 Yunyu Du
Yunyu Du Tieying Li2
Tieying Li2
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 15 November 2023
Sec. Cancer Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1295349
This article is part of the Research Topic Double-Edged Swords: Important Factors Connecting Metabolic Disorders and Cancer Development - From Basic Research to Translational Applications, volume II View all 12 articles
Cancer treatment still encounters challenges, such as side effects and drug resistance. The tripartite-motif (TRIM) protein family is widely involved in regulation of the occurrence, development, and drug resistance of tumors. MG53, a member of the TRIM protein family, shows strong potential in cancer therapy, primarily due to its E3 ubiquitin ligase properties. The classic membrane repair function and anti-inflammatory capacity of MG53 may also be beneficial for cancer prevention and treatment. However, MG53 appears to be a key regulatory factor in impaired glucose metabolism and a negative regulatory mechanism in muscle regeneration that may have a negative effect on cancer treatment. Developing MG53 mutants that balance the pros and cons may be the key to solving the problem. This article aims to summarize the role and mechanism of MG53 in the occurrence, progression, and invasion of cancer, focusing on the potential impact of the biological function of MG53 on cancer therapy.
The tripartite-motif (TRIM) family is characterized by a really interesting new gene (RING) finger domain, one or two B-box domains, and a coiled coil domain (1). Tripartite domains are highly conserved among TRIM proteins and hence perform similar functions in cellular processes (2). The vast majority of TRIM proteins contain RING finger domains in their N-terminal regions and seem to participate mostly in ubiquitination (3). B-box domains may exist solely in TRIM proteins and may mediate protein–protein interactions (1, 4). The coiled coil domain has been proven to mediate homo-oligomeric and hetero-oligomeric interactions given that self-association via this domain is believed to play a critical role in catalytic activity of TRIM proteins (5). The variation in the C-terminal domain contributes to the diverse functions of TRIM proteins.
About 80 TRIM protein genes have been identified in humans (6). Many diseases have been shown to be associated with TRIM proteins. These diseases include metabolic and neurodegenerative diseases, viral infections, and cancers (7–10). The role of TRIM proteins in cancer has received more attention. As a result of structural differences, TRIM proteins act as oncogenes and tumor suppressors in different cancers (11). However, the relationship between some members of TRIM proteins and cancer remains unexplored (10).
TRIM72, also known as Mitsugumin 53 (MG53), is secreted by muscle tissues and is a TRIM family protein derived from an immunoproteomics pool (12). The C-terminal of MG53 contains PRY and SPRY domains, which are the most common domains in TRIM proteins (4, 13). These domains can recognize specific partner proteins, thus acting as protein-interacting modules (14). As a typical E3 ubiquitin ligase, MG53 was initially found to participate in damage repair in skeletal muscle cells, and its key feature of membrane repair in a variety of organ injuries was later confirmed (12, 13). MG53 overexpression can inhibit systemic insulin response and subsequently cause metabolic issues (15). However, other researchers take a completely opposing position (16). Evidence suggesting that MG53 may perform an anticancer role in cancers, such as hepatocellular carcinoma, colorectal carcinoma, tongue cancer, and nonsmall cell lung cancer (NSCLC), has recently emerged (17–20). In this review, we summarize the roles and mechanisms of MG53 in a variety of cancers and discuss the possible contribution of the diverse biological functions of MG53 in cancer.
Colorectal cancer is the second most common cause of cancer deaths worldwide and is expected to cause 1.2 million deaths by 2030 (21, 22). Considering that most patients with colorectal cancer progress slowly over many years, colorectal cancer is usually curable if diagnosed at an early stage (23). Screening for colorectal cancer requires the development of sensitive biomarkers in peripheral blood. Many members of the TRIM protein family have been reported to act as oncogenic and tumor-suppressive factors in gastrointestinal cancers via different signaling pathways (24). In addition, TRIM47 may be an effective diagnostic marker for predicting colorectal cancer (25).
The gene and protein levels of MG53 were considerably lower in colon cancer tissues than in healthy colon tissues, and the same results were found in the serum of patients with colon cancer (26). In colon cancer and normal colon tissues, MG53 may be expressed and secreted by stromal cells instead of normal colon or colon cancer cells, and serum MG53 levels are negatively correlated with colon cancer stage and metastasis, suggesting that the low MG53 levels in the serum of patients with colon cancer may be due to local tissue lesions (26). Low levels of MG53 in focal tissues have also been suggested to account for the poor prognosis of stage II colon carcinoma (27). Under colorectal carcinogen induction, MG53 knockout mice present more severe tumor progression than wild-type mice, whereas mice with MG53 overexpression have relatively good colorectal structure and function (19). MG53 has also been shown to inhibit the proliferation of colorectal cancer cells in an in vitro study. And this study found that MG53, as an E3 ubiquitin ligase capable of targeting cyclin D1, induces its ubiquitination-dependent degradation to inhibit the proliferation of gastrointestinal cancer cells by arresting the cell cycle at the G1 phase (28). In addition, MG53 acts differently on different anticancer drugs. MG53 and pabocinib inhibit the proliferation of colon cancer cells synergistically, and MG53 could partially ameliorate drug resistance (19). The safety of recombinant human MG53 (rhMG53) has been validated in a mouse model of colorectal cancer (28). Although rhMG53 do not affect the doxorubicin sensitivity of resistant colorectal cancer cells (SW620/AD300), it inhibits the proliferation of colorectal cancer cells. Moreover, in mouse tumor xenograft models of colorectal adenocarcinoma with multidrug resistance, the combination of doxorubicin and rhMG53 appeared to be more effective than doxorubicin or rhMG53 alone (28).
Although vaccination and antiviral therapy have reduced the incidence of hepatocellular carcinoma, the incidence and mortality rates of this malignancy continue to increase in many regions of the world (29). In hepatocellular carcinoma, the expression of numerous TRIM proteins tends to be altered and has been shown to be correlated with diagnosis, treatment, and prognosis (30). TRIM proteins appear to be involved in the survival, growth, aerobic glycolysis, immune infiltration, and invasion of hepatocellular carcinoma cells (31–34).
The mRNA expression of MG53 was detected in human hepatocellular carcinoma and normal human hepatocyte cell lines. In patients with hepatocellular carcinoma, the high expression of MG53 may be associated with poor overall survival (35). However, one study has shown that the gene and protein expression levels of MG53 have been suggested to be drastically lower in hepatocellular carcinoma tissue than in matched noncancerous liver tissue (17). MG53 regulates the ubiquitination and degradation of RAC1, a small GTPase with oncogenic function, this effect, in turn, inhibits the malignant progression of hepatocellular carcinoma and improves the resistance of hepatocellular carcinoma to sorafenib treatment by blocking the RAC1/MAPK signaling pathway (17).
Although the application of precision medicine in NSCLC treatment has advanced considerably over the past decade, the 5-year survival rate of patients with metastatic NSCLC remains less than 5% due to multiple drug resistance mechanisms (36, 37). Some TRIM proteins may contribute to NSCLC or resistance to targeted drugs (38–44), whereas others have completely opposite functions (45–47).
MG53 is downregulated in metastatic tumors from patients with NSCLC relative to in nonmetastatic tumors, and MG53 knockout promotes the growth and metastasis of lung tumors in mice (48, 49). G3BP2, a protein associated with the formation of multiple tumors, was upregulated in the cytosol of tumor cells from patients with NSCLC relative to in nontumor cells. Circulating levels of MG53 appear to influence the proliferation and migration of NSCLC cells directly via G3BP2. Instead of performing classical ubiquitination-dependent degradation functions, the amino terminus of MG53 physically interacts with G3BP2 and enhances its nuclear translocation, which may be a key mechanism by which MG53 inhibits the G3BP2-mediated formation of lung cancer tumors and stress granules (20, 50). Furthermore, an in vitro study showed that rhMG53 inhibited the formation of stress granules and potentiated the cytotoxic effect of cisplatin on human NSCLC cells (20).
MG53 appears to have an ameliorative effect on multiple types of cancer. However, many TRIM proteins have inconsistent effects on different cancers. A three-dimensional growth system study reported that MG53 dramatically suppressed the proliferation, invasion, and colony formation of tongue cancer cells (18). Knocking down MG53 in tongue cancer cells resulted in a remarkable increase in the phosphorylation of AKTSer308 and AKTThr473. Animal studies showed that in mice, knocking out MG53 also accelerated the progression of tongue cancer (18). O6-methylguanine DNA methyl transferase (MGMT) is an important target in cancer therapy because it blocks the beneficial effects of chemotherapy on tumor cells (51). The RING structural domain of MG53 interacts with the N-terminal region of MGMT and regulates the ubiquitination-dependent degradation of MGMT. Human uveal melanoma cells have higher MGMT levels and lower MG53 levels than normal human pigment epithelium cells. MG53 overexpression in uveal melanoma cells contributes to improved chemoresistance to dacarbazine treatment (52). MG53 is downregulated in the tumor tissue of patients with breast cancer relative to in paired adjacent nontumor tissue and is also downregulated in many breast cancer cell lines relative to in normal human mammary cell lines. In vivo and in vitro, MG53 inhibits breast cancer progression likely because it can inhibit the activation of the PI3K/Akt/mTOR pathway and reduce lactate levels through protein phosphatase 3 catalytic subunit α (53). One study analyzed ubiquitin-related genes in The Cancer Genome Atlas cohort and found that MG53 was correlated strongly with the grade, stage, and T stage of clear cell renal cell carcinoma. However, the expression of MG53 in patients with clear cell renal cell carcinoma remains to be confirmed (54).
