 Xiaodong Li
Xiaodong Li Fanguang Meng
Fanguang Meng Hankang Wang
Hankang Wang Liwei Sun
Liwei Sun Shulin Chang1,2
Shulin Chang1,2 Guijie Li
Guijie Li Feng Chen
Feng Chen- 1Department of Radiology, The First Affiliated Hospital of Shandong First Medical University and Shandong Provincial Qianfoshan Hospital, Shandong Medicine and Health Key Laboratory of Abdominal Medicine Imaging, Jinan, China
- 2Graduate School, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, China
Ferroptosis is a type of controlled cell death caused by lipid peroxidation, which results in the rupture of the cell membrane. ferroptosis has been repeatedly demonstrated over the past ten years to be a significant factor in a number of diseases. The liver is a significant iron storage organ, thus ferroptosis will have great potential in the treatment of liver diseases. Ferroptosis is particularly prevalent in HCC. In the opening section of this article, we give a general summary of the pertinent molecular mechanisms, signaling pathways, and associated characteristics of ferroptosis. The primary regulating mechanisms during ferroptosis are then briefly discussed, and we conclude by summarizing the development of a number of novel therapeutic strategies used to treat HCC in recent years. Ferroptosis is a crucial strategy for the treatment of HCC and offers new perspectives on the treatment of liver cancer.
1 Introduction
Hepatocellular carcinoma (HCC) is the most frequent malignant tumor of the liver, with the highest occurrence among people with chronic liver disease (1) and a dismal 5-year survival rate. Although the number of therapeutic options for HCC has gradually increased in recent decades, including surgical resection, immunotherapy, targeted therapy, or combination therapy, overall survival and prognosis remain limited due to the lack of specific symptoms in the early stages of the disease and the loss of therapeutic opportunities (2). As a result, it is critical to investigate the pathophysiology of hepatocellular carcinoma as well as the regulation of signaling pathways in order to uncover new therapeutic targets and develop safe and effective therapeutic choices.
Ferroptosis is an iron-dependent controlled cell death caused by polyunsaturated fatty acid-containing phospholipids (PL-PUFA) in cell membranes that differentiates from other types of cell death by iron-dependent accumulation of lipid peroxides and redox imbalance (3). Numerous investigations have found that numerous diseases, including neurological diseases, autoimmune diseases, liver diseases, and malignancies, are intimately associated to ferroptosis (4). During carcinogenesis, ferroptosis prevents the formation and proliferation of HCC cells. However, HCC cells create specialized defensive mechanisms to prevent ferroptosis and hence continue tumor growth. The cell death mode process is changed by targeting ferroptosis-related regulatory components. We outline the ferroptosis-related pathways and important influencing factors in this research in order to give new therapeutic methods for patients.
2 The mechanism of ferroptosis
The rise in ROS surpasses the regulating capacity of glutathione (GSH) and GPX 4, resulting in lipid peroxidation (5). The morphological aspects of ferroptosis are characterized by changes in mitochondrial morphology and mitochondrial cristae structure, such as mitochondrial atrophy, increased membrane density, decreased mitochondrial cristae, and outer mitochondrial membrane rupture. In addition, unlike other cell deaths, ferroptosis cells do not demonstrate reduced cell size, nucleus abnormalities, chromatin aggregation, or cytoskeletal disintegration (3, 6–8). At the cellular level, ferroptosis is triggered by the inhibition of cell membrane translocation factors such as cystine/glutamate translocator (also known as the system Xc-) or the activation of transferrins, as well as the blockade of intracellular antioxidant enzymes such as glutathione peroxidase 4 (GPX4) (3, 9). In this article, a synopsis discusses the physiology of ferroptosis and summarizes the relevant targets and mechanisms of action (Figure 1).
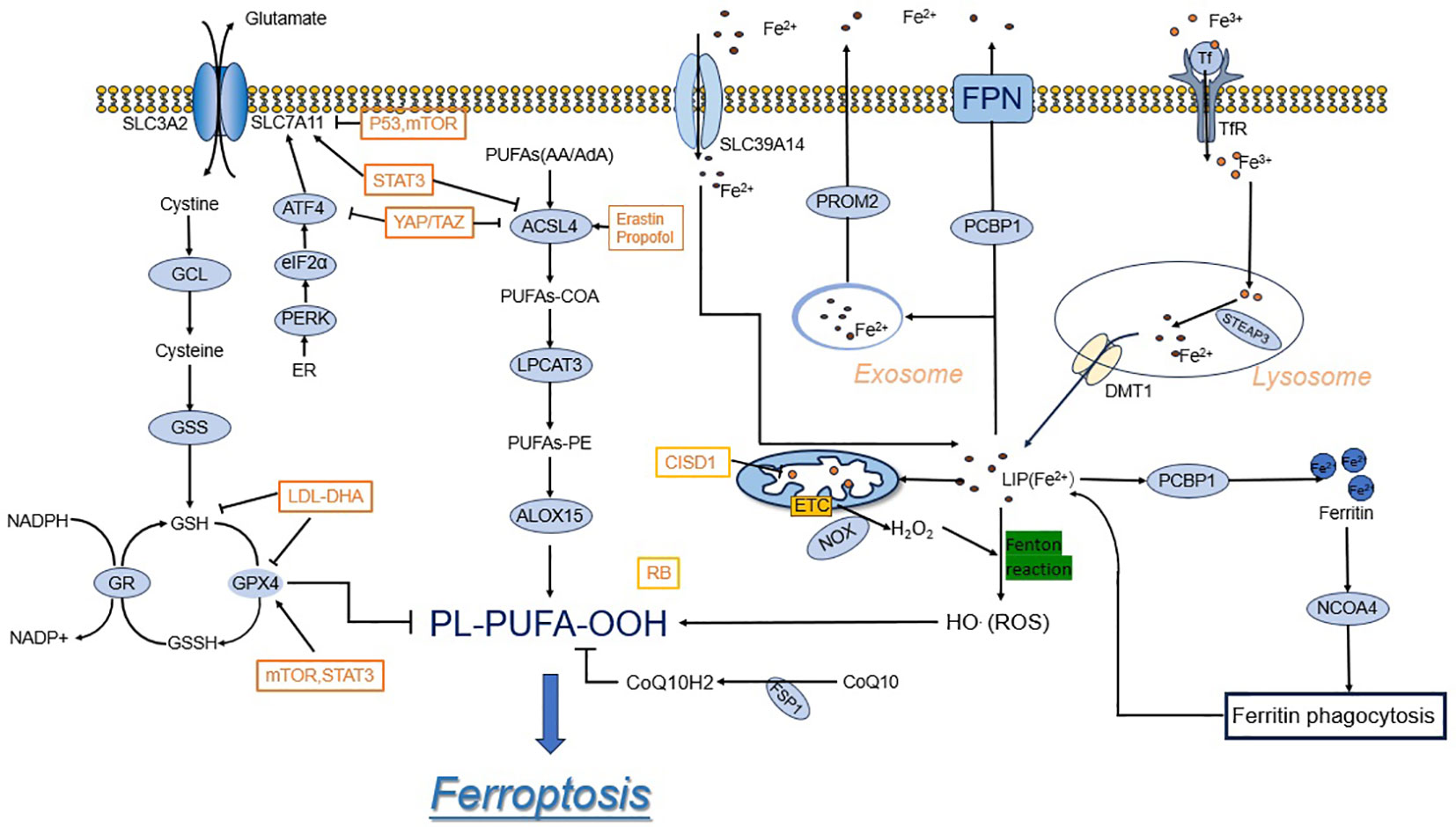
Figure 1 Ferroptosis mechanisms and regulations. System xc- imports cystine, which is reduced in the cell to the amino acid cysteine. Cysteine and glutamate are used in the biosynthesis of reduced glutathione, which is in turn used by GPX4 to reduce reactive PUFA phospholipid hydroperoxides (PUFA-PL-OOH) to non-reactive and non-lethal PUFA phospholipid alcohols (PUFA-PL-OH). Iron is present in LIP in the form of Fe2+, which promotes Ferroptosis. In the top panel, acetyl-CoA is used to make free PUFAs, which are activated by ACSL4, LPCAT3, and ACSL1 to generate PUFA-PLs.
2.1 Reactive oxygen species metabolism
The peroxidation of lipids is an advantageous feedback chain reaction initiated by free radicals; these ROS, including superoxide (O2), peroxide (H2O2 and ROOH), and free radicals (HO and RO), catalyze polyunsaturated fatty acid oxidation (10). Mitochondria are the main producer of intracellular ROS, i.e., partial reduction inside the respiratory chain of the mitochondria (11, 12). Additionally, ROS are produced by enzymes in the endoplasmatic reticulum, peroxisomes, and the plasma membrane (such as NOX and NADPH oxidases), and additionally non-enzymatically via the Fenton reaction (13, 14). Under the catalyst effect of Fe2+, hydrogen peroxide creates hydroxyl radicals (HO), which is the foundation of free radical lipid peroxidation, known as the Fenton reaction. The two most common ROS that impact lipids are hydroxyl radicals (HO) and hydroperoxides (ROO) (15).
The GSH/GPX 4 axis prevents iron-catalyzed apoptosis, whereas cysteine (cys), an inhibitory amino acid in GSH synthesis that blocks cys input via SLC7A11, causes ferroptosis by lowering GSH levels.
2.1.1 System Xc-
Solute carrier family 7 member 11 (SLC7A11, catalytic subunit) and solute carrier family 3 member 2 (SLC3A2, regulatory subunit) make up System Xc- (3). The cell membrane’s System Xc- maintains redox equilibrium by absorbing external cys and excreting it in a proportion equal to one to intracellular glu (16). Cysteine is readily transformed to cysteine after entering the cell via System Xc-. Cysteine is an inhibitory amino acid that inhibits GSH production. GSH is a widespread endogenous antioxidant that exists in both reduced (GSH) and oxidized (GSSG) forms, and the GSH/GSSG ratio shows the level of cellular oxidative stress. Ferroptosis was accelerated by lowering intracellular cysteine levels and hence GSH concentration, which increased lipid peroxidation. Furthermore, SLC7A11 enhances cystine absorption and glutathione production, which protects cells from oxidative stress and ferroptosis. SLC7A11 is regulated by two transcription factors, nuclear factor erythroid 2-related factor 2 (NRF2) and activator of transcription 4 (ATF4), according to extensive research.
