 Sabine Richard
Sabine Richard Juan Ren
Juan Ren Frédéric Flamant
Frédéric Flamant
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 03 October 2023
Sec. Thyroid Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1256877
This article is part of the Research Topic The Role of Thyroid Hormones in Vertebrate Development, volume II View all 13 articles
Thyroid hormone (TH) signaling plays a major role in mammalian brain development. Data obtained in the past years in animal models have pinpointed GABAergic neurons as a major target of TH signaling during development, which opens up new perspectives to further investigate the mechanisms by which TH affects brain development. The aim of the present review is to gather the available information about the involvement of TH in the maturation of GABAergic neurons. After giving an overview of the kinds of neurological disorders that may arise from disruption of TH signaling during brain development in humans, we will take a historical perspective to show how rodent models of hypothyroidism have gradually pointed to GABAergic neurons as a main target of TH signaling during brain development. The third part of this review underscores the challenges that are encountered when conducting gene expression studies to investigate the molecular mechanisms that are at play downstream of TH receptors during brain development. Unravelling the mechanisms of action of TH in the developing brain should help make progress in the prevention and treatment of several neurological disorders, including autism and epilepsy.
Thyroid hormone (TH) signaling plays a major role in mammalian brain development (1). Any alteration in TH economy during brain development – be it TH synthesis, TH transport or activity of TH receptors (TRs) – is likely to induce long-lasting and irreversible defects, ranging from mild intellectual disability to profound physical and mental impairments. This has been known for a long time, but the precise underlying mechanisms are still unknown. Data obtained in the past years in animal models have pinpointed the GABAergic system as a major target of TH signaling during development (2, 3). GABAergic neurons, which use GABA (gamma-aminobutyric acid) as a neurotransmitter, are the chief inhibitory neurons in the vertebrate central nervous system. Alteration in the development of the GABAergic system is known to be associated with neurological disorders such as intellectual disability, autism spectrum disorder (ASD), and epilepsy (4). On the other hand, thyroid dysfunction is often associated with some of these neurological disorders, such as anxiety and seizure susceptibility (5, 6). Wiens and Trudeau (7) have previously reviewed in vitro and in vivo evidence in rodents, indicating that alterations in TH signaling may affect the GABAergic system in several ways: GABA synthesis and metabolism, GABA release and reuptake, GABA receptor expression and function, etc. However, the molecular and cellular mechanisms by which TH impacts GABAergic neurons are unknown. The aim of the present review is to gather the available information about the direct involvement of TH in GABAergic neuron maturation.
TH (either its most active form T3: 3,3’,5-triiodo-L-thyronine, or its less active precursor T4: thyroxine) binds to nuclear receptors, which are transcription factors regulating gene expression. Two genes (THRA and THRB in humans, Thra and Thrb in rodents) encode the TRα1, TRβ1 and TRβ2 nuclear receptors. While TRα1 is present at all developmental stages in many cell types and in all the rodent brain areas, TRβ1 mRNA appears later and TRβ2 is restricted to few brain areas (8). However, alternate splicing of Thra mRNA also generates the TRα2 mRNA, which encodes a non-receptor protein. Notably, in the brain, TRα2 is more abundant than TRα1 (9). Knowing that direct measurement of TR protein concentrations is difficult, due to poor antibody specificity and very low abundance of TR proteins, the respective abundance of the different TR isotypes in the different cell types remains unclear. In humans, old studies of TH binding suggest that TRβ1 is the predominant receptor in the human fetal brain (10).
More convincingly than TR expression patterns, both human and mouse genetics clearly demonstrate that TRα1 mediates crucial, significant actions of TH on early brain development, while TRβ1 and TRβ2 appear to be necessary for more specific and discreet steps of brain development (11–14). Patients who bear mutations in THRA are very likely to present significant neurological disorders such as epilepsy, motor incoordination or impaired cognitive function (15, 16). THRB mutations alter the regulation of the hypothalamo-pituitary-thyroid axis, increasing the circulating level of TH, which may in turn alter neurodevelopment. Neurocognitive impairment has been frequently associated with mutations in THRB, but the consequences on brain function appear to be less dramatic than those induced by THRA mutations (17, 18).
In the present paper, we will retrace how rodent models have helped uncover the critical role of TH/TR signaling in the differentiation of GABAergic neurons. Deciphering the network of TH/TR target genes in GABAergic neurons during brain development appears as a promising way of generating novel approaches to alleviate various neurodevelopmental disorders associated with GABAergic dysfunction, including epilepsy and ASD. Moreover, identifying TH target genes in GABAergic neurons during brain development is likely to contribute to a more general understanding of TH action in the brain.
Disruption of TH signaling may arise from very different causes affecting various aspects of TH molecular landscape (19). In mammals, TH is synthesized in the thyroid gland from tyrosine and iodine. In the blood and cerebrospinal fluid, TH is partly bound to distributor proteins, such as transthyretin, thyroxine-binding globulin, albumin and lipoproteins (20). The main source of TH for the fetus is from the mother throughout gestation, since even when the fetal thyroid starts to synthesize TH, the maternal TH remains the main source of circulating fetal TH (21). Transfer of maternal TH to the fetal brain involves crossing the placental and blood-brain barriers. TH entry into the brain is facilitated by specific transmembrane transporters, with monocarboxylate transporter 8 (Mct8) and organic anion transporter polypeptide 1c1 (Oatp1c1) playing a prominent role in TH brain transport in rodents (1). Around 80% of the T3 that is present in the brain derives from local deiodination of T4, mediated by type 2 iodothyronine deiodinase (Dio2), while the rest originates from the circulation (22). Deiodinase 3 (Dio3) degrades T4 and T3 into inactive metabolites and is thus the major physiological TH inactivator (23). As a consequence of the multiplicity of molecular actors involved from maternal TH synthesis to regulation of the expression of TH target genes, a wide variety of causes may affect thyroid hormone economy in the fetal brain (24): iodine deficiency, insufficient TH synthesis by the thyroid gland, problems in blood and cerebrospinal fluid transport of TH, abnormalities in the placental barrier, defects in membrane transport proteins, altered enzymatic activity of deiodinases, defects in cytosolic TH binding proteins, and last but not least, defects in TH receptor activity. The consequences of such defects have been previously described in the specialized literature. From this array of causes, TH deficiency during fetal and postnatal development may cause diverse kinds of neurological impairment (1). As a matter of example, we have chosen to mention here three emblematic diseases linked to poor TH signaling during development, one linked to a lack of hormone, the second to a deficit in TH membrane transporters and the third to a deficit in TH receptors.
● Congenital hypothyroidism, defined in humans as partial or complete TH deficiency at birth, can start early during development and represents the major cause of preventable intellectual disability in the world. If not detected and treated early, congenital hypothyroidism can have devastating effects on neurocognitive function. The clinical spectrum goes from mere developmental delay to persistent intellectual disability, hearing and speech impairment, psychomotor impairment, with the most severe condition being historically known as cretinism (25, 26). A major factor influencing the degree of severity of the symptoms is the time at which TH deficiency occurs, relative to brain developmental steps. In children with normal thyroid function born to hypothyroid mothers, TH starts to be synthesized shortly before birth, which allows partial recuperation after the initial developmental delay. In hypothyroid children born to euthyroid mothers, the maternal TH contribution during the latter part of gestation also provides partial compensation for the inadequate fetal TH supply. By contrast, hypothyroidism that stems in utero and that extends throughout childhood has more severe consequences (27). Newborn screening, accompanied by T4 replacement therapy, has been efficiently implemented for decades in several countries, but most newborns worldwide remain away from such protocols of screening and treatment (28). Moreover, even under T4 treatment, significant impairment in clinical and cognitive scores may persist in children with congenital hypothyroidism (29).
