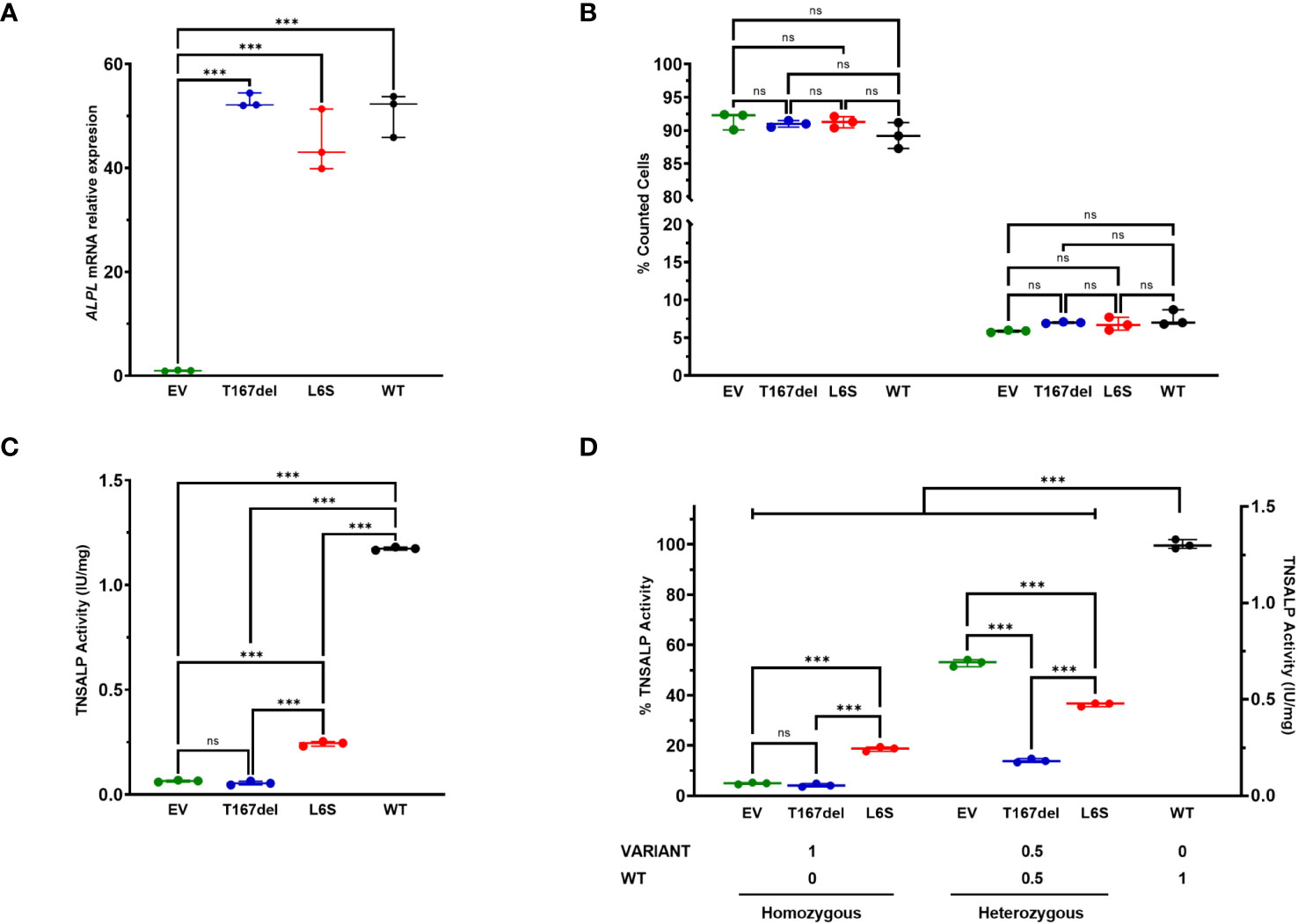
Figure 4 In vitro characterization of TNSALP activity of studied variants. Results are expressed as mean with standard deviation. ANOVA was used for comparisons between groups (***p<0.001). (A) shows relative mRNA expression after overexpression of new ALPL gene variants in HEK293T cells. The results were normalized using the housekeeping RPL13. (B) shows cell viability results obtained by flow cytometry. The results of survival and apoptosis were represented as a percentage concerning the total count of the cells in each culture. (C) shows TNSALP activity determination of the new ALPL variants. The quantitative results of the ALP assay were expressed in International Units per milligram of protein (IU/mg). (D) shows determination of the dominant negative effect of the study variants. Determinations were made by TNSALP activity and normalized per mg of protein. ns: not significant.
Cell survival was not compromised in any of the cases. Figure 4B shows that all populations had high survival rates (92-89%) and low apoptosis rates (5-7%). Furthermore, no statistically significant differences were observed between any of the study groups.
TNSALP activity in transfected cells showed a decrease in two variants compared to the WT protein. As shown in Figure 4C, the T167del variant had null activity, with no significant differences with respect to EV whereas the L6S variant had a statistically significant higher activity compared to EV and the T167del mutation.
