 Nicole A. Teaney
Nicole A. Teaney Nicole E. Cyr
Nicole E. Cyr- 1Stonehill College, Neuroscience Program, Easton, MA, United States
- 2Stonehill College, Department of Biology, Easton, MA, United States
Forkhead box O (FoxO) proteins are transcription factors that mediate many aspects of physiology and thus have been targeted as therapeutics for several diseases including metabolic disorders such as type 2 diabetes mellitus (T2D). The role of FoxO1 in metabolism has been well studied, but recently FoxO1’s potential for diabetes prevention and therapy has been debated. For example, studies have shown that increased FoxO1 activity in certain tissue types contributes to T2D pathology, symptoms, and comorbidities, yet in other tissue types elevated FoxO1 has been reported to alleviate symptoms associated with diabetes. Furthermore, studies have reported opposite effects of active FoxO1 in the same tissue type. For example, in the liver, FoxO1 contributes to T2D by increasing hepatic glucose production. However, FoxO1 has been shown to either increase or decrease hepatic lipogenesis as well as adipogenesis in white adipose tissue. In skeletal muscle, FoxO1 reduces glucose uptake and oxidation, promotes lipid uptake and oxidation, and increases muscle atrophy. While many studies show that FoxO1 lowers pancreatic insulin production and secretion, others show the opposite, especially in response to oxidative stress and inflammation. Elevated FoxO1 in the hypothalamus increases the risk of developing T2D. However, increased FoxO1 may mitigate Alzheimer’s disease, a neurodegenerative disease strongly associated with T2D. Conversely, accumulating evidence implicates increased FoxO1 with Parkinson’s disease pathogenesis. Here we review FoxO1’s actions in T2D conditions in metabolic tissues that abundantly express FoxO1 and highlight some of the current studies targeting FoxO1 for T2D treatment.
1 Introduction
T2D is a global health concern. A recent study reported that T2D accounts for more than 90% of the 529 million diabetes cases in 2021 (1). Moreover, the incidence of T2D is expected to increase in adults and children over the next thirty years. T2D is characterized by insulin resistance and hyperglycemia. Consequently, its treatments target these metabolic states. FoxO1 is a transcription factor that mediates many cellular functions including inflammation, oxidative stress (OS), cell proliferation, apoptosis, and autophagy which contribute to the regulation of insulin signaling and glycemia. However, whether increased FoxO1 transcriptional activity exacerbates or alleviates T2D appears to be tissue-type specific and may depend on cellular conditions (2, 3). Recent studies have called attention to these FoxO1 controversies (4, 5). The current review describes FoxO1’s actions in T2D conditions in metabolic tissues that highly express FoxO1 and highlights some of the current progress in this field.
2 FoxO1 structure, function, and regulation
FoxO1 (also forkhead in rhabdomyosarcomam (FKHR)) is one of four mammalian isoforms belonging to the FoxO transcription factor family. The others include FoxO3, FoxO4, and FoxO6 (5–7). These FoxO proteins are mainly localized in the nucleus and exert their actions through their ability to regulate the transcription of specific genes. However, they can also be found in the cytoplasm where they are considered inactive. Although their primary action occurs in the nucleus, new evidence indicates that FoxO proteins can be localized to the mitochondria where they have varying effects on mitochondrial function (8–10). FoxO1 structure, as depicted in Figure 1, is similar to the other mammalian FoxO isoforms. For example, FoxO1, FoxO3, FoxO4, and FoxO6 each contain a highly conserved winged‐helix or forkhead DNA‐binding domain (DBD) and bind to the consensus DNA sequence 5′‐TTGTTTAC‐3′ (11, 12). Their remaining functional domains consist of a nuclear localization signal (NLS), a nuclear export signal (NES), and a transactivation domain (TAD). FoxO amino acid composition varies slightly among species. FoxO1 consists of about 655 amino acids, FoxO3 has about 670 amino acids, Foxo4 has about 505 amino acids, and FoxO6 has about 490 amino acids with the greatest amino acid differences among these proteins occurring in the TAD (6). FoxO1, FoxO3 and FoxO4 are ubiquitously expressed (13–17). For example, FoxO1 is predominantly expressed in adipose tissue but also abundantly found in other metabolic tissues such as liver, skeletal muscle, pancreas, and brain. FoxO3 is primarily expressed in liver and FoxO4 is largely expressed in muscle. In contrast, expression of FoxO6 is mostly concentrated in the brain (18).
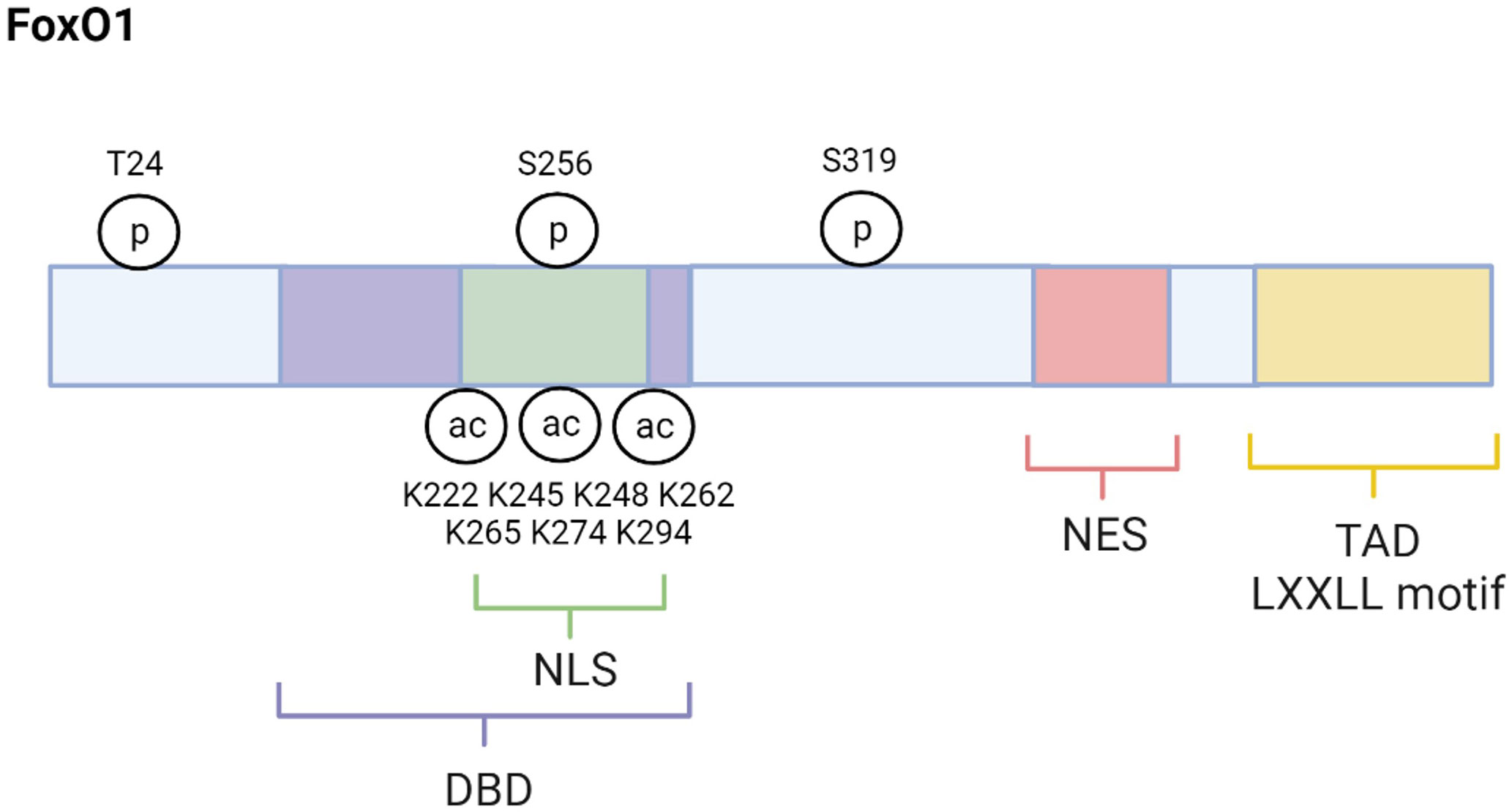
Figure 1 FoxO1 structure and regulatory sites. FoxO1 structure is composed of several functional domains, including DNA-binding domain (DBD), nuclear location signal (NLS), nuclear export signal (NES), and transactivation domain (TAD). The major regulatory sites discussed in this review are also shown. Created with BioRender.com.
FoxO1, FoxO3, FoxO4, and FoxO6 control some of the same cellular functions by regulating genes involved in the cell cycle, OS, inflammation, glucose metabolism, and autophagy (6, 19). However, whole-body knockout studies in mice reveal a unique role in specific cellular functions for each FoxO isoform. For example, lack of FoxO1 is lethal on embryonic day 10.5 due to vasculature defects indicating an important role for FoxO1 in angiogenesis during development (20). FoxO3 has been shown to regulate reproduction as FoxO3 null female mice exhibit impaired ovarian follicular development and infertility. While one study reported no phenotypic differences between FoxO4 null mice and wild-type mice (20), another showed that FoxO4 knockout mice were at a higher risk of developing colitis when challenged with increased inflammation due to elevated levels of NF-κB, which is a transcription factor that mediates the immune response. These results suggest that FoxO4 regulates gut inflammation and health (21). Salih et al. (22) demonstrated that FoxO6 is a key mediator of memory consolidation and controls the transcription of synaptic proteins in hippocampal neurons involved in long-term memory.
Activity of all FoxOs depends on post-translational modifications. While processes such as ubiquitination, methylation, and glycosylation can alter FoxOs, phosphorylation and acetylation are the principal post-translational modifications regulating FoxO activity (5, 6). Phosphorylation strongly regulates FoxO activity as it affects FoxO’s cellular localization, thus transcriptional capacity. There are several kinases that phosphorylate FoxO proteins and cause these proteins to be shuttled out of the nucleus into the cytoplasm where they are inactive. These FoxO inactivating kinases include AKT (Protein kinase B), extracellular signal- regulated kinase (ERK), and cyclin-dependent kinase (CDK) 1/2 (23–26). In contrast, phosphorylation by the FoxO activating kinases such as adenosine monophosphate-activated (AMPK), Jun- N-terminal kinase (JNK), mammalian sterile 20-like kinase (MST1), and Protein Kinase RNA-Like ER Kinase (PERK) result in nuclear localization and nuclear retention of FoxO proteins which increases their transcriptional activity (27–30).
Many physiological conditions regulate FoxO activating and inactivating kinases including conditions that are altered in the pathogenesis and pathology of T2D. For example, JNK is a FoxO-activating kinase known to increase in response to elevated inflammation and OS, which are conditions that contribute to the development of T2D (28, 31). JNK phosphorylates FoxO4 specifically at T447 and T451 which promotes its nuclear transport and activity (28). Although FoxO1 is also phosphorylated and translocated into the nucleus by JNK the exact mechanism has not yet been described (32). Activation of FoxOs by JNK increases the transcription of certain antioxidants like catalase and manganese superoxide dismutase (MnSOD, or SOD2) to combat OS (28, 31). Therefore, the post-translational phosphorylation of FoxOs by JNK helps to protect against OS and T2D. In contrast to JNK, phosphorylation of FoxO1, FoxO3, and FoxO4 by the active, phosphorylated AKT (pAKT) causes the exportation and sequestration of these FoxO1 proteins into the cytoplasm (33). In particular, AKT phosphorylates FoxO1 at T24, S256, and S319 sites (23) (Figure 1). AKT phosphorylation of FoxO3 occurs at T32, S253, and S315 (34, 35). T28, S193, and S258 were identified as putative sites for AKT phosphorylation of FoxO4, but studies indicate that insulin stimulated AKT phosphorylates FoxO4 specifically at S193, and S258 (36, 37). Unlike the other FoxO isoforms, FoxO6 is less sensitive to AKT-mediated inactivation because it contains T26 and S184 as AKT phosphorylation sites (38). In fact, research indicates that AKT suppression of FoxO6 activity is independent of cytoplasmic translocation.
One cellular condition in which pAKT is enhanced is when insulin levels are high postprandially. Insulin binding to its receptor recruits insulin receptor substrate (IRS), which in turn, activates PI3K (phosphatidylinositol-3 kinase) which activates AKT (Figure 2). The phosphorylation of FoxO proteins by pAKT has been well studied and shown to augment 14-3-3 protein binding to phosphorylated serine and threonine residues on pFoxO, which causes a conformational change that exposes a leucine-dense NES and subsequently recruits nuclear export factor Exportin 1 to shuttle FoxOs to the cytoplasm (39–44) (Figure 2). Furthermore, 14-3-3 binding covers the FoxO NLS, which inhibits nuclear reentry (45, 46). Therefore, FoxO activity is reduced in the fed state. In contrast, during insulin resistance conditions, insulin’s activation of PI3K/AKT signaling is compromised. Consequently, AKT-mediated FoxO inactivation is reduced and FoxO activity increases in T2D (Figure 3).
