
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 23 November 2023
Sec. Pituitary Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1305606
 Sunita M. C. De Sousa1,2,3*
Sunita M. C. De Sousa1,2,3* Angeline Shen4,5Christopher J. Yates4,5
Angeline Shen4,5Christopher J. Yates4,5 Roderick Clifton-Bligh6,7,8Stephen Santoreneos9James King10
Roderick Clifton-Bligh6,7,8Stephen Santoreneos9James King10 John Toubia11
John Toubia11 Giampaolo Trivellin12,13Andrea G. Lania12,13
Giampaolo Trivellin12,13Andrea G. Lania12,13 Constantine A. Stratakis14,15,16
Constantine A. Stratakis14,15,16 David J. Torpy1,3
David J. Torpy1,3 Hamish S. Scott3,17
Hamish S. Scott3,17Introduction: Germline loss-of-function variants in PAM, encoding peptidylglycine α-amidating monooxygenase (PAM), were recently discovered to be enriched in conditions of pathological pituitary hypersecretion, specifically: somatotrophinoma, corticotrophinoma, and prolactinoma. PAM is the sole enzyme responsible for C-terminal amidation of peptides, and plays a role in the biosynthesis and regulation of multiple hormones, including proopiomelanocortin (POMC).
Methods: We performed exome sequencing of germline and tumour DNA from 29 individuals with functioning pituitary adenomas (12 prolactinomas, 10 thyrotrophinomas, 7 cyclical Cushing’s disease). An unfiltered analysis was undertaken of all PAM variants with population prevalence <5%.
Results: We identified five coding, non-synonymous PAM variants of interest amongst seven individuals (six germline, one somatic). The five variants comprised four missense variants and one truncating variant, all heterozygous. Each variant had some evidence of pathogenicity based on population prevalence, conservation scores, in silico predictions and/or prior functional studies. The yield of predicted deleterious PAM variants was thus 7/29 (24%). The variants predominated in individuals with thyrotrophinomas (4/10, 40%) and cyclical Cushing’s disease (2/7, 29%), compared to prolactinomas (1/12, 8%).
Conclusion: This is the second study to demonstrate a high yield of suspected loss-of-function, predominantly germline, PAM variants in individuals with pathological pituitary hypersecretion. We have extended the association with corticotrophinoma to include the specific clinical entity of cyclical Cushing’s disease and demonstrated a novel association between PAM variants and thyrotrophinoma. PAM variants might act as risk alleles for pituitary adenoma formation, with a possible genotype-phenotype relationship between truncating variants and altered temporal secretion of cortisol.
Pituitary adenomas arise from endocrine cells of the anterior pituitary. The World Health Organization classifies pituitary adenomas, now also referred to as pituitary neuroendocrine tumours, by transcription factor expression and hormone immunohistochemistry categories that relate to the cell of origin. The major pituitary adenoma lineages are: PIT1 (lactotroph, somatotroph, and thyrotroph tumours), TPIT (corticotroph tumours), SF1 (gonadotroph tumours), and no distinct cell lineage (null-cell and plurihormonal tumours) (1).
The genes that have been hitherto implicated in pituitary tumorigenesis typically respect the cell of origin paradigm. In the germline setting, AIP variants and X-chromosome microduplications involving GPR101 are strongly associated with somatotrophinomas, whilst variants in MEN1, PRKAR1A and the SDHx genes (SDHA, SDHB, SDHC, SDHD, SDHAF2) are associated with all pituitary adenoma subtypes but with a predilection for lactotroph hyperplasia/adenomas. In the somatic pituitary adenoma setting, the principal driver mutations are GNAS and USP8 variants, each found in approximately half of somatototrophinomas and corticotrophinomas, respectively (2).
PAM is a newly implicated gene in pituitary tumorigenesis, with the princeps study by Trivellin et al. earlier this year demonstrating loss-of-function PAM variants across hypersecretory pituitary adenoma subtypes in patients from different centres in United States and Europe (3). Out of 299 individuals with sporadic pituitary adenomas and 18 kindreds with familial isolated pituitary adenomas, they identified seven PAM variants with deleterious effects on protein expression and/or function. Some variants were shared between individuals. The phenotypes of cases with these PAM variants comprised familial gigantism, sporadic somatotrophinoma, and paediatric corticotrophinoma. The investigators proceeded to analyse genotype/phenotype data from the UK Biobank (UKBB) and found that PAM variants were enriched in patients with ICD-10 codes associated with sellar lesions.
PAM is a 974-amino acid (longest isoform), 25-exon gene which encodes peptidylglycine α-amidating monooxygenase (PAM), a bifunctional enzyme responsible for C-terminal amidation of peptides. The resultant amidated peptides can be significantly more biologically potent than their unmodified precursors (4). Through this and other mechanisms, PAM is essential to the biosynthesis and regulated processing and secretion of multiple pituitary and hypothalamic peptide hormones in addition to many neuropeptides. PAM contains two enzymatic domains – peptidylglycine α-hydroxylating monooxygenase (PHM) and peptidyl-α-hydroxyglycine α-amidating lyase (PAL) – which act sequentially to generate C-terminal amidated peptides (3, 5). PAM expression in the pituitary gland is demonstrated in the GTEx (https://gtexportal.org) and Human Protein Atlas (https://www.proteinatlas.org) databases, and this has been confirmed by dedicated pituitary studies (3, 6). To date, the Trivellin et al. study is the only evidence of a role for PAM variants in pituitary tumorigenesis (3). By contrast, PAM variants have an established role in being associated with increased risk of type 2 diabetes mellitus (T2DM) (4).
To further investigate the relationship between PAM and functioning pituitary adenomas, we searched for PAM variants in an independent Australian cohort of individuals with prolactinoma, thyrotrophinoma or cyclical Cushing’s disease. We hypothesised that PAM may act as a tumour suppressor gene like other pituitary tumorigenesis genes such as MEN1 and AIP (2), with a somatic second-hit involving the wild-type (WT) allele in accordance with the Knudson two-hit hypothesis (7). This cohort of patients had been recruited for whole exome sequencing (WES) of paired tumour and germline DNA because of the paucity of known driver mutations in prolactinomas, thyrotrophinomas and the specific subset of corticotrophinomas associated with cyclical Cushing’s disease.
The study cohort comprised 29 unrelated patients with functioning pituitary adenomas (12 prolactinomas, 10 thyrotrophinomas, 7 cyclical Cushing’s disease) that had been surgically resected at tertiary referral pituitary centres in Australia: Royal Adelaide Hospital in South Australia, Royal Melbourne Hospital in Victoria, and Royal North Shore Hospital in New South Wales. None of the study subjects were part of the original cohort published by Trivellin et al. (3). Cyclical Cushing’s disease was defined by biochemical documentation of episodic cortisol excess interspersed by periods of normal or low cortisol secretion (8). Clinical and genetic characteristics of some study participants have been previously published (9–11). Pathogenic germline variants in established pituitary predisposition genes (AIP, CDKN1B, MEN1, PRKAR1A, SDHA, SDHB, SDHC and SDHD) had been excluded in all individuals.