● Allan-Herndon-Dudley syndrome is a rare X-linked disease that affects human males with mutations in the Slc16a2 gene encoding Mct8, which is critically needed for TH to enter the human brain. Allan-Herndon-Dudley patients have a shortened life expectancy and present with physical and intellectual disability, speech deficits and severe neurological abnormalities, including, in some cases, epilepsy (30–33). Histopathological analyses have revealed that hypomyelination is the most salient feature of the brains of Allan-Herndon-Dudley patients. Notably, at the level of the cerebral cortex, López-Espíndola et al. (34) have shown that this syndrome is associated with a reduction in the numbers of parvalbumin (PV)-expressing neurons, a category of GABAergic interneurons which play pivotal roles in cortical development and function. In addition, Allan-Herndon-Dudley patients’ cerebellum displays abnormal differentiation of Purkinje cells, which are GABAergic projection neurons (34).
● Resistance to thyroid hormone receptor alpha (RTHα) is another rare disease due to mutations in THRA, the gene coding for TRα1. The clinical features of patients with RTHα are quite heterogeneous. Delayed milestones in the development of motor and speech abilities are the most common neurological symptoms (15). Notably, among the 40 reported cases to date, three had epilepsy (35–37), which significantly outweighs the incidence of epilepsy in the general population [4-10 per 1,000 people (38)]. Moreover, it has been suggested that the proportion of THRA mutations was higher in ASD patients than in the general population. Testing this possibility, Kalikiri and colleagues (39) made the astonishing discovery of seven novel THRA mutations, all likely to be pathological, in a small cohort of 30 patients with ASD in India. This adds to another case of ASD with a THRA mutation, which was previously discovered in Canada (40).
The variety of neurological symptoms associated with TH signaling defects is explained by the fact that TH influences a wide panel of cellular processes in the developing brain, such as neurogenesis, neuronal migration, neuronal and glial cell differentiation, myelination and synaptogenesis (1). Accordingly, many genes have been found to be under direct or indirect regulation by TH (41), and deciphering the precise mechanisms underlying TH action will necessarily involve isolating direct from indirect effects.
The timing of neurodevelopmental stages differs significantly between humans and rodents, rodent early post-natal stages roughly corresponding to the end of the second trimester of human pregnancy (24). However, as major steps in brain development are conserved between humans and rodents, our understanding of the role of TH in the developing brain has greatly benefited from rodent studies. Seminal work undertaken to decipher the actions of TH in the developing brain involved rat models of congenital hypothyroidism. In the past decades, genetic tools have allowed to develop mouse models precisely designed to dissect the effects of TH in specific cells of the brain. The following sections will focus on these models and their contribution to our understanding of the role of TH in GABAergic neuron development.
The morphological consequences of congenital hypothyroidism have been extensively studied and include alterations in cortical lamination, high density of hippocampal neurons, poor differentiation of the gray-white matter boundary and delayed cerebellar development (1). The following paragraphs will focus on the effects of developmental hypothyroidism on GABAergic neurons.
The relative simplicity of the microanatomy of the cerebellar cortex, as well as its strong TH signaling dependency, make it an excellent model to study the neurodevelopmental function of TH. The proliferation and migration of granule cells, which represent the vast majority of the neuronal population in the cerebellum, are stimulated by contacts with a monolayer of GABAergic projection neurons, called Purkinje cells. Congenital hypothyroidism dramatically affects the morphological maturation of Purkinje cells. The growth, dendrite arborization and dendrite spine number of Purkinje cells are markedly decreased in hypothyroid rats (42–44). The cerebellum also contains GABAergic interneurons, called basket, stellate and Golgi cells. Early studies of rat cerebellum have shown that congenital hypothyroidism delayed the postnatal increase in GABA receptor density (45), and lowered the final number of basket cells (42). More recently, Manzano et al. (46) have reported that hypothyroid rats at postnatal day (PND) 16 exhibited a decreased number of Golgi cells, as well as a delayed disappearance of the precursors of cerebellar GABAergic interneurons. Moreover, they found that on PND8, the proliferation of GABAergic interneuron precursors in cerebellar white matter was reduced in hypothyroid rats. Thus, several components of the GABAergic system are impaired in the hypothyroid rat cerebellum.
In 1996, Berbel et al. (47) were the first to describe the impact of severe congenital hypothyroidism on a subset of cortical GABAergic inhibitory neurons expressing the calcium-binding protein parvalbumin (PV). They described a striking reduction in PV-positive terminals in the neocortex of adult hypothyroid rats (47). Ten years later, Gilbert et al. (48) further showed that moderate degrees of TH insufficiency during development were sufficient to induce a significant reduction of PV fiber staining and PV cell body count in rat cortex and hippocampus at PND23. By contrast, hypothyroid rats have been shown to exhibit an increased density of calretinin neurons, another GABAergic interneuron subtype, in the dentate gyrus of the hippocampus (48). These effects were in part irreversible, since returning to a euthyroid state in adulthood only allowed partial recovery (48, 49). By means of cross-fostering and hormonal replacement studies, Gilbert et al. (48) also emphasized that the developmental window over which TH insufficiency occurred was determinant, the first postnatal weeks appearing as the most critical stages for TH influence on PV expression in the cortex and hippocampus. However, TH insufficiency that spanned the prenatal and postnatal period produced more profound deficits in PV staining than postnatal insufficiency alone. Of note, adult-onset hypothyroidism did not appear to impact the expression of PV in the cortex and hippocampus (48). Last but not least, the number of GABAergic neurons in the cortex and hippocampus was not altered by congenital hypothyroidism, indicating that the decrease in PV staining resulted from an alteration in phenotypic expression of PV, rather than neuronal loss (48). This was later confirmed by showing that hypothyroid rats did not differ from controls in the number of cells that expressed a GABA-synthesizing enzyme, GAD67 [glutamic acid decarboxylase 67 aka GAD1; (50)]. Intriguingly though, the expression of another GAD isotype, GAD65 (aka GAD2), was significantly reduced in both neuronal somata and processes in the hippocampus of the same hypothyroid rats (50). In a separate study, it was also found that the protein levels of GAD67 were lower in the medial prefrontal cortex of hypothyroid, compared to control, rats (51). In rats, but not in mice (52), a defect in neuronal migration causes heterotopia, i.e. the accumulation of gray matter in the corpus callosum (53). Of note, a few GABAergic neurons have been identified within the heterotopia, even though they constitute a minority of the heterotopic cells.
Electrophysiology studies of the dentate gyrus of the hippocampus in adult rats that were hypothyroid during development, have revealed that alterations in GABAergic interneuron populations were associated with functional deficits in inhibitory synaptic transmission (48, 54). Accordingly, in the hippocampus of hypothyroid rats at PND15, there was a near 80% reduction in KCC2 protein, a neuron-specific K+/Cl- cotransporter that is a key player in determining the response of excitatory neurons to GABAergic neurotransmission (50).
Mutations of proteins of the TH signaling pathway, notably TRs and TH transporters, often cause defects in GABAergic neuron differentiation in the cerebellum, cortex, hippocampus and other brain regions (Table 1).