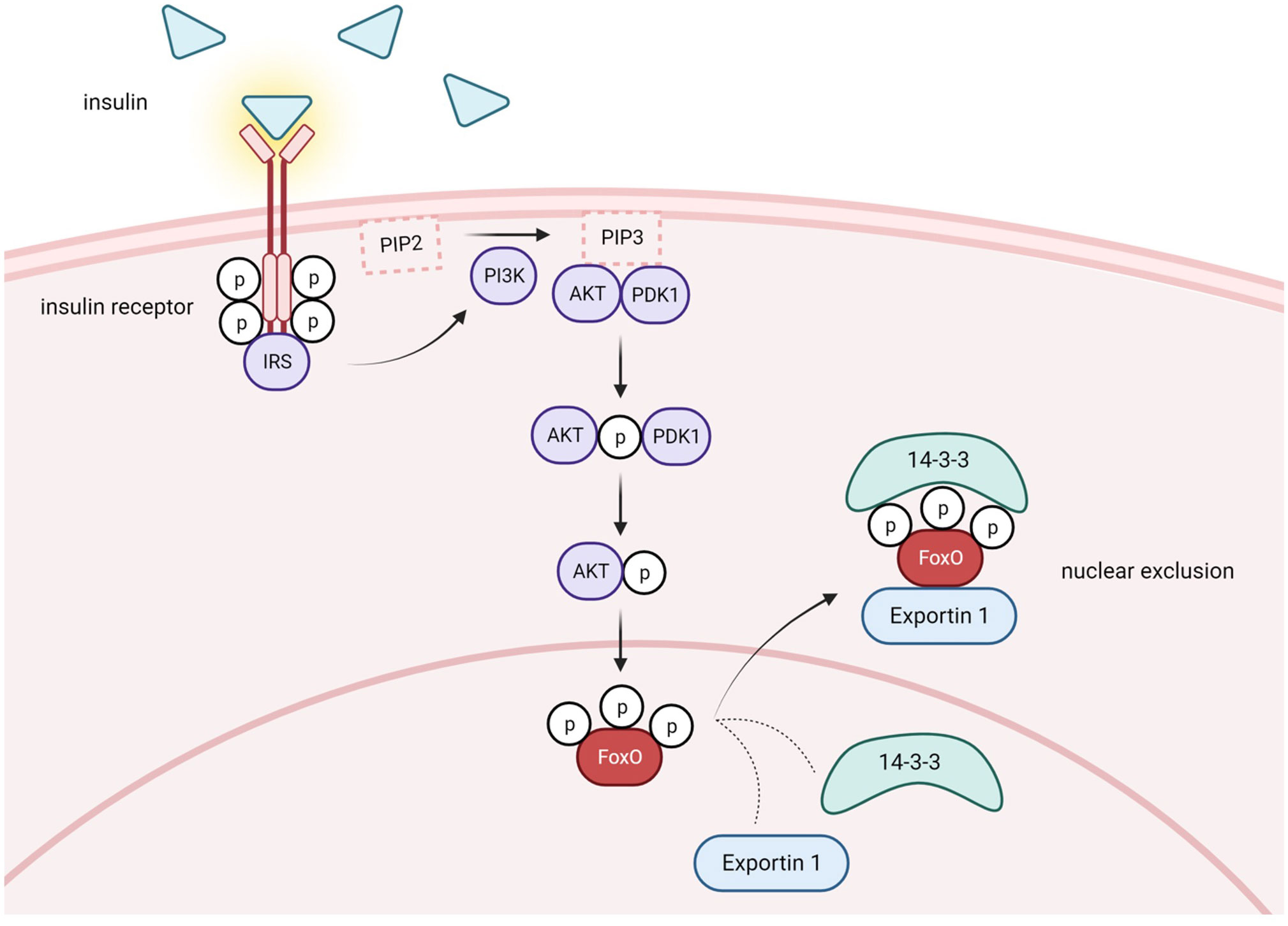
Figure 2 Postprandial conditions lead to FoxO nuclear exclusion via PI3K/AKT pathway activation. Under healthy conditions in the postprandial state, insulin is released from pancreatic B-cells and binds to insulin receptors in several different tissues. Activation of the receptor tyrosine-kinases leads to recruitment of insulin receptor substrate (IRS) and initiation of the PI3K/AKT signaling cascade. PI3K phosphorylates phosphatidylinositol 4,5-bisphosphate (PIP2), generating phosphatidylinositol-3,4,5-triphosphate (PIP3). Subsequently, PDK1 phosphorylates and activates AKT. Phosphorylated AKT phosphorylates FoxO, which promotes recruitment of Exportin 1 and nuclear exclusion. 14-3-3 protein is also recruited and covers FoxO's NLS, inhibiting nuclear reentry. Activation of the PI3K/AKT pathway causes cytoplasmic retention and decreases the transcriptional capacity of FoxO. Created with BioRender.com.
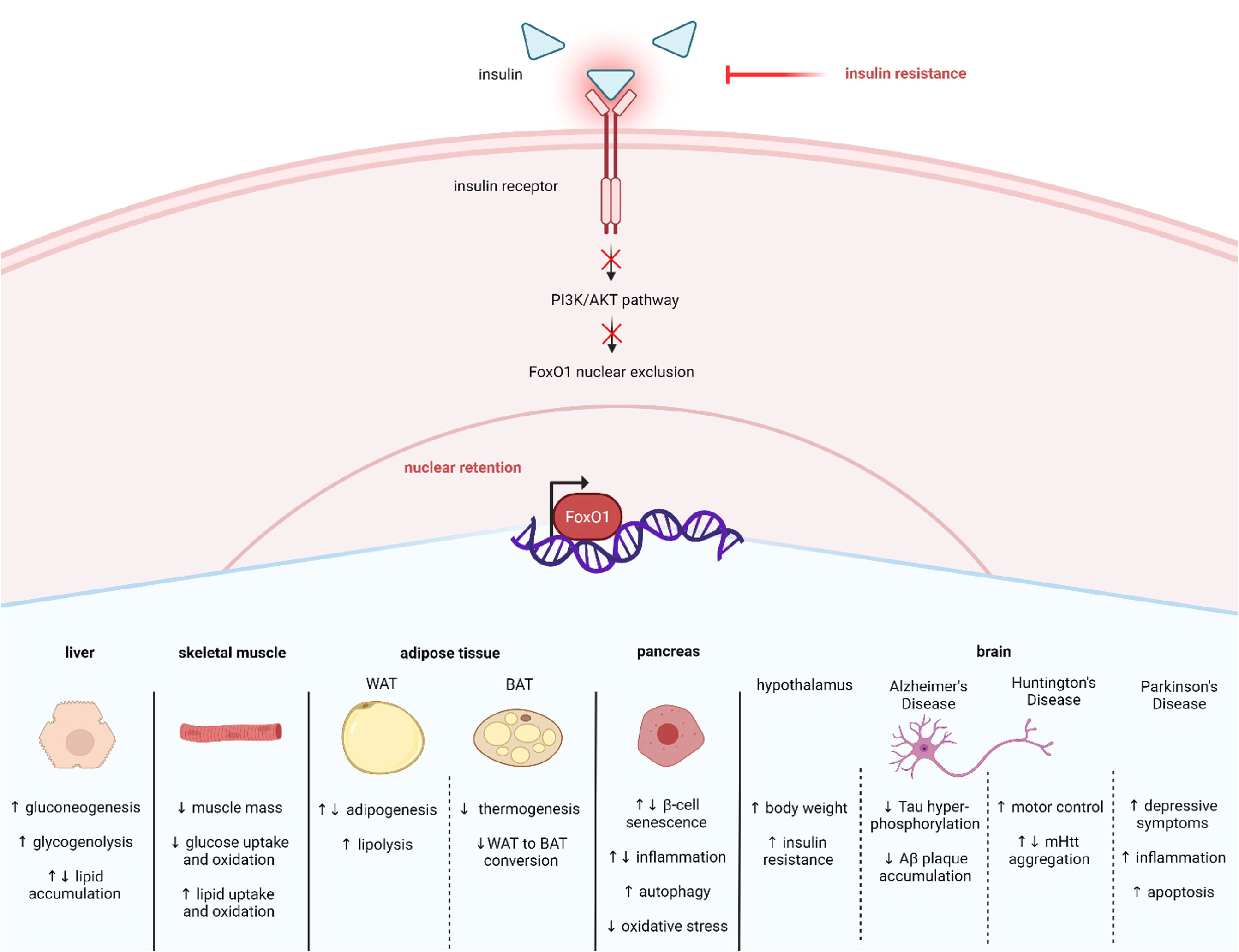
Figure 3 Insulin resistance affects many tissues via FoxO1 nuclear retention. Insulin resistance in T2D results in less activation of the PI3K/AKT pathway. Decreased insulin induced PI3K/AKT signaling enhances FoxO1 nuclear retention and activity, which mediates many cellular responses across tissue types, including liver, skeletal muscle, adipose, pancreas, and brain. Created with BioRender.com.
In addition to phosphorylation, acetylation is an important post-translational modification that affects FoxO activity. Histone acetyltransferases, like CREB-binding protein (CBP) and p300, acetylate FoxOs at the K222, K245, K248, K262, K265, K274 and K294 residues, which decreases DNA binding affinity and inhibits transcriptional activity (47, 48) (Figure 1). Furthermore, acetylation of FoxOs increases their sensitivity to inactivation via phosphorylation and nuclear exclusion (47). Interestingly, FoxO inactivation by CBP/p300 is enhanced during OS conditions which can lead to a decrease in FoxO1 upregulation of antioxidants like MnSOD and put the cell at risk for apoptosis (49, 50). However, there are mechanisms to increase FoxO1 activity during OS. For example, JNK activates FoxO proteins even under conditions of low OS (28). Furthermore, the inactivation of FoxOs by CBP/p300 acetylation can be reversed by deacetylating and thus re-activating FoxOs (49). Histone deacetylases like the NAD+-dependent Sirtuins, including the mammalian Sirtuin 1 (SIRT1), deacetylate FoxOs (51). Research reveals that SIRT1 deacetylation of FoxOs during OS can reverse the CBP/p300 effects on FoxO inhibition. For example, SIRT1 deacetylation and activation of FOXOs upregulates antioxidants and genes involved in DNA repair to mitigate OS and thus help to prevent the development of OS-related diseases like T2D (49, 52, 53).
SIRT1 also plays a significant role in regulating glucose and lipid metabolism (54, 55). Furthermore, SIRT1 is altered in metabolic conditions like T2D. For example, studies have reported that SIRT1 levels are lower in serum and muscle cells of patients with T2D, suggesting that loss of SIRT1 activity may contribute to T2D pathology (56, 57). Consistent with this idea, other studies have shown that increasing SIRT1 can alleviate T2D symptoms. For example, overexpression of SIRT1 in the liver of diabetic db/db mice improves insulin resistance and glucose tolerance (58). SIRT1’s actions are exerted in part by regulating FoxO1. Specifically, SIRT1 interacts with FoxO1 at its LXXLL motif located at amino acids 459–463 which enables deacetylation to occur (51) (Figure 1). One mechanism through which SIRT1 improves T2D symptoms is by deacetylating and thus activating FoxO1 in adipocytes and increasing FoxO1’s interaction with its coactivator CCAAT/enhancer-binding protein α (C/EBPα) to upregulate the transcription of the insulin-sensitizing hormone adiponectin (59). However, SIRT1 can also act to mitigate T2D by enhancing AKT-mediated FoxO1 inactivation in tissues like liver and skeletal muscle (60, 61). Therefore, increased SIRT1 can improve T2D symptoms by either activating or inactivation FoxO1. The current review focuses on FoxO1 regulation in T2D within specific metabolic tissues, including liver, skeletal muscle, adipose tissue, pancreatic β-cells, and brain (Figure 3).
3 FoxO1 and T2D – liver
FoxO proteins are known to mediate glucose metabolism. In the liver, glucose is largely produced from glycogen, the stored form of carbohydrates, by the process of glycogenolysis. However, glucose can also be synthesized in the liver from other sources such as pyruvate, glycerol, lactate, and amino acids through gluconeogenesis, especially during conditions when glycogen is depleted such as a prolonged fast (62). Activation of FoxOs promotes hepatic glucose production by increasing the expression of glucose 6-phosphatase (G6PC), fructose 1,6-bisphosphatase, and phosphoenolpyruvate carboxykinase (PEPCK) which are enzymes involved in gluconeogenesis (63, 64). The enzyme G6PC catalyzes the conversion of glucose-6-phosphate to glucose in gluconeogenesis as well as in glycogenolysis. Therefore, FoxOs increase hepatic glucose levels via both pathways. FoxO’s control of gluconeogenesis and glycogenolysis is mainly regulated by insulin signaling. For example, in the fed state, insulin levels increase and stimulate the PI3K/AKT pathway, which will export FoxO out of the nucleus and reduce FoxO’s transcriptional activity (Figure 2). Thus, FoxO inactivation contributes to insulin’s ability to lower blood glucose levels by decreasing hepatic glucose production and output. In contrast, during conditions when insulin signaling is diminished, such as in fasting and insulin resistance states, AKT-mediated nuclear exclusion of FoxO decreases, which causes an overall increase in FoxO transcriptional activity to elevate hepatic glucose production and output (Figure 3). The nutrient sensor SIRT1 has been shown to facilitate AKT-mediated effects on FoxO1 and gluconeogenesis. For example, liver-specific SIRT1 deficiency increases AKT which suppresses FoxO1 causing the downregulation of G6PC and PEPCK which decreases gluconeogenesis and lowers blood glucose levels (60). Liver-specific genetic studies indicate that while FoxO3 may contribute, FoxO1 is the predominant isoform regulating gluconeogenesis (17). In addition, studies reveal that hepatic FoxO1 plays a role in T2D since liver-specific FoxO1 activation in transgenic mice increases glucose levels and impairs glucose tolerance (65). Furthermore, loss of hepatic FoxO1 function improves glycemia, mitochondrial function, and T2D symptoms in diabetic mice (17, 64, 66, 67).