In accordance with the current evidence, MG53 appears to be beneficial for delaying the progression of various cancers and improving resistance to some anticancer drugs in in vitro and animal models (Figure 1). Available studies suggest that the antitumor effect of MG53 may be mainly derived from its role as an E3 ubiquitin ligase. However, the current evidence for specific cancer types remains insufficient and lacks mechanism research. Further safety verification is required for the application of rhMG53.
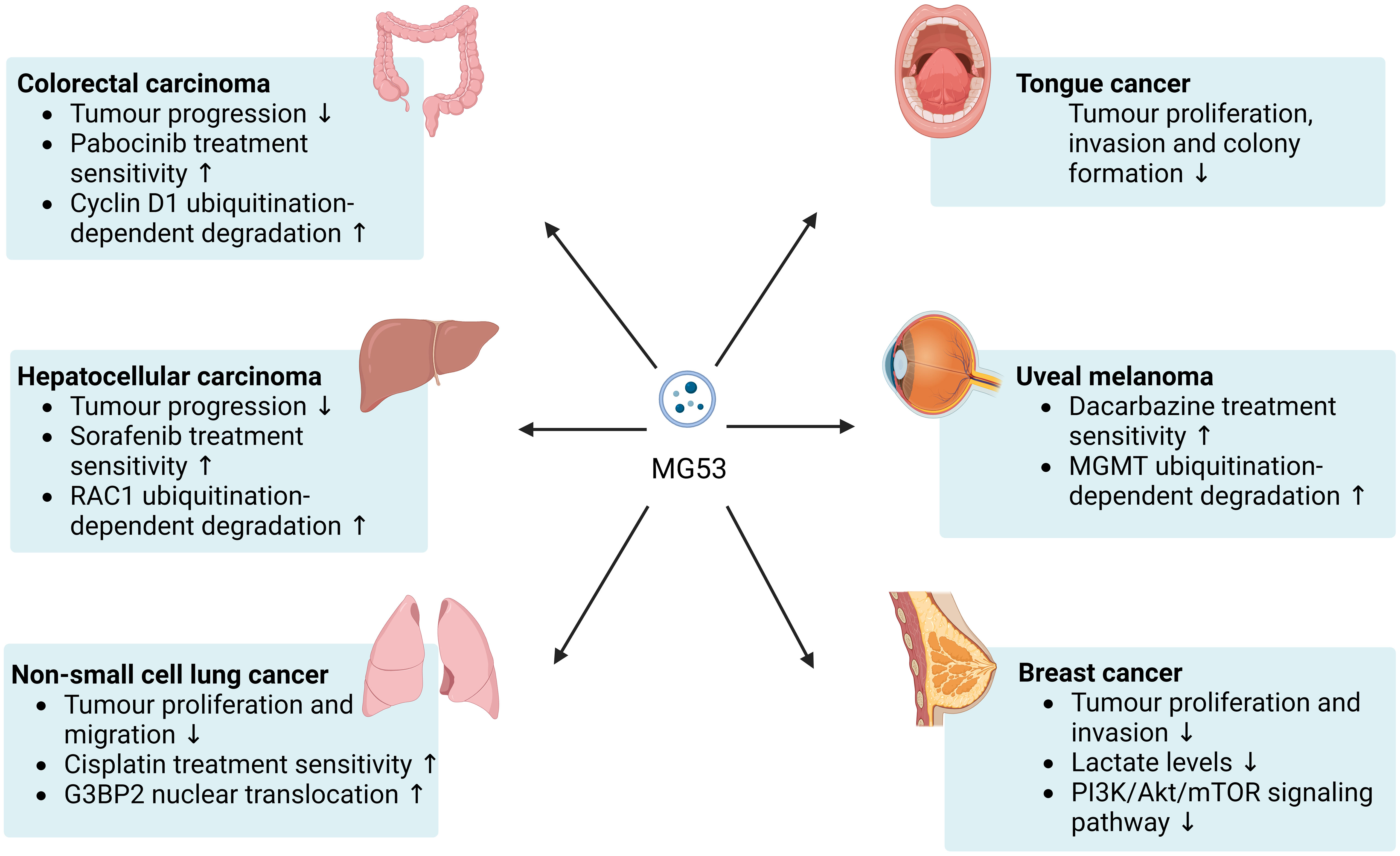
Figure 1 Beneficial effects of MG53 on cancer. MG53 can inhibits the progression of a broad range of cancers and helps improve the therapeutic sensitivity of numerous anticancer drugs. The antitumor capacity of MG53 may be mainly attributed to its E3 ubiquitin ligase properties.
During cancer progression and treatment, many organs suffer varying degrees of tissue damage from the tumor, cancer complications, and treatment side effects, all of which are related to plasma membrane damage and may accelerate cancer progression (55–57). MG53 was initially well known for its function in the repair of muscle cell membranes. Evidence showing that MG53 can participate in the repair of various cell membranes and promote tissue regeneration has emerged with the deepening of research (13, 58). MG53 secreted by skeletal muscles is transported in the circulatory system in the form of vesicles and participates in muscle cell membrane repair. The failure of MG53-mediated membrane damage repair may cause certain skeletal muscle diseases (12, 59–61). In addition, the pathological processes of myocardial injury and cancer are intertwined, and heart failure induced by anticancer therapy has become a key focus in cardiac oncology research (62, 63). Evidence also suggests that cancer and ischemia–reperfusion injury share common pathways also exists (64).
The occurrence of acute and chronic kidney injury is strongly associated with the development of kidney cancer, and early intervention for kidney injury is an effective means of kidney cancer prevention (65). Moreover, the presence of acute kidney injury is fairly prevalent in patients with cancer. The management strategies for acute kidney injury differ in accordance with predisposing factors. For example, immunotherapy-induced acute kidney injury is influenced by tumor type and treatment modality (66, 67). When renal proximal tubular epithelium cells experience acute injury, such as mechanical or chemical damage, MG53 rapidly translocates to the injured site to form a repair patch. By contrast, in injured renal proximal tubular epithelium cells with MG53 knockout, the defect in membrane repair function leads to rapid death of cells. MG53 knockout mice exhibit tubulointerstitial defects and show more severe renal injury than wild-type mice during ischemia–reperfusion. In animals, the preadministration of rhMG53 alleviates cisplatin or iodine contrast agent-induced acute kidney injury (68, 69). In chronic kidney disease, MG53 provides benefits by controlling inflammation and promoting mitochondrial autophagy (70, 71).
Chronic lung injury, such as chronic obstructive pulmonary disease, is strongly associated with the development of lung cancer, and this mechanistic overlap has attracted increasing attention (72, 73). Chemotherapy, surgery, medication treatment for lung cancer, and even treatment for other types of cancer can lead to lung injury (74–77). Chronic moderate liver injury tends to induce hepatic cell carcinogenesis rather than hepatocellular senescence, which can inhibit carcinogenesis (78). In several models of lung injury, MG53 shows reparative effects on pulmonary epithelial cells. Animals lacking MG53 exhibit increased susceptibility to injury induced by various factors, and rhMG53 can protect lung tissue from lung injury. MG53 may execute its membrane repair function by coregulating the endocytosis of alveolar epithelial cells with caveolin 1 (79–85).
The liver is susceptible to the effects of drugs, such as conventional chemotherapy drugs, small-molecule-targeting drugs, including multikinase inhibitors, or immune checkpoint inhibitors, all of which can induce varying degrees of liver injury (86–88). With the widespread application of immune checkpoint inhibitors in liver tumor therapy, the relationship between checkpoint inhibitors and liver safety has received increased attention (89). Although hepatocytes do not express MG53 mRNA, circulating MG53 leads to the ubiquitination-dependent degradation of RIPK3, which inhibits the phosphorylation and membrane translocation of MLKL and thus alleviates acetaminophen-induced hepatocyte injury (90, 91). MG53 can also ameliorate oxidative stress and hepatocyte death induced by hepatic ischemia–reperfusion through interaction with dysferlin (92).
Overall, the plasma membrane repair function of MG53 has considerable potential for application in cancer prevention and treatment. Current research focuses on the association between MG53 and the progression of tumor tissues, whereas only a few studies have investigated the contribution of plasma membrane repair by MG53 to cancer treatment. However, the fact that excessive membrane repair contributes to cancer cell invasion is also important to consider when using rhMG53 (93). For example, the annexin family, which participates in membrane repair together with MG53, is overexpressed in invasive cancer cells and promotes the plasma membrane repair of cancer cells. Inhibiting Annexin-mediated repair is beneficial for inducing cancer cell death (94–98).