The antioxidant response is mediated by NRF 2, a master transcription factor. NRF 2 interacts with kelch-like ECH-associated protein-1 (KEAP1) and targets KEAP1-Cullin3-mediated polyubiquitination and proteasomal degradation under non-stress settings (17). Oxidative stress inducers, such as oxidizing agents and electrophilic chemicals, cause KEAP1 oxidation, which inhibits NRF2 degradation by the KEAP1-Cullin3 ubiquitin ligase complex. The stabilized NRF2 then translocates into the nucleus, attaches to antioxidant response elements (AREs) in gene promoters, and regulates the transcription of target genes involved in antioxidant responses and cellular redox maintenance. Several antioxidant response elements (AREs) were discovered after analyzing the SLC7A11 promoter. Further investigation revealed that pro-electronic reagents and other cellular stressors stimulate SLC 7A 11 expression by tethering NRF 2 to antioxidant response elements (AREs) in the SLC 7A 11 promoter (18). As consequence, NRF2 overexpression increases the expression of SLC7A11 as well as other antioxidant target genes, resulting in enhanced GSH production (19). SLC7A11 is thus one of the NRF 2 transcriptional targets mediating antioxidant responses. ATF 4 is a transcription factor that controls redox homeostasis, amino acid metabolism, and ER stress (20). Unlike NRF 2, which is stabilized in response to stress, ATF 4 mRNA translation is accelerated under varied stress circumstances. Eukaryotic initiation factor 2α (eIF2α) is a regulator that comes before ATF4. eIF2α phosphorylation stimulates ATF4 mRNA translation and increases ATF4 protein levels (20). General control non-derepressible-2 (GCN2) is an upstream kinase of eIF2α that is triggered by amino acid deprivation. As a result, amino acid deficiency stimulates GCN 2, which phosphorylates and inactivates eIF2α, resulting in enhanced ATF4 protein production. ATF4 then attaches to amino acid response elements (AARE) in gene promoters, regulating transcription of genes involved in amino acid metabolism and stress response (21). Specifically, ATF4 binds to the AARE in the SLC7A11 promoter, causing SLC7A11 expression (22).
Normally, ATF4 and NRF2 work together to control gene expression. For instance, under stressful circumstances, ATF4 and NRF2 interact at the SLC7A11 promoter and work together to synergistically control SLC7A11 transcription (23). Additionally, under certain situations that cause iron-catalyzed apoptosis, p53 can accelerate iron apoptosis by suppressing SLC7A11 expression, whereas p53 deficiency-induced increases in SLC7A11 decrease ferroptosis (24). ATF3, another transcription factor from the ATF/CREB family, has been found to bind to the SLC7A11 promoter and restrict its expression independently of p53, and ATF 3 accelerates erastin-induced ferroptosis by down-regulating SLC7A11 levels (25). ATF3 primarily regulates SLC7A11 transcription under basal conditions and has no effect on stress-induced SLC7A11 expression. In summary, various stress circumstances enhance SLC7A11 transcription in part via ATF4 and/or NRF2, whereas p53 and ATF3 mostly suppress SLC7A11 expression under normal conditions. Thus, these transcription factors change cellular vulnerability to ferroptosis by regulating SLC7A11 expression.
2.1.2 GPX4/GSH
Ursini et al. discovered GPX4 as a selenoenzyme in 1985, making it the sole member of the PL-OOH scavenger family capable of turning harmful lipid hydroperoxides into harmless phosphatidyl alcohols (26). GPX4’s primary role as a major antioxidant peroxidase is to use GSH as a cofactor to resist lipid peroxidation and hence safeguard membrane integrity. GPX4 interacts with the polar head of phospholipids, allowing it to bind to bilayer membranes, according to structural and biochemical investigations (27). GSH is utilized as a cofactor by GPX4 to convert membrane lipid peroxides into harmless lipid alcohols (28). GPX4 uses two GSH molecules as donors to reduce a phospholipid hydroperoxide (PL-OOH) molecule to an alcohol molecule, producing GSSG, which can be converted to GSH utilizing NADPH/H+ and glutathione reductase (29). Overall, the resistance to ferroptosis of GPX4 requires catalytic selenocysteine residues and depletion of GSH, and inhibition of peroxide accumulation is achieved by GSR/GSH (glutathione disulfide reductase) and NADPH/H+ recirculation. GPX4 protects cells from oxidative damage by lowering thymine hydroperoxides, cholesterol hydroperoxides, and fatty acid hydroperoxides in addition to utilizing GSH to remove harmful PL-OOH (30, 31). Reactive oxygen species (ROS) build up in cells and trigger ferroptosis when GPX4 is suppressed (32). GPX4 might perform a special protective role in growth and development. In a mouse model, Yant et al. showed that gpx 4-/- embryos died in utero at mid-gestation (33, 34). The antioxidant function of GSH, which is mostly made up of glutamate, cysteine, and glycine, is primarily found in mammalian cells. These amino acids have sulfhydryl structures that can be oxidized, which enables GSH to shield cells from oxidative stress damage. The selenoenzyme GPX4 functions as a cofactor for GSH, which causes ferroptosis by inhibiting GPX4 synthesis from scratch (35). While altering the enzymatic activity of GPX4, glutamine-cysteine synthase activity, cysteine concentration, and GSH feedback inhibition directly control GSH production. On the one hand, GPX4 is transcriptionally regulated. By boosting GSH cell levels, nuclear factor erythroid-2-related factor 2 (NRF2) enhances GPX4 activity (36). To maintain redox equilibrium and promote carcinogenesis, oncogenic KRAS stimulates the Nrf2 antioxidant system (37). Through metabolic reprogramming, NRF2 activation imparts chemoresistance in KRAS-driven cancer cells (38). NRF2 inhibits lipid peroxidation and ferroptosis by activating antioxidant-responsive element (ARE)-containing genes involved in GSH production, glutamine metabolism, and iron metabolism (36). GSL2 is a mitochondrial aminotransferase and a direct transcriptional target of P53, which binds to the corresponding element in the GLS2 gene and induces transcription of GLS2, which inhibits ferroptosis by increasing cellular antioxidant capacity by promoting the production of GSH and NADPH (39, 40), but has no direct effect on GPX4 expression through binding the GPX4 promoter (41). Meanwhile, as previously described, cystine uptake was reduced after SLC7A11 was inhibited as a direct target of P53, which in turn affected GSH biosynthesis (24).
On the other hand, different chemicals influence GPX4 synthesis at the post-transcriptional stage. In general, GPX4 homeostasis is affected by both selenocysteine biosynthesis/uptake (35, 42) and GSH synthesis (43, 44),implying that GPX4 is important in regulating ferroptosis. SLC7A11 overexpression is detected in many human malignancies, and inhibiting the SLC7A11-GSH axis has been shown to have an anticancer effect (45). As a result, the cyst(e) ine/GSH/GPX4 axis is crucial for avoiding lipid peroxidation and ferroptosis. Cancerous tumors require significant levels of cysteine and GSH to counteract the increased intracellular ROS and nutritional dependency required for the enhanced action of SLC7A11 (46). A lack of intracellular cysteine, GSH, or SLC7A11 suppression reduces GPX4 activity, which can enhance lipid peroxidation and trigger ferroptosis (47).
2.2 Lipid peroxidation
Lipid peroxidation is a biomarker of ferroptosis (48). By changing the lipid composition of cellular membranes, abnormal lipid metabolism increases lipid peroxidation and promotes ferroptosis (49). During ferroptosis, acyl-CoA synthetase long-chain family member 4 (ACSL 4), lysophosphatidylcholine acyltransferase 3 (LPCAT 3), and arachidonic acid 15-lipoxygenase (ALOX 15) are required for lipid peroxidation. Arachidonic acid (AA), epinephrine (AdA), and phosphatidylethanolamines (PE), which are necessary for ferroptosis, have been found by quantitative lipidomics (50). ACSL4 encodes for the formation of proteins that link acetyl coenzyme A to AA during lipid synthesis to generate long-chain polyunsaturated lipoyl coenzyme A (AA-CoA). In the presence of LPCAT3, AA-CoA then interacts with PE to generate PE-AA, which then enters the lipid oxidation pathway. In the presence of LOX, PE-AA produces phospholipid peroxides. The process described above happens when ACSL4 and LPCAT3 dope PUFA (for example, arachidonic acid, AA) into membrane-localized lipids, which must be present in a membrane-bound milieu for PUFA to be lethal following peroxidation (51). Phospholipid peroxide accumulation depletes intracellular COQ10 and GSH on the one hand while increasing cellular vulnerability to ferroptosis on the other. Thus, it is clear that lipid anabolism works as a precursor to and regulator of lipid oxidation metabolism (52).
2.2.1 ACSL 4/LPCAT 3
A synthetase long-chain family (ACSL) is a major enzyme involved in lipid metabolism in vivo, notably facilitating the production of fatty acids with carbon chains ranging from 12 to 20 (53). In mammalian cells, there are five isozymes (ACSL 1, ACSL 3, ACSL4, ACSL5, ACSL6), and different isozymes play different roles in different tissues. ACSL4 is the key enzyme catalyzing the activation of long-chain fatty acids in ferroptosis, and its aberrant expression is closely associated with a number of biological responses, including steroidogenesis, inflammatory responses, cell death, and immune activation responses (54). The endoplasmic reticulum (ER) and the outer mitochondrial membrane (OMM) are the primary locations of the ACSL4 protein. In particularly, ACSL4 plays a crucial role in regulating ferroptosis, and its overexpression boosts the synthesis of -pufa6 PUFA and encourages ferroptosis (51).
Meanwhile, ACSL4 is regulated by multiple factors, such as integrin α6β4-mediated inhibition of ACSL 4 expression by Src and signal transducer and activator of transcription 3 (STAT3) or activation of the androgen receptor (55). It’s noteworthy to observe that the presence of free AA can alter the levels of ACSL4 by encouraging its ubiquitination and proteasomal destruction (56). The transcription of ACSL4 is started by a cAMP-response element binding (CREB) site found in the proximal promoter region of the gene (57). By encouraging ferroptosis by releasing YAP’s activity, the ACSL4 gene can begin to transcribe (58). In HCC cells, where ACSL4 is overexpressed and is associated with lower overall survival and disease-free survival (DFS) in patients with the disease, ACSL4 also plays a significant role. Aspirin and sorafenib together, as shown by Xia et al., synergistically triggered apoptosis in HCC cells by inhibiting ACSL4 expression (59).