Insulin decreases hepatic glucose production via the AKT/FoxO1 pathway and increases lipogenesis largely via activation of mechanistic target of rapamycin complex 1 (mTORC1), which activates the major lipid regulator sterol regulatory element binding protein 1c (SREBP1c) (68, 69). However, evidence indicates that FoxO1 also contributes to insulin’s control of lipid metabolism in the liver. For example, microsomal triglyceride transfer protein (MTP) and apolipoprotein apoC-III are proteins that associate with very low-density lipoprotein (VLDL) and regulate triglyceride transport. FoxO1 directly upregulates the transcription of both MTP and apoC-III (70, 71) (see Figure 2). Insulin stimulated AKT suppression of FoxO1 decreases MTP and apoC-III. Conversely, reduced activation of the PI3K/AKT pathway increases FoxO1 activity along with the production of MTP and apoC-III. Moreover, FoxO1 overactivation was found to cause VLDL accumulation in the liver, elevated plasma triglyceride levels, and an increased the risk of atherosclerosis (70, 71). However, conflicting results have been reported from studies using transgenic mice with overactive FoxO1 expression in the liver. For example, some studies show that elevated hepatic FoxO1 increases de novo lipogenesis and triglyceride levels, yet another study found that constitutively active hepatic FoxO1 decreases triglyceride and cholesterol levels (65, 72). Liver-specific knockout of FoxO1 and FoxO3 independently indicates that FoxO3 is the more substantial regulator of lipogenesis as FoxO3 deletion increased the expression of lipogenic enzymes. However, simultaneous deletion of FoxO1 and FoxO3 revealed a synergistic increase in the transcription of the lipogenic enzyme glucokinase (Gck), which lead to hepatic steatosis (17). Further research has demonstrated that FoxO1 associates with the corepressor SIN3 Transcription Regulator Family Member A (SIN3A) to downregulate Gck expression and decrease lipogenesis (73). These results suggest that in conditions in which FoxO1 activity is enhanced like during insulin resistance, FoxO1 should decrease lipid synthesis. Given that insulin resistance is often associated with hyperglycemia and hyperlipidemia, a pathogenic state termed “selective insulin resistance”, researchers have suggested that other regulators may play a more prominent role regulating lipid metabolism in T2D (74, 75). While FoxO1’s role in regulating gluconeogenesis is well established, more research is needed to elucidate FoxO1’s role in lipid metabolism.
Since FoxO1 contributes to hyperglycemia in T2D, research has focused on targeting drugs that inhibit hepatic FoxO1 as potential T2D therapeutics (e.g. 76). For example, the selective FoxO1 inhibitor AS1842856 reduces fasting plasma glucose levels in db/db diabetic mice. More recent studies have identified candidate treatments that mitigate T2D symptoms by enhancing AKT-mediated suppression of FoxO1. For example, the traditional Chinese herbal medicine, Simiao Wan, lowers blood glucose levels by enhancing AKT expression and decreasing FoxO1 expression (77). The dietary fiber Oat β-D-glucan has been shown to decrease blood glucose levels and enhance insulin sensitivity (78). Recently, Guo et al. (79) demonstrated that Oat β-D-glucan exerts its actions in part by lowing gluconeogenesis via increased PI3K/AKT and reduced FoxO1. Vitamin D deficiency has been associated with T2D and Vitamin D supplementation improves insulin sensitivity in insulin resistant individuals (80–83). Yuan et al. (84) demonstrated that Vitamin D3 directly upregulates hepatic SIRT1 transcription which activates AKT to inactivate FoxO1 to decrease gluconeogenesis and improve glycemia. Similarly, the nicotinamide metabolite N1-Methylnicotinamide (MNAM) lowers blood glucose and hepatic lipid accumulation by increasing SIRT1 and pAKT and decreasing FoxO1 activity (85). The bioactive lipid, prostaglandin E2 (PGE2) also reduces gluconeogenesis and ameliorates T2D symptoms by inhibiting FoxO1 activity (86). SW03329 is a small molecule that enhances PGE2 levels by inhibiting the PGE2-degrading enzyme 15-hydroxyprostaglandin dehydrogenase. SW033291 reduces gluconeogenesis, blood glucose, serum triglycerides and body weight in diabetic mice. These actions of SW033291on gluconeogenesis are mediated by increased AKT inactivation of FoxO1. The role of n3-polyunsaturated fatty acids (n3-PUFAs) on diabetes and nonalcoholic fatty liver disease (NAFLD) has received considerable attention, and a recent meta-review analysis concluded that n3-PUFAs act to prevent and treat T2D (87). Further research demonstrated that n3-PUFAs reduce hepatic FoxO1 expression to improve insulin sensitivity as well as glycemia and lipid profiles of diabetic mice (88). This study also showed that metformin, the popular first-line T2D drug, lowers FoxO1 expression in diabetic mice. Guo et al. (89) described the mechanism by which metformin acts on FoxO1 to reduce hepatic glucose production and blood glucose levels, which was not by regulating insulin signaling, but rather by affecting glucagon signaling. Glucagon is a hyperglycemic hormone that activates Protein Kinase A (PKA) to elevate gluconeogenesis and thus blood glucose levels. PKA acts as a FoxO1 activating kinase importing and retaining FoxO1 in the nucleus to increase gluconeogenesis (90). Metformin was found to lower blood glucose by inhibiting glucagon induced PKA activity causing a decrease in FoxO1 and gluconeogenesis. Altogether, these results advocate for the use of hepatic FoxO1 inhibition in T2D treatment.
4 FoxO1 and T2D – skeletal muscle
FoxO1 regulates muscle mass and glucose metabolism in skeletal muscle. For example, FoxO1 overexpression in transgenic mice results in reduced body weight and muscle mass as well as impaired glucose tolerance (91). FoxO1 upregulates the ubiquitin ligase atrogin-1 (also known as MAFbx) which enhances muscle atrophy (92). Furthermore, insulin-like growth factor (IGF) activation of the PI3K/AKT pathway suppresses FoxO1 and FoxO3 action on atrogin-1, which promotes muscle mass. Combined loss of FoxO1 and FoxO4 function in skeletal muscle increases the expression of atrogin-1 and causes muscle hypertrophy (93). Furthermore, evidence suggests that FoxO1 may also play a role in muscle atrophy during T2D. Hyperglycemia in T2D increases the levels of Advanced Glycation End Products (AGEs), which are toxic and exacerbate T2D symptoms (94). Du et al. (95) found that elevated AGE increases skeletal muscle atrophy by increasing OS and endoplasmic reticulum (ER) stress, which is caused by the accumulation of unfolded or misfolded proteins. ER stress activates PERK, which in turn activates FoxO1 to increase atrogin-1. SiRNA silencing of FoxO1 rescued the AGE effect on muscle atrophy signifying that AGE production in T2D may promote muscle wasting via FoxO1. Additionally, FoxO1 has been shown to decrease muscle mass by upregulating the ubiquitin ligase MuRF1 and the liposomal protease cathepsin L (96, 97). Skeletal muscle specific knockout studies further support a role for FoxOs in mediating muscle atrophy. For example, one study showed that the collective deletion of skeletal muscle FoxO1, FoxO3, and FoxO4 upregulates the SMART (Specific of Muscle Atrophy and Regulated by Transcription) group of ubiquitin ligases to protect against fasting-induced muscle wasting (98). Likewise, another study demonstrated that insulin and IGF act to increase muscle mass through their negative regulation of FoxO activity (99). This study showed that while insulin is the primary mediator of muscle protein synthesis, insulin and IGF work synergistically to increase muscle mass. Moreover, muscle specific knockout of both insulin and IGF (MIGIRKO) depletes muscle mass in a FoxO-dependent manner as simultaneous deletion of FoxO1, FoxO3, and FoxO4 in MIGIRKO mice reversed the loss of muscle mass. Notably, ageing patients with T2D have significantly reduced muscle mass, which is associated with increased mortality (100–102). A recent study showed that skeletal muscle specific knockout of AKT1 and AKT2 causes sarcopenia, the progressive reduction in muscle strength and mass, and insulin resistance both of which were largely reversed by additional knockout of FoxO1 (103). Future studies should determine whether targeting FoxO1 inactivation could treat T2D-induced muscle wasting.
The increase in blood glucose that characterizes T2D is largely caused by decreased insulin-stimulated glucose uptake into skeletal muscle via GLUT4 glucose transporters (104, 105). Evidence suggests that FoxO1 and FoxO3 play a role in insulin’s regulation of GLUT4. For example, Lundell et al. (106) found that decreased expression of either FoxO1 or FoxO3 in skeletal muscle reduces GLUT4 protein levels and glucose uptake. Furthermore, attenuation of FoxO3, and not FoxO1, decreased glycogen content indicating that FoxO3 is important for the control of glucose storage in skeletal muscle. Further studies suggest that both AKT and SIRT1 regulate FoxO1’s effects on muscle mass and GLUT4 (107). Elevated SIRT1 during cardiac or skeletal muscle hypertrophy has been associated with increased AKT levels and activity and decreased FoxO1 (61, 107–109). For example, Gombos et al. (61) found that experimentally induced skeletal muscle hypertrophy enhances SIRT1 and AKT levels, decreases FoxO1 levels, and increases GLUT4 levels. Altogether, studies show that increased skeletal muscle FoxO1 acts to elevate blood glucose by decreasing GLUT4 levels and thus reducing glucose uptake.
Reduced glucose uptake and increased lipid accumulation in skeletal muscle contributes to the shift from glucose oxidation to lipid oxidation observed in T2D (110–112). FoxO1 has been shown to mediate lipid metabolism in skeletal muscle. For example, a study using the C2C12 skeletal muscle cell line reported that FoxO1 upregulates lipoprotein lipase (LPL), which is an enzyme that hydrolyses triglycerides into fatty acids (113). In addition, FoxO1 alters the cellular localization of the fatty acid translocase CD36 to enhance fatty acid uptake (111). Specifically, overactivation of FoxO1, by impeding AKT-induced FoxO1 inactivation, increases CD36 localization in the plasma membrane along with fatty acid uptake into C2C12 skeletal muscle cells. Blocking AKT-mediated FoxO1 inactivation also promotes fatty acid oxidation. In addition to fatty acid oxidation, insulin activation of AKT regulates glucose oxidation in skeletal muscle. For example, insulin augments glucose oxidation by strongly suppressing pyruvate dehydrogenase kinase (PDK) 4, which is an enzymes that inhibits the conversion of pyruvate to acetyl-CoA thereby reducing glucose oxidation and ATP production (114). However, low insulin signaling conditions like fasting and diabetes increases skeletal muscle PDK4 (115). For example, rats induced with diabetes present with increased PDK activity in skeletal muscle as well as lower glucose oxidation, and insulin reverses these effects (116, 117). FoxO1 mediates insulin’s action on PDK4. For example, FoxO1 has been shown to directly upregulate PDK4 transcription in skeletal muscle (118). Furthermore, stimulation of acute insulin resistance in rats increases PDK4 expression, decreases the active phosphorylated form of AKT, and increases the inactive phosphorylated form of FoxO1 (112). Overall, these results suggest that increased FoxO1 activity in insulin resistance contributes to the decrease in skeletal muscle glucose uptake and utilization along with the increase in fatty acid uptake and oxidation.
Recent studies demonstrate that pharmacological inhibition of FoxO1 improves muscle mass (119). For example, treatment with the FoxO1 specific inhibitor AS1842856 partially reverses the loss of fast-twitch muscle mass caused by AKT1/2 deletion (103). Furthermore, FoxO1 inhibition also mitigates insulin resistance. For example, the traditional Chinese herbal treatment Geniposide, which is an iridoid glycoside found in Gardenia fruits, improves glucose utilization in skeletal muscle by downregulating FoxO1 and PDK4 expression (120). Similarly, the Chinese herbal formula Yunpi Heluo (YPHL) decoction was found to increase SIRT1 and decrease FoxO1 in skeletal muscle of diabetic Zucker rats. YPHL treatment in these rats also improves glycemia and insulin sensitivity, and lowers cholesterol, triglyceride levels, and body weight (121). Vitamin D is known to alleviate T2D symptoms (80–83). Studies indicate that Vitamin D3 improves T2D by targeting hepatic FoxO1 suppression (84). Recent work shows that lack of the vitamin D receptor in skeletal muscle increases FoxO1 activity and causes insulin resistance and glucose intolerance (83). Thus, the beneficial effects of Vitamin D on insulin sensitivity may also act via FoxO1 inactivation in skeletal muscle. Taken together, these results suggest that FoxO1 inhibition in skeletal muscle may help treat T2D.
5 FoxO1 and T2D – adipose tissue
FoxO1 is densely expressed in white adipose tissue (WAT), where it controls adipocyte differentiation from preadipocytes. FoxO1 decreases adipogenesis by acting as a transrepressor of peroxisome proliferator-activated receptor gamma (PPARγ) and as a transactivator of the cell cycle inhibitor p21 (122–124). However, siRNA silencing of FoxO1 as well as FoxO1 inhibition with the selective antagonist AS1842856 inhibits adipogenesis (125, 126). FoxO1’s complicated regulation of adipocyte differentiation is timing dependent and reflects its role in regulating the cell cycle (123). Despite this complexity, targeting FoxO1 in WAT for T2D treatment has proven effective. For example, reducing FoxO1 activity in insulin-impaired mice increases insulin sensitivity, and decreasing FoxO1 activity specifically in WAT enhances insulin sensitivity and glucose tolerance (122, 127, 128). Recent evidence reveals that regulation of adipocyte FoxO1 by 3′-phosphoinositide-dependent kinase 1 (PDK1) plays an important role in metabolic disease. PDK1 is a kinase involved in insulin signaling. Insulin binding its receptor activates PI3K, which phosphorylates AKT directly and indirectly through facilitating AKT activation by other kinases like PDK1. For example, PI3K increases phosphatidylinositol (3-5)-trisphosphate (PIP3) which recruits PDK1 to the plasma membrane and promotes the interaction between PDK1 and AKT (129, 130). PDK1 phosphorylates AKT specifically at Thr308 (Figure 2). FoxO1 activity is decreased with increased PDK1/AKT signaling. Adipocyte-specific PDK1 knockout (A-PDK1KO) mice manifest symptoms of metabolic disease including insulin resistance, glucose intolerance, and hepatic steatosis that resembles the phenotype generated by knocking out the insulin receptor in adipocytes (131). These results emphasize the importance of PDK1 in insulin’s metabolic actions in WAT. Furthermore, these A-PDK1KO metabolic changes were reversed in mice with a combined deletion of PDK1 and FoxO1 indicating that FoxO1 activity was required for the development of insulin resistance, glucose intolerance, and hepatic steatosis in mice lacking PDK1 (132). These data suggest that inhibiting FoxO1 activity during insulin resistance when PDK1 signaling is reduced may help to reverse T2D symptoms. Other agents have been shown to mitigate T2D symptoms via FoxO1 inhibition in WAT. For example, the nonsteroidal anti-inflammatory drug-activated gene-1 (NAG-1) exerts its anti-diabetic effects by reducing adipocyte FoxO1 (133). Similarly, serum- and glucocorticoid-inducible kinase 1 (SGK1) increases AKT phosphorylation of FoxO1 which increases glucose uptake in adipocytes, and thus improves insulin sensitivity (134). Photobiomodulation (PBMT) is a non-invasive therapy that uses light sources like low-level lasers to stimulate cell proliferation, alter signaling pathways, decrease inflammation, and relieve pain in conditions such as diabetes (135). PBMT may improve T2D by inhibiting FoxO1. For example, a recent study demonstrated that PBMT via abdominal laser treatment enhances AKT inactivation of FoxO1 which decreases free fatty acid production and release from adipocytes of obese and diabetic mice (136). Knockdown of prolyl 4-hydroxylase subunit alpha 3 (P4HA3) with siRNA increases pAKT and pFoxO1, which decreases the transcriptional capacity of FoxO1. Consequently, P4HA3 inhibits adipogenesis, decreases fasting blood glucose levels, improves insulin resistance, and decreases body weight in diabetic mice (137). Altogether, these studies demonstrate that FoxO1 inhibition in WAT may alleviate T2D symptoms.