Regarding the measurements of the TNSALP activity in the co-transfections, lower activity is observed in those in which the mutations were used compared to those that used EV (Figure 4D). In heterozygosity, EV: WT presents a percentage of activity of 53.06%, which represents half of the total activity obtained in cells transfected in homozygosis with the WT plasmid, while L6S: WT and T167del: WT in heterozygosis presented an average activity of 36.33. % and 13.97% respectively. These results confirm that although both variants generate a DNE, the T167del variant exerts a greater effect than the L6S variant on the WT monomer.
The expression of TNSALP in the cell membrane was analyzed by flow cytometry in homozygosity and heterozygosity of each variant.
Figure 5A shows the gating process for the selection of those cells that expressed TNSALP on the surface of the cell membrane for the homozygous and heterozygous. Figure 5B shows cells that expressed TNSALP on the surface under homozygous conditions. The cell population with the WT variant obtained a significantly higher percentage of cells that expressed TNSALP on the surface (18.43%), followed by the cell population that expressed the L6S variant (3.7%), while the cells that expressed the T167del variant (1.16%) did not present differences in the percentage of cells that expressed TNSALP compared to EV (0.53%). Figure 5C shows the amount of TNSALP on the cell surface of the different homozygous conditions. The WT variant expressed a significantly higher amount of surface protein (0.749) than the other variants. There were no differences between the populations that expressed the T167del (0.27) and L6S (0.24) variants; however, the cells that expressed T167del showed a greater amount of TNSALP on the surface than those cells that expressed EV (0.17).
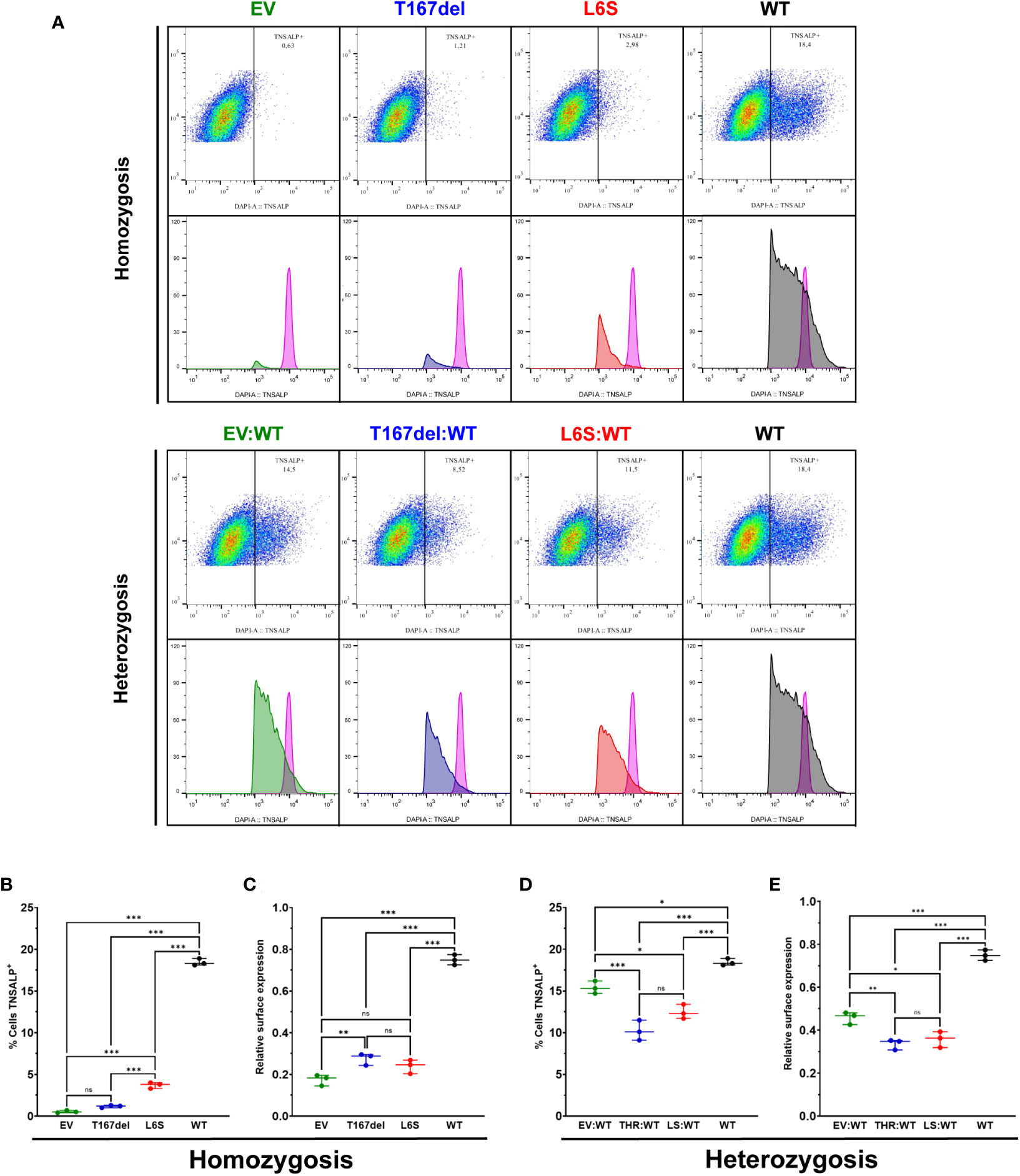
Figure 5 Determination of TNSALP localization on the cell surface of HEK293T cells. (A) shows an example of flow cytometry gating and histogram in cells homozygous and heterozygous for TNSALP variants. The internal control is represented by the histogram in pink. (B, C) show the percentage of TNSALP positive cells (B) and TNSALP relative expression on cell surface (C) in homozygosis. Results are expressed as mean with standard deviation. ANOVA was used for comparisons between groups (***p<0.001, **p<0.01, *p<0.05). (D, E) show the percentage of TNSALP positive cells (D) and TNSALP relative expression on cell surface (E) in heterozygosis. Results are expressed as mean with standard deviation. ANOVA was used for comparisons between groups (***p<0.001, **p<0.01, *p<0.05). ns: not significant.