LPCAT3 is a member of the LPLAT family of lysophospholipid acyltransferases. Among them, LPCAT3 dopes PUFA-CoA into phospholipids primarily in the endoplasmic reticulum (ER). A haploid gene screen for LPCAT3 found it in cells exposed to non-apoptotic cell death-inducing small compounds (60). The discovery of genes involved in lipid metabolism, such as ACSL4, during this screening lends credence to the notion that PUFA metabolism is fundamental to ferroptosis. In mouse lung epithelial (MLE) cells and mouse embryonic cells, Lpcat3 knockdown results in resistance to RSL3-induced ferroptosis, which may demonstrate the crucial function of LPCAT3 in ferroptosis (50, 60).
2.2.2 ALOX
Lipid peroxidation, which is mediated by either cytochrome P450 oxidoreductase (POR) (61) or lipoxygenase (ALOX) (62, 63), is the last stage in the generation of lipid peroxides. Humans have six ALOX genes, including ALOX5, ALOX12, ALOX12B, ALOX15, ALOX15B, and ALOXE3, which influence ferroptosis in different ways depending on the situation (64). ALOX12, for example, promotes TP53-dependent iron apoptosis in specific cancer cells (61). In bronchial epithelial cells, renal epithelial cells, and neurons, ALOX15 binds to phosphatidylethanolamine-binding protein 1 (PEBP1) and triggers RSL3-induced ferroptosis (65). ALOX5 is involved in ferroptosis triggered by erastin or RSL3 (66), which can be suppressed by its binding protein microsomal glutathione S-transferase I (MGSTl) (67).
2.3 Iron metabolism
Iron is a trace element that exists in two forms in the human body: divalent iron (Fe2+, absorbable) and trivalent iron (Fe3+, non-absorbable) (68, 69). The liver is the primary organ in charge of iron metabolism, delivering and regulating the iron required for metabolism throughout the body, and there are two pathways for iron uptake: transferrin-bound iron (TBI) and non-transferrin-bound iron (NTBI). Under normal conditions, Fe2+ is largely imported by membrane iron transport protein (FPN) on the basolateral membrane of the duodenum, where it is oxidized to Fe3+ and then collected by transferrin (Tf) in plasma (70). Transferrin is a plasma glycoprotein generated by hepatocytes and transported to organs throughout the body, where it chelates circulating Fe3+ safely (70). The iron-loaded transferrin is then recognized by the cell membrane receptor TfR1 and translocated into endosomes via the receptor-mediated endocytosis pathway, where ferric ions are released from the transferrin in acidic endosomes and then reduced to Fe2+ by the enzyme prostate 3 metal reductase (STEAP 3) (71). But in conditions of iron overload, plasma transferrin’s iron-binding capability is exceeded, resulting in NTBI. Through the presence of divalent metal transporter protein 1 (DMT1, also known as soluble protein carrier [SLC11A2]) and ZRT,IRT-related protein 14 (ZIP14, also known as SLC39A14), Fe2+ can be transferred into the cytoplasm as an unstable iron pool (LIP) (72). To balance the redox state and intracellular iron homeostasis, excess intracellular Fe2+ can be translocated out of the cell via the carrier SLC40A1 (73, 74). The remainder of the iron leaves the cell via membrane iron transport protein (FPN). Ferritin is a cytosolic protein with two subunits (FTL and FTH1) that serves as the primary protein for iron storage (as Fe3+) (75). Iron metabolism can regulate ferroptosis via a variety of methods. Excess iron is retained in ferritin during iron overload, where it binds nuclear receptor coactivator 4 (NOCA4). NOCA4 attaches to ferritin and is destroyed by ferritin phagocytosis (76), eventually releasing free iron to participate in the Fenton reaction, which generates ROS and increases the accumulation of intracellular lipid peroxides (LPOs) and ferroptosis (77) (Figure 2).
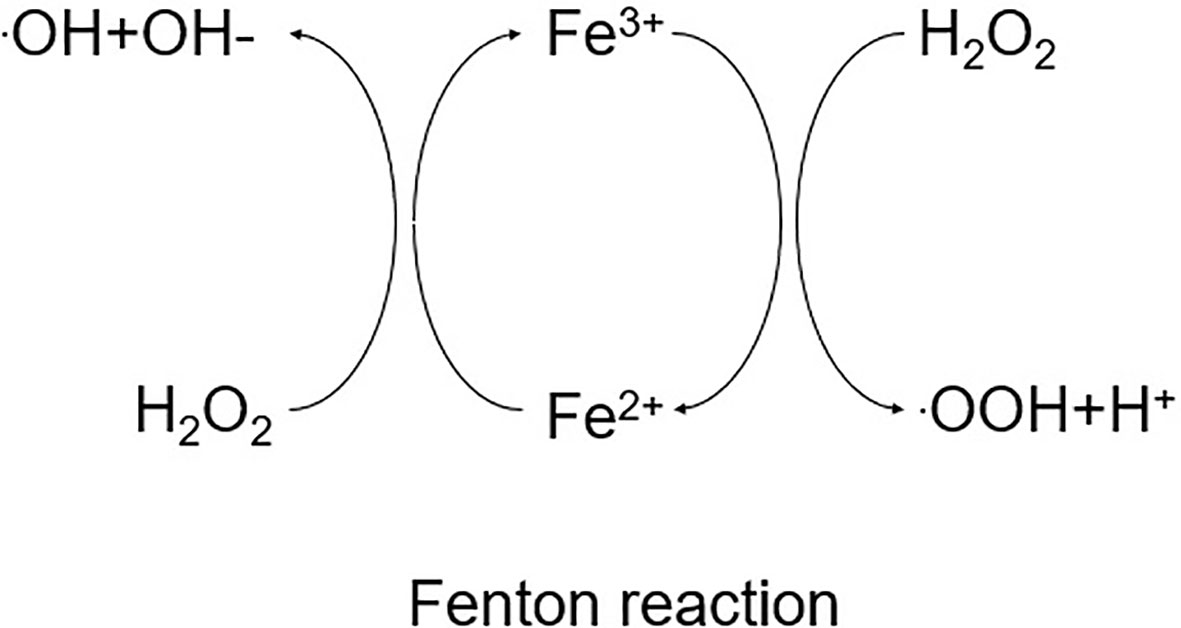
Figure 2 Schematic diagram of the Fenton reaction.
Several studies in recent years have shown that silencing TRFC (the gene encoding TFR1) inhibits erastin-induced iron apoptosis and prevents LIP accumulation, whereas the heme oxygenase HO-1 increases intracellular free iron levels and promotes heme degradation, which speeds up the ferroptosis process (78, 79). Furthermore, inhibiting iron response element binding protein 2 (IREB2) using shRNA changes the expression of various iron genes (e.g., TRFC, FTH1, and FTL), affecting iron absorption, metabolism, and storage (3). LIP is coordinated in the cytoplasm by a complex buffer of both small and large molecules, the main proteins of which are poly-r(C)-binding protein 1 (PCBP1) and PCBP2. They are required for iron chaperone action and are in charge of iron transport to ferritin. PCBP2 has also been demonstrated to preferentially interact with the iron transporter proteins divalent metallotransporter1 (DMT1) and membrane iron transporter protein to increase their iron import and export activities (80, 81). Sun et al. (78) previously established that heat shock protein beta-1 (HSPB1) is a negative regulator of ferroptosis in cancer cells. In cancer cells, erastin, a particular iron apoptosis-inducing chemical, increases heat shock factor 1 (HSF1)-dependent HSPB1 expression. HSF1 and HSPB1 knockdown promotes erastin-induced ferroptosis, but heat shock preconditioning and HSPB1 overexpression prevent erastin-induced ferroptosis. The activation of HSPB1 by protein kinase C decreased ferroptosis by lowering iron-mediated generation of lipid reactive oxygen species. Iron can also be exported by prominin-2 (prom 2)-mediated multivesicular bodies (MVB) of iron-containing proteins, and exosomes improve resistance to ferroptosis by depleting cells of iron and their potential to trigger lipid peroxidation (82). Under oxidative stress conditions, NRF2 separates from Keap1 and activates the expression of downstream target genes containing antioxidant response elements (ARE), such as iron heavy chain protein (FTH1) and iron light chain protein (FTL), promoting GSH synthesis and regenerating NADPH to exceed ROS production (36, 83). NRF2 stimulates the production of heme oxygenase-1 (HO-1) and the accumulation of Fe2+ in LIP as well as the release of iron from hemoglobin, hence inducing ferroptosis.
3 Mechanisms of ferroptosis regulation
There are at least three major regulatory mechanisms that play important roles in the ferroptosis process-the antioxidant regulator NRF2, the transsulfuration pathway, and mechanistic target of the rapamycin (mTOR) (84).
3.1 Inhibition of NRF2 triggers ferroptosis
Among the numerous mechanisms that regulate ferroptosis, NRF2 is a significant regulator of the antioxidant system, and its regulation of SLC7A11 has already been discussed. (Figure 3) Higher levels of oxidative stress are caused by excessive metabolic and proliferative loads in cancer cells (85). A transcription factor called NFE2-related factor 2 (NRF2) combines information from stressed cells and controls different transcriptional elements (86). NRF2, however, is no longer ubiquitinated and degraded in response to elevated oxidative stress or mutations in KEAP1, CUL3, or NRF2; this enables translated NRF2 to be translocated to the nucleus and stimulate transcription of genes with antioxidant response elements (AREs) (87, 88). NRF 2 transcriptionally induces the expression of several antioxidant defense proteins involved in iron and ROS metabolism, such as SOD1, GPX4, glutathione synthetase (GCL and GSS), HO-1, and FTH 1 (86–88). Under conditions of oxidative stress, NRF2 associates with Keap1 and activates the expression of downstream target genes containing AREs, such as ferric heavy chain proteins (FTH1) and ferric light chain proteins (FTL), which encourages GSH synthesis and regenerates NADPH to compete with ROS production (36, 83). A NRF2’s downstream antioxidant enzyme, NAD(P)H-quinone oxidoreductase 1 (NQO1), catalyzes the conversion of quinone to hydroquinone while lowering the formation of semi-hydroquinone and ROS. Meanwhile, NRF2 activated heme oxygenase-1 (HO-1) expression, which encouraged Fe2+ buildup in LIP and iron release from hemoglobin, as well as iron apoptosis development. A study of the drug resistance mechanism of sorafenib in hepatocellular carcinoma (HCC) cells discovered the first evidence that NFE2L2 inhibits ferroptosis (87). Sorafenib and erastin can activate the NFE2L2 pathway, resulting in the expression of antioxidant genes such as quinone oxidoreductase 1 (NQO1) and metallothionein 1G (MT1G) in HCC cells (87, 89). ABCC5, an ATP-binding cassette (ABC) protein, is a member of the ATP-dependent transporter family (90). ABCC5’s primary biological role is to expel xenobiotics and certain endogenous metabolites from cells. Sorafenib upregulates ABCC5 via the PI3K/Akt/Nrf2 pathway, which inhibits lipid peroxidation-mediated ferroptosis and promotes cancer growth, resulting in acquired sorafenib resistance in human HCC cells, according to Huang et al (91). Furthermore, ABCC5 overexpression increased SLC7A11 expression, decreased lipid peroxidation, and increased intracellular GSH levels, resulting in reduced ferroptosis (91). NRF2 activation increases the expression of the metallothionein-1 (MT1) family’s MT1G mRNA. MT1G knockdown enhances glutathione (GSH) depletion and lipid peroxidation, resulting in iron apoptosis (92). The cytosolic pro-oxidant quiescent sulfhydryl oxidase 1 (QSOX1) also enhanced ferroptosis in HCC cells by inhibiting NRF2. GSTZ1 loss in HCC can activate NRF2-associated pathways, according to recent research. GSTZ1 is thought to act as a tumor suppressor in HCC due to its reported downregulation, which results in a worse prognosis for patients and increased carcinogenesis (93, 94). Li et al. demonstrated that GSTZ1 downregulation resulted in decreased GSH and elevated ROS levels, which activated NRF2-associated pathways (94). CDGSH iron sulfur domain 2 (CISD2) is a protein that contains iron and sulfur. Sorafenib-induced ferroptosis in resistant cells was boosted by inhibiting CISD2 via ferritinophagy or the p62-Keap1-NRF2 pathway (95). Sorafenib-induced ferroptosis and cell death is aided by the downregulation of complexin II (CPLX2) and haloperidol (a sigma receptor 1 antagonist) (96, 97).