FoxO1 also regulates lipolysis by directly upregulating the lipolytic enzyme adipose triglyceride lipase (ATGL) (123). Deacetylation of FoxO1 by SIRT1 augments this response (138). Adipose-specific knockdown of Sirtuin 6 increases the acetylated form of FoxO1 causing an decrease in FoxO1 activity and downregulation of ATGL, which results in adipocyte hypertrophy (without hyperplasia), and promotes insulin resistance in obese fed mice (139). Adipocyte hypertrophy has received attention as an important factor inciting metabolic dysfunction via increased inflammation which was also reported in this study (140). Additionally, WAT ATGL is the first identified biosynthetic enzyme of mammalian fatty acid esters of hydroxy fatty acids (FAHFAs) some of which have anti-diabetic actions further highlighting FoxO1 and ATGL as therapeutic targets for T2D treatment (141). More research is needed to determine whether FoxO1’s regulation of lipolysis and FAHFAs via AGTL could prove effective in T2D treatment.
Brown adipose tissue (BAT) is a target for T2D treatment because of its ability to increase basal metabolic rate and decrease body weight. FoxO1 impedes BAT function by downregulating PPARγ coactivator (PGC)-1α and uncoupling protein 1 (UCP1) (128). FoxO1 also blocks the conversion of WAT to BAT by upregulating transcription factor EB (TFEB) (142) (Figure 3). The role of FoxO1 in regulating WAT and BAT depends on timing and insulin signaling. For example, Homan et al. (143) showed that knocking out FoxO1, FoxO3, and FoxO4 in adipocytes during early development of mice lacking the insulin and IGF receptors increases BAT mass and function, partially increases WAT, intensifies hyperinsulinemia, and improves hepatic insulin sensitivity. Future research should characterize the specific roles of each FoxO and determine whether these changes are dependent on developmental timing. Pharmacological intervention with the FoxO1 inhibitor AS1842856 decreases TFEB and increases UCP1 to increase brown adipocyte differentiation and function. Furthermore, Zhuang et al. (137) demonstrated that metformin inhibits autophagy and promotes BAT differentiation in adipose-derived stem cells by FoxO1 inactivation. Taken together, evidence supports FoxO1 inhibition in BAT as an attractive target for T2D therapeutics.
6 FoxO1 and T2D – pancreatic β-cells
Pancreatic β-cells produce and secrete insulin. These cells are particularly sensitive to the development of insulin resistance. Indeed, early insulin resistance induces a compensatory response in which insulin production and secretion is enhanced in part due to β-cell hyperplasia. Enhanced insulin signaling during compensatory hyperinsulinemia increases the activity of pancreatic duodenal homeobox 1 (PDX1) which is a transcription factor that augments β-cells proliferation (144). Kulkarni et al. (144) showed that PDX1 deficiency impairs insulin’s compensatory response on β-cell mass. Further work demonstrated that FoxO1 links insulin and PDX1 action on β-cells proliferation (145). FoxO1 suppresses the transcriptional activity of PDX1 (146, 147). Mechanistically, compensatory hyperinsulinemia increases pancreatic PI3K/AKT signaling, which decreases FoxO1 activity and causes an increase in PDX1 activity and β-cell mass (145). However, if this response fails to compensate and insulin resistance persists, β-cell proliferation will decrease along with insulin secretion and T2D will develop. In this state of advanced insulin resistance, the decrease in insulin lowers PI3K/AKT signaling and increases FoxO1 activity which impairs PDX1-mediated β-cell growth further reducing insulin secretion and contributing to T2D pathogenesis.
In addition, microRNA (miRNA) studies have signified a role for FoxO1 in T2D. For example, FoxO1 upregulates miR-802, which is elevated in diabetic mouse pancreatic islets and contributes to β-cell dysfunction by decreasing insulin secretion (148). Li et al. (149) showed that miR-233 is upregulated in pancreatic cells of diabetic mice and humans and improves β-cell proliferation and function via FoxO1 suppression. Similarly, an irisin-induced increase in miR-133a-3p downregulates FoxO1 to protect β-cells from pyroptosis (150). Pharmacological inhibition of β-cell FoxO1 also ameliorates T2D symptoms. For example, the anti-allergic drug and FoxO1 inhibitor tranilast improves glucose tolerance in diabetic mice and reverses palmitic acid (PA)-induced reduction in β-cell insulin secretion (151). Similarly, glucagon-like peptide-1 receptor agonists (GLP-1RA) mimic incretin hormones, which increase insulin secretion and β-cell proliferation through PI3K/AKT-mediated nuclear exclusion of FoxO1 (152, 153). For example, the GLP-1RAs liraglutide and EXf inactivate FoxO1 causing increased β-cell proliferation in a PI3K-dependent manner (154, 155). Additionally, the isoflavone puerarin increases GLP-1R activation of AKT, decreases FoxO1 activity, and improves glucose tolerance (156, 157). Vitamin D also improves insulin secretion via reduced FoxO1 protein levels which reduces β-cell ferroptosis (158). Furthermore, Zhang et al. (159) reversed the negative effects of β-cell senescence using the Saccharina japonica derivative fuco-manno-glucuronogalactan (SFGG) in insulin secreting MIN6 cells. SFGG anti-senescence effects are regulated by pAKT-mediated FoxO1 suppression suggesting that FoxO1 contributes to the T2D pathogenesis by accelerating β-cell ageing. FoxO1 deficiency may also contribute to β-cell senescence and T2D as knockout of the proliferation mediator SMAD7 reduced FoxO1 which enhanced β-cell ageing and caused diabetes suggesting that there may be an optimal range of FoxO1 activity (160).
FoxO1 may help to prevent T2D under conditions like OS and inflammation which are associated with T2D pathogenesis. For example, FoxO1 protects β-cells from OS by upregulating antioxidants and by interfering with Carbohydrate Response Element-binding Protein (Chrebp) to combat DNA damage and apoptosis (2, 5, 19, 161, 162). In contrast to AKT, phosphorylation of FoxO1 by inflammatory pathways like c-Jun N-terminal kinase (JNK) augments nuclear entry of FoxO1 to increase insulin transcription via upregulation of NeuroD and MafA transcription factors (163, 164). Moreover, studies indicates that as insulin resistance and OS progresses, FoxO1 acts to combat these conditions but over time its protein levels decline causing β-cell dedifferentiation and marking a molecular switch to T2D pathology (165, 166). Furthermore, the GLP-1RA exenatide decreases inflammation and boosts β-cell function by improving the balance between CD4+ T-helper 17 (Th17) cells and regulatory T-cells (Tregs) (167). The FoxO1 inhibitor AS1842856 reversed these effects indicating that FoxO1 activity may be important for T17/Treg balance and reduced inflammation. FoxO1 also safeguards β-cells during hypoxia by regulating autophagy (168). For example, liraglutide protects INS-1 cells from PA-induced injury by enhancing autophagy and cell survival through elevated FoxO1 activity (169). Similarly, Liang et al. (170) demonstrated that FoxO1 stimulates autophagy and enhances survival in cells under T2D-induced hypoxia.
FoxO1 knockout studies have examined its dual role in T2D. For example, FoxO1 deletion in mouse β-cells causes mitochondrial dysfunction, cell dedifferentiation, and hyperglycemia (171, 172). Kobayashi et al. (173) showed that knocking out FoxO1 in the pancreas increases β-cell mass, but this was not the case when FoxO1 was deleted specifically from β-cells. Furthermore, glucose intolerance in db/db diabetic mice was worsened when FoxO1 was knocked out in the pancreas as well as when FoxO1 was knocked out explicitly in β-cells. These data show that while FoxO1 inhibits β-cell proliferation, it can also improve glucose tolerance. Therefore, activating FoxO1 could prove effective. For example, L-Methionine (L-Met) regulates FoxO1 by altering the bivalent domain histone methylation marks H3K27me3 and H3K4me3 to increase FoxO1-mediated upregulation of the insulin transcription factor MafA and mitigate type 1 diabetes mellitus in rats (174). Given that Obex®, a supplement containing L-Met, caused weight loss and improved insulin homeostasis in overweight and obese participants of a double−blind, randomized, controlled phase III clinical trial, future studies should explore how altering bivalent domain controls FoxO1 regulation of β-cells during T2D (175).
7 FoxO1 and T2D – brain
FoxO1 mediates the hypothalamic control of metabolism by integrating signals from peripheral regulators like ghrelin, insulin, and leptin (176). Ghrelin-activated FoxO1 in the hypothalamic arcuate nucleus (ARC) induces hyperphagia by upregulating the orexigenic peptide Agouti-related protein and downregulating proopiomelanocortin (POMC) from which the anorexigenic peptide alpha melanocyte stimulating hormone (α-MSH) is derived (177). FoxO1 also decreases the POMC processing enzyme Carboxypeptidase E (Cpe) further reducing α-MSH (178). FoxO1 ablation in POMC neurons increases CPE and α-MSH resulting in reduced food intake and protection from diet-induced obesity (DIO) weight gain. In contrast, FoxO1 overexpression in hypothalamus and pancreas increases food intake and impairs glucose tolerance and insulin secretion (179). Leptin and insulin reverse FoxO1 action by enhancing AKT inactivation of FoxO1 (33, 180). However, leptin and insulin signaling is compromised in obesity and T2D in the ARC (181). Therefore, pharmacologically inactivating hypothalamic FoxO1 could treat these conditions. Indeed, central inhibition of FoxO1 directly or via SIRT1 inhibition reduces body weight and improves insulin sensitivity in DIO rodents (182, 183). Furthermore, actions of the orexigenic melanin-concentrating hormone (MCH) in the ARC that promote hyperphagia, adipocyte lipid storage, and glucose intolerance are dependent on SIRT1 and FoxO1 activation (184). Overall, evidence suggests that FoxO1 inhibition in the hypothalamus has the potential to treat obesity induced T2D.
Because of FoxO1’s role in regulating brain metabolism, it may link metabolic dysfunction and neurodegenerative disease (185). For example, neurodegenerative diseases like Alzheimer’s disease (AD), Huntington’s disease (HD), and Parkinson’s disease (PD) are worsened by T2D (186–188), and FoxO1 expression is elevated in each of these conditions (189–193). Furthermore, increased FoxO1 has been shown to reduce pathological AD features such as tau hyperphosphorylation and amyloid beta (Aβ) plaques (190, 194–196). A recent study screened FoxO1 activators as potential therapeutics and demonstrated that one, Compound D, decreases Aβ1-40 and Aβ1-42 in SH-SY5Y cells (197). Additionally, exercise, an AD intervention, increases circulating FoxO1 in African American men with mild cognitive impairment (198), and upregulates FoxO1 to improve AD symptoms in early-onset AD mice (199). Interestingly, the ani-aging protein klotho improves AD symptoms by either activating (200, 201) or inactivating FoxO1 (202). Conflicting results of FoxO1 have also been reported in HD studies. For example, Li et al. (203) showed that AKT-mediated FoxO1 inactivation increases pathogenic mutant Huntington (mHtt) protein aggregates, yet another study showed that increasing FoxO1-dependent expression of autophagy, mitochondrial, and antioxidants genes improves HD symptoms including increased neuronal survival and enhanced motor control (204). Most studies of PD demonstrate that increased FoxO1 contributes to its pathology via increased inflammation and apoptosis (205, 206). Evidence also suggests that FoxO1 mediates the depressive symptoms of PD (207). More research is needed to better understand FoxO1’s role in the pathology of neurodegenerative diseases.
8 Conclusion and prospective
Insulin resistance in T2D increases FoxO1 activation in metabolic tissues including liver, skeletal muscle, adipose tissue, pancreas, and brain, which underscores FoxO1’s potential clinical relevance. However, recent evidence in these tissues and others like heart and kidney (208, 209) highlights controversy and indicates that inactivating FoxO1 could either mitigate or exacerbate T2D pathology (2–5). Knowing a patient’s history and risk for certain diseases may be critical. For example, FoxO1 appears to play opposing roles in AD and PD pathology at least under certain conditions. Furthermore, FoxO1 can act as a tumor suppressor and thus activating FoxO1 may help treat certain cancers, yet FoxO1 activation worsens stroke pathology (210, 211). Therefore, targeting the optimal tissue for activation or inhibition of FoxO1 and developing strategies for tissue-specific delivery of FoxO1 therapeutics could effectively treat T2D and minimize adverse side effects. Likewise, this approach could extend to targeting FoxO1 for treatment of other diseases.
Author contributions
NC: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. NT: Writing – original draft, Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. NT was supported with National Science Foundation (NSF), Strand 2: S-STEM: Design & Dev -Type 1 Award number: 1643763, and the Endocrine Society Summer Research Fellowship. NT and NC were supported by the Stonehill Undergraduate Research Experience (SURE) award.
