 Xiaoqin Wu1
Xiaoqin Wu1 Rakesh K. Arya
Rakesh K. Arya Laura E. Nagy
Laura E. Nagy
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 15 December 2023
Sec. Molecular and Structural Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1267996
This article is part of the Research TopicMolecular aspects/mechanisms in obesity and liver diseasesView all 9 articles
Background: The RIP1-RIP3-MLKL-mediated cell death pathway is associated with progression of non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH). Previous work identified a critical role for MLKL, the key effector regulating necroptosis, but not RIP3, in mediating high fat diet-induced liver injury in mice. RIP1 and RIP3 have active N-terminus kinase domains essential for activation of MLKL and subsequent necroptosis. However, little is known regarding domain-specific roles of RIP1/RIP3 kinase in liver diseases. Here, we hypothesized that RIP1/RIP3 kinase activity are required for the development of high fat diet-induced liver injury.
Methods: Rip1K45A/K45A and Rip3K51A/K51A kinase-dead mice on a C57BL/6J background and their littermate controls (WT) were allowed free access to a diet high in fat, fructose and cholesterol (FFC diet) or chow diet.
Results: Both Rip1K45A/K45A and Rip3K51A/K51A mice were protected against FFC diet-induced steatosis, hepatocyte injury and expression of hepatic inflammatory cytokines and chemokines. FFC diet increased phosphorylation and oligomerization of MLKL and hepatocyte death in livers of WT, but not in Rip3K51A/K51A, mice. Consistent with in vivo data, RIP3 kinase deficiency in primary hepatocytes prevented palmitic acid-induced translocation of MLKL to the cell surface and cytotoxicity. Additionally, loss of Rip1 or Rip3 kinase suppressed FFC diet-mediated formation of crown-like structures (indicators of dead adipocytes) and expression of mRNA for inflammatory response genes in epididymal adipose tissue. Moreover, FFC diet increased expression of multiple adipokines, including leptin and plasminogen activator inhibitor 1, in WT mice, which was abrogated by Rip3 kinase deficiency.
Discussion: The current data indicate that both RIP1 and RIP3 kinase activity contribute to FFC diet-induced liver injury. This effect of RIP1 and RIP3 kinase deficiency on injury is consistent with the protection of Mlkl-/- mice from high fat diet-induced liver injury, but not the reported lack of protection in Rip3-/- mice. Taken together with previous reports, our data suggest that other domains of RIP3 likely counteract the effect of RIP3 kinase in response to high fat diets.
Upon activation of death receptor signaling, receptor-interacting protein kinases 1 (RIP1) and 3 (RIP3) play critical roles in the classical pathway of necroptotic cell death, leading to phosphorylation of mixed lineage kinase domain-like (MLKL), the effector molecule in necroptosis. Canonical RIP1-RIP3-MLKL-mediated necroptosis is implicated in multiple human diseases, including in the progression of non-alcohol-associated fatty liver/steatohepatitis (NAFL/NASH) (1–3). Studies using RIP1 kinase inhibitors and RIP1 kinase dead mice have consistently identified the important role of RIP1 kinase activity in multiple dietary models of fatty liver disease. For example, RIPA-56, a highly specific inhibitor of RIPK1, reduced high fat diet (HFD)-induced liver steatosis and injury partly through an increase in mitochondrial respiration (4). Another recent study found that genetic inhibition of RIP1 kinase reduced hepatic cell death and inflammation and alleviated hepatic steatosis, liver damage and fibrosis in both HFD and methionine-choline deficient (MCD) dietary models (5).
In contrast, RIP3, the direct target of RIP1 kinase, plays differential roles in multiple dietary models of fatty liver disease. RIP3 expression is not detectable in healthy hepatocytes (6–8), associated with epigenetic suppression (6). However, expression is detected in hepatocytes in patients with alcohol-associated liver disease (9) and in murine hepatocytes in response to chronic ethanol feeding (9, 10) and FFC-diet induced liver injury (8), but not in response to choline deficient high fat or 60% high fat diets (6). Rip3-/- mice are protected from ethanol (9, 10) and MCD diet-induced liver injury (11); while Rip3 deficiency did not protect mice in two different models of high-fat diet induced liver injury: 1) the traditional high-fat diet (HFD) model (12) and 2) the Western diet model utilizing a diet high in fat, fructose and cholesterol (FFC diet) (8, 13). Interestingly, Rip3 knockout mice on a combined choline deficient (CD)-HFD diet developed more pronounced glucose intolerance and adipose tissue inflammation and liver injury than WT littermates (14). Taken together, previous work indicates that the roles of RIP1 and RIP3 in murine models of NAFL/NASH are complex and not completely understood.
RIP1 and RIP3 have an active N-terminus kinase domain essential for activation of MLKL in the classical necroptotic pathway. In addition to regulating cell death, RIPK1 and RIPK3 kinases are implicated in the regulation of inflammatory responses. For example, both RIP1 and RIP3 kinases promote sustained activation of ERK, cFos and NFκB required for acute inflammatory response (15). Moreover, RIP3 kinase activity is essential for TLR-induced NLRP3 activation in the absence of both IAPs and caspase-8 (16). We have previously reported that FFC diets increase the phosphorylation, oligomerization and translocation of MLKL to the plasma membrane. Mlkl-deficient are protected from FFC diet-induced tissue injury, but, surprisingly, Rip3-deficient mice are not protected (8, 13). These studies suggest that MLKL can be activated by RIP3- independent mechanisms and/or that there are domain-specific roles of RIP1/RIP3 kinase in liver diseases.
Here we hypothesized that the kinase domain in RIP1 and RIP3 are required for development of FFC diet-induced liver injury. Rip1 (Rip1 KDKI, Rip1K45A/K45A) and Rip3 (Rip3 KDKI, Rip3K51A/K51A) kinase dead knock-in mice and littermate (wild type, WT) controls were fed chow or FFC diet for 12 weeks. FFC diet increased the phosphorylation and oligomerization of MLKL in livers of WT, but not in Rip3K51A/K51A, mice. Importantly, Rip1K45A/K45A and Rip3K51A/K51A mice were protected against FFC diet-induced liver injury and inflammatory responses in adipose. Together, these data indicate that RIP1 and RIP3 kinase activity contributes to FFC diet-induced injury. This is consistent with the protection of Mlkl-/- mice from Western diet-induced liver injury, but not the reported lack of protection in Rip3-/- mice. Taken together with previous reports, our data suggest that other domains of RIP3 likely counter the role of RIP3 kinase in response to FFC diet.
All procedures using animals were approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Rip1K45A/K45A and Rip3K51A/K51A mice were originally generated by GlaxoSmithKline. Briefly, Rip1K45A/K45A and Rip3K51A/K51A mice were generated through homologous recombination, utilizing targeting construct that mutated the catalytic lysine residue to alanine (K45A and K51A, respectively), resulting in the complete elimination of kinase activity (17, 18). Both strains are viable and fertile backcrossed to a C57BL/6J background and WT controls were wild-type littermates.
Male mice (5-6 weeks of age) were allowed free access to a chow diet (Chow) containing 6% fat/13.0 kJ/g (#2918, Teklad Mills, Madison, WI) or FFC diet (AIN-76A Western diet, #5342, TestDiet, St. Louis, MO) for 12 weeks. The FFC diet contained 40% energy as fat (12% saturated fatty acid, 0.2% cholesterol) with fructose and glucose added to the water (42 g/L final concentration).
Food intake was similar between genotypes in response to FFC diet. As expected, body and liver weights were higher with FFC feeding in all genotypes compared to chow-fed mice. Rip1K45A/K45A and Rip3K51A/K51A mice gained 12% and 11% less, respectively, on the FFC diet compared to WT (Supplementary Figure S1).
Plasma fractions were isolated using the plasma membrane protein extraction kit (ab65400, Abcam). Liver tissues were resuspended in homogenization buffer and lysed using a Dounce homogenizer (50-60 strokes). Homogenates were centrifuged to obtain the cytosolic fraction as well as the 10,000g pellet (total membrane fraction). The pellet was further purified according to the manufacturer’s instructions to obtain plasma membrane enriched proteins. Additional Materials and Methods are provided the Supplementary Material.