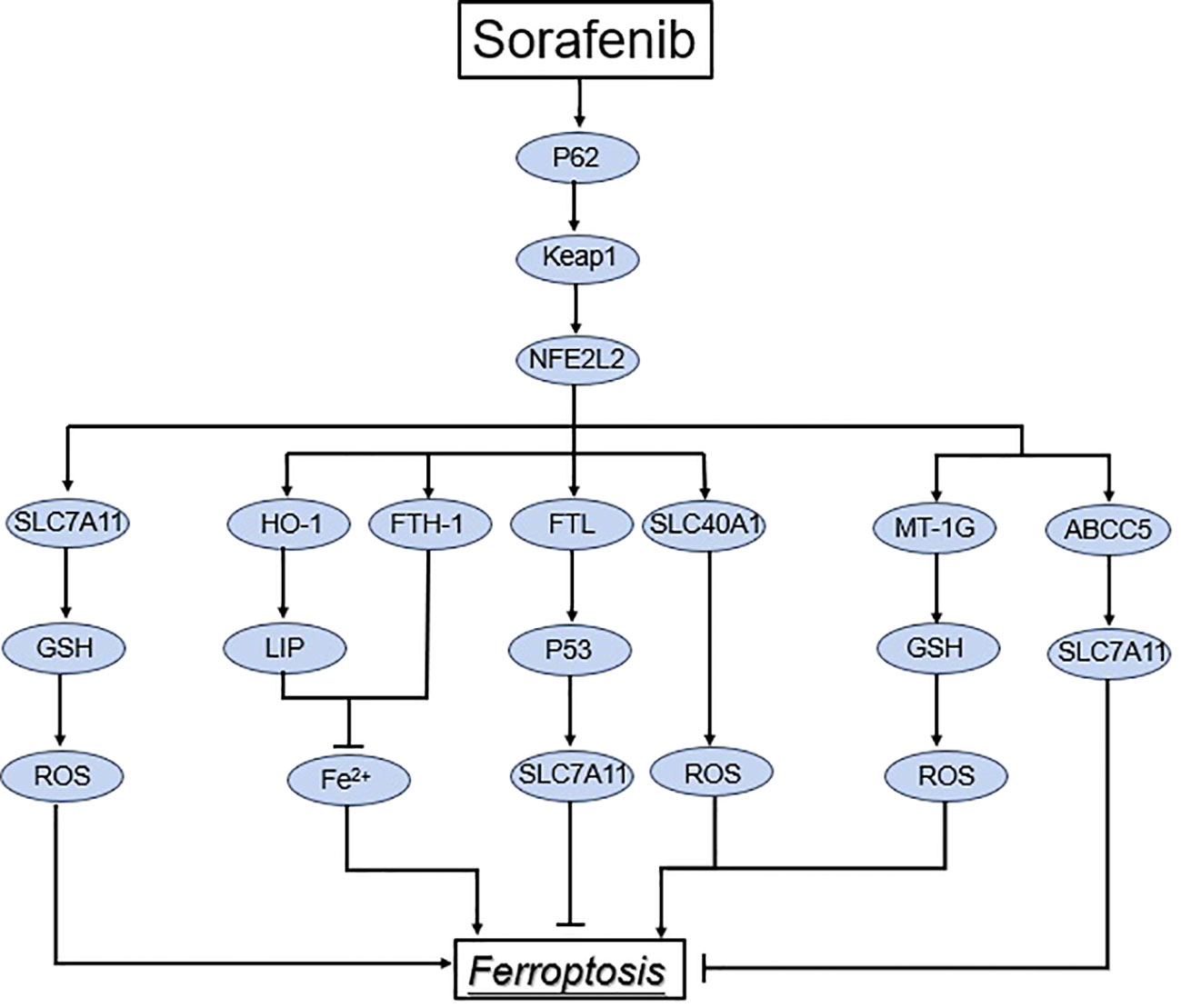
Figure 3 Regulation of the NRF2 pathway in ferroptosis. Under normal conditions, keap1 in association with CUL3 induce proteasomal degradation of NRF2. However, under stress conditions, keap1 cysteine residues are altered, leading to NRF2 dissociation from Keap1. As a result, NRF2 translocates to the nucleus and transactivates genes involved in the suppression of lipid peroxidation and ferroptosis.
3.2 The transsulfuration pathway
When the System Xc-GSH-GPX4 pathway is blocked, the transsulfuration pathway acts as an alternative antioxidant process to limit lipid peroxidation. The transsulfuration (TSS) route promotes ferroptosis resistance by generating cys from methionine, which is negatively controlled by cysteyl-tRNA synthetase 1 (CARS1) (84, 98). Methionine is converted to S-adenosylmethionine (SAM) by methionine adenosyltransferase (MAT), which then creates S-adenosylhomocysteine (SAH) via TSS.SAH is converted to homocysteine (HCY), a precursor of cysteine, by S-adenosylhomocysteine hydrolase (SAHH) (99, 100). Homocysteine, a metabolite of the transsulfur pathway, has been found to promote iron apoptosis rather than promote iron resistance (101). As opposed to serving as a source of cysteine, this shows that homocysteine can act independently (84). In hepatocellular carcinoma (HCC) cells, post-translational activation of cystathionine -synthase (CBS) under TNF-induced oxidative stress increases TSS activity through a proteolytically cleaved and highly active form of CBS, increasing cystathionine and GSH production (102), which in turn promotes tumor progression and inhibits ferroptosis. Using either CBS knockdown or the cystathionase (CTH) inhibitor propargylglycine (PAG) to block the TSS pathway significantly increases ferroptosis in tumor cells (98).
While mTORC2 primarily promotes cell proliferation and survival through the phosphorylation of AKT, many tumors need mTORC1 to maintain their ability to control protein synthesis and cell growth, as well as to coordinate nutrient and energy supply to ensure that they undergo unrestricted cell division (103). Mammalian target of rapamycin complex 2 (mTORC2; also known as mechanotarget of rapamycin complex 2) is a serine/threonine kinase made up of several protein components, including mTOR, and has the ability to phosphorylate a number of downstream targets to integrate upstream growth factor stimulation with cellular processes (such as cell survival) (104). By modifying SLC7A11, the mammalian target of rapamycin (mTOR) complex family of proteins controls iron apoptosis (46). High cell density, on the other hand, inhibits mTORC1 and encourages SLC7A11 lysosomal degradation while improving SLC7A11 protein stability by lowering lysosomal degradation (46). Additionally, via phosphorylating SLC7A11 at serine 26, mTORC2 reduces the functioning of the SLC7A11 transporter (105). According to Zhang et al. (106), the mTOR pathway increases GPX4 protein synthesis and sterol response element binding protein (SREBP)-mediated lipogenesis to encourage resistance to ferroptosis (107). Hepcidin transcription regulation is linked to the pathways that react to mitogen stimulation and nutritional status through components of Ras/RAF MAPK and mTOR signaling. Additionally, mTOR is crucial for iron metabolism. In murine primary hepatocytes, mTOR and low doses of RAF inhibitors promoted ferredoxin mRNA expression (108).
3.3 Mechanistic target of the rapamycin
Particularly, when ACSL4 was overexpressed, mTOR phosphorylation greatly increased, and when ACSL4 was downregulated, mTOR phosphorylation significantly decreased. Rapamycin therapy also preserved the function of ACSL4 overexpression in increasing cell proliferation and preventing cell death (109). Additionally, according to Chen et al. (110), curcumin promoted iron apoptosis in colorectal cancer by controlling the expression of vital iron apoptosis indicators Fer 1, SLC7A11, GSH, MDA, and ROS through the PI3K/mTOR pathway. Recent studies have shown that the mTOR-related signaling system can either promote or inhibit ferroptosis in hepatocellular carcinoma cells (111, 112). For instance, HU et al. (113) found that miR-21-5p controlled the AKT/mTOR signaling pathway through MELK in vivo, reducing iron apoptosis in hepatocellular carcinoma cells. They did this by using a subcutaneous tumorigenic model in nude mice. Additionally, by phosphorylating p62, mTORC1 facilitates the binding of p62 and Keap1, which causes Keap1 to degrade and NRF2 to accumulate (107).
4 Ferroptosis in HCC therapy
A variety of ferroptosis inducers exist in hepatocellular carcinoma therapy. In the following narrative, we summarize the compounds that have become more popular in HCC therapy in the last few years, which are able to inhibit HCC cells in different pathways from ferroptosis, and may play a great role in the future of hepatocellular carcinoma therapy (Table 1).