Acknowledgments
We thank Dr. John McCoy for providing helpful feedback on an early draft of the manuscript. Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ong KL, Stafford LK, McLaughlin SA, Boyko EJ, Vollset SE, Smith AE, et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the global burden of disease study 2021. Lancet (2023) 402:203–34. doi: 10.1016/S0140-6736(23)01301-6
2. Benchoula K, Arya A, Parhar IS, Hwa WE. Foxo1 signaling as a therapeutic target for type 2 diabetes and obesity. Eur J Pharmacol (2021) 891:173758. doi: 10.1016/j.ejphar.2020.173758
3. Nathanael J, Suardana P, Vianney YM, Dwi Putra SE. The role of Foxo1 and its modulation with small molecules in the development of diabetes mellitus: A review. Chem Biol Drug Design (2022) 99(2):344–61. doi: 10.1111/cbdd.13989
4. Brunetti A, Arcidiacono B, Foti DP, Semple RK. Editorial: Transcriptional regulation of glucose metabolism: Gaps and controversies. Front Endocrinol (Lausanne) (2019) 10:629. doi: 10.3389/fendo.2019.00629
5. Marchelek-Mysliwiec M, Nalewajska M, Turoń-Skrzypińska A, Kotrych K, Dziedziejko V, Sulikowski T, et al. The role of forkhead box O in pathogenesis and therapy of diabetes mellitus. Int J Mol Sci (2022) 23(19):11611. doi: 10.3390/ijms231911611
6. Calissi G, Lam EW-F, Link W. Therapeutic strategies targeting Foxo transcription factors. Nat Rev Drug Discov (2021) 20(1):21–38. doi: 10.1038/s41573-020-0088-2
7. Gui T, Burgering BM. Foxos: masters of the equilibrium. FEBS J (2022) 289(24):7918–39. doi: 10.1111/febs.16221
8. Lettieri-Barbato D, Ioannilli L, Aquilano K, Ciccarone F, Rosina M, Ciriolo MR. Foxo1 localizes to mitochondria of adipose tissue and is affected by nutrient stress. Metabolism (2019) 95:84–92. doi: 10.1016/j.metabol.2019.04.006
9. Celestini V, Tezil T, Russo L, Fasano C, Sanese P, Forte G, et al. Uncoupling foxo3a mitochondrial and nuclear functions in cancer cells undergoing metabolic stress and chemotherapy. Cell Death Dis (2018) 9(2):231. doi: 10.1038/s41419-018-0336-0
10. Caballero-Caballero A, Engel T, Martinez-Villarreal J, Sanz-Rodriguez A, Chang P, Dunleavy M, et al. Mitochondrial localization of the forkhead box class O transcription factor Foxo 3a in brain. J Neurochem (2013) 124(6):749–56. doi: 10.1111/jnc.12133
11. Furuyama T, Nakazawa T, Nakano I, Mori N. Identification of the differential distribution patterns of mrnas and consensus binding sequences for mouse daf-16 homologues. Biochem J (2000) 349(2):629–34. doi: 10.1042/0264-6021:3490629
12. Kaestner KH, Knöchel W, Martínez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev (2000) 14(2):142–6. doi: 10.1101/gad.14.2.142
13. Obsil T, Obsilova V. Structure/function relationships underlying regulation of Foxo transcription factors. Oncogene (2008) 27(16):2263–75. doi: 10.1038/onc.2008.20
14. Pang W-J, Yu T-Y, Bai L, Yang Y-J, Yang G-S. Tissue expression of porcine Foxo1 and its negative regulation during primary preadipocyte differentiation. Mol Biol Rep (2009) 36:165–76. doi: 10.1007/s11033-007-9163-6
15. Hoekman MF, Jacobs FM, Smidt MP, Burbach JPH. Spatial and temporal expression of Foxo transcription factors in the developing and adult murine brain. Gene Expr Patterns (2006) 6(2):134–40. doi: 10.1016/j.modgep.2005.07.003
16. Furuyama T, Yamashita H, Kitayama K, Higami Y, Shimokawa I, Mori N. Effects of aging and caloric restriction on the gene expression of Foxo1, 3, and 4 (Fkhr, Fkhrl1, and Afx) in the rat skeletal muscles. Microsc Res Tech (2002) 59(4):331–4. doi: 10.1002/jemt.10213
17. Zhang K, Li L, Qi Y, Zhu X, Gan B, DePinho RA, et al. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology (2012) 153(2):631–46. doi: 10.1210/en.2011-1527
18. Jacobs FM, van der Heide LP, Wijchers PJ, Burbach JPH, Hoekman MF, Smidt MP. Foxo6, a novel member of the Foxo class of transcription factors with distinct shuttling dynamics. J Biol Chem (2003) 278(38):35959–67. doi: 10.1074/jbc.M302804200
19. Link W, Fernandez-Marcos PJ. Foxo transcription factors at the interface of metabolism and cancer. Int J Cancer (2017) 141(12):2379–91. doi: 10.1002/ijc.30840
20. Hosaka T, Biggs WH III, Tieu D, Boyer AD, Varki NM, Cavenee WK, et al. Disruption of forkhead transcription factor (Foxo) family members in mice reveals their functional diversification. Proc Natl Acad Sci (2004) 101(9):2975–80. doi: 10.1073/pnas.0400093101
21. Zhou W, Cao Q, Peng Y, Zhang QJ, Castrillon DH, DePinho RA, et al. Foxo4 inhibits nf-Kb and protects mice against colonic injury and inflammation. Gastroenterology (2009) 137(4):1403–14. doi: 10.1053/j.gastro.2009.06.049
22. Salih DA, Rashid AJ, Colas D, de la Torre-Ubieta L, Zhu RP, Morgan AA, et al. Foxo6 regulates memory consolidation and synaptic function. Genes Dev (2012) 26(24):2780–801. doi: 10.1101/gad.208926.112
23. Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member Fkhr by protein kinase B. J Biol Chem (1999) 274(24):17179–83. doi: 10.1074/jbc.274.24.17179
24. Williams KA, Zhang M, Xiang S, Hu C, Wu J-Y, Zhang S, et al. Extracellular signal-regulated kinase (Erk) phosphorylates histone deacetylase 6 (Hdac6) at serine 1035 to stimulate cell migration. J Biol Chem (2013) 288(46):33156–70. doi: 10.1074/jbc.M113.472506
25. Liu P, Kao T, Huang H. Cdk1 promotes cell proliferation and survival via phosphorylation and inhibition of Foxo1 transcription factor. Oncogene (2008) 27(34):4733–44. doi: 10.1038/onc.2008.104
26. Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. Cdk2-dependent phosphorylation of Foxo1 as an apoptotic response to DNA damage. Science (2006) 314(5797):294–7. doi: 10.1126/science.1130512
27. You S, Li H, Hu Z, Zhang W. Eif 2α Kinases perk and Gcn 2 act on Foxo to potentiate Foxo activity. Genes Cells (2018) 23(9):786–93. doi: 10.1111/gtc.12625
28. Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, Bos JL, et al. Foxo transcription factor activation by oxidative stress mediated by the small gtpase ral and jnk. EMBO J (2004) 23(24):4802–12. doi: 10.1038/sj.emboj.7600476
29. Saline M, Badertscher L, Wolter M, Lau R, Gunnarsson A, Jacso T, et al. Ampk and akt protein kinases hierarchically phosphorylate the N-terminus of the Foxo1 transcription factor, modulating interactions with 14-3-3 proteins. J Biol Chem (2019) 294(35):13106–16. doi: 10.1074/jbc.RA119.008649
30. Yuan Z, Lehtinen MK, Merlo P, Villén J, Gygi S, Bonni A. Regulation of neuronal cell death by mst1-Foxo1 signaling. J Biol Chem (2009) 284(17):11285–92. doi: 10.1074/jbc.M900461200
31. Nemoto S, Finkel T. Redox regulation of forkhead proteins through a P66shc-dependent signaling pathway. Science (2002) 295(5564):2450–2. doi: 10.1126/science.1069004
32. Weng Q, Liu Z, Li B, Liu K, Wu W, Liu H. Oxidative stress induces mouse follicular granulosa cells apoptosis via Jnk/Foxo1 pathway. PloS One (2016) 11(12):e0167869. doi: 10.1371/journal.pone.0167869
33. Biggs WH III, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor Fkhr1. Proc Natl Acad Sci (1999) 96(13):7421–6. doi: 10.1073/pnas.96.13.7421
34. Dobson M, Ramakrishnan G, Ma S, Kaplun L, Balan V, Fridman R, et al. Bimodal regulation of Foxo3 by akt and 14-3-3. Biochim Biophys Acta (BBA)-Mol Cell Res (2011) 1813(8):1453–64. doi: 10.1016/j.bbamcr.2011.05.001
35. Singh A, Ye M, Bucur O, Zhu S, Tanya Santos M, Rabinovitz I, et al. Protein phosphatase 2a reactivates Foxo3a through a dynamic interplay with 14-3-3 and Akt. Mol Biol Cell (2010) 21(6):1140–52. doi: 10.1091/mbc.e09-09-0795
36. De Ruiter ND, Burgering BM, Bos JL. Regulation of the forkhead transcription factor Afx by Ral-dependent phosphorylation of threonines 447 and 451. Mol Cell Biol (2001) 21(23):8225–35. doi: 10.1128/MCB.21.23.8225-8235.2001
37. Kops GJ, Ruiter N, De Vries-Smits AM, Powell DR, Bos JL, Burgering BMT. Direct control of the forkhead transcription factor Afx by protein kinase B. Nature (1999) 398(6728):630–4. doi: 10.1038/19328
38. van der Heide LP, Jacobs FM, Burbach JPH, Hoekman MF, Smidt MP. Foxo6 transcriptional activity is regulated by thr26 and ser184, independent of nucleo-cytoplasmic shuttling. Biochem J (2005) 391(3):623–9. doi: 10.1042/BJ20050525
39. Yang X-J, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell (2008) 31(4):449–61. doi: 10.1016/j.molcel.2008.07.002
40. Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, et al. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. cell (1999) 96(6):857–68. doi: 10.1016/S0092-8674(00)80595-4
41. Brunet A, Kanai F, Stehn J, Xu J, Sarbassova D, Frangioni JV, et al. 14-3-3 transits to the nucleus and participates in dynamic nucleocytoplasmic transport. J Cell Biol (2002) 156(5):817–28. doi: 10.1083/jcb.200112059
42. Stade K, Ford CS, Guthrie C, Weis K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell (1997) 90(6):1041–50. doi: 10.1016/s0092-8674(00)80370-0
43. Asada S, Daitoku H, Matsuzaki H, Saito T, Sudo T, Mukai H, et al. Mitogen-activated protein kinases, erk and P38, phosphorylate and regulate Foxo1. Cell Signal (2007) 19(3):519–27. doi: 10.1016/j.cellsig.2006.08.015
44. Nakae J, Park B-C, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor Fkhr on serine 253 through a wortmannin-sensitive pathway. J Biol Chem (1999) 274(23):15982–5. doi: 10.1074/jbc.274.23.15982
45. Brownawell AM, Kops GJ, Macara IG, Burgering BM. Inhibition of nuclear import by protein kinase B (Akt) regulates the subcellular distribution and activity of the forkhead transcription factor Afx. Mol Cell Biol (2001) 21(10):3534–46. doi: 10.1128/MCB.21.10.3534-3546.2001
46. Rena G, Prescott AR, Guo S, Cohen P, Unterman TG. Roles of the forkhead in rhabdomyosarcoma (Fkhr) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem J (2001) 354(3):605–12. doi: 10.1042/0264-6021:3540605
47. Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci (2005) 102(32):11278–83. doi: 10.1073/pnas.0502738102
48. Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates Foxo1 function independent of its subcellular localization. J Biol Chem (2010) 285(35):27396–401. doi: 10.1074/jbc.M110.140228
49. van der Horst A, Tertoolen LG, de Vries-Smits LM, Frye RA, Medema RH, Burgering BM. Foxo4 is acetylated upon peroxide stress and deacetylated by the longevity protein Hsir2sirt1. J Biol Chem (2004) 279(28):28873–9. doi: 10.1074/jbc.M401138200
50. Mortuza R, Chen S, Feng B, Sen S, Chakrabarti S. High glucose induced alteration of sirts in endothelial cells causes rapid aging in a P300 and Foxo regulated pathway. PloS One (2013) 8(1):e54514. doi: 10.1371/journal.pone.0054514
51. Nakae J, Cao Y, Daitoku H, Fukamizu A, Ogawa W, Yano Y, et al. The lxxll motif of murine forkhead transcription factor Foxo1 mediates Sirt1-dependent transcriptional activity. J Clin Invest (2006) 116(9):2473–83. doi: 10.1172/JCI25518
52. Hasegawa K, Wakino S, Yoshioka K, Tatematsu S, Hara Y, Minakuchi H, et al. Sirt1 protects against oxidative stress-induced renal tubular cell apoptosis by the bidirectional regulation of catalase expression. Biochem Biophys Res Commun (2008) 372(1):51–6. doi: 10.1016/j.bbrc.2008.04.176
53. Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, et al. Stress-dependent regulation of Foxo transcription factors by the sirt1 deacetylase. science (2004) 303(5666):2011–5. doi: 10.1126/science.109463
54. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of Pgc-1α and Sirt1. Nature (2005) 434(7029):113–8. doi: 10.1038/nature03354
55. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, MaChado de Oliveira R, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing Ppar-Γ. Nature (2004) 429(6993):771–6. doi: 10.1038/nature02583
56. Fröjdö S, Durand C, Molin L, Carey AL, El-Osta A, Kingwell BA, et al. Phosphoinositide 3-kinase as a novel functional target for the regulation of the insulin signaling pathway by Sirt1. Mol Cell Endocrinol (2011) 335(2):166–76. doi: 10.1016/j.mce.2011.01.008