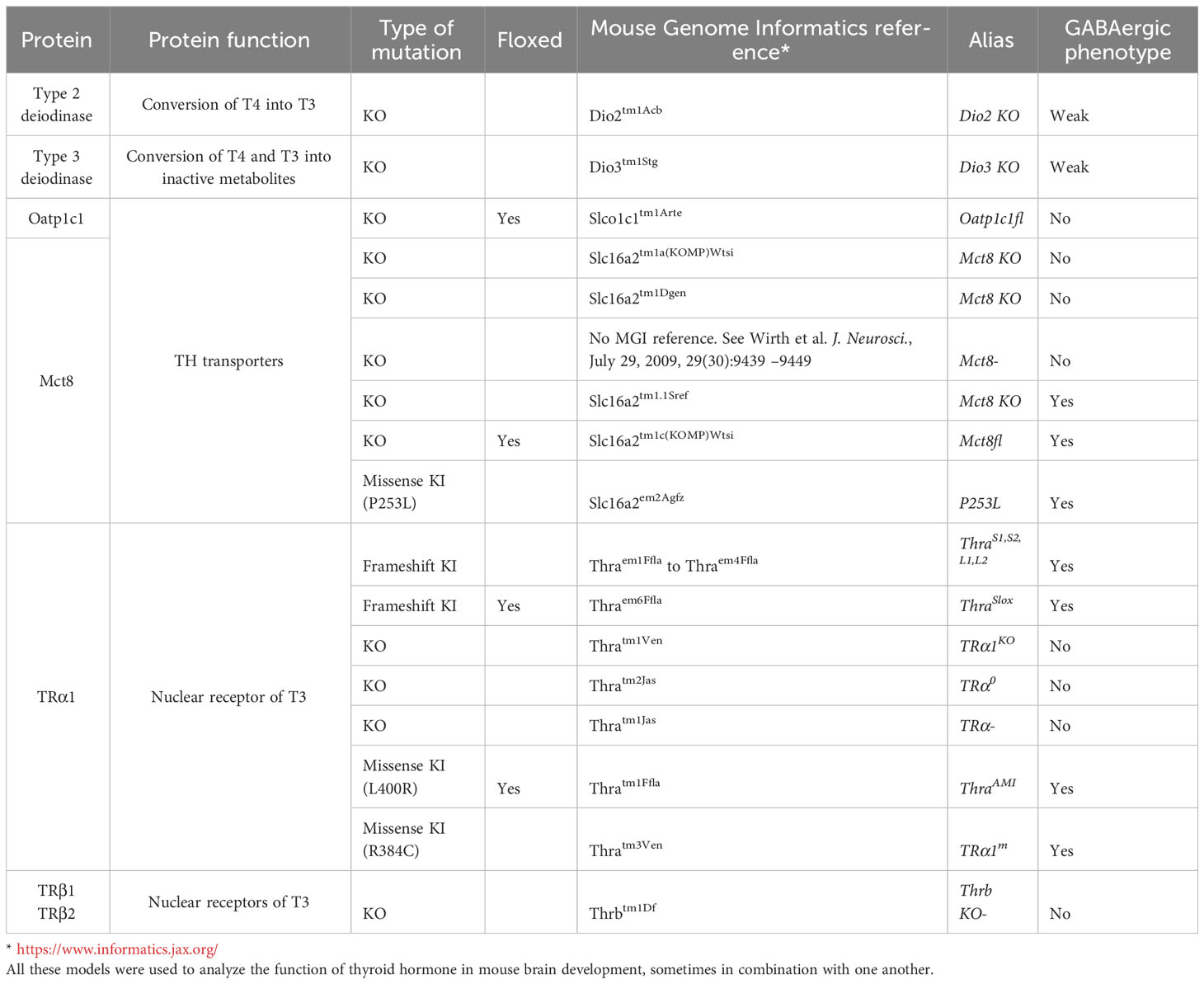
Table 1 Mouse models with gene mutations that are cited in the present paper.
In several instances, it was found that mutant mice expressing a Thra or Thrb knock-in mutation exhibited stronger phenotypes than mice in which Thra or Thrb had been knocked out. This results from a number of reasons, notably that (12)some of the knock-in mutated alleles encoding for TRs exert dominant-negative activity: they prevent the normal function of intact TRs that are still present in cells. Therefore, some germline Thra knock-in mutations have particularly drastic effects on brain development, even in heterozygous mice (55). Indeed, dominant-negative receptors constitutively interact with corepressor proteins, and thus permanently repress the expression of TR target genes, whether TH is present or not. Moreover, it has been observed that Thra knock-in mutations, encoding for dominant-negative forms of TRα1, do not only affect the transcription of TRα1 target genes, but also induce a repression of known TRβ1 target genes, thus strengthening the impact on the resulting phenotype (56). However, the main explanation for the mild neurological consequences of KOs is that getting rid of TRs does not only eliminate TH-induced activation of gene expression, but also eliminates the transcriptional repression mediated by unliganded or mutant receptors (12, 57).
Mice with a dominant-negative Thrb allele exhibit severe neurological deficiencies, notably a marked impairment in balance and coordination, and profound defects in cerebellar development, notably in the number and arborization of cerebellar Purkinje cells (58, 59). By contrast, no reduction in Purkinje cell number was found in Thrb KO mice, which appeared to exhibit normal neurological development, with the exception of a loss of auditory function (60, 61).
In the cerebellum of TRα1 KO mice, there was no apparent defect in granule cell migration, nor in Purkinje cell morphology (12), but further analyses revealed a reduced number of GABAergic interneuron precursors between PND4 and PND10, a reduced rate of proliferation of GABAergic interneuron precursors in the white matter at PND6 as well as a reduced expression of a GABAergic transporter (GAT-1) at PND11 (46)
The development of the cerebellum was also found to be significantly impaired in Dio3-/- mice, in which the intracellular TH content is increased (62). Notably, Dio3-/- mice exhibited accelerated expansion of the molecular layer, which contains the dendritic tree of Purkinje cells. However, the number of Purkinje cells did not differ between Dio3-/- and control mice. Notably, the expansion of the molecular layer follows a normal timing in Dio3-/- TRα1-/- double KO (DKO) mice, indicating a role for TRα1 in mediating the action of TH on Purkinje cell maturation.
In knock-in mice expressing TRα1R384C, which are characterized by a 10-fold reduction in the affinity of TRα1 to TH, there was an overall delay in the development of the cerebellum. Cerebellar Purkinje cells showed a delayed, but otherwise normal, arborization (2). At PND9, the expression of PV, calbindin and calretinin was lower in mice expressing TRα1R384C than in control mice, but these differences were normalized a few days later (63). By PND21, the structure of the cerebellum was similar in mice expressing TRα1R384C and in control littermates. Notwithstanding, adult mice expressing TRα1R384C showed reduced motor performance on the Rotarod. T3 treatment during PND10-PND35 resulted in complete normalization of their locomotor behavior as adults. By contrast, T3 treatment in adults did not improve performance (2), indicating the existence of a specific time window for the action of TH/TRα1 signaling on brain development.
A series of mouse models mimicking human THRA mutations resulted in various degrees of alteration of the molecular functions of TRα1 (64). However, these mice exhibited little defects in cerebellar histology, the most notable defect being a slight reduction in the density of PV-expressing GABAergic interneurons in the molecular layer. The mild phenotype of these mice with frameshifts produced by genome editing contrasts with the severity of the phenotype of previously used Thra knock-in mice. Further investigations have revealed that the elimination of alternate splicing in these knock-in mice increased the expression level of the mutated TRα1 receptor and the severity of the phenotype (64).
In mice lacking both TH transporters, Mct8 and Oatp1c1 (Mct8/Oatp1c1 DKO mice), TH signaling in the brain is significantly reduced and the arborization of the dendritic tree of Purkinje cells is significantly delayed, due a defect in intraneuronal transport of TH (65).
Finally, Amano et al. (66) have reported transient postnatal cerebellar defects, including alterations in granule cell migration and in Purkinje cell electrophysiological properties, in a mouse model of hypothyroidism, Duoxa-/- KO mice, which lack a dual oxidase that is essential for thyroid hormone synthesis. In particular, despite the fact that cerebellar histology returned to normal on postnatal day 25, motor coordination was still impaired at that age in Duoxa-/- mice, suggesting irreversible behavioral defects in these mice.
