 Avinaash V. Maharaj
Avinaash V. Maharaj- Centre for Endocrinology, William Harvey Research Institute, Queen Mary University of London (QMUL), London, United Kingdom
Familial Glucocorticoid Deficiency encompasses a broad spectrum of monogenic recessive disorders that theoretically solely abrogate cortisol biosynthesis. In reality, delineating clear genotype-phenotype correlations in this disorder is made complicated by marked phenotypic heterogeneity even within kindreds harbouring identical variants. Phenotypes range from isolated glucocorticoid insufficiency to cortisol deficiency plus a variety of superimposed features including salt-wasting and hypoaldosteronism, primary hypothyroidism, hypogonadism and growth defects. Furthermore, mutation type, domain topology and perceived enzyme activity do not always predict disease severity. Given the high burden of disease and implications of a positive diagnosis, genetic testing is crucial in the management of patients warranting detailed delineation of genomic variants including viable functional studies.
1 Introduction
1.1 Historical origins of Familial Glucocorticoid Deficiency
Unsubstantiated reports of Addisonian-like familial disease associated with hyperpigmentation were recorded from as early as 1900 but the disorder first came to prominence in 1959 when Shepard and colleagues described a unique form of hypocortisolaemia in two siblings with preserved mineralocorticoid function (1). Over subsequent decades, the phenotypic spectrum evolved to include a distinct constellation of features including hyperpigmentation, recurrent hypoglycaemia, seizures, high plasma deoxycorticosterone and tall stature (2–9). Initially designated as hereditary adrenocortical unresponsiveness to adrenocorticotropin (ACTH) (4), this syndrome was eventually termed Familial Glucocorticoid Deficiency (FGD).
1.2 Physiological regulation of cortisol production
Higher brain centres regulate the synthesis of endogenous cortisol through an intricate negative feedback pathway governed by the tropic Corticotrophin releasing factor (CRF) secreted by the hypothalamo-adenohypophyseal portal system (Figure 1). CRF preferentially binds to the CRF type 1 G-protein coupled receptor (CRF-1R) and acts as a secretagogue to potentiate ACTH release from the anterior pituitary. ACTH then acts as an agonist for the melanocortin 2 receptor (MC2R), expressed in adrenocortical cells of the adrenal zona fasciculata, thereby producing cortisol via modulation of adenylate cyclase/protein kinase A signalling. Cortisol mediates the negative feedback loop via the centrally expressed glucocorticoid receptor. Glucocorticoid mediated feedback may be divided into: (i) non-genomic rapid inhibition of glutamate release at the hypothalamic paraventricular nucleus via endocannabinoid synthesis, (ii) genomic prefrontal limbic regulation and, (iii) destabilization of hypothalamic pituitary adrenocortical axis-activating neuropeptide mRNA (10). Loss of feedback regulation forms the basis of ACTH resistance syndromes of which defects in MC2R were the first to be elucidated.
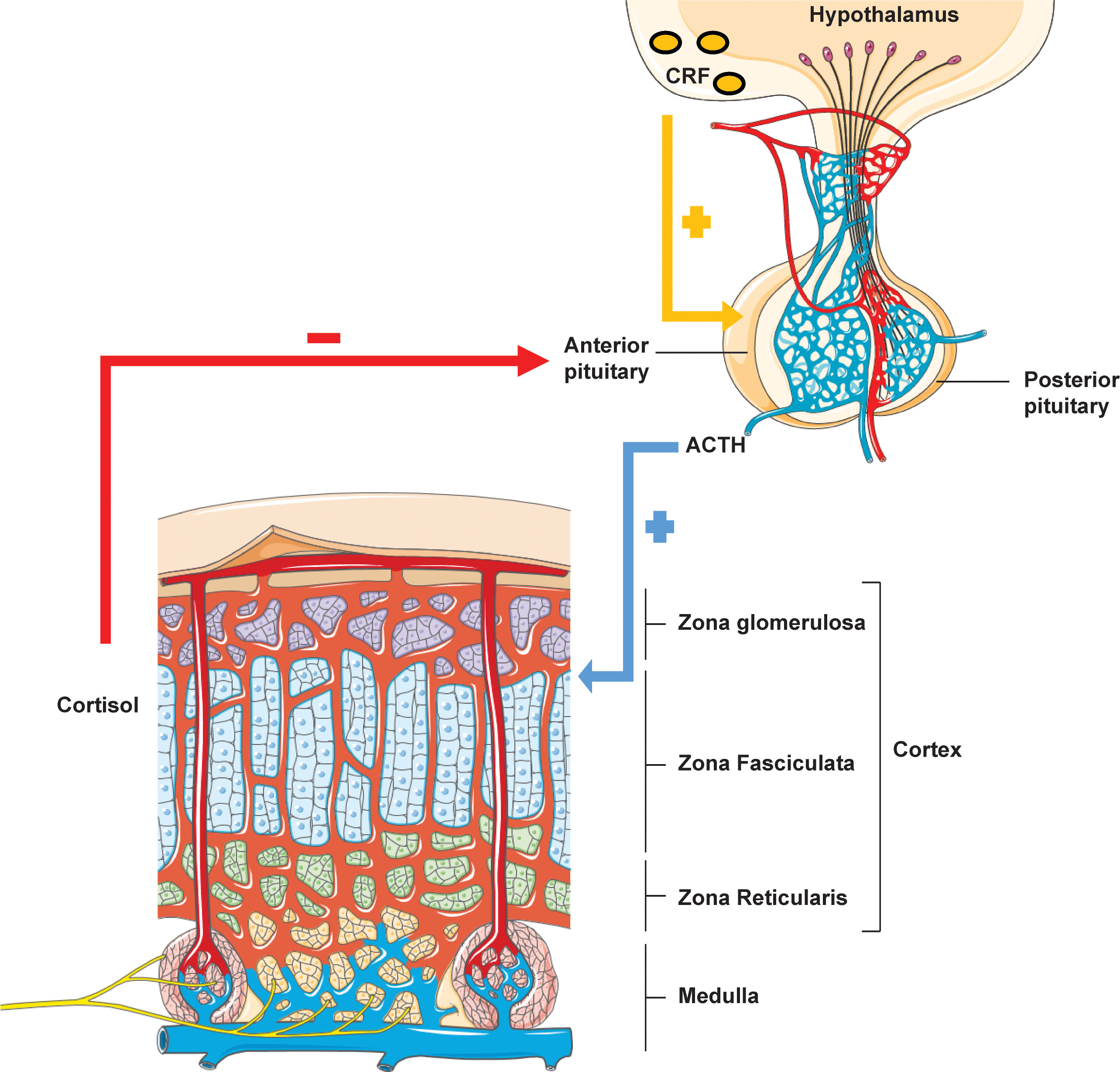
Figure 1 Hypothalamic-pituitary-adrenal axis. Rhythmicity of the HPA axis is driven by hypothalamic tropic factor CRH which stimulates anterior pituitary ACTH production. ACTH acts on the adrenal zona fasciculata to mediate glucocorticoid biosynthesis. Cortisol itself then exerts feedback inhibition of both ACTH and CRH via a negative feedback loop.
2 Familial glucocorticoid deficiency (GCCD1/FGD Type 1) – melanocortin-2 receptor (MC2R) defects (OMIM #202200)
The ACTH receptor is a seven transmembrane domain receptor encoded by the MC2R gene, which maps onto the short arm of chromosome 18 and consists of 2 exons, the first of which is non-coding/untranslated whilst the latter encodes the full sequence of the receptor. Following isolation and sub-cloning of the human ACTH receptor in 1992 (11), the first reported kindreds with primary adrenal insufficiency secondary to defects in MC2R were characterised by Clark et al. and Tsigos et al. in 1993 (12, 13). Since these initial reports, around 48 mutations have been described in association with FGD (Human Gene Mutation Database http://www.hgmd.cf.ac.uk) (14), the majority of which are missense/nonsense variants (Figure 2) in addition to small genomic deletions and insertions. Variants in MC2R account for about 25% of FGD cases, the majority of which are non-conservative, single amino acid substitutions that likely abrogate cyclic adenosine monophosphate (cAMP) generation and impair receptor trafficking (17–19).