57. Bartoli-Leonard F, Wilkinson F, Schiro A, Inglott FS, Alexander M, Weston R. Suppression of Sirt1 in diabetic conditions induces osteogenic differentiation of human vascular smooth muscle cells via runx2 signalling. Sci Rep (2019) 9(1):878. doi: 10.1038/s41598-018-37027-2
58. Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, et al. Hepatic overexpression of sirt1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J (2011) 25(5):1664. doi: 10.1096/fj.10-173492
59. Qiao L, Shao J. Sirt1 regulates adiponectin gene expression through Foxo1-C/enhancer-binding protein A Transcriptional complex. J Biol Chem (2006) 281(52):39915–24. doi: 10.1074/jbc.M607215200
60. Wang R-H, Kim H-S, Xiao C, Xu X, Gavrilova O, Deng C-X. Hepatic Sirt1 deficiency in mice impairs mtorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest (2011) 121(11):4477–90. doi: 10.1172/JCI46243
61. Gombos Z, Koltai E, Torma F, Bakonyi P, Kolonics A, Aczel D, et al. Hypertrophy of rat skeletal muscle is associated with increased Sirt1/Akt/Mtor/S6 and suppressed Sestrin2/Sirt3/Foxo1 levels. Int J Mol Sci (2021) 22(14):7588. doi: 10.3390/ijms22147588
63. O-Sullivan I, Zhang W, Wasserman DH, Liew CW, Liu J, Paik J, et al. Foxo1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun (2015) 6(1):7079. doi: 10.1038/ncomms8079
64. Matsumoto M, Pocai A, Rossetti L, DePinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab (2007) 6(3):208–16. doi: 10.1016/j.cmet.2007.08.006
65. Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, et al. Foxo1 regulates multiple metabolic pathways in the liver: effects on gluconeogenic, glycolytic, and lipogenic gene expression. J Biol Chem (2006) 281(15):10105–17. doi: 10.1074/jbc.M600272200
66. Altomonte J, Richter A, Harbaran S, Suriawinata J, Nakae J, Thung SN, et al. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am J Physiol-Endocrinol Metab (2003) 285(4):E718–E28. doi: 10.1152/ajpendo.00156.2003
67. Cheng Z, Guo S, Copps K, Dong X, Kollipara R, Rodgers JT, et al. Foxo1 integrates insulin signaling with mitochondrial function in the liver. Nat Med (2009) 15(11):1307–11. doi: 10.1038/nm.2049
68. Foretz M, Guichard C, Ferré P, Foufelle F. Sterol regulatory element binding protein-1c is a major mediator of insulin action on the hepatic expression of glucokinase and lipogenesis-related genes. Proc Natl Acad Sci (1999) 96(22):12737–42. doi: 10.1073/pnas.96.22.12737
69. Xiong X, Tao R, DePinho RA, Dong XC. Deletion of hepatic Foxo1/3/4 genes in mice significantly impacts on glucose metabolism through downregulation of gluconeogenesis and upregulation of glycolysis. PloS One (2013) 8(8):e74340. doi: 10.1371/journal.pone.0074340
70. Kamagate A, Qu S, Perdomo G, Su D, Kim DH, Slusher S, et al. Foxo1 mediates insulin-dependent regulation of hepatic Vldl production in mice. J Clin Invest (2008) 118(6):2347–64. doi: 10.1172/JCI32914
71. Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, et al. Foxo1 mediates insulin action on apoc-iii and triglyceride metabolism. J Clin Invest (2004) 114(10):1493–503. doi: 10.1172/JCI19992
72. Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor Foxo1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest (2006) 116(9):2464–72. doi: 10.1172/JCI27047
73. Langlet F, Haeusler RA, Lindén D, Ericson E, Norris T, Johansson A, et al. Selective inhibition of Foxo1 activator/repressor balance modulates hepatic glucose handling. Cell (2017) 171(4):824–35.e18. doi: 10.1016/j.cell.2017.09.045
74. Ferré P, Phan F, Foufelle F. Srebp-1c and lipogenesis in the liver: an update. Biochem J (2021) 478(20):3723–39. doi: 10.1042/BCJ20210071
75. Brown MS, Goldstein JL. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab (2008) 7(2):95–6. doi: 10.1016/j.cmet.2007.12.009
76. Nagashima T, Shigematsu N, Maruki R, Urano Y, Tanaka H, Shimaya A, et al. Discovery of novel forkhead box O1 inhibitors for treating type 2 diabetes: improvement of fasting glycemia in diabetic Db/Db mice. Mol Pharmacol (2010) 78(5):961–70. doi: 10.1124/mol.110.065714
77. Xia T, Xu W-J, Hu Y-N, Luo Z-Y, He W, Liu C-S, et al. Simiao wan and its ingredients alleviate type 2 diabetes mellitus via Irs1/Akt2/Foxo1/Glut2 signaling. Front Nutr (2023) 9:1012961. doi: 10.3389/fnut.2022.1012961
78. Biörklund M, Van Rees A, Mensink R, Önning G. Changes in serum lipids and postprandial glucose and insulin concentrations after consumption of beverages with B-glucans from oats or barley: A randomised dose-controlled trial. Eur J Clin Nutr (2005) 59(11):1272–81. doi: 10.1038/sj.ejcn.1602240
79. Guo H, Wu H, Hou Y, Hu P, Du J, Cao L, et al. Oat B-D-glucan ameliorates type ii diabetes through Tlr4/Pi3k/Akt mediated metabolic axis. Int J Biol Macromol (2023) 249:126039. doi: 10.1016/j.ijbiomac.2023.126039
80. González-Molero I, Rojo-Martínez G, Morcillo S, Gutiérrez-Repiso C, Rubio-Martín E, Almaraz MC, et al. Vitamin D and incidence of diabetes: A prospective cohort study. Clin Nutr (2012) 31(4):571–3. doi: 10.1016/j.clnu.2011.12.001
81. Pittas AG, Nelson J, Mitri J, Hillmann W, Garganta C, Nathan DM, et al. Plasma 25-hydroxyvitamin D and progression to diabetes in patients at risk for diabetes: an ancillary analysis in the diabetes prevention program. Diabetes Care (2012) 35(3):565–73. doi: 10.2337/dc11-1795
82. Naharci I, Bozoglu E, Kocak N, Doganci S, Doruk H, Serdar M. Effect of vitamin D on insulin sensitivity in elderly patients with impaired fasting glucose. Geriatr Gerontol Int (2012) 12(3):454–60. doi: 10.1111/j.1447-0594.2011.00791.x
83. Nazarian S, Peter JVS, Boston RC, Jones SA, Mariash CN. Vitamin D3 supplementation improves insulin sensitivity in subjects with impaired fasting glucose. Trans Res (2011) 158(5):276–81. doi: 10.1016/j.trsl.2011.05.002
84. Yuan Q, Zhang R, Sun M, Guo X, Yang J, Bian W, et al. Sirt1 mediates vitamin D deficiency-driven gluconeogenesis in the liver via Mtorc2/Akt signaling. J Diabetes Res (2022) 2022:1–16. doi: 10.1155/2022/1755563
85. Zhang J, Chen Y, Liu C, Li L, Li P. N 1-methylnicotinamide improves hepatic insulin sensitivity via activation of Sirt1 and inhibition of Foxo1 acetylation. J Diabetes Res (2020) 2020:1–11. doi: 10.1155/2020/1080152
86. Liang M, Wang L, Wang W. The 15-hydroxyprostaglandin dehydrogenase inhibitor sw033291 ameliorates abnormal hepatic glucose metabolism through Pge2–Ep4 receptor–Akt signaling in a type 2 diabetes mellitus mouse model. Cell Signal (2023) 108:110707. doi: 10.1016/j.cellsig.2023.110707
87. Xiao Y, Zhang Q, Liao X, Elbelt U, Weylandt KH. The effects of omega-3 fatty acids in type 2 diabetes: A systematic review and meta-analysis. Prostaglandins Leukot Essent Fatty Acids (2022) 182:102456. doi: 10.1016/j.plefa.2022.102456
88. Ramadan NM, Elmasry K, Elsayed HRH, El-Mesery A, Eraky SM. The Hepatoprotective Effects of N3-Polyunsaturated Fatty Acids against Non-Alcoholic Fatty Liver Disease in Diabetic Rats through the Foxo1/Pparα/Gabarapl1 Signalling Pathway. Life Sci (2022) 311:121145. doi: 10.1016/j.lfs.2022.121145
89. Guo X, Li X, Yang W, Liao W, Shen JZ, Ai W, et al. Metformin targets Foxo1 to control glucose homeostasis. Biomolecules (2021) 11(6):873. doi: 10.3390/biom11060873
90. Wu Y, Pan Q, Yan H, Zhang K, Guo X, Xu Z, et al. Novel mechanism of Foxo1 phosphorylation in glucagon signaling in control of glucose homeostasis. Diabetes (2018) 67(11):2167–82. doi: 10.2337/db18-0674
91. Kamei Y, Miura S, Suzuki M, Kai Y, Mizukami J, Taniguchi T, et al. Skeletal muscle Foxo1 (Fkhr) transgenic mice have less skeletal muscle mass, down-regulated type I (Slow twitch/red muscle) fiber genes, and impaired glycemic control*[Boxs]. J Biol Chem (2004) 279(39):41114–23. doi: 10.1074/jbc.M400674200
92. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase Atrogin-1 and cause skeletal muscle atrophy. Cell (2004) 117(3):399–412. doi: 10.1016/S0092-8674(04)00400-3
93. Reed SA, Sandesara PB, Senf SM, Judge AR. Inhibition of Foxo transcriptional activity prevents muscle fiber atrophy during cachexia and induces hypertrophy. FASEB J (2012) 26(3):987. doi: 10.1096/fj.11-189977
94. Goh S-Y, Cooper ME. The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab (2008) 93(4):1143–52. doi: 10.1210/jc.2007-1817
95. Du H, Ma Y, Wang X, Zhang Y, Zhu L, Shi S, et al. Advanced glycation end products induce skeletal muscle atrophy and insulin resistance via activating ros-mediated er stress Perk/Foxo1 signaling. Am J Physiol-Endocrinol Metab (2023) 324(3):E279–E87. doi: 10.1152/ajpendo.00218.2022
96. Yamazaki Y, Kamei Y, Sugita S, Akaike F, Kanai S, Miura S, et al. The cathepsin L gene is a direct target of Foxo1 in skeletal muscle. Biochem J (2010) 427(1):171–8. doi: 10.1042/BJ20091346
97. Waddell DS, Baehr LM, Van Den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, et al. The glucocorticoid receptor and Foxo1 synergistically activate the skeletal muscle atrophy-associated Murf1 gene. Am J Physiol-Endocrinol Metab (2008) 295(4):E785–E97. doi: 10.1152/ajpendo.00646.2007
98. Milan G, Romanello V, Pescatore F, Armani A, Paik J-H, Frasson L, et al. Regulation of autophagy and the ubiquitin–proteasome system by the Foxo transcriptional network during muscle atrophy. Nat Commun (2015) 6(1):6670. doi: 10.1038/ncomms7670
99. O’Neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, Fentz J, et al. Insulin and igf-1 receptors regulate Foxo-mediated signaling in muscle proteostasis. J Clin Invest (2016) 126(9):3433–46. doi: 10.1172/JCI86522
100. Kim KS, Park KS, Kim MJ, Kim SK, Cho YW, Park SW. Type 2 diabetes is associated with low muscle mass in older adults. Geriatr Gerontol Int (2014) 14:115–21. doi: 10.1111/ggi.12189
101. Leenders M, Verdijk LB, van der Hoeven L, Adam JJ, Van Kranenburg J, Nilwik R, et al. Patients with type 2 diabetes show a greater decline in muscle mass, muscle strength, and functional capacity with aging. J Am Med Dir Assoc (2013) 14(8):585–92. doi: 10.1016/j.jamda.2013.02.006
102. Miyake H, Kanazawa I, Tanaka K-i, Sugimoto T. Low skeletal muscle mass is associated with the risk of all-cause mortality in patients with type 2 diabetes mellitus. Ther Adv Endocrinol Metab (2019) 10:2042018819842971. doi: 10.1177/20420188198429
103. Sasako T, Umehara T, Soeda K, Kaneko K, Suzuki M, Kobayashi N, et al. Deletion of skeletal muscle Akt1/2 causes osteosarcopenia and reduces lifespan in mice. Nat Commun (2022) 13(1):5655. doi: 10.1038/s41467-022-33008-2
104. Garvey WT, Maianu L, Zhu J-H, Brechtel-Hook G, Wallace P, Baron AD. Evidence for defects in the trafficking and translocation of glut4 glucose transporters in skeletal muscle as a cause of human insulin resistance. J Clin Invest (1998) 101(11):2377–86. doi: 10.1172/JCI1557
105. DeFronzo RA, Gunnarsson R, Björkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (Type ii) diabetes mellitus. J Clin Invest (1985) 76(1):149–55. doi: 10.1172/JCI111938
106. Lundell LS, Massart J, Altıntaş A, Krook A, Zierath JR. Regulation of glucose uptake and inflammation markers by Foxo1 and Foxo3 in skeletal muscle. Mol Metab (2019) 20:79–88. doi: 10.1016/j.molmet.2018.09.011
107. Lee D, Goldberg AL. Sirt1 protein, by blocking the activities of transcription factors Foxo1 and Foxo3, inhibits muscle atrophy and promotes muscle growth. J Biol Chem (2013) 288(42):30515–26. doi: 10.1074/jbc.M113.489716
108. Sundaresan NR, Pillai VB, Wolfgeher D, Samant S, Vasudevan P, Parekh V, et al. The deacetylase Sirt1 promotes membrane localization and activation of akt and Pdk1 during tumorigenesis and cardiac hypertrophy. Sci Signal (2011) 4(182):ra46–ra. doi: 10.1126/scisignal.2001465