Inflammatory response is an important defense mechanism of the body, but it can also promote the formation of tumor microenvironment and tumor promotion, especially chronic inflammation (99). Anti-inflammatory therapy targeting inflammation-related factors such as nuclear factor-κB (NF-κB) plays an important role in cancer control (100). TRIM proteins are widely involved in regulating inflammatory responses and MG53 appears to have anti-inflammatory effects in multiple tissues (101).
MG53 interacts with the p65 subunit of NF-κB and thereby inhibits the nuclear translocation of NF-κB, which in turn alleviates inflammatory responses in kidney, nervous system and airway (70, 102, 103). After infection of macrophages or mice with virus, MG53 attenuates inflammatory response by decreasing type I interferon levels (104). In mice with Duchenne muscular dystrophy, MG53 appears to enhance mitochondrial autophagy, thereby reducing nucleotide oligomerization domain-like receptor protein 3 (NLRP3) inflammasomes and suppressing chronic inflammation in skeletal muscles (105). Similarly, it was emphasised that MG53 may improve neuroinflammation by decreasing NLRP3 inflammasomes in a study using human umbilical cord mesenchymal stem cells and mice (106).
MG53 can ameliorate inflammation in many disease models, but its role in carcinogenic inflammation, inflammation caused by cancer and inflammation triggered by cancer treatment remains to be investigated.
Furthermore, other biological functions of MG53 may be beneficial for cancer therapy. For example, angiogenesis is an important target for cancer treatment, and MG53 inhibits angiogenesis in vivo and in vitro by decreasing focal adhesion kinase phosphorylation and blocking the Src/Akt/ERK1/2 signaling pathway (107, 108). Peroxisome proliferator-activated receptor-α (PPARα) agonists have a role in anti-tumor therapy and MG53 attenuates inflammatory responses in cardiomyocytes by upregulating PPARα expression (109, 110). However, there is too little relevant evidence to demonstrate that these functions of MG53 are beneficial for cancer therapy.
Insulin resistance is a key factor in the occurrence and development of cancer, and a substantial proportion of patients with cancer have insulin resistance (111–114). Impaired glucose tolerance is also strongly associated with long-term cancer risk and is an important risk factor for cancer-related death (115–119). During cancer treatment, the blood glucose and insulin levels of patients must be monitored to learn about the insulin resistance induced by therapeutic measures and thus adjust the treatment protocol promptly (120).
The relationship between MG53 and insulin resistance has long been controversial. Some studies have suggested that MG53 induces insulin resistance through multiple pathways, including targeting insulin receptor substrate 1 (IRS-1), insulin receptors (IRs), and AMP-activated protein kinase for ubiquitin-dependent degradation, promoting the expression of peroxisome proliferator-activated receptor-α and its target genes to facilitate myocardial lipid uptake and thereby leading to lipid accumulation and toxicity, and binding to the extracellular structural domains of IRs to inhibit receptors allosterically (15, 121–124). In addition, the direct application of rhMG53 may exacerbate insulin resistance, and the protective effect of MG53 on myocardial cells may be counteracted by its adverse metabolism. Two mutants of rhMG53, rhMG53-C14A and rhMG53-S255A, can eliminate adverse effects on metabolism while retaining the membrane repair function of rhMG53 (121, 125–128).
However, MG53 expression is inconsistent in various models of metabolic disorders, and neither the ablation nor overexpression of MG53 in wild-type and db/db mice has been noted to alter insulin signaling. Additionally, in rats, the repeated intravenous administration of rhMG53 does not seem to affect glucose metabolism (16, 129–133). Indeed, the lack of IRS-1 does not immediately give rise to diabetes because strong compensatory mechanisms exist between different IR subtypes (134, 135).
The aforementioned controversy may be attributed to the overlooked role of MG53 in pancreatic β-cells. In the absence of global insulin resistance, the IRs of pancreatic β-cells can inhibit high glucose-induced insulin secretion and their knockout can promote insulin secretion and improve glucose tolerance. However, this regulatory function of the IRs to β-cells does not occur in the presence of global insulin resistance (136). High glucose and insulin levels can promote the secretion of MG53 in striated muscle, and MG53 can induce the ubiquitination-dependent degradation and inactivation of IRs (15, 121). Under the assumption that MG53 can affect the function of pancreatic β-cells through IRs, MG53 overexpression would have a complicated effect on glucose metabolism in healthy and insulin-resistant humans (Figure 2). A cohort study involving 283 subjects supports our hypothesis. This study found that although serum MG53 levels appeared to be unrelated to insulin resistance, subjects with impaired glucose metabolism had remarkably higher circulating levels of MG53 than healthy subjects. Furthermore, circulating levels of MG53 were found to be an independent risk factor for the development of type 2 diabetes rather than a simple disease marker, and elevated circulating levels of MG53 represent the diminished function of β-cells (137).
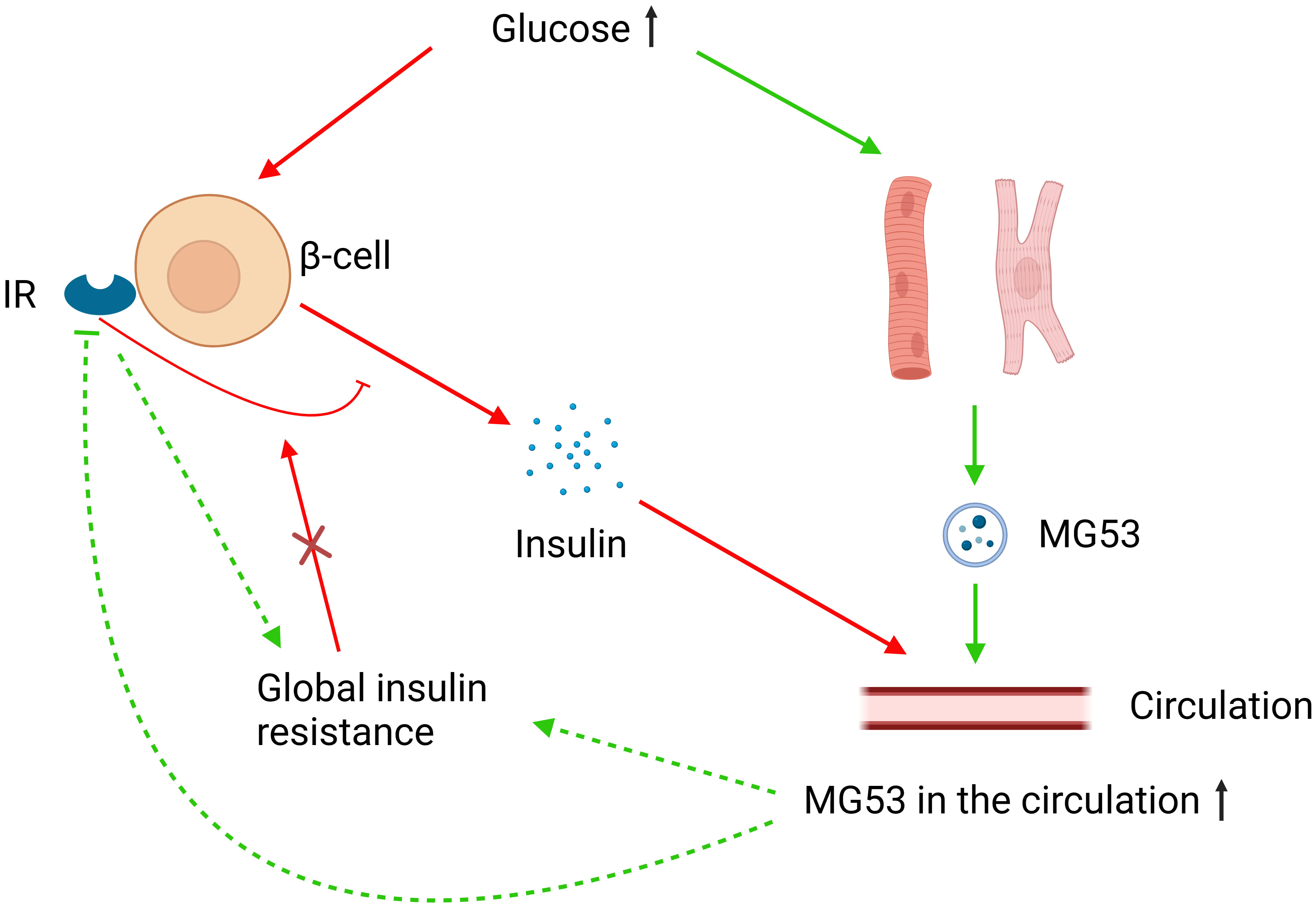
Figure 2 MG53 and glucose metabolism. Elevated glucose levels increase MG53 secretion from muscle tissue, and excess MG53 can lead to global insulin resistance through the inhibition of IRs or other pathways. High glucose also stimulates insulin secretion from pancreatic β-cells, where IRs play an inhibitory role. However, in the case of global insulin resistance, this inhibitory function of IRs fails. MG53 may have different effects on insulin secretion and therefore glucose metabolism in different severities of insulin resistance.