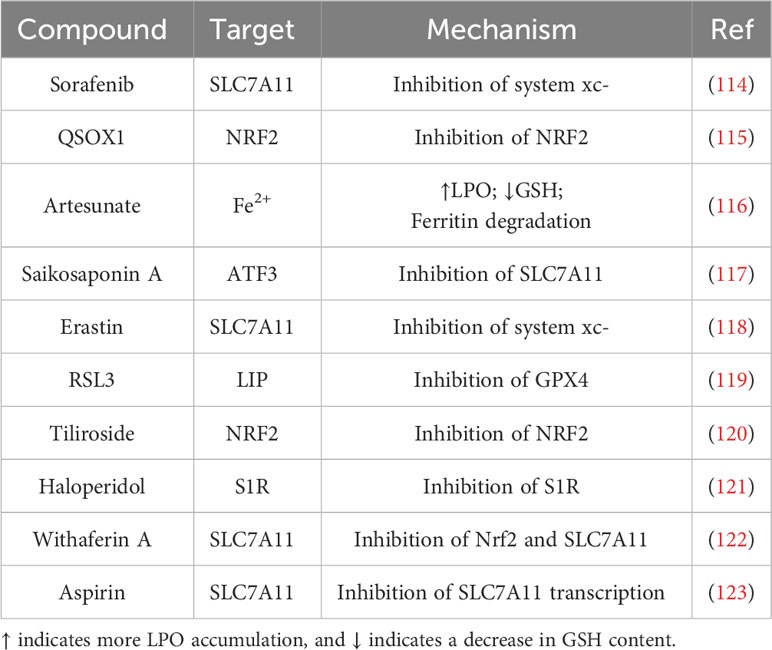
Table 1 Ferroptosis inducer and their major mode of action in HCC.
4.1 Nanoparticles
HCC is difficult to detect in the early stages of clinical work due to examination methods and clinical manifestations limitations. Once diagnosed, it progresses into advanced hepatocellular carcinoma that is extremely resistant to treatment and has unsatisfactory therapeutic results. Therefore, the need for innovative therapeutic approaches is urgent, and in recent years, targeted or chemotherapeutic drug delivery approaches using nanoparticles have started to emerge. Tang et al. (124) discovered that iron-catalyzed apoptosis might be induced by the degradation of manganese-silica nanoparticles themselves (through breakage of Mn-Obond). Manganese-silica nanoparticles loaded with sorafenib were created and combined with the drug’s effects. Significant anti-tumor effects were produced when applied to HCC cells. Similar to the above, Liu et al. (125) created an iron-apoptosis-associated therapy for hepatocellular cancer using a sorafenib-loaded MIL-101 (Fe) nanomedicine that was delivered in conjunction with iRGD. The neurociliin-1 (NRP-1) receptors are largely found in tumor and vascular tissues. The iRGD peptide preferentially binds to these receptors, activating the endocytosis transport route and facilitating drug penetration into tumor tissues (125). This procedure dramatically raised lipid peroxidation and malondialdehyde levels, increased auto-induced iron apoptosis in HepG 2 cells, and lowered glutathione and glutathione peroxidase 4 (GPX4) levels. This combination (125) have several promising characteristics including drug-loading, controllable release, peroxidase activity, biocompatibility, and T2 magnetic resonance imaging ability. One of the primary palliative treatment options for advanced hepatocellular carcinoma is transcatheter arterial chemoembolization (TACE). However, TACE creates a hypoxic microenvironment in the tumor that reduces the sensitivity of chemotherapy and plays a significant role in the unpredictable treatment outcome (126). Gelatin was used as the wall material for the microspheres during the preparation of adriamycin/triiron tetraoxide composite gelatin microspheres (ADM/Fe3O4-MS), and it was confirmed (127) that it is possible to use uniformly sized gelatin microspheres coloaded with-Fe3O4 nanoparticles and adriamycin to increase the effectiveness of TACE in the treatment of HCC.
4.2 Phytochemicals
Active substances derived from plants are referred to as phytochemicals. Examples include polysaccharides, polyphenols, and alkaloids. Numerous studies have demonstrated the exceptional anti-tumor efficacies of numerous phytochemicals, including baicalin and curcumin, with less adverse effects when compared to other chemotherapy medicines (128). Numerous phytochemicals have been employed as ferroptosis modulators in HCC cells to far. By encouraging iron apoptosis through the increase of ER stress and the production of PEBP1/15-LO, DHA can limit the progression of HCC (129, 130). Additionally, Heteronemin and Artesunate increased ROS and promoted ferritin degradation to induce ferroptosis, respectively, to enhance the anticancer activity of sorafenib against HCC (131). An isolated substance from Lonergania japonica called lonerganine has anti-infective properties. Lonerganine was originally used to treat HCC by Jin et al. (132). Metabolomics and a mouse xenograft model both supported the findings that lobelia alkaloids reduced tumor volume and weight and prevented hepatocyte invasion and migration. By inhibiting GPX4 and GSS, solasonine increased the amounts of lipid ROS in HepG2 cells to produce these effects (132).
4.3 Chemotherapy
Despite the advent of novel and rising HCC therapeutic techniques during the last two decades, chemotherapy remains the anticancer treatment pioneer. It is employed as a first-line therapy method for the majority of advanced cancers (133), but drug resistance poses a significant obstacle to tumor treatment. Sorafenib blocks systemic XC-mediated cystine input, causing endoplasmic reticulum stress, GSH depletion, and iron-dependent lipid ROS buildup (114). Nrf2 and metallothionein-1G (MT-1G) are two critical variables in sorafenib-resistant HCC (87). Sorafenib causes the production of P62, which inhibits NRF2 degradation and promotes the inactivation of KEAP1, followed by the activation of heme oxygenase-1 (HO-1), quinone oxidoreductase-1, and ferritin heavy chain-1, which enhances NRF2 nuclear localization. Sorafenib stimulates NRF2 expression in HCC cells, which boosts MT-1G expression, which prevents ferroptosis by inhibiting GSH depletion, which is one of the pathways associated in sorafenib resistance (87). Inhibiting any of the aforementioned pathways might thus theoretically accelerate ferroptosis in HCC cells. An iron-sulfur protein is CDGSH iron-sulfur structural domain 2 (CISD 2). CISD 2 inhibition improved sorafenib-induced iron apoptosis in drug-resistant cells via inhibiting ferritin phagocytosis or the p62-Keap1-NRF2 pathway (95).
Metformin is commonly used to treat diabetes, and its involvement in malignancies has been studied extensively. Metformin has been shown (134) to induce iron apoptosis and increase ROS levels in hepatocellular carcinoma cells via ATF4/STAT3, thereby decreasing hepatocellular carcinoma cell resistance to sorafenib. Meanwhile, metformin used with other medications to treat malignancies has produced impressive effects. Tang et al. (135) demonstrated that metformin mixed with sorafenib promoted iron apoptosis via the p62-Keap1-Nrf2/HO1 signaling pathway, inhibiting the value-added of HCC cells.
Unfortunately, chemotherapy alone is often unsatisfactory, with intrinsic and acquired resistance to conventional chemotherapeutic agents and targeted drugs developing in a short period of time. This largely limits the efficacy of the drugs and the survival of the patients. Therefore, the use of combination therapy is a viable path. A meta-analysis by Li et al. (136) demonstrated that the efficacy of combination therapy was superior to that of monotherapy. Interventional therapy, which promotes ischemic necrosis of tumor tissues by blocking the blood-supplying arteries of the tumor, has become the optimal choice for patients with advanced unresectable hepatocellular carcinoma. Drug-eluting microspheres have been widely used in the treatment of hepatocellular carcinoma for the simultaneous effects of embolization of blood vessels supplying tumors and sustained release of chemotherapeutic agents (137). However, in a hypoxic environment, vascular endothelial growth factor and HIF-1α are activated in tumor cells, increasing tumor neovascularization and the likelihood of metastasis. Ionizing radiation from radiation therapy also induced HIF-1α accumulation, which resulted in nuclear translocation of SLC7A11 and inhibited ferroptosis in HCC (138). In this situation, combination therapy shows unique advantages, CPT (an NRF2 inhibitor) (139), anti-angiogenic drugs (140), immune drugs (PD-1 inhibitors), etc. (141) in conjunction with chemotherapeutic drugs can reduce the resistance of tumor tissues.
4.4 ncRNA
Noncoding RNA (ncRNA) studies have revealed that ncRNAs such as microRNAs, long noncoding RNAs (lncRNAs), and circular RNAs play important roles in the carcinogenesis and progression of HCC. These non-coding RNAs are involved in tumorigenesis and progression and affect ferroptosis (142). Two homeoboxes Through activities on the Fork head Box M1 (FoxM1) protein, A pseudogene 8 (DUXAP8), a pseudogenederived lncRNA, may promote pancreatic cancer, non-small-cell lung cancer, and HCC. LncRNA DUXAP 8 is overexpressed in hepatocellular carcinoma and is associated with a poor prognosis, possibly contributing to sorafenib resistance by suppressing ferroptosis (143). Shi and colleagues (143) explored whether DUXAP 8 reduced HCC vulnerability to sorafenib-induced ferroptosis by acting on SLC7A11. Through binding to SLC7A11, DUXAP 8 decreases membrane translocation and facilitates sorting of depalmitoylated SLC7A11 to lysosomes. Thus, suppressing DUXAP8 accelerates iron mortality in HCC, and combining it with sorafenib improves treatment efficacy in patients with advanced HCC. The above results were demonstrated in CDX models at the same time. According to a recent study (144), the lncRNA HEPFAL acts on SLC7A11 and reduces its expression, resulting in a decrease in the amount of cystine transported into the cell and, as a result, a decrease in the intracellular GSH level, which effectively inhibits the iron apoptosis function of GPX4 and, ultimately, iron apoptosis in tumor cells. In the meantime, the PI3K-AKT-mTOR signaling pathway is involved in this process. Mechanistically, it could be because PI3K activates AKT via downstream molecules, and AKT activates mTOR by phosphorylation. mTORC1 is activated by AKT and governs ferroptosis through modulating a number of molecules, including SLC7A11, GPX4, and ferroportin, primarily via NRF2. In conclusion, these should be investigated further as additional research uncover the significance of RNA in ferroptosis in HCC.