109. Koltai E, Bori Z, Chabert C, Dubouchaud H, Naito H, Machida S, et al. Sirt1 may play a crucial role in overload-induced hypertrophy of skeletal muscle. J Physiol (2017) 595(11):3361–76. doi: 10.1113/JP273774
110. Felber J-P, Meyer H, Curchod B, Iselin H, Rousselle J, Maeder E, et al. Glucose storage and oxidation in different degrees of human obesity measured by continuous indirect calorimetry. Diabetologia (1981) 20:39–44. doi: 10.1007/BF00253814
111. Bastie CC, Nahlé Z, McLoughlin T, Esser K, Zhang W, Unterman T, et al. Foxo1 stimulates fatty acid uptake and oxidation in muscle cells through Cd36-dependent and-independent mechanisms. J Biol Chem (2005) 280(14):14222–9. doi: 10.1074/jbc.M413625200
112. Kim YI, Lee FN, Choi WS, Lee S, Youn JH. Insulin regulation of skeletal muscle Pdk4 Mrna expression is impaired in acute insulin-resistant states. Diabetes (2006) 55(8):2311–7. doi: 10.2337/db05-1606
113. Kamei Y, Mizukami J, Miura S, Suzuki M, Takahashi N, Kawada T, et al. A forkhead transcription factor Fkhr up-regulates lipoprotein lipase expression in skeletal muscle. FEBS Lett (2003) 536(1-3):232–6. doi: 10.1016/S0014-5793(03)00062-0
114. Lee FN, Zhang L, Zheng D, Choi WS, Youn JH. Insulin suppresses Pdk-4 expression in skeletal muscle independently of plasma Ffa. Am J Physiol-Endocrinol Metab (2004) 287(1):E69–74. doi: 10.1152/ajpendo.00461.2003
115. Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes (1999) 48(8):1593–9. doi: 10.2337/diabetes.48.8.1593
116. Feldhoff PW, Arnold J, Oesterling B, Vary TC. Insulin-induced activation of pyruvate dehydrogenase complex in skeletal muscle of diabetic rats. Metabolism (1993) 42(5):615–23. doi: 10.1016/0026-0495(93)90221-9
117. Fuller SJ, Randle PJ. Reversible phosphorylation of pyruvate dehydrogenase in rat skeletal-muscle mitochondria. Effects of starvation and diabetes. Biochem J (1984) 219(2):635–46. doi: 10.1042/bj2190635
118. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor Foxo1 (Fkhr)-dependent induction of Pdk4 gene expression in skeletal muscle during energy deprivation. Biochem J (2003) 375(2):365–71. doi: 10.1042/BJ20030022
119. Vilchinskaya N, Altaeva E, Lomonosova Y. Gaining insight into the role of Foxo1 in the progression of disuse-induced skeletal muscle atrophy. Adv Biol Regul (2022) 85:100903. doi: 10.1016/j.jbior.2022.100903
120. Li Y, Pan H, Zhang X, Wang H, Liu S, Zhang H, et al. Geniposide improves glucose homeostasis via regulating Foxo1/Pdk4 in skeletal muscle. J Agric Food Chem (2019) 67(16):4483–92. doi: 10.1021/acs.jafc.9b00402
121. Mao Z-J, Xia W-S, Chai F. Yunpi heluo decoction attenuates insulin resistance by regulating Sirt1-Foxo1 autophagy pathway in skeletal muscle of zucker diabetic fatty rats. J Ethnopharmacol (2021) 270:113828. doi: 10.1016/j.jep.2021.113828
122. Nakae J, Kitamura T, Kitamura Y, Biggs WH, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev Cell (2003) 4(1):119–29. doi: 10.1016/s1534-5807(02)00401-x
123. Ioannilli L, Ciccarone F, Ciriolo MR. Adipose tissue and Foxo1: bridging physiology and mechanisms. Cells (2020) 9(4):849. doi: 10.3390/cells9040849
124. Fan W, Imamura T, Sonoda N, Sears DD, Patsouris D, Kim JJ, et al. Foxo1 transrepresses peroxisome proliferator-activated receptor Γ Transactivation, coordinating an insulin-induced feed-forward response in adipocytes. J Biol Chem (2009) 284(18):12188–97. doi: 10.1074/jbc.M808915200
125. Munekata K, Sakamoto K. Forkhead transcription factor Foxo1 is essential for adipocyte differentiation. In Vitro Cell Dev Biol-Anim (2009) 45:642–51. doi: 10.1007/s11626-009-9230-5
126. Zou P, Liu L, Zheng L, Liu L, Stoneman RE, Cho A, et al. Targeting Foxo1 with as1842856 suppresses adipogenesis. Cell Cycle (2014) 13(23):3759–67. doi: 10.4161/15384101.2014.965977
127. Nakae J, Biggs WH, Kitamura T, Cavenee WK, Wright CV, Arden KC, et al. Regulation of insulin action and pancreatic B-cell function by mutated alleles of the gene encoding forkhead transcription factor Foxo1. Nat Genet (2002) 32(2):245–53. doi: 10.1038/ng890
128. Nakae J, Cao Y, Oki M, Orba Y, Sawa H, Kiyonari H, et al. Forkhead transcription factor Foxo1 in adipose tissue regulates energy storage and expenditure. Diabetes (2008) 57(3):563–76. doi: 10.2337/db07-0698
129. Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol (1997) 7(4):261–9. doi: 10.1016/s0960-9822(06)00122-9
130. Frech M, Andjelkovic M, Ingley E, Reddy KK, Falck JR, Hemmings BA. High affinity binding of inositol phosphates and phosphoinositides to the pleckstrin homology domain of Rac/protein kinase B and their influence on kinase activity. J Biol Chem (1997) 272(13):8474–81. doi: 10.1074/jbc.272.13.8474
131. Imi Y, Ogawa W, Hosooka T. Insulin resistance in adipose tissue and metabolic diseases. Diabetol Int (2023) 14(2):119–24. doi: 10.1007/s13340-022-00616-8
132. Hosooka T, Hosokawa Y, Matsugi K, Shinohara M, Senga Y, Tamori Y, et al. The Pdk1-Foxo1 signaling in adipocytes controls systemic insulin sensitivity through the 5-lipoxygenase–leukotriene B4 axis. Proc Natl Acad Sci (2020) 117(21):11674–84. doi: 10.1073/pnas.1921015117
133. Lertpatipanpong P, Lee J, Kim I, Eling T, Oh SY, Seong JK, et al. The anti-diabetic effects of Nag-1/Gdf15 on Hfd/Stz-induced mice. Sci Rep (2021) 11(1):15027. doi: 10.1038/s41598-021-94581-y
134. Zhang M, Chen H, Liu Ms, Zhu Ky, Hao Y, Zhu Dl, et al. Serum-and glucocorticoid-inducible kinase 1 promotes insulin resistance in adipocytes via degradation of insulin receptor substrate 1. Diabetes/Metab Res Rev (2021) 37(4):e3451. doi: 10.1002/dmrr.3451
135. Dompe C, Moncrieff L, Matys J, Grzech-Leśniak K, Kocherova I, Bryja A, et al. Photobiomodulation—Underlying mechanism and clinical applications. J Clin Med (2020) 9(6):1724. doi: 10.3390/jcm9061724
136. Gong L, Zou Z, Huang L, Guo S, Xing D. Photobiomodulation therapy decreases free fatty acid generation and release in adipocytes to ameliorate insulin resistance in type 2 diabetes. Cell Signal (2020) 67:109491. doi: 10.1016/j.cellsig.2019.109491
137. Zhuang L, Li C, Hu X, Yang Q, Pei X, Jin G. High expression of P4ha3 in obesity: A potential therapeutic target for type 2 diabetes. Braz J Med Biol Res (2022) 55:1–12. doi: 10.1590/1414-431X2022e11741
138. Chakrabarti P, English T, Karki S, Qiang L, Tao R, Kim J, et al. Sirt1 controls lipolysis in adipocytes via Foxo1-mediated expression of Atgl. J Lipid Res (2011) 52(9):1693–701. doi: 10.1194/jlr.M014647
139. Kuang J, Zhang Y, Liu Q, Shen J, Pu S, Cheng S, et al. Fat-specific sirt6 ablation sensitizes mice to high-fat diet–induced obesity and insulin resistance by inhibiting lipolysis. Diabetes (2017) 66(5):1159–71. doi: 10.2337/db16-1225
140. Nunn ER, Shinde AB, Zaganjor E. Weighing in on adipogenesis. Front Physiol (2022) 13:821278. doi: 10.3389/fphys.2022.821278
141. Patel R, Santoro A, Hofer P, Tan D, Oberer M, Nelson AT, et al. Atgl is a biosynthetic enzyme for fatty acid esters of hydroxy fatty acids. Nature (2022) 606(7916):968–75. doi: 10.1038/s41586-022-04787-x
142. Liu L, Tao Z, Zheng LD, Brooke JP, Smith CM, Liu D, et al. Foxo1 interacts with transcription factor eb and differentially regulates mitochondrial uncoupling proteins via autophagy in adipocytes. Cell Death Discov (2016) 2(1):1–8. doi: 10.1038/cddiscovery.2016.66
143. Homan EP, Brandão BB, Softic S, El Ouaamari A, O’Neill BT, Kulkarni RN, et al. Differential roles of foxo transcription factors on insulin action in brown and white adipose tissue. J Clin Invest (2021) 131(19):1–16. doi: 10.1172/JCI143328
144. Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. Pdx-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest (2004) 114(6):828–36. doi: 10.1172/JCI21845
145. Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, Wright CV, et al. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic B Cell growth. J Clin Invest (2002) 110(12):1839–47. doi: 10.1172/JCI200216857
146. Buteau J, Spatz ML, Accili D. Transcription factor Foxo1 mediates glucagon-like peptide-1 effects on pancreatic B-cell mass. Diabetes (2006) 55(5):1190–6. doi: 10.1074/jbc.M508510200
147. Kawamori D, Kaneto H, Nakatani Y, Matsuoka T-a, Matsuhisa M, Hori M, et al. The forkhead transcription factor Foxo1 bridges the Jnk pathway and the transcription factor Pdx-1 through its intracellular translocation. J Biol Chem (2006) 281(2):1091–8. doi: 10.1038/s41467-020-15529-w
148. Zhang F, Ma D, Zhao W, Wang D, Liu T, Liu Y, et al. Obesity-induced overexpression of Mir-802 impairs insulin transcription and secretion. Nat Commun (2020) 11(1):1822. doi: 10.1038/s41467-020-15529-w
149. Li Y, Deng S, Peng J, Wang X, Essandoh K, Mu X, et al. Microrna-223 is essential for maintaining functional B-cell mass during diabetes through inhibiting both Foxo1 and Sox6 pathways. J Biol Chem (2019) 294(27):10438–48. doi: 10.1074/jbc.RA119.007755
150. Tan A, Li T, Yang J, Yu J, Chen H. Irisin attenuates pyroptosis in high glucose-induced pancreatic beta cells via the mir-133a-3p/Foxo1 axis. Endokrynologia Polska (2023) 74:277–84. doi: 10.5603/EP.a2023.0035
151. Choi H-E, Kim DY, Choi MJ, Kim JI, Kim O-H, Lee J, et al. Tranilast protects pancreatic B-cells from palmitic acid-induced lipotoxicity via Foxo-1 inhibition. Sci Rep (2023) 13(1):101. doi: 10.1038/s41598-022-25428-3
152. Holz GG, Chepurny OG. Diabetes outfoxed by Glp-1? Science’s STKE (2005) 2005(268):pe2–pe. doi: 10.1126/stke.2682005pe2
153. Mayendraraj A, Rosenkilde MM, Gasbjerg LS. Glp-1 and gip receptor signaling in beta cells–a review of receptor interactions and co-stimulation. Peptides (2022), 151:170749. doi: 10.1016/j.peptides.2022.170749
154. Fang D, Huang Z, Guan H, Liu J, Yao B, Xiao H, et al. The Akt/Foxo1/P27 pathway mediates the proliferative action of liraglutide in B Cells. Mol Med Rep (2012) 5(1):233–8. doi: 10.3892/mmr.2011.607
155. Hou G-j, Li C-n, Huan Y, Liu S-n, Liu Q, Liu M-z, et al. The Pi3k/Akt1-Foxo1 translocation pathway mediates Exf effects on nit-1 cell survival. Exp Clin Endocrinol Diabetes (2017) 125(10):669–76. doi: 10.1055/s-0043-117048
156. Yang L, Yao D, Yang H, Wei Y, Peng Y, Ding Y, et al. Puerarin protects pancreatic B-cells in obese diabetic mice via activation of Glp-1r signaling. Mol Endocrinol (2016) 30(3):361–71. doi: 10.1210/me.2015-1213
157. Bastien-Dionne P-O, Valenti L, Kon N, Gu W, Buteau J. Glucagon-like peptide 1 inhibits the sirtuin deacetylase Sirt1 to stimulate pancreatic B-cell mass expansion. Diabetes (2011) 60(12):3217–22. doi: 10.2337/db11-0101
158. Ding Y, Wu Q. 1, 25d/Vdr inhibits pancreatic B Cell ferroptosis by downregulating Foxo1 expression in diabetes mellitus. Cell Signal (2023) 105:110564. doi: 10.1016/j.cellsig.2022.110564
159. Zhang W, Wu N, Wang H, Mao G, Yan X, Zhang F, et al. Sulfated fuco-manno-glucuronogalactan alleviates pancreatic beta cell senescence via Pi3k/Akt/Foxo1 pathway. Int J Biol Macromol (2023) 236:123846. doi: 10.1074/jbc.M116.770032
160. Xiao X, Chen C, Guo P, Zhang T, Fischbach S, Fusco J, et al. Forkhead box protein 1 (Foxo1) inhibits accelerated B Cell aging in pancreas-specific Smad7 mutant mice. J Biol Chem (2017) 292(8):3456–65. doi: 10.1074/jbc.M116.770032
161. Zhang T, Kim DH, Xiao X, Lee S, Gong Z, Muzumdar R, et al. Foxo1 plays an important role in regulating B-cell compensation for insulin resistance in male mice. Endocrinology (2016) 157(3):1055–70. doi: 10.1210/en.2015-1852
162. Kibbe C, Chen J, Xu G, Jing G, Shalev A. Foxo1 competes with carbohydrate response element-binding protein (Chrebp) and inhibits thioredoxin-interacting protein (Txnip) transcription in pancreatic beta cells. J Biol Chem (2013) 288(32):23194–202. doi: 10.1111/j.1463-1326.2007.00782.x
