 Asma Allouch1
Asma Allouch1 Mashael Al-Shafai
Mashael Al-Shafai Atiyeh M. Abdallah
Atiyeh M. Abdallah
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Endocrinol., 26 April 2024
Sec. Reproduction
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1289333
This article is part of the Research TopicSearching for Causes of Infertility: From Pathophysiologic Mechanisms to Therapeutic StrategiesView all 8 articles
Introduction: Premature ovarian insufficiency (POI) is a primary cause of infertility with variable clinical manifestations. POI is a multifactorial disease with both environmental and known genetic etiologies, but data on the genetic variations associated with POI in the Middle East and North Africa (MENA) region are scarce. The aim of this study was to systematically review all known genetic causes of POI in the MENA region.
Methods: The PubMed, Science Direct, ProQuest, and Embase databases were searched from inception to December 2022 for all reports of genetic variants associated with POI in the MENA region. Clinical and genetic data were collected from eligible articles, and ClinVar and PubMed (dbSNP) were searched for variants.
Results: Of 1,803 studies, 25 met the inclusion criteria. Fifteen studies were case-control studies and ten were case reports representing 1,080 non-syndromic POI patients in total. Seventy-nine variants in 25 genes associated with POI were reported in ten MENA countries. Of the 79 variants, 46 were rare and 33 were common variants. Of the 46 rare variants, 19 were pathogenic or likely pathogenic according to ACMG classification guidelines and ClinVar. No clear phenotype-genotype association was observed. Male family members carrying pathogenic variants also had infertility problems.
Discussion: To our best knowledge, this is the first systematic review of the genetic variants associated with POI in the MENA region. Further functional studies are needed to assess the disease-causing molecular mechanisms of these variants. Knowledge of the genetic basis of POI in the Middle East could facilitate early detection of the condition and thus early implementation of therapeutic interventions, paving the way for precision medicine options in specific populations.
Primary ovarian insufficiency (POI) is a complex and significant medical condition characterized by the abrupt and permanent cessation of menstrual periods in women under the age of 40(PMID 36630623). This condition leads to a unique physiological state, characterized by hypergonadotropic hypoestrogenic amenorrhea, which in turn is a consequence of the depletion of the ovarian follicular pool. POI can manifest itself either before puberty, referred to as primary amenorrhea, or after the onset of menstrual cycles, termed secondary amenorrhea. (PMID 32934798).
Studying POI is of paramount importance as it has far-reaching implications for the affected individuals’ reproductive health, general well-being, and long-term health outcomes. Not only does it signify the loss of fertility, but it also raises concerns about hormonal imbalances, bone health, and cardiovascular risks. Additionally, POI may be a symptom of underlying genetic, autoimmune, or environmental factors that can impact a woman’s overall health. A deeper understanding of POI is essential for early diagnosis, targeted treatment, and support to enhance the quality of life for those affected. This introduction sets the stage for a more comprehensive exploration of the multifaceted aspects of POI, emphasizing its importance in both clinical and research contexts. (PMID33427510).
According to the European Society of Human Reproduction and Embryology (ESHRE), the diagnostic criteria for POI are age, cycle irregularities for four months or more (oligo/amenorrhea), and elevated follicle-stimulating hormone (FSH) levels (>25 IU/L) (1). POI affects around 1% of women aged between 35 and 40 years and 0.1% of women younger than 30 years worldwide. Fifteen percent of patients report a family history of POI, underscoring the strong genetic etiology of the condition (2). Indeed, POI is multifactorial and clinically heterogeneous, and most POI cases are idiopathic or spontaneous, with genetic, autoimmune, and environmental factors contributing to its pathoetiology. Additionally, it has been shown that syndromic POI accounts for 10-20% of POI cases: Turner syndrome in 4-5% of cases and Fragile X syndrome (FMR1 permutations) in 3-15% (3). Twin studies have estimated that the heritability of POI is 53-71%, and up to 40% of POI cases are reported to have genetic causes. This systematic review focuses on reported non-syndromic POI cases, where the majority of variants identified in non-syndromic POI from consanguineous families have been detected in genes important for meiosis, homologous recombination, as well as DNA damage and repair (4).
Relatively recent next-generation (NGS) and whole-exome sequencing (WES) efforts of large POI families have identified new genetic variants associated with the disease (5). However, few have been functionally validated as causative (6). According to the Online Mendelian Inheritance in Man (OMIM) database, BNC1, NOBOX, and NR5A1 show autosomal dominant inheritance; FANCM, GDF9, HFM1, HSF2BP, MCM9, MRAPS22, MSH4, MSH5, STAG3, SPIDR, SYCE1, and ZSWIM7 show autosomal recessive inheritance; and BMP15 shows X-linked inheritance in non-syndromic POI. However, POI genetics is believed to be much more complex than participation of these genes alone (7).
To our best knowledge, this is the first systematic review of the genetic variations associated with non-syndromic POI in the MENA region. Knowledge about the genetic basis on POI in the Middle East paves the way for implementation of precision medicine strategies in this population.
This search strategy for this review followed the Preferred Reporting Items for Systematic Review and Meta-Analysis (PRISMA) guidelines (8). The primary objective of this study was to determine the genetic variations associated with POI in populations across the MENA region. There are several MENA region definitions but, in this review, we followed the Michael and Staley definition, which includes the Arab countries (Qatar, Saudi Arabia, Somalia, Sudan, Syria, Tunisia, United Arab Emirates, Algeria, Bahrain, Djibouti, Egypt, Iraq, Jordan, Kuwait, Lebanon, Libya, Mauritania, Morocco, Oman, Palestine and Yemen) in addition to Turkey and Iran (9). The secondary objective was to report the variability in clinical phenotypes observed in patients diagnosed with POI.
The PubMed, Science Direct, ProQuest, and Scopus databases were searched from inception to October 2022 using key phrases constructed to include both primary and secondary outcomes. The keywords included the terms “primary ovarian insufficiency” OR “premature ovarian failure” AND “North African and Middle Eastern countries” AND “mutation”, “gene”, “variant”, or “polymorphism”. The abstracts and titles of all retrieved articles were initially screened, and articles satisfying the inclusion criteria were then thoroughly evaluated and included in the final analysis.
Two researchers evaluated the retrieved records, and discrepancies was resolved through a consensus with the senior author. The total number of hits from each database was documented. Research articles meeting the following inclusion criteria according to the PICOS statement (Table 1) were fully assessed: 1) publication in a peer-reviewed journal; 2) population from a MENA country according to the Michael and Staley definition (9); and 3) the article explored genetic variants associated with POI. Any research papers that lacked genetic variants associated with POI, studied a population not from a MENA country, or that were reviews, books, protocols, or guidelines were excluded (Table 1). After eliminating duplicates, titles and abstracts of the remaining articles were reviewed, and any record not matching the inclusion criteria was excluded. The full text of these eligible papers was assessed to collect relevant records. All assessments were performed by two researchers with the assistance of the senior author to resolve any disagreements. The screening and selection process in shown in Figure 1.
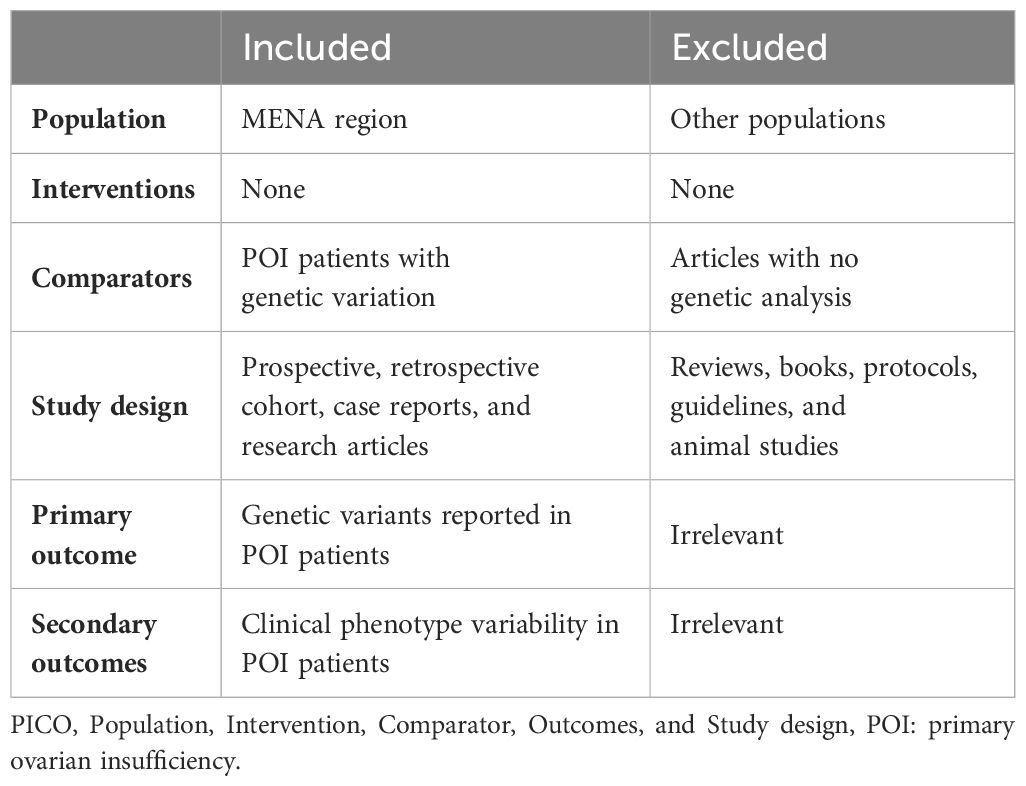
Table 1 Inclusion and exclusion criteria according to the PICOS statement*.