5 Concluding remarks and future perspectives
There is mounting evidence that ferroptosis activation or inhibition has a significant impact on a variety of illnesses and could be used to treat HCC. In recent years, an increasing variety of medicines and newly created materials have been used in experimental animals with good efficacy. In the meantime, the formation of ferroptosis resistance during treatment is a new issue. Several regulatory mechanisms in the ferroptosis process protect cells from ferroptosis inhibition. Because a single ferroptosis pathway is more likely to trigger resistance to ferroptosis, multiple pathways should be coupled to promote ferroptosis in tumor cells at the same time. Furthermore, addressing an ferroptosis factor that causes damage to normal tissues, such as kidney injury and neurotoxicity, should not be disregarded. Creating various drug delivery channels is also an option, as is improving drug delivery, drug biodistribution, and pharmacokinetics.
Author contributions
XL: Writing – original draft, Writing – review & editing. FM: Writing – review & editing. HW: Writing – review & editing. LS: Writing – review & editing. SC: Writing – review & editing. FC: Writing – review & editing. GL: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Natural Science Foundation of Shandong Province (grant no. ZR2019BH041 to FC and no. ZR2023MH120 to GL), the Nature Science Foundation of China (grant no. 81803008 to FC) and the cultivating fund of the First hospital of Shandong First Medical University (grant no. QYPY2021NSFC0616 to FC).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Röcken C, Carl-McGrath S. Pathology and pathogenesis of hepatocellular carcinoma. Digestive Dis (Basel Switzerland) (2001) 19(4):269–78. doi: 10.1159/000050693
2. Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: Looking outside the box. J Hepatol (2020) 72(2):342–52. doi: 10.1016/j.jhep.2019.09.010
3. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
4. Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: A novel anti-tumor action for cisplatin. Cancer Res Treat (2018) 50(2):445–60. doi: 10.4143/crt.2016.572
5. Qiu Y, Cao Y, Cao W, Jia Y, Lu N. The application of ferroptosis in diseases. Pharmacol Res (2020) 159:104919. doi: 10.1016/j.phrs.2020.104919
6. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ (2016) 23(3):369–79. doi: 10.1038/cdd.2015.158
7. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell (2003) 3(3):285–96. doi: 10.1016/s1535-6108(03)00050-3
8. Doll S, Conrad M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life (2017) 69(6):423–34. doi: 10.1002/iub.1616
9. Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol (2021) 18(5):280–96. doi: 10.1038/s41571-020-00462-0
10. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol (2014) 10(1):9–17. doi: 10.1038/nchembio.1416
11. Tang D, Kroemer G. Ferroptosis. Curr Biol CB (2020) 30(21):R1292–r7. doi: 10.1016/j.cub.2020.09.068
12. Sassetti E, Clausen MH, Laraia L. Small-molecule inhibitors of reactive oxygen species production. J Med Chem (2021) 64(9):5252–75. doi: 10.1021/acs.jmedchem.0c01914
13. Papa S, Martino PL, Capitanio G, Gaballo A, De Rasmo D, Signorile A, et al. The oxidative phosphorylation system in mammalian mitochondria. Adv Exp Med Biol (2012) 942:3–37. doi: 10.1007/978-94-007-2869-1_1
14. Cheng Z, Li Y. What is responsible for the initiating chemistry of iron-mediated lipid peroxidation: an update. Chem Rev (2007) 107(3):748–66. doi: 10.1021/cr040077w
15. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell (2019) 35(6):830–49. doi: 10.1016/j.ccell.2019.04.002
16. Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem (1999) 274(17):11455–8. doi: 10.1074/jbc.274.17.11455
17. Sykiotis GP, Bohmann D. Stress-activated cap'n'collar transcription factors in aging and human disease. Sci Signal (2010) 3(112):re3. doi: 10.1126/scisignal.3112re3
18. Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, et al. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem (2002) 277(47):44765–71. doi: 10.1074/jbc.M208704200
19. Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, et al. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci (2003) 23(8):3394–406. doi: 10.1523/jneurosci.23-08-03394.2003
20. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep (2016) 17(10):1374–95. doi: 10.15252/embr.201642195
21. Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol metabolism: TEM (2009) 20(9):436–43. doi: 10.1016/j.tem.2009.05.008
22. Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun (2004) 325(1):109–16. doi: 10.1016/j.bbrc.2004.10.009
23. Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, et al. Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol (2014) 34(18):3421–34. doi: 10.1128/mcb.00221-14
24. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
25. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ (2020) 27(2):662–75. doi: 10.1038/s41418-019-0380-z
26. Ursini F, Maiorino M, Gregolin C. The selenoenzyme phospholipid hydroperoxide glutathione peroxidase. Biochim Biophys Acta (1985) 839(1):62–70. doi: 10.1016/0304-4165(85)90182-5
27. Cozza G, Rossetto M, Bosello-Travain V, Maiorino M, Roveri A, Toppo S, et al. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radical Biol Med (2017) 112:1–11. doi: 10.1016/j.freeradbiomed.2017.07.010
28. Weaver K, Skouta R. The selenoprotein glutathione peroxidase 4: from molecular mechanisms to novel therapeutic opportunities. Biomedicines (2022) 10(4):891. doi: 10.3390/biomedicines10040891
29. Lin X, Ping J, Wen Y, Wu Y. The mechanism of ferroptosis and applications in tumor treatment. Front Pharmacol (2020) 11:1061. doi: 10.3389/fphar.2020.01061
30. Maiorino M, Thomas JP, Girotti AW, Ursini F. Reactivity of phospholipid hydroperoxide glutathione peroxidase with membrane and lipoprotein lipid hydroperoxides. Free Radical Res Commun (1991) 12-13 Pt 1:131–5. doi: 10.3109/10715769109145777
31. Thomas JP, Maiorino M, Ursini F, Girotti AW. Protective action of phospholipid hydroperoxide glutathione peroxidase against membrane-damaging lipid peroxidation. In situ reduction of phospholipid and cholesterol hydroperoxides. J Biol Chem (1990) 265(1):454–61. doi: 10.1016/s0021-9258(19)40252-4
32. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10(1):1617. doi: 10.1038/s41467-019-09277-9
33. Yant LJ, Ran Q, Rao L, Van Remmen H, Shibatani T, Belter JG, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radical Biol Med (2003) 34(4):496–502. doi: 10.1016/s0891-5849(02)01360-6
34. Imai H, Hirao F, Sakamoto T, Sekine K, Mizukura Y, Saito M, et al. Early embryonic lethality caused by targeted disruption of the mouse PHGPx gene. Biochem Biophys Res Commun (2003) 305(2):278–86. doi: 10.1016/s0006-291x(03)00734-4
35. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell (2018) 172(3):409–22.e21. doi: 10.1016/j.cell.2017.11.048
36. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
37. Mukhopadhyay S, Vander Heiden MG, McCormick F. The Metabolic Landscape of RAS-Driven Cancers from biology to therapy. Nat Cancer (2021) 2(3):271–83. doi: 10.1038/s43018-021-00184-x
38. Mukhopadhyay S, Goswami D, Adiseshaiah PP, Burgan W, Yi M, Guerin TM, et al. Undermining glutaminolysis bolsters chemotherapy while NRF2 promotes chemoresistance in KRAS-driven pancreatic cancers. Cancer Res (2020) 80(8):1630–43. doi: 10.1158/0008-5472.Can-19-1363
39. Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, et al. Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci USA (2010) 107(16):7461–6. doi: 10.1073/pnas.1002459107
40. Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci USA (2010) 107(16):7455–60. doi: 10.1073/pnas.1001006107
41. Liu J, Zhang C, Wang J, Hu W, Feng Z. The Regulation of Ferroptosis by Tumor Suppressor p53 and its Pathway. Int J Mol Sci (2020) 21(21):8387. doi: 10.3390/ijms21218387
42. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell (2019) 177(5):1262–79.e25. doi: 10.1016/j.cell.2019.03.032
43. Anderton B, Camarda R, Balakrishnan S, Balakrishnan A, Kohnz RA, Lim L, et al. MYC-driven inhibition of the glutamate-cysteine ligase promotes glutathione depletion in liver cancer. EMBO Rep (2017) 18(4):569–85. doi: 10.15252/embr.201643068
44. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
45. Liu J, Xia X, Huang P. xCT: A critical molecule that links cancer metabolism to redox signaling. Mol Ther J Am Soc Gene Ther (2020) 28(11):2358–66. doi: 10.1016/j.ymthe.2020.08.021
46. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell (2021) 12(8):599–620. doi: 10.1007/s13238-020-00789-5
47. Li S, Lu Z, Sun R, Guo S, Gao F, Cao B, et al. The role of SLC7A11 in cancer: friend or foe? Cancers (2022) 14(13):3059. doi: 10.3390/cancers14133059
48. Tang Z, Xu Z, Zhu X, Zhang J. New insights into molecules and pathways of cancer metabolism and therapeutic implications. Cancer Commun (London England) (2021) 41(1):16–36. doi: 10.1002/cac2.12112
49. Harayama T, Riezman H. Understanding the diversity of membrane lipid composition. Nat Rev Mol Cell Biol (2018) 19(5):281–96. doi: 10.1038/nrm.2017.138
50. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
51. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol (2017) 13(1):91–8. doi: 10.1038/nchembio.2239
52. Gao M, Deng J, Liu F, Fan A, Wang Y, Wu H, et al. Triggered ferroptotic polymer micelles for reversing multidrug resistance to chemotherapy. Biomaterials (2019) 223:119486. doi: 10.1016/j.biomaterials.2019.119486