The study was approved by the local institutional research committees (Melbourne Health: HREC/16/MH/132; Royal Adelaide Hospital: SSA/18/CALHN/445). All participants provided written informed consent to participate in the study.
Germline and tumour DNA was obtained from each patient, apart from a single patient in whom tumour DNA was not available.
Fresh blood samples were obtained from each patient for extraction of germline DNA from peripheral blood leucocytes. Operative tumour specimens were retrieved for somatic DNA extraction. Tumour specimens had either been stored as fresh frozen or formalin-fixed paraffin-embedded (FFPE) tissue. Both tumour and germline DNA were extracted using commercially available kits (Qiagen and Bioline) according to manufacturer protocols. FFPE samples were deparaffinised and additional DNA repair steps were performed using uracil-N-glycosylase to enzymatically remove formalin-induced cytosine deamination artefacts.
Next generation sequencing (NGS) of germline and tumour DNA was performed using whole-exome capture (Roche NimbleGen SeqCap EZ MedExome v3.0 in 27 cases; IDT xGen Exome v2 in 2 cases) sequenced on the Illumina NextSeq or NovaSeq Sequencing System.
Variant calling was undertaken at the Australian Cancer Research Facility (ACRF) of the Centre for Cancer Biology, SA Pathology (Adelaide, Australia). The Burrows-Wheeler Alignment tool, BWA-MEM, was used to align short reads to human reference assembly GRCh37/hg19 (version b37+decoy). Variants up to a size of approx. 50 bp were called using Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.4 (12).
Raw WES data were filtered for variants in PAM with minor allele frequency (MAF) below 5.0% in the general population, noting that one of the key variants of interest (p.Asp563Gly) in the Trivellin et al. study has a global MAF of 4.2% (gnomAD v4.0.0) and yet still exhibited significant reductions in both PHM and PAL activity (3).
The final set of PAM variants of interest was confirmed by manual inspection of raw sequencing data in Integrated Genomics Viewer (IGV). Pathogenicity predictions were made using in silico tools such as Combined Annotation Dependent Depletion (CADD) and Genomic Evolutionary Rate Profiling (GERP). Evolutionary conservation of the PAM variants was derived from protein sequence alignments of the following sequences from UniProt and Clustal Omega: human PAM-1 (P19021-5), chimpanzee PAM-1 (A0A2I3SM67-1), rat PAM-1 (P14925-1), Aplysia PAM-1 (Q9NJI4-1), Drosophila PHM (O01404-1) and PAL2 (Q9W1L5-1), and Chlamydomonas PAM (A0A0S2C767-1), as previously described (3). 3D models of WT and mutant PAM proteins were generated using HOPE, an automated program that analyses the structural and functional effects of point mutations using a range of information sources including calculations on the 3D coordinates of the protein by using WHAT IF Web services, sequence annotations from the UniProt database, predictions by DAS services, and the AlphaFold Protein Structure Database (AlphaFold DB, https://alphafold.ebi.ac.uk) (13–15).
We investigated potential PAM somatic variants in all patients as described above. In cases with a germline PAM variant, we also screened for loss of heterozygosity (LOH) by comparing variant frequencies in germline vs. tumour DNA.
To investigate a specific link between PAM variants and cyclical Cushing’s disease, we sought to identify cases of cyclical hypercortisolism amongst the previously described cohort published by Trivellin et al. (3). We hence examined clinical data from the subset of patients in the Trivellin et al. cohort derived from the National Institutes of Health (NIH). These patients were originally recruited by the NIH in accordance with the NIH research protocol, 97-CH-0076 (ClinicalTrials.gov: NCT00001595).
Amongst 29 patients with functioning pituitary adenomas, we identified 10 coding, non-synonymous single nucleotide variants in PAM that were uncommon (MAF <5.0%), of adequate quality, and not located in a low complexity region. Of these 10 variants, four were discarded due to low coverage (total depth <10X), and one (p.Glu491Asp) was not further studied due to a previous likely benign classification based on functional studies and other data (3).
The five remaining variants comprised four missense variants and one truncating variant as outlined in Table 1. The five variants of interest were found in seven individuals, all in the heterozygous state and confirmed by inspection of raw sequencing data. The variant was germline in six cases and somatic in the remaining case. Amongst the six germline cases, the variant was present in both the germline DNA and tumour DNA in five patients, whilst the tumour status was unknown in the remaining case as tumour DNA was unavailable. All five variants had a predicted or proven deleterious effect on protein function as detailed below. The final yield of uncommon, predicted deleterious PAM variants was thus 7/29 (24%). The variants predominated in individuals with thyrotrophinomas (4/10, 40%) and cyclical Cushing’s disease (2/7, 29%), compared to individuals with prolactinomas (1/12, 8%).
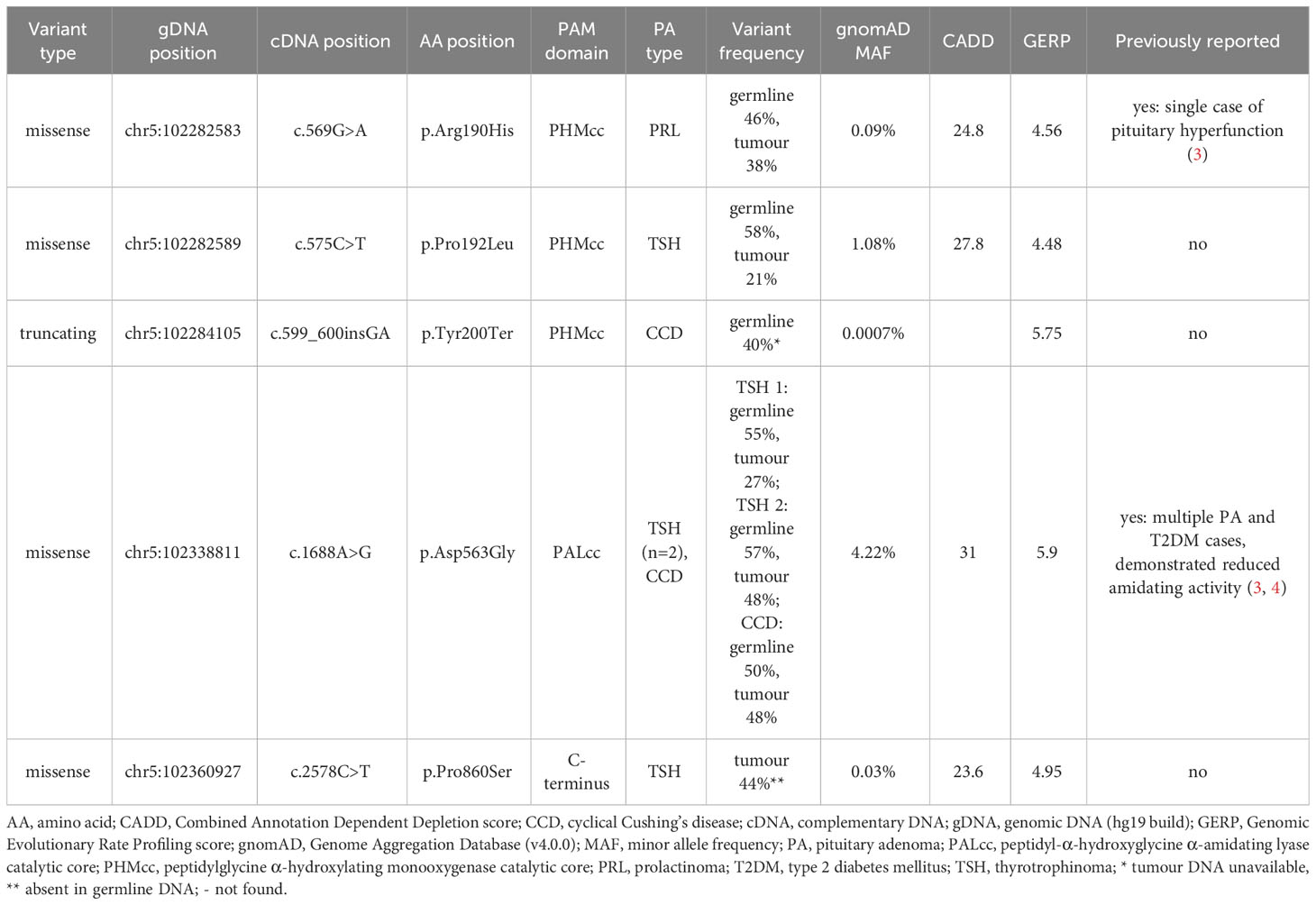
Table 1 Molecular characteristics of the five predicted deleterious PAM variants.