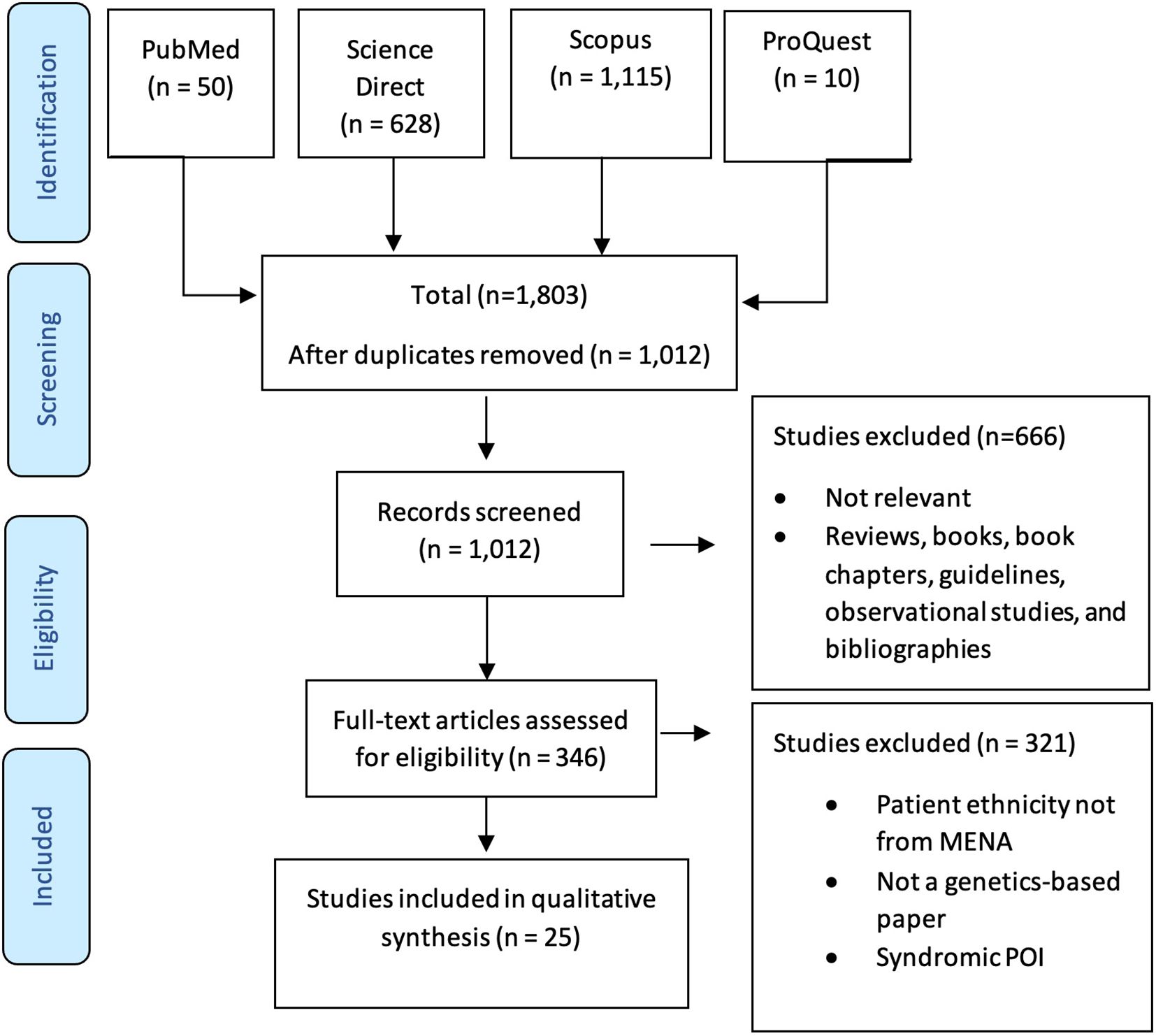
Figure 1 The screening and selection process used in this study according to PRISMA guidelines.
Risk of bias in eligible articles was assessed by two researchers using the Quality Assessment of Diagnostic Accuracy Studies-2 (QUADAS-2) tool (10), which rates sections as “low risk”, “high risk”, or “unclear risk”. Discrepancies were resolved with the senior author opinion. Data items were extracted from both tables and text of eligible articles and compiled in a Microsoft Excel spreadsheet. Two researchers reviewed data accuracy. Gene name, variant, sample size, genetic test used, number of patients tested, proportion with the causative variant, zygosity, consanguinity, gender, and phenotypic information were extracted. (PMID25741868).
Several databases have been consulted to further investigate all the retrieved genetic variants from the 25 included studies. Namely, NCBI ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), NCBI (dbSNP) (https://www.ncbi.nlm.nih.gov/snp/), and Genome Aggregation Database (gnomAD) to obtain the minor allele frequency (MAF) (https://gnomad.broadinstitute.org/). Based on the minor allele frequency in gnomAD, retrieved variants were categorized into rare and common variations, where rare variants were found in less than or equal to 1% of gnomAD population (MAF ≤ 0.01), and common variants were defined as having allele frequency higher than 1% (MAF > 0.01) (PMID: 35117776). Further, to interpret the retrieved variants, the standards and guidelines for the clinical Interpretation of genetic variants by the American College of Medical Genetics and the Association for Molecular Pathology (ACMG/AMP) of 2015 were followed (via https://wintervar.wglab.org/). The ACMG/AMP guidelines entail classifying genetic variants into five categories by integrating several of the typical forms of variant evidence (e.g., population data, computational data, functional data, segregation data), these categories are: Pathogenic (P), Likely Pathogenic (LP), Variant of Uncertain Significance (VUS), Likely Benign (LB), or Benign (B).(PMID: 25741868).
Our search yielded 1,803 studies, of which 1,012 remained after removal of duplicates. The remaining articles were subjected to primary screening by title, abstract, and PICOS assessment for eligibility according to the inclusion and exclusion criteria. Of the screened articles, 346 were eligible for full assessment. Of these, 315 articles were either not genetic studies, patient ethnicity was not from the MENA region, or the study included syndromic POI. Therefore, 25 articles were eligible for inclusion in this review. Table 2 summarizes the included articles.
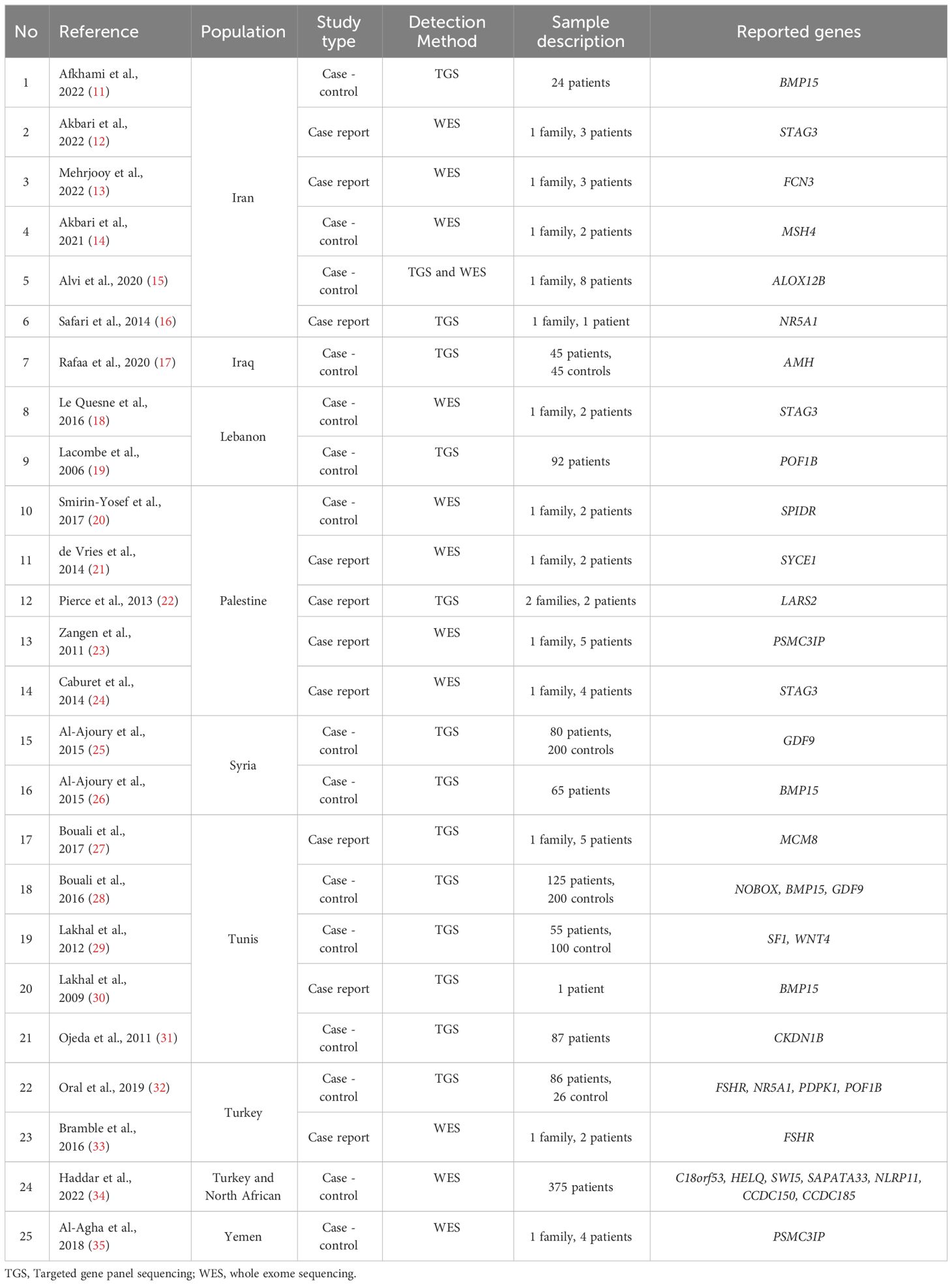
Table 2 Description of the included articles with genetic variants reported in the MENA populations.
The risk of bias assessment in the four domains of the QUADAS-2 tool is shown in Figure 2. The patient selection domain had the highest risk of bias, with 25% of studies showing a high risk of bias. This was expected, as most of the included articles were case reports with preselected participants. The remaining three domains had mainly a low risk of bias.
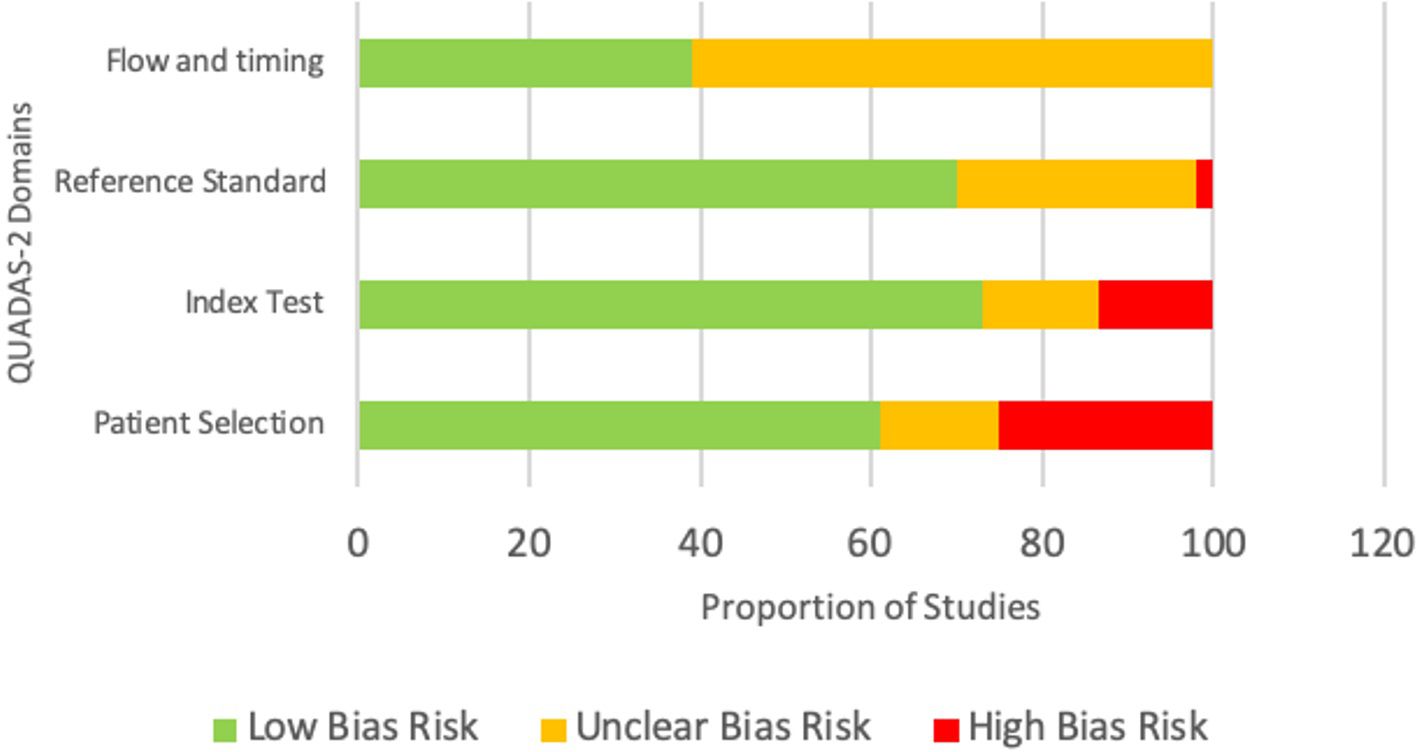
Figure 2 Risk of bias assessment using the QUADAS-2 tool.
In the 25 included articles, there were 1,080 non-syndromic POI participants from 10 familial/case report studies and 15 case-control studies. In total, seventy-nine POI-associated variants were reported in 25 genes. Consanguinity was observed in all the family studies. Genetic testing was through direct gene sequencing (n= 6), targeted gene panel sequencing (n=7), whole genome sequencing (N=2), whole exome sequencing (n=6), or homozygosity mapping and exome sequencing (n= 2). In most cases, the genotype was confirmed by Sanger sequencing.
The 79 POI-causing variants were reported from ten MENA countries: Tunisia, Yemen, Iran, Turkey, Palestine, Lebanon, Syria, Morocco, Iraq, and in Arab Israelis. No POI cases were reported from Algeria, Jordan, Bahrain, Qatar, Mauritania, Libya, Sudan, Somalia, Egypt, Oman, the Comoros Islands, Emirates, Djibouti, Afghanistan, and Cyprus. Table 2 lists the genes reported in the included articles.
Of the 79 variants, 46 variants were rare, while 33 were common polymorphisms. Six polymorphisms were shared in GDF9 (c.447C>T, c.546G>T) in Tunisian and Syrian patients. In BMP15, c.-9C>G and c. 308 A>G were shared among Tunisian and Iranian patients and c.852C>T was shared among Tunisian and Syrian patients. Finally, c.538G>A was shared among all three cohorts.
Of the 46 rare variants, 19 were reported pathogenic or likely pathogenic according to ACMG/AMP guidelines and the ClinVar database (Table 3). In the ClinVar database, seven variants were reported as pathogenic, nine as likely pathogenic, one as a variant of uncertain significance (VUS), and two had no data. However, following the ACMG/AMP guidelines, only two of the 19 variants were found as pathogenic, three as likely pathogenic, seven as VUS, and seven had no data. Most of these variants were detected in the patients in homozygous state (79%, 15 variants), while four variants (21%) were seen in the heterozygous state. Table 4 shows variants reported as VUS by either ACMG/AMP or ClinVar or both. Figure 3 shows the geographical distribution of POI-associated variants in the MENA region.
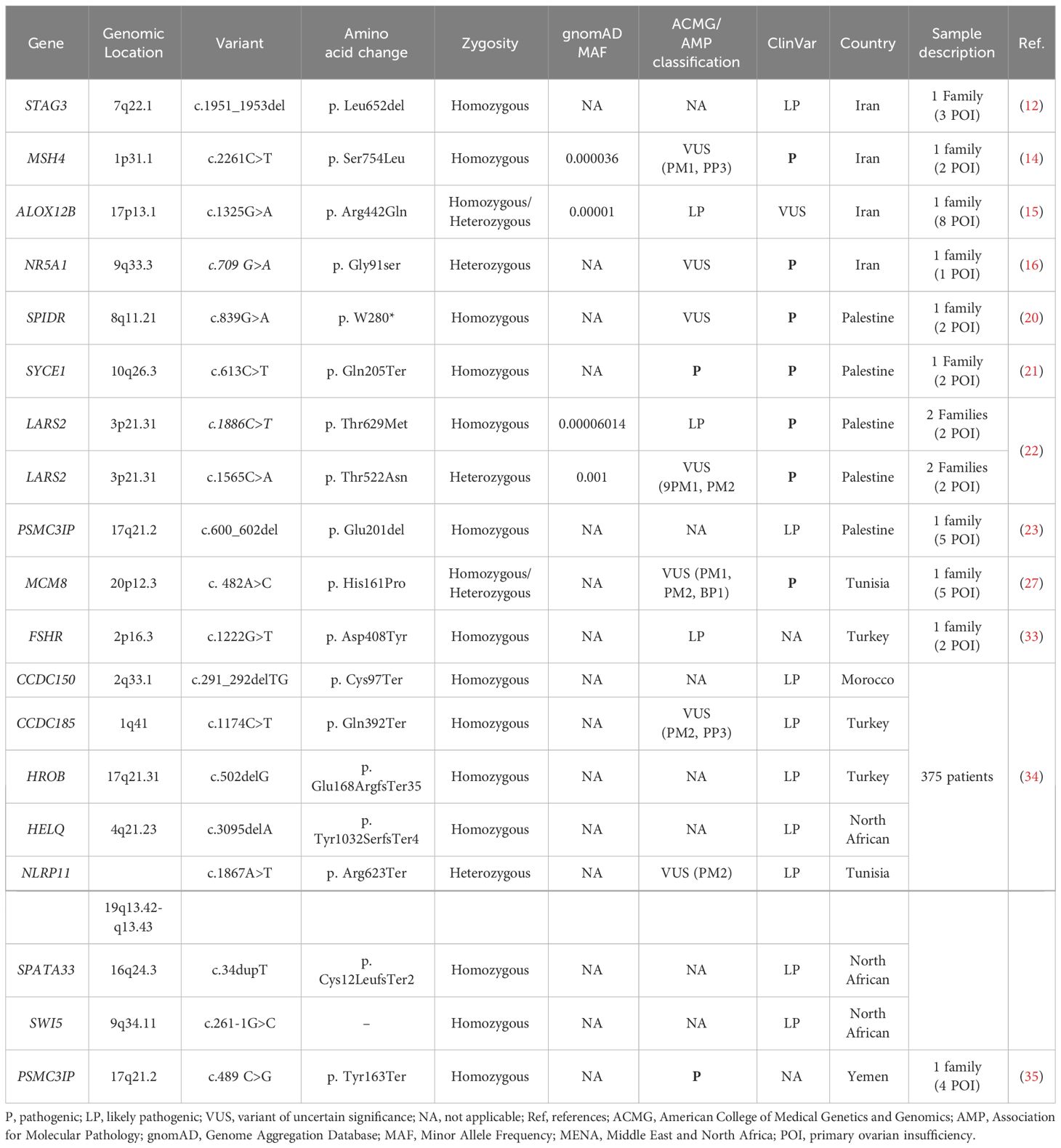
Table 3 Pathogenic and likely pathogenic variants reported in POI patients in MENA populations according to ACMG/AMP criteria and ClinVar.
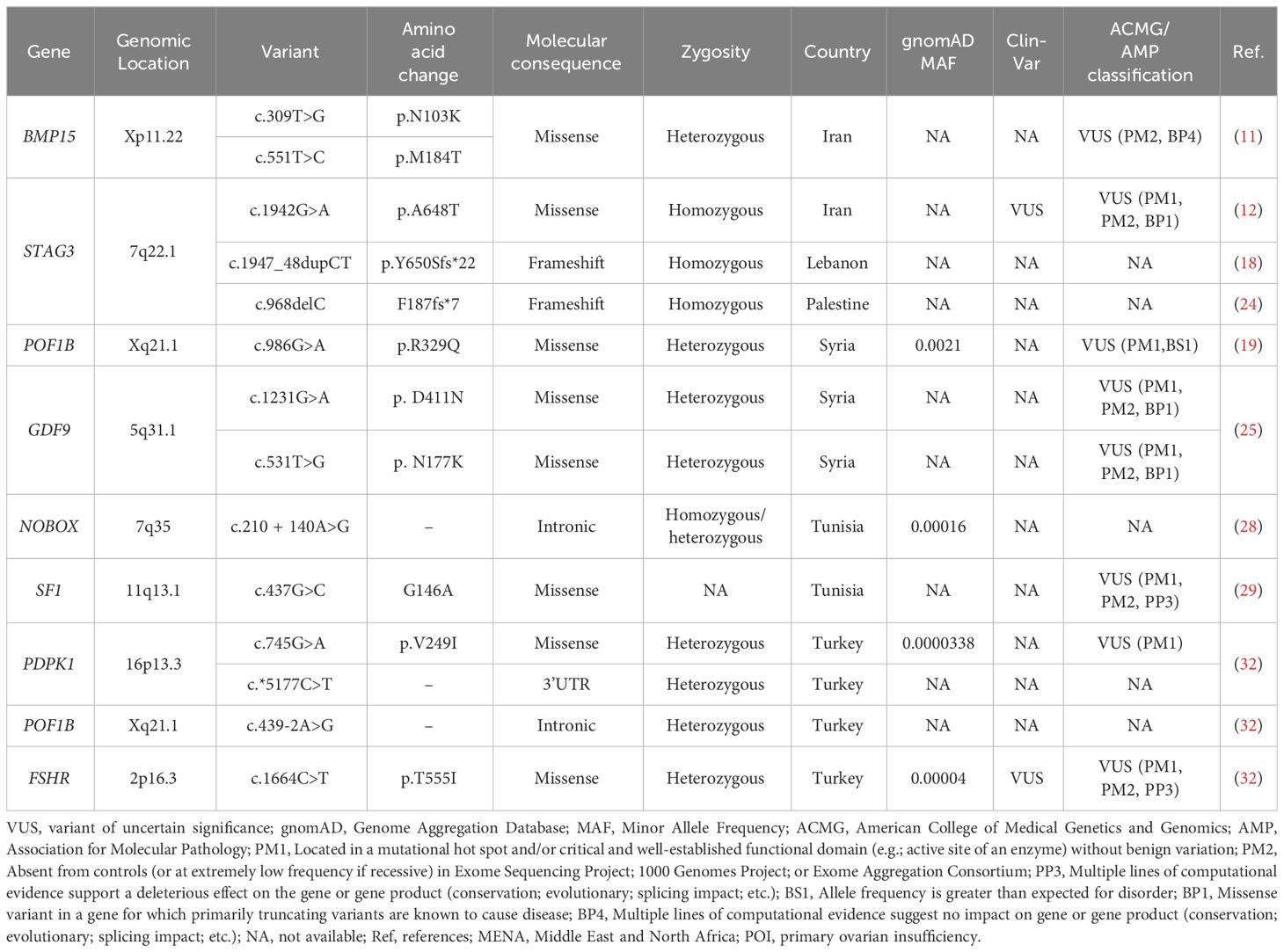
Table 4 Rare variants of uncertain significance reported in POI patients in MENA populations.