The above evidence suggests that MG53 has important implications for glucose metabolic disorders although the relationship between MG53 and insulin resistance is controversial. However, current research remains insufficient to elucidate its underlying mechanisms. Additional robust evidence is needed to explain the mechanism underlying the involvement of MG53 in glucose metabolism and validate the safety of rhMG53 in patients with metabolic disorders and cancer.
The potential adverse effects of MG53 on cancer cachexia must also be considered when applying rhMG53 in cancer therapy. Cancer cachexia, a common syndrome among patients with cancer, is characterized primarily by the loss of muscle tissue and inadequately relieved by nutritional means (138, 139). Changes in factors related to protein metabolism during cancer progression or treatment led to an imbalance between protein synthesis and degradation, resulting in muscle tissue reduction (140, 141). Reduced muscle mass in cancer cachexia is partly attributed to suppression of the anabolic signaling pathway induced by insulin-like growth factor1 (140). Cardiac atrophy and fibrosis in cancer cachexia are associated with the activated transforming growth factor-β (TGF-β)-mediated SMAD2/3 catabolic signaling pathway (142–145). MG53 inhibits the IGF-induced IRS-1/PI (3)K/Akt pathway, which is the best-characterized mechanism in cardiac and skeletal muscle myogenesis, through the ubiquitin-dependent degradation of IRS-1 (125, 139, 146, 147). Caveolin-1 plays an antifibrotic role in multiple organs and reduces cardiac fibrosis by repressing the TGF-β/Smad2 pathway (148, 149). MG53 can inhibit the expression of caveolin-1, thereby promoting TGF-β1/SMAD2-induced myocardial fibrosis (150). The activation of signal transducers and activator of transcription 3 (STAT3) has been implicated in promoting the progression of many cancers as well as exacerbating the loss of skeletal muscle tissue in cancer cachexia (151, 152). MG53 overexpression promotes the phosphorylation of STAT3, which thereby induces cardiac fibrosis, and its effect on cardiac lesions in cancer cachexia remains to be investigated (153).
Current research strongly suggests that MG53 is an important cancer therapy target, despite its potentially negative effects. Future researches should be focused on elucidating the mechanism of MG53’s role in cancer, glucose metabolism, and myogenesis, and on this basis, attempts should be made to retain the cancer therapeutic ability of MG53 while removing its side effects. MG53 mutants that retain membrane repair function without impairing glucose metabolism have been developed by eliminating the E3 ubiquitin ligase property of MG53 (128). In cancer therapy, however, the E3 ubiquitin ligase function of MG53 seems to play a crucial role. For the ubiquitination-dependent degradation of different proteins, MG53 may need to be activated at different sites, which could be the key to solving this problem (123, 128).
In summary, discussing the possible negative effects of MG53 on cancer treatment, especially in the context of varying degrees of insulin resistance and across gender, is urgently needed. If adverse effects are evident, developing safe mutants of MG53 may be a win-win approach.
The TRIM protein family has always been an important therapeutic target for cancer treatment, and in recent years, the role of MG53 in cancer has gradually been recognized. We found that almost all the evidence indicates that MG53 has a strong inhibitory effect on the progression of cancer and may serve as a biomarker for cancer. However, due to the lack of clinical research, the effect of MG53 on human cancer is actually undetermined. Furthermore, current research appears to overlook the contribution of the membrane repair function and anti-inflammatory properties of MG53 to cancer and does not discuss the potential adverse effects of MG53 on cancer treatment. Therefore, the safety of rhMG53 also needs further discussion. From the perspective of the biological functions of MG53, MG53 may still be a double-edged sword in cancer treatment and further research is needed to comprehensively investigate its role in cancer.
YD: Writing – original draft, Writing – review & editing. TL: Writing – review & editing. MY: Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Natural Science Foundation of China (Grant no. 31371205).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Nisole S, Stoye JP, Saib A. TRIM family proteins: retroviral restriction and antiviral defense. Nat Rev Microbiol (2005) 3(10):799–808. doi: 10.1038/nrmicro1248
2. Li Y, Wu H, Wu W, Zhuo W, Liu W, Zhang Y, et al. Structural insights into the TRIM family of ubiquitin E3 ligases. Cell Res (2014) 24(6):762–5. doi: 10.1038/cr.2014.46
3. Meroni G, Diez-Roux G. TRIM/RBCC, a novel class of 'single protein RING finger' E3 ubiquitin ligases. Bioessays. (2005) 27(11):1147–57. doi: 10.1002/bies.20304
4. Esposito D, Koliopoulos MG, Rittinger K. Structural determinants of TRIM protein function. Biochem Soc Trans (2017) 45(1):183–91. doi: 10.1042/BST20160325
5. Koliopoulos MG, Esposito D, Christodoulou E, Taylor IA, Rittinger K. Functional role of TRIM E3 ligase oligomerization and regulation of catalytic activity. EMBO J (2016) 35(11):1204–18. doi: 10.15252/embj.201593741
6. Hatakeyama S. TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci (2017) 42(4):297–311. doi: 10.1016/j.tibs.2017.01.002
7. Wan T, Li X, Li Y. The role of TRIM family proteins in autophagy, pyroptosis, and diabetes mellitus. Cell Biol Int (2021) 45(5):913–26. doi: 10.1002/cbin.11550
8. Zhu Y, Afolabi LO, Wan X, Shim JS, Chen L. TRIM family proteins: roles in proteostasis and neurodegenerative diseases. Open Biol (2022) 12(8):220098. doi: 10.1098/rsob.220098
9. Khan R, Khan A, Ali A, Idrees M. The interplay between viruses and TRIM family proteins. Rev Med Virol (2019) 29(2):e2028. doi: 10.1002/rmv.2028
10. Zhao G, Liu C, Wen X, Luan G, Xie L, Guo X. The translational values of TRIM family in pan-cancers: From functions and mechanisms to clinics. Pharmacol Ther (2021) 227:107881. doi: 10.1016/j.pharmthera.2021.107881
11. Huang N, Sun X, Li P, Liu X, Zhang X, Chen Q, et al. TRIM family contribute to tumorigenesis, cancer development, and drug resistance. Exp Hematol Oncol (2022) 11(1):75. doi: 10.1186/s40164-022-00322-w
12. Cai C, Masumiya H, Weisleder N, Matsuda N, Nishi M, Hwang M, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol (2009) 11(1):56–64. doi: 10.1038/ncb1812
13. Li Z, Wang L, Yue H, Whitson BA, Haggard E, Xu X, et al. MG53, A tissue repair protein with broad applications in regenerative medicine. Cells (2021) 10(1):122. doi: 10.3390/cells10010122
14. Woo JS, Imm JH, Min CK, Kim KJ, Cha SS, Oh BH. Structural and functional insights into the B30.2/SPRY domain. EMBO J (2006) 25(6):1353–63. doi: 10.1038/sj.emboj.7600994
15. Wu H-K, Zhang Y, Cao C-M, Hu X, Fang M, Yao Y, et al. Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation. (2019) 139(7):901–14. doi: 10.1161/CIRCULATIONAHA.118.037216
16. Wang Q, Bian Z, Jiang Q, Wang X, Zhou X, Park KH, et al. MG53 does not manifest the development of diabetes in db/db mice. Diabetes. (2020) 69(5):1052–64. doi: 10.2337/db19-0807
17. Ma X, Ma X, Zhu L, Zhao Y, Chen M, Li T, et al. The E3 ubiquitin ligase MG53 inhibits hepatocellular carcinoma by targeting RAC1 signaling. Oncogenesis. (2022) 11(1):40. doi: 10.1038/s41389-022-00414-6
18. Yin W, Liu Y, Bian Z. MG53 inhibits the progression of tongue cancer cells through regulating PI3K-AKT signaling pathway: evidence from 3D cell culture and animal model. Small (Weinheim an der Bergstrasse Germany). (2019) 15(8):e1805492. doi: 10.1002/smll.201805492
19. Fang M, Wu H-K, Pei Y, Zhang Y, Gao X, He Y, et al. E3 ligase MG53 suppresses tumor growth by degrading cyclin D1. Signal Transduction Targeted Ther (2023) 8(1):263. doi: 10.1038/s41392-023-01458-9
20. Li H, Lin P-H, Gupta P, Li X, Zhao SL, Zhou X, et al. MG53 suppresses tumor progression and stress granule formation by modulating G3BP2 activity in non-small cell lung cancer. Mol Cancer. (2021) 20(1):118. doi: 10.1186/s12943-021-01418-3
21. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol (2019) 16(12):713–32. doi: 10.1038/s41575-019-0189-8
22. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. (2017) 66(4):683–91. doi: 10.1136/gutjnl-2015-310912
23. Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet (2014) 383(9927):1490–502. doi: 10.1016/S0140-6736(13)61649-9
24. Eberhardt W, Haeussler K, Nasrullah U, Pfeilschifter J. Multifaceted roles of TRIM proteins in colorectal carcinoma. Int J Mol Sci (2020) 21(20):7532. doi: 10.3390/ijms21207532
25. Liang Q, Tang C, Tang M, Zhang Q, Gao Y, Ge Z. TRIM47 is up-regulated in colorectal cancer, promoting ubiquitination and degradation of SMAD4. J Exp Clin Cancer Res (2019) 38(1):159. doi: 10.1186/s13046-019-1143-x
26. Chen Z, Yin X, Li K, Chen S, Li H, Li Y, et al. Serum levels of TRIM72 are lower among patients with colon cancer: identification of a potential diagnostic marker. Tohoku J Exp Med (2018) 245(1):61–8. doi: 10.1620/tjem.245.61
27. Fernandez-Acenero MJ, Cruz M, Sastre-Varela J, Casal JI, Nieto MAC, Del Puerto-Nevado L, et al. TRIM72 immunohistochemical expression can predict relapse in colorectal carcinoma. Pathol Oncol Res (2020) 26(2):861–5. doi: 10.1007/s12253-019-00629-w
28. Gupta P, Li H, Zhang G-N, Barbuti AM, Yang Y, Lin P-H, et al. MG53 inhibits cellular proliferation and tumor progression in colorectal carcinoma. Int J Biol Sci (2022) 18(14):5221–9. doi: 10.7150/ijbs.67869
29. Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. (2021) 7(1):6. doi: 10.1038/s41572-020-00240-3
30. Lu K, Pan Y, Huang Z, Liang H, Ding ZY, Zhang B. TRIM proteins in hepatocellular carcinoma. J BioMed Sci (2022) 29(1):69. doi: 10.1186/s12929-022-00854-7
31. Liu Y, Tao S, Liao L, Li Y, Li H, Li Z, et al. TRIM25 promotes the cell survival and growth of hepatocellular carcinoma through targeting Keap1-Nrf2 pathway. Nat Commun (2020) 11(1):348. doi: 10.1038/s41467-019-14190-2
32. Ge Y, Zhao R, Li B, Xiao B, Zhou L, Zuo S. Aerobic glycolysis and tumor progression of hepatocellular carcinoma are mediated by ubiquitin of P53 K48-linked regulated by TRIM37. Exp Cell Res (2022) 421(2):113377. doi: 10.1016/j.yexcr.2022.113377
33. Cao J, Su B, Peng R, Tang H, Tu D, Tang Y, et al. Bioinformatics analysis of immune infiltrates and tripartite motif (TRIM) family genes in hepatocellular carcinoma. J Gastrointest Oncol (2022) 13(4):1942–58. doi: 10.21037/jgo-22-619
34. Zhang Z, Xu C, Zhang X, Huang L, Zheng C, Chen H, et al. TRIM11 upregulation contributes to proliferation, invasion, and EMT of hepatocellular carcinoma cells. Oncol Res (2017) 25(5):691–9. doi: 10.3727/096504016X14774897404770
35. Dai W, Wang J, Wang Z, Xiao Y, Li J, Hong L, et al. Comprehensive analysis of the prognostic values of the TRIM family in hepatocellular carcinoma. Front Oncol (2021) 11:767644. doi: 10.3389/fonc.2021.767644
36. Boumahdi S, de Sauvage FJ. The great escape: tumor cell plasticity in resistance to targeted therapy. Nat Rev Drug Discovery (2020) 19(1):39–56. doi: 10.1038/s41573-019-0044-1
37. Arbour KC, Riely GJ. Systemic therapy for locally advanced and metastatic non-small cell lung cancer: A review. Jama. (2019) 322(8):764–74. doi: 10.1001/jama.2019.11058
38. Liang M, Wang L, Sun Z, Chen X, Wang H, Qin L, et al. E3 ligase TRIM15 facilitates non-small cell lung cancer progression through mediating Keap1-Nrf2 signaling pathway. Cell Commun Signal (2022) 20(1):62. doi: 10.1186/s12964-022-00875-7
39. Zhong T, Zhang J, Liu X, Li H. TRIM17-mediated ubiquitination and degradation of RBM38 promotes cisplatin resistance in non-small cell lung cancer. Cell Oncol (Dordr). (2023). 46(5):1493–1507. doi: 10.21203/rs.3.rs-2164253/v1
40. Jiang J, Ren H, Xu Y, Wudu M, Wang Q, Liu Z, et al. TRIM67 promotes the proliferation, migration, and invasion of non-small-cell lung cancer by positively regulating the notch pathway. J Cancer. (2020) 11(5):1240–9. doi: 10.7150/jca.38286
41. Pan X, Chen Y, Shen Y, Tantai J. Knockdown of TRIM65 inhibits autophagy and cisplatin resistance in A549/DDP cells by regulating miR-138-5p/ATG7. Cell Death Dis (2019) 10(6):429. doi: 10.1038/s41419-019-1660-8
42. Zhan W, Han T, Zhang C, Xie C, Gan M, Deng K, et al. TRIM59 promotes the proliferation and migration of non-small cell lung cancer cells by upregulating cell cycle related proteins. PloS One (2015) 10(11):e0142596. doi: 10.1371/journal.pone.0142596
43. Zhang J, Xu Z, Yu B, Xu J, Yu B. Tripartite motif containing 35 contributes to the proliferation, migration, and invasion of lung cancer cells in vitro and in vivo. Biosci Rep (2020) 40(4). doi: 10.1042/BSR20200065
44. Luo Q, Lin H, Ye X, Huang J, Lu S, Xu L. Trim44 facilitates the migration and invasion of human lung cancer cells via the NF-kappaB signaling pathway. Int J Clin Oncol (2015) 20(3):508–17. doi: 10.1007/s10147-014-0752-9
45. Yu B, Zhou Y, He J. TRIM13 inhibits cell proliferation and induces autophagy in lung adenocarcinoma by regulating KEAP1/NRF2 pathway. Cell Cycle (Georgetown Tex). (2023) 22(12):1496–513. doi: 10.1080/15384101.2023.2216504
46. Xu L, Wu Q, Zhou X, Wu Q, Fang M. TRIM13 inhibited cell proliferation and induced cell apoptosis by regulating NF-kappaB pathway in non-small-cell lung carcinoma cells. Gene. (2019) 715:144015. doi: 10.1016/j.gene.2019.144015
47. Wang N, Zhang T. Downregulation of microRNA-135 promotes sensitivity of non-small cell lung cancer to gefitinib by targeting TRIM16. Oncol Res (2018) 26(7):1005–14. doi: 10.3727/096504017X15144755633680
48. Chen S, Sanjana NE, Zheng K, Shalem O, Lee K, Shi X, et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell. (2015) 160(6):1246–60. doi: 10.1016/j.cell.2015.02.038
49. Chow RD, Wang G, Ye L, Codina A, Kim HR, Shen L, et al. In vivo profiling of metastatic double knockouts through CRISPR-Cpf1 screens. Nat Methods (2019) 16(5):405–8. doi: 10.1038/s41592-019-0371-5
50. Jin G, Zhang Z, Wan J, Wu X, Liu X, Zhang W. G3BP2: structure and function. Pharmacol Res (2022) 186:106548. doi: 10.1016/j.phrs.2022.106548