In 2008, Wallis et al. (63) provided a detailed histological study of different subtypes of cortical GABAergic interneurons in knock-in mice expressing TRα1R384C. Consistent with what had previously been described in hypothyroid rats, they found a developmental delay in the appearance of PV immunoreactive neurons in these mutant mice. An electrophysiological investigation of the PND19-PND21 cortex of mouse pups expressing TRα1R384C revealed a 10-fold reduction in fast spiking neurons compared to controls. This was in line with the results of the immunohistochemical study, since many cortical PV immunoreactive cells are fast spiking neurons. At adult stages though, the density in PV neurons did not significantly differ between mutant and control mice (63, 67).
PV-expressing neurons were not the only GAABAergic neuron subtype found to be impacted by impaired TH signaling. Indeed, in mice expressing TRα1R384C, the density of calretinin-positive neurons in the cortex was significantly increased in adult mutant mice, compared to control mice. Regarding calbindin immunoreactivity, the authors described different results depending on the cortical layers under study: while mutant mice exhibited a lower density of calbindin-positive cells in layers II-III, there were no significant differences between mutant and control mice in layers IV-VI. The population of cortical somatostatin-positive neurons did not differ between mutant and control mice. Moreover, the total number of GABAergic cells in the cortex, as assessed by GAD67 immunoreactivity, did not differ significantly between mutant and control mice, indicating that the proliferation and migration of cortical GABAergic neuron precursors was unaffected by the mutation. As a whole, these results indicated that impaired TH/TRα1 signaling impacted the maturation of several populations of cortical GABAergic neurons, but the effects differed depending on the subtype of GABAergic neuron under study (63).
In an attempt to rescue the expression of PV in young mice expressing TRα1R384C, Wallis et al. (63) treated mutant mouse pups with TH between PND11 and PND13, but this failed to induce the expression of PV in the short term. By contrast, PV expression was restored in PND14 mice expressing TRα1R384C that were exposed to high levels of TH from around birth. This suggests that TH/TRα1 signaling does not directly regulate PV expression, but rather influences the cell maturation process in a broader way.
Blocking TH entry into the brain also severely compromises the differentiation of cortical GABAergic interneurons. Indeed, Mayerl et al. (65) have recorded significantly reduced PV and Gad67 immunoreactivity (Gad67 being used as a marker of all GABAergic interneurons) in the somatosensory cortex of 12-day old and adult Mct8/Oatp1c1 DKO mice, indicating that these defects were not transient, but permanent. In addition, they observed in the somatosensory cortex of adult, but not 12-day old, mice, a significant increase in the density of calretinin neurons. These results are congruent with those obtained in mice expressing TRα1R384C, in which the affinity of TRα1 for TH is significantly reduced (63). In agreement with the previous results, in Mct8/Dio2 DKO mice, PV expression in cortical neurons was also found to be significantly reduced until adulthood (67). Moreover, several classes of GABAergic interneurons were found to be affected in mice expressing a mutated Mct8 transporter (P253L) mimicking a mutation found in human patients: in the cortex of these mice at adult stage, a decreased density of PV-, calbindin- and GAD65/67-positive neurons, as well as an increased density of calretinin-positive neurons, were reported (68).
TRα1-/- mice showed reduced PV perisomatic terminals on hippocampal CA1 pyramidal neurons, compared to controls (69). These structural defects were associated with poor performance in hippocampal-dependent behavioral tasks.
Likewise, in adult mice expressing TRα1R384C dominant negative receptor, the number of PV-positive cell somata and the density of PV-positive terminals in the CA1 region of the hippocampus were found to be significantly reduced, compared to control mice (2). Fast-spiking PV-expressing interneurons are involved in the generation of rhythmic network oscillations in the gamma frequency range, which play an important role in higher processes in the brain, such as learning, memory, cognition and perception (70, 71). Extracellular field recordings from the stratum pyramidale in hippocampal slices (63) showed that the gamma oscillation frequency (20–80 Hz) was significantly lower in mutant mice expressing TRα1R384C, compared to controls, which was congruent with the reduced number of PV-expressing neurons (63). Moreover, hippocampal pyramidal neurons from mice expressing TRα1R384C showed hypoexcitablility, compared to those of control mice (72). The notable impairments in the maturation of GABAergic neurons in knock-in mice expressing TRα1R384C led the authors to suspect that these mice might be more susceptible to epilepsy than control mice. Unexpectedly, they were found to present a marked resistance to pentylenetetrazole-induced seizures, compared to control mice (72). Accordingly, pentylenetetrazole induced a significant increase in neuronal activity in the hippocampus of control mice, but not of mice expressing TRα1R384C. This phenotype was likely due to altered chloride homeostasis in principal neurons of mutant mice. In normal mouse neurodevelopment, GABAergic transmission is excitatory at early postnatal stages. During the second and third weeks of life, changes in the expression of chloride channels in principal neurons lead GABAergic transmission to switch from excitatory to inhibitory (73). Since Hadjab-Lallemend and colleagues (72) have found that mice expressing TRα1R384C exhibited an imbalance in chloride channel subtypes in principal neurons, it is suspected that in these mice GABAergic transmission is maintained in an immature state, i.e. excitatory, until adulthood. Exposure to high levels of TH during both embryonic and postnatal developmental periods combined but not in adulthood, allowed to normalize the seizure behavior observed in these mutant mice (72).
The brain defects observed in mice expressing TRα1R384C were also accompanied by significant changes in hippocampal-dependent behavior, indicative of increased anxiety and impaired memory (2). Interestingly, most of these behavioral defects, together with the structural defects in the hippocampus, could be reversed by exogenous administration of a high dose of TH for 12 days in adulthood. By contrast, the anxiety and memory defects observed in adulthood could not be prevented by an early TH treatment (between PND10 and PND35) (2). The latter results on mice expressing TRα1R384C are at odds with the clinical observation that many of the defects induced by altered TH signaling on brain development are irreversible unless they are treated early in life. They underscore the complexity of TH action in the brain, and the necessity to get a better knowledge of the timely action of TH in different brain regions.
Intriguingly, a short-term treatment of knock-in mice expressing TRα1R384C with a GABA receptor antagonist (pentylenetetrazol) rescued their memory performance, and this was accompanied by histological and electrophysiological changes reflecting an increase in the local excitatory drive in the CA1 region of the hippocampus (74).
In mice expressing a mutated Mct8 transporter (P253L), histological analysis of the hippocampus revealed defects in GABAergic interneuron populations that were similar to those previously described in the cortex: decreased density of PV-, calbindin- and GAD65/67-positive neurons, as well as increased density of calretinin-positive neurons (68).
Mittag et al. (75, 76) have described a population of PV-expressing neurons in the mouse anterior hypothalamus, which requires prenatal signaling via both TRα1 and TRβ isoforms for proper development. These neurons are involved in the central autonomic control of blood pressure and heart rate, and are also temperature-sensitive. As hypothyroidism in humans is associated with bradycardia (77), it is conceivable that these effects are mediated by PV-expressing hypothalamic neurons. However, to our knowledge, there is no evidence that this population of PV-expressing neurons is GABAergic, as Laing et al. (78) have recently reported that anterior hypothalamic PV-expressing neurons in mice are glutamatergic.
Rodent models of hypothyroidism as well as classical knock-in and KO mouse lines, as reviewed in the preceding paragraphs, have shed light on the complex influence of thyroid hormone on brain development, with GABAergic neurons appearing as particularly sensitive to impaired TH signaling. The advent of conditional mutagenesis, allowing to alter TH signaling in specific cell types, has allowed to get more insight into the brain cell types in which TH has a direct action.