Characteristics of the individuals carrying the predicted deleterious PAM variants are provided in Table 2.
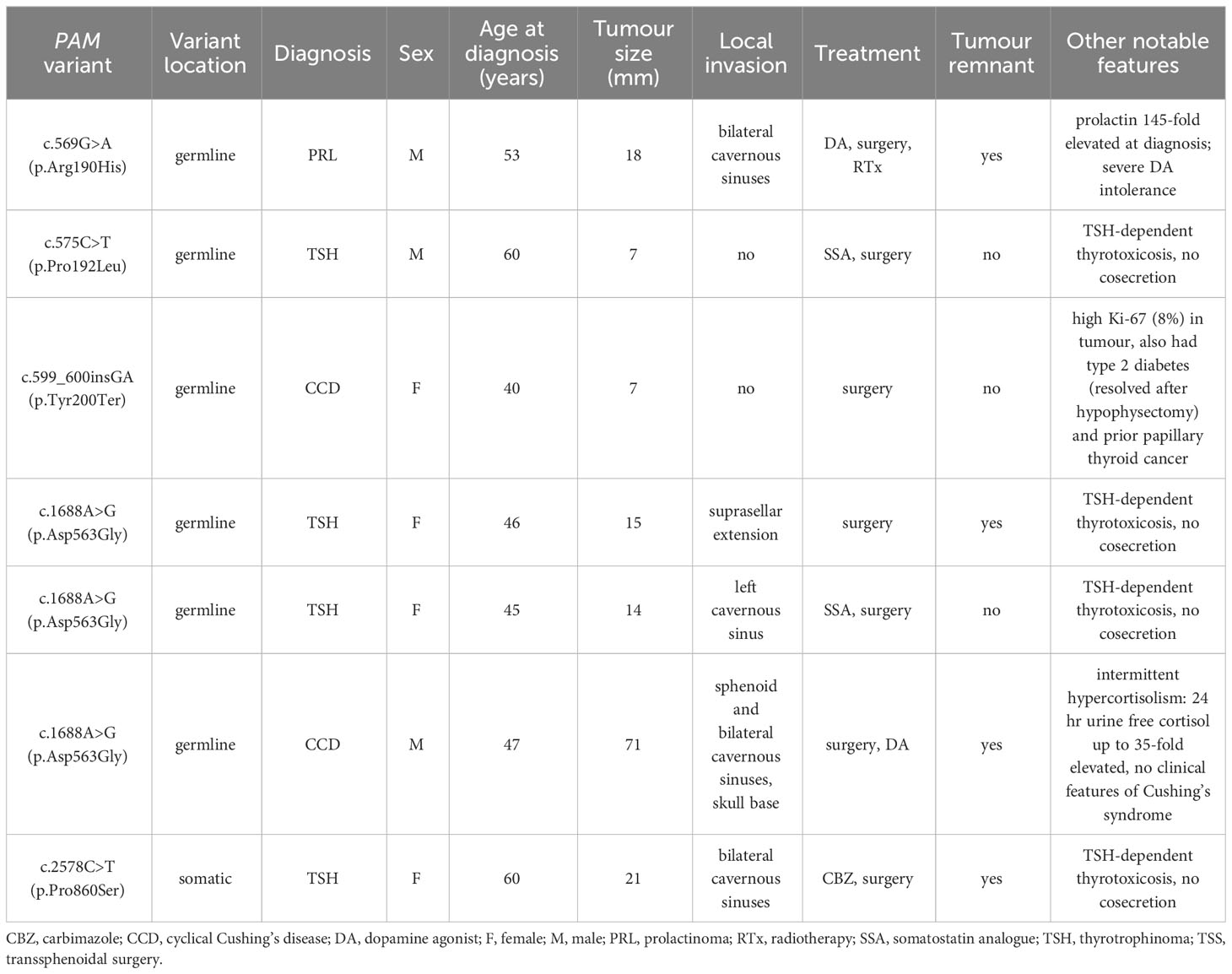
Table 2 Clinical characteristics of the seven patients with predicted deleterious PAM variants.
As shown in Table 1, all five PAM variants of interest had high GERP scores (>2). The four missense variants additionally had high CADD scores (>20). The truncating variant (p.Tyr200Ter) is a 2 bp frameshift insertion which is predicted to introduce a premature stop codon immediately at the same position and is likely to result in nonsense mediated decay as it is situated in exon 8.