51. Bai P, Fan T, Sun G, Wang X, Zhao L, Zhong R. The dual role of DNA repair protein MGMT in cancer prevention and treatment. DNA Repair (Amst). (2023) 123:103449. doi: 10.1016/j.dnarep.2023.103449
52. Li X, Yang C, Luo N, Yang Y, Guo Y, Chen P, et al. Ubiquitination and degradation of MGMT by TRIM72 increases the sensitivity of uveal melanoma cells to Dacarbazine treatment. Cancer biomark (2022) 34(2):275–84. doi: 10.3233/CBM-210345
53. Wang Z, Li H, Wang H, Li X, Zhang Q, Wang H, et al. TRIM72 exerts antitumor effects in breast cancer and modulates lactate production and MCT4 promoter activity by interacting with PPP3CA. Anticancer Drugs (2022) 33(5):489–501. doi: 10.1097/CAD.0000000000001304
54. Wu Y, Zhang X, Wei X, Feng H, Hu B, Deng Z, et al. Development of an individualized ubiquitin prognostic signature for clear cell renal cell carcinoma. Front Cell Dev Biol (2021) 9:684643. doi: 10.3389/fcell.2021.684643
55. Ammirante M, Shalapour S, Kang Y, Jamieson CAM, Karin M. Tissue injury and hypoxia promote Malignant progression of prostate cancer by inducing CXCL13 expression in tumor myofibroblasts. Proc Natl Acad Sci United States America. (2014) 111(41):14776–81. doi: 10.1073/pnas.1416498111
56. He Y, Zha J, Wang Y, Liu W, Yang X, Yu P. Tissue damage-associated "danger signals" influence T-cell responses that promote the progression of preneoplasia to cancer. Cancer Res (2013) 73(2):629–39. doi: 10.1158/0008-5472.CAN-12-2704
57. Wu X-Z. Cancer and chronic tissue injury: abnormal repair tissue or functional repair tissue? Med Hypotheses (2006) 67(3):676–7. doi: 10.1016/j.mehy.2006.03.022
58. Cai C, Masumiya H, Weisleder N, Pan Z, Nishi M, Komazaki S, et al. MG53 regulates membrane budding and exocytosis in muscle cells. J Biol Chem (2009) 284(5):3314–22. doi: 10.1074/jbc.M808866200
59. Weisleder N, Takeshima H, Ma J. Mitsugumin 53 (MG53) facilitates vesicle trafficking in striated muscle to contribute to cell membrane repair. Communicative Integr Biol (2009) 2(3):225–6. doi: 10.4161/cib.2.3.8077
60. McElhanon KE, Young N, Hampton J, Paleo BJ, Kwiatkowski TA, Beck EX, et al. Autoantibodies targeting TRIM72 compromise membrane repair and contribute to inflammatory myopathy. J Clin Invest (2020) 130(8):4440–55. doi: 10.1172/JCI131721
61. Wang C, Wang H, Wu D, Hu J, Wu W, Zhang Y, et al. A novel perspective for burn-induced myopathy: Membrane repair defect. Sci Rep (2016) 6:31409. doi: 10.1038/srep31409
62. Tocchetti CG, Ameri P, de Boer RA, D'Alessandra Y, Russo M, Sorriento D, et al. Cardiac dysfunction in cancer patients: beyond direct cardiomyocyte damage of anticancer drugs: novel cardio-oncology insights from the joint 2019 meeting of the ESC Working Groups of Myocardial Function and Cellular Biology of the Heart. Cardiovasc Res (2020) 116(11):1820–34. doi: 10.1093/cvr/cvaa222
63. Scott JM, Nilsen TS, Gupta D, Jones LW. Exercise therapy and cardiovascular toxicity in cancer. Circulation. (2018) 137(11):1176–91. doi: 10.1161/CIRCULATIONAHA.117.024671
64. Nemeth DV, Baldini E, Sorrenti S, D'Andrea V, Bellini MI. Cancer metabolism and ischemia-reperfusion injury: two sides of the same coin. J Clin Med (2022) 11(17):5096. doi: 10.3390/jcm11175096
65. Peired AJ, Lazzeri E, Guzzi F, Anders HJ, Romagnani P. From kidney injury to kidney cancer. Kidney Int (2021) 100(1):55–66. doi: 10.1016/j.kint.2021.03.011
66. Bridoux F, Cockwell P, Glezerman I, Gutgarts V, Hogan JJ, Jhaveri KD, et al. Kidney injury and disease in patients with hematological Malignancies. Nat Rev Nephrol. (2021) 17(6):386–401. doi: 10.1038/s41581-021-00405-7
67. Liu F, Wang Z, Li X, Zhang Z, Yang Y, Chen J, et al. Comparative risk of acute kidney injury among cancer patients treated with immune checkpoint inhibitors. Cancer Commun (Lond). (2023) 43(2):214–24. doi: 10.1002/cac2.12396
68. Duann P, Li H, Lin P, Tan T, Wang Z, Chen K, et al. MG53-mediated cell membrane repair protects against acute kidney injury. Sci Trans Med (2015) 7(279):279ra36. doi: 10.1126/scitranslmed.3010755
69. Liu C, Hu Y-H, Han Y, Wang Y-B, Zhang Y, Zhang X-Q, et al. MG53 protects against contrast-induced acute kidney injury by reducing cell membrane damage and apoptosis. Acta pharmacologica Sinica. (2020) 41(11):1457–64. doi: 10.1038/s41401-020-0420-8
70. Li H, Duann P, Li Z, Zhou X, Ma J, Rovin BH, et al. The cell membrane repair protein MG53 modulates transcription factor NF-κB signaling to control kidney fibrosis. Kidney Int (2022) 101(1):119–30. doi: 10.1016/j.kint.2021.09.027
71. Lijie G, Yueyue Z, Nan Z, Ling W, Xuan W, Weijie Y. Mitsugumin 53 promotes mitochondrial autophagy through regulating Ambra1 expression in C2C12 myoblast cells. Cell Biol Int (2019) 43(3):290–8. doi: 10.1002/cbin.11097
72. Gosens R, Giangreco A, Sahai E, Chambers RC. Mechanistic overlap between chronic lung injury and cancer: ERS Lung Science Conference 2017 report. Eur Respir Rev (2017) 26(144):170060. doi: 10.1183/16000617.0060-2017
73. Kitamura J, Uemura M, Kurozumi M, Sonobe M, Manabe T, Hiai H, et al. Chronic lung injury by constitutive expression of activation-induced cytidine deaminase leads to focal mucous cell metaplasia and cancer. PloS One (2015) 10(2):e0117986. doi: 10.1371/journal.pone.0117986
74. Bernchou U, Christiansen RL, Asmussen JT, Schytte T, Hansen O, Brink C. Extent and computed tomography appearance of early radiation induced lung injury for non-small cell lung cancer. Radiother Oncol (2017) 123(1):93–8. doi: 10.1016/j.radonc.2017.02.001
75. Alam N, Park BJ, Wilton A, Seshan VE, Bains MS, Downey RJ, et al. Incidence and risk factors for lung injury after lung cancer resection. Ann Thorac Surg (2007) 84(4):1085–91. doi: 10.1016/j.athoracsur.2007.05.053
76. Shibaki R, Ozawa Y, Noguchi S, Murakami Y, Takase E, Azuma Y, et al. Impact of pre-existing interstitial lung abnormal shadow on lung injury development and severity in patients of non-small cell lung cancer treated with osimertinib. Cancer Med (2022) 11(20):3743–50. doi: 10.1002/cam4.4750
77. Satoh T, Gemma A, Kudoh S, Sakai F, Yamaguchi K, Watanabe T, et al. Incidence and clinical features of drug-induced lung injury in patients with advanced colorectal cancer receiving cetuximab: results of a prospective multicenter registry. Jpn J Clin Oncol (2014) 44(11):1032–9. doi: 10.1093/jjco/hyu128
78. Wang C, Chen WJ, Wu YF, You P, Zheng SY, Liu CC, et al. The extent of liver injury determines hepatocyte fate toward senescence or cancer. Cell Death Dis (2018) 9(5):575. doi: 10.1038/s41419-018-0622-x
79. Jia Y, Chen K, Lin P, Lieber G, Nishi M, Yan R, et al. Treatment of acute lung injury by targeting MG53-mediated cell membrane repair. Nat Commun (2014) 5:4387. doi: 10.1038/ncomms5387
80. Li H, Rosas L, Li Z, Bian Z, Li X, Choi K, et al. MG53 attenuates nitrogen mustard-induced acute lung injury. J Cell Mol Med (2022) 26(7):1886–95. doi: 10.1111/jcmm.16917
81. Cong X, Nagre N, Herrera J, Pearson AC, Pepper I, Morehouse R, et al. TRIM72 promotes alveolar epithelial cell membrane repair and ameliorates lung fibrosis. Respir Res (2020) 21(1):132. doi: 10.1186/s12931-020-01384-2