ThraAMI allele encodes for a dominant-negative version of TRα1 (TRα1L400R), which is expressed only in cells where Cre recombinase is present (55). Ubiquitous expression of TRα1L400R was shown to induce a severe phenotype, leading to death around the 3rd or 4th week of life (55). The same level of severity was observed when TRα1L400R was expressed exclusively in brain cells (79). A detailed histological analysis of the cerebellum in these mice has revealed profound alterations in neuronal and glial differentiation, which were reminiscent of congenital hypothyroidism, including a strong reduction in the size and density of Purkinje cell arborization, a delay in GABAergic interneuron maturation, a delay in the migration of granule cell progenitors and abnormal Bergmann glia maturation (80). Crossing ThraAMI mice with mice expressing Cre in specific cerebellar cell types allowed to carry out a genetic dissection of the effects of TH in the developing cerebellum (81). The principal targets of TH in the cerebellum proved to be Purkinje cells, GABAergic interneurons, oligodendrocyte precursor cells and Bergmann glia (79, 82). Strikingly, the migration of granule cell precursors was altered when TH signaling was blocked specifically in Bergmann glia, or in Purkinje cells and GABAergic interneurons, but not in the least when TH signaling was blocked in granule cells themselves. Similar observations have been made in mice expressing a dominant-negative TRβ receptor in cerebellar Purkinje cells. Indeed, these mutant mice exhibited delayed Purkinje cell dendrite arborization, as well as delayed granule cell migration (83). Collectively, these results indicate that the defect in radial migration of granule cell precursors, which is a typical hallmark of the hypothyroid cerebellum, is not a cell-autonomous consequence of the lack of TH signaling, but rather results from an alteration of granule cell precursor environment (79).
Mice expressing a mutated dominant negative TRα1 receptor in all GABAergic neurons (either TRα1L400R or TRα1E395fs401X) were found to present epileptic seizures as early as 11 days of age (3). At two weeks of age, the maturation of GABAergic neurons of different types (PV-, somatostatin-, NPY- or calretinin-expressing cells) appeared to be severely impaired, in the cerebellum as well as in the cortex, hippocampus and striatum. In particular, the density of PV-expressing neurons was drastically reduced in all these brain areas. Most of these mice died before the end of the 4th week of life. The mice that survived until adulthood exhibited signs of hyperactivity and the defects in GABAergic neurons were still present. Notably, there was no normalization of PV expression over time (3). This was the first demonstration that TH signaling has a cell-autonomous effect influencing the maturation of GABAergic neurons, and that this developmental effect has lifelong consequences.
Mice expressing a dominant-negative TRβ receptor in cerebellar Purkinje cells were found to exhibit significant impairment in altered long-term synaptic plasticity at parallel fiber–Purkinje cell synapses in adulthood, even though there was no abnormality in the morphology or basal properties of these synapses at this age (84). These results stress the importance of TH action during neural development in establishing proper cerebellar function in adulthood, even if cerebellar morphology appears to be normal.
Conditional mutagenesis was also used to abolish TH transporter expression specifically in progenitors of PV interneurons [Mct8 fl/fl; Oatp1c1 fl/fl; Nkx2.1Cre mice (85)]. This induced a reduction in the density of PV+ interneurons, as well as an increase in the density of calretinin-positive neurons in the somatosensory cortex of 12-day old pups. These results clearly point to PV-expressing neurons as direct targets of TH signaling during development. However, cell numbers normalized in the adult conditional KO mice, whereas these changes were sustained at later time points when the same transporters were knocked out ubiquitously (Mct8/Oatp1c1 DKO mice), indicating that the influence of TH on PV neuron maturation relies not only on cell-autonomous effects, but also on TH signaling in other cell types (85). As Sonic hedgehog (Shh) signaling pathway in the medial ganglionic eminence is known to play a key role in determining the fate of PV neuron progenitors (86), the level of activation of this pathway was assessed in ubiquitous Mct8/Oatp1c1 DKO mice and in conditional Mct8 fl/fl; Oatp1c1 fl/fl; Nkx2.1Cre mice. At early stages of brain development, i.e. E12.5, it was found that Shh signaling was significantly reduced in the medial ganglionic eminence of Mct8/Oatp1c1 DKO mice, but not in conditional Mct8 fl/fl; Oatp1c1 fl/fl; Nkx2.1Cre mice. In other words, Shh pathway in PV neuron progenitors was impacted when TH transporters were knocked out ubiquitously, but not when they were knocked out specifically in PV neuron progenitors. This indicates that non-cell autonomous mechanisms must relay the influence of TH on Shh signaling pathway in PV neuron progenitors of the medial ganglionic eminence (85).
As a conclusion, the current understanding is that what was initially found in the cerebellum also holds true in the rest of the brain: TH acts directly on a limited number of cell types, notably GABAergic neurons, but its influence propagates to other cell types through intercellular communication, notably via neurotrophins (79, 82).
Since RNA-seq has advantageously replaced microarray analysis, a growing number of datasets of gene expression linked to TH signaling has accumulated [reviews in (87) and (41)]. These results are theoretically suitable for identifying genes which are putative TR target genes in GABAergic neurons and identifying the molecular mechanisms that lead from TH stimulation to neuronal maturation. However, this remains a difficult task. To start with, although early studies have shown that a few genes, notably Hr and Klf9, are T3-responsive in many cell types (88, 89), more recent studies have mainly demonstrated that the repertoire of TH-responsive genes widely varies across cell types and brain areas (41). Thus, it finally appears that the overlap between sets of TR target genes in different types of cells might be limited. As regards developing GABAergic neurons, this implies that TH might not play the same role in cortical fast-spiking parvalbumin neurons, striatal medium spiny neurons or cerebellar Purkinje cells, to mention a few.
In spite of continuous advances in gene expression analysis techniques, identifying true TR target genes in developing GABAergic neurons remains challenging, for a number of reasons. One major issue is to handle the extreme cellular heterogeneity of the brain. As GABAergic neurons represent a minority of the cell population in most brain areas, the response to TH in GABAergic neurons is often masked by the response to TH in other cell types. The striatum is a favorable exception, as it is mainly populated by GABAergic medium spiny neurons (3, 90). For brain areas where GABAergic neurons are less abundant, RNA sequencing can be advantageously coupled to cell sorting in order to analyze gene expression levels in a specific cell type.
Several criteria should be fulfilled for a gene to be considered as a direct TR target gene. First, if one considers that TRs are essentially transcription activators, their target genes are expected to be down-regulated in the brain of hypothyroid mice or in the brain of mice carrying mutations that impair TH signaling (74, 91–93). Gene expression analyses in a variety of mouse models with impaired thyroid hormone signaling have confirmed that TR KO mice have an attenuated phenotype compared to hypothyroid mice, which is in agreement with a potent role of unliganded TRs in the repression of gene expression (13).
Second, when comparing gene expression levels between different conditions in a given brain region, one must take into account that cell composition may differ between conditions. In the analyses of mixed cell populations, like the whole cortex, whole striatum (3) or primary neuronal cell cultures prepared from fetal cortex (94) or from post-natal cerebellum (95), a decrease in the abundance of a GABAergic neuron-specific mRNA caused by hypothyroidism or by a genetic mutation is not sufficient to conclude that TH directly regulates the transcription of this gene within GABAergic neurons. An alternative explanation is that the long-term alteration of TH signaling has modified the composition of the cell population in hypothyroid/mutant mice, and that GABAergic neurons are under-represented in the brain area under study, when compared to control mice. A way to circumvent this problem is to analyze gene expression levels a few hours after treating mice with TH, and combine these results with those obtained in hypothyroid and mutant mouse groups. Considering genes that are upregulated shortly after TH treatment and downregulated in mice with impaired TH signaling tightens the analysis around potential TR target genes, while avoiding secondary effects due to tissue reorganization.