53. Kuwata H, Hara S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins other Lipid Mediators (2019) 144:106363. doi: 10.1016/j.prostaglandins.2019.106363
54. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell (2022) 40(4):365–78.e6. doi: 10.1016/j.ccell.2022.02.003
55. Brown CW, Amante JJ, Goel HL, Mercurio AM. The α6β4 integrin promotes resistance to ferroptosis. J Cell Biol (2017) 216(12):4287–97. doi: 10.1083/jcb.201701136
56. Kan CF, Singh AB, Stafforini DM, Azhar S, Liu J. Arachidonic acid downregulates acyl-CoA synthetase 4 expression by promoting its ubiquitination and proteasomal degradation. J Lipid Res (2014) 55(8):1657–67. doi: 10.1194/jlr.M045971
57. Orlando U, Cooke M, Cornejo Maciel F, Papadopoulos V, Podestá EJ, Maloberti P. Characterization of the mouse promoter region of the acyl-CoA synthetase 4 gene: role of Sp1 and CREB. Mol Cell Endocrinol (2013) 369(1-2):15–26. doi: 10.1016/j.mce.2013.01.016
58. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Publisher Correction: Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature (2019) 572(7770):E20. doi: 10.1038/s41586-019-1480-0
59. Xia H, Lee KW, Chen J, Kong SN, Sekar K, Deivasigamani A, et al. Simultaneous silencing of ACSL4 and induction of GADD45B in hepatocellular carcinoma cells amplifies the synergistic therapeutic effect of aspirin and sorafenib. Cell Death Discov (2017) 3:17058. doi: 10.1038/cddiscovery.2017.58
60. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol (2015) 10(7):1604–9. doi: 10.1021/acschembio.5b00245
61. Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, et al. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol (2019) 21(5):579–91. doi: 10.1038/s41556-019-0305-6
62. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol (2020) 16(3):302–9. doi: 10.1038/s41589-020-0472-6
63. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell (2021) 81(2):355–69.e10. doi: 10.1016/j.molcel.2020.11.024
64. Liu J, Kang R, Tang D. Signaling pathways and defense mechanisms of ferroptosis. FEBS J (2022) 289(22):7038–50. doi: 10.1111/febs.16059
65. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell (2017) 171(3):628–41.e26. doi: 10.1016/j.cell.2017.09.044
66. Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy (2021) 17(4):948–60. doi: 10.1080/15548627.2020.1739447
67. Kuang F, Liu J, Xie Y, Tang D, Kang R. MGST1 is a redox-sensitive repressor of ferroptosis in pancreatic cancer cells. Cell Chem Biol (2021) 28(6):765–75.e5. doi: 10.1016/j.chembiol.2021.01.006
68. Lakhal-Littleton S. Mechanisms of cardiac iron homeostasis and their importance to heart function. Free Radical Biol Med (2019) 133:234–7. doi: 10.1016/j.freeradbiomed.2018.08.010
69. Bi Y, Ajoolabady A, Demillard LJ, Yu W, Hilaire ML, Zhang Y, et al. Dysregulation of iron metabolism in cardiovascular diseases: From iron deficiency to iron overload. Biochem Pharmacol (2021) 190:114661. doi: 10.1016/j.bcp.2021.114661
70. Hino K, Yanatori I, Hara Y, Nishina S. Iron and liver cancer: an inseparable connection. FEBS J (2022) 289(24):7810–29. doi: 10.1111/febs.16208
71. Kawabata H. Transferrin and transferrin receptors update. Free Radical Biol Med (2019) 133:46–54. doi: 10.1016/j.freeradbiomed.2018.06.037
72. Zhao N, Gao J, Enns CA, Knutson MD. ZRT/IRT-like protein 14 (ZIP14) promotes the cellular assimilation of iron from transferrin. J Biol Chem (2010) 285(42):32141–50. doi: 10.1074/jbc.M110.143248
73. Mayr R, Griffiths WJ, Hermann M, McFarlane I, Halsall DJ, Finkenstedt A, et al. Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology (2011) 140(7):2056–63, 63.e1. doi: 10.1053/j.gastro.2011.02.064
74. Li J, Liu J, Xu Y, Wu R, Chen X, Song X, et al. Tumor heterogeneity in autophagy-dependent ferroptosis. Autophagy (2021) 17(11):3361–74. doi: 10.1080/15548627.2021.1872241
75. Munro HN, Linder MC. Ferritin: structure, biosynthesis, and role in iron metabolism. Physiol Rev (1978) 58(2):317–96. doi: 10.1152/physrev.1978.58.2.317
76. Rochette L, Dogon G, Rigal E, Zeller M, Cottin Y, Vergely C. Lipid peroxidation and iron metabolism: two corner stones in the homeostasis control of ferroptosis. Int J Mol Sci (2022) 24(1):449. doi: 10.3390/ijms24010449
77. Daher R, Manceau H, Karim Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. Presse medicale (Paris France 1983) (2017) 46(12 Pt 2):e272–e8. doi: 10.1016/j.lpm.2017.10.006
78. Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X, et al. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene (2015) 34(45):5617–25. doi: 10.1038/onc.2015.32
79. Chiang SK, Chen SE, Chang LC. A dual role of heme oxygenase-1 in cancer cells. Int J Mol Sci (2018) 20(1):39. doi: 10.3390/ijms20010039
80. Yanatori I, Richardson DR, Toyokuni S, Kishi F. The iron chaperone poly(rC)-binding protein 2 forms a metabolon with the heme oxygenase 1/cytochrome P450 reductase complex for heme catabolism and iron transfer. J Biol Chem (2017) 292(32):13205–29. doi: 10.1074/jbc.M117.776021
81. Yanatori I, Richardson DR, Imada K, Kishi F. Iron export through the transporter ferroportin 1 is modulated by the iron chaperone PCBP2. J Biol Chem (2016) 291(33):17303–18. doi: 10.1074/jbc.M116.721936
82. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell (2019) 51(5):575–86.e4. doi: 10.1016/j.devcel.2019.10.007
83. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxidants Redox Signal (2018) 29(17):1756–73. doi: 10.1089/ars.2017.7176
84. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell (2022) 185(14):2401–21. doi: 10.1016/j.cell.2022.06.003
85. Zhao L, Zhou X, Xie F, Zhang L, Yan H, Huang J, et al. Ferroptosis in cancer and cancer immunotherapy. Cancer Commun (London England) (2022) 42(2):88–116. doi: 10.1002/cac2.12250
86. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer (2012) 12(8):564–71. doi: 10.1038/nrc3278
87. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatol (Baltimore Md) (2016) 63(1):173–84. doi: 10.1002/hep.28251
88. Anandhan A, Dodson M, Schmidlin CJ, Liu P, Zhang DD. Breakdown of an ironclad defense system: the critical role of NRF2 in mediating ferroptosis. Cell Chem Biol (2020) 27(4):436–47. doi: 10.1016/j.chembiol.2020.03.011
89. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatol (Baltimore Md) (2016) 64(2):488–500. doi: 10.1002/hep.28574
90. Jedlitschky G, Burchell B, Keppler D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J Biol Chem (2000) 275(39):30069–74. doi: 10.1074/jbc.M005463200
91. Huang W, Chen K, Lu Y, Zhang D, Cheng Y, Li L, et al. ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia (New York NY) (2021) 23(12):1227–39. doi: 10.1016/j.neo.2021.11.002
92. Houessinon A, François C, Sauzay C, Louandre C, Mongelard G, Godin C, et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol Cancer (2016) 15(1):38. doi: 10.1186/s12943-016-0526-2
93. Yang F, Li J, Deng H, Wang Y, Lei C, Wang Q, et al. GSTZ1-1 deficiency activates NRF2/IGF1R axis in HCC via accumulation of oncometabolite succinylacetone. EMBO J (2019) 38(15):e101964. doi: 10.15252/embj.2019101964
94. Li J, Wang Q, Yang Y, Lei C, Yang F, Liang L, et al. GSTZ1 deficiency promotes hepatocellular carcinoma proliferation via activation of the KEAP1/NRF2 pathway. J Exp Clin Cancer Res CR (2019) 38(1):438. doi: 10.1186/s13046-019-1459-6
95. Li Y, Xu B, Ren X, Wang L, Xu Y, Zhao Y, et al. Inhibition of CISD2 promotes ferroptosis through ferritinophagy-mediated ferritin turnover and regulation of p62-Keap1-NRF2 pathway. Cell Mol Biol Lett (2022) 27(1):81. doi: 10.1186/s11658-022-00383-z
96. Li H, Zhao J, Zhong XL, Xu PY, Du LJ, Fang P, et al. CPLX2 regulates ferroptosis and apoptosis through NRF2 pathway in human hepatocellular carcinoma cells. Appl Biochem Biotechnol (2023) 195(1):597–609. doi: 10.1007/s12010-022-04135-9
97. Bai T, Wang S, Zhao Y, Zhu R, Wang W, Sun Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem Biophys Res Commun (2017) 491(4):919–25. doi: 10.1016/j.bbrc.2017.07.136
98. Hayano M, Yang WS, Corn CK, Pagano NC, Stockwell BR. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ (2016) 23(2):270–8. doi: 10.1038/cdd.2015.93
99. Cao J, Chen X, Jiang L, Lu B, Yuan M, Zhu D, et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat Commun (2020) 11(1):1251. doi: 10.1038/s41467-020-15109-y
100. Chen H, Liu S, Ji L, Wu T, Ji Y, Zhou Y, et al. Folic acid supplementation mitigates alzheimer's disease by reducing inflammation: A randomized controlled trial. Mediators Inflamm (2016) 2016:5912146. doi: 10.1155/2016/5912146
101. Wallis KF, Morehead LC, Bird JT, Byrum SD, Miousse IR. Differences in cell death in methionine versus cysteine depletion. Environ Mol mutagenesis (2021) 62(3):216–26. doi: 10.1002/em.22428
102. Zou CG, Banerjee R. Tumor necrosis factor-alpha-induced targeted proteolysis of cystathionine beta-synthase modulates redox homeostasis. J Biol Chem (2003) 278(19):16802–8. doi: 10.1074/jbc.M212376200
103. Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol (2019) 21(1):63–71. doi: 10.1038/s41556-018-0205-1
104. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 169(2):361–71. doi: 10.1016/j.cell.2017.03.035
105. Gu Y, Albuquerque CP, Braas D, Zhang W, Villa GR, Bi J, et al. mTORC2 regulates amino acid metabolism in cancer by phosphorylation of the cystine-glutamate antiporter xCT. Mol Cell (2017) 67(1):128–38.e7. doi: 10.1016/j.molcel.2017.05.030
106. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun (2021) 12(1):1589. doi: 10.1038/s41467-021-21841-w
107. Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA (2020) 117(49):31189–97. doi: 10.1073/pnas.2017152117
108. Mleczko-Sanecka K, Roche F, da Silva AR, Call D, D'Alessio F, Ragab A, et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood (2014) 123(10):1574–85. doi: 10.1182/blood-2013-07-515957
109. Hou J, Jiang C, Wen X, Li C, Xiong S, Yue T, et al. ACSL4 as a potential target and biomarker for anticancer: from molecular mechanisms to clinical therapeutics. Front Pharmacol (2022) 13:949863. doi: 10.3389/fphar.2022.949863
110. Chen M, Tan AH, Li J. Curcumin represses colorectal cancer cell proliferation by triggering ferroptosis via PI3K/akt/mTOR signaling. Nutr Cancer (2023) 75(2):726–33. doi: 10.1080/01635581.2022.2139398
111. Huang SC, Chen YM, Hu YY, Shi YJ, Xiao QW, Li Z, et al. Downregulation of MCF2L promoted the ferroptosis of hepatocellular carcinoma cells through PI3K/mTOR pathway in a rhoA/rac1 dependent manner. Dis Markers (2022) 2022:6138941. doi: 10.1155/2022/6138941
112. Tong Q, Qin W, Li ZH, Liu C, Wang ZC, Chu Y, et al. SLC12A5 promotes hepatocellular carcinoma growth and ferroptosis resistance by inducing ER stress and cystine transport changes. Cancer Med (2023) 12(7):8526–41. doi: 10.1002/cam4.5605
113. Hu Z, Li L, Li M, Zhang X, Zhang Y, Ran J, et al. miR-21-5p inhibits ferroptosis in hepatocellular carcinoma cells by regulating the AKT/mTOR signaling pathway through MELK. J Immunol Res (2023) 2023:8929525. doi: 10.1155/2023/8929525
114. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife (2014) 3:e02523. doi: 10.7554/eLife.02523
115. Sun J, Zhou C, Zhao Y, Zhang X, Chen W, Zhou Q, et al. Quiescin sulfhydryl oxidase 1 promotes sorafenib-induced ferroptosis in hepatocellular carcinoma by driving EGFR endosomal trafficking and inhibiting NRF2 activation. Redox Biol (2021) 41:101942. doi: 10.1016/j.redox.2021.101942
116. Li S, He Y, Chen K, Sun J, Zhang L, He Y, et al. RSL3 drives ferroptosis through NF-κB pathway activation and GPX4 depletion in glioblastoma. Oxid Med Cell Longev (2021) 2021:2915019. doi: 10.1155/2021/2915019
117. Lan T, Wang W, Zeng XX, Tong YH, Mao ZJ, Wang SW. Saikosaponin A triggers cell ferroptosis in hepatocellular carcinoma by inducing endoplasmic reticulum stress-stimulated ATF3 expression. Biochem Biophys Res Commun (2023) 674:10–8. doi: 10.1016/j.bbrc.2023.06.086
118. Lippmann J, Petri K, Fulda S, Liese J. Redox modulation and induction of ferroptosis as a new therapeutic strategy in hepatocellular carcinoma. Trans Oncol (2020) 13(8):100785. doi: 10.1016/j.tranon.2020.100785
119. Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z, et al. HCAR1/MCT1 regulates tumor ferroptosis through the lactate-mediated AMPK-SCD1 activity and its therapeutic implications. Cell Rep (2020) 33(10):108487. doi: 10.1016/j.celrep.2020.108487
120. Yang C, Lu T, Liu M, Yuan X, Li D, Zhang J, et al. Tiliroside targets TBK1 to induce ferroptosis and sensitize hepatocellular carcinoma to sorafenib. Phytomed Int J phytother phytopharmaco (2023) 111:154668. doi: 10.1016/j.phymed.2023.154668
121. Bai T, Lei P, Zhou H, Liang R, Zhu R, Wang W, et al. Sigma-1 receptor protects against ferroptosis in hepatocellular carcinoma cells. J Cell Mol Med (2019) 23(11):7349–59. doi: 10.1111/jcmm.14594
122. Zhang Y, Li Y, Qiu Q, Chen Z, Du Y, Liu X. MITD1 deficiency suppresses clear cell renal cell carcinoma growth and migration by inducing ferroptosis through the TAZ/SLC7A11 pathway. Oxid Med Cell Longev (2022) 2022:7560569. doi: 10.1155/2022/7560569
123. Wang YF, Feng JY, Zhao LN, Zhao M, Wei XF, Geng Y, et al. Aspirin triggers ferroptosis in hepatocellular carcinoma cells through restricting NF-κB p65-activated SLC7A11 transcription. Acta pharmacologica Sin (2023) 44(8):1712–24. doi: 10.1038/s41401-023-01062-1
124. Tang H, Chen D, Li C, Zheng C, Wu X, Zhang Y, et al. Dual GSH-exhausting sorafenib loaded manganese-silica nanodrugs for inducing the ferroptosis of hepatocellular carcinoma cells. Int J pharmaceutics (2019) 572:118782. doi: 10.1016/j.ijpharm.2019.118782
125. Liu X, Zhu X, Qi X, Meng X, Xu K. Co-administration of iRGD with sorafenib-loaded iron-based metal-organic framework as a targeted ferroptosis agent for liver cancer therapy. Int J nanomed (2021) 16:1037–50. doi: 10.2147/ijn.S292528
126. Martin SP, Fako V, Dang H, Dominguez DA, Khatib S, Ma L, et al. PKM2 inhibition may reverse therapeutic resistance to transarterial chemoembolization in hepatocellular carcinoma. J Exp Clin Cancer Res CR (2020) 39(1):99. doi: 10.1186/s13046-020-01605-y
127. Chen M, Li J, Shu G, Shen L, Qiao E, Zhang N, et al. Homogenous multifunctional microspheres induce ferroptosis to promote the anti-hepatocarcinoma effect of chemoembolization. J nanobiotechnol (2022) 20(1):179. doi: 10.1186/s12951-022-01385-x
128. Choudhari AS, Mandave PC, Deshpande M, Ranjekar P, Prakash O. Phytochemicals in cancer treatment: from preclinical studies to clinical practice. Front Pharmacol (2019) 10:1614. doi: 10.3389/fphar.2019.01614
129. Wang Z, Li M, Liu Y, Qiao Z, Bai T, Yang L, et al. Dihydroartemisinin triggers ferroptosis in primary liver cancer cells by promoting and unfolded protein response−induced upregulation of CHAC1 expression. Oncol Rep (2021) 46(5):240. doi: 10.3892/or.2021.8191
130. Cui Z, Wang H, Li S, Qin T, Shi H, Ma J, et al. Dihydroartemisinin enhances the inhibitory effect of sorafenib on HepG2 cells by inducing ferroptosis and inhibiting energy metabolism. J Pharmacol Sci (2022) 148(1):73–85. doi: 10.1016/j.jphs.2021.09.008
131. Li ZJ, Dai HQ, Huang XW, Feng J, Deng JH, Wang ZX, et al. Artesunate synergizes with sorafenib to induce ferroptosis in hepatocellular carcinoma. Acta pharmacologica Sin (2021) 42(2):301–10. doi: 10.1038/s41401-020-0478-3
132. Jin M, Shi C, Li T, Wu Y, Hu C, Huang G. Solasonine promotes ferroptosis of hepatoma carcinoma cells via glutathione peroxidase 4-induced destruction of the glutathione redox system. Biomedicine pharmacother = Biomed pharmacotherapie (2020) 129:110282. doi: 10.1016/j.biopha.2020.110282
133. D'Alterio C, Scala S, Sozzi G, Roz L, Bertolini G. Paradoxical effects of chemotherapy on tumor relapse and metastasis promotion. Semin Cancer Biol (2020) 60:351–61. doi: 10.1016/j.semcancer.2019.08.019
134. Hu Z, Zhao Y, Li L, Jiang J, Li W, Mang Y, et al. Metformin promotes ferroptosis and sensitivity to sorafenib in hepatocellular carcinoma cells via ATF4/STAT3. Mol Biol Rep (2023) 50(8):6399–413. doi: 10.1007/s11033-023-08492-4
135. Tang K, Chen Q, Liu Y, Wang L, Lu W. Combination of metformin and sorafenib induces ferroptosis of hepatocellular carcinoma through p62-keap1-nrf2 pathway. J Cancer (2022) 13(11):3234–43. doi: 10.7150/jca.76618
136. Li H, Wu Z, Chen J, Su K, Guo L, Xu K, et al. External radiotherapy combined with sorafenib has better efficacy in unresectable hepatocellular carcinoma: a systematic review and meta-analysis. Clin Exp Med (2023) 23(5):1537–49. doi: 10.1007/s10238-022-00972-4
137. Huang J, Huang W, Zhan M, Guo Y, Liang L, Cai M, et al. Drug-eluting bead transarterial chemoembolization combined with FOLFOX-based hepatic arterial infusion chemotherapy for large or huge hepatocellular carcinoma. J hepatocellular Carcinoma (2021) 8:1445–58. doi: 10.2147/jhc.S339379
138. Shang Y, Luo M, Yao F, Wang S, Yuan Z, Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell Signal (2020) 72:109633. doi: 10.1016/j.cellsig.2020.109633
139. Chen F, Wang H, Zhu J, Zhao R, Xue P, Zhang Q, et al. Camptothecin suppresses NRF2-ARE activity and sensitises hepatocellular carcinoma cells to anticancer drugs. Br J Cancer (2017) 117(10):1495–506. doi: 10.1038/bjc.2017.317
140. Su K, Guo L, Ma W, Wang J, Xie Y, Rao M, et al. PD-1 inhibitors plus anti-angiogenic therapy with or without intensity-modulated radiotherapy for advanced hepatocellular carcinoma: A propensity score matching study. Front Immunol (2022) 13:972503. doi: 10.3389/fimmu.2022.972503
141. Kong R, Wang N, Han W, Bao W, Lu J. IFNγ-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells. J leukocyte Biol (2021) 110(2):301–14. doi: 10.1002/jlb.3ma1220-815rrr
142. Chen S, Zhao Y. Circular RNAs: Characteristics, function, and role in human cancer. Histol histopathol (2018) 33(9):887–93. doi: 10.14670/hh-11-969
143. Shi Z, Li Z, Jin B, Ye W, Wang L, Zhang S, et al. Loss of LncRNA DUXAP8 synergistically enhanced sorafenib induced ferroptosis in hepatocellular carcinoma via SLC7A11 de-palmitoylation. Clin Trans Med (2023) 13(6):e1300. doi: 10.1002/ctm2.1300
Keywords: ferroptosis, hepatocellular carcinoma, chemotherapy, drug resistance, therapy
Citation: Li X, Meng F, Wang H, Sun L, Chang S, Li G and Chen F (2024) Iron accumulation and lipid peroxidation: implication of ferroptosis in hepatocellular carcinoma. Front. Endocrinol. 14:1319969. doi: 10.3389/fendo.2023.1319969
Received: 12 October 2023; Accepted: 12 December 2023;
Published: 04 January 2024.
Edited by:
Dragos Cretoiu, Carol Davila University of Medicine and Pharmacy, RomaniaReviewed by:
Paul D. Kennedy, Cayman Chemical Company, United StatesYunwei Han, The Affiliated Hospital of Southwest Medical University, China
Copyright © 2024 Li, Meng, Wang, Sun, Chang, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Feng Chen, ZmVuZ3hpYW82NTU2QDEyNi5jb20=; Guijie Li, c2R1bGdqQDE2My5jb20=