Four of the five variants correspond to the critical catalytic domains of PAM as depicted in Figure 1. The p.Arg190His PHMcc variant has previously been reported in association with unspecified pituitary hyperfunction in the UKBB (3). The nearby p.Pro192Leu variant, also situated in the PHMcc, is located at the same residue as a variant of interest (p.Pro192Arg) in a UKBB case with a diagnosis of syndrome of inappropriate secretion of antidiuretic hormone (3), but it is relatively frequent in the general population with a maximum population MAF of 1.4% in Europeans. The truncating variant (p.Tyr200Ter) is also situated in the PHMcc. The p.Asp563Gly PALcc variant was the only variant found in multiple patients. This variant is relatively frequent in the general population, and was not significantly more frequent in our cohort (global gnomAD MAF 4.2% vs. 5.2%, p-value non-significant by 2-tailed Fisher’s exact test). However, p.Asp563Gly is one of the key PAM risk alleles in T2DM (4), and has been demonstrated repeatedly to cause significant reductions in total amidating activity, PHM activity and PAL activity (3, 4). The remaining variant (p.Pro860Ser) is situated adjacent to the transmembrane domain in the C-terminal end of PAM. Whilst this is a non-catalytic region, a nearby variant (p.Leu856Pro) was found in association with Cushing’s disease in UKBB (3), and the juxtamembrane region encompassing these variants has been shown in a rat model to influence pH-dependent aggregation of PAM in secretory granules (16).
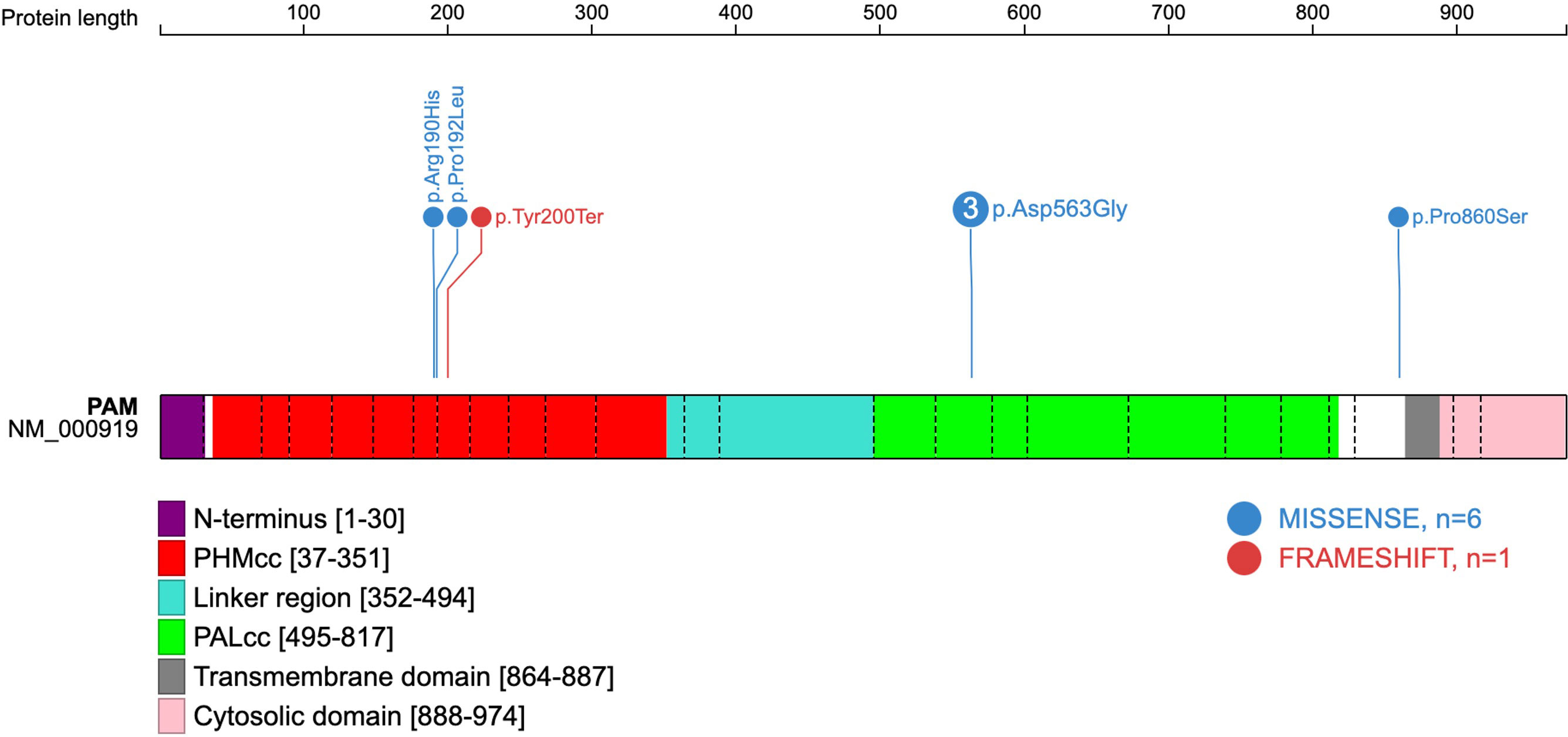
Figure 1 Schematic representation of the human PAM protein (PAM-1 isoform NM_000919.3). The domains have been previously deduced, and the schematic gene representation has been adapted from reference (3), with inclusion of the five predicted deleterious variants found amongst seven patients of the present cohort. Exon boundaries are indicated by dotted lines and codon numbers are shown. PHMcc, catalytic core of PHM; PALcc, catalytic core of PAL.
Although functional assays were not available to verify loss-of-function effects by the variants in our cohort, the missense variants were located in well-conserved positions with deleterious in silico predictions, and the truncating variant is a definitive null variant. Evolutionary conservation of the five PAM variants is demonstrated in Figure 2. The predicted crystal structures from the missense variants are shown in Figure 3.