82. Whitson BA, Mulier K, Li H, Zhou X, Cai C, Black SM, et al. MG53 as a novel therapeutic protein to treat acute lung injury. Military Med (2021) 186(Suppl 1):339–45. doi: 10.1093/milmed/usaa313
83. Nagre N, Cong X, Ji H-L, Schreiber JM, Fu H, Pepper I, et al. Inhaled TRIM72 protein protects ventilation injury to the lung through injury-guided cell repair. Am J Respir Cell Mol Biol (2018) 59(5):635–47. doi: 10.1165/rcmb.2017-0364OC
84. Nagre N, Wang S, Kellett T, Kanagasabai R, Deng J, Nishi M, et al. TRIM72 modulates caveolar endocytosis in repair of lung cells. Am J Physiol Lung Cell Mol Physiol (2016) 310(5):L452–L64. doi: 10.1152/ajplung.00089.2015
85. Kim SC, Kellett T, Wang S, Nishi M, Nagre N, Zhou B, et al. TRIM72 is required for effective repair of alveolar epithelial cell wounding. Am J Physiol Lung Cell Mol Physiol (2014) 307(6):L449–L59. doi: 10.1152/ajplung.00172.2014
86. Bahirwani R, Reddy KR. Drug-induced liver injury due to cancer chemotherapeutic agents. Semin Liver Dis (2014) 34(2):162–71. doi: 10.1055/s-0034-1375957
87. Houron C, Danielou M, Mir O, Fromenty B, Perlemuter G, Voican CS. Multikinase inhibitor-induced liver injury in patients with cancer: A review for clinicians. Crit Rev Oncol Hematol (2021) 157:103127. doi: 10.1016/j.critrevonc.2020.103127
88. De Martin E, Michot JM, Papouin B, Champiat S, Mateus C, Lambotte O, et al. Characterization of liver injury induced by cancer immunotherapy using immune checkpoint inhibitors. J Hepatol (2018) 68(6):1181–90. doi: 10.1016/j.jhep.2018.01.033
89. Colombo M, Lleo A. Is liver injury an affordable risk of immune checkpoint inhibitor therapy for cancer? Gastroenterology (2018) 155(6):2021–3. doi: 10.1053/j.gastro.2018.11.016
90. Han Y, Black S, Gong Z, Chen Z, Ko J-K, Zhou Z, et al. Membrane-delimited signaling and cytosolic action of MG53 preserve hepatocyte integrity during drug-induced liver injury. J Hepatology. (2022) 76(3):558–67. doi: 10.1016/j.jhep.2021.10.017
91. Jaeschke H, Umbaugh DS. Protection against acetaminophen-induced liver injury with MG53: Muscle-liver axis and necroptosis. J Hepatology. (2022) 77(2):560–2. doi: 10.1016/j.jhep.2022.02.027
92. Yao W, Li H, Han X, Chen C, Zhang Y, Tai WL, et al. MG53 anchored by dysferlin to cell membrane reduces hepatocyte apoptosis which induced by ischaemia/reperfusion injury in vivo and in vitro. J Cell Mol Med (2017) 21(10):2503–13. doi: 10.1111/jcmm.13171
93. Dias C, Nylandsted J. Plasma membrane integrity in health and disease: significance and therapeutic potential. Cell Discovery (2021) 7(1):4. doi: 10.1038/s41421-020-00233-2
94. Demonbreun AR, Quattrocelli M, Barefield DY, Allen MV, Swanson KE, McNally EM. An actin-dependent annexin complex mediates plasma membrane repair in muscle. J Cell Biol (2016) 213(6):705–18. doi: 10.1083/jcb.201512022
95. Han R. Muscle membrane repair and inflammatory attack in dysferlinopathy. Skeletal muscle. (2011) 1(1):10. doi: 10.1186/2044-5040-1-10
96. Lauritzen SP, Boye TL, Nylandsted J. Annexins are instrumental for efficient plasma membrane repair in cancer cells. Semin Cell Dev Biol (2015) 45:32–8. doi: 10.1016/j.semcdb.2015.10.028
97. Heitmann ASB, Zanjani AAH, Klenow MB, Mularski A, Sønder SL, Lund FW, et al. Phenothiazines alter plasma membrane properties and sensitize cancer cells to injury by inhibiting annexin-mediated repair. J Biol Chem (2021) 297(2):101012. doi: 10.1016/j.jbc.2021.101012
98. Bouvet F, Ros M, Bonedeau E, Croissant C, Frelin L, Saltel F, et al. Defective membrane repair machinery impairs survival of invasive cancer cells. Sci Rep (2020) 10(1):21821. doi: 10.1038/s41598-020-77902-5
99. Greten FR, Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
100. Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol (2015) 12(10):584–96. doi: 10.1038/nrclinonc.2015.105
101. Yang L, Xia H. TRIM proteins in inflammation: from expression to emerging regulatory mechanisms. Inflammation. (2021) 44(3):811–20. doi: 10.1007/s10753-020-01394-8
102. Guan F, Zhou X, Li P, Wang Y, Liu M, Li F, et al. MG53 attenuates lipopolysaccharide-induced neurotoxicity and neuroinflammation via inhibiting TLR4/NF-κB pathway in vitro and in vivo. Prog Neuropsychopharmacol Biol Psychiatry (2019) 95:109684. doi: 10.1016/j.pnpbp.2019.109684
103. Tan S, Li M, Song X. MG53 alleviates airway inflammatory responses by regulating nuclear factor-κB pathway in asthmatic mice. Allergol Immunopathol (Madr). (2023) 51(4):175–81. doi: 10.15586/aei.v51i4.880
104. Sermersheim M, Kenney AD, Lin P-H, McMichael TM, Cai C, Gumpper K, et al. MG53 suppresses interferon-β and inflammation via regulation of ryanodine receptor-mediated intracellular calcium signaling. Nat Commun (2020) 11(1):3624. doi: 10.1038/s41467-020-17177-6
105. Wu M, Li H, He J, Liang J, Liu Y, Zhang W. TRIM72 Alleviates Muscle Inflammation in mdx Mice via Promoting Mitophagy-Mediated NLRP3 Inflammasome Inactivation. Oxid Med Cell longevity. (2023) 2023:8408574. doi: 10.1155/2023/8408574
106. Ma S, Wang Y, Zhou X, Li Z, Zhang Z, Wang Y, et al. MG53 Protects hUC-MSCs against Inflammatory Damage and Synergistically Enhances Their Efficacy in Neuroinflammation Injured Brain through Inhibiting NLRP3/Caspase-1/IL-1β Axis. ACS Chem Neurosci (2020) 11(17):2590–601. doi: 10.1021/acschemneuro.0c00268
107. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. (2000) 407(6801):249–57. doi: 10.1038/35025220
108. Dong J, Zhou H, Li Y, Li R, Chen N, Zheng Y, et al. MG53 inhibits angiogenesis through regulating focal adhesion kinase signaling. J Cell Mol Med (2021) 25(15):7462–71. doi: 10.1111/jcmm.16777
109. Zeng W, Yin X, Jiang Y, Jin L, Liang W. PPARα at the crossroad of metabolic-immune regulation in cancer. FEBS J (2022) 289(24):7726–39. doi: 10.1111/febs.16181
110. Han X, Chen D, Liufu N, Ji F, Zeng Q, Yao W, et al. MG53 protects against sepsis-induced myocardial dysfunction by upregulating peroxisome proliferator-activated receptor-α. Oxid Med Cell longevity. (2020) 2020:7413693. doi: 10.1155/2020/7413693
111. Chiefari E, Mirabelli M, La Vignera S, Tanyolaç S, Foti DP, Aversa A, et al. insulin resistance and cancer: in search for a causal link. I Int J Mol Sci (2021) 22(20):11137. doi: 10.3390/ijms222011137
112. Godsland IF. Insulin resistance and hyperinsulinemia in the development and progression of cancer. Clin Sci (London Engl 1979). (2009) 118(5):315–32. doi: 10.1042/CS20090399
113. Dev R, Bruera E, Dalal S. Insulin resistance and body composition in cancer patients. Ann Oncol (2018) 29(suppl_2):ii18–26. doi: 10.1093/annonc/mdx815
114. Kim D-S, Scherer PE. Obesity, diabetes, and increased cancer progression. Diabetes Metab J (2021) 45(6):799–812. doi: 10.4093/dmj.2021.0077
115. He S, Wang J, Shen X, Qian X, An Y, Gong Q, et al. Cancer and its predictors in Chinese adults with newly diagnosed diabetes and impaired glucose tolerance (IGT): a 30-year follow-up of the Da Qing IGT and Diabetes Study. Br J Cancer. (2022) 127(1):102–8. doi: 10.1038/s41416-022-01758-x
116. Dankner R, Chetrit A, Segal P. Glucose tolerance status and 20 year cancer incidence. Israel Med Assoc J IMAJ. (2007) 9(8):592–6.