Cells in heterozygosity had a significant increase in the percentage of cells expressing TNSALP on the cell surface. The EV: WT variant (15.4%) presented a percentage of cells that expressed TNSALP significantly higher than those cells heterozygous for the T167del: WT (10.23%) and L6S: WT (12.46%) variants; however, they presented a lower percentage of cells expressing TNSALP than cells homozygous for the WT variant (18.43%) (Figure 5D). Regarding antigenic density, the homozygous cells that expressed the WT variant (0.749) expressed a significantly higher amount of protein on the surface than those cells that expressed the variants in heterozygosis. Heterozygous cells that expressed EV: WT (0.46) showed significantly more surface protein than those cells that expressed the T167del:WT (0.336) and L6S:WT (0.358) variants (Figure 5E).
In this study, two new, previously undescribed variants in the ALPL gene have been identified in two 15-year-old male patients, leading to the genetic screening of the patients’ relatives for better understanding and personalized management for affected families.
In silico predictions concluded that T167del is probably pathogenic while L6S does not present a clear consensus. The results of the alignment analysis indicate that the Met1, Leu6 and Leu8 amino acids for L6S variant and 165TTTR168 amino acids for T167del variant are highly conserved across all species included in this analysis (Supplementary Figure S1). These results besides 3D modeling (Figures 3B, C) suggest that these residues may play a major role in the protein function. We suggest that Met1, Leu6 and Leu8 amino acids in L6S variant could be involved in the cellular localization of the protein agreeing with the results obtained by Silvent et al. (35). For T167del variant T167 could play an important role as a stabilizer of the active site, since we found that it has a structural function providing stability to E332, a direct Mg2+ ligand that is part of the catalytic center.
Other mutations have been identified in the same positions as L6S variant (c.17T>A; L6*) (36) and T167del variant (c.500C>T; T167M) (37–39). For the first one, this variant is considered pathogenic; however, the effect of the mutation is not comparable to our identified variant since c.17T>A gives rise to a nonsense mutation resulting in a 6 amino acid peptide (36). For the second one, it has been associated with a severe pathogenic phenotype of HPP being described in patients with severe childhood HPP (37), patients with non-lethal perinatal HPP (38) and adult HPP (39). Based on our results and previous scientific literature, T167del variant has been classified as pathogenic.
The results of the in vitro TNSALP activity are consistent with the results observed at the clinical level. P1 presented significantly higher serum ALP activity (73 and 42 IU/L) than P2 (45 and 38 IU/L) in concordance with the in vitro results where a substantial decrease in TNSALP activity was observed in T167del compared to L6S (Figure 4C). The suspicion that both variants might be pathogenic is reinforced by the determination of their negative dominance over the WT monomer (Figure 4D). In both cases, TNSALP activity was lower than that obtained in the control group. The fact that the L6S allele presents a moderate dominance over the WT allele is consistent with the serum ALP activity of P1 being close to the reference range. Furthermore, it has been described that mutations with higher DNE are usually found in the crown domain, homodimeric interphase, or the area of the active center (26). This agrees with the results obtained for T167del (located close to the active center of the protein) which showed more severe DNE.
Our results showed retention in the efflux of protein to the membrane both in homozygosity and in heterozygosity due to the drastic reduction both in the amount of protein in the membrane and in the number of cells expressing TNSALP. In this context, the use of flow cytometry is a very useful tool to obtain relevant explanations about TNSALP cellular localization. In the case of the L6S variant, the same mutation has been described in gap junction protein beta 1, producing its accumulation at the intracellular level (40). We have observed a decrease not only in the percentage of cells that express TNSALP but also in the amount of protein in the membrane. Thus, the amino acid L6 could play an important role in the process of exporting TNSALP to the cell membrane which could explain the low antigenic density of TNSALP although the percentage of cells expressing this protein is higher. In the case of the T167del variant, the underlying mechanisms in the alteration of the cellular localization of the protein are unknown to date. However, due to the pathogenicity of the mutation, we suggest that the mutated protein could be taken to the proteasome for its complete degradation, which would explain the low activity and antigenic density of both homozygous and heterozygous.