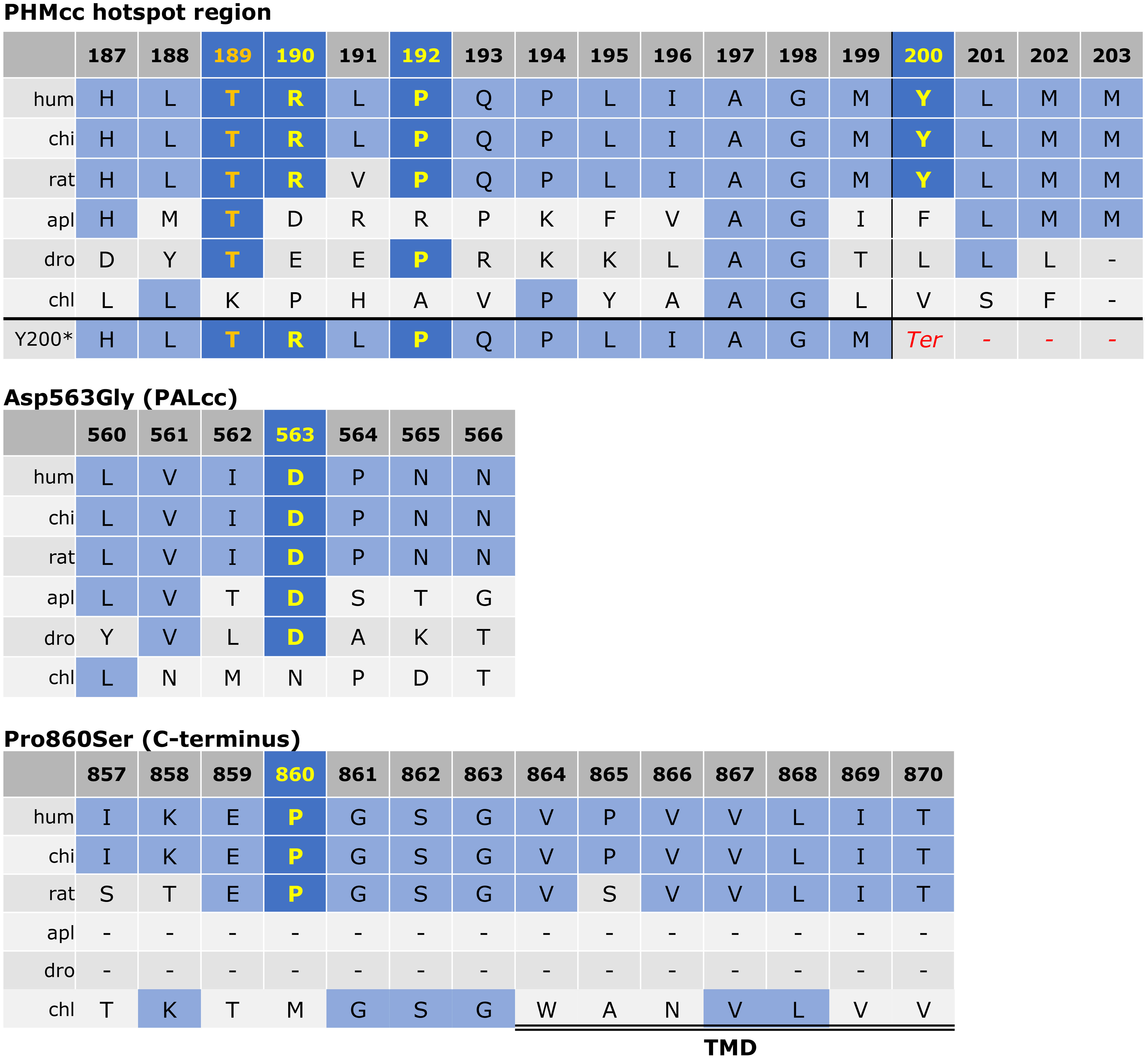
Figure 2 Protein sequence alignment for PAM variants of interest. As previously described (3), the alignment was derived via the Clustal Omega program with default settings and the UniProt alignment tool. The conserved residues of interest are shown in yellow. The Thr189 residue was used as an engineered inactive mutant by Trivellin et al. (3) and is shown in orange for reference. The other variants (p.Arg190His, p.Pro192Leu, p.Tyr200Ter, p.Asp563Gly, and p.Pro860Ser) were observed in our cohort. For the p.Tyr200Ter variant, the predicted frameshift is visible in the last row with introduction of a premature stop codon shown in red. The start of the transmembrane domain (TMD) adjacent to p.Pro860Ser is indicated by the double line. PHMcc, catalytic core of PHM; PALcc, catalytic core of PAL.
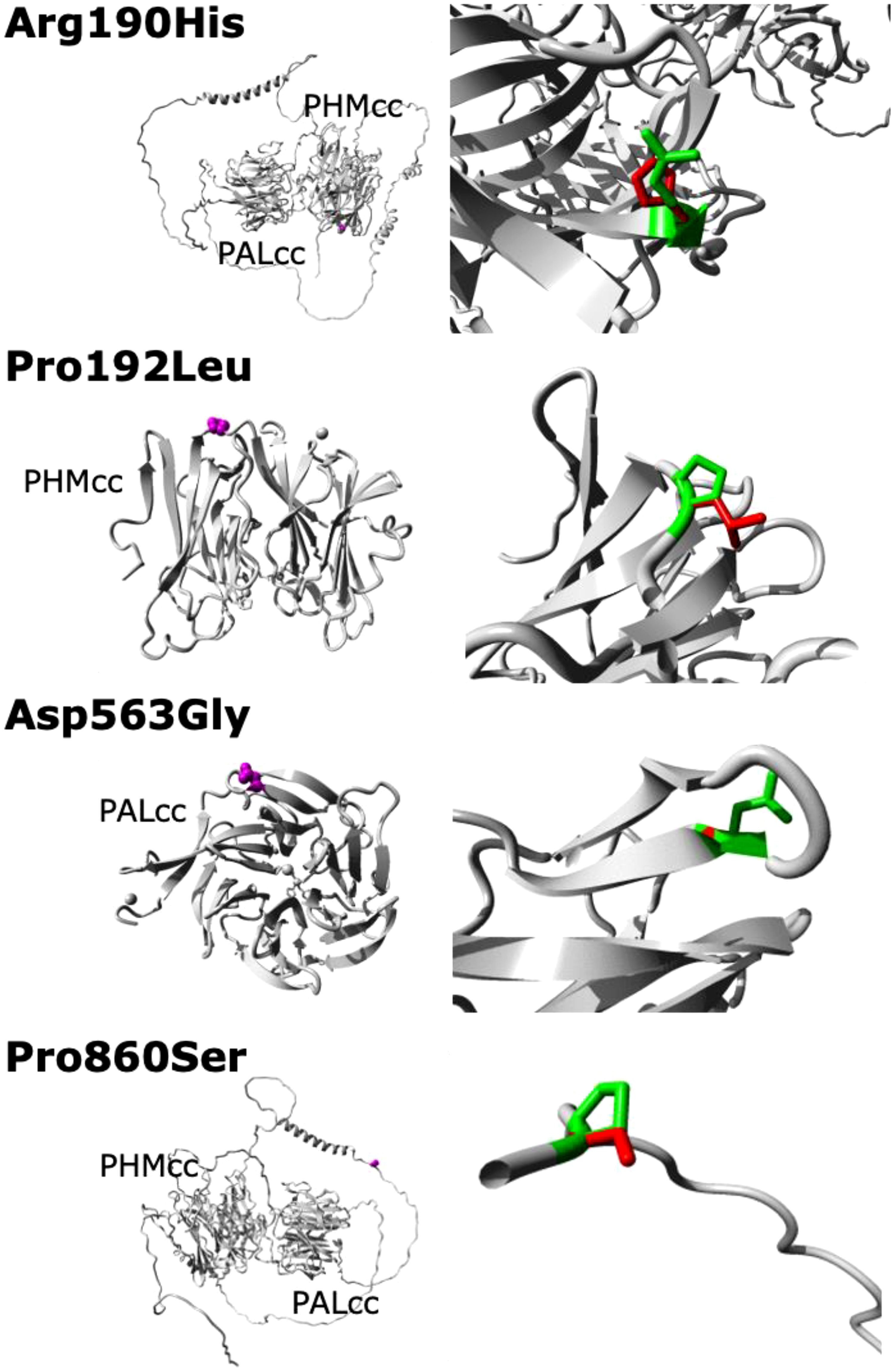
Figure 3 Predicted crystal structures of the WT and missense mutant PAM proteins. No structural data are available for full-length PAM; in particular, no structures are available for the linker region between PHMcc and PALcc or for the region between PALcc and the transmembrane domain (these regions are shown here as unfolded, except for two helical regions in the linker region). The left panels show an overview of the mutant proteins in ribbon-presentation, with the proteins coloured grey and the side chains of the mutated residues coloured magenta and shown as small spheres. The right panels show magnified views of the mutant resides, with the side chains of the WT and mutant residues coloured green and red, respectively. PHMcc, catalytic core of PHM; PALcc, catalytic core of PAL.