A third difficulty in determining the direct influence of TH/TR signaling on the transcription of a given gene in GABAergic neurons is to rule out effects that are downstream of TH signaling. Indeed, in many genetically modified animals as well as in models of pharmacologically-induced hypothyroidism, general growth and development are significantly affected, so that it is likely that there are additional factors, secondary to TH signaling disruption, that contribute to the neurological status and neuroanatomical integrity. Thus, changes in gene expression that are recorded in GABAergic neurons may be secondary to an extracellular event, such as induction of neurotrophin secretion by T3 by neighboring cells (96). Primary cell cultures constitute a way to reduce interactions between neighboring cell types (94, 95). One of the most powerful methods to investigate what is going on in a specific cell type in vivo relies on Cre/loxP recombination, which allows the mutation of specific genes in specific cell types. This approach was used to study gene expression in the striatum of ThraAMI/gn mice, in which the expression of the dominant-negative TRα1L400R mutant receptor selectively abrogates response to TH in GABAergic neurons. As a whole, the putative direct TR target genes identified in that study did not highlight a specific pathway, but rather illustrated that TH signaling in GABAergic neurons is likely to affect a wide variety of functions such as cellular interactions, axon pathfinding or electrical and synaptic activity of the cell (3).
Finally, the cell-autonomous response of gene expression, as identified by RNA sequencing data from cell type-specific mutant mouse models, is not a full demonstration for a TR-mediated transactivation. Indeed, the effect of TH on a given gene can also be secondary to an intracellular event. For example, it can result from the TH-induced upregulation of a transcription activator. Although a time-course analysis following short-term TH treatment helps to recognize genuine TR target genes, one of the best current indications for a direct transcriptional activation relies on chromatin analysis. In the striatum, the expression of a tagged TRα1 expressed only in GABAergic neurons has allowed to address the occupancy of chromatin at a genome-wide scale. This has led to the conclusion that, although thousands of genomic sites are occupied by TRα1, the number of genes that are transcriptionally activated by the ligand-activated receptor is surprisingly small (3). Atac-seq analysis may be used to identify TH-induced changes in chromatin compaction, which indirectly inform of TR occupancy. One of the main advantages of this technique is that it can be efficiently implemented even when starting with small cell numbers (97).
Up to now, the analysis of TR target genes has failed to provide a unified picture of the influence of TH in GABAergic neurons. However, it is clear that many genes identified as TH-responsive in primary cultures of cortical neurons are related to the radial migration and terminal differentiation of cortical GABAergic interneurons (94). Gene expression analyses have also provided interesting working hypotheses, some of which have been tested in in vitro systems. Thus, it has been shown that a crosstalk between the signaling pathways mediated by TH on the one hand, and by αvβ3 integrin on the other hand, seems to play an important role in postnatal dendritic arborization of Purkinje cells (98). Another notable example is the demonstration that an up-regulation of Klf9 by TH is a key event in the postnatal loss of the regenerative capacity that Purkinje cells display after axotomy (99).
When analyzing genome-wide datasets, a special attention has often been paid to the Pvalb gene, which encodes PV. Indeed, histological analyses have consistently shown a reduction in PV expression in rodent models with altered TH signaling (see section 3 in the present review). In a microarray analysis conducted in the cortex and striatum of 21-day-old hypothyroid mouse pups, Pvalb came out as one of the most strongly downregulated genes (13). Even subclinical hypothyroidism was shown to induce a large fold decrease in Pvalb mRNA expression in 14-day-old rats (100). Moreover, Pvalb mRNA levels in the cortex and striatum were significantly lower in TRα1-/- and TRα1-/- TRβ1-/- mouse pups than in control mice. By contrast, Pvalb mRNA levels were not affected in TRβ1-/- mice, suggesting a predominant implication of TRα1 in mediating the effects of TH on PV expression (13). Finally, in ThraE403X/E403X mice, which express the first Thra mutation that was discovered in a patient, RNA-sequencing analysis has shown that genes such as Flywch2, Pvalb, and Syt2, which are preferentially expressed in PV-expressing neurons, were downregulated compared to control mice (93). As a conclusion, Pvalb expression is significantly reduced in mouse models with altered TH signaling, but up to now there is no convincing evidence that Pvalb is a direct target gene of TRs.
Even if the rodent brain is widely used as a model to decipher what is going on in the human brain, the brains of rodents and primates differ in several ways and studies in the human brain, when available, are extremely valuable to help translating the rodent data to the clinic. Notably, in the primate cortex, GABAergic neurons account for about 20% of the total neuron population, whereas in rodents, this percentage is about 15%. This difference is mainly due to an increase in the calretinin-expressing interneuron population. It is thought that the increased interneuron population is related to the increased associative functions and connectivity of the primate cortex, compared to the rodent cortex (101). Another major difference between the mouse and human brains relates to TH transporters in the blood-brain-barrier: in mice, both Mct8 and Oatp1c1 play a role in TH entry into the brain, whereas Oatp1c1 is not present in the human blood-brain-barrier. This explains why disruption of the Mct8 gene in mice does not result in neurological impairment, while it has severe consequences in humans (101, 102).
Datamining in single cell RNA-seq studies was performed for the human fetal cortex at gestational weeks 16–18, equivalent to mid gestation in rodents (103). Although a similar analysis has not been performed in mice, it seems that the expression pattern of the main components of TH signaling is not the same in human and rodents. In particular, some cells of the human GABAergic lineage express THRB at this early stage, whereas it appears that Thrb expression is induced at later stages in the mouse brain (8). More precisely in humans, THRB expression is predominant in the subpopulation of GABAergic neuron progenitors migrating from the caudal ganglionic eminence and in calretinin-expressing interneurons that derive from these progenitors. This raises the interesting possibility that THRB mutations selectively alter calretinin-expressing GABAergic interneurons in the human cortex. Moreover, SLC16A2 (encoding Mct8 transporter) and THRA show widespread expression in most human cortical cell types.
GABAergic neurons, and notably PV-expressing neurons, are a main target of TH signaling during brain development. Although the precise instrumental role of TH in these neurons remains elusive, a widely accepted working hypothesis considers that TH promotes the transition from the embryonic to adult pattern of gene expression in the brain (94, 104). For example, TH is involved in triggering the loss of axon regenerative capacity in Purkinje cells (99), such loss of axon regenerative capacity being a hallmark of brain maturation in rodents (105). At a wider scale, it is tempting to speculate that TH is involved in the regulation of critical periods of heightened plasticity in the brain (106). In agreement with such hypothesis, blocking TR signaling specifically in GABAergic neurons was found to significantly impair the development of perineuronal nets, which constitute a specialized extracellular matrix enwrapping mature PV-expressing neurons (3). Thus, TH signaling might trigger the setting-up of perineuronal nets, which stabilizes neuronal networks after taking into account the input from environmental stimuli (107). Such mechanisms are critical for proper brain development.
GABAergic neurons are fundamental for maintaining the balance between excitation and inhibition throughout the brain (108). In particular, PV-expressing neurons play key roles in the coordination of neuronal networks and associated oscillations (70). Thus, as impaired TH signaling during brain development significantly affects GABAergic neurons in general, and PV-expressing neurons in particular, this may account for many of the neurological disorders seen in patients with impaired TH signaling. Indeed, as was previously mentioned, studies that were carried out in in the last ten years have revealed that patients with THRA mutations display a high risk of epilepsy and ASD (Section 2 of the present review).