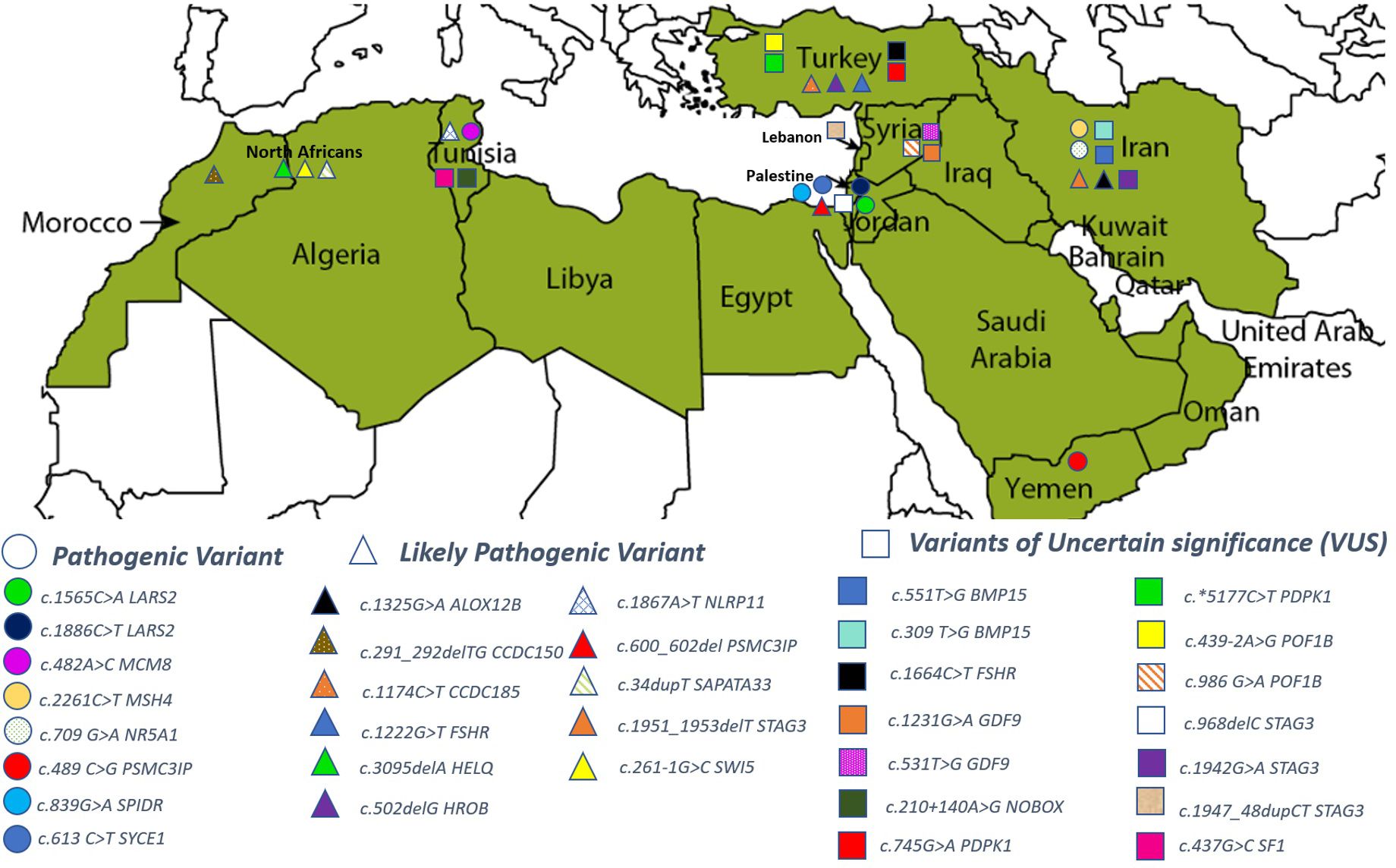
Figure 3 Geographical distribution of POI-associated genetic variants in the MENA region.
Table 5 details the phenotype-genotype correlations in the 1,080 patients from the 25 included articles. High FSH and LH levels were seen in all POI patients. All patients presented with normal karyotyping and negative FMR1 premutation, so met eligibility criteria for unexplained non-syndromic POI. All patients presented with either primary or secondary amenorrhea. Where pelvic ultrasound data were available, most subjects had atrophic ovaries with diminished follicle reserve, small uteri, and bilateral streak gonads. Three studies report infertility in 3 males, all of whom were carriers of the pathogenic variants reported (PSMC3IP, NR5A1, and STAG3 variants). No clear phenotype-genotype correlations were observed.
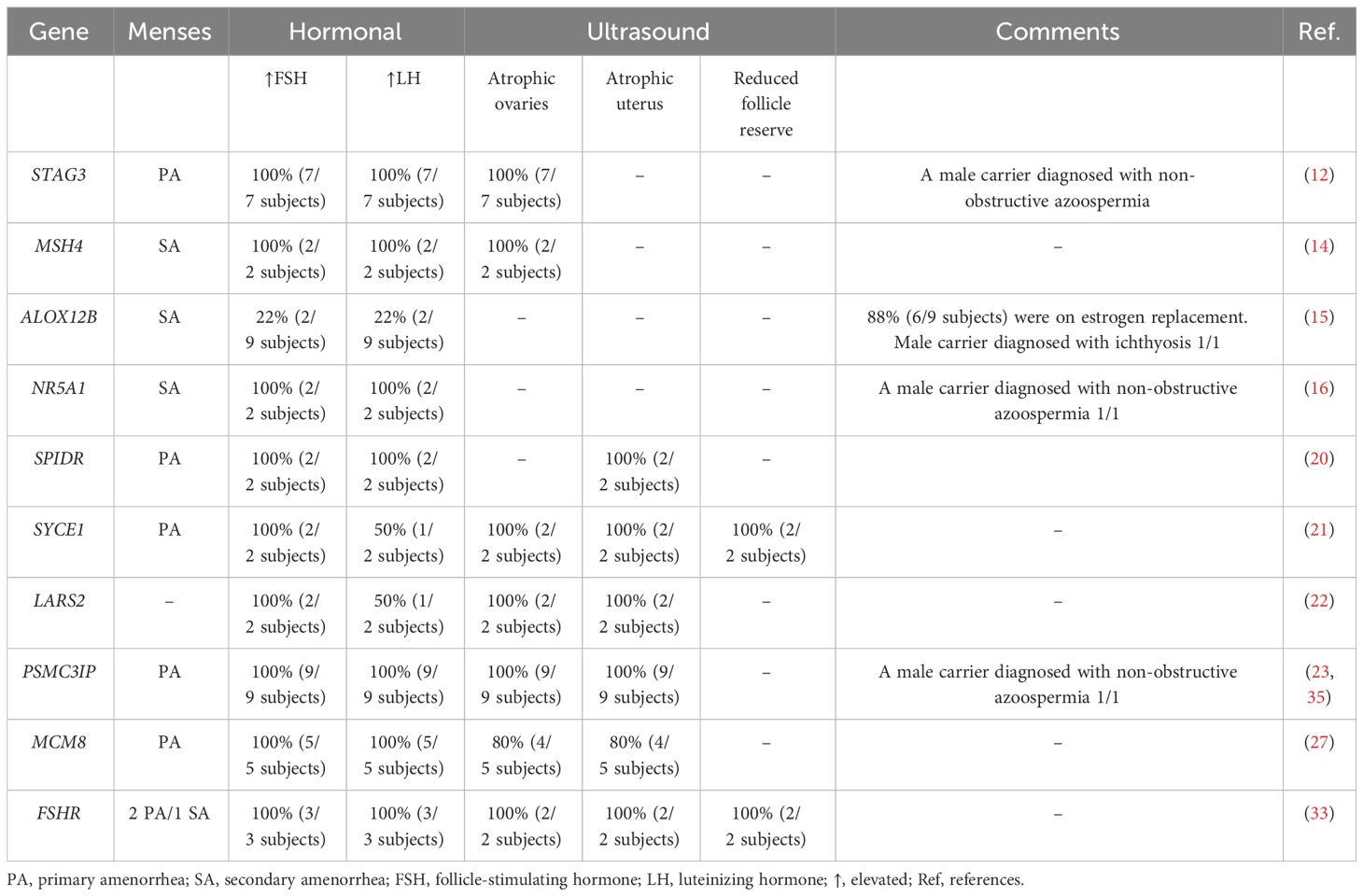
Table 5 Genotype-phenotype correlations reported for the pathogenic and likely pathogenic variants in the MENA region.
Over 80 gene variants have been reported to be causative for POI worldwide (36). In this systematic review conducted on the MENA populations, we captured 79 genetic variants in 25 genes from 25 eligible studies comprising 1,080 cases. Forty-six variants were rare, while 33 were common polymorphisms. Among the 46 rare variants, 19 were pathogenic or likely pathogenic according to ACMG/AMP guidelines and the ClinVar database. Among the 25 reported genes, BMP15 and STAG3 were most frequently reported in MENA populations, with BMP15 reported in four articles and STAG3 in three articles. The most clinically relevant findings included the pathogenic variants in SYCE1, LARS2, MCM8, and NR5A1 and the first report of pathogenic PSMC3IP variants in association with POI in two Arab cases (24, 26). The segregation of pathogenic loss of function variants in PSMC3IP, NR5A1, and STAG3 with POI in 13 female carriers and non-obstructive azoospermia (NOA) in three male carriers in the studied pedigrees suggested a significant role of these variants in infertility in both genders (13, 17, 24, 36).
PSMC3IP encodes PSMC3-interacting protein or HOP2, whose loss of function is associated with ovarian dysgenesis and insufficiency. PSMC3IP plays a role in meiosis and DNA repair (37). In animal models, PSMC3IP knockout results in extreme loss of gametogenesis (38). The pathogenic variant in PSMC3IP (c.600_602del, p.Glu201del) in POI was first reported in 2011 in five females in a Palestinian family, all of whom were homozygous carriers with primary amenorrhea and undetectable ovaries (23). This remained the only report until 2018, when a homozygous stop-gain variant (c.489C>G,p.Tyr163Ter) was reported in four affected Yemini sisters and their brother (35), the sisters presenting with primary amenorrhea, delayed puberty, and very low AMH levels and their affected brother presenting with NOA. This prompted a thorough evaluation of PSMC3IP function. In 2019, compound heterozygous variants (c.496_497delCT, p.Arg166Alafs; c.430_431insGA, p.Leu144Ter) were detected in a woman in a French cohort of 33 POI patients (39). A Fujian woman with POI presented with the same phenotype of primary amenorrhea and similarly harbored a compound heterozygous variant (c.597 + 1G>T and c.268G>C) (40). A woman of Italian origin with secondary amenorrhea was also recently reported to possess biallelic variants (c.206_208delAGA and c.189 G>T) in PSMC3IP, thus expanding the phenotypic spectrum of PSMC3IP in association with POI. Interestingly, the sister of the affected female in this latter study also carried both variants but presented with normal menses and hormonal levels except for low AMH and infertility (41).
STAG3 encodes a subunit of cohesin, a protein complex that functions in chromosome segregation during meiosis (24). STAG3 variants are well established in POI, with reports in over 16 different patients from unrelated families (37). Interestingly, a recent study of familial infertility identified a homozygous mutation in a POI female and her NOA brother (42). Similarly, a study of an Iranian family of one POI female and her two NOA brothers identified double homozygous mutations in STAG3, one of which was predicted to be likely pathogenic according to ClinVar (c.1951_1953del, p. Leu652del) and the other a VUS according to ACMG/AMP guidelines (c.1942G>A, p.Ala648Thr) (12). We additionally found two VUSs in STAG3 (c.1947_48dupCT, p. Y650Sfs*22; c.968delC, p. F187fs*7) in Lebanese and Palestinian patients (18, 24). Additionally, a likely homozygous pathogenic variant (c.1222G>T, p. Asp408Tyr) and a heterozygous VUS (c.1664C > T, p.Thr555Ile) in FSHR were identified in Turkish probands (32, 33). FSHR mutations are implicated with ovarian dysgenesis and POI in an autosomal recessive manner, with over 19 published research articles and case reports (37).
The pathogenic variants reported in ClinVar are SYCE1, LARS2, MCM8, and NR5A1. SYCE1 encodes synaptonemal complex central element protein 1 (SYCP1), an element of the synaptonemal complex, which binds to a homolog associated in meiosis (synapsis) before crossover (43). Each homolog connected through SCYP1 contains SYCE1 and SYCE2, which are crucial for synaptonemal complex assembly and completion of oogenesis (44). Several studies have showed that female infertility can be caused by a failure in building a synaptonemal complex, a potential causative of primary ovarian insufficiency (45). The SYCE1 c.613C>T nucleotide change was the first to be reported to cause POI in two daughters from a Palestinian family in 2014 (21). A year later, the c.197-2 A > G variant was reported in association with azoospermia in two first cousins originally from Iran (46).
A mutation in LARS2, which encodes mitochondrial leucyl-tRNA synthetase, is also potentially causative for POI. LARS2 is clearly associated with molecular defects in mitochondrial function that contribute to the development of POI (47). The LARS2 variant p.Thr522Asn has been reported in three individuals from Palestine with severe hearing loss and POI associated with Perrault syndrome (22, 48). Interestingly, in vitro functional studies of the p. Thr522Asn variant show that the mutation causes a significant decrease in protein activity, which is probably responsible for its deleterious impact (22, 49). Moreover, minichromosomal maintenance protein-8 (MCM8) is known to be an initiator of eukaryotic genome replication. Indeed, MCM8 plays a vital role in meiosis and DNA double-stranded break repair, and it contributes to menopause (50). In 2017, the homozygous c.482A>C mutation in MCM8 was first reported to be deleterious, segregating in a Tunisian family with several affected siblings with POI. In vitro analyses in the same study showed that MCM8 knockout results in small gonads and disrupted follicle development, so may be related to the primary ovarian insufficiency phenotype (27). Another study showed that MCM8 frameshift mutations are pathogenic and may directly cause POI (51). Finally, NR5A1 is associated with a wide spectrum of phenotypes including sex determination, XY sex gonadal dysgenesis, adrenocortical insufficiency, and ovarian failure (52). p.Gly91Ser was the first NR5A1 variant to be reported in two siblings, a man with azoospermia and his sister with POI (16). NR5A1 is likely to play a pivotal role in regulating ovary and testis genes, but there has yet to be a functional study to confirm its role (53).
Beyond the exploration of causative mutations linked to Primary Ovarian Insufficiency (POI), numerous studies have delved into the investigation of common polymorphisms within Middle Eastern and North African (MENA) populations. Among these, some of the most frequently reported common polymorphisms are found in genes such as NOBOX, with 15 distinct variants (PMID 26848058), BMP15 with 11 variants(PMID 3523244), GDF9 featuring 4 variants(PMID 16278619), AMH contributing 2 variants(PMID 33344660), CKDN1B with 1 variant(PMID21575944), and FCN3 presenting a single variant(PMID 34641644). To provide a more comprehensive perspective, the genes NOBOX, BMP15, GDF9, AMH, CKDN1B, and FCN3 each play crucial roles in ovarian function and reproductive health. NOBOX is a key player in early folliculogenesis, orchestrating the development of ovarian follicles. BMP15 and GDF9 are essential for promoting the growth and maturation of ovarian follicles, vital to produce mature eggs. AMH, on the other hand, helps assess ovarian reserve and regulates the development of ovarian follicles. CKDN1B, though less directly tied to ovarian function, influences cell cycle regulation and may have subtle effects on ovarian health. In contrast, FCN3, while not a direct participant in ovarian function, is related to the immune system, and variations in immune-related genes can indirectly affect reproductive health. Genetic variants in these genes can lead to diverse effects on ovarian function, potentially contributing to conditions such as Primary Ovarian Insufficiency (POI) and impacting fertility in Middle Eastern and North African populations (PMID 24782009).
Four of the common BMP15 variants observed in MENA populations (c.-9C>G, c.308A>G, c.538G>A, and c.852C>T) have also been reported in Indian and Brazilian patients. Three of these (c.-9C>G, c.308A>G, and c.852C>T) were significantly associated with ovarian failure (54, 55): the c.-9C>G variant was associated with anovulation or infertility in a Spanish cohort of polycystic ovarian syndrome (PCOS) patients (55) but not in Chinese patients (56). One of the GDF9 variants found in POI patients from MENA have also been reported in different cohorts with different ancestries, namely GDF9 c.447C>T, which was a risk factor for POI in an Indian cohort (54) but not in Chinese Hui or Mexican patients (56, 57). The presence of common POI polymorphisms in the same genes that contain rare disease-causing mutations is an interesting finding, as it indicates a hereditary predisposition and may help in defining the genetic role of these genes in developing the disease.
POI demands early diagnosis to prevent irreversible damage to a patient’s fertility. According to recent studies, timely identification of POI is crucial, as it enables healthcare professionals to implement appropriate interventions, thereby preserving reproductive capabilities (58–60). Genetic testing assumes a pivotal role in this process, as it helps identify any underlying genetic abnormalities that contribute to POI. By understanding the genetic variants of the condition, healthcare providers can offer tailored treatment options and inform patients about their reproductive prognosis and potential risks. Hormone replacement therapy (HRT) is one possible intervention to manage hormonal imbalances and alleviate associated symptoms in POI, thereby potentially restoring fertility. Additionally, fertility preservation techniques, such as oocyte or embryo cryopreservation, provide an opportunity for future conception. Early diagnosis coupled with genetic testing and the availability of suitable interventions like HRT and fertility preservation are extremely important for mitigating the impact of POI and ensuring that patients receive appropriate support for their fertility and overall well-being (61).
Technological advances have facilitated the discovery of several new genes that may cause POI, partially explaining its heterogeneity. Several case studies have identified potential candidate genes that might cause POI, and, in many cases, this was supported by familial segregation analysis. Further functional studies are required to assess the underlying molecular mechanisms and pathways responsible for POI. Knowledge of the genetic basis of POI in the MENA region is expected to facilitate early diagnosis of POI patients and thus the early implementation of therapeutic interventions, paving the way for precision medicine options in specific patient populations.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
AA: Data curation, Investigation, Methodology, Writing – original draft. TA-B: Data curation, Investigation, Methodology, Writing – original draft. MA-S: Conceptualization, Resources, Supervision, Validation, Visualization, Writing – review & editing. AMA: Conceptualization, Formal analysis, Funding acquisition, Project administration, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. APC for this publication was covered by College of Health Sciences, Qatar University. The findings achieved herein are solely the responsibility of the authors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. European Society for Human R, Embryology Guideline Group on POI, Webber L, Davies M, Anderson R, Bartlett J, et al. ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod (2016). 31(5):926–37. doi: 10.1093/humrep/dew027
2. Rudnicka E, Kruszewska J, Klicka K, Kowalczyk J, Grymowicz M, Skorska J, et al. Premature ovarian insufficiency - aetiopathology, epidemiology, and diagnostic evaluation. Prz Menopauzalny (2018). 17(3):105–8. doi: 10.5114/pm.2018.78550
3. Man L, Lustgarten Guahmich N, Vyas N, Tsai S, Arazi L, Lilienthal D, et al. Ovarian reserve disorders, can we prevent them? A review. Int J Mol Sci (2022). 23(23). doi: 10.3390/ijms232315426
4. Snieder H, MacGregor AJ, Spector TD. Genes control the cessation of a woman’s reproductive life: a twin study of hysterectomy and age at menopause. J Clin Endocrinol Metab (1998). 83(6):1875–80. doi: 10.1210/jcem.83.6.4890
5. Luo W, Ke H, Tang S, Jiao X, Li Z, Zhao S, et al. Next-generation sequencing of 500 POI patients identified novel responsible monogenic and oligogenic variants. J Ovarian Res (2023). 16(1):39. doi: 10.1186/s13048-023-01104-6
6. Ferrarini E, De Marco G, Orsolini F, Gianetti E, Benelli E, Fruzzetti F, et al. Characterization of a novel mutation V136L in bone morphogenetic protein 15 identified in a woman affected by POI. J Ovarian Res (2021). 14(1):85. doi: 10.1186/s13048-021-00836-7
7. Qin Y, Jiao X, Simpson JL, Chen ZJ. Genetics of primary ovarian insufficiency: new developments and opportunities. Hum Reprod Update (2015). 21(6):787–808. doi: 10.1093/humupd/dmv036
8. Page MJ, Moher D, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. PRISMA 2020 explanation and elaboration: updated guidance and exemplars for reporting systematic reviews. BMJ (2021). 372:n160. doi: 10.1136/bmj.n160
9. Al-Haidose A, Yassin MA, Ahmed MN, Kunhipurayil HH, Al-Harbi AA, Aljaberi MA, et al. Distinct clinical and prognostic features of myelodysplastic syndrome in patients from the Middle East, North Africa, and beyond: A systemic review. J Clin Med (2023). 12(8):2832. doi: 10.3390/jcm12082832
10. Whiting PF, Rutjes AW, Westwood ME, Mallett S, Deeks JJ, Reitsma JB, et al. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med (2011). 155(8):529–36. doi: 10.7326/0003-4819-155-8-201110180-00009
11. Afkhami F, Shahbazi S, Farzadi L, Danaei S. Novel bone morphogenetic protein 15 (BMP15) gene variants implicated in premature ovarian insufficiency. Reprod Biol Endocrinol (2022). 20(1):42. doi: 10.1186/s12958-022-00913-6
12. Akbari A, Zoha Tabatabaei S, Salehi N, Padidar K, Almadani N, Ali Sadighi Gilani M, et al. Novel STAG3 variant associated with primary ovarian insufficiency and non-obstructive azoospermia in an Iranian consanguineous family. Gene (2022). 821:146281. doi: 10.1016/j.gene.2022.146281
13. Mehrjooy S, Nikbakht R, Mohammadi Asl J, Ghadiri A, Ghandil P. Utilization of whole exome sequencing in non-syndromic premature ovarian failure: ficolin-3 gene mutation in an Iranian family. Iran BioMed J (2021). 25(6):441–6. doi: 10.52547/ibj.25.6.441
14. Akbari A, Padidar K, Salehi N, Mashayekhi M, Almadani N, Sadighi Gilani MA, et al. Rare missense variant in MSH4 associated with primary gonadal failure in both 46, XX and 46, XY individuals. Hum Reprod (2021). 36(4):1134–45. doi: 10.1093/humrep/deaa362
15. Alavi A, Darki F, Bidgoli MMR, Zare-Abdollahi D, Moini A, Shahshahani MM, et al. Mutation in ALOX12B likely cause of POI and also ichthyosis in a large Iranian pedigree. Mol Genet Genomics (2020). 295(4):1039–53. doi: 10.1007/s00438-020-01663-z
16. Safari S, Zare-Abdollahi D, Mirfakhraie R, Ghafouri-Fard S, Pouresmaeili F, Movafagh A, et al. An Iranian family with azoospermia and premature ovarian insufficiency segregating NR5A1 mutation. Climacteric (2014). 17(3):301–3. doi: 10.3109/13697137.2013.847079
17. Rafaa TA, Suleiman AA, Dawood MF, Al-Rawi AM. Association of two single nucleotide polymorphisms rs10407022 and rs3741664 with the risk of primary ovarian insufficiency in a sample of Iraqi women. Mol Biol Res Commun (2020). 9(4):141–4. doi: 10.22099/mbrc.2020.36371.1477
18. Le Quesne Stabej P, Williams HJ, James C, Tekman M, Stanescu HC, Kleta R, et al. STAG3 truncating variant as the cause of primary ovarian insufficiency. Eur J Hum Genet (2016). 24(1):135–8. doi: 10.1038/ejhg.2015.107
19. Lacombe A, Lee H, Zahed L, Choucair M, Muller JM, Nelson SF, et al. Disruption of POF1B binding to nonmuscle actin filaments is associated with premature ovarian failure. Am J Hum Genet (2006). 79(1):113–9. doi: 10.1086/505406
20. Smirin-Yosef P, Zuckerman-Levin N, Tzur S, Granot Y, Cohen L, Sachsenweger J, et al. A biallelic mutation in the homologous recombination repair gene SPIDR is associated with human gonadal dysgenesis. J Clin Endocrinol Metab (2017). 102(2):681–8. doi: 10.1210/jc.2016-2714
21. de Vries L, Behar DM, Smirin-Yosef P, Lagovsky I, Tzur S, Basel-Vanagaite L. Exome sequencing reveals SYCE1 mutation associated with autosomal recessive primary ovarian insufficiency. J Clin Endocrinol Metab (2014). 99(10):E2129–32. doi: 10.1210/jc.2014-1268
22. Pierce SB, Gersak K, Michaelson-Cohen R, Walsh T, Lee MK, Malach D, et al. Mutations in LARS2, encoding mitochondrial leucyl-tRNA synthetase, lead to premature ovarian failure and hearing loss in Perrault syndrome. Am J Hum Genet (2013). 92(4):614–20. doi: 10.1016/j.ajhg.2013.03.007
23. Zangen D, Kaufman Y, Zeligson S, Perlberg S, Fridman H, Kanaan M, et al. XX ovarian dysgenesis is caused by a PSMC3IP/HOP2 mutation that abolishes coactivation of estrogen-driven transcription. Am J Hum Genet (2011). 89(4):572–9. doi: 10.1016/j.ajhg.2011.09.006
24. Caburet S, Arboleda VA, Llano E, Overbeek PA, Barbero JL, Oka K, et al. Mutant cohesin in premature ovarian failure. N Engl J Med (2014). 370(10):943–9. doi: 10.1056/NEJMoa1309635
25. Al-Ajoury R, Kassem E, Al-Halabi B, Moassess F, Al-Achkar W. Mutations analysis of the growth differentiation factor 9 gene in Syrian women with ovarian failure. Int J Hum Genet (2015). 15(3):139–44. doi: 10.1080/09723757.2015.11886261
26. Al-ajoury R, Kassem E, Al-halabi B, Moassess F, Al-achkar W. Investigation of some genetic variations in BMP15 accompanied with premature ovarian failure (POF) in Syrian women. Middle East Fertility Soc J (2015). 20(2):91–6. doi: 10.1016/j.mefs.2014.02.005
27. Bouali N, Francou B, Bouligand J, Imanci D, Dimassi S, Tosca L, et al. New MCM8 mutation associated with premature ovarian insufficiency and chromosomal instability in a highly consanguineous Tunisian family. Fertil Steril (2017). 108(4):694–702. doi: 10.1016/j.fertnstert.2017.07.015
28. Bouali N, Francou B, Bouligand J, Lakhal B, Malek I, Kammoun M, et al. NOBOX is a strong autosomal candidate gene in Tunisian patients with primary ovarian insufficiency. Clin Genet (2016). 89(5):608–13. doi: 10.1111/cge.12750