Values shown in all figures represent the means ± SEM. Analysis of variance was performed using the general linear model’s procedure (SAS, Carey, IN). Data were log-transformed as necessary to obtain a normal distribution. Follow-up comparisons were made by least square means testing. P values of less than 0.05 were considered significant. Values with different superscripts are significantly different from each other, P <0.05.
RIP1, upstream of RIP3 in the pathway of necroptosis, is a multitasking molecule harboring distinct roles with kinase-dependent/independent functions (19, 20). In order to study whether RIP1 kinase activity is required in a dietary model of liver injury, Rip1K45A/K45A mice and WT were allowed free access to chow or FFC diet for 12 weeks. FFC diet feeding increased phosphorylation of MLKL in liver of WT, but not Rip1K45A/K45A, mice (Figure 1A). Consistent with a previous study in a different dietary model of NAFL/NASH (5), Rip1K45A/K45A mice were protected from FFC diet-induced liver injury. Absence of Rip1 kinase prevented FFC diet-mediated hepatic steatosis (Figure 1B), increased activity of ALT/AST in the circulation (Figure 1C), hepatic triglyceride accumulation (Figure 1C) and pro-inflammatory responses (Figure 1D).
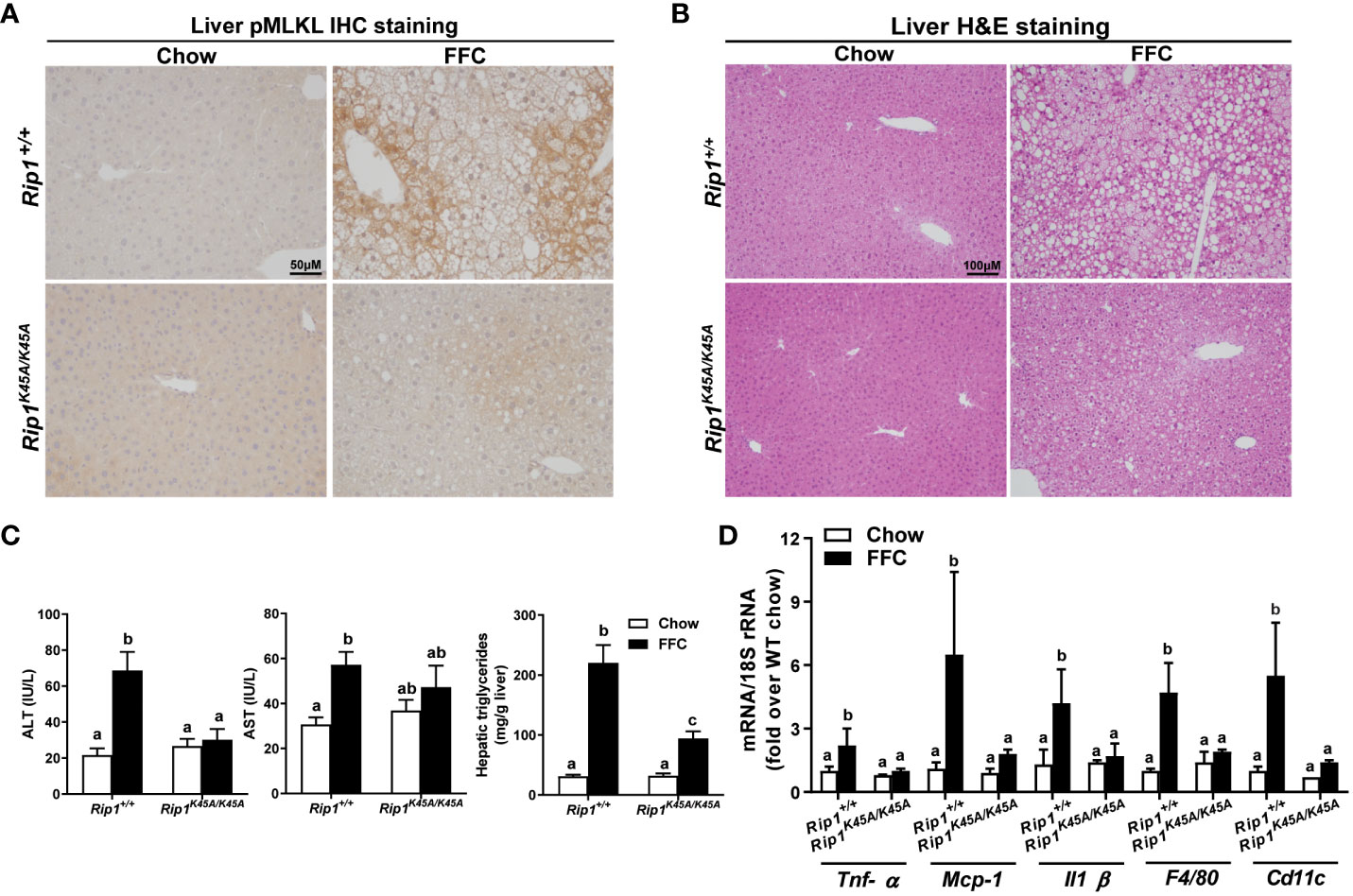
Figure 1 RIP1 kinase activity was required for FFC-induced phosphorylation of MLKL and liver injury. Rip1+/+ and Rip1K45A/K45A mice were allowed free access to chow or FFC diet for 12 weeks. (A) Immunohistochemistry staining for pMLKL in paraffin-embedded liver sections. Images were acquired using a 10X objective. Representative images are shown from n=5-6 per group. (B) Hematoxylin and eosin (H&E) staining of liver sections. Images were acquired at 10X magnification. (C) ALT/AST concentration in plasma and hepatic triglyceride content in liver homogenates. (D) mRNA expression of genes of interest in livers was detected by qRT-PCR. Values with different alphabetical superscripts are significantly different from each other, n = 5-6 per group. p <0.05, assessed by ANOVA.
Immunohistochemical analysis revealed that FFC diet feeding also increased phosphorylation of MLKL in liver of WT, but not Rip3K51A/K51A, mice (Figure 2A). Since RIP3 is the proximal kinase in the phosphorylation of MLKL, we also investigated the subcellular localization of MLKL in WT and Rip3K51A/K51A mice in response to FFC diet. FFC diet induced the translocation and oligomerization of MLKL at the plasma membrane in WT, but not Rip3 kinase deficient, mice (Figure 2B), indicating that the functional activation of MLKL in response to FFC diet requires RIP3 kinase activity. Rip3K51A/K51A mice were also protected from FFC diet-induced liver injury. Absence of Rip3 kinase prevented FFC diet-mediated hepatic steatosis (Figure 2C), increased activity of ALT/AST in the circulation (Figure 2D) and accumulation of hepatic triglycerides (Figure 2D). Expression of mRNA for a number of pro-inflammatory chemokines and cytokines (Figure 3A) and immune cell markers, the acute phase protein Crp-1 and phagocytic-related genes (Figure 3B) were also increase in WT, but not Rip3 kinase deficient, mice. In contrast, expression of mRNA for the anti-inflammatory cytokine Il-10 was higher in Rip3K51A/K51A mice compared to WT mice in response to FFC diet (Figure 3A).