Besides being highly sensitive to altered TH signaling, PV-expressing neurons appear as an important node in many neurodevelopmental disorders, including ASD and epilepsy. ASD is a multifactorial neurodevelopmental disorder that encompasses a complex and heterogeneous set of traits. One unifying explanation for the complexity of ASD may lie in the disruption of the balance between excitatory and inhibitory circuits during critical periods of development (109), which echoes our current understanding of TH action in GABAergic circuits. Moreover, post-mortem studies of the cerebral cortex of ASD patients have revealed that the number of PV-expressing interneurons was decreased, and that Pvalb was the most strongly downregulated gene, compared to control patients (110, 111). Finally, Berbel and collaborators (112) have highlighted that brain morphological changes observed in mouse models of developmental hypothyroidism, such as alterations in cortical lamination, high neuronal density in several hippocampal layers, poor differentiation of the gray-white matter boundary or neuronal heterotopias, resembled the brain lesions of children with autism. The same authors noted that a large number of genes that have been found to be TH-regulated at the transcriptional level in rodent cerebral cortex have also been found to be mutated in ASD patients (112, 113).
As for ASD, epilepsy encompasses a group of multifactorial diseases, suggesting that diverse genetic or environmental insults may impair common pathways, leading in the end to symptoms of epilepsy. Again, PV-expressing neurons might be at the crossroads of these common pathways. Indeed, impaired development or function of PV-expressing interneurons has been associated with some genetic forms of epilepsy in humans (114). As a consequence, PV-expressing neurons have been identified as a critical target of therapeutic approaches in epilepsy (115).
Collectively, the convergence of symptoms between hypothyroid, epileptic and ASD patients suggests that common pathways involving PV-expressing neurons might underlie these pathologies of brain development. Such convergence might partly be linked to comorbidities that contribute independently to the overt pathology, but in any case, improving our understanding of the role of TH in the development of the GABAergic system, notably in PV-expressing neurons, should help make progress in the prevention and treatment of several neurological disorders. In addition, deciphering the network of TH target genes in the brain may help detect pharmacological or chemical agents that are likely to disrupt TH signaling, and give an insight on subtle neurological insults that may result from exposure to such TH system-disrupting chemicals (116). However, there is still a long way to go before we understand the precise molecular mechanisms underlying TH action in the brain. Notably, the huge diversity of GABAergic neurons (117) makes it difficult to depict a unified view of the mechanisms of action of TH in these neurons. It is hoped that the advent of single-cell RNA sequencing (118) and of spatio-temporal transcriptomics (119) will help untangling the role of TH signaling in each GABAergic neuron subtype, in each brain region, at all developmental stages.
FF: Writing – original draft, Writing – review and editing. SR: Writing – original draft, Writing – review and editing. JR: Writing – review and editing.
JR was supported by grants of the China Scholarship Council and ENS de Lyon, within the framework of the PROSFER program (Program of Sino-French Education for Research). Research in our group is funded by the European Union’s Horizon 2020 research and innovation program, under grant agreement no. 825753 (ERGO) and by the French Agence Nationale de la Recherche (Hypothyro project, ANR 22-CE14-0026-01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bernal J. Thyroid hormones in brain development and function. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, editors. Endotext. South Dartmouth (MA: MDText.com, Inc (2015). p. 1–62.
2. Venero C, Guadaño-Ferraz A, Herrero AI, Nordström K, Manzano J, de Escobar GM, et al. Anxiety, memory impairment, and locomotor dysfunction caused by a mutant thyroid hormone receptor alpha1 can be ameliorated by T3 treatment. Genes Dev (2005) 19(18):2152–63. doi: 10.1101/gad.346105
3. Richard S, Guyot R, Rey-Millet M, Prieux M, Markossian S, Aubert D, et al. A pivotal genetic program controlled by thyroid hormone during the maturation of GABAergic neurons. iScience (2020) 23(3):100899. doi: 10.1016/j.isci.2020.100899
4. Braat S, Kooy RF. The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron (2015) 86(5):1119–30. doi: 10.1016/j.neuron.2015.03.042
5. Sait Gönen M, Kisakol G, Savas Cilli A, Dikbas O, Gungor K, Inal A, et al. Assessment of anxiety in subclinical thyroid disorders. Endocr J (2004) 51(3):311–5. doi: 10.1507/endocrj.51.311
6. Andersen SL, Laurberg P, Wu CS, Olsen J. Maternal thyroid dysfunction and risk of seizure in the child: a Danish nationwide cohort study. J Pregnancy (2013) 2013:636705. doi: 10.1155/2013/636705
7. Wiens SC, Trudeau VL. Thyroid hormone and gamma-aminobutyric acid (GABA) interactions in neuroendocrine systems. Comp Biochem Physiol A Mol Integr Physiol (2006) 144(3):332–44. doi: 10.1016/j.cbpa.2006.01.033
8. Bradley DJ, Towle HC, Scott Young IIIW. Spatial and temporal expression of α- and β-thyroid hormone receptor mRNAs, including the β2 subtype, in the developing mammalian nervous system. J Neurosci (1992) 12(6):2288–302. doi: 10.1523/JNEUROSCI.12-06-02288.1992
9. Minakhina S, Bansal S, Zhang A, Brotherton M, Janodia R, De Oliveira V, et al. A direct comparison of thyroid hormone receptor protein levels in mice provides unexpected insights into thyroid hormone action. Thyroid (2020) 30(8):1193–204. doi: 10.1089/thy.2019.0763
10. Bernal J, Pekonen F. Ontogenesis of the nuclear 3, 5, 3’-triiodothyronine receptor in the human fetal brain. Endocrinology (1984) 114(2):677–9. doi: 10.1210/endo-114-2-677
11. Itoh Y, Esaki T, Kaneshige M, Suzuki H, Cook M, Sokoloff L, et al. Brain glucose utilization in mice with a targeted mutation in the thyroid hormone α or β receptor gene. Proc Natl Acad Sci U S A (2001) 98(17):9913–8. doi: 10.1073/pnas.171319498
12. Morte B, Manzano J, Scanlan T, Vennström B, Bernal J. Deletion of the thyroid hormone receptor α1 prevents the structural alterations of the cerebellum induced by hypothyroidism. Proc Natl Acad Sci (2002) 99(6):3985–9. doi: 10.1073/pnas.062413299
13. Gil-Ibañez P, Morte B, Bernal J. Role of thyroid hormone receptor subtypes alpha and beta on gene expression in the cerebral cortex and striatum of postnatal mice. Endocrinology (2013) 154(5):1940–7. doi: 10.1210/en.2012-2189
14. Flamant F, Gauthier K, Richard S. Genetic investigation of thyroid hormone receptor function in the developing and adult brain. Curr Top Dev Biol (2017) 125:303–35. doi: 10.1016/bs.ctdb.2017.01.001
15. Erbas IM, Demir K. The clinical spectrum of resistance to thyroid hormone alpha in children and adults. J Clin Res Pediatr Endocrinol (2021) 13(1):1–14. doi: 10.4274/jcrpe.galenos.2020.2019.0190