Tumour DNA was not available from the patient with the truncating PAM variant (cyclical Cushing’s disease). Another patient only exhibited their PAM variant in tumour DNA (thyrotrophinoma), whilst their germline DNA was WT at this allele. In the other five patients with predicted deleterious PAM variants, persistent heterozygosity of the variant of interest was observed in tumour DNA as shown in Table 1, thereby excluding LOH.
The NIH cohort comprised patients from around the world. Inclusion in the NIH protocol required patients to have a documented period of at least 6 months of consistent hypercortisolism in order to permit the subsequent NIH diagnostic testing algorithm. Thus, cyclical hypercortisolism with cycles of less than 6 months was specifically excluded by the study design. There was accordingly no evidence of cyclicity amongst patients with PAM variants and Cushing’s disease in the NIH cohort.
Germline genetic variation is traditionally considered to account for only 5% of pituitary adenomas (2). Herein we report predicted deleterious PAM variants in 7/29 (24%) individuals with functioning pituitary adenomas, with higher rates in the setting of thyrotrophinoma (4/10, 40%) and cyclical Cushing’s disease (2/7, 29%) compared to prolactinoma (1/12, 8%). The five observed PAM variants each exhibit some evidence of pathogenicity based on population prevalence, conservation scores, in silico predictions and/or prior functional studies. Our findings corroborate the seminal study by Trivellin et al. linking deleterious PAM variants with a variety of functioning pituitary adenomas, namely: somatotrophinoma, paediatric corticotrophinoma, and prolactinoma (3). We now demonstrate the first cases of PAM variants associated with cyclical Cushing’s disease and thyrotrophinomas. The discovery of PAM as a pituitary tumorigenesis gene is striking in its strong relationship with a mixture of pituitary adenoma subtypes across PIT1 and TPIT lineages.
Although the relationship between PAM and pituitary tumorigenesis is new, PAM has been intimately associated with pituitary function since discovery of the protein in 1982 as the porcine pituitary enzyme responsible for C-terminal amide formation and hence its critical involvement in the production of α-MSH from POMC (17). PAM is now known to be essential for the generation of all other amidated peptides, such as oxytocin and vasopressin, and other processes, including secretory granule biogenesis and retrograde signalling from the granule lumen to nucleus (5). C-terminal amidation is achieved by sequential activity of the two catalytic domains of PAM. Firstly, PHMcc functions as a monooxygenase with copper and ascorbate to α-hydroxylate the substrate (glycine-extended peptide) into an intermediate (hydroxyglycine-extended peptide). Subsequently, PALcc functions as a lyase to cleave the N-C bond of the intermediate and produce the final amidated product (5, 18). Supporting a mechanistic role for PAM variants in pituitary tumorigenesis, several rare variants were identified in the PHMcc and PALcc regions in our study and in the Trivellin et al. study cohort and their subsequent UKBB analysis (3). In the T2DM setting, the key PAM risk alleles (rs78408340, p.Ser539Trp; rs35658696, p.Asp563Gly) are also situated in the PALcc (4).
There appears to be a particular mutational hotspot within PHMcc involving the Arg190 and Pro192 residues that were mutated in our cohort and in the Trivellin et al. study, and the Thr189 residue which was used as an engineered inactive mutant by Trivellin et al. (6% PHM activity induced by Thr189Ile compared to WT) (3). This may be explained by the role of this region in copper reduction. This region of PHMcc (Arg190-Pro194) is the only direct connection of the N-terminus region of PHMcc to the C-terminus region of PHMcc. Communication between the N- and C-terminii of PHMcc is critical in allowing the transfer of copper between the two sites, which is in turn required for substrate hydroxylation (19). Hence, variants in this region might be expected to hinder essential conformational transitions and disrupt protein function.
Our finding of a PAM truncating variant (p.Tyr200Ter) in the setting of cyclical Cushing’s disease is particularly noteworthy as this is only the second truncating PAM variant to be associated with pituitary hypersecretion (3). Apart from PRKAR1A variants in the subset of cyclical Cushing’s syndrome related to primary pigmented nodular adrenocortical disease (20) and a case study raising a putative link between AHR and cyclical Cushing’s disease (9), there are currently no genes implicated in cyclical Cushing’s syndrome. The null nature of the PAM truncating variant in our patient with cyclical pituitary Cushing’s, and the existing data supporting a role between PAM variants and pituitary tumorigenesis, together favour a true mechanistic association between cyclical Cushing’s disease and the variant. In addition, another patient in our cohort with cyclical Cushing’s disease harboured the established loss-of-function Asp563Gly variant.
The single truncating PAM variant (p.His778fs) in the original study by Trivellin et al. also had Cushing’s disease (3). In light of our findings, this case and other NIH-recruited cases of Cushing’s disease with PAM variants in the original study were reviewed by the authors for hypercortisolism cyclicity. As patients in the NIH cohort were required to have a 6-month period of sustained hypercortisolism, none of the NIH cases had cyclical Cushing’s disease with cycles of less than 6 months duration, although we cannot exclude the possibility of cycles longer than 6 months duration. In any case, the carrier parent of the child with Cushing’s disease and the p.His778fs variant was found to have abnormal circadian rhythm of cortisol secretion with a high midnight serum cortisol despite being clinically unaffected (3).
How PAM variants might lead to cyclical Cushing’s disease and altered cortisol diurnal rhythm is yet to be elucidated. Since PAM is involved in the downstream amidation of ACTH leading to α-MSH synthesis (5, 17), and α-MSH exerts negative feedback on corticotrophin-releasing hormone (CRH) in rodents (21, 22), PAM could be considered to be an indirect negative regulator of ACTH activity. In addition, PAM might alter the timing of negative feedback on ACTH through its roles in secretory granule biogenesis and communication between secretory granules and the nucleus. Temporal disturbances in the hypothalamic-pituitary-adrenal (HPA) axis might also be an indirect consequence of the effect of PAM on a host of other amidated neuropeptides functioning in the hypothalamus and throughout the body, with synthesis of such amidated peptides expected to be reduced by deleterious PAM variants. Supporting the notion of PAM-mediated restraint of ACTH excess, there is a tendency towards greater PAM mRNA expression in silent corticotrophinomas compared to functioning corticotrophinomas (23). Taken together, PAM appears to be an indirect negative regulator of ACTH, and mutant PAM may thus lead to ACTH excess, but it remains unclear how mutant PAM might cause periodic disturbances in ACTH production, which is what would be expected if it were the underlying cause of cyclical hypercortisolism and abnormal cortisol diurnal rhythm. Future studies looking into whether PAM expression is light-entrained and exploring the temporal relationships between PAM and ACTH expression may help determine if PAM is a clock gene capable of influencing circadian and infradian ACTH variations.
Our study also demonstrates a novel association between PAM variants and thyrotrophinoma, with PAM variants found in 4/10 thyrotrophinoma patients. The variant was germline in three cases, with two of these cases harbouring the established loss-of-function p.Asp563Gly variant and the other harbouring the p.Pro192Leu variant situated in the PHMcc with high scores by CADD (27.8) and GERP (4.48) predicting a deleterious effect. The remaining variant was purely somatic, situated in the C-terminus and with a slightly lower CADD score of 23.6. If the latter variant is disregarded, the finding of PAM variants in 30% of thyrotrophinoma cases remains striking given the rarity of thyrotrophinomas and the lack of any known driver mutations for this pituitary adenoma subtype in the few genomic studies that have included thyrotrophinomas (24, 25).
The relatively high MAFs of some of the PAM variants in the pituitary adenoma setting coupled with the predicted or proven deleterious nature of some of these variants, suggest that PAM variants might act as pituitary adenoma risk alleles (26). This would be in keeping with the negative family histories in most pituitary adenoma cases carrying germline PAM variants (3). PAM variants have similarly been found to act as risk alleles for T2DM rather than causing monogenic diabetes (4). As pituitary tumours in general are highly prevalent (10% detection rate on MRI of healthy controls (27)) and infrequently familial (5% of all pituitary adenoma cases (2)), it is plausible that genetic predisposition to the condition relates more commonly to risk alleles than highly penetrant monogenic conditions. Even in the case of AIP variants, which are the most prevalent cause of familial pituitary tumours, the penetrance of pituitary adenomas is only 15-30% (2). Whether there are genotype-phenotype correlations amongst PAM variants in the pituitary setting remains to be seen. Truncating PAM variants may be specifically associated with altered temporal secretion of cortisol given that the only two reported truncating variants have been associated with loss of diurnal rhythm of cortisol secretion (3) and now with cyclical Cushing’s disease.
As in the study by Trivellin et al. (3), we did not observe LOH in the tumours of our patients with germline PAM variants. This may be explained by somatic second-hits that are not identifiable within the limits of the genetic testing methodologies used, such that PAM may still act as a tumour suppressor like other genes involved in pituitary tumorigenesis, such as MEN1 and AIP (2). Alternatively, PAM variants may cause pituitary tumorigenesis through haploinsufficiency. There are animal studies demonstrating the effects of PAM haploinsufficiency, with PAM+/- mice demonstrating altered levels of several amidated peptides (18).
The predicted deleterious PAM variants found in this Australian cohort complement the findings from the original cohort of patients from the Unites States and Europe (3). Independently demonstrating a relationship between gene variants and a disease in different cohorts is a critical component of the ClinGen Clinical Validity Classification system (28). These data may thus help confirm the gene-disease relationship between PAM and functioning pituitary adenomas. There are, however, limitations in our study. The study was initially designed to search for driver mutations and hence focused on sequencing of tumour-germline DNA pairs from individuals, without the additional ethical approval needed to sequence and phenotype family members which might have otherwise helped elucidate the role of the germline PAM variants found here. In addition, our study was relatively small, and employed only WES, which will identify most sequence variants and some copy number variation but miss other important genetic aberrations. Finally, although we have depicted the expected crystal structure changes induced by the missense PAM variants identified in our cohort, these predictions are based on incomplete data. Structural data do not exist for full-length PAM; in particular, we have had to include low probability data for the linker and cytosolic regions of PAM.
We anticipate that further independent pituitary adenoma cohorts will be examined for PAM variants, which will help to confirm the proposed gene-disease relationship, and hopefully solve unanswered questions, such as whether PAM is a traditional tumour suppressor or rather an haploinsufficient gene, and precisely how PAM inactivation results in pituitary hormone hypersecretion. Future directions of research should include PAM sequencing in independent cohorts of thyrotrophinomas, and further exploration of the relationship between PAM and POMC, including temporal effects, to determine how PAM variants might produce cyclical Cushing’s disease and abnormal cortisol diurnal rhythm.
The datasets presented in this article are not readily available because of patient confidentiality. Requests to access the datasets should be directed to A/Prof Sunita De Sousa.
The studies involving humans were approved by Melbourne Health: HREC/16/MH/132; Royal Adelaide Hospital: SSA/18/CALHN/445. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
SD: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing. AS: Conceptualization, Funding acquisition, Investigation, Writing – review & editing. CY: Conceptualization, Funding acquisition, Writing – review & editing. RC-B: Investigation, Writing – review & editing. SS: Investigation, Writing – review & editing. JK: Investigation, Writing – review & editing. JT: Data curation, Formal Analysis, Writing – review & editing. GT: Data curation, Writing – review & editing. AL: Writing – review & editing. CS: Data curation, Writing – review & editing. DT: Conceptualization, Investigation, Supervision, Writing – review & editing. HS: Conceptualization, Methodology, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. SD is supported by the Royal Adelaide Hospital Mary Overton Early Career Research Fellowship, the Royal Australasian College of Physicians Fellows Research Establishment Fellowship, and the Endocrine Society of Australia Postdoctoral Award. Some of the original DNA sequencing was undertaken with support from a Royal Adelaide Hospital Health Services Charitable Gifts Board grant. CS is funded by the Foundation of Research & Technology Hellas (FORTH).
We thank Jinghua Feng, Paul Wang, Andreas Schreiber, David Lawrence, Milena Babic, Rosalie Kenyon and other staff at SA Pathology for technical support in DNA extraction and sequencing and bioinformatic analysis. We are also indebted to Prof Betty A. Eipper and Prof Richard E. Mains for their valuable manuscript suggestions.