163. Buteau J, Accili D. Regulation of pancreatic B-cell function by the forkhead protein Foxo1. Diabetes Obes Metab (2007) 9:140–6. doi: 10.1016/j.cmet.2005.08.004
164. Kitamura YI, Kitamura T, Kruse J-P, Raum JC, Stein R, Gu W, et al. Foxo1 Protects against Pancreatic B Cell Failure through Neurod and Mafa Induction. Cell Metab (2005) 2(3):153–63. doi: 10.1016/j.cmet.2005.08.004
165. Kim-Muller JY, Fan J, Kim YJR, Lee S-A, Ishida E, Blaner WS, et al. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic B Cells in diabetic mice. Nat Commun (2016) 7(1):12631. doi: 10.1038/ncomms12631
166. Son J, Accili D. Reversing pancreatic B-cell dedifferentiation in the treatment of type 2 diabetes. Exp Mol Med (2023) 55:1652–8. doi: 10.1038/s12276-023-01043-8
167. Xu Q, Zhang X, Li T, Shao S. Exenatide regulates th17/treg balance via Pi3k/Akt/Foxo1 pathway in Db/Db mice. Mol Med (2022) 28(1):144. doi: 10.1186/s10020-022-00574-6
168. Li X, Wan T, Li Y. Role of Foxo1 in regulating autophagy in type 2 diabetes mellitus. Exp Ther Med (2021) 22(1):1–8. doi: 10.3892/etm.2021.10139
169. Li X-D, He S-S, Wan T-T, Li Y-B. Liraglutide protects palmitate-induced ins-1 cell injury by enhancing autophagy mediated via Foxo1. Mol Med Rep (2021) 23(2):1–. doi: 10.3892/mmr.2020.11786
170. Liang R, Liu N, Cao J, Liu T, Sun P, Cai X, et al. Hif-1α/Foxo1 axis regulated autophagy is protective for B Cell survival under hypoxia in human islets. Biochim Biophys Acta (BBA)-Mol Basis Dis (2022) 1868(5):166356. doi: 10.1016/j.bbadis.2022.166356
171. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic B Cell dedifferentiation as a mechanism of diabetic B Cell failure. Cell (2012) 150(6):1223–34. doi: 10.1016/j.cell.2012.07.029
172. Fan J, Du W, Kim-Muller JY, Son J, Kuo T, Larrea D, et al. Cyb5r3 links Foxo1-dependent mitochondrial dysfunction with B-cell failure. Mol Metab (2020) 34:97–111. doi: 10.1016/j.molmet.2019.12.008
173. Kobayashi M, Kikuchi O, Sasaki T, Kim H-J, Yokota-Hashimoto H, Lee Y-S, et al. Foxo1 as a double-edged sword in the pancreas: analysis of pancreas-and B-cell-specific Foxo1 knockout mice. Am J Physiol-Endocrinol Metab (2012) 302(5):E603–E13. doi: 10.1152/ajpendo.00469.2011
174. Navik U, Rawat K, Tikoo K. L-methionine prevents B-cell damage by modulating the expression of arx, mafa and regulation of Foxo1 in type 1 diabetic rats. Acta Histochemica (2022) 124(1):151820. doi: 10.1016/j.acthis.2021.151820
175. Cabrera-Rode E, Cubas-Dueñas I, Acosta JR, Hernández JC, González AIC, Calero TMG, et al. Efficacy and safety of obex® in overweight and obese subjects: A randomised, double-blind, placebo-controlled clinical trial. BMC Complement Med Ther (2023) 23(1):1–15. doi: 10.1186/s12906-023-03847-7
176. Peng S, Li W, Hou N, Huang N. A review of Foxo1-regulated metabolic diseases and related drug discoveries. Cells (2020) 9(1):184. doi: 10.3390/cells9010184
177. Toorie AM, Nillni EA. Minireview: central Sirt1 regulates energy balance via the melanocortin system and alternate pathways. Mol Endocrinol (2014) 28(9):1423–34. doi: 10.1210/me.2014-1115
178. Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, et al. The obesity susceptibility gene Cpe links Foxo1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med (2009) 15(10):1195–201. doi: 10.1038/nm.2026
179. Kim H-J, Kobayashi M, Sasaki T, Kikuchi O, Amano K, Kitazumi T, et al. Overexpression of Foxo1 in the Hypothalamus and pancreas causes obesity and glucose intolerance. Endocrinology (2012) 153(2):659–71. doi: 10.1210/en.2011-1635
180. Plum L, Belgardt BF, Brüning JC. Central insulin action in energy and glucose homeostasis. J Clin Invest (2006) 116(7):1761–6. doi: 10.1172/JCI29063
181. Casado ME, Collado-Pérez R, Frago LM, Barrios V. Recent advances in the knowledge of the mechanisms of leptin physiology and actions in neurological and metabolic pathologies. Int J Mol Sci (2023) 24(2):1422. doi: 10.3390/ijms24021422
182. Ropelle ER, Pauli JR, Prada P, Cintra DE, Rocha GZ, Moraes JC, et al. Inhibition of hypothalamic foxo1 expression reduced food intake in diet-induced obesity rats. J Physiol (2009) 587(10):2341–51. doi: 10.1113/jphysiol.2009.170050
183. Cyr NE, Steger JS, Toorie AM, Yang JZ, Stuart R, Nillni EA. Central Sirt1 regulates body weight and energy expenditure along with the pomc-derived peptide A-Msh and the processing enzyme Cpe production in diet-induced obese male rats. Endocrinology (2015) 156(3):961–74. doi: 10.1210/en.2014-1970
184. Al-Massadi O, Quiñones M, Clasadonte J, Hernandez-Bautista R, Romero-Picó A, Folgueira C, et al. Mch regulates Sirt1/Foxo1 and reduces pomc neuronal activity to induce hyperphagia, adiposity, and glucose intolerance. Diabetes (2019) 68(12):2210–22. doi: 10.2337/db19-0029
185. Du S, Zheng H. Role of Foxo transcription factors in aging and age-related metabolic and neurodegenerative diseases. Cell Biosci (2021) 11:1–17. doi: 10.1186/s13578-021-00700-7
186. Hu W, Yang Z, Yang W, Han M, Xu B, Yu Z, et al. Roles of forkhead box O (Foxo) transcription factors in neurodegenerative diseases: A panoramic view. Prog Neurobiol (2019) 181:101645. doi: 10.1016/j.pneurobio.2019.101645
187. Ramasubbu K, Devi Rajeswari V. Impairment of insulin signaling pathway Pi3k/Akt/Mtor and insulin resistance induced ages on diabetes mellitus and neurodegenerative diseases: A perspective review. Mol Cell Biochem (2023) 478(6):1307–24. doi: 10.1007/s11010-022-04587-x
188. Cullinane PW, de Pablo Fernandez E, König A, Outeiro TF, Jaunmuktane Z, Warner TT. Type 2 diabetes and Parkinson’s disease: A focused review of current concepts. Movement Disord (2023) 38(2):162–77. doi: 10.1002/mds.29298
189. Ezkurdia A, Ramírez MJ, Solas M. Metabolic syndrome as a risk factor for Alzheimer’s disease: A focus on insulin resistance. Int J Mol Sci (2023) 24(5):4354. doi: 10.3390/ijms24054354
190. Michailidis M, Moraitou D, Tata DA, Kalinderi K, Papamitsou T, Papaliagkas V. Alzheimer’s disease as type 3 diabetes: common pathophysiological mechanisms between Alzheimer’s disease and type 2 diabetes. Int J Mol Sci (2022) 23(5):2687. doi: 10.3390/ijms23052687
191. Nichols E, Steinmetz JD, Vollset SE, Fukutaki K, Chalek J, Abd-Allah F, et al. Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: an analysis for the global burden of disease study 2019. Lancet Public Health (2022) 7(2):e105–e25. doi: 10.1016/S2468-2667(21)00249-8
192. Liu L, Bai J, Liu F, Xu Y, Zhao M, Zhao C, et al. Cross-talking pathways of forkhead box O1 (Foxo1) are involved in the pathogenesis of Alzheimer’s disease and Huntington’s disease. Oxid Med Cell Longev (2022) 2022:1–14. doi: 10.1155/2022/7619255
193. Dumitriu A, Latourelle JC, Hadzi TC, Pankratz N, Garza D, Miller JP, et al. Gene expression profiles in Parkinson disease prefrontal cortex implicate Foxo1 and genes under its transcriptional regulation. PloS Genet (2012) 8(6):e1002794. doi: 10.1371/journal.pgen.1002794
194. Sajan M, Hansen B, Ivey IIIR, Sajan J, Ari C, Song S, et al. Brain insulin signaling is increased in insulin-resistant states and decreases in Foxos and Pgc-1α and increases in Aβ1–40/42 and phospho-tau may abet Alzheimer development. Diabetes (2016) 65(7):1892–903. doi: 10.2337/db15-1428
195. Zhang W, Bai S, Yang J, Zhang Y, Liu Y, Nie J, et al. Foxo1 overexpression reduces Aβ Production and tau phosphorylation in vitro. Neurosci Lett (2020) 738:135322. doi: 10.1016/j.neulet.2020.135322
196. Zhang W, Shan-Shan B, Zhang Q, Ru-Ling S, He-Cheng W, You-Cai L, et al. Physalin B reduces Aβ Secretion through down-regulation of bace1 expression by activating Foxo1 and inhibiting Stat3 phosphorylation. Chin J Nat Med (2021) 19(10):732–40. doi: 10.1016/S1875-5364(21)60090-0
197. Lv MT, Wang HC, Meng XW, Shi YT, Zhang YM, Shan LL, et al. In silico and in vitro analyses of a novel Foxo1 agonist reducing Aβ Levels via downregulation of bace1. CNS Neurosci Ther (2023). doi: 10.1111/cns.14140
198. Bedada FB, Ntekim OE, Nwulia EO, Fungwe TV, Nadarajah SR, Obisesan TO. Exercise training-increased Fbxo32 and Foxo1 in a gender-dependent manner in mild cognitively impaired African Americans: gems-1 study. Front Aging Neurosci (2021) 13:641758. doi: 10.3389/fnagi.2021.641758
199. Zhao N, Zhang X, Li B, Wang J, Zhang C, Xu B. Treadmill exercise improves Pink1/Parkin-mediated mitophagy activity against Alzheimer’s disease pathologies by upregulated Sirt1-Foxo1/3 axis in app/Ps1 mice. Mol Neurobiol (2023) 60(1):277–91. doi: 10.1007/s12035-022-03035-7
200. Kuang X, Chen Y-S, Wang L-F, Li Y-J, Liu K, Zhang M-X, et al. Klotho upregulation contributes to the neuroprotection of ligustilide in an Alzheimer’s disease mouse model. Neurobiol Aging (2014) 35(1):169–78. doi: 10.1016/j.neurobiolaging.2013.07.019
201. Zhou H-J, Zeng C-Y, Yang T-T, Long F-Y, Kuang X, Du J-R. Lentivirus-mediated klotho up-regulation improves aging-related memory deficits and oxidative stress in senescence-accelerated mouse prone-8 mice. Life Sci (2018) 200:56–62. doi: 10.1016/j.lfs.2018.03.027
202. Yossef RR, Al-Yamany MF, Saad MA, El-Sahar AE. Neuroprotective effects of vildagliptin on drug induced Alzheimer’s disease in rats with metabolic syndrome: role of hippocampal klotho and Akt signaling pathways. Eur J Pharmacol (2020) 889:173612. doi: 10.1016/j.ejphar.2020.173612
203. Li L, Sun Y, Zhang Y, Wang W, Ye C. Mutant huntingtin impairs pancreatic B-cells by recruiting Irs-2 and disturbing the Pi3k/Akt/Foxo1 signaling pathway in Huntington’s disease. J Mol Neurosci (2021) 71:2646–58. doi: 10.1007/s12031-021-01869-9
204. Vidal RL, Figueroa A, Court FA, Thielen P, Molina C, Wirth C, et al. Targeting the Upr Transcription Factor Xbp1 Protects against Huntington’s Disease through the Regulation of Foxo1 and Autophagy. Hum Mol Genet (2012) 21(10):2245–62. doi: 10.1093/hmg/dds040
205. Elangovan A, Venkatesan D, Selvaraj P, Pasha MY, Babu HWS, Iyer M, et al. Mirna in Parkinson’s disease: from pathogenesis to theranostic approaches. J Cell Physiol (2023) 238(2):329–54. doi: 10.3892/ijmm.2019.4426
206. Lun P, Ji T, Wan D-H, Liu X, Chen X-D, Yu S, et al. Hottip downregulation reduces neuronal damage and microglial activation in Parkinson’s disease cell and mouse models. Neural Regen Res (2022) 17(4):887. doi: 10.4103/1673-5374.322475
207. Li Y, Jiao Q, Du X, Jiang H. Sirt1/Foxo1-associated mao-a upregulation promotes depressive-like behavior in transgenic mice expressing human A53t A-synuclein. ACS Chem Neurosci (2020) 11(22):3838–48. doi: 10.1021/acschemneuro.0c00628
208. Menghini R, Casagrande V, Iuliani G, Rizza S, Mavilio M, Cardellini M, et al. Metabolic aspects of cardiovascular diseases: is Foxo1 a player or a target? Int J Biochem Cell Biol (2020) 118:105659. doi: 10.1016/j.biocel.2019.105659
209. Wang Y, He W. Improving the dysregulation of Foxo1 activity is a potential therapy for alleviating diabetic kidney disease. Front Pharmacol (2021) 12:630617. doi: 10.3389/fphar.2021.630617
210. Guo S, Mangal R, Dandu C, Geng X, Ding Y. Role of forkhead box protein O1 (Foxo1) in stroke: A literature review. Aging Dis (2022) 13(2):521. doi: 10.14336/AD.2021.0826
Keywords: FoxO1, type 2 diabetes, insulin resistance, Akt, metabolism
Citation: Teaney NA and Cyr NE (2023) FoxO1 as a tissue-specific therapeutic target for type 2 diabetes. Front. Endocrinol. 14:1286838. doi: 10.3389/fendo.2023.1286838
Received: 31 August 2023; Accepted: 06 October 2023;
Published: 23 October 2023.
Edited by:
Jean-François Tanti, INSERM U1065 Centre Méditerranéen de Médecine Moléculaire, FranceReviewed by:
Tarik Issad, Centre National de la Recherche Scientifique (CNRS), FranceEric Hajduch, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2023 Teaney and Cyr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole E. Cyr, bmN5ckBzdG9uZWhpbGwuZWR1