16. Moran C, Chatterjee K. Resistance to thyroid hormone due to defective thyroid receptor alpha. Best Pract Res Clin Endocrinol Metab (2015) 29(4):647–57. doi: 10.1016/j.beem.2015.07.007
17. Ferrara AM, Onigata K, Ercan O, Woodhead H, Weiss RE, Refetoff S. Homozygous thyroid hormone receptor beta-gene mutations in resistance to thyroid hormone: three new cases and review of the literature. J Clin Endocrinol Metab (2012) 97(4):1328–36. doi: 10.1210/jc.2011-2642
18. Concolino P, Costella A, Paragliola RM. Mutational landscape of resistance to thyroid hormone beta (RTHβ). Mol Diagnosis Ther (2019) 23:353–68. doi: 10.1007/s40291-019-00399-w
19. Krude H, Kuhnen P, Biebermann H. Treatment of congenital thyroid dysfunction: Achievements and challenges. Best Pract Res Clin Endocrinol Metab (2015) 29(3):399–413. doi: 10.1016/j.beem.2015.04.004
20. Alshehri B, D’Souza DG, Lee JY, Petratos S, Richardson SJ. The diversity of mechanisms influenced by transthyretin in neurobiology: development, disease and endocrine disruption. J Neuroendocrinol (2015) 27(5):303–23. doi: 10.1111/jne.12271
21. Barez-Lopez S, Obregon MJ, Bernal J, Guadano-Ferraz A. Thyroid hormone economy in the perinatal mouse brain: implications for cerebral cortex development. Cereb Cortex (2018) 28(5):1783–93. doi: 10.1093/cercor/bhx088
22. Crantz FR, Silva JE, Larsen PR. An analysis of the sources and quantity of 3,5,3′-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology (1982) 110(2):367–75. doi: 10.1210/endo-110-2-367
23. Luongo C, Trivisano L, Alfano F, Salvatore D. Type 3 deiodinase and consumptive hypothyroidism: a common mechanism for a rare disease. Front Endocrinol (Lausanne) (2013) 4:115. doi: 10.3389/fendo.2013.00115
24. Richard S, Flamant F. Regulation of T3 availability in the developing brain: the mouse genetics contribution. Front Endocrinol (Lausanne) (2018) 9:265. doi: 10.3389/fendo.2018.00265
25. Hulse A. Congenital hypothyroidism and neurological development. J Child Psychol Psychiat (1983) 24(4):629–35. doi: 10.1111/j.1469-7610.1983.tb00139.x
26. Prezioso G, Giannini C, Chiarelli F. Effect of thyroid hormones on neurons and neurodevelopment. Horm Res Paediatr (2018) 90(2):73–81. doi: 10.1159/000492129
27. Gilbert ME, Rovet J, Chen Z, Koibuchi N. Developmental thyroid hormone disruption: prevalence, environmental contaminants and neurodevelopmental consequences. Neurotoxicology (2012) 33(4):842–52. doi: 10.1016/j.neuro.2011.11.005
28. Kopel J. A global perspective on newborn congenital hypothyroidism screening. Proc (Bayl Univ Med Cent) (2020) 33(1):137–9. doi: 10.1080/08998280.2019.1668715
29. Persani L. Rescue of neurological development in congenital hypothyrodism: we should leave no stone unturned. J Clin Endocrinol Metab (2021) 106(12):e5267–e5269. doi: 10.1210/clinem/dgab487
30. Schwartz CE, May MM, Carpenter NJ, Rogers RC, Martin J, Bialer MG, et al. Allan-Herndon-Dudley syndrome and the monocarboxylate transporter 8 (MCT8) gene. Am J Hum Genet (2005) 77(1):41–53. doi: 10.1086/431313
31. Remerand G, Boespflug-Tanguy O, Tonduti D, Touraine R, Rodriguez D, Curie A, et al. Expanding the phenotypic spectrum of Allan-Herndon-Dudley syndrome in patients with SLC16A2 mutations. Dev Med Child Neurol (2019) 61(12):1439–47. doi: 10.1111/dmcn.14332
32. Grijota-Martinez C, Barez-Lopez S, Gomez-Andres D, Guadano-Ferraz A. MCT8 deficiency: the road to therapies for a rare disease. Front Neurosci (2020) 14:380. doi: 10.3389/fnins.2020.00380
33. Groeneweg S, van Geest FS, Abaci A, Alcantud A, Ambegaonkar GP, Armour CM, et al. Disease characteristics of MCT8 deficiency: an international, retrospective, multicentre cohort study. Lancet Diabetes Endocrinol (2020) 8(7):594–605. doi: 10.1016/S2213-8587(20)30153-4
34. López-Espíndola D, Morales-Bastos C, Grijota-Martinez C, Liao XH, Lev D, Sugo E, et al. Mutations of the thyroid hormone transporter MCT8 cause prenatal brain damage and persistent hypomyelination. J Clin Endocrinol Metab (2014) 99(12):E2799–804. doi: 10.1210/jc.2014-2162
35. Moran C, Schoenmakers N, Agostini M, Schoenmakers E, Offiah A, Kydd A, et al. An adult female with resistance to thyroid hormone mediated by defective thyroid hormone receptor alpha. J Clin Endocrinol Metab (2013) 98(11):4254–61. doi: 10.1210/jc.2013-2215
36. van Gucht AL, Meima ME, Zwaveling-Soonawala N, Visser WE, Fliers E, Wennink JM, et al. Resistance to thyroid hormone alpha in an 18-month-old girl: clinical, therapeutic, and molecular characteristics. Thyroid (2016) 26(3):338–46. doi: 10.1089/thy.2015.0463
37. Le Maire A, Bouhours-Nouet N, Soamalala J, Mirebeau-Prunier D, Paloni M, Guee L, et al. Two novel cases of resistance to thyroid hormone due to THRA mutation. Thyroid (2020) 30(8):1217–21. doi: 10.1089/thy.2019.0602
38. World Health Organization. Epilepsy 2023 (2023). Available at: https://www.who.int/news-room/fact-sheets/detail/epilepsy.
39. Kalikiri MK, Mamidala MP, Rao AN, Rajesh V. Analysis and functional characterization of sequence variations in ligand binding domain of thyroid hormone receptors in autism spectrum disorder (ASD) patients. Autism Res (2017) 10(12):1919–28. doi: 10.1002/aur.1838
40. Yuen RKC, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, et al. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med (2015) 21:185. doi: 10.1038/nm.3792
41. Zekri Y, Guyot R, Flamant F. An atlas of thyroid hormone receptors’ Target genes in mouse tissues. Int J Mol Sci (2022) 23(19):1444. doi: 10.3390/ijms231911444
42. Nicholson JL, Altman J. The effects of early hypo- and hyperthyroidism on the development of rat cerebellar cortex. I. Cell proliferation and differentiation. Brain Res (1972) 44:13–23. doi: 10.1016/0006-8993(72)90362-9
43. Koibuchi N, Chin WW. Thyroid hormone action and brain development. Trends Endocrinol Metab (2000) 11(4):123–8. doi: 10.1016/S1043-2760(00)00238-1
44. Li JQ, Wang X, Yan YQ, Wang KW, Qin DK, Xin ZF, et al. The effects on fetal brain development in the rat of a severely iodine deficient diet derived from an endemic area: observations on the first generation. Neuropathol Appl Neurobiol (1986) 12(3):261–76. doi: 10.1111/j.1365-2990.1986.tb00139.x
45. Patel AJ, Smith RM, Kingsbury AE, Hunt A, Balázs R. Effects of thyroid state on brain development: muscarinic acetylcholine and GABA receptors. Brain Res (1980) 198:389–402. doi: 10.1016/0006-8993(80)90752-0
46. Manzano J, Cuadrado M, Morte B, Bernal J. Influence of thyroid hormone and thyroid hormone receptors in the generation of cerebellar gamma-aminobutyric acid-ergic interneurons from precursor cells. Endocrinology (2007) 148(12):5746–51. doi: 10.1210/en.2007-0567
47. Berbel P, Marco P, Cerezo JR, DeFelipe J. Distribution of parvalbumin immunoreactivity in the neocortex of hypothyroid adult rats. Neurosci Lett (1996) 204:65–8. doi: 10.1016/0304-3940(96)12318-1
48. Gilbert ME, Sui L, Walker MJ, Anderson W, Thomas S, Smoller SN, et al. Thyroid hormone insufficiency during brain development reduces parvalbumin immunