 Yifan Zhang
Yifan Zhang Jun Ma2
Jun Ma2 Min Hu
Min Hu Xiaoxi Wei
Xiaoxi Wei- 1Department of Orthodontics, Hospital of Stomatology Jilin University, Changchun, Jilin, China
- 2Department of Oral Anatomy and Physiology, Hospital of Stomatology Jilin University, Changchun, Jilin, China
Bone homeostasis, depending on the balance between bone formation and bone resorption, is responsible for maintaining the proper structure and function of the skeletal system. As an important group of transcription factors, retinoic acid receptor-related orphan receptors (RORs) have been reported to play important roles in bone homeostasis by regulating the transcription of target genes in skeletal cells. On the other hand, the dysregulation of RORs often leads to various skeletal diseases such as osteoporosis, rheumatoid arthritis (RA), and osteoarthritis (OA). Herein, we summarized the roles and mechanisms of RORs in skeletal diseases, aiming to provide evidence for potential therapeutic strategies.
1 Introduction
Bone homeostasis is an essential dynamic process of bone modeling and remodeling (1), which requires accurate regulation by a variety of transcription factors as well as signaling pathways (2, 3). Retinoic acid receptor-related orphan receptors (RORs) are ligand-dependent transcription factors and can regulate osteogenesis and osteoclastogenesis in bone metabolic disorders such as osteoporosis (4). In addition, the RORs, particularly RORγ, mediate the inflammatory response and lead to bone destruction in inflammatory bone diseases such as rheumatoid arthritis (RA) and osteoarthritis (OA) (5). In this review, we discussed the effect of RORs on maintaining bone homeostasis and summarized prospective therapeutic strategies targeting RORs for common bone diseases.
2 The RORs
RORs are a subgroup of the thyroid hormone receptor and belong to the orphan nuclear receptors (NRs) (6). The RORs contain three members: RORα (same as RORA, NR1F1), RORβ (same as RORB, NR1F2), and RORγ (same as RORC, NR1F3). It has been reported that each member has several isoforms that differ only at their N-terminus, and these isoforms are expressed in different mammalian species. Specifically, RORα contains RORα1–4 in humans, but only RORα1 and RORα4 are expressed in mice. Both RORβ and RORγ generate two isoforms (RORβ1/RORβ2 and RORγ1/RORγ2, RORγ2 is also known as RORγt) in humans and mice (7) (Figure 1A).
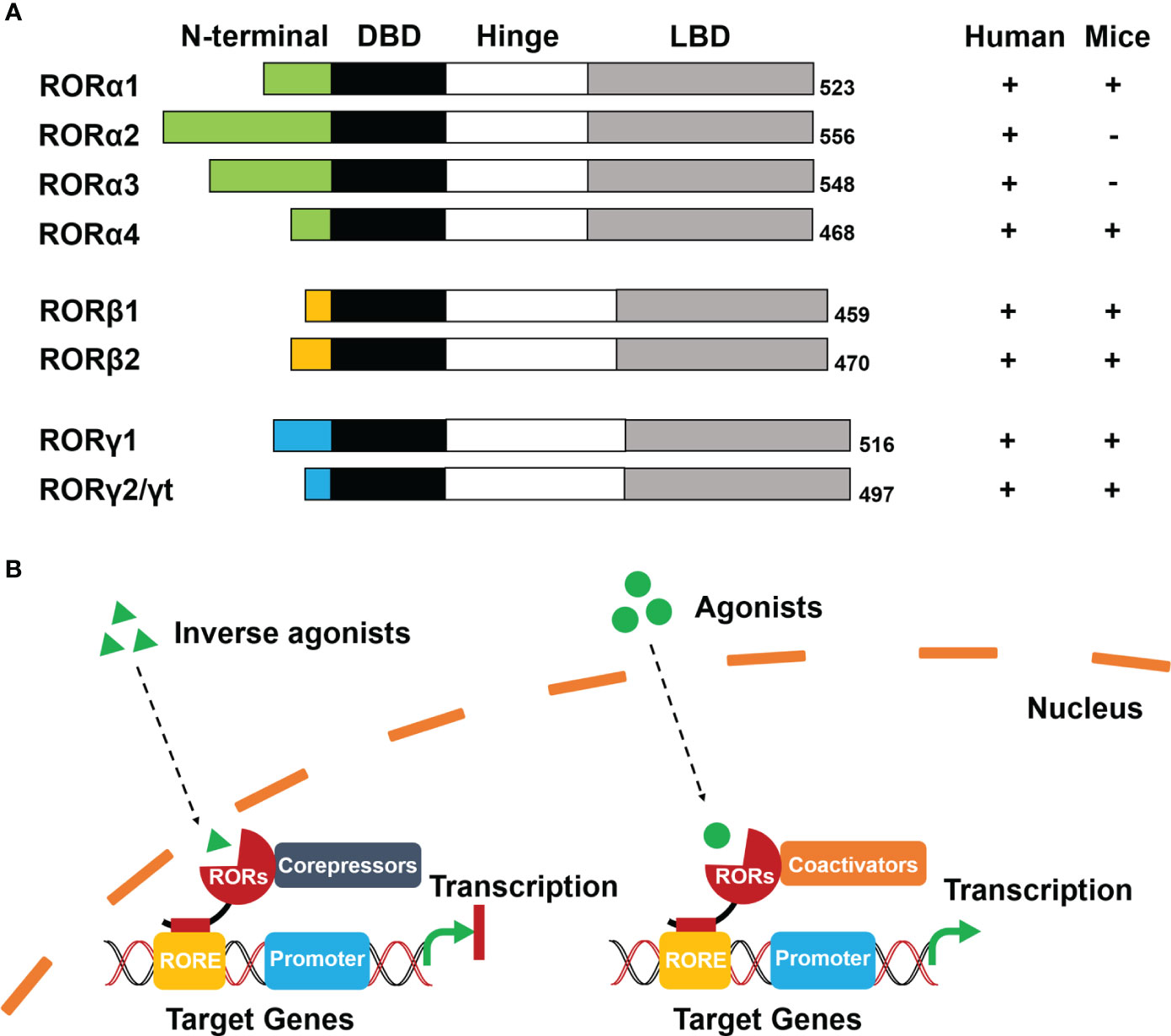
Figure 1 Schematic diagram of structure and ligand-dependent mechanism of RORs. (A) Structure: Four functional domains constitute RORs, including a distinct N-terminal domain, a highly conserved DNA-binding domain (DBD), a ligand-binding domain (LBD), and a hinge domain. The numbers at the tail represent the total number of encoded amino acids in the corresponding ROR isoforms. (B) Mechanism: Inverse agonists trans-activate RORs to recruit co-repressors and inhibit transcription of target genes (left). Agonists activate RORs to recruit co-activators and initiate transcription of target genes (right).
The modular structure of RORs is typical of NRs and consists of four functional domains: a distinctive N-terminal domain, a highly conserved DNA-binding domain (DBD), a ligand-binding domain (LBD), and a hinge domain (Figure 1A). Differences in sequence of the N-terminal domain determine the isoforms of RORs. ROR response elements (ROREs) have an AGGTCA consensus motif with an A/T-rich region. With two highly conserved zinc finger motifs, The DBD is responsible for recognizing the AGGTCA consensus region in ROREs as a monomer. The LBD is the most important functional domain that is crucial for ligand-binding and facilitates the recruitment of cofactors in transcriptional regulation. The hinge domain linking DBD with LBD stabilizes the RORs’ protein structure (8).
Even though evidence has indicated that RORs can be self-activated without any ligands or other potential stimulators, RORs transcriptional regulation is primarily ligand-dependent (6, 7). Cholesterol and its metabolites were firstly identified as natural ligands by crystallography studies (8). Melatonin was once considered as an endogenous ligand of RORβ. Although it could bind to RORβ directly, the result was not reproducible and therefore has been withdrawn (9). Apart from natural ligands, synthetic ligands have made significant progress (10). According to conformational changes of RORs, the ligands include agonists, antagonists, and inverse agonists (11).
RORs interact with co-repressors as well as co-activators and function as repressors or activators of gene transcription respectively. When inverse agonists enter the nucleus, RORs can be trans-activated and recruit co-repressors, such as the nuclear receptor co-repressor (NCoR) and the silencing mediator for retinoid and thyroid hormone receptor (SMRT) (4). The co-repressor complexes then combine with the RORE of target genes that subsequently prevent the promoter initiation and suppress the transcription. In contrast, agonists activate RORs with conformational changes resulting in the dissociation of co-repressor complexes and the recruitment of co-activators, such as the p160 steroid receptor co-activator (SRC) family, p300, and BRG2 complex (6, 12). The formation of co-activator complexes eventually facilitates transcriptional regulation by enhancing the promoter of target genes (Figure 1B).
The RORs serve functions in several physiological processes such as cell division and differentiation, circadian rhythm, metabolism, and immune regulation (12). ROR family members and their isoforms exhibit a distinct tissue-specific pattern of expression and play different roles in pathological processes. Specifically, RORα is widely distributed in multiple tissues, including the liver, skeletal muscle, skin, and adipose, which is mainly associated with circadian rhythm abnormalities, tumorigenesis, and metabolic diseases. In addition, RORα is highly expressed in the pre-hypertrophic and hypertrophic chondrocytes of the growth plate, suggesting that RORα may be a regulator of hypertrophic differentiation of chondrocytes and contribute to endochondral ossification (13, 14). RORβ is expressed restrictedly in the brain, retina, bone, and pineal gland, and is essential for retinal cell survival and circadian rhythm regulation (4). Moreover, the two isoforms of RORγ differ in the expression with distinct N-terminus. RORγ1 is expressed in various tissues such as skeletal muscle tissue, liver, and kidney, while RORγ2 (same as RORγt) is exclusively expressed in immune cells and plays prominent roles in thymocyte development (12).
3 Role of RORs in skeletal diseases
Skeletal diseases, such as osteoporosis, rheumatoid arthritis, and osteoarthritis, significantly impair individuals’ health and quality of life. Patients may suffer from long-term pain, limitation of movement, and hypofunction. Studies have found that RORs play an important role in bone metabolism and maintenance of bone homeostasis. Interestingly, ROR family members play different roles in regulating osteogenesis. Most studies believe that RORα mainly promotes osteogenesis. On the contrary, RORβ inhibits osteogenic differentiation. RORγ regulates bone balance by participating in the inflammatory response and stimulating osteoclastic differentiation. Herein, we summarize the role of RORs in common skeletal diseases.
3.1 Osteoporosis
Osteoporosis is one of the most common bone metabolic disorders, characterized by bone loss and microstructural changes, which in turn increase the risk of fractures (15, 16). Osteoporosis occurs when the rate of bone resorption consistently exceeds the rate of bone formation. RORs affect the occurrence and progression of osteoporosis by regulating the proliferation and differentiation of skeletal cells and multiple osteogenic signaling pathways.
3.1.1 RORα in osteoporosis
Studies have shown that RORα positively regulates osteogenic differentiation in vitro and in vivo. Spontaneous mutations of the Rorα gene were identified in a staggered (sg) mouse strain. Homozygous staggerer (sg/sg) mice showed limited skeletal development with a reduction in long bone thickness and bone mineral density (17, 18). Application of cholesterol sulfate, a RORα agonist, prevented bone mass loss in the inflammation-mediated or ovariectomy (OVX) -mediated osteoporosis models (19). Studies in goats have also confirmed a substantial association between RORα and growth traits including height and bone length (20). In vitro evidence suggested that RORα was highly expressed in human mesenchymal stem cells (hMSCs) and that the expression level increased with osteogenic differentiation (17, 21).
Several upstream signaling pathways are mediated by RORα in osteogenic differentiation (Figure 2A). (1) Circadian clock system: Brain and muscle ARNT-like protein 1 (Bmal1) knockout mice exhibited osteoporosis (22). Further studies found that BMAL1 blocked the Wnt/β-catenin signaling pathway by inhibiting the expression of RORα, thereby preventing the osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs) (23). (2) Oxidative stress: It has been reported that oxidative stress is a key trigger of osteoporosis. OSGIN2, an inducer of oxidative stress, prevented osteogenic differentiation of BMSCs by inhibiting RORα expression (24, 25). (3) DNA methylation: Histone methylation has been recognized as an important modulator in the osteogenic differentiation of MSCs (26). RORα mediated DNA methyltransferase 1 (DNMT1) to control the DNA methylation of MSCs, thereby suppressing chondrogenic and osteogenic differentiation (27). (4) Estrogen: Estrogen plays a significant role in regulating bone metabolism (28). Estradiol is one of the Estrogen’s natural products, which could activate RORα and promote osteoblast differentiation (29).