117. Saydah SH, Loria CM, Eberhardt MS, Brancati FL. Abnormal glucose tolerance and the risk of cancer death in the United States. Am J Epidemiol (2003) 157(12):1092–100. doi: 10.1093/aje/kwg100
118. Harding JL, Soderberg S, Shaw JE, Zimmet PZ, Pauvaday V, Kowlessur S, et al. All-cause cancer mortality over 15 years in multi-ethnic Mauritius: the impact of diabetes and intermediate forms of glucose tolerance. Int J Cancer. (2012) 131(10):2385–93. doi: 10.1002/ijc.27503
119. Hirakawa Y, Ninomiya T, Mukai N, Doi Y, Hata J, Fukuhara M, et al. Association between glucose tolerance level and cancer death in a general Japanese population: the Hisayama Study. Am J Epidemiol (2012) 176(10):856–64. doi: 10.1093/aje/kws178
120. Ariaans G, de Jong S, Gietema JA, Lefrandt JD, de Vries EGE, Jalving M. Cancer-drug induced insulin resistance: innocent bystander or unusual suspect. Cancer Treat Rev (2015) 41(4):376–84. doi: 10.1016/j.ctrv.2015.02.007
121. Song R, Peng W, Zhang Y, Lv F, Wu H-K, Guo J, et al. Central role of E3 ubiquitin ligase MG53 in insulin resistance and metabolic disorders. Nature. (2013) 494(7437):375–9. doi: 10.1038/nature11834
122. Park JS, Lee H, Choi BW, Ro S, Lee D, Na JE, et al. An MG53-IRS1-interaction disruptor ameliorates insulin resistance. Exp Mol Med (2018) 50(6):1–12. doi: 10.1038/s12276-018-0099-9
123. Jiang P, Ren L, Zhi L, Yu Z, Lv F, Xu F, et al. Negative regulation of AMPK signaling by high glucose via E3 ubiquitin ligase MG53. Mol Cell (2021) 81(3):629–637.e5. doi: 10.1016/j.molcel.2020.12.008
124. Liu F, Song R, Feng Y, Guo J, Chen Y, Zhang Y, et al. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor α. Circulation. (2015) 131(9):795–804. doi: 10.1161/CIRCULATIONAHA.114.012285
125. Yi J-S, Park JS, Ham Y-M, Nguyen N, Lee N-R, Hong J, et al. MG53-induced IRS-1 ubiquitination negatively regulates skeletal myogenesis and insulin signalling. Nat Commun (2013) 4:2354. doi: 10.1038/ncomms3354
126. Lee H, Park J-J, Nguyen N, Park JS, Hong J, Kim S-H, et al. MG53-IRS-1 (Mitsugumin 53-insulin receptor substrate-1) interaction disruptor sensitizes insulin signaling in skeletal muscle. J Biol Chem (2016) 291(52):26627–35. doi: 10.1074/jbc.M116.754424
127. Feng H, Shen H, Robeson MJ, Wu Y-H, Wu H-K, Chen G-J, et al. MG53 E3 ligase-dead mutant protects diabetic hearts from acute ischemic/reperfusion injury and ameliorates diet-induced cardiometabolic damage. Diabetes. (2022) 71(2):298–314. doi: 10.2337/db21-0322
128. Lv F, Wang Y, Shan D, Guo S, Chen G, Jin L, et al. Blocking MG53S255 Phosphorylation Protects Diabetic Heart From Ischemic Injury. Circ Res (2022) 131(12):962–76. doi: 10.1161/CIRCRESAHA.122.321055
129. Ma H, Liu J, Bian Z, Cui Y, Zhou X, Zhou X, et al. Effect of metabolic syndrome on mitsugumin 53 expression and function. PloS One (2015) 10(5):e0124128. doi: 10.1371/journal.pone.0124128
130. Bian Z, Wang Q, Zhou X, Tan T, Park KH, Kramer HF, et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat Commun (2019) 10(1):4659. doi: 10.1038/s41467-019-12483-0
131. Philouze C, Turban S, Cremers B, Caliez A, Lamarche G, Bernard C, et al. MG53 is not a critical regulator of insulin signaling pathway in skeletal muscle. PloS One (2021) 16(2):e0245179. doi: 10.1371/journal.pone.0245179
132. Zhuang L, Bassel-Duby R, Olson EN. Secreted MG53 from striated muscle impairs systemic insulin sensitivity. Circulation. (2019) 139(7):915–7. doi: 10.1161/CIRCULATIONAHA.118.038387
133. Yang S, Zhao H, Xu K, Qian Y, Wu M, Yang T, et al. Evaluation of common variants in MG53 and the risk of type 2 diabetes and insulin resistance in Han Chinese. SpringerPlus. (2016) 5(1):612. doi: 10.1186/s40064-016-2218-1
134. Tsuruzoe K, Emkey R, Kriauciunas KM, Ueki K, Kahn CR. Insulin receptor substrate 3 (IRS-3) and IRS-4 impair IRS-1- and IRS-2-mediated signaling. Mol Cell Biol (2001) 21(1):26–38. doi: 10.1128/MCB.21.1.26-38.2001
135. Laustsen PG, Michael MD, Crute BE, Cohen SE, Ueki K, Kulkarni RN, et al. Lipoatrophic diabetes in Irs1(-/-)/Irs3(-/-) double knockout mice. Genes Dev (2002) 16(24):3213–22. doi: 10.1101/gad.1034802
136. Skovsø S, Panzhinskiy E, Kolic J, Cen HH, Dionne DA, Dai X-Q, et al. Beta-cell specific Insr deletion promotes insulin hypersecretion and improves glucose tolerance prior to global insulin resistance. Nat Commun (2022) 13(1):735. doi: 10.1038/s41467-022-28039-8
137. Bianchi C, Raggi F, Rossi C, Frontoni S, Bonadonna RC, Del Prato S, et al. MG53 marks poor beta cell performance and predicts onset of type 2 diabetes in subjects with different degrees of glucose tolerance. Diabetes Metab (2022) 48(2):101292. doi: 10.1016/j.diabet.2021.101292
138. Argilés JM, Stemmler B, López-Soriano FJ, Busquets S. Inter-tissue communication in cancer cachexia. Nat Rev Endocrinol (2018) 15(1):9–20. doi: 10.1038/s41574-018-0123-0
139. Argilés JM, Busquets S, Stemmler B, López-Soriano FJ. Cancer cachexia: understanding the molecular basis. Nat Rev Cancer. (2014) 14(11):754–62. doi: 10.1038/nrc3829
140. Setiawan T, Sari IN, Wijaya YT, Julianto NM, Muhammad JA, Lee H, et al. Cancer cachexia: molecular mechanisms and treatment strategies. J Hematol Oncol (2023) 16(1):54. doi: 10.1186/s13045-023-01454-0
141. Yeom E, Yu K. Understanding the molecular basis of anorexia and tissue wasting in cancer cachexia. Exp Mol Med (2022) 54(4):426–32. doi: 10.1038/s12276-022-00752-w
142. Schmidt SF, Rohm M, Herzig S, Berriel Diaz M. Cancer cachexia: more than skeletal muscle wasting. Trends Cancer. (2018) 4(12):849–60. doi: 10.1016/j.trecan.2018.10.001
143. Lima JDCC, Simoes E, de Castro G, Morais MRPT, de Matos-Neto EM, Alves MJ, et al. Tumour-derived transforming growth factor-β signalling contributes to fibrosis in patients with cancer cachexia. J cachexia sarcopenia muscle. (2019) 10(5):1045–59. doi: 10.1002/jcsm.12441
144. Hagg A, Kharoud S, Goodchild G, Goodman CA, Chen JL, Thomson RE, et al. TMEPAI/PMEPA1 is a positive regulator of skeletal muscle mass. Front Physiol (2020) 11:560225. doi: 10.3389/fphys.2020.560225
145. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest (2017) 127(10):3770–83. doi: 10.1172/JCI94753
146. Lee CS, Yi JS, Jung SY, Kim BW, Lee NR, Choo HJ, et al. TRIM72 negatively regulates myogenesis via targeting insulin receptor substrate-1. Cell Death differentiation. (2010) 17(8):1254–65. doi: 10.1038/cdd.2010.1
147. Ham Y-M, Mahoney SJ. Compensation of the AKT signaling by ERK signaling in transgenic mice hearts overexpressing TRIM72. Exp Cell Res (2013) 319(10):1451–62. doi: 10.1016/j.yexcr.2013.02.016
148. Shihata WA, Putra MRA, Chin-Dusting JPF. Is there a potential therapeutic role for caveolin-1 in fibrosis? Front Pharmacol (2017) 8:567. doi: 10.3389/fphar.2017.00567
149. Wu S-J, He R-L, Zhao L, Yu X-Y, Jiang Y-N, Guan X, et al. Cardiac-specific overexpression of caveolin-1 in rats with ischemic cardiomyopathy improves arrhythmogenicity and cardiac remodeling. Can J Cardiol (2023) 39(1):73–86. doi: 10.1016/j.cjca.2022.10.005
150. Zhang M, Wang H, Wang X, Bie M, Lu K, Xiao H. MG53/CAV1 regulates transforming growth factor-β1 signaling-induced atrial fibrosis in atrial fibrillation. Cell Cycle (Georgetown Tex). (2020) 19(20):2734–44. doi: 10.1080/15384101.2020.1827183
151. Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharmacol Rev (2020) 72(2):486–526. doi: 10.1124/pr.119.018440
152. Martin A, Gallot YS, Freyssenet D. Molecular mechanisms of cancer cachexia-related loss of skeletal muscle mass: data analysis from preclinical and clinical studies. J cachexia sarcopenia muscle. (2023) 14(3):1150–67. doi: 10.1002/jcsm.13073
Keywords: MG53, cancer, glucose metabolism, membrane repair, insulin resistance
Citation: Du Y, Li T and Yi M (2023) Is MG53 a potential therapeutic target for cancer? Front. Endocrinol. 14:1295349. doi: 10.3389/fendo.2023.1295349
Received: 16 September 2023; Accepted: 01 November 2023;
Published: 15 November 2023.
Edited by:
Conghui Yao, Harvard Medical School, United StatesReviewed by:
Ping Guo, Steadman Philippon Research Institute, United StatesCopyright © 2023 Du, Li and Yi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Muqing Yi, bXVxaW5neWlAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.