Regarding clinical manifestations, in Family 1 there is a clear vitamin D deficiency in patients affected by L6S variant. Vitamin D deficiency is highly prevalent in HPP patients and should be treated with vitamin D supplements to avoid secondary hyperparathyroidism (41) as observed in II-2a. I-2a had tooth loss at an early age and a series of symptoms different from the pathognomonic symptoms of HPP such as digestive abnormalities (chronic diarrhea, esophagitis and diverticulitis) or inflammatory diseases (arteritis and polymyalgia rheumatica). II-2a presented chronic gastritis as a digestive symptom and giant cell tumor and hyperparathyroidism while II-3a suffered from neurological disorders such as fibromyalgia, immune disorders such as spondyloarthritis with dyscarthrosis, psoriatic arthritis and neutrophilia; skeletal disorders such as pelvic dysmetria and HBP (Table 3). P1 inherited some of these manifestations such as digestive disorders, vitamin D deficiency and HBP at an early age (Table 1). Regarding Family 2, both carriers of new T167del variant from Family 2 showed vitamin D deficiency. II-1b presented early tooth loss consistent with a phenotype of odontohypophosphatasia (Table 3) while P2 suffered from inverted psoriasis and Crohn’s disease inherited from II-2b (Table 1). The in vitro results of T167del variant besides to the immune disturbances, suggest that this HPP-related genotype could contribute to a worse prognosis of the comorbidities. In this context, some symptoms such as fibromyalgia (21, 39, 42, 43), digestive affections (44) or HBP (43) have been slightly linked to HPP; however, these complications as a symptom related to HPP have not been explored in depth to date. Some studies suggest that digestive alterations may be associated with disturbances in the immune system (45, 46). This hypothesis is strengthened by the clinical manifestations of P2 who has been diagnosed with Crohn’s disease and inverse psoriasis. Overall, 50% of the patients studied had some type of autoimmune/inflammatory disease in two unrelated families. This finding is reinforced by a recent study published by our research group, in which a prevalence of immune diseases in HPP patients of 37.5% was observed. This prevalence represents an incidence of autoimmune diseases 4-10 times higher than that described for the general population (3-8%). Autoimmune diseases found in the patients participants in this study mainly affect bones, skin and circulatory system (6).
Our hypothesis is that some of the patients´ symptoms in the context of inflammatory process, could be related to the role of TNSALP beyond bone metabolism. In this context, increased TNSALP activity during episodes of late-onset sepsis suggests its immunomodulatory role (47). In this line, TNSALP has ectonucleotidase capacity, hydrolyzing extracellular ATP or LPS that act on Toll-like receptor 4 which is responsible for the activation of the innate immune system by macrophages releasing proinflammatories cytokines as interleukins 6 and 8 (48). These molecules are also degraded by intestinal alkaline phosphatase (IAP), which has been described as a protein involved in the development of inflammatory bowel disease. In fact, both isoenzymes, seem to have some cross-talk, since mutations in gene encoding IAP (called ALPI) lead to upregulation of TNSALP expression in intestinal tissue (49). This suggests that there could be some type of compensation of alkaline phosphatase activity when one of these enzymes presents loss of function. Moreover, TNSALP also regulates purinergic signaling, whereby extracellular ATP and ADP trigger inflammation through nucleotide receptors. TNSALP breaks down ATP and ADP into AMP and adenine, halting inflammation and promoting anti-inflammatory responses through adenine receptors. Thus, TNSALP has been associated with protecting against inflammation in diseases and favoring intestinal microbial populations by its role in ATP/ADP hydrolysis (13). On the other hand, TNSALP has been reported to be expressed in phagocytes (50), neutrophils (51) and T lymphocytes. A study in mice showed that TNSALP is required for the complete stimulation of T lymphocytes and T-cell-dependent colitis (10). Given the known role of TNSALP in modulating inflammation and the immune response, a deficiency in TNSALP activity may have some involvement in dysregulation of the immune system and for hence, could contribute to the worsening of inflammatory diseases found in many cases, in HPP patients. In this line some clinical trials where TNSALP is being used as a treatment for acute kidney injury associated with sepsis have been tested (52).
The computational 3D modeling and alignment linked to clinical results and functional analyses, suggest that L6S variant could be classified as likely pathogenic associated with a mild HPP phenotype. Regarding T167del variant, all the data collected suggest the classification of this variant as likely pathogenic with a moderate phenotype.
These results highlight the importance of establishing HPP as a systemic pathology, not only related to bone mineralization disturbances. Currently, digestive and autoimmune disorders are considered as independent processes in HPP patients; however, our findings reveal that these processes could be related to ALP deficiency. However, the etiology of these comorbidities remains elusive. Additional investigations are necessary to comprehend the pathogenic mechanisms associated with HPP-related complications fully. In this context, it is essential to increase the cohort of patients to corroborate and reinforce the potential correlation between TNSALP levels and the worsening of digestive, autoinflammatory, and/or autoimmune manifestations. Another limitation of the study lies in the utilization of a predictive 3D model obtained through alpha fold. A recent publication has presented a crystallized structure of TNSALP (53), offering valuable insights for forthcoming research endeavors. On the other hand, although it is difficult to establish a gene-phenotypic relationship of each variant described in HPP due to the participation of several external factors that enhance phenotypic variability, it is important to identify and characterize new variants that serve as a starting point for future research and patient management.
In conclusion, we have identified two new previously undescribed variants that produce clinical manifestations of HPP more related to systemic diseases than to bone disorders. Considering the results shown in this study linked to previous scientific evidence, we suggest low ALP activity could be related to worsening of inflammatory/autoimmune disorders in HPP patients. In this study we show that both new identified mutations could be classified as likely pathogenic and have a DNE that affects both enzymatic activity and cell location through overexpression in HEK293T cells.
Leiden Open Variation Database (LOVD), https://databases.lovd.nl/shared/genes/ALPL (May 12, 2022); The Genome Aggregation Database (gnomAD), https://gnomad.broadinstitute.org/gene/ENSG00000162551?dataset=gnomad_r3 (May 12, 2022); ALPL mutation Database https://alplmutationdatabase.jku.at/ (Nov 16, 2023); Kyoto Encyclopedia of Genes and Genomes (KEGG), https://www.genome.jp/entry/3.1.3.1 (June 24, 2022); ClinVar, https://www.ncbi.nlm.nih.gov/clinvar/ (May 12, 2022); PROVEAN, https://www.jcvi.org/research/provean (May 27, 2022); MutPred, http://mutpred.mutdb.org/(May 27, 2022); http://mutpred2.mutdb.org/mutpredindel/ (May 27, 2022); Mutation Taster https://www.mutationtaster.org/ (May 28,2022); Combined annotation-dependent depletion (CADD), https://cadd.gs.washington.edu/score (June 15, 2022); mutation significance cut-off (MSC), http://pec630.rockefeller.edu:8080/MSC/ (June 15, 2022); UniProt, https://www.uniprot.org/ (June 23, 2022); AlphaFold2_advanced, https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb (June 23, 2022); VarSome, https://varsome.com/ (June 29, 2023); Human Protein Atlas, https://www.proteinatlas.org/ENSG00000162551-ALPL/tissue#expression_cluster (November 01, 2023).
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by Research Ethics Committee of Granada Center (Project ID: 0777-M1-20 approved on 8 May 2019). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
LM-H: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – original draft, Writing – review & editing. MM-T: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing, Resources. RS-D: Formal Analysis, Investigation, Methodology, Writing – review & editing. ÁJ-O: Formal Analysis, Investigation, Methodology, Software, Writing – review & editing. FA-V: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – review & editing. TG-C: Data curation, Investigation, Validation, Writing – review & editing. VC-B: Data curation, Investigation, Methodology, Validation, Writing – review & editing. SG-S: Investigation, Methodology, Validation, Writing – review & editing. JG-V: Conceptualization, Data curation, Validation, Writing – review & editing. CG-F: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing. BG-F: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Instituto de Salud Carlos III grant PI21/01069 co-funded by the European Regional Development Fund (FEDER), by Junta de Andalucı́a grant PI-0268-2019 and by CIBER of Frailty and Healthy Aging (CIBERFES;CB16/10/00475). In addition, SG-S, CG-F, TG-C and BG-F are funded by grants from the Instituto de Salud Carlos III (FI19/00118, CD20/00022, CM21/00221 and CP22/00022). VC-B is funded by Junta de Andalucı́a grant (RH-0141-2020).
This article is part of the doctoral thesis entitled “Autoimmune and gastrointestinal alterations associated with Hypophosphatasia: Role of the intestinal microbiome and miRNome” which is framed within the Biomedicine doctoral program of the University of Granada (UGR). Figure 1 was drawn by using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1320516/full#supplementary-material
1. Villa-Suárez JM, García-Fontana C, Andújar-Vera F, González-Salvatierra S, de Haro-Muñoz T, Contreras-Bolívar V, et al. Hypophosphatasia: A unique disorder of bone mineralization. Int J Mol Sci (2021) 22:4303. doi: 10.3390/ijms22094303
2. Whyte MP. Hypophosphatasia - aetiology, nosology, pathogenesis, diagnosis and treatment. Nat Rev Endocrinol (2016) 12:233–46. doi: 10.1038/nrendo.2016.14
3. Mornet E, Yvard A, Taillandier A, Fauvert D, Simon-Bouy B. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet (2011) 75:439–45. doi: 10.1111/j.1469-1809.2011.00642.x
4. García-Fontana C, Villa-Suárez JM, Andújar-Vera F, González-Salvatierra S, Martínez-Navajas G, Real PJ, et al. Epidemiological, clinical and genetic study of hypophosphatasia in A spanish population: identification of two novel mutations in the alpl gene. Sci Rep (2019) 9:9569. doi: 10.1038/s41598-019-46004-2
5. Mornet E, Taillandier A, Domingues C, Dufour A, Benaloun E, Lavaud N, et al. Hypophosphatasia: a genetic-based nosology and new insights in genotype-phenotype correlation. Eur J Hum Genet (2021) 29:289–99. doi: 10.1038/s41431-020-00732-6
6. González-Cejudo T, Villa-Suárez JM, Ferrer-Millán M, Andújar-Vera F, Contreras-Bolívar V, Andreo-López MC, et al. Mild hypophosphatasia may be twice as prevalent as previously estimated: an effective clinical algorithm to detect undiagnosed cases. Clin Chem Lab Med CCLM (2023) 62(1):128–37. doi: 10.1515/cclm-2023-0427
7. Fenn JS, Lorde N, Ward JM, Borovickova I. Hypophosphatasia. J Clin Pathol (2021) 74:635–40. doi: 10.1136/jclinpath-2021-207426
8. Le Du M-H, Millan JL. Structural evidence of functional divergence in human alkaline phosphatases. J Biol Chem (2002) 277:49808–14. doi: 10.1074/jbc.M207394200
9. Millán JL, Whyte MP. Alkaline phosphatase and hypophosphatasia. Calcif Tissue Int (2016) 98:398–416. doi: 10.1007/s00223-015-0079-1
10. Hernández-Chirlaque C, Gámez-Belmonte R, Ocón B, Martínez-Moya P, Wirtz S, Sánchez de Medina F, et al. Tissue non-specific alkaline phosphatase expression is needed for the full stimulation of T cells and T cell-dependent colitis. J Crohns Colitis (2017) 11:857–70. doi: 10.1093/ecco-jcc/jjw222
11. Cruz T, Gleizes M, Balayssac S, Mornet E, Marsal G, Millán JL, et al. Identification of altered brain metabolites associated with TNAP activity in a mouse model of hypophosphatasia using untargeted NMR-based metabolomics analysis. J Neurochem (2017) 140:919–40. doi: 10.1111/jnc.13950
12. Narisawa S, Yadav MC, Millán JL. In vivo overexpression of tissue-nonspecific alkaline phosphatase increases skeletal mineralization and affects the phosphorylation status of osteopontin. J Bone Miner Res Off J Am Soc Bone Miner Res (2013) 28:1587–98. doi: 10.1002/jbmr.1901
13. Rader BA. Alkaline phosphatase, an unconventional immune protein. Front Immunol (2017) 8:897. doi: 10.3389/fimmu.2017.00897
14. Goettsch C, Strzelecka-Kiliszek A, Bessueille L, Quillard T, Mechtouff L, Pikula S, et al. TNAP as a therapeutic target for cardiovascular calcification: a discussion of its pleiotropic functions in the body. Cardiovasc Res (2022) 118:84–96. doi: 10.1093/cvr/cvaa299
15. Beck-Nielsen SS, Brusgaard K, Rasmussen LM, Brixen K, Brock-Jacobsen B, Poulsen MR, et al. Phenotype presentation of hypophosphatemic rickets in adults. Calcif Tissue Int (2010) 87:108–19. doi: 10.1007/s00223-010-9373-0
16. Rathbun JC. Hypophosphatasia; a new developmental anomaly. Am J Dis Child 1911 (1948) 75:822–31. doi: 10.1001/archpedi.1948.02030020840003
17. Uday S, Matsumura T, Saraff V, Saito S, Orimo H, Högler W. Tissue non-specific alkaline phosphatase activity and mineralization capacity of bi-allelic mutations from severe perinatal and asymptomatic hypophosphatasia phenotypes: Results from an in vitro mutagenesis model. Bone (2019) 127:9–16. doi: 10.1016/j.bone.2019.05.031
18. Barvencik F, Beil FT, Gebauer M, Busse B, Koehne T, Seitz S, et al. Skeletal mineralization defects in adult hypophosphatasia–a clinical and histological analysis. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA (2011) 22:2667–75. doi: 10.1007/s00198-011-1528-y
19. Dahir KM, Seefried L, Kishnani PS, Petryk A, Högler W, Linglart A, et al. Clinical profiles of treated and untreated adults with hypophosphatasia in the Global HPP Registry. Orphanet J Rare Dis (2022) 17:277. doi: 10.1186/s13023-022-02393-8
20. Martins L, Rodrigues TL, Ribeiro MM, Saito MT, Giorgetti APO, Casati MZ, et al. Novel ALPL genetic alteration associated with an odontohypophosphatasia phenotype. Bone (2013) 56:390–7. doi: 10.1016/j.bone.2013.06.010
21. Colazo JM, Hu JR, Dahir KM, Simmons JH. Neurological symptoms in hypophosphatasia. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA (2019) 30:469–80. doi: 10.1007/s00198-018-4691-6
22. Cundy T, Michigami T, Tachikawa K, Dray M, Collins JF, Paschalis EP, et al. Reversible deterioration in hypophosphatasia caused by renal failure with bisphosphonate treatment. J Bone Miner Res Off J Am Soc Bone Miner Res (2015) 30:1726–37. doi: 10.1002/jbmr.2495
23. Koga M, Kinoshita Y, Kato H, Kobayashi H, Shinoda Y, Nangaku M, et al. Massive calcification around large joints in a patient subsequently diagnosed with adult-onset hypophosphatasia. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA (2022) 33:505–9. doi: 10.1007/s00198-021-06145-5
24. Lia-Baldini AS, Muller F, Taillandier A, Gibrat JF, Mouchard M, Robin B, et al. A molecular approach to dominance in hypophosphatasia. Hum Genet (2001) 109:99–108. doi: 10.1007/s004390100546
25. Fauvert D, Brun-Heath I, Lia-Baldini A-S, Bellazi L, Taillandier A, Serre J-L, et al. Mild forms of hypophosphatasia mostly result from dominant negative effect of severe alleles or from compound heterozygosity for severe and moderate alleles. BMC Med Genet (2009) 10:51. doi: 10.1186/1471-2350-10-51
26. Del Angel G, Reynders J, Negron C, Steinbrecher T, Mornet E. Large-scale in vitro functional testing and novel variant scoring via protein modeling provide insights into alkaline phosphatase activity in hypophosphatasia. Hum Mutat (2020) 41:1250–62. doi: 10.1002/humu.24010
27. McKiernan FE, Shrestha LK, Berg RL, Fuehrer J. Acute hypophosphatasemia. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA (2014) 25:519–23. doi: 10.1007/s00198-013-2447-x
28. Colantonio DA, Kyriakopoulou L, Chan MK, Daly CH, Brinc D, Venner AA, et al. Closing the gaps in pediatric laboratory reference intervals: a CALIPER database of 40 biochemical markers in a healthy and multiethnic population of children. Clin Chem (2012) 58:854–68. doi: 10.1373/clinchem.2011.177741
29. Riancho-Zarrabeitia L, García-Unzueta M, Tenorio JA, Gómez-Gerique JA, Ruiz Pérez VL, Heath KE, et al. Clinical, biochemical and genetic spectrum of low alkaline phosphatase levels in adults. Eur J Intern Med (2016) 29:40–5. doi: 10.1016/j.ejim.2015.12.019
30. Itan Y, Shang L, Boisson B, Ciancanelli MJ, Markle JG, Martinez-Barricarte R, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods (2016) 13:109–10. doi: 10.1038/nmeth.3739
31. Okonechnikov K, Golosova O, Fursov M, UGENE team. Unipro UGENE: a unified bioinformatics toolkit. Bioinforma Oxf Engl (2012) 28:1166–7. doi: 10.1093/bioinformatics/bts091
32. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. ColabFold: making protein folding accessible to all. Nat Methods (2022) 19:679–82. doi: 10.1038/s41592-022-01488-1
33. Pettersen EF, Goddard TD, Huang CC, Meng EC, Couch GS, Croll TI, et al. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci Publ Protein Soc (2021) 30:70–82. doi: 10.1002/pro.3943
34. Lopez-Perez D, Redruello-Romero A, Garcia-Rubio J, Arana C, Garcia-Escudero LA, Tamayo F, et al. In obese patients with type 2 diabetes, mast cells in omental adipose tissue decrease the surface expression of CD45, CD117, CD203c, and fcϵRI. Front Endocrinol (2022) 13:818388. doi: 10.3389/fendo.2022.818388
35. Silvent J, Gasse B, Mornet E, Sire J-Y. Molecular evolution of the tissue-nonspecific alkaline phosphatase allows prediction and validation of missense mutations responsible for hypophosphatasia. J Biol Chem (2014) 289:24168–79. doi: 10.1074/jbc.M114.576843
36. Taillandier A, Cozien E, Muller F, Merrien Y, Bonnin E, Fribourg C, et al. Fifteen new mutations (-195C>T, L-12X, 298-2A>G, T117N, A159T, R229S, 997 + 2T>A, E274X, A331T, H364R, D389G, 1256delC, R433H, N461I, C472S) in the tissue-nonspecific alkaline phosphatase (TNSALP) gene in patients with hypophosphatasia. Hum Mutat (2000) 15:293. doi: 10.1002/(SICI)1098-1004(200003)15:3<293::AID-HUMU11>3.0.CO;2-Q
37. Whyte MP, Zhang F, Wenkert D, McAlister WH, Mack KE, Benigno MC, et al. Hypophosphatasia: validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone (2015) 75:229–39. doi: 10.1016/j.bone.2015.02.022
38. Ligutić I, Barisić I, Anticević D, Vrdoljak J. Hypophosphatasia: report of two affected girls with spontaneous improvement of skeletal defects. Lijec Vjesn (2005) 127:288–92.