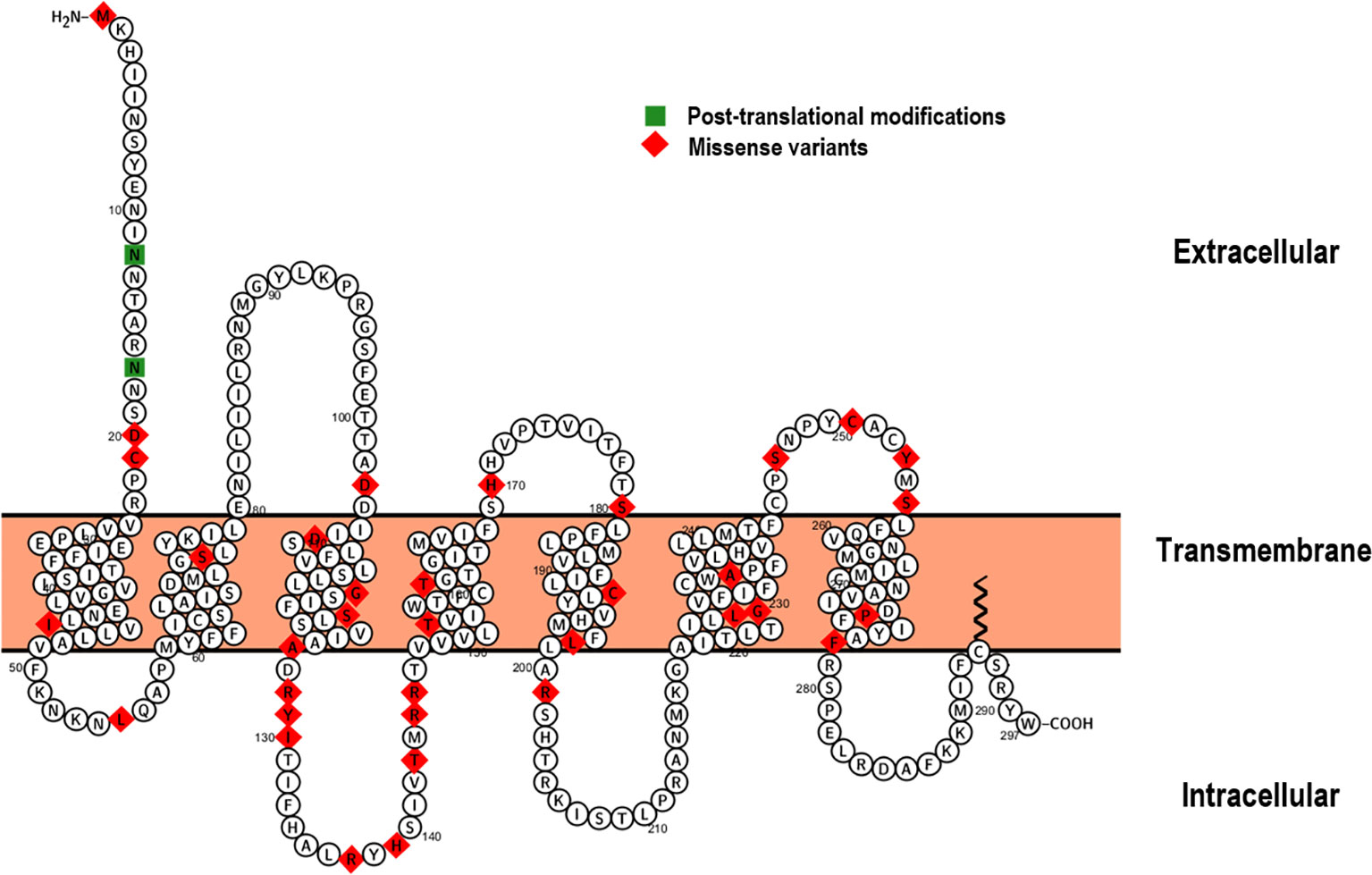
Figure 2 Proteoform structure of the ACTH receptor (15). The topological (extracellular, transmembrane and cytoplasmic) domains are outlined and sites of missense variation highlighted (red). The majority of MC2R variants causing FGD Type 1 are associated with defective receptor trafficking and mainly occur within the transmembrane domains (16). Sites of post-translational modifications are indicated in green.
Classically in FGD Type 1, patients are mineralocorticoid replete however, in clinical practice, the picture may be less unambiguous. Several reports have alluded to mild disruptions of the renin-angiotensin-aldosterone axis in patients with MC2R defects, in whom early glucocorticoid replacement may mask aldosterone insufficiency (20, 21). Furthermore, several patients with homozygous frameshift truncating variants have presented in the neonatal period with transient salt wasting (20). Interestingly Mc2r-/- mice demonstrate reduced serum aldosterone levels (22). This disparity may reflect the fact that the majority of human subjects harbour missense mutations that retain some degree of enzyme activity. Patients also invariably present with early and significant hyperpigmentation due to the action of markedly increased ACTH on the Melanocortin 1 receptor (MC1R). This association was clearly demonstrable in a patient with FGD harbouring homozygous missense variants in both MC2R and MC1R and lacking the ‘classic’ hyperpigmented phenotype (23).
An often overlooked but increasingly recognised feature of FGD Type 1 is hypothyroidism (18, 24–29). Patient phenotypes range from transient neonatal hypothyroidism that normalise with short term thyroxine replacement to subclinical hypothyroidism and persistent thyroid hypo-function (28, 29). Despite assertions that elevated ACTH levels may inhibit thyroid stimulating hormone release (30), the exact mechanism underlying the incidence of hypothyroidism in patients with FGD remain to be elucidated. Interestingly, another feature recognised in patients with ACTH receptor defects is reduced adrenal androgen production or lack of adrenarche. Weber et al. demonstrated consistently sub-optimal serum DHEAS levels in 6 patients with MC2R variants (31). This model of ‘functional’ ACTH deficiency suggests that ACTH at least partially regulates adrenarche given that patients with central ACTH deficiency (hypopituitarism) also exhibit low levels of adrenal androgens (31–34).
Tall stature is an inconsistent but historically common feature of FGD type 1 despite an unaffected Growth hormone-IGF-1 axis (35). Although the exact mechanism underlying this phenotype is unclear, it is likely theorised to be due to the sustained effect of markedly elevated Adrenocorticotropin levels on the growth plate (35, 36). This also correlates to a later median age at diagnosis (2.0 years) when compared to FGD Type 2 (37). ACTH has been shown in vitro to increase rat chondrocyte progenitor cell proliferation and matrix production (38). Interestingly, growth trajectories return to normal once glucocorticoid treatment is instituted. Mc2r knockout mice do not recapitulate this trait and demonstrate similar body lengths to wild-type littermates (22). There is however little genotype phenotype correlation and disease severity and onset is highly variable (39).
3 Familial glucocorticoid deficiency (GCCD2/FGD Type 2) – melanocortin-2 receptor accessory protein (MRAP) (OMIM #607398)
After initial characterisation of loss of function defects in MC2R as a primary cause of FGD, it became apparent that other genetic factors were involved in pathogenesis of FGD given the existence of several families with an isolated adrenal insufficiency phenotype and negative MC2R genomic screening. In vitro expression studies by Noon et al. revealed that a fluorescent tagged MC2R cDNA clone was able to traffic to the cell membranes of adrenocorticotropin resistant murine adrenocortical tumour cells but remained restricted to the endoplasmic reticulum in non-adrenal cells. This suggested that an adrenal derived co-factor was necessary for MC2R expression (40, 41) (Figure 3).
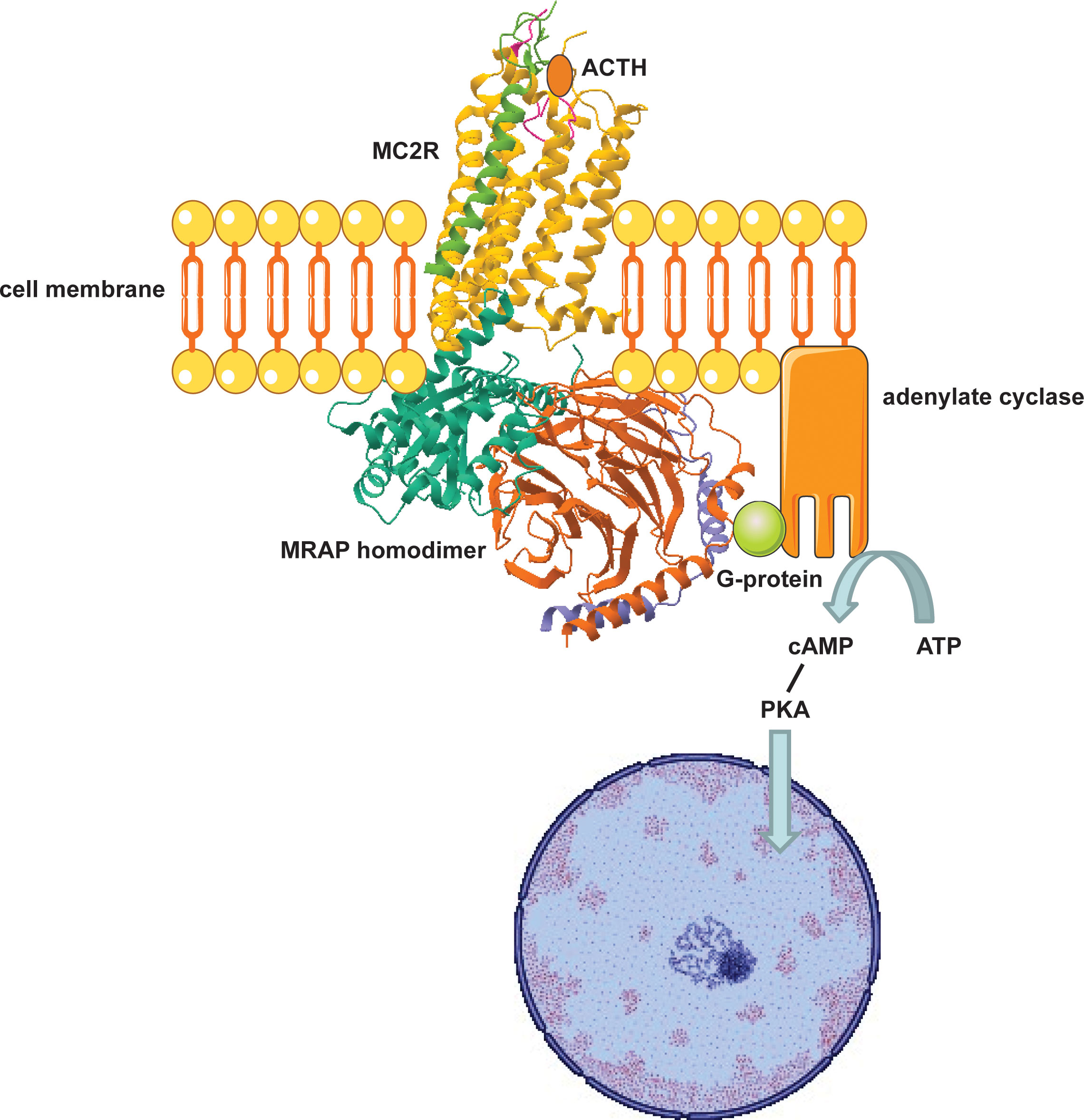
Figure 3 Adrenocortical cell surface MC2R-MRAP complex mediates glucocorticoid biosynthesis. MRAP traffics MC2R to the cell surface where ligand binding (ACTH) induces a G-protein coupled increase in cAMP which phosphorylates Protein Kinase A (PKA). PKA then induces transcription of specific intra-nuclear factors that mobilise STAR activity and expression of enzymes involved in steroidogenesis.
In 2005, Metherell et al. conducted autozygosity mapping in a single family in which runs of homozygosity identified a 2.2 Mbp region. Within this region, the adrenally expressed chromosome 21 open reading frame 61 (C21orf61) gene previously encoding a fat-associated low-molecular-weight protein, was identified. Variants in this gene, retitled the melanocortin 2 receptor accessory protein (MRAP), were subsequently identified in several families with FGD (42). Variants in MRAP (FGD Type 2) account for 20-25% of FGD cases, with around 15 mutations being described since initial characterisation of this gene (37, 42–45). The majority of variants are splice site/nonsense that ultimately lead to a truncated and non-functional receptor. Patients generally present at an earlier age (median age of 0.08 years at diagnosis) with normal stature when compared to subjects with MC2R variants (37, 45, 46). This suggests reduced exposure to the unfettered actions of ACTH on the growth plate seen in FGD Type 1. Mrap-/- mice phenocopy the isolated glucocorticoid deficiency and normal mineralocorticoid function of human subjects. Interestingly, the adrenals from knockout mice are small with indistinct cortical zonation and dysregulated accumulation of WNT4/β-catenin (47).
4 Partial loss of function mutations in steroidogenic acute regulatory protein (STAR) and cytochrome P450 side chain cleavage enzyme (CYP11A1)
4.1 STAR (GCCD3, OMIM #609197)
Defects in STAR disrupt steroidogenesis globally resulting in classic congenital lipoid adrenal hyperplasia (CLAH). Patients invariably present with hyper-reninaemic hypoaldosteronism in the setting of hypocortisolaemia, enlarged adrenals due to progressive lipid deposition and gonadal insufficiency. A two hit model has been proposed to account for pathogenesis of CLAH (Figure 4); the first ‘hit’ being lack of STAR and the second being cholesterol mediated oxidative damage. In STAR deficient adrenocortical and testicular Leydig cells, steroidogenesis is markedly diminished with the exception of minimal residual STAR-independent steroid biosynthesis. The primary biosynthetic defect leads to compensatory increases in ACTH and LH which promote LDL receptor mediated cholesterol uptake and de novo synthesis. Progressive lipid accumulation leads to mitochondrial oxidative damage and cellular stress ultimately abolishing residual STAR-independent steroidogenic capacity (48, 49). Early fetal destruction of Leydig cell integrity in 46,XY subjects leads to lack of testosterone and feminized external genitalia (50). The fetal ovary, on the contrary, is relatively preserved until puberty when gonadotrophin stimulation leads to cholesterol accumulation.
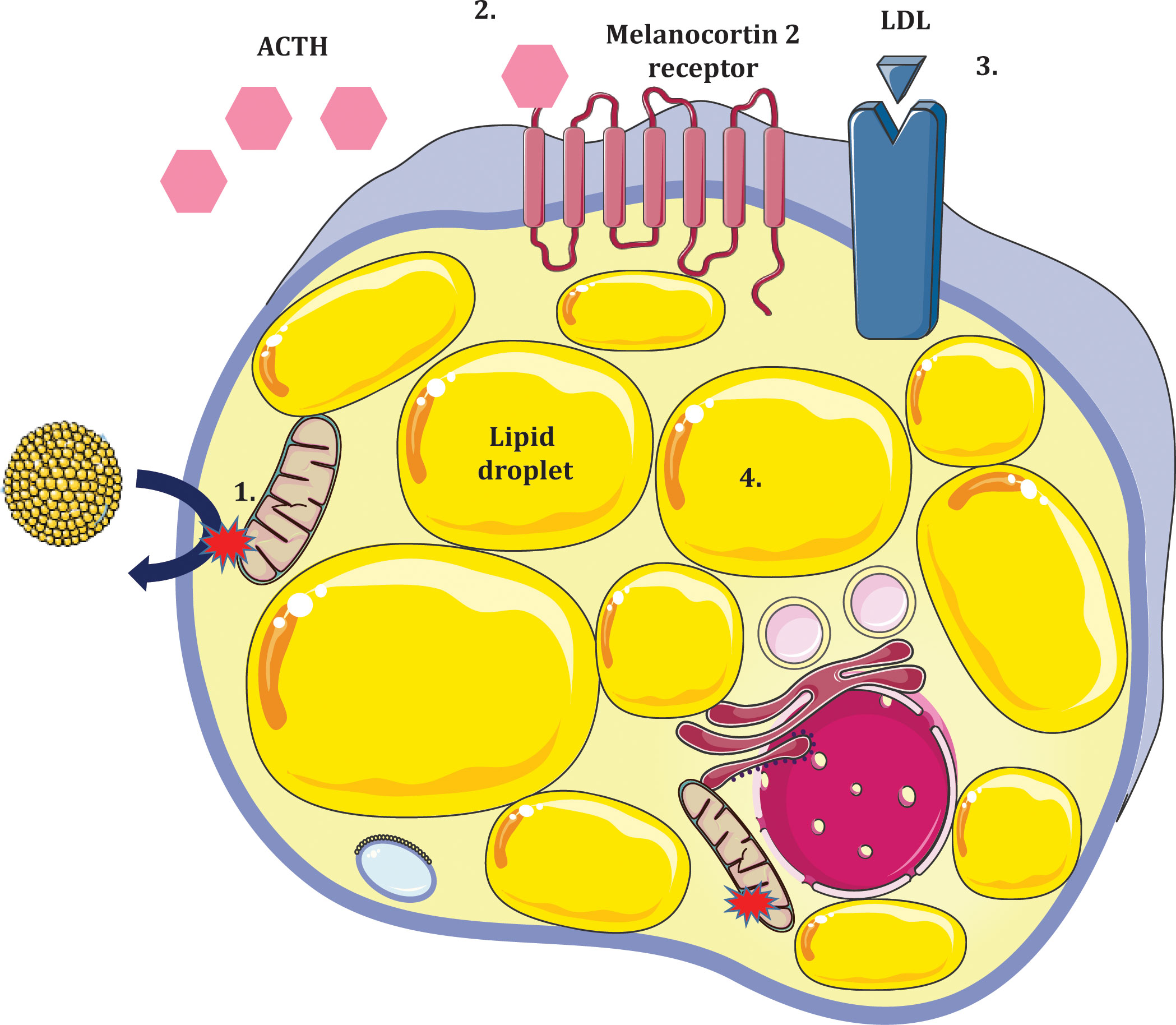
Figure 4 Two ‘hit’ model of lipoid congenital adrenal hyperplasia due to deficiency of STAR in adrenocortical cells. 1. Absence of STAR prevents cholesterol import to the inner mitochondrial membrane (first hit). Some residual STAR-independent steroidogenesis occurs but relative hypocortisolaemia ensues. 2. Loss of negative feedback to the anterior pituitary results in increased ACTH production. 3. Steroidogenic stimuli promote expressivity of LDL receptors which increase adrenocortical cholesterol uptake. 4. Accumulation of intracellular lipids causes oxidative damage and cellular stress (second hit) further impeding steroidogenic potential leading to hyperplastic adrenals.
In some patients, mutations in STAR are associated with retention of up to 20% of wild type activity and a mild phenotype. These partial loss of function variants result in non-classic CLAH (NCLAH), often indistinguishable from FGD due to an isolated adrenal phenotype (51–55). Age at presentation is highly variable, ranging from 18 months to adulthood (51, 56). NCLAH cortisol deficient patients exhibit normal external genitalia and consonant pubertal development however, primary gonadal failure may occur progressively over time (51, 53).
Expression of a mutant Star protein (N47-StAR) in knockout mice (Star−/−) gave rise to a partial loss of function phenotype. When compared to wild type transgenic mice, Star−/−N47Tg mice had lower basal and stimulated corticosterone levels. Star−/−N47Tg mice had normal external genitalia but exhibited progressive gonadal insufficiency with aging (57).
4.2 Cholesterol side-chain cleavage enzyme, P450scc (CYP11A1) (OMIM #613743)
CYP11A1, a mitochondrial monooxygenase regulates placental production of progesterone required for maintenance of gestational viability (48, 58). Absolute deficiency of this enzyme was previously thought to be incompatible with term gestation i.e. embryonically lethal, however, several patients with side chain cleavage enzyme deficiency have been reported. It is postulated that in these instances, persistence of the corpus luteum throughout pregnancy compensates for the placentation defect (59). Severe loss of function mutations in CYP11A1 produce a phenotype not dissimilar to that of CLAH due to STAR deficiency. Primary adrenal failure in combination with 46, XY sex reversal characterise these patients although unlike STAR deficient subjects, adrenal size is unaffected.
However, like STAR, partial loss of function mutations can produce a phenotype consistent with non-classic CLAH (60). Furthermore, partial inactivating mutations in CYP11A1 have been implicated in development of an isolated glucocorticoid deficiency. Parajes et al. (2011) described a homozygous mutation (p.R451W) in two 46, XY male siblings who presented with primary adrenal insufficiency and normal external genitalia without hypogonadism (61). Unlike severe enzyme deficiency which presents within the first 10 days of life, partial loss of function variants in CYP11A1 are associated with later ages of onset ranging from 6 months to 15 years (62–65).
Maharaj et al. (64) described a heterozygous missense variant (rs6161, c.940G>A, p.Glu314Lys) in CYP11A1, previously designated benign, in a cohort of 19 probands (13 families) with isolated glucocorticoid deficiency. The variant occurred in compound heterozygosity with a second gene disrupting allelic hit in CYP11A1 in 17 probands and in combination with two synonymous single nucleotide variants (p.Thr330= and p.Ser391=) in two probands. In vitro splicing assays demonstrated exon skipping due to aberrant splicing for these three variants. The impact of the rs6161 variant on splicing was further corroborated by several subsequent groups (66–69).
Cyp11a1-/- mice do not survive weaning and demonstrate marked corticosterone and aldosterone deficiency when compared to wild-type litter mates. Interestingly, corticosteroid administration prolonged survival until adulthood. Cyp11a1-/- XY males exhibit sex reversal with feminization of external genitalia and disorganization of internal genitalia. Similar to human counterparts, knockout adrenals are small however they demonstrate progressive lipid droplet accumulation (70).
5 FGD-like syndrome due to altered cellular redox status (NNT, TXNRD2)
5.1 Nicotinamide nucleotide transhydrogenase (NNT) (GCCD4, OMIM #614736)
Single nucleotide polymorphism genotyping and targeted exome sequencing of consanguineous kindreds with adrenal insufficiency identified variants in the gene encoding the inner mitochondrial membrane enzyme, nicotinamide nucleotide transhydrogenase (NNT) (71). The initial 15 probands characterised by Meimaridou et al. presented with a classical FGD phenotype indistinguishable from mutations in MC2R and MRAP. Patients were diagnosed before age 40 months with biochemical evidence of marked hypocortisolaemia, elevated ACTH and normal renin and aldosterone levels. A significant number of patients (8 out of 15) presented with sequelae of hypoglycaemia. A further study noted that some patients were mineralocorticoid deficient highlighting a degree of phenotypic variability (46, 72).
Lentiviral knockdown of NNT in an H295R (adrenocortical cancer) cell line demonstrated increased mitochondrial reactive oxygen species generation in the knockdown condition with a lowered reduced glutathione (GSH) to oxidized glutathione (GSSG) ratio. This was indicative of an altered cellular redox balance with subsequent perturbations in adrenal steroidogenesis (71, 73). A reduced GSH : GSSG ratio has also been seen in other in vitro models of adrenal disease where oxidative stress is implicated in the pathogenic mechanism, including TXNRD2 and AAAS-KD H295R adrenocortical cell lines (74, 75). Similarly, metabolic profiling of glucocorticoid deficient fdx1b−/− zebrafish revealed significant alterations to glutathione metabolism and biochemically lowered GSH to GSSG ratios in ferredoxin null zebrafish larvae (76).
Heterozygous loss-of-function NNT mutations have been linked to left ventricular non-compaction (LVNC) in two probands and their families. Nnt deficient zebrafish cardiomyocytes demonstrated reduced proliferation and contractility leading to cardiac oedema. In vivo, co-injection of wild-type human NNT mRNA was able to rescue the cardiac oedema phenotype whilst mutagenized human NNT ORF constructs failed to rescue morpholinos-induced cardiac dysfunction (77). Roucher-Boulez et al. subsequently identified a novel homozygous NNT mutation (p.R379*) in a single patient who exhibited features of glucocorticoid insufficiency with progressive left ventricular hypertrophy (78).
C57BL/6J mice possess an intrinsic inactivating mutation in Nnt resulting in an untranslated protein. Interestingly, C57BL/6J Nnt-null mice do not develop cardiomyopathy (79) but they do have a lower adrenal reserve with attenuated corticosterone levels, both basally and following ACTH provocation. Adrenal histology revealed a markedly disorganized zona fasciculata with increased apoptosis (71). Mitochondria isolated from Nnt null mice exhibited an increased oxidized/reduced glutathione ratio and impaired ability to metabolize organic peroxide suggesting that loss of Nnt leads to redox imbalance (80). At protein level, Nnt null mice exhibited a 65% reduction in expression of Cyp11a1, which catalyses the rate limiting step of steroidogenesis (73).
5.2 Thioredoxin reductase 2 (TXNRD2) (GCCD5, OMIM #617825)
Amongst the genetic causes of FGD, TXNRD2 is perhaps the most enigmatic. Until recently, only one consanguineous kindred was known to harbour a deleterious homozygous variant in TXNRD2 in association with a phenotype of glucocorticoid insufficiency. In 2014, Prasad et al. characterised 7 individuals from a Kashmiri kindred who were found to have a stop gain variant, p.Y447*, associated with loss of TXNRD2 (74, 81). The age of presentation was highly variable ranging from 0.1 to 10.8 years (74). Of the glucocorticoid deficient family members genotyped, only one had co-morbid heart defects precipitating cardiac failure; cardiomyopathy being a recognised feature of TXNDR2 haploinsufficiency (82).