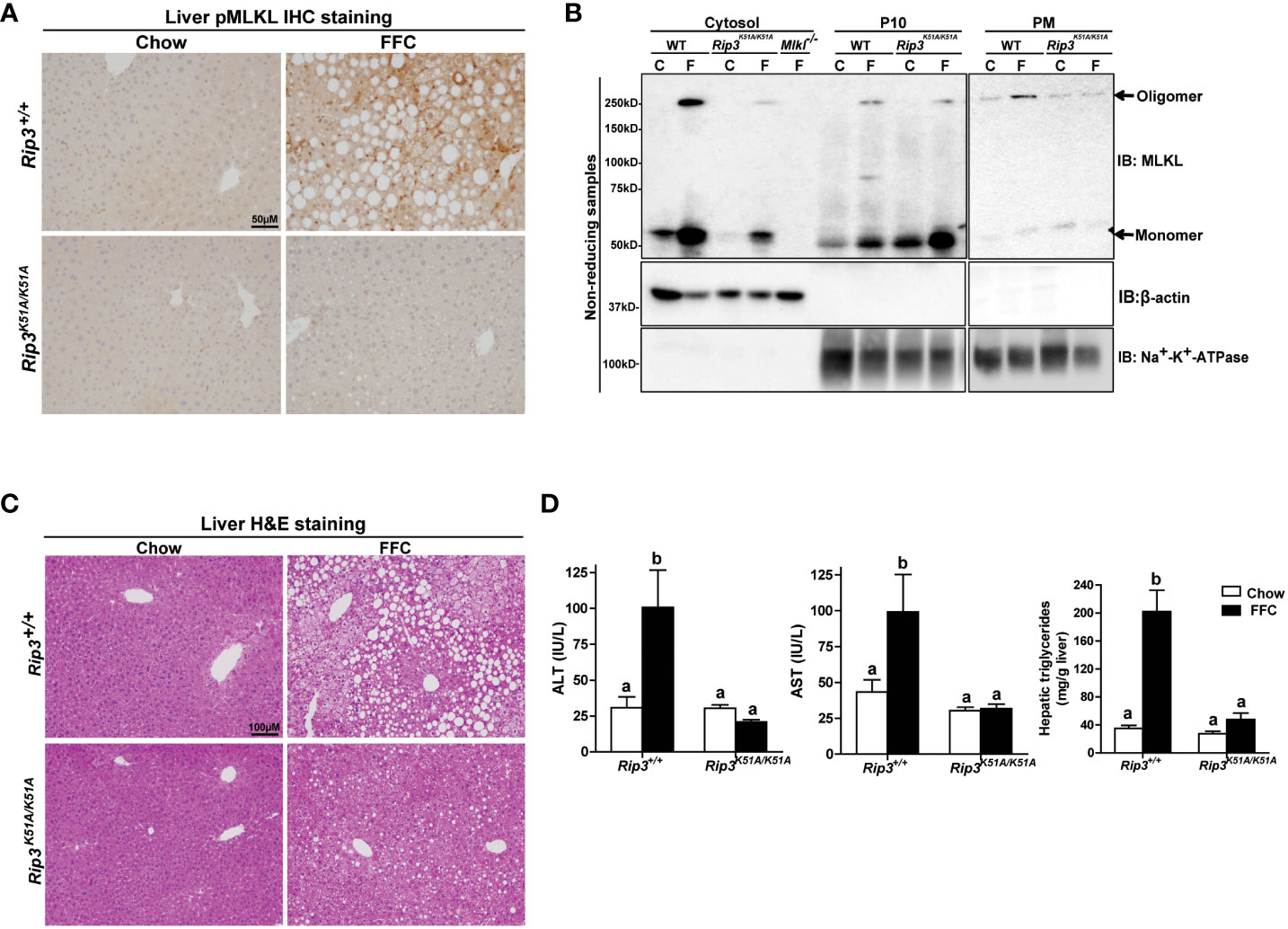
Figure 2 RIP3 kinase activity was required for FFC-induced phosphorylation and oligomerization of MLKL in mouse liver and liver injury. Rip3+/+ and Rip3K51A/K51A mice were allowed free access to chow or FFC diet for 12 weeks. (A) Immunohistochemistry staining for pMLKL in paraffin-embedded liver sections. Images were acquired using a 10X objective. Representative images are shown from n=5-6 per group. (B) Subcellular fractions (cytosol, P10 (10,000 g pellet) and plasma membrane (PM) were isolated from liver, proteins separated by SDS-PAGE under non-reducing condition, and immunoblotted (IB) with anti-MLKL antibody. Enrichment of sub-cellular fractions was verified by probing for Na+/K+-ATPase (plasma and intracellular membrane marker) and β-actin (cytosolic marker). Blots are representative from n = 3 for PM and 6 for cytosol and P10 independent experiments. Western blots were semi-quantified on Image J. Values for MLKL oligomer in the P10 fraction were 0.05 ± 0.02a for chow-fed and 0.32 ± 0.14b for FFC-fed in Rip3+/+ and 0.05 ± 0.03a for chow-fed and 0.12 ± 0.04a for FFC-fed in Rip3K51A/K51A (p<0.05). Values for MLKL oligomer in the PM fraction were 0.07 ± 0.03a for chow-fed and 0.24 ± 0.06b for FFC-fed in Rip3+/+ and 0.05 ± 0.02a for chow-fed and 0.07 ± 0.02a for FFC-fed in Rip3K51A/K51A (p<0.05). (C) Hematoxylin and eosin (H&E) staining of liver sections. Images were acquired at 10X magnification. (D) ALT/AST concentration in plasma and hepatic triglyceride content in liver homogenates. Values represent means ± SEM. Values with different alphabetical superscripts are significantly different from each other, n = 5-6 per group. p <0.05, assessed by ANOVA.
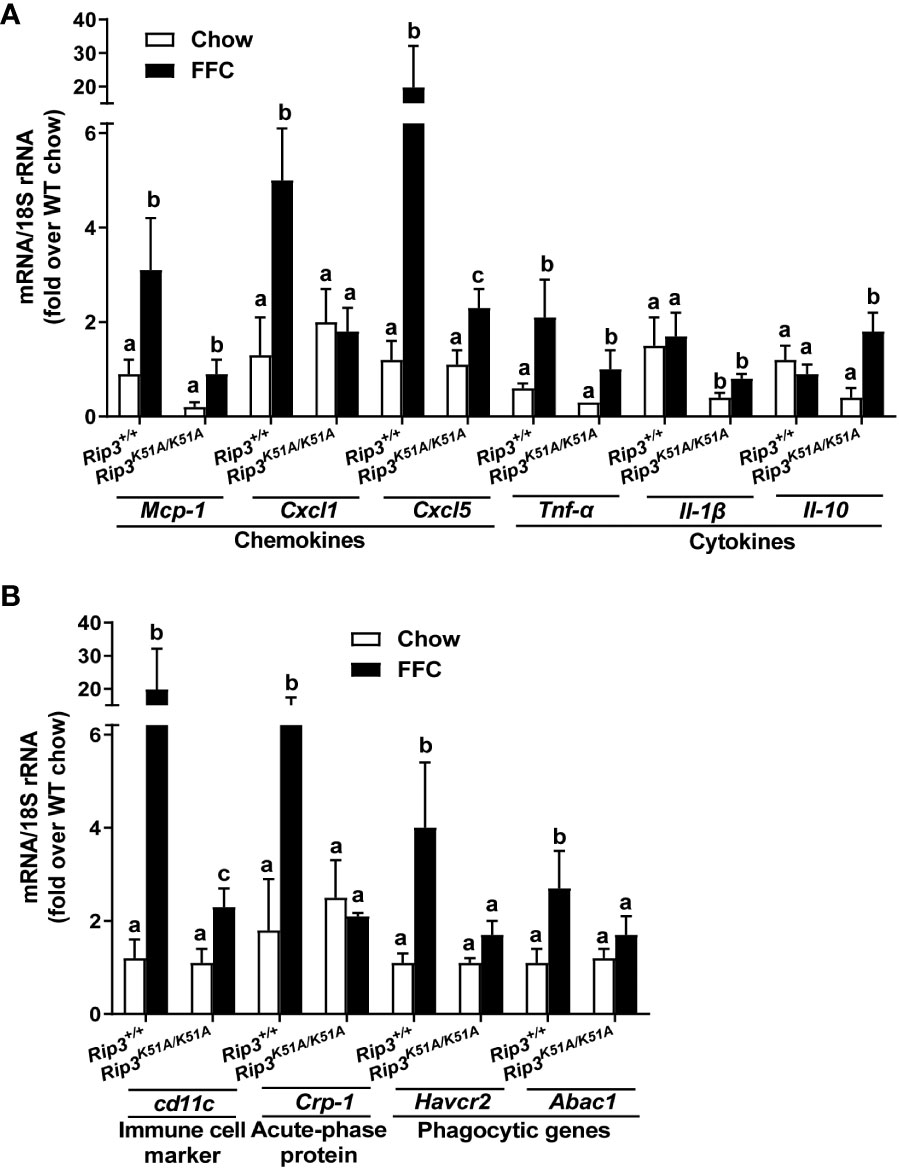
Figure 3 RIP3 kinase activity contributed to FFC-induced liver inflammatory responses. Rip3+/+ and Rip3K51A/K51A mice were allowed free access to chow or FFC diet for 12 weeks. (A) Expression of chemokine (Mcp-1, Cxcl1 and Cxcl5) and cytokine (Tnf-α, Il-1β and Il-10) mRNA in livers. (B) Expression of immune cell marker Cd11c, acute-phase protein Crp-1, and phagocytic gene (Havcr2, Abca1) mRNA in livers. Expression was normalized to 18S rRNA and expressed as the percent increase over chow-fed controls. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 5-6 per group. p <0.05, assessed by ANOVA.
Insulin resistance is a common feature of NAFL/NASH (21). FFC diet increased fasting glucose concentrations in WT and Rip3K51A/K51A mice compared to chow-fed mice independently of genotype (Figure 4A). However, fasting insulin concentrations and calculated Homeostatic Model Assessment for Insulin Resistance (HOMA-IR) were increased with FFC feeding in WT, but not Rip3K51A/K51A, mice (Figure 4A). Plasma indicators of lipid homeostasis were also differentially impacted by genotype. FFC diet increased plasma cholesterol and TG in WT mice; this increase in plasma cholesterol, but not TG, was reduced in Rip3K51A/K51A mice (Figure 4B). Rip3 kinase deficiency also prevented FFC diet-induced accumulation of multiple lipogenesis-related genes, including Fabp4, Fas, Mgat, Pparγ and Srebp-1c (Figure 4C). Together, these data indicate that the kinase activity of RIP3 contributes to FFC diet-induced metabolic perturbations and liver injury.
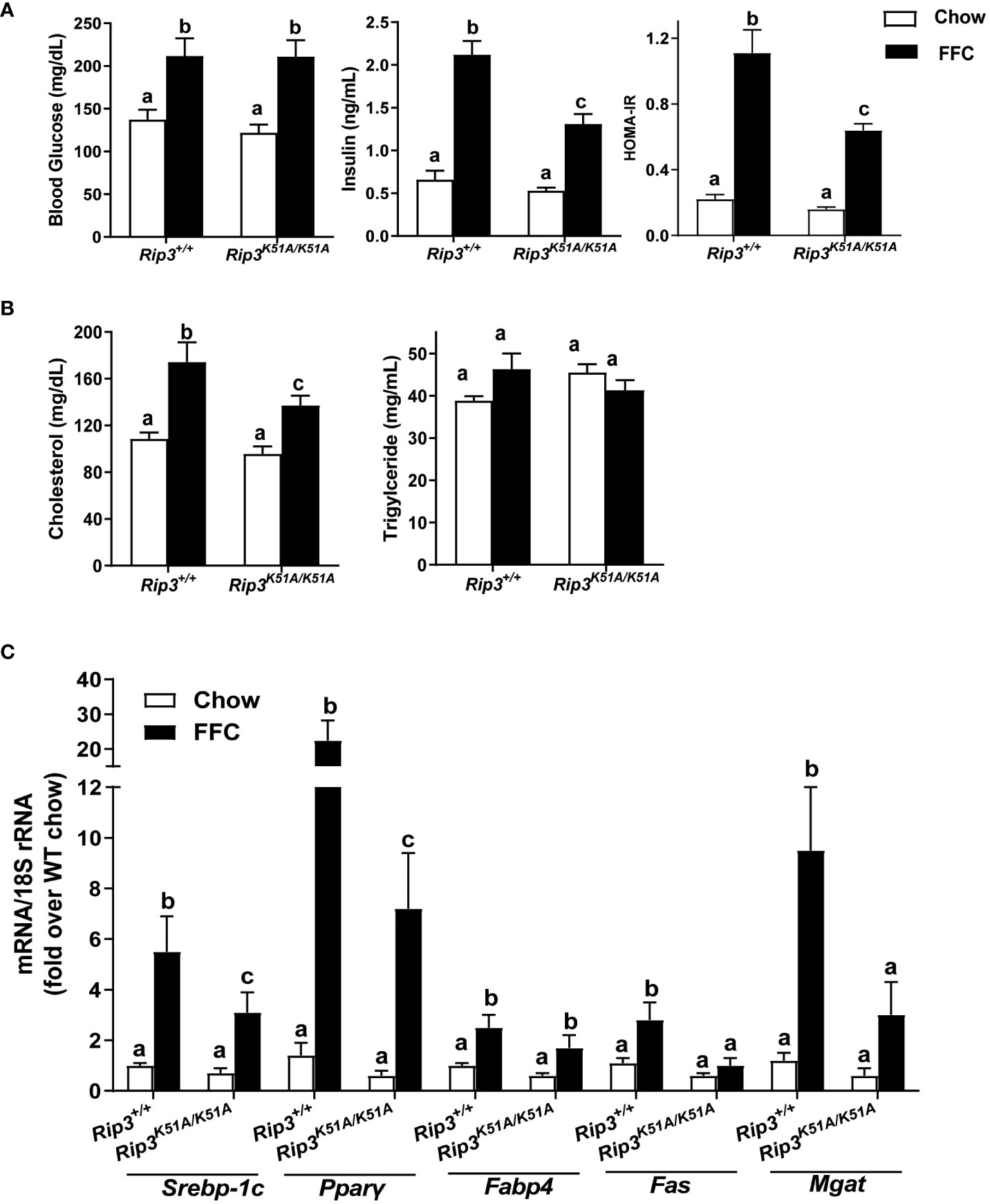
Figure 4 RIP3 kinase activity was involved in FFC-induced metabolic disturbances and regulation of genes involved in lipogenesis. Rip3+/+ and Rip3K51A/K51A mice were allowed free access to chow or FFC diet for 12 weeks. (A) Plasma concentrations of blood glucose, insulin and calculated HOMA-IR, as well as (B) cholesterol and triglycerides in plasma from Rip3+/+ and Rip3K51A/K51A mice. (C) Expression of lipogenesis-related mRNA including Srebp-1c, Pparγ, Fabp4, Fas and Mgat, in the liver was determined by qRT-PCR. Expression was normalized to 18S rRNA and expressed as the percent increase over chow-fed controls. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 5-6 per group. p <0.05, assessed by ANOVA.
Hepatocellular death was assessed by both TUNEL and M30 staining in order to distinguish apoptotic and necroptotic cell death; TUNEL staining recognizes both necroptotic/necrotic and apoptotic cells (22) while M30 staining, which detects caspase-cleaved cytokeratin 18, is an indicator of hepatocyte apoptosis (23). Interestingly, while there was no difference in M30+ cells between genotypes in response to FFC diet, the number of TUNEL+ hepatocytes was higher in WT compared to Rip3 kinase deficient mice after FFC feeding (Figures 5A, B); in contrast, the number of TUNEL+ non-parenchymal cells (NPCs) was similar between WT and Rip3K51A/K51A mice after FFC feeding (Figure 5A).