29. Lakhal B, Ben-Hadj-Khalifa S, Bouali N, Braham R, Hatem E, Saad A. Mutational screening of SF1 and WNT4 in Tunisian women with premature ovarian failure. Gene (2012). 509(2):298–301. doi: 10.1016/j.gene.2012.08.007
30. Lakhal B, Laissue P, Braham R, Elghezal H, Saad A, Fellous M, et al. A novel BMP15 variant, potentially affecting the signal peptide, in a familial case of premature ovarian failure. Clin Endocrinol (Oxf) (2009). 71(5):752–3. doi: 10.1111/j.1365-2265.2009.03571.x
31. Ojeda D, Lakhal B, Fonseca DJ, Braham R, Landolsi H, Mateus HE, et al. Sequence analysis of the CDKN1B gene in patients with premature ovarian failure reveals a novel mutation potentially related to the phenotype. Fertil Steril (2011). 95(8):2658–60 e1. doi: 10.1016/j.fertnstert.2011.04.045
32. Oral E, Toksoy G, Sofiyeva N, Celik HG, Karaman B, Basaran S, et al. Clinical and genetic investigation of premature ovarian insufficiency cases from Turkey. J Gynecol Obstet Hum Reprod (2019). 48(10):817–23. doi: 10.1016/j.jogoh.2019.04.007
33. Bramble MS, Goldstein EH, Lipson A, Ngun T, Eskin A, Gosschalk JE, et al. A novel follicle-stimulating hormone receptor mutation causing primary ovarian failure: a fertility application of whole exome sequencing. Hum Reprod (2016). 31(4):905–14. doi: 10.1093/humrep/dew025
34. Heddar A, Ogur C, Da Costa S, Braham I, Billaud-Rist L, Findikli N, et al. Genetic landscape of a large cohort of Primary Ovarian Insufficiency: New genes and pathways and implications for personalized medicine. EBioMedicine (2022). 84:104246. doi: 10.1016/j.ebiom.2022.104246
35. Al-Agha AE, Ahmed IA, Nuebel E, Moriwaki M, Moore B, Peacock KA, et al. Primary ovarian insufficiency and azoospermia in carriers of a homozygous PSMC3IP stop gain mutation. J Clin Endocrinol Metab (2018). 103(2):555–63. doi: 10.1210/jc.2017-01966
36. Golezar S, Ramezani Tehrani F, Khazaei S, Ebadi A, Keshavarz Z. The global prevalence of primary ovarian insufficiency and early menopause: a meta-analysis. Climacteric (2019). 22(4):403–11. doi: 10.1080/13697137.2019.1574738
37. Franca MM, Mendonca BB. Genetics of ovarian insufficiency and defects of folliculogenesis. Best Pract Res Clin Endocrinol Metab (2022). 36(1):101594. doi: 10.1016/j.beem.2021.101594
38. Petukhova GV, Romanienko PJ, Camerini-Otero RD. The Hop2 protein has a direct role in promoting interhomolog interactions during mouse meiosis. Dev Cell (2003). 5(6):927–36. doi: 10.1016/s1534-5807(03)00369-1
39. Yang X, Touraine P, Desai S, Humphreys G, Jiang H, Yatsenko A, et al. Gene variants identified by whole-exome sequencing in 33 French women with premature ovarian insufficiency. J Assist Reprod Genet (2019). 36(1):39–45. doi: 10.1007/s10815-018-1349-4
40. Mei L, Huang L, Huang Y, Wu X, He H, He X, et al. Two novel biallelic mutations in PSMC3IP in a patient affected by premature ovarian insufficiency. Mol Med Rep (2022). 25(2). doi: 10.3892/mmr.2021.12561
41. Sirchia F, Giorgio E, Cucinella L, Valente EM, Nappi RE. Biallelic mutations in PSMC3IP are associated with secondary amenorrhea: expanding the spectrum of premature ovarian insufficiency. J Assist Reprod Genet (2022). 39(5):1177–81. doi: 10.1007/s10815-022-02471-7
42. Jaillard S, McElreavy K, Robevska G, Akloul L, Ghieh F, Sreenivasan R, et al. STAG3 homozygous missense variant causes primary ovarian insufficiency and male non-obstructive azoospermia. Mol Hum Reprod (2020). 26(9):665–77. doi: 10.1093/molehr/gaaa050
43. Costa Y, Speed R, Ollinger R, Alsheimer M, Semple CA, Gautier P, et al. Two novel proteins recruited by synaptonemal complex protein 1 (SYCP1) are at the centre of meiosis. J Cell Sci (2005). 118(Pt 12):2755–62. doi: 10.1242/jcs.02402
44. Iwai T, Yoshii A, Yokota T, Sakai C, Hori H, Kanamori A, et al. Structural components of the synaptonemal complex, SYCP1 and SYCP3, in the medaka fish Oryzias latipes. Exp Cell Res (2006). 312(13):2528–37. doi: 10.1016/j.yexcr.2006.04.015
45. Hamer G, Gell K, Kouznetsova A, Novak I, Benavente R, Hoog C. Characterization of a novel meiosis-specific protein within the central element of the synaptonemal complex. J Cell Sci (2006). doi: 10.1242/jcs.03182
46. Maor-Sagie E, Cinnamon Y, Yaacov B, Shaag A, Goldsmidt H, Zenvirt S, et al. Deleterious mutation in SYCE1 is associated with non-obstructive azoospermia. J Assist Reprod Genet (2015). 32(6):887–91. doi: 10.1007/s10815-015-0445-y
47. Neyroud A-S, Rudinger-Thirion J, Frugier M, Riley L, Bidet M, Akloul L, et al. LARS2 variants can present as premature ovarian insufficiency in the absence of overt hearing loss. Eur J Hum Genet (2022). doi: 10.1038/s41431-022-01252-1
48. Demain LA, Urquhart JE, O’Sullivan J, Williams SG, Bhaskar SS, Jenkinson EM, et al. Expanding the genotypic spectrum of Perrault syndrome. Clin Genet (2017). 91(2):302–12. doi: 10.1111/cge.12776
49. Riley LG, Rudinger-Thirion J, Schmitz-Abe K, Thorburn DR, Davis RL, Teo J, et al. LARS2 variants associated with hydrops, lactic acidosis, sideroblastic anemia, and multisystem failure. JIMD Rep (2016). 28:49–57. doi: 10.1007/8904_2015_515
50. Huang JW, Acharya A, Taglialatela A, Nambiar TS, Cuella-Martin R, Leuzzi G, et al. MCM8IP activates the MCM8-9 helicase to promote DNA synthesis and homologous recombination upon DNA damage. Nat Commun (2020). 11(1):2948. doi: 10.1038/s41467-020-16718-3
51. Zhang YX, He WB, Xiao WJ, Meng LL, Tan C, Du J, et al. Novel loss-of-function mutation in MCM8 causes premature ovarian insufficiency. Mol Genet Genomic Med (2020). 8(4):e1165. doi: 10.1002/mgg3.1165
52. Faienza MF, Chiarito M, Baldinotti F, Canale D, Savino C, Paradies G, et al. NR5A1 gene variants: variable phenotypes, new variants, different outcomes. Sex Dev (2019). 13(5-6):258–63. doi: 10.1159/000507411
53. Lourenco D, Brauner R, Lin L, De Perdigo A, Weryha G, Muresan M, et al. Mutations in NR5A1 associated with ovarian insufficiency. N Engl J Med (2009). 360(12):1200–10. doi: 10.1056/NEJMoa0806228
54. Dixit H, Rao LK, Padmalatha V, Kanakavalli M, Deenadayal M, Gupta N, et al. Mutational screening of the coding region of growth differentiation factor 9 gene in Indian women with ovarian failure. Menopause (2005). 12(6):749–54. doi: 10.1097/01.gme.0000184424.96437.7a
55. Gonzalez A, Ramirez-Lorca R, Calatayud C, Mendoza N, Ruiz A, Saez ME, et al. Association of genetic markers within the BMP15 gene with anovulation and infertility in women with polycystic ovary syndrome. Fertil Steril (2008). 90(2):447–9. doi: 10.1016/j.fertnstert.2007.06.083
56. Ma L, Chen Y, Mei S, Liu C, Ma X, Li Y, et al. Single nucleotide polymorphisms in premature ovarian failure-associated genes in a Chinese Hui population. Mol Med Rep (2015). 12(2):2529–38. doi: 10.3892/mmr.2015.3762
57. Juarez-Rendon KJ, Garcia-Ortiz JE. “Evaluation of four genes associated with primary ovarian insufficiency in a cohort of Mexican women”. J Assist Reprod Genet (2018). 35(8):1483–8. doi: 10.1007/s10815-018-1232-3
58. Huang QY, Chen SR, Chen JM, Shi QY, Lin S. Therapeutic options for premature ovarian insufficiency: an updated review. Reprod Biol Endocrinol (2022). 20(1):28. doi: 10.1186/s12958-022-00892-8
59. Liu H, Wei X, Sha Y, Liu W, Gao H, Lin J, et al. Whole-exome sequencing in patients with premature ovarian insufficiency: early detection and early intervention. J Ovarian Res (2020). 13(1):114. doi: 10.1186/s13048-020-00716-6
60. Na J, Kim GJ. Recent trends in stem cell therapy for premature ovarian insufficiency and its therapeutic potential: a review. J Ovarian Res (2020). 13(1):74. doi: 10.1186/s13048-020-00671-2
Keywords: primary ovarian insufficiency, Arab, MENA, genetic variant, systematic review
Citation: Allouch A, Al-Barazenji T, Al-Shafai M and Abdallah AM (2024) The landscape of genetic variations in non-syndromic primary ovarian insufficiency in the MENA region: a systematic review. Front. Endocrinol. 14:1289333. doi: 10.3389/fendo.2023.1289333
Received: 06 September 2023; Accepted: 01 December 2023;
Published: 26 April 2024.
Edited by:
Marta Olszewska, Polish Academy of Sciences, PolandReviewed by:
Abdulsamed Kükürt, Kafkas University, TürkiyeCopyright © 2024 Allouch, Al-Barazenji, Al-Shafai and Abdallah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atiyeh M. Abdallah, YWFiZGFsbGFoQHF1LmVkdS5xYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.