Sibbing et al. identified two heterozygous mutations in TXNRD2 (p.G375R and p.A59T) in 3 individuals with dilated cardiomyopathy (DCM). When cell survival was used as a marker of Txnrd2 function, neither mutant construct was able to rescue Txnrd2 function in Txnrd2-/- GSH depleted mouse embryonic fibroblasts in contrast to a wild type construct (83).
In 2022, a new study highlighted a novel homozygous missense variant in TXNRD2 (c.1081G>A, p.V361M) in a proband with glucocorticoid and gonadal insufficiency but normal cardiac function (84). This adds credence to the impact of TXNRD2 as a player in the pathogenesis of isolated glucocorticoid deficiency and further broadens the phenotypic continuum of this disorder.
6 FGD-like syndrome due to defective DNA replication (Minichromosome maintenance 4, MCM4) (OMIM #609981)
In 2012, dual reports of partial MCM4 deficiency in 14 probands from the Irish Traveller population highlighted a unique syndrome that encompassed adrenal insufficiency, intrauterine and postnatal growth restriction, microcephaly and natural killer cell (NK) deficiency (85, 86). The variant highlighted (and to date, the only variant associated with this disorder) was a homozygous splice site substitution c.71-2A>G leading to a single base cDNA insertion c.70_71insG and frameshift truncation, p.P24Rfs*4. Unlike other forms of FGD, the adrenal phenotype was relatively mild with onset of adrenal insufficiency often in late childhood. In healthy control derived peripheral lymphocytes, two MCM4 isoforms are detectable at 96KDa and 85KDa. In patient cells, only the minor isoform is present. This smaller protein is touted to lack the N-terminal MCM4 domain and may partially rescue patient phenotype (85, 86).
Complete loss of Mcm4 in mice is lethal. Hypomorphic Mcm4Chaos3/–Mcm3+/– mice are viable and demonstrate abnormal adrenal morphology. The adrenal capsule is thinned with significant numbers of non-steroidogenic Cyp11a1/Cyp11b1 negative cells that are on the contrary, Gata-4 and Gli1-positive. This remodelled adrenal cortex demonstrates a lack of steroidogenic output in keeping with human disease (85, 87).
7 Disorder of sphingolipid metabolism due to SGPL1 deficiency (OMIM #617575) leading to primary adrenal insufficiency and steroid resistant nephrotic syndrome
Until recently, inborn errors of sphingolipid metabolism due to single enzyme defects within the sphingolipid pathway have been characterised by their predominantly neurological phenotype. Sphingosine-1 phosphate lyase insufficiency syndrome (SPLIS) due to defects in SGPL1, which coordinates the final degradative step in the sphingolipid pathway, is a newly described multi-systemic disorder, in which adrenal failure features prominently (88–90).
Sphingolipid synthesis involves a series of tightly regulated, enzyme-catalysed steps that initiate in the endoplasmic reticulum from non-sphingolipid precursors to biosynthesis of higher order complex glycosphingolipids within the Golgi apparatus. Despite the diversity within the biosynthetic pathway, sphingolipid metabolism begins with a common entry point and exit via a single degradative pathway. This common initial step involves the coupling of cytosolic serine and palmitoyl CoA to 3-ketodihydrosphingosine through the action of serine palmitoyltransferase (SPT) whilst SGPL1 executes the penultimate step of the metabolic pathway, catalytic cleavage of sphingosine-1 phosphate (S1P), into 2E-hexadecanal and phosphoethanolamine (91, 92). SGPL1 is the major modulator of S1P signalling (93). Under normal physiological conditions, S1P is largely pro-proliferative, suppressing the pro-apoptotic actions of ceramide however loss of function mutations in SGPL1 result in a pathological accumulation of S1P which studies have shown to be associated with induction of apoptosis.
In 2017, Prasad et al. (88) described loss of function human mutations in SGPL1 and a novel syndrome characterised by primary adrenal insufficiency. Extra-adrenal phenotypic features included steroid-resistant nephrotic syndrome, hypothyroidism, ichthyosis, neurodevelopmental delay, hypogonadism and lymphopenia. Mass spectrometric analysis of plasma sphingolipids in one patient and heterozygous parents revealed elevated ceramides and S1P levels when compared to age and sex matched controls suggesting that the underlying multi-system pathology in these patients may be due to organ-specific cytosolic accumulation of sphingolipid intermediates. Correspondingly, Lovric et al. described a cohort of 7 families who were found on next generation sequencing to harbour 9 unique, recessive mutations in SGPL1 (89). Several studies have subsequently corroborated these initial findings and expanded the phenotypic spectrum to include microcephaly, sensorineural deafness, and progressive neurological deterioration (94). A further neurological phenotype has been described in siblings bearing compound heterozygous loss of function mutations in SGPL1, involving axonal mononeuropathy giving rise to Charcot Marie Tooth-like disease (95).
Sgpl1-/- mice exhibit early postnatal mortality but those that survive demonstrate disrupted adrenal morphology. Adrenocortical zonation is disordered and cells of the zona fasciculata have reduced expression of steroidogenic enzymes and contain fewer lipid droplets when compared to wild type mice (88). Electron microscopy of Sgpl1-/- kidneys demonstrated foot process effacement and absent slit diaphragms (89). Tamoxifen-inducible Sgpl1-ablated (SPLFlox/Flox Cre+) mice with partial lyase deficiency, interestingly, demonstrated glomerulopathy with progressive proteinuria and markedly increased intra-renal S1P levels (96). These mice additionally showed dermal irritation and hyperkeratosis whilst other phenotypic features of Sgpl1 silencing were less evident suggesting that some degree of wild type activity may be protective against developing multi-systemic disease.
8 Conclusion
From initial descriptions of defects in the ACTH receptor to disorders of sphingolipid metabolism, the genomic landscape of FGD has dramatically transformed over the last three decades. Pending the discovery of novel genes implicated in pathogenesis of glucocorticoid deficient disorders, the likelihood of oligogenic inheritance is augmented in the diagnosis of unsolved FGD cases. One report suggests that digenic, tri-allelic inheritance of variants in both STAR and CYP11A1 account for an isolated case of adrenal failure (97). Oligogenic heterozygosity is increasingly pertinent particularly in cases of haplotype ambiguity. Given the increasing accessibility of next generation sequencing techniques, the heritability of adrenal insufficiency-related phenotypes may be more easily uncovered allowing earlier patient intervention with significantly disease modulating impacts.
Author contributions
AM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The images generated in this article were created using modified images obtained from Servier free medical art (https://smart.servier.com/).
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Shepard TH, Landing BH, Mason DG. Familial Addison’s disease; case reports of two sisters with corticoid deficiency unassociated with hypoaldosteronism. AMA J Dis Child (1959) 97:154–62. doi: 10.1001/archpedi.1959.02070010156002
2. Thistlethwaite D, Darling JA, Fraser R, Mason PA, Rees LH, Harkness RA. Familial glucocorticoid deficiency. Studies of diagnosis and pathogenesis. Arch Dis Child (1975) 50:291–7. doi: 10.1136/adc.50.4.291
3. Migeon CJ, Kenny FM, Kowarski A, Snipes CA, Spaulding JS, Finkelstein JW, et al. The syndrome of congenital adrenocortical unresponsiveness to ACTH. Report of six cases. Pediatr Res (1968) 2:501–13. doi: 10.1203/00006450-196811000-00008
4. Franks RC, Nance WE. Hereditary adrenocortical unresponsiveness to acth. Pediatrics (1970) 45:43–8. doi: 10.1542/peds.45.1.43
5. Moshang T, Rosenfield RL, Bongiovanni AM, Parks JS, Amrhein JA. Familial glucocorticoid insufficiency. J Pediatr (1973) 82:821–6. doi: 10.1016/S0022-3476(73)80073-3
6. Stempfel RS, Engel FL. A congenital, familial syndrome of adrenocortical insufficiency without hypoaldosteronism. J Pediatr (1960) 57:443–51. doi: 10.1016/S0022-3476(60)80252-1
7. Williams HE, Freeman M. Primary familial addison’s disease. J Paediatrics Child Health (1965) 1:93–7. doi: 10.1111/j.1440-1754.1965.tb02588.x
8. Kelch RP, Kaplan SL, Biglieri EG, Daniels GH, Epstein CJ, Grumbach MM. Hereditary adrenocortical unresponsiveness to adrenocorticotropic hormone. J Pediatr (1972) 81:726–36. doi: 10.1016/S0022-3476(72)80093-3
9. Kershnar AK, Roe TF, Kogut MD. Adrenocorticotropic hormone unresponsiveness: Report of a girl with excessive growth and review of 16 reported cases. J Pediatr (1972) 80:610–9. doi: 10.1016/S0022-3476(72)80058-1
10. Herman JP, McKlveen JM, Solomon MB, Carvalho-Netto E, Myers B. Neural regulation of the stress response: glucocorticoid feedback mechanisms. Braz J Med Biol Res (2012) 45:292–8. doi: 10.1590/S0100-879X2012007500041
11. Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science (1992) 257:1248–51. doi: 10.1126/science.1325670
12. Clark AJ, McLoughlin L, Grossman A. Familial glucocorticoid deficiency associated with point mutation in the adrenocorticotropin receptor. Lancet (1993) 341:461–2. doi: 10.1016/0140-6736(93)90208-X
13. Tsigos C, Arai K, Hung W, Chrousos GP. Hereditary isolated glucocorticoid deficiency is associated with abnormalities of the adrenocorticotropin receptor gene. J Clin Invest (1993) 92(5):2458–61. doi: 10.1172/JCI116853
14. Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, et al. Human gene mutation database (HGMD®): 2003 update. Hum Mutat (2003) 21:577–81. doi: 10.1002/humu.10212
15. Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics (2014) 30:884–6. doi: 10.1093/bioinformatics/btt607
16. Chung TT, Webb TR, Chan LF, Cooray SN, Metherell LA, King PJ, et al. The majority of ACTH receptor (MC2R) mutations found in Familial Glucocorticoid Deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J Clin Endocrinol Metab (2008) 93:4948–54. doi: 10.1210/jc.2008-1744
17. Chung TT, Webb TR, Chan LF, Cooray SN, Metherell LA, King PJ, et al. The majority of adrenocorticotropin receptor (Melanocortin 2 receptor) mutations found in familial glucocorticoid deficiency type 1 lead to defective trafficking of the receptor to the cell surface. J Clin Endocrinol Metab (2008) 93:4948–54. doi: 10.1210/jc.2008-1744
18. Mohammed I, Haris B, Hussain K. A novel homozygous MC2R variant leading to type-1 familial glucocorticoid deficiency. J Endocr Soc (2022) 6:bvac058. doi: 10.1210/jendso/bvac058
19. Fridmanis D, Roga A, Klovins J. ACTH receptor (MC2R) specificity: what do we know about underlying molecular mechanisms. Front Endocrinol (2017) 8:13. doi: 10.3389/fendo.2017.00013
20. Lin L, Hindmarsh PC, Metherell LA, Alzyoud M, Al-Ali M, Brain CE, et al. Severe loss-of-function mutations in the adrenocorticotropin receptor (ACTHR, MC2R) can be found in patients diagnosed with salt-losing adrenal hypoplasia. Clin Endocrinol (Oxf) (2007) 66:205–10. doi: 10.1111/j.1365-2265.2006.02709.x
21. Chan LF, Metherell LA, Krude H, Ball C, O’Riordan SMP, Costigan C, et al. Homozygous nonsense and frameshift mutations of the ACTH receptor in children with familial glucocorticoid deficiency (FGD) are not associated with long-term mineralocorticoid deficiency. Clin Endocrinol (Oxf) (2009) 71:171–5. doi: 10.1111/j.1365-2265.2008.03511.x
22. Chida D, Nakagawa S, Nagai S, Sagara H, Katsumata H, Imaki T, et al. Melanocortin 2 receptor is required for adrenal gland development, steroidogenesis, and neonatal gluconeogenesis. Proc Natl Acad Sci USA (2007) 104:18205–10. doi: 10.1073/pnas.0706953104
23. Turan S, Hughes C, Atay Z, Guran T, Haliloglu B, Clark AJL, et al. An atypical case of familial glucocorticoid deficiency without pigmentation caused by coexistent homozygous mutations in MC2R (T152K) and MC1R (R160W). J Clin Endocrinol Metab (2012) 97:E771. doi: 10.1210/jc.2011-2414
24. Mazur A, Koehler K, Schuelke M, Skunde M, Ostański M, Huebner A. Familial glucocorticoid deficiency type 1 due to a novel compound heterozygous MC2R mutation. Hormone Res (2008) 69:363–8. doi: 10.1159/000117393
25. al Kandari HM, Katsumata N, al Alwan I, al Balwi M, Rasoul MSA. Familial glucocorticoid deficiency in five Arab kindreds with homozygous point mutations of the ACTH receptor (MC2R): genotype and phenotype correlations. Horm Res Paediatr (2011) 76:165–71. doi: 10.1159/000328035
26. Liu S, Zeng T, Luo C, Peng D, Xu X, Liu Q, et al. A rare homozygous variant of MC2R gene identified in a Chinese family with familial glucocorticoid deficiency type 1: A case report. Front Endocrinol (2023) 14:1113234. doi: 10.3389/fendo.2023.1113234
27. Mathew RP, Kovacs WJ. Short stature in a patient with familial glucocorticoid deficiency. J Pediatr Endocrinol Metab (2011) 24:569–71. doi: 10.1515/jpem.2011.203
28. Heshmatzad K, Mahdieh N, Rabbani A, Didban A, Rabbani B. The genetic perspective of familial glucocorticoid deficiency: in silico analysis of two novel variants. Int J Endocrinol (2020) 2020:e2190508. doi: 10.1155/2020/2190508
29. Abuduxikuer K, Li Z-D, Xie X-B, Li Y-C, Zhao J, Wang J-S. Novel melanocortin 2 receptor variant in a chinese infant with familial glucocorticoid deficiency type 1, case report and review of literature. Front Endocrinol (Lausanne) (2019) 10:359. doi: 10.3389/fendo.2019.00359
30. Fortier C. Interrelationships in the control of ACTH and TSH secretion.,”. In: Lissák K, editor. Hormones and brain function. Boston, MA: Springer US (1973). p. 93–103. doi: 10.1007/978-1-4684-2007-4_9
31. Weber A, Clark AJL, Perry LA, Honour JW, Savage MO. Diminished adrenal androgen secretion in familial glucocorticoid deficiency implicates a significant role for ACTH in the induction of adrenarche. Clin Endocrinol (1997) 46:431–7. doi: 10.1046/j.1365-2265.1997.1580969.x
32. Reiter EO, Fuldauer VG, Root AW. Secretion of the adrenal androgen, dehydroepiandrosterone sulfate, during normal infancy, childhood, and adolescence, in sick infants, and in children with endocrinologic abnormalities. J Pediatr (1977) 90:766–70. doi: 10.1016/s0022-3476(77)81244-4
33. Ilondo MM, Vanderschueren-Lodeweyckx M, Vlietinck R, Pizarro M, Malvaux P, Eggermont E, et al. Plasma androgens in children and adolescents. Part II. A longitudinal study in patients with hypopituitarism. Horm Res (1982) 16:78–95. doi: 10.1159/000179487
34. Witchel SF, Pinto B, Burghard AC, Oberfield SE. Update on adrenarche. Curr Opin Pediatr (2020) 32:574–81. doi: 10.1097/MOP.0000000000000928
35. Elias LLK, Huebner A, Metherell LA, Canas A, Warne GL, Bitti MLM, et al. Tall stature in familial glucocorticoid deficiency. Clin Endocrinol (2000) 53:423–30. doi: 10.1046/j.1365-2265.2000.01122.x
36. Imamine H, Mizuno H, Sugiyama Y, Ohro Y, Sugiura T, Togari H. Possible relationship between elevated plasma ACTH and tall stature in familial glucocorticoid deficiency. Tohoku J Exp Med (2005) 205:123–31. doi: 10.1620/tjem.205.123
37. Chung T-TLL, Chan LF, Metherell LA, Clark AJL. Phenotypic characteristics of familial glucocorticoid deficiency (FGD) type 1 and 2. Clin Endocrinol (Oxf) (2010) 72:589–94. doi: 10.1111/j.1365-2265.2009.03663.x
38. Evans JF, Niu Q-T, Canas JA, Shen C-L, Aloia JF, Yeh JK. ACTH enhances chondrogenesis in multipotential progenitor cells and matrix production in chondrocytes. Bone (2004) 35:96–107. doi: 10.1016/j.bone.2004.03.015
39. Weiss RE, Refetoff S. Genetic diagnosis of endocrine disorders. Cambridge, Massachusetts: Academic Press (2015). p. 462.