CS and GT are named in a patent involving the GPR101 molecule and its function. CS is a paid consultant for ELPEN, Lundbeck and Sterotherapeutics pharmaceutical companies and holds additional patents on the PRKAR1A and PDE11A molecules and their function. All other authors declare no competing interests.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO classification of pituitary tumors. Endocr Pathol (2022) 33(1):6–26. doi: 10.1007/s12022-022-09703-7
2. De Sousa SMC, Lenders NF, Lamb LS, Inder WJ, McCormack A. Pituitary tumours: molecular and genetic aspects. J Endocrinol (2023) 257(3):e220291. doi: 10.1530/JOE-22-0291
3. Trivellin G, Daly AF, Hernández-Ramírez LC, Araldi E, Tatsi C, Dale RK, et al. Germline loss-of-function PAM variants are enriched in subjects with pituitary hypersecretion. Front Endocrinol (Lausanne). (2023) 14:1166076. doi: 10.3389/fendo.2023.1166076
4. Thomsen SK, Raimondo A, Hastoy B, Sengupta S, Dai XQ, Bautista A, et al. Type 2 diabetes risk alleles in PAM impact insulin release from human pancreatic β-cells. Nat Genet (2018) 50(8):1122–31. doi: 10.1038/s41588-018-0173-1
5. Kumar D, Mains RE, Eipper BA. 60 YEARS OF POMC: From POMC and α-MSH to PAM, molecular oxygen, copper, and vitamin C. J Mol Endocrinol (2016) 56(4):T63–76. doi: 10.1530/JME-15-0266
6. Steel JH, Martínez A, Springall DR, Treston AM, Cuttitta F, Polak JM. Peptidylglycine alpha-amidating monooxygenase (PAM) immunoreactivity and messenger RNA in human pituitary and increased expression in pituitary tumours. Cell Tissue Res (1994) 276(1):197–207. doi: 10.1007/BF00354800
7. Berger AH, Knudson AG, Pandolfi PP. A continuum model for tumour suppression. Nature (2011) 476(7359):163–9. doi: 10.1038/nature10275
8. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing's syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab (2008) 93(5):1526–40. doi: 10.1210/jc.2008-0125
9. De Sousa SMC, Manavis J, Feng J, Wang P, Schreiber AW, Scott HS, et al. A putative role for the aryl hydrocarbon receptor (AHR) gene in a patient with cyclical Cushing's disease. BMC endocrine Disord (2020) 20(1):18–. doi: 10.1186/s12902-020-0495-8
10. De Sousa SMC, Wang PPS, Santoreneos S, Shen A, Yates CJ, Babic M, et al. The genomic landscape of sporadic prolactinomas. Endocrine pathology. (2019) 30(4):318–28. doi: 10.1007/s12022-019-09587-0
11. Shen A, Wang P, De Sousa SMC, Torpy DJ, Scott H, King J, et al. MON-LB48 the genomic landscape of sporadic thyrotrophinomas. J Endocr Soc (2020) 4(Suppl 1):MON-LB48. doi: 10.1210/jendso/bvaa046.2282
12. Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinf (2013) 43:11.0.1–33. doi: 10.1002/0471250953.bi1110s43
13. Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinf (2010) 11:548. doi: 10.1186/1471-2105-11-548
14. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res (2022) 50(D1):D439–d44. doi: 10.1093/nar/gkab1061
15. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021) 596(7873):583–9. doi: 10.1038/s41586-021-03819-2
16. Bell-Parikh LC, Eipper BA, Mains RE. Response of an integral granule membrane protein to changes in pH. J Biol Chem (2001) 276(32):29854–63. doi: 10.1074/jbc.M103936200
17. Bradbury AF, Finnie MD, Smyth DG. Mechanism of C-terminal amide formation by pituitary enzymes. Nature (1982) 298(5875):686–8. doi: 10.1038/298686a0
18. Yin P, Bousquet-Moore D, Annangudi SP, Southey BR, Mains RE, Eipper BA, et al. Probing the production of amidated peptides following genetic and dietary copper manipulations. PloS One (2011) 6(12):e28679. doi: 10.1371/journal.pone.0028679
19. Prigge ST, Mains RE, Eipper BA, Amzel LM. New insights into copper monooxygenases and peptide amidation: structure, mechanism and function. Cell Mol Life Sci (2000) 57(8-9):1236–59. doi: 10.1007/PL00000763
20. Powell AC, Stratakis CA, Patronas NJ, Steinberg SM, Batista D, Alexander HR, et al. Operative management of Cushing syndrome secondary to micronodular adrenal hyperplasia. Surgery (2008) 143(6):750–8. doi: 10.1016/j.surg.2008.03.022
21. Tozawa F, Suda T, Dobashi I, Ohmori N, Kasagi Y, Demura H. Central administration of alpha-melanocyte-stimulating hormone inhibits corticotropin-releasing factor release in adrenalectomized rats. Neurosci Lett (1994) 174(1):117–9. doi: 10.1016/0304-3940(94)90133-3
22. Botte DA, Noronha IL, Malheiros DM, Peixoto TV, de Mello SB. Alpha-melanocyte stimulating hormone ameliorates disease activity in an induced murine lupus-like model. Clin Exp Immunol (2014) 177(2):381–90. doi: 10.1111/cei.12336
23. García-Martínez A, Cano DA, Flores-Martínez A, Gil J, Puig-Domingo M, Webb SM, et al. Why don't corticotroph tumors always produce Cushing's disease? Eur J Endocrinol (2019) 181(3):351–61. doi: 10.1530/EJE-19-0338
24. Sapkota S, Horiguchi K, Tosaka M, Yamada S, Yamada M. Whole-exome sequencing study of thyrotropin-secreting pituitary adenomas. J Clin Endocrinol Metab (2017) 102(2):566–75. doi: 10.1210/jc.2016-2261
25. Song ZJ, Reitman ZJ, Ma ZY, Chen JH, Zhang QL, Shou XF, et al. The genome-wide mutational landscape of pituitary adenomas. Cell Res (2016) 26(11):1255–9. doi: 10.1038/cr.2016.114
26. Lebo M, Steeves M, Benson K, Conlin L, Ganapathi M, Jobanputra V, et al. O31: Risk allele evidence curation, classification, and reporting: Recommendations from the ClinGen Low Penetrance/Risk Allele Working Group*. Genet Med Open (2023) 1(1):100457. doi: 10.1016/j.gimo.2023.100457
27. Hall WA, Luciano MG, Doppman JL, Patronas NJ, Oldfield EH. Pituitary magnetic resonance imaging in normal human volunteers: occult adenomas in the general population. Ann Internal Med (1994) 120(10):817–20. doi: 10.7326/0003-4819-120-10-199405150-00001
Keywords: peptidylglycine α-amidating monooxygenase, whole exome sequencing, Cushing’s disease, prolactinoma, thyrotrophinoma, pituitary adenomas
Citation: De Sousa SMC, Shen A, Yates CJ, Clifton-Bligh R, Santoreneos S, King J, Toubia J, Trivellin G, Lania AG, Stratakis CA, Torpy DJ and Scott HS (2023) PAM variants in patients with thyrotrophinomas, cyclical Cushing’s disease and prolactinomas. Front. Endocrinol. 14:1305606. doi: 10.3389/fendo.2023.1305606
Received: 02 October 2023; Accepted: 06 November 2023;
Published: 23 November 2023.
Edited by:
Hidenori Fukuoka, Kobe University, JapanReviewed by:
Ning-Ai Liu, Cedars Sinai Medical Center, United StatesCopyright © 2023 De Sousa, Shen, Yates, Clifton-Bligh, Santoreneos, King, Toubia, Trivellin, Lania, Stratakis, Torpy and Scott. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sunita M. C. De Sousa, U3VuaXRhLkRlU291c2FAc2EuZ292LmF1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.