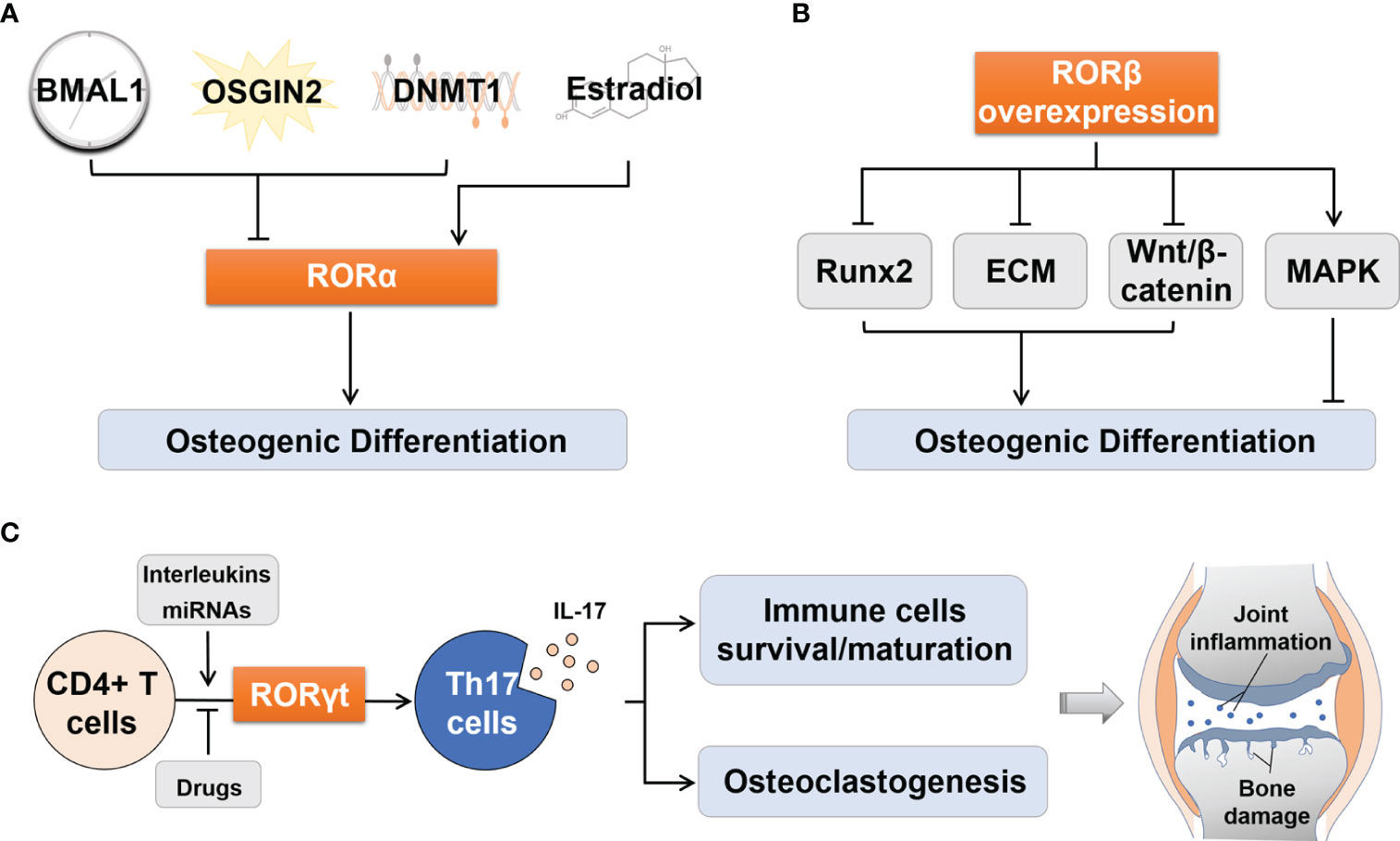
Figure 2 Regulation of osteogenic differentiation by RORα and RORβ and of Th17 cell differentiation by RORγt. (A) Upstream effectors including BMAL1, OSGIN2 and DNMT1 could suppressed osteogenic differentiation by inhibiting RORα, while Estradiol activated RORα to promote osteogenic differentiation. (B) RORβ overexpression inhibited osteogenic differentiation by inhibiting Runx2 transcription, disrupting ECM synthesis, inhibiting Wnt/β-catenin pathway, and promoting proliferation through the MAPK pathway. (C) RORγt regulates the process of differentiation from CD4+ T cells to Th17 cells, and the consequent IL-17 secretion. The latter can influence the survival and maturation of immune cells and osteoclastogenesis, which thus aggravates joint synovial inflammation and bone damage in RA patients. BMAL1, Brain and muscle ARNT-like protein 1; DNMT1, DNA methyltransferase 1; Runx2, Runt-related transcription factor 2; ECM, Extracellular matrix; MAPK, Mitogen-activated protein kinase; Th17, T helper 17; IL-17, Interleukin 17; miRNAs, MicroRNAs.
There are several main mechanisms underlying the downstream pathway of RORα. One is directly promoting the transcription of osteogenic differentiation marker genes, such as alkaline phosphatase (ALP), collagen type I (COL I), bone sialoprotein (Bsp), and dentin matrix protein 1 (DMP1) (17, 21, 30). The second is activating osteogenic differentiation signaling pathways, such as Wnt/β-catenin and BMP2/Smad1/5/9 pathways (17, 29). Furthermore, RORα can associate with a variety of cell types and influence the microenvironment directly or indirectly. For instance, in the diabetic bone defect model, underexpression of RORα in macrophages inhibited the transcription of serum C-C motif chemokine 3 (Ccl3) and interleukin-6 (Il-6), which led to the inability of macrophages to convey migration and aggregation signals to BMSCs, ultimately leading to diabetic bone aplasia (31).
However, it has also been reported that RORα negatively regulates osteogenic differentiation. For example, transient overexpression of RORα suppressed osteocalcin (Ocn) transcription induced by vitamin D3 and consequently inhibited osteogenesis (17). In addition, bisphenol A (BPA), a ROR agonist, could promote bone resorption (32). The controversial results may be due to the time and extent of RORα expression in two different ways of both bone formation and resorption in bone metabolism.
3.1.2 RORβ in osteoporosis
In contrast to RORα, RORβ negatively regulates osteogenic differentiation and promotes the progression of postmenopausal or age-related osteoporosis. Deletion of the Rorβ gene could prevent bone loss in aged mice (33). RORβ expression was significantly increased with age in the BMSCs from postmenopausal women, which was consistent with the mouse models of age-related osteoporosis (34, 35). Data on overexpression or knockdown of RORβ in osteoblastic MC3T3-E1 cells also suggested its negative effect on osteogenic differentiation (33, 36).
The following mechanisms may explain the negative effects of RORβ on osteogenic differentiation (Figure 2B). (1) Runt-related transcription factor 2 (Runx2) is an essential transcription factor that drives the phenotypic expression of osteoblasts. Its absence can lead to a complete loss of ossifying capacity (37). When the expression of RORβ was up-regulated in MC3T3-E1 cells, transcription of Runx2 and its target genes Ocn and osterix (Osx) were significantly inhibited (34). (2) Transforming growth factor β (TGF-β) and bone morphogenetic protein (BMP) control the formation of extracellular matrix (ECM) and provide a framework for bone mineral deposition (38). Studies have shown that overexpression of RORβ upregulated the expression of these cytokines and then interfered with ECM synthesis, thereby inhibiting bone mineralization (36). (3) RORβ down-regulated the Wnt/β-catenin signaling pathway by inhibiting the activities of Wnt downstream target genes transcription factor 7 (Tcf7) and osteoprotegerin (Opg), which disrupted the balance between bone formation and bone resorption, leading to bone loss (33). (4) RORβ could stimulate osteoblast proliferation through the mitogen-activated protein kinase (MAPK) pathway and prevent osteoblasts from exiting the cell cycle, thereby delaying osteogenic differentiation (36). In addition, RORβ has been identified as a molecular target of miR-219a-5p in recent years, which is involved in the development of osteoporosis (39).
3.1.3 RORγ in osteoporosis
Most studies on the effect of RORγ on osteoporosis were limited to RORγ2 (RORγt). The main mechanism is to act as a specific transcription factor of immune cells, especially T helper cell 17 (Th17), and mediate the differentiation of Th17 cells to secret interleukin 17 (IL-17), thereby enhancing inflammatory response (5). In recent years, it has been found that T cell-mediated inflammation plays an important role in the progression of osteoporosis (40, 41). In human and mouse models, RORγt affected bone loss in postmenopausal osteoporosis caused by estrogen deficiency (42, 43). The production of IL-17, mediated by RORγt, directly stimulated osteoclastogenesis by upregulating the expression of RANKL (44, 45). IL-27 inhibited the differentiation of Th17 cells in OVX mice by inhibiting the transcription factor RORγt (46). In addition, as one of the areas of scientific interest in recent years, intestinal flora plays a key role in regulating bone homeostasis through the gut-bone axis (47). The treatment of Lactobacillus rhamnosus GG (LGG) stimulated Th17 cell differentiation with the alteration of RORγt expression, and had an advantage in osteogenic promotion, which may account for the amelioration of osteoporosis in OVX model rats (48, 49). Another popular probiotic, Bifidobacterium longum (BL), appeared its immunomodulatory potential to the Th17 cell differentiation that suppressed osteoclastogenesis in postmenopausal osteoporosis (50).
3.2 Rheumatoid arthritis
Rheumatoid arthritis is a systemic autoimmune disease characterized by initial synovial inflammation followed by cartilage degeneration and subchondral bone destruction (51, 52). The promotion of osteoclast formation and differentiation is the main reason for bone erosion (53). Immune cells, especially Th17 cells, are activated and concentrated in the inflamed synovium, contributing to persistent joint inflammation and bone erosion in RA (54). Among RORs, many studies indicated the roles of RORα and RORγt in inflammatory diseases like RA and OA, whereas little has been done on RORβ.
3.2.1 RORα in rheumatoid arthritis
RORα is involved in the inflammatory response stage of RA and the regulation of osteoclast activity in bone destruction. RORα is expressed in Th17 cells and the expression is elevated in collagen-induced arthritis (CIA) mice (55). RORα can act as a transcription factor to mediate the differentiation of Th17 cells. The expression of RORα and RORγt together promoted the differentiation of Th17 and significantly up-regulated the expression of IL-17, thereby promoting inflammatory response (56). However, some studies have also reported that RORα is a negative regulator of inflammatory response. Compared with controls, the expression level of RORα in osteoblasts of RA patients was lower (30). The RORα agonist, cholesterol sulfate, could reduce joint inflammation and bone destruction in CIA mice by inhibiting RORγt expression and Th17 cell differentiation (57). Differences in the disease itself and the immune microenvironment determine the cellular response to any stimulus, which may help to partially explain the different mechanisms by which RORα regulates inflammation in Th17 cell differentiation. In addition, RORα is involved in the process of bone resorption in RA. RORα agonists prevented osteoclastogenesis and induced osteoclast apoptosis in CIA mice (57). However, direct depletion of Rorα did not affect the agonist-ligand-mediated inhibition of osteoclast differentiation (19). This suggested that RORα agonists might be involved in RA osteoclast survival and differentiation in a RORα-independent manner.
3.2.2 RORγ in rheumatoid arthritis
RORγt, as a lineage-specific transcription factor of Th17 cells, regulates the differentiation of CD4+ T cells into Th17 cells, then promotes the production of IL-17, and finally aggravates joint inflammation and bone destruction in RA (58) (Figure 2C). In RA patients, the aggravation of inflammation is closely related to the increase in the proportion of Th17 cells and the expression levels of RORγt and IL-17 (5, 59).
Several upstream signals including cytokines, RA drugs, and microRNAs (miRNAs), can regulate RORγt expression and Th17 cell differentiation. Specifically, pro-inflammatory factors, such as IL-6 (60), IL-23 (61), and tumor necrosis factor-α (TNF-α) (62), transmitted inflammatory signals to Th17 cells. Some natural and synthetic RA drugs, such as digoxin and pioglitazone, reduced the expression levels of RORγt and IL-17, thereby alleviating inflammation and preventing further bone destruction (63–67). miRNAs regulating RORγt expression, such as miRNA-16 (68), miRNA-301a-3p (69), miRNA-34a (70), and miRNA-146a (71, 72), were also involved in RORγt-mediated regulation of Th17 cell differentiation during RA progression. In addition, the RORγt-CCR6-CCL20 axis might be involved in the directional migration of Th17 cells toward synovial inflammation (73). Furthermore, epigenetic modifications can regulate RORγt activity through post-translational mechanisms such as ubiquitination and acetylation (74). During the early stages of RA activity, DNA methylation of RORγt DNA sequences was enhanced (75). TNF-α inhibitors could attenuate the histone acetylation modification on the RORγt promoter and reduce its expression in Th17 cells (62).
In RA, upstream cytokines stimulated signal transduction and activation transcription factor 3 (STAT3) into the nucleus and initiated RORγt activation (61, 65). RORγt and STAT3 could bind in the IL-17 promoter region and thus regulate the production of IL-17. In the downstream pathways, IL-17 overexpression promoted the survival and maturation of fibroblasts and immune cells in the synovium, thereby aggravating synovial inflammation [90], which ran through the whole process of RA (76). At the same time, increased IL-17 levels might promote osteoclast generation through the RANKL/OPG signaling pathway, thereby aggravating subchondral bone erosion (77). Metallothionein-1 (MT-1) inhibited Th17 differentiation by reducing the expression of STAT3 and RORγt, and significantly suppressed synovial inflammatory response and subsequent bone destruction (78).
3.3 Osteoarthritis
OA is a chronic degenerative arthritis that affects all joints and is characterized by cartilage degradation, subchondral bone remodeling, and bone mineralization (79). Different from RA, where synovial inflammation is the main pathological change, cartilage degeneration is the pathological center of OA (80). The molecular mechanisms underlying OA pathophysiology are poorly understood. OA was previously considered to be a non-inflammatory joint disease. However, recent studies have identified proinflammatory cytokines and immune inflammatory cells as key mediators of cartilage damage in OA (81). OA and RA share some risk factors and pathogenic features, and they both exhibit highly inflammatory features driven by CD4+ T cells (82). The expression of IL-17 in the synovial tissue of OA and RA patients was increased. This eventually led to increased joint inflammation and bone erosion (83).
It has also been suggested that metabolic disorders may also be an important mechanism leading to this disease. RORα can significantly affect the progression of OA through cartilage-specific cholesterol metabolism. In the CH25H-CYP7B1-RORα axis of cholesterol metabolism, RORα acted as a substrate for cholesterol hydroxylase (CH25H and CYP7B1) (84). High cholesterol and its metabolites could directly activate RORα in chondrocytes, which could bind to the promoter of cartilage matrix catabolic factors and activate its transcription, leading to cartilage damage (85, 86). In addition, RORα accelerated cartilage matrix degradation through the IL-6/STAT3 signaling pathway (87, 88).
4 Therapy strategies targeting RORs in skeletal diseases
Therapeutic applications targeting RORs have developed along with the identification of ROR ligands including agonists, inverse agonists, and antagonists. Specifically, ROR agonists enhance the transcription levels of target genes by either promoting the recruitment of coactivator complexes or preventing the recruitment of corepressor complexes, whereas ROR inverse agonists do the opposite. ROR antagonists reduce the transcription by preventing the recruitment of any complexes (11, 89).
Since the first ROR agonist was identified, RORs have been investigated as a potential therapeutic target for various diseases (90, 91). Among bone diseases, inflammatory arthropathy has received the most attention. Treatment with RORα agonist cholesterol sulfate prevented osteoclast osteogenesis in CIA mice and protected against bone loss in postmenopausal osteoporosis (19). A dual inverse agonist of RORβ and RORγt, N-(5-(arylcarbonyl)thiazol-2-yl)amides, has been identified to exhibit therapeutic potential in CIA mice (92). Compared with RORα and RORβ, the treatment targeting RORγt is more commonly used in inflammatory and autoimmune arthropathies.
Several natural compounds have been identified as RORγt-specific modulators that can be used in the treatment of autoimmune diseases. Digoxin was the first RORγt inhibitor reported to reduce inflammation in CIA mice (66), however, its disadvantages were obvious cytotoxicity and limited therapeutic index. In addition, ursolic acid had anti-RORγt activity, but it could also activate glucocorticoid receptors as a side effect (93). To solve these problems, the synthesis of selective RORγt small molecule modulators has become a promising treatment for autoimmune diseases such as RA (94). In vivo data showed that amide drugs had good clinical efficacy as effective RORγt inhibitors (95). Phenylenediamine derivative of RORγt inhibitor reduced the severity of arthritis symptoms in mice (96). Moreover, imidazolopyridine analogs, a RORγt selective inverse agonist, alleviated the pathological symptoms of adjuvant arthritis (AIA) in rats (97, 98). Recently, three azacyclic inverse RORγt agonists were identified by structure-activity relationship studies, which showed biologically similar efficacy in preclinical models of RA (99).
5 Discussion and conclusion
In summary, as transcription factors, RORs directly regulate osteogenic differentiation and osteoclastogenesis and associate with other factors or signals to indirectly mediate the destruction of cartilage and bone by immune-inflammatory responses. In terms of selective molecular modulators, RORγ has made remarkable progress in autoimmune diseases. However, on the pharmacological front, there are concerns about the safety and efficacy of existing small-molecule drugs targeting RORγ. In addition, the effects of administration form and ROR binding mode on clinical efficacy remain to be further studied.
Author contributions
YZ: Conceptualization, Writing – original draft. JM: Conceptualization, Funding acquisition, Writing – review & editing. XB: Writing – review & editing. MH: Writing – review & editing. XW: Conceptualization, Funding acquisition, Writing – original draft, Writing – review & editing, Supervision.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China [grant number NSFC-82001083]; the Natural Science Foundation of Jilin Province for Youths [grant number YDZJ2023ZYTS468].
Acknowledgments
We kindly thank Jingzheng Yi, MD, for her assistance in grammar correction and polishing the language.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Siddiqui JA, Partridge NC. Physiological bone remodeling: systemic regulation and growth factor involvement. Physiology (2016) 31(3):233–45. doi: 10.1152/physiol.00061.2014
3. Chan WCW, Tan Z, To MKT, Chan D. Regulation and role of transcription factors in osteogenesis. Int J Mol Sci (2021) 22(11):5445. doi: 10.3390/ijms22115445
4. Feng S, Xu S, Wen Z, Zhu Y. Retinoic acid-related orphan receptor RORβ, circadian rhythm abnormalities and tumorigenesis. Int J Mol Med (2015) 35(6):1493–500. doi: 10.3892/ijmm.2015.2155
5. Leipe J, Grunke M, Dechant C, Reindl C, Kerzendorf U, Schulze-Koops H, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum (2010) 62(10):2876–85. doi: 10.1002/art.27622
6. Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl receptor Signaling (2009) 7(1):7003. doi: 10.1621/nrs.07003
7. Cook DN, Kang HS, Jetten AM. Retinoic acid-related orphan receptors (RORs): regulatory functions in immunity, development, circadian rhythm, and metabolism. Nucl receptor Res (2015) 2:101185. doi: 10.11131/2015/101185
8. Ladurner A, Schwarz PF, Dirsch VM. Natural products as modulators of retinoic acid receptor-related orphan receptors (RORs). Natural Product Rep (2021) 38(4):757–81. doi: 10.1039/d0np00047g
9. Ma H, Kang J, Fan W, He H, Huang F. ROR: nuclear receptor for melatonin or not? Molecules (2021) 26(9):2693. doi: 10.3390/molecules26092693
10. Lu L, Sun N, Wang Y. Development and therapeutic potential of allosteric retinoic acid receptor-related orphan receptor γt (RORγt) inverse agonists for autoimmune diseases. Eur J Medicinal Chem (2023) 258:115574. doi: 10.1016/j.ejmech.2023.115574
11. Bronner SM, Zbieg JR, Crawford JJ. RORγ antagonists and inverse agonists: a patent review. Expert Opin Ther patents (2017) 27(1):101–12. doi: 10.1080/13543776.2017.1236918
12. Jetten AM, Ueda E. The ROR nuclear orphan receptor subfamily: critical regulators of multiple biological processes. Prog Nucleic Acid Res Mol Biol (2001) 69:205–47. doi: 10.1016/s0079-6603(01)69048-2
13. Woods A, James CG, Wang G, Dupuis H, Beier F. Control of chondrocyte gene expression by actin dynamics: a novel role of cholesterol/Ror-α signalling in endochondral bone growth. J Cell Mol Med (2009) 13(9b):3497–516. doi: 10.1111/j.1582-4934.2009.00684.x
14. Yan Q, Feng Q, Beier F. Reduced chondrocyte proliferation, earlier cell cycle exit and increased apoptosis in neuronal nitric oxide synthase-deficient mice. Osteoarthritis Cartilage (2012) 20(2):144–51. doi: 10.1016/j.joca.2011.11.014
15. Akkawi I, Zmerly H. Osteoporosis: current concepts. Joints (2018) 6(02):122–7. doi: 10.1055/s-0038-1660790
16. Johnston CB, Dagar M. Osteoporosis in older adults. Med Clinics (2020) 104(5):873–84. doi: 10.1016/j.mcna.2020.06.004
17. Meyer T, Kneissel M, Mariani J, Fournier B. In vitro and in vivo evidence for orphan nuclear receptor RORα function in bone metabolism. Proc Natl Acad Sci (2000) 97(16):9197–202. doi: 10.1073/pnas.150246097
18. Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, et al. Disruption of the nuclear hormone receptor RORα in staggerer mice. Nature (1996) 379(6567):736–9. doi: 10.1038/379736a0
19. Park JH, Lee J, Lee GR, Kwon M, Lee HI, Kim N, et al. Cholesterol sulfate inhibits osteoclast differentiation and survival by regulating the AMPK–Sirt1–NF-κB pathway. J Cell Physiol (2023) 238(9):2063–75. doi: 10.1002/jcp.31064
20. Zhou Q, Hu H, Yang Y, Kang Y, Lan X, Wu X, et al. Insertion/deletion (Indel) variant of the goat RORA gene is associated with growth traits. Anim Biotechnol (2022) 34(7):2175–82. doi: 10.1080/10495398.2022.2078980
21. Miyamoto S, Cooper L, Watanabe K, Yamamoto S, Inoue H, Mishima K, et al. Role of retinoic acid-related orphan receptor-α in differentiation of human mesenchymal stem cells along with osteoblastic lineage. Pathobiology (2010) 77(1):28–37. doi: 10.1159/000272952
22. Samsa WE, Vasanji A, Midura RJ, Kondratov RV. Deficiency of circadian clock protein BMAL1 in mice results in a low bone mass phenotype. Bone (2016) 84:194–203. doi: 10.1016/j.bone.2016.01.006
23. Zheng J, Zhang L, Tan Z, Zhao Q, Wei X, Yang Y, et al. Bmal1-and Per2-mediated regulation of the osteogenic differentiation and proliferation of mouse bone marrow mesenchymal stem cells by modulating the Wnt/β-catenin pathway. Mol Biol Rep (2022) 49(6):4485–501. doi: 10.1007/s11033-022-07292-6
24. Kimball JS, Johnson JP, Carlson DA. Oxidative stress and osteoporosis. J Bone Joint Surg (2021) 103(15):1451–61. doi: 10.2106/JBJS.20.00989
25. Shuai Y, Liu B, Rong L, Shao B, Chen B, Jin L. OSGIN2 regulates osteogenesis of jawbone BMSCs in osteoporotic rats. BMC Mol Cell Biol (2022) 23(1):22. doi: 10.1186/s12860-022-00423-8
26. Zhao L, Yin B, Ma Q, Shi Y, Wang C, Ye L. The involvement of histone methylation in the osteogenic differentiation of mesenchymal stem cells. Curr Stem Cell Res Ther (2021) 16(8):915–23. doi: 10.2174/1574888X16666210203110354
27. Chen T, Liu J, Li Z, Lai P, Zhang S, Qu J, et al. DNMT1 is a negative regulator of osteogenesis. Biol Open (2022) 11(3):bio058534. doi: 10.1242/bio.058534
28. Cauley JA. Estrogen and bone health in men and women. Steroids (2015) 99:11–5. doi: 10.1016/j.steroids.2014.12.010
29. Min H-Y, Son H-E, Jang W-G. Estradiol-induced RORα expression positively regulates osteoblast differentiation. Steroids (2019) 149:108412. doi: 10.1016/j.steroids.2019.05.004
30. Benderdour M, Fahmi H, Beaudet F, Fernandes JC, Shi Q. Nuclear receptor retinoid-related orphan receptor α1 modulates the metabolic activity of human osteoblasts. J Cell Biochem (2011) 112(8):2160–9. doi: 10.1002/jcb.23141
31. Shen Y, Tang Q, Wang J, Zhou Z, Yin Y, Zhang Y, et al. Targeting RORα in macrophages to boost diabetic bone regeneration. Cell Proliferation (2023) 56(10):e13474. doi: 10.1111/cpr.13474
32. Karunarathne WAHM, Choi YH, Park SR, Lee C-M, Kim G-Y. Bisphenol A inhibits osteogenic activity and causes bone resorption via the activation of retinoic acid-related orphan receptor α. J Hazard Mater (2022) 438:129458. doi: 10.1016/j.jhazmat.2022.129458
33. Farr JN, Weivoda MM, Nicks KM, Fraser DG, Negley BA, Onken JL, et al. Osteoprotection through the deletion of the transcription factor Rorβ in mice. J Bone Mineral Res (2018) 33(4):720–31. doi: 10.1002/jbmr.3351
34. Roforth MM, Liu G, Khosla S, Monroe DG. Examination of nuclear receptor expression in osteoblasts reveals Rorβ as an important regulator of osteogenesis. J Bone Mineral Res (2012) 27(4):891–901. doi: 10.1002/jbmr.1502
35. Syed FA, Mödder UI, Roforth M, Hensen I, Fraser DG, Peterson JM, et al. Effects of chronic estrogen treatment on modulating age-related bone loss in female mice. J Bone Mineral Res (2010) 25(11):2438–46. doi: 10.1002/jbmr.129
36. Roforth MM, Khosla S, Monroe DG. Identification of Rorβ targets in cultured osteoblasts and in human bone. Biochem Biophys Res Commun (2013) 440(4):768–73. doi: 10.1016/j.bbrc.2013.10.006
37. Komori T. Regulation of proliferation, differentiation and functions of osteoblasts by Runx2. Int J Mol Sci (2019) 20(7):1694. doi: 10.3390/ijms20071694
38. Wang J, Guo F, Chen G, Sun J, Tang Q, Chen L. Spatial-temporal patterns and inflammatory factors of bone matrix remodeling. Stem Cells Int (2021) 2021:1–15. doi: 10.1155/2021/4307961
39. Aquino-Martinez R, Farr JN, Weivoda MM, Negley BA, Onken JL, Thicke BS, et al. miR-219a-5p regulates Rorβ during osteoblast differentiation and in age-related bone loss. J Bone Mineral Res (2019) 34(1):135–44. doi: 10.1002/jbmr.3586
40. Bultink IE, Lems WF. Osteoarthritis and osteoporosis: what is the overlap? Curr Rheumatol Rep (2013) 15:1–8. doi: 10.1007/s11926-013-0328-0
41. Srivastava RK, Dar HY, Mishra PK. Immunoporosis: immunology of osteoporosis—role of T cells. Front Immunol (2018) 9:657. doi: 10.3389/fimmu.2018.00657
42. Geusens PP, van den Bergh JP. Osteoporosis and osteoarthritis: shared mechanisms and epidemiology. Curr Opin Rheumatol (2016) 28(2):97–103. doi: 10.1097/BOR.0000000000000256
43. Im G-I, Kim M-K. The relationship between osteoarthritis and osteoporosis. J Bone mineral Metab (2014) 32:101–9. doi: 10.1007/s00774-013-0531-0
44. Tyagi AM, Srivastava K, Mansoori MN, Trivedi R, Chattopadhyay N, Singh D. Estrogen deficiency induces the differentiation of IL-17 secreting Th17 cells: a new candidate in the pathogenesis of osteoporosis. PloS One (2012) 7(9):e44552. doi: 10.1371/journal.pone.0044552
45. Zhao R. Immune regulation of bone loss by T h17 cells in oestrogen-deficient osteoporosis. Eur J Clin Invest (2013) 43(11):1195–202. doi: 10.1111/eci.12158
46. Shukla P, Mansoori MN, Kakaji M, Shukla M, Gupta SK, Singh D. Interleukin 27 (IL-27) alleviates bone loss in estrogen-deficient conditions by induction of early growth response-2 gene. J Biol Chem (2017) 292(11):4686–99. doi: 10.1074/jbc.M116.764779
47. Zaiss MM, Jones RM, Schett G, Pacifici R. The gut-bone axis: how bacterial metabolites bridge the distance. J Clin Invest (2019) 129(8):3018–28. doi: 10.1172/JCI128521
48. Guo M, Liu H, Yu Y, Zhu X, Xie H, Wei C, et al. Lactobacillus rhamnosus GG ameliorates osteoporosis in ovariectomized rats by regulating the Th17/Treg balance and gut microbiota structure. Gut Microbes (2023) 15(1):2190304. doi: 10.1080/19490976.2023.2190304
49. Sapra L, Dar HY, Bhardwaj A, Pandey A, Kumari S, Azam Z, et al. Lactobacillus rhamnosus attenuates bone loss and maintains bone health by skewing Treg-Th17 cell balance in Ovx mice. Sci Rep (2021) 11(1):1807. doi: 10.1038/s41598-020-80536-2
50. Sapra L, Shokeen N, Porwal K, Saini C, Bhardwaj A, Mathew M, et al. Bifidobacterium longum ameliorates ovariectomy-induced bone loss via enhancing anti-osteoclastogenic and immunomodulatory potential of regulatory B Cells (Bregs). Front Immunol (2022) 13:875788. doi: 10.3389/fimmu.2022.875788
51. Scott DL, Wolfe F, Huizinga TW. Rheumatoid arthritis. Lancet (2010) 376(9746):1094–108. doi: 10.1016/S0140-6736(10)60826-4
52. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. New Engl J Med (2011) 365(23):2205–19. doi: 10.1056/NEJMra1004965
53. Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y, et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med (2006) 203(12):2673–82. doi: 10.1084/jem.20061775
54. Auréal M, Machuca-Gayet I, Coury F. Rheumatoid arthritis in the view of osteoimmunology. Biomolecules (2020) 11(1):48. doi: 10.3390/biom11010048
55. Fan Z-D, Zhang Y-Y, Guo Y-H, Huang N, Ma H-H, Huang H, et al. Involvement of P2X7 receptor signaling on regulating the differentiation of Th17 cells and type II collagen-induced arthritis in mice. Sci Rep (2016) 6(1):35804. doi: 10.1038/srep35804
56. Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors RORα and RORγ. Immunity (2008) 28(1):29–39. doi: 10.1016/j.immuni.2007.11.016
57. Park J-S, Moon S-J, Lim M-A, Byun J-K, Hwang S-H, Yang S, et al. Retinoic acid receptor-related receptor alpha ameliorates autoimmune arthritis via inhibiting of Th17 cells and osteoclastogenesis. Front Immunol (2019) 10:2270. doi: 10.3389/fimmu.2019.02270
58. Hwang ES. Transcriptional regulation of T helper 17 cell differentiation. Yonsei Med J (2010) 51(4):484–91. doi: 10.3349/ymj.2010.51.4.484
59. van Baarsen LG, Lebre MC, van der Coelen D, Aarrass S, Tang MW, Ramwadhdoebe TH, et al. Heterogeneous expression pattern of interleukin 17A (IL-17A), IL-17F and their receptors in synovium of rheumatoid arthritis, psoriatic arthritis and osteoarthritis: possible explanation for nonresponse to anti-IL-17 therapy? Arthritis Res Ther (2014) 16(4):1–10. doi: 10.1186/s13075-014-0426-z
60. Hu S, Guo P, Wang Z, Zhou Z, Wang R, Zhang M, et al. Down-regulation of A3AR signaling by IL-6-induced GRK2 activation contributes to Th17 cell differentiation. Exp Cell Res (2021) 399(2):112482. doi: 10.1016/j.yexcr.2021.112482
61. Samuels JS, Holland L, López M, Meyers K, Cumbie WG, McClain A, et al. Prostaglandin E2 and IL-23 interconnects STAT3 and RoRγ pathways to initiate Th17 CD4+ T-cell development during rheumatoid arthritis. Inflammation Res (2018) 67:589–96. doi: 10.1007/s00011-018-1153-8
62. Lin Y-C, Lin Y-C, Wu C-C, Huang M-Y, Tsai W-C, Hung C-H, et al. The immunomodulatory effects of TNF-α inhibitors on human Th17 cells via RORγt histone acetylation. Oncotarget (2017) 8(5):7559. doi: 10.18632/oncotarget.13791
63. Feng W, Wan X, Fan S, Liu C-Z, Zheng X-X, Liu Q-P, et al. Mechanism underlying the action of Duanteng-Yimu Tang in regulating Treg/Th17 imbalance and anti-rheumatoid arthritis. Heliyon (2023) 9(5):e15867. doi: 10.1016/j.heliyon.2023.e15867
64. Hua D, Yang J, Meng Q, Ling Y, Wei Q, Wang Z, et al. Soufeng sanjie formula alleviates collagen-induced arthritis in mice by inhibiting Th17 cell differentiation. Chin Med (2021) 16(1):1–15. doi: 10.1186/s13020-021-00448-9
65. Chen H-L, Lin SC, Li S, Tang K-T, Lin C-C. Alantolactone alleviates collagen-induced arthritis and inhibits Th17 cell differentiation through modulation of STAT3 signalling. Pharm Biol (2021) 59(1):132–43. doi: 10.1080/13880209.2021.1876102
66. Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature (2011) 472(7344):486–90. doi: 10.1038/nature09978
67. Koufany M, Chappard D, Netter P, Bastien C, Weryha G, Jouzeau JY, et al. The peroxisome proliferator–activated receptor γ Agonist pioglitazone preserves bone microarchitecture in experimental arthritis by reducing the interleukin-17–dependent osteoclastogenic pathway. Arthritis Rheum (2013) 65(12):3084–95. doi: 10.1002/art.38130
68. Wu Y-H, Liu W, Xue B, Zhang L, Liu X-Y, Liu B, et al. Upregulated expression of microRNA-16 correlates with Th17/Treg cell imbalance in patients with rheumatoid arthritis. DNA Cell Biol (2016) 35(12):853–60. doi: 10.1089/dna.2016.3349
69. Tang X, Yin K, Zhu H, Tian J, Shen D, Yi L, et al. Correlation between the expression of microRNA-301a-3p and the proportion of Th17 cells in patients with rheumatoid arthritis. Inflammation (2016) 39:759–67. doi: 10.1007/s10753-016-0304-8
70. Xie M, Wang J, Gong W, Xu H, Pan X, Chen Y, et al. NF-κB-driven miR-34a impairs Treg/Th17 balance via targeting Foxp3. J Autoimmun (2019) 102:96–113. doi: 10.1016/j.jaut.2019.04.018
71. Tavasolian F, Hosseini AZ, Soudi S, Naderi M. miRNA-146a improves immunomodulatory effects of MSC-derived exosomes in rheumatoid arthritis. Curr Gene Ther (2020) 20(4):297–312. doi: 10.2174/1566523220666200916120708
72. Liu M, Ren T, Lin Z, Hua M. Upregulated miR-146a expression in peripheral blood relates to Th17 and treg imbalance in elder rheumatoid arthritis patients. Lifestyle Genomics (2022) 15(3):98–106. doi: 10.1159/000525112
73. Kaneko S, Kondo Y, Yokosawa M, Furuyama K, Segawa S, Tsuboi H, et al. The RORγt-CCR6-CCL20 axis augments Th17 cells invasion into the synovia of rheumatoid arthritis patients. Modern Rheumatol (2018) 28(5):814–25. doi: 10.1080/14397595.2017.1416923
74. Kumar R, Theiss AL, Venuprasad K. RORγt protein modifications and IL-17-mediated inflammation. Trends Immunol (2021) 42(11):1037–50. doi: 10.1016/j.it.2021.09.005
75. Huang Y, Wang H, Ba X, Chen Z, Wang Y, Qin K, et al. Decipher manifestations and Treg/Th17 imbalance in multi-staging rheumatoid arthritis and correlation with TSDR/RORC methylation. Mol Immunol (2020) 127:1–11. doi: 10.1016/j.molimm.2020.08.002
76. Lubberts E, Koenders MI, Oppers-Walgreen B, Van Den Bersselaar L, Coenen-de Roo CJ, Joosten LA, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum (2004) 50(2):650–9. doi: 10.1002/art.20001
77. Sun X, Feng X, Tan W, Lin N, Hua M, Wei Y, et al. Adiponectin exacerbates collagen-induced arthritis via enhancing Th17 response and prompting RANKL expression. Sci Rep (2015) 5(1):11296. doi: 10.1038/srep11296
78. Sun J, Li L, Li L, Ding L, Liu X, Chen X, et al. Metallothionein-1 suppresses rheumatoid arthritis pathogenesis by shifting the Th17/Treg balance. Eur J Immunol (2018) 48(9):1550–62. doi: 10.1002/eji.201747151
79. Glyn-Jones S, Palmer A. Osteoarthritis. Lancet (2015) 386(9991):376–87. doi: 10.1016/S0140-6736(14)60802-3
80. Tateiwa D, Yoshikawa H, Kaito T. Cartilage and bone destruction in arthritis: pathogenesis and treatment strategy: a literature review. Cells (2019) 8(8):818. doi: 10.3390/cells8080818
81. Wojdasiewicz P, Poniatowski ŁA, Szukiewicz D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflammation (2014) 2014:561459. doi: 10.1155/2014/561459
82. Li Y-s, Luo W, Zhu S-a, Lei G-h. T cells in osteoarthritis: alterations and beyond. Front Immunol (2017) 8:356. doi: 10.3389/fimmu.2017.00356
83. Li X, Xiao S, Li F, Fang K, Wen J, Gong H. Max interacting protein 1 induces IL-17-producing T helper/regulatory T imbalance in osteoarthritis by upregulating tectonic family member 2. Tissue Cell (2022) 78:101906. doi: 10.1016/j.tice.2022.101906
84. Choi W-S, Lee G, Song W-H, Koh J-T, Yang J, Kwak J-S, et al. The CH25H–CYP7B1–RORα axis of cholesterol metabolism regulates osteoarthritis. Nature (2019) 566(7743):254–8. doi: 10.1038/s41586-019-0920-1
85. Zhang W, Robertson WB, Zhao J, Chen W, Xu J. Emerging trend in the pharmacotherapy of osteoarthritis. Front Endocrinol (2019) 10:431. doi: 10.3389/fendo.2019.00431
86. Li X, Zhang L, Shi X, Liao T, Zhang N, Gao Y, et al. MicroRNA-10a-3p improves cartilage degeneration by regulating CH25H-CYP7B1-RORα mediated cholesterol metabolism in knee osteoarthritis rats. Front Pharmacol (2021) 12:690181. doi: 10.3389/fphar.2021.690181
87. Liang T, Chen T, Qiu J, Gao W, Qiu X, Zhu Y, et al. Inhibition of nuclear receptor RORα attenuates cartilage damage in osteoarthritis by modulating IL-6/STAT3 pathway. Cell Death Dis (2021) 12(10):886. doi: 10.1038/s41419-021-04170-0
88. Zhu Y-S, Yan H, Mo T-T, Zhang J-N, Jiang C. Identification of diagnostic markers in synovial tissue of osteoarthritis by weighted gene coexpression network. Biochem Genet (2023) 61(5):2056–75. doi: 10.1007/s10528-023-10359-z
89. Pandya VB, Kumar S, Sachchidanand, Sharma R, Desai RC. Combating autoimmune diseases with retinoic acid receptor-related orphan receptor-γ (RORγ or RORc) inhibitors: hits and misses. J medicinal Chem (2018) 61(24):10976–95. doi: 10.1021/acs.jmedchem.8b00588
90. Cyr P, Bronner SM, Crawford JJ. Recent progress on nuclear receptor RORγ modulators. Bioorganic medicinal Chem Lett (2016) 26(18):4387–93. doi: 10.1016/j.bmcl.2016.08.012
91. Kumar N, Solt LA, Conkright JJ, Wang Y, Istrate MA, Busby SA, et al. The benzenesulfoamide T0901317 [N-(2, 2, 2-trifluoroethyl)-N-[4-[2, 2, 2-trifluoro-1-hydroxy-1-(trifluoromethyl) ethyl] phenyl]-benzenesulfonamide] is a novel retinoic acid receptor-related orphan receptor-α/γ inverse agonist. Mol Pharmacol (2010) 77(2):228–36. doi: 10.1124/mol.109.060905
92. Gege C, Schlüter T, Hoffmann T. Identification of the first inverse agonist of retinoid-related orphan receptor (ROR) with dual selectivity for RORβ and RORγt. Bioorganic Medicinal Chem Lett (2014) 24(22):5265–7. doi: 10.1016/j.bmcl.2014.09.053
93. Xu T, Wang X, Zhong B, Nurieva RI, Ding S, Dong C. Ursolic acid suppresses interleukin-17 (IL-17) production by selectively antagonizing the function of RORγt protein. J Biol Chem (2011) 286(26):22707–10. doi: 10.1074/jbc.C111.250407
94. Zeng J, Li M, Zhao Q, Chen M, Zhao L, Wei S, et al. Small molecule inhibitors of RORγt for Th17 regulation in inflammatory and autoimmune diseases. J Pharm Anal (2023) 13(6):545–62. doi: 10.1016/j.jpha.2023.05.009
95. Wang Y, Cai W, Zhang G, Yang T, Liu Q, Cheng Y, et al. Discovery of novel N-(5-(arylcarbonyl) thiazol-2-yl) amides and N-(5-(arylcarbonyl) thiophen-2-yl) amides as potent RORγt inhibitors. Bioorganic medicinal Chem (2014) 22(2):692–702. doi: 10.1016/j.bmc.2013.12.021
96. Ji J, Dou H, Li X, Song Y, Li X, Li E, et al. Novel benzenediamine derivative FC99 ameliorates zymosan-induced arthritis by inhibiting RORγt expression and Th17 cell differentiation. Acta Biochim Biophys Sin (2014) 46(10):829–36. doi: 10.1093/abbs/gmu074
97. Guendisch U, Weiss J, Ecoeur F, Riker JC, Kaupmann K, Kallen J, et al. Pharmacological inhibition of RORγt suppresses the Th17 pathway and alleviates arthritis in vivo. PloS One (2017) 12(11):e0188391. doi: 10.1371/journal.pone.0188391
98. Ortiz MA, Piedrafita FJ, Nefzi A. 1, 5-disubstituted acylated 2-amino-4, 5-dihydroimidazoles as a new class of retinoic acid receptor–related orphan receptor (ROR) inhibitors. Int J Mol Sci (2022) 23(8):4433. doi: 10.3390/ijms23084433
Keywords: RORs, bone homeostasis, osteoporosis, rheumatoid arthritis, osteoarthritis
Citation: Zhang Y, Ma J, Bao X, Hu M and Wei X (2023) The role of retinoic acid receptor-related orphan receptors in skeletal diseases. Front. Endocrinol. 14:1302736. doi: 10.3389/fendo.2023.1302736
Received: 27 September 2023; Accepted: 23 October 2023;
Published: 08 November 2023.
Edited by:
Ting Zheng, Hospital for Special Surgery, United StatesReviewed by:
Kenji Hata, Osaka University, JapanCopyright © 2023 Zhang, Ma, Bao, Hu and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoxi Wei, eGlhb3hpd0BqbHUuZWR1LmNu