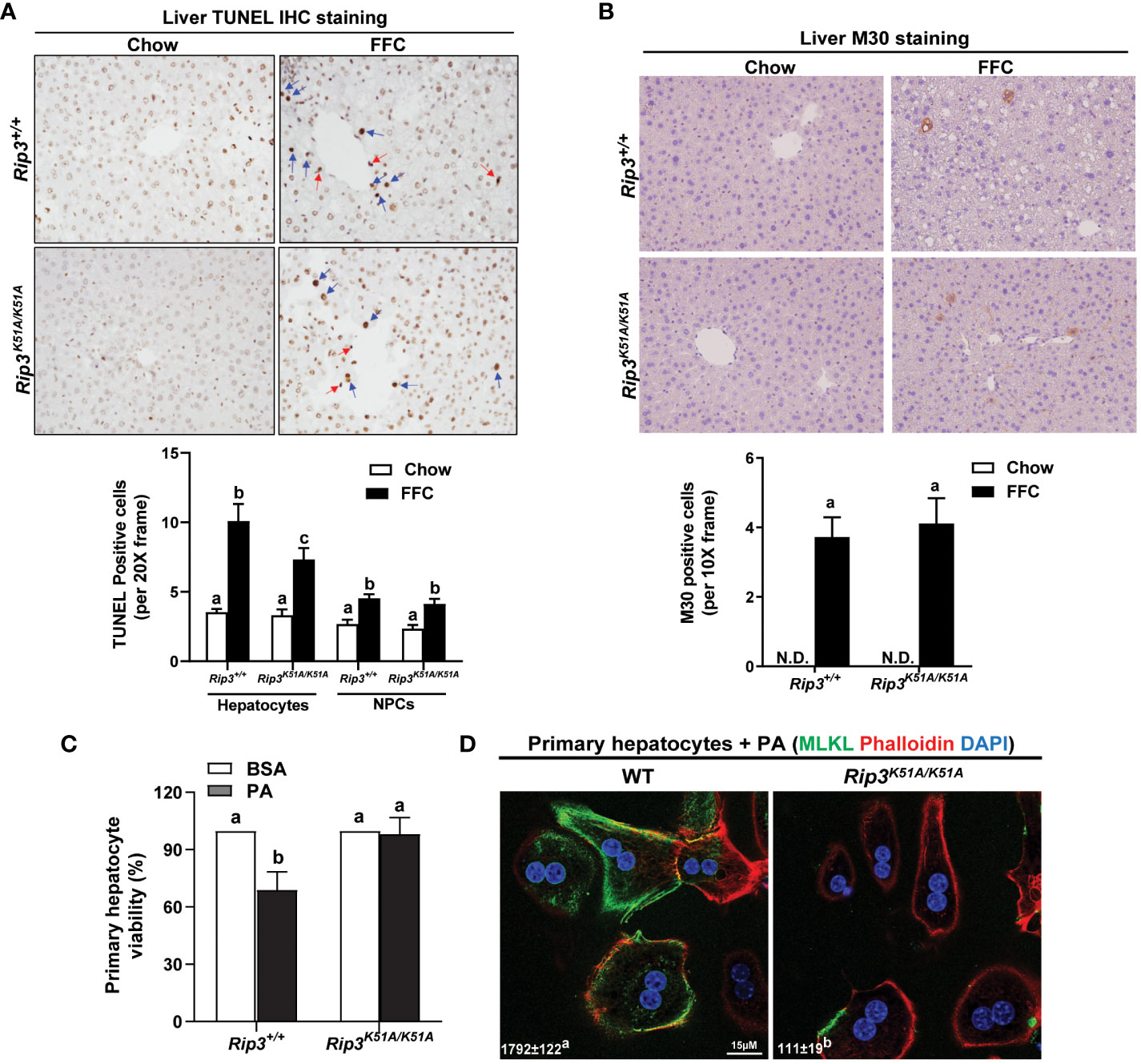
Figure 5 Effect of RIP3 kinase activity in FFC-induced cell death in mouse liver and PA-mediated cytotoxicity in primary hepatocytes. Rip3+/+ and Rip3K51A/K51A mice were allowed free access to chow or FFC diet for 12 weeks. (A, B) Paraffin-embedded livers were de-paraffinized followed by quantitative analyses of (A) TUNEL or (B) M30 staining. Images were acquired using 20X or 10X objective. The blue arrows indicate TUNEL-positive hepatocytes and red arrows indicate TUNEL-positive NPCs. TUNEL-positive hepatocytes and NPCs were counted and expressed as the total number of cells per 20X frame and M30 positive cells were quantified per 10X frame, n = 5-6 per group. (C, D) Primary hepatocytes were isolated from chow-fed WT and Rip3K51A/K51A mice and then exposed to 500 µM palmitic acid (PA) for 24h. (C) MTS assay was performed to determine PA-induced hepatotoxicity and (D) location of MLKL at the cell surface (phalloidin: F-actin) in response to PA visualized by confocal microscopy. n=4 independent isolations. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 3 per group. p <0.05, assessed by ANOVA. N.D., not detectable. TUNEL, Terminal deoxynucleotidyl transferase dUTP nick end labeling.
To further assess the contribution of RIP3 kinase activity to hepatocyte death, in vivo lipotoxicity was modeled by exposing hepatocytes to PA in vitro. PA-induced hepatocyte death is mediated in both caspase- dependent and independent mechanisms (8, 12). Here, Rip3 kinase deficiency prevented PA-mediated hepatoxicity (Figure 5C). Consistent with in vivo data, challenge of hepatocytes with PA increased the co-localization of MLKL with the cell surface marker F-actin (phalloidin) in hepatocytes from WT, but not Rip3K51A/K51A, mice (Figure 5D). These data suggest that the kinase activity of RIP3 is involved in regulating FFC diet-induced hepatocyte death and PA-mediated cytotoxicity and MLKL translocation to the membrane.
Liver-adipose crosstalk plays a critical role in the pathogenesis of NAFLD (24). Adipose tissue dysfunction is associated with adipose inflammation and perturbed secretome (25). Therefore, we investigated whether RIP1 and/or RIP3 kinase impacted FFC diet-mediated adipose inflammation. Crown-like structures, clusters of dead or dying adipocytes surrounded by infiltrating immune cells, are a hallmark of obesity and metabolic syndrome (26). FFC diet increased adipocyte size and the numbers of crown like structures in WT, but not in Rip1K45A/K45A or Rip3K51A/K51A, mice (Figure 6A/C). Consistent with the morphologic data, FFC diet increased expression of mRNA for cytokines (Tnf-α and Mcp-1), immune cell markers (F4/80 and Cd11c) and complement receptor C3ar to a greater degree in WT compared to Rip1K45A/K45A or Rip3K51A/K51A mice (Figure 6B/D).
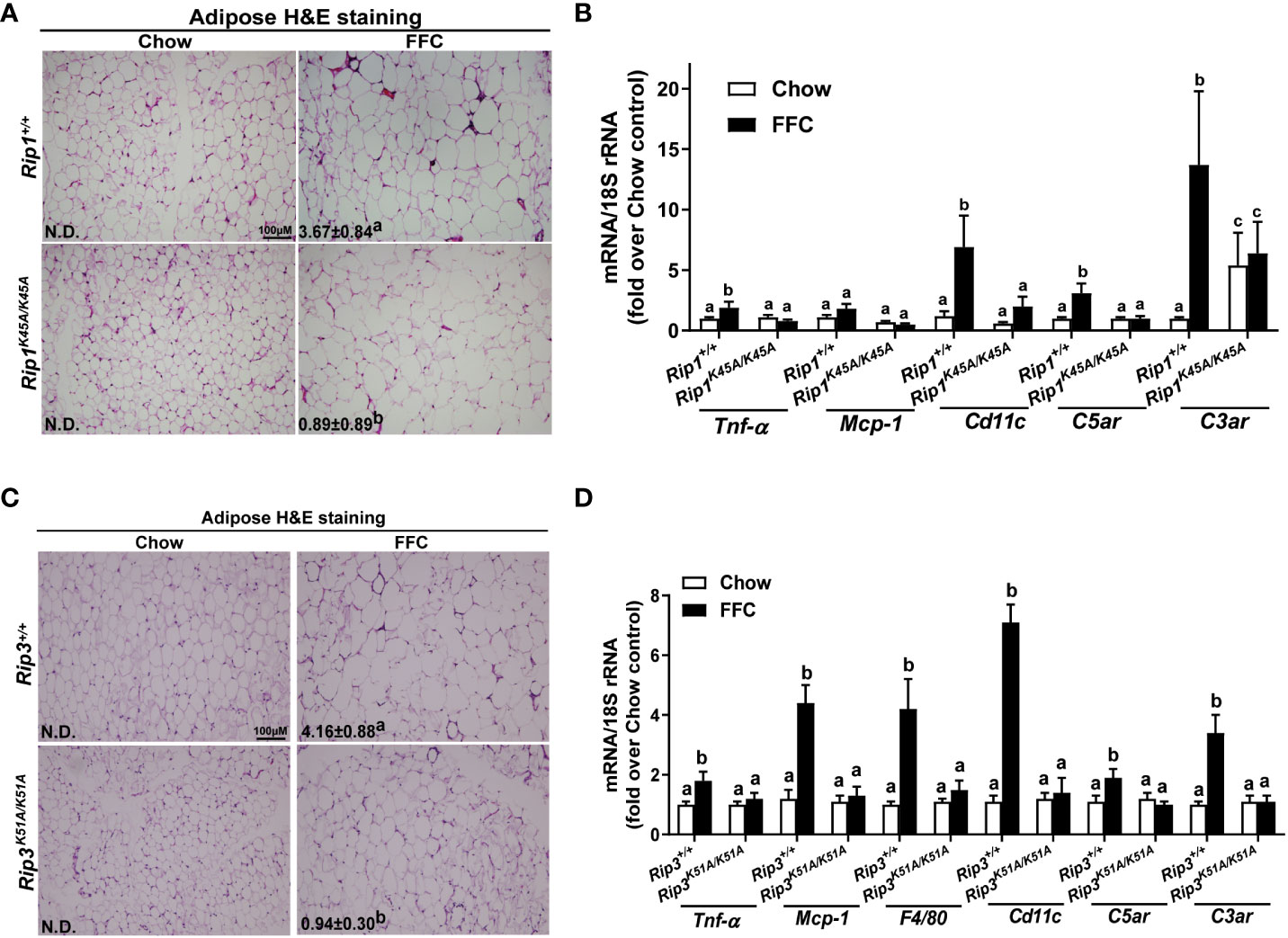
Figure 6 Absence of Rip1 or Rip3 kinase activity prevented FFC diet-induced inflammatory responses in the adipose. Mice were allowed free access to chow or FFC diet for 12 weeks and epididymal white adipose tissue collected and analyzed. (A/C) H&E staining of epididymal white adipose tissues and quantification of crown-like structures per 10X frame in (A) Rip1+/+ and Rip1K45A/K45A mice and (C) Rip3+/+ and Rip3K51A/K51A mice. (B/D) Expression of genes of interest were measured by qRT-PCR in adipose tissue from (B) Rip1+/+ and Rip1K45A/K45A mice and (D) Rip3+/+ and Rip3K51A/K51A mice. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 5-6 per group. p <0.05, assessed by ANOVA.
To further characterize the role of RIP3 kinase in generating the adipose secretome induced by FFC diet, an adipokine array was used to assess the content of multiple adipokines in circulation. Of the 38 adipokines detected (Figure 7A), leptin and plasminogen activator inhibitor-1 (PAI-1) were increased the most in FFC diet-fed WT mice; this response was abrogated in Rip3K51A/K51A mice (Figure 7B). qPCR confirmed that Rip3 kinase deficiency prevented FFC diet-induced expression of mRNA for Leptin and Serpine 1, the gene coding for PAI-1(Figure 7C). In previous studies, RIP3 can interact with PI3K/AKT signaling in mediating cellular events, including TNF-mediated cell death (27) and vascular smooth muscle cell growth (28). Moreover, TSC1/mTOR was found to control RIPK3-dependent necroptosis in intestinal inflammation and cancer (29). Therefore, we speculate that RIP3 kinase might be involved in the regulation of signaling, such as PI3K/AKT and mTOR pathways, required for leptin production and secretion from adipose tissue, both at baseline and in response to FFC diet feeding. Overall, deficiency of Rip3 kinase attenuates FFC diet-induced adipose inflammation and accumulation of adipokines in the circulation.
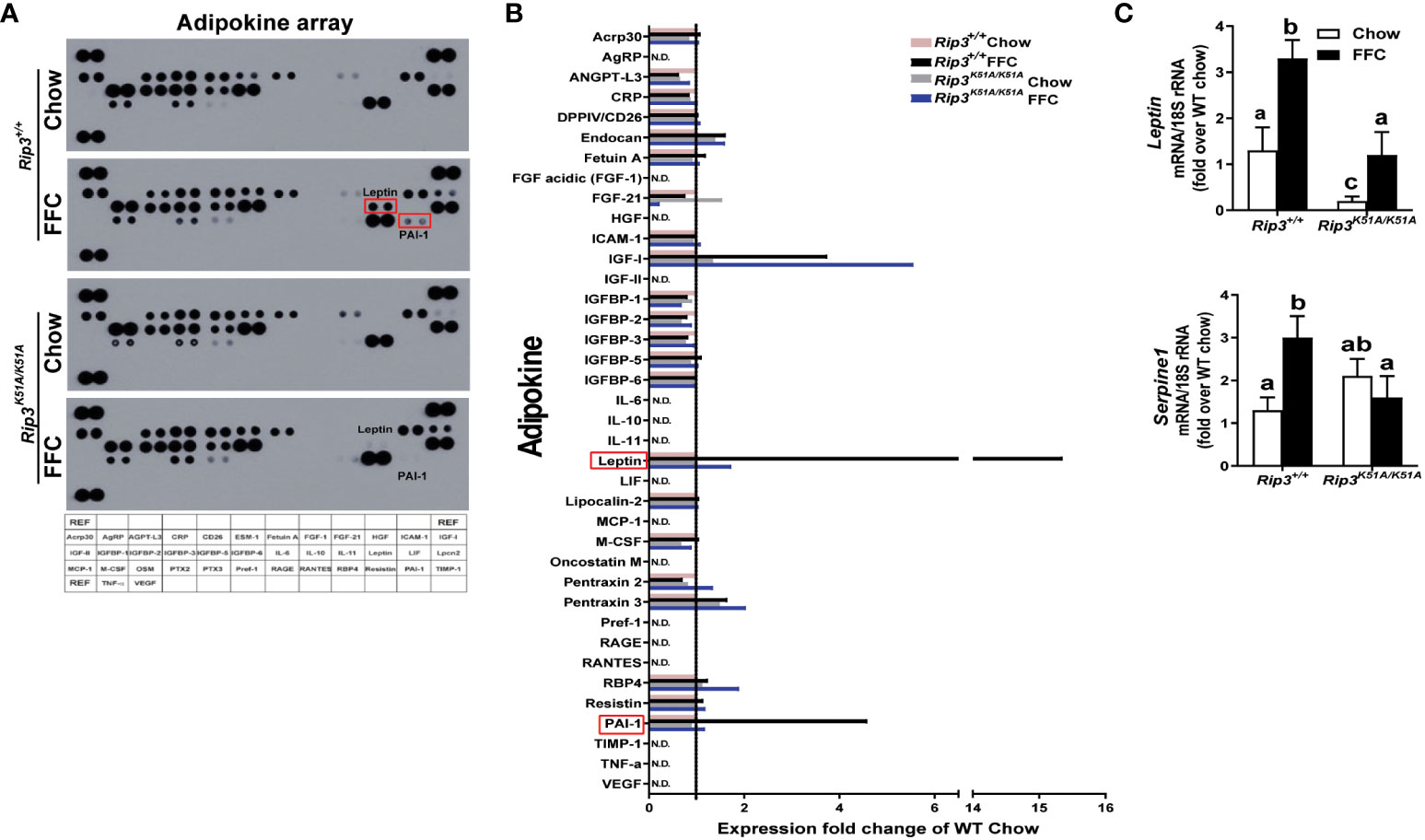
Figure 7 Plasma adipokine profiles in epididymal adipose from Rip3 kinase deficient mice. (A) Mice were allowed free access to chow or FFC diet for 12 weeks and epididymal white adipose tissue collected and analyzed. (A) Plasma adipokine profiles were assess using an adipokine array. (B) Relative density was calculated using Image J; data were normalized to reference spots, and each experimental group was expressed as fold change of chow-fed Rip3+/+ group. (C) Expression of mRNA for Leptin and Serpine-1 (PAI-1 gene) was normalized to 18S rRNA. Values represent means ± SEM. Values with different superscripts are significantly different from each other, n = 6 per group. p <0.05, assessed by ANOVA.
The prevalence of obesity has steadily increased over the past few decades and has become a global health concern. Obesity is highly associated with the development of NAFL/NASH, with up to 70-80% of obese individuals having some degree of fatty liver disease (30). It is crucial to identify potential therapeutic targets in order to prevent disease progression at early stages of injury. In the current study, we found that both RIP1 and RIP3 kinases contribute to FFC diet-induced injury by affecting liver and adipose tissue functions in mice. Therefore, therapeutics designed to inhibit RIP1 and RIP3 kinase activity in hepatocytes would likely be beneficial in reducing liver injury associated with consumption of high fat diets and associated obesity.
The contribution of RIP1 and RIP3 to metabolic liver disease is complex and likely dependent on both domain-specific and cell-specific functional activity. RIP3 protein is composed of a C-terminal receptor-interacting protein homotypic interaction motif (RHIM) and an N-terminal kinase domain. The RHIM domain and nearby regions mediate the assembly of RIP3 oligomers, providing a scaffold for the RIP3 kinase domain to self-phosphorylate and/or phosphorylate its substrate MLKL (31). Emerging evidence indicates that RIP3 and MLKL play differential roles in different dietary models of NAFL/NASH, including the FFC model (8, 13). Current data indicated that Rip3 kinase deficiency prevented FFC diet-induced liver injury. This is consistent with the protection of Mlkl-/- mice from FFC diet-induced liver injury, but not the lack of protection in Rip3-/- mice on this same diet. Taken together, these data suggest that other domains of RIP3 might counteract the effect of RIP3 kinase in response to high fat diets. Indeed, RHIM domain interactions with other RHIM domain-containing proteins, including RIP1, ZBP1 and TRIF, are a vital component of the signalling cascade and can also be involved in cell survival and death pathways in certain scenarios, particularly during viral and bacterial pathogen infection (32). The RHIM domain specific roles of RIP1 and RIP3 in NAFL/NASH need further investigation.
The necroptotic pathway is tightly regulated by multiple post-translational modifications, including phosphorylation and ubiquitination in different domains. These modifications play differential roles in cell survival and death pathways. For example, A20 inhibits K63-linked ubiquitination of RIP3 at K5 and disrupts the formation of necroptotic RIP1-RIP3 complexes (33). In addition, E3 ubiquitin ligases, including PELI1 and CHIP (targeting K363 in the intermediate domain) (34–36), and TRIM25 (targeting K501) (37), can induce K48-linked ubiquitylation of RIP3, leading to its proteasomal degradation. Apart from phosphorylation and ubiquitination, O-GlcNAc transferase (OGT)-mediated O-GlcNAcylation of RIP3 on T467 prevents RIP1/3 hetero- and RIP3/3 homo-interaction, thus dampening inflammation and necroptosis (38). Taken together these data demonstrate that different modifications in multiple domains of RIP3 can modify the role of RIP3 kinase in triggering necroptosis.
Given the distinct differences in the effect of Rip3 deficiency and Rip3 kinase inactive mutant in response to FFC feeding, we speculate that other kinases may become dominant and activate MLKL when RIP3 is completely absent. For example, in response to serum and amino acid deprivation, CAMK2/CaMKII, instead of RIP3, phosphorylates MLKL on the same sites (T357/S358 human; S345 murine) as RIP3 phosphorylation to facilitate autophagic flux (39). Members of the TAM (Tyro3, Axl, and Mer) family of receptor tyrosine kinases are also implicated in other phosphorylation sites of MLKL, include Y376 (human) and Y363 (murine) (40). In addition, recent studies reveal the role of MLKL ubiquitination in its necroptotic and non-necroptotic functions (41–43). Thus the emerging evidence indicates that the molecular mechanisms by which MLKL is activated in different conditions and cells in the presence/absence of RIP3 are complex and multifaceted.
In addition to domain-specific functions, it is also likely that cell-specific functions of RIP1-RIP3-MLKL play an important role in metabolic liver disease. For example, the RIP1-RIP3-MLKL-axis in immune cells plays a crucial role in the development of metabolic liver disease. A previous study demonstrated that RIP1 kinase activity in hematopoietic-derived macrophages contributes to hepatic inflammation and fibrosis progression in NASH (5). We also recently reported that macrophage-derived MLKL is essential for maintaining immune cell homeostasis and macrophage phagocytic function in alcohol-associated metabolic liver disease (44). While there was no difference in death of non-parenchymal cells in the liver, which include liver resident and infiltrating innate immune cells, between WT and Rip3K51A/K51A mice after FFC feeding, we cannot exclude the possibility that the RIP1 kinase-RIP3 kinase-MLKL-axis in hepatic immune cells plays a crucial role in the development of NAFL/NASH. Further studies utilizing cell-specific knockouts in the RIP1-RIP3-MLKL pathway would be beneficial to explore cell-specific roles of necroptotic cell death pathway in murine models of dietary-induced NAFL/NASH.
Domain-specific and cell-specific roles for RIP1-RIP3 may also be relevant to the impact of high fat diets and obesity in adipose tissue. RIP3 and pMLKL are induced in visceral white adipose tissue (WAT) in patients with obesity and are positively associated with body mass index and perturbed metabolic serum markers, such as HbA1c and insulin (14). However, the role of RIP3 in adipose tissue function is complex and poorly understood. Rip3 deficiency does not affect white and beige adipocyte differentiation (45). RIP3 is thought to maintain adipose tissue homeostasis and dampen inflammation by inhibiting caspase-8-dependent apoptosis of adipocytes, associated with a prevention of glucose intolerance, in the choline deficient-HFD dietary model of obesity (14). However, bone marrow transplant studies revealed that immune cell-derived RIP3 in adipose tissue does not mediate the metabolic phenotype of choline deficient-HFD-fed model (14), suggesting a cell-specific role for RIP3 in non-myeloid cells, such as adipocytes, rather than immune cells, in adipose tissue. A domain-specific function of RIP3 is also indicated, since Rip3-/- mice are more susceptible to HFD and FFC diet-induced inflammatory responses in white adipose tissue (8, 12), but, in the current study, when only the kinase activity of RIP1 or RIP3 was eliminated, FFC diet-induced adipocyte death and adipose tissue inflammation were prevented (Figure 6).
In summary, Rip1 and Rip3 kinase deficiency prevented activation of MLKL and protected mice from FFC diet-induced liver injury and metabolic perturbations. Therefore, the RIP1 kinase-RIP3 kinase-MLKL-axis may serve as a potential target of small-molecule therapeutics by inhibiting pro-necroptotic pathway in the treatment of NAFL/NASH.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
The animal study was approved by Cleveland Clinic Institutional Animal Care and Use Committee. The study was conducted in accordance with the local legislation and institutional requirements.
XW: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Writing – original draft, Writing – review & editing. RA: Investigation, Methodology, Writing – review & editing. EH: Methodology, Writing – review & editing. MM: Project administration, Supervision, Writing – review & editing. LN: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by NIH grants; P50 AA024333 (LN), R01 AA027456 (LN), U01 AA026938 (LN), R01 AA023722 (LN), K99 AA029146 (XW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1267996/full#supplementary-material
1. Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol (2017) 18:127–36. doi: 10.1038/nrm.2016.149
2. Kolbrink B, von Samson-Himmelstjerna FA, Murphy JM, Krautwald S. Role of necroptosis in kidney health and disease. Nat Rev Nephrol (2023) 19, 300–314. doi: 10.1038/s41581-022-00658-w
3. Shojaie L, Iorga A, Dara L. Cell death in liver diseases: A review. Int J Mol Sci (2020) 21:9682. doi: 10.3390/ijms21249682
4. Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F, et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J Hepatol (2020) 72:627–35. doi: 10.1016/j.jhep.2019.11.008
5. Tao L, Yi Y, Chen Y, Zhang H, Orning P, Lien E, et al. RIP1 kinase activity promotes steatohepatitis through mediating cell death and inflammation in macrophages. Cell Death Differ (2021) 28:1418–33. doi: 10.1038/s41418-020-00668-w
6. Preston SP, Stutz MD, Allison CC, Nachbur U, Gouil Q, Tran BM, et al. Epigenetic silencing of RIPK3 in hepatocytes prevents MLKL-mediated necroptosis from contributing to liver pathologies. Gastroenterology (2022) 163:1643–1657 e14. doi: 10.1053/j.gastro.2022.08.040
7. Dara L, Johnson H, Suda J, Win S, Gaarde W, Han D, et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology (2015) 62:1847–57. doi: 10.1002/hep.27939
8. Wu X, Poulsen KL, Sanz-Garcia C, Huang E, McMullen MR, Roychowdhury S, et al. MLKL-dependent signaling regulates autophagic flux in a murine model of non-alcohol-associated fatty liver and steatohepatitis. J Hepatol (2020) 73:616–27. doi: 10.1016/j.jhep.2020.03.023
9. Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology (2013) 57:1773–83. doi: 10.1002/hep.26200
10. Wang S, Ni HM, Dorko K, Kumer SC, Schmitt TM, Nawabi A, et al. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget (2016) 7:17681–98. doi: 10.18632/oncotarget.6893
11. Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med (2014) 6:1062–74. doi: 10.15252/emmm.201403856
12. Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology (2016) 64:1518–33. doi: 10.1002/hep.28676
13. Wu X, Nagy LE. MLKL contributes to Western diet-induced liver injury through inhibiting autophagy. Autophagy (2020) 16:1351–2. doi: 10.1080/15548627.2020.1760624
14. Gautheron J, Vucur M, Schneider AT, Severi I, Roderburg C, Roy S, et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat Commun (2016) 7:11869. doi: 10.1038/ncomms11869
15. Najjar M, Saleh D, Zelic M, Nogusa S, Shah S, Tai A, et al. RIPK1 and RIPK3 kinases promote cell-death-independent inflammation by toll-like receptor 4. Immunity (2016) 45:46–59. doi: 10.1016/j.immuni.2016.06.007
16. Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun (2015) 6:6282. doi: 10.1038/ncomms7282
17. Kaiser WJ, Daley-Bauer LP, Thapa RJ, Mandal P, Berger SB, Huang C, et al. RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc Natl Acad Sci U.S.A. (2014) 111:7753–8. doi: 10.1016/j.molcel.2014.10.021
18. Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell (2014) 56:481–95. doi: 10.1016/j.molcel.2014.10.021
19. Degterev A, Ofengeim D, Yuan J. Targeting RIPK1 for the treatment of human diseases. Proc Natl Acad Sci U.S.A. (2019) 116:9714–22. doi: 10.1073/pnas.1901179116
20. Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol (2014) 76:129–50. doi: 10.1146/annurev-physiol-021113-170259
21. Bugianesi E, McCullough AJ, Marchesini G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology (2005) 42:987–1000. doi: 10.1002/hep.20920
22. Shindo R, Ohmuraya M, Komazawa-Sakon S, Miyake S, Deguchi Y, Yamazaki S, et al. Necroptosis of intestinal epithelial cells induces type 3 innate lymphoid cell-dependent lethal ileitis. iScience (2019) 15:536–51. doi: 10.1016/j.isci.2019.05.011
23. Ausch C, Buxhofer-Ausch V, Olszewski U, Hinterberger W, Ogris E, Schiessel R, et al. Caspase-cleaved cytokeratin 18 fragment (M30) as marker of postoperative residual tumor load in colon cancer patients. Eur J Surg Oncol (2009) 35:1164–8. doi: 10.1016/j.ejso.2009.02.007
24. Azzu V, Vacca M, Virtue S, Allison M, Vidal-Puig A. Adipose tissue-liver cross talk in the control of whole-body metabolism: implications in nonalcoholic fatty liver disease. Gastroenterology (2020) 158:1899–912. doi: 10.1053/j.gastro.2019.12.054
25. Chait A, den Hartigh LJ, Adipose Tissue Distribution. Inflammation and its metabolic consequences, including diabetes and cardiovascular disease. Front Cardiovasc Med (2020) 7:22. doi: 10.3389/fcvm.2020.00022
26. Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, et al. Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res (2008) 49:1562–8. doi: 10.1194/jlr.M800019-JLR200
27. Hu S, Chang X, Zhu H, Wang D, Chen G. PI3K mediates tumor necrosis factor induced-necroptosis through initiating RIP1-RIP3-MLKL signaling pathway activation. Cytokine (2020) 129:155046. doi: 10.1016/j.cyto.2020.155046
28. Li Q, Li G, Lan X, Zheng M, Chen KH, Cao CM, et al. Receptor interacting protein 3 suppresses vascular smooth muscle cell growth by inhibition of the phosphoinositide 3-kinase-Akt axis. J Biol Chem (2010) 285:9535–44. doi: 10.1074/jbc.M109.071332
29. Xie Y, Zhao Y, Shi L, Li W, Chen K, Li M, et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and cancer. J Clin Invest (2020) 130:2111–28. doi: 10.1172/JCI133264
30. Lazarus JV, Mark HE, Anstee QM, Arab JP, Batterham RL, Castera L, et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol (2022) 19:60–78. doi: 10.1038/s41575-021-00523-4
31. Murphy JM, Vince JE. Post-translational control of RIPK3 and MLKL mediated necroptotic cell death. F1000Res (2015) 4. doi: 10.12688/f1000research.7046.1
32. Riebeling T, Kunzendorf U, Krautwald S. The role of RHIM in necroptosis. Biochem Soc Trans (2022) 50:1197–205. doi: 10.1042/BST20220535
33. Onizawa M, Oshima S, Schulze-Topphoff U, Oses-Prieto JA, Lu T, Tavares R, et al. The ubiquitin-modifying enzyme A20 restricts ubiquitination of the kinase RIPK3 and protects cells from necroptosis. Nat Immunol (2015) 16:618–27. doi: 10.1038/ni.3172
34. Choi SW, Park HH, Kim S, Chung JM, Noh HJ, Kim SK, et al. PELI1 selectively targets kinase-active RIP3 for ubiquitylation-dependent proteasomal degradation. Mol Cell (2018) 70:920–935 e7. doi: 10.1016/j.molcel.2018.05.016
35. Seo J, Lee EW, Sung H, Seong D, Dondelinger Y, Shin J, et al. CHIP controls necroptosis through ubiquitylation- and lysosome-dependent degradation of RIPK3. Nat Cell Biol (2016) 18:291–302. doi: 10.1038/ncb3314
36. Lee SB, Kim JJ, Han SA, Fan Y, Guo LS, Aziz K, et al. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat Cell Biol (2019) 21:940–51. doi: 10.1038/s41556-019-0356-8
37. Mei P, Xie F, Pan J, Wang S, Gao W, Ge R, et al. E3 ligase TRIM25 ubiquitinates RIP3 to inhibit TNF induced cell necrosis. Cell Death Differ (2021) 28:2888–99. doi: 10.1038/s41418-021-00790-3
38. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, et al. O-glcNAc transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity (2019) 50:576–590 e6. doi: 10.1016/j.immuni.2019.01.007
39. Zhan Q, Jeon J, Li Y, Huang Y, Xiong J, Wang Q, et al. CAMK2/CaMKII activates MLKL in short-term starvation to facilitate autophagic flux. Autophagy (2022) 18:726–44. doi: 10.1080/15548627.2021.1954348
40. Najafov A, Mookhtiar AK, Luu HS, Ordureau A, Pan H, Amin PP, et al. TAM kinases promote necroptosis by regulating oligomerization of MLKL. Mol Cell (2019) 75:457–468 e4. doi: 10.1016/j.molcel.2019.05.022
41. Garcia LR, Tenev T, Newman R, Haich RO, Liccardi G, John SW, et al. Ubiquitylation of MLKL at lysine 219 positively regulates necroptosis-induced tissue injury and pathogen clearance. Nat Commun (2021) 12:3364. doi: 10.1038/s41467-021-23474-5
42. Liu Z, Dagley LF, Shield-Artin K, Young SN, Bankovacki A, Wang X, et al. Oligomerization-driven MLKL ubiquitylation antagonizes necroptosis. EMBO J (2021) 40:e103718. doi: 10.15252/embj.2019103718
43. Yoon S, Bogdanov K, Wallach D. Site-specific ubiquitination of MLKL targets it to endosomes and targets Listeria and Yersinia to the lysosomes. Cell Death Differ (2022) 29:306–22. doi: 10.1038/s41418-021-00924-7
44. Wu X, Fan X, McMullen MR, Miyata T, Kim A, Pathak V, et al. Macrophage-derived MLKL in alcohol-associated liver disease: Regulation of phagocytosis. Hepatology (2023) 77:902–19. doi: 10.1002/hep.32612
Keywords: RIP1 kinase, RIP3 kinase, cell death, NAFLD, obesity, FFC diet
Citation: Wu X, Arya RK, Huang E, McMullen MR and Nagy LE (2023) Receptor-interacting protein 1 and 3 kinase activity are required for high-fat diet induced liver injury in mice. Front. Endocrinol. 14:1267996. doi: 10.3389/fendo.2023.1267996
Received: 27 July 2023; Accepted: 28 November 2023;
Published: 15 December 2023.
Edited by:
Elena Piccinin, University of Bari Aldo Moro, ItalyReviewed by:
Shengmin Yan, Tulane University, United StatesCopyright © 2023 Wu, Arya, Huang, McMullen and Nagy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura E. Nagy, bmFneUwzQGNjZi5vcmc=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.