39. Braunstein NA. Multiple fractures, pain, and severe disability in a patient with adult-onset hypophosphatasia. Bone Rep (2016) 4:1–4. doi: 10.1016/j.bonr.2015.10.005
40. Tsai P-C, Yang D-M, Liao Y-C, Chiu T-Y, Kuo H-C, Su Y-P, et al. Clinical and biophysical characterization of 19 GJB1 mutations. Ann Clin Transl Neurol (2016) 3:854–65. doi: 10.1002/acn3.347
41. Bianchi ML. Hypophosphatasia: an overview of the disease and its treatment. Osteoporos Int J Establ Result Coop Eur Found Osteoporos Natl Osteoporos Found USA (2015) 26:2743–57. doi: 10.1007/s00198-015-3272-1
42. Collmann H, Mornet E, Gattenlöhner S, Beck C, Girschick H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv Syst ChNS Off J Int Soc Pediatr Neurosurg (2009) 25:217–23. doi: 10.1007/s00381-008-0708-3
43. Pierpont EI, Simmons JH, Spurlock KJ, Shanley R, Sarafoglou KM. Impact of pediatric hypophosphatasia on behavioral health and quality of life. Orphanet J Rare Dis (2021) 16:80. doi: 10.1186/s13023-021-01722-7
44. Martins L, de Almeida AB, Dos Santos EJL, Foster BL, MaChado RA, Kantovitz KR, et al. A novel combination of biallelic ALPL mutations associated with adult hypophosphatasia: A phenotype-genotype association and computational analysis study. Bone (2019) 125:128–39. doi: 10.1016/j.bone.2019.05.005
45. Kandulski A, Malfertheiner P. Gastroesophageal reflux disease—from reflux episodes to mucosal inflammation. Nat Rev Gastroenterol Hepatol (2012) 9:15–22. doi: 10.1038/nrgastro.2011.210
46. Hait EJ, McDonald DR. Impact of gastroesophageal reflux disease on mucosal immunity and atopic disorders. Clin Rev Allergy Immunol (2019) 57:213–25. doi: 10.1007/s12016-018-8701-4
47. Pettengill M, Matute JD, Tresenriter M, Hibbert J, Burgner D, Richmond P, et al. Human alkaline phosphatase dephosphorylates microbial products and is elevated in preterm neonates with a history of late-onset sepsis. PloS One (2017) 12:e0175936. doi: 10.1371/journal.pone.0175936
48. Chow JC, Young DW, Golenbock DT, Christ WJ, Gusovsky F. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J Biol Chem (1999) 274:10689–92. doi: 10.1074/jbc.274.16.10689
49. Parlato M, Charbit-Henrion F, Pan J, Romano C, Duclaux-Loras R, Le Du M-H, et al. Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol Med (2018) 10:e8483. doi: 10.15252/emmm.201708483
50. Shanmugham LN, Petrarca C, Castellani ML, Symeonidou I, Frydas S, Vecchiet J, et al. IL-1beta induces alkaline phosphatase in human phagocytes. Arch Med Res (2007) 38:39–44. doi: 10.1016/j.arcmed.2006.05.016
51. Li H, Zhao Y, Li W, Yang J, Wu H. Critical role of neutrophil alkaline phosphatase in the antimicrobial function of neutrophils. Life Sci (2016) 157:152–7. doi: 10.1016/j.lfs.2016.06.005
52. Heemskerk S, Masereeuw R, Moesker O, Bouw MPWJM, van der Hoeven JG, Peters WHM, et al. Alkaline phosphatase treatment improves renal function in severe sepsis or septic shock patients. Crit Care Med (2009) 37:417–423, e1. doi: 10.1097/CCM.0b013e31819598af
Keywords: hypophosphatasia, tissue non-specific alkaline phosphatase, autoimmune diseases, gastrointestinal disorders, metabolic disease
Citation: Martínez-Heredia L, Muñoz-Torres M, Sanabria-de la Torre R, Jiménez-Ortas Á, Andújar-Vera F, González-Cejudo T, Contreras-Bolívar V, González-Salvatierra S, Gómez-Vida JM, García-Fontana C and García-Fontana B (2024) Systemic effects of hypophosphatasia characterization of two novel variants in the ALPL gene. Front. Endocrinol. 14:1320516. doi: 10.3389/fendo.2023.1320516
Received: 12 October 2023; Accepted: 04 December 2023;
Published: 03 January 2024.
Edited by:
Licy L. Yanes Cardozo, University of Mississippi Medical Center, United StatesReviewed by:
Daniel Liedtke, Julius Maximilian University of Würzburg, GermanyCopyright © 2024 Martínez-Heredia, Muñoz-Torres, Sanabria-de la Torre, Jiménez-Ortas, Andújar-Vera, González-Cejudo, Contreras-Bolívar, González-Salvatierra, Gómez-Vida, García-Fontana and García-Fontana. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manuel Muñoz-Torres, bW10QG1hbXV0by5lcw==; Cristina García-Fontana, Y2dmb250YW5hQHVnci5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.