40. Noon LA, Franklin JM, King PJ, Goulding NJ, Hunyady L, Clark AJ. Failed export of the adrenocorticotrophin receptor from the endoplasmic reticulum in non-adrenal cells: evidence in support of a requirement for a specific adrenal accessory factor. J Endocrinol (2002) 174:17–25. doi: 10.1677/joe.0.1740017
41. Schimmer BP, Kwan WK, Tsao J, Qiu R. Adrenocorticotropin-resistant mutants of the Y1 adrenal cell line fail to express the adrenocorticotropin receptor. J Cell Physiol (1995) 163:164–71. doi: 10.1002/jcp.1041630119
42. Metherell LA, Chapple JP, Cooray S, David A, Becker C, Rüschendorf F, et al. Mutations in MRAP, encoding a new interacting partner of the ACTH receptor, cause familial glucocorticoid deficiency type 2. Nat Genet (2005) 37:166–70. doi: 10.1038/ng1501
43. Chen C, Zhou R, Fang Y, Jiang L, Liang L, Wang C. Neonatal presentation of familial glucocorticoid deficiency with a MRAP mutation: A case report. Mol Genet Metab Rep (2016) 9:15–7. doi: 10.1016/j.ymgmr.2016.09.003
44. Smith C, Maharaj AV, Qamar Y, Read J, Williams J, Chan L, et al. In vitro splicing assay proves the pathogenicity of intronic variants in MRAP. J Endocrine Soc (2021) 5:A85–6. doi: 10.1210/jendso/bvab048.172
45. Modan-Moses D, Ben-Zeev B, Hoffmann C, Falik-Zaccai TC, Bental YA, Pinhas-Hamiel O, et al. Unusual presentation of familial glucocorticoid deficiency with a novel MRAP mutation. J Clin Endocrinol Metab (2006) 91:3713–7. doi: 10.1210/jc.2006-0687
46. Hasenmajer V, Ferrigno R, Minnetti M, Pellegrini B, Isidori AM, Lenzi A, et al. Rare forms of genetic paediatric adrenal insufficiency: Excluding congenital adrenal hyperplasia. Rev Endocr Metab Disord (2023) 24:345–63. doi: 10.1007/s11154-023-09784-7
47. Novoselova TV, Hussain M, King PJ, Guasti L, Metherell LA, Charalambous M, et al. MRAP deficiency impairs adrenal progenitor cell differentiation and gland zonation. FASEB J (2018) 32(11):6186–96. doi: 10.1096/fj.201701274RR
48. Miller WL. MECHANISMS IN ENDOCRINOLOGY: Rare defects in adrenal steroidogenesis. Eur J Endocrinol (2018) 179:R125–41. doi: 10.1530/EJE-18-0279
49. Bens S, Mohn A, Yüksel B, Kulle AE, Michalek M, Chiarelli F, et al. Congenital lipoid adrenal hyperplasia: functional characterization of three novel mutations in the STAR gene. J Clin Endocrinol Metab (2010) 95:1301–8. doi: 10.1210/jc.2009-1176
50. Liu J, Dai H-M, Guang G-P, Hu W-M, Jin P. Clinical and functional analyses of the novel STAR c.558C>A in a patient with classic lipoid congenital adrenal hyperplasia. Front Genet (2023) 14:1096454. doi: 10.3389/fgene.2023.1096454
51. Metherell LA, Naville D, Halaby G, Begeot M, Huebner A, Nürnberg G, et al. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J Clin Endocrinol Metab (2009) 94:3865–71. doi: 10.1210/jc.2009-0467
52. Burget L, Parera LA, Fernandez-Cancio M, Gräni R, Henzen C, Flück CE. A rare cause of primary adrenal insufficiency due to a homozygous Arg188Cys mutation in the STAR gene. Endocrinology Diabetes Metab Case Rep (2018) 2018:EDM-18-0003. doi: 10.1530/EDM-18-0003
53. Flück CE, Pandey AV, Dick B, Camats N, Fernández-Cancio M, Clemente M, et al. Characterization of novel stAR (Steroidogenic acute regulatory protein) mutations causing non-classic lipoid adrenal hyperplasia. PloS One (2011) 6:e20178. doi: 10.1371/journal.pone.0020178
54. Luo Y, Bai R, Wang Z, Zhu X, Xing J, Li X. STAR mutations causing non-classical lipoid adrenal hyperplasia manifested as familial glucocorticoid deficiency. Mol Med Rep (2020) 22:681–6. doi: 10.3892/mmr.2020.11140
55. Baker BY, Lin L, Kim CJ, Raza J, Smith CP, Miller WL, et al. Nonclassic congenital lipoid adrenal hyperplasia: a new disorder of the steroidogenic acute regulatory protein with very late presentation and normal male genitalia. J Clin Endocrinol Metab (2006) 91:4781–5. doi: 10.1210/jc.2006-1565
56. Zhang T, Ma X, Wang J, Jia C, Wang W, Dong Z, et al. Clinical and molecular characterization of thirty Chinese patients with congenital lipoid adrenal hyperplasia. J Steroid Biochem Mol Biol (2021) 206:105788. doi: 10.1016/j.jsbmb.2020.105788
57. Sasaki G, Ishii T, Jeyasuria P, Jo Y, Bahat A, Orly J, et al. Complex role of the mitochondrial targeting signal in the function of steroidogenic acute regulatory protein revealed by bacterial artificial chromosome transgenesis in vivo. Mol Endocrinol (2008) 22:951–64. doi: 10.1210/me.2007-0493
58. Pikuleva IA, Waterman MR. Cytochromes p450: roles in diseases. J Biol Chem (2013) 288:17091–8. doi: 10.1074/jbc.R112.431916
59. Rumsby G, Woodward GM. . Disorders of steroidogenesis: guide to steroid profiling and biochemical diagnosis. Manhattan, New York City: Springer International Publishing (2019). doi: 10.1007/978-3-319-96364-8
60. Sahakitrungruang T, Tee MK, Blackett PR. Miller WL. Partial defect in the cholesterol side-chain cleavage enzyme P450scc (CYP11A1) resembling nonclassic congenital lipoid adrenal hyperplasia. J Clin Endocrinol Metab (2011) 96:792–8. doi: 10.1210/jc.2010-1828
61. Parajes S, Kamrath C, Rose IT, Taylor AE, Mooij CF, Dhir V, et al. A novel entity of clinically isolated adrenal insufficiency caused by a partially inactivating mutation of the gene encoding for P450 side chain cleavage enzyme (CYP11A1). J Clin Endocrinol Metab (2011) 96:E1798–1806. doi: 10.1210/jc.2011-1277
62. Hiort O, Holterhus P-M, Werner R, Marschke C, Hoppe U, Partsch C-J, et al. Homozygous disruption of P450 side-chain cleavage (CYP11A1) is associated with prematurity, complete 46,XY sex reversal, and severe adrenal failure. J Clin Endocrinol Metab (2005) 90:538–41. doi: 10.1210/jc.2004-1059
63. Tee MK, Abramsohn M, Loewenthal N, Harris M, Siwach S, Kaplinsky A, et al. Varied clinical presentations of seven patients with mutations in CYP11A1 encoding the cholesterol side-chain cleavage enzyme, P450scc. J Clin Endocrinol Metab (2013) 98:713–20. doi: 10.1210/jc.2012-2828
64. Maharaj A, Buonocore F, Meimaridou E, Ruiz-Babot G, Guasti L, Peng H-M, et al. Predicted benign and synonymous variants in CYP11A1 cause primary adrenal insufficiency through missplicing. J Endocr Soc (2019) 3:201–21. doi: 10.1210/js.2018-00130
65. Buonocore F, McGlacken-Byrne SM, del Valle I, Achermann JC. Current insights into adrenal insufficiency in the newborn and young infant. Front Pediatr (2020) 8:619041. doi: 10.3389/fped.2020.619041
66. Goursaud C, Mallet D, Janin A, Menassa R, Tardy-Guidollet V, Russo G, et al. Aberrant splicing is the pathogenicity mechanism of the p.Glu314Lys variant in CYP11A1 gene. Front Endocrinol (Lausanne) (2018) 9:491. doi: 10.3389/fendo.2018.00491
67. Lara-Velazquez M, Perdomo-Pantoja A, Blackburn PR, Gass JM, Caulfield TR, Atwal PS. A novel splice site variant in CYP11A1 in trans with the p.E314K variant in a male patient with congenital adrenal insufficiency. Mol Genet Genomic Med (2017) 5:781–7. doi: 10.1002/mgg3.322
68. Kolli V, Kim H, Torky A, Lao Q, Tatsi C, Mallappa A, et al. Characterization of the CYP11A1 nonsynonymous variant p.E314K in children presenting with adrenal insufficiency. J Clin Endocrinol Metab (2019) 104:269–76. doi: 10.1210/jc.2018-01661
69. Kallali W, Gray E, Mehdi MZ, Lindsay R, Metherell LA, Buonocore F, et al. Long-term outcome of partial P450 side-chain cleavage enzyme deficiency in three brothers: the importance of early diagnosis. Eur J Endocrinol (2020) 182:K15–24. doi: 10.1530/EJE-19-0696
70. Hu M-C, Hsu N-C, El Hadj NB, Pai C-I, Chu H-P, Wang C-KL, et al. Steroid deficiency syndromes in mice with targeted disruption of Cyp11a1. Mol Endocrinol (2002) 16:1943–50. doi: 10.1210/me.2002-0055
71. Meimaridou E, Kowalczyk J, Guasti L, Hughes CR, Wagner F, Frommolt P, et al. Mutations in NNT encoding nicotinamide nucleotide transhydrogenase cause familial glucocorticoid deficiency. Nat Genet (2012) 44:740–2. doi: 10.1038/ng.2299
72. Jazayeri O, Liu X, van Diemen CC, Bakker-van Waarde WM, Sikkema-Raddatz B, Sinke RJ, et al. A novel homozygous insertion and review of published mutations in the NNT gene causing familial glucocorticoid deficiency (FGD). Eur J Med Genet (2015) 58:642–9. doi: 10.1016/j.ejmg.2015.11.001
73. Meimaridou E, Goldsworthy M, Chortis V, Fragouli E, Foster PA, Arlt W, et al. NNT is a key regulator of adrenal redox homeostasis and steroidogenesis in male mice. J Endocrinol (2018) 236:13–28. doi: 10.1530/JOE-16-0638
74. Prasad R, Chan LF, Hughes CR, Kaski JP, Kowalczyk JC, Savage MO, et al. Thioredoxin Reductase 2 (TXNRD2) mutation associated with familial glucocorticoid deficiency (FGD). J Clin Endocrinol Metab (2014) 99:E1556–1563. doi: 10.1210/jc.2013-3844
75. Prasad R, Metherell LA, Clark AJ, Storr HL. Deficiency of ALADIN impairs redox homeostasis in human adrenal cells and inhibits steroidogenesis. Endocrinology (2013) 154:3209–18. doi: 10.1210/en.2013-1241
76. Weger M, Weger BD, Görling B, Poschet G, Yildiz M, Hell R, et al. Glucocorticoid deficiency causes transcriptional and post-transcriptional reprogramming of glutamine metabolism. EBioMedicine (2018) 36:376–89. doi: 10.1016/j.ebiom.2018.09.024
77. Bainbridge MN, Davis EE, Choi W-Y, Dickson A, Martinez HR, Wang M, et al. Loss of function mutations in NNT are associated with left ventricular noncompaction. Circ Cardiovasc Genet (2015) 8:544–52. doi: 10.1161/CIRCGENETICS.115.001026
78. Roucher-Boulez F, Mallet-Motak D, Samara-Boustani D, Jilani H, Ladjouze A, Souchon P-F, et al. NNT mutations: a cause of primary adrenal insufficiency, oxidative stress and extra-adrenal defects. Eur J Endocrinol (2016) 175:73–84. doi: 10.1530/EJE-16-0056
79. Williams JL, Paudyal A, Awad S, Nicholson J, Grzesik D, Botta J, et al. Mylk3 null C57BL/6N mice develop cardiomyopathy, whereas Nnt null C57BL/6J mice do not. Life Sci Alliance (2020) 3(4):e201900593. doi: 10.26508/lsa.201900593
80. Ronchi JA, Figueira TR, Ravagnani FG, Oliveira HCF, Vercesi AE, Castilho RF. A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic Biol Med (2013) 63:446–56. doi: 10.1016/j.freeradbiomed.2013.05.049
81. Buonocore F, Maharaj A, Qamar Y, Koehler K, Suntharalingham JP, Chan LF, et al. Genetic analysis of pediatric primary adrenal insufficiency of unknown etiology: 25 years’ Experience in the UK. J Endocrine Soc (2021) 5:bvab086. doi: 10.1210/jendso/bvab086
82. Rajapreyar I, Sinkey R, Pamboukian SV, Tita A. Did a shared thioredoxin-reductase gene mutation lead to maternal peripartum cardiomyopathy and fatal dilated cardiomyopathy in her son? A case report. Case Rep Women’s Health (2020) 26:e00196. doi: 10.1016/j.crwh.2020.e00196
83. Sibbing D, Pfeufer A, Perisic T, Mannes AM, Fritz-Wolf K, Unwin S, et al. Mutations in the mitochondrial thioredoxin reductase gene TXNRD2 cause dilated cardiomyopathy. Eur Heart J (2011) 32:1121–33. doi: 10.1093/eurheartj/ehq507
84. Patjamontri S, Lucas-Herald A, McMillan M, Prasad R, Metherell L, McGowan R, et al. Thioredoxin reductase 2 (TXNRD2) variant as A cause of micropenis, undescended testis and selective glucocorticoid deficiency. ESPE Abstracts. Bioscientifica (2022). doi: 10.1159/000535528
85. Hughes CR, Guasti L, Meimaridou E, Chuang C-H, Schimenti JC, King PJ, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest (2012) 122:814–20. doi: 10.1172/JCI60224
86. Gineau L, Cognet C, Kara N, Lach FP, Dunne J, Veturi U, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest (2012) 122:821–32. doi: 10.1172/JCI61014
87. Chuang C-H, Wallace MD, Abratte C, Southard T, Schimenti JC. Incremental genetic perturbations to MCM2-7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PloS Genet (2010) 6:e1001110. doi: 10.1371/journal.pgen.1001110
88. Prasad R, Hadjidemetriou I, Maharaj A, Meimaridou E, Buonocore F, Saleem M, et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. J Clin Invest (2017) 127:942–53. doi: 10.1172/JCI90171
89. Lovric S, Goncalves S, Gee HY, Oskouian B, Srinivas H, Choi W-I, et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. J Clin Invest (2017) 127:912–28. doi: 10.1172/JCI89626
90. Janecke AR, Xu R, Steichen-Gersdorf E, Waldegger S, Entenmann A, Giner T, et al. Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Hum Mutat (2017) 38:365–72. doi: 10.1002/humu.23192
91. Gault CR, Obeid LM, Hannun YA. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol (2010) 688:1–23. doi: 10.1007/978-1-4419-6741-1_1
92. Kumar A, Byun H-S, Bittman R, Saba JD. The sphingolipid degradation product trans-2-hexadecenal induces cytoskeletal reorganization and apoptosis in a JNK-dependent manner. Cell Signal (2011) 23:1144–52. doi: 10.1016/j.cellsig.2011.02.009
93. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol (2012) 22:50–60. doi: 10.1016/j.tcb.2011.09.003
94. Maharaj A, Kwong R, Williams J, Smith C, Storr H, Krone R, et al. A retrospective analysis of endocrine disease in sphingosine-1-phosphate lyase insufficiency: case series and literature review. Endocrine Connections (2022) 11(8):e220250. doi: 10.1530/EC-22-0250
95. Atkinson D, Nikodinovic Glumac J, Asselbergh B, Ermanoska B, Blocquel D, Steiner R, et al. Sphingosine 1-phosphate lyase deficiency causes Charcot-Marie-Tooth neuropathy. Neurology (2017) 88:533–42. doi: 10.1212/WNL.0000000000003595
96. Schümann J, Grevot A, Ledieu D, Wolf A, Schubart A, Piaia A, et al. Reduced activity of sphingosine-1-phosphate lyase induces podocyte-related glomerular proteinuria, skin irritation, and platelet activation. Toxicol Pathol (2015) 43:694–703. doi: 10.1177/0192623314565650
Keywords: adrenocorticotropin, steroidogenesis, hypocortisolaemia, zona fasciculata, multi-systemic
Citation: Maharaj AV (2023) Familial Glucocorticoid Deficiency: the changing landscape of an eponymous syndrome. Front. Endocrinol. 14:1268345. doi: 10.3389/fendo.2023.1268345
Received: 27 July 2023; Accepted: 28 November 2023;
Published: 21 December 2023.
Edited by:
Rosario Ferrigno, AORN Santobono-Pausilipon, ItalyReviewed by:
Angela Huebner, University Hospital Carl Gustav Carus, GermanyIwona Ben-Skowronek, Medical University of Lublin, Poland
Copyright © 2023 Maharaj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Avinaash V. Maharaj, YS52Lm1haGFyYWpAcW11bC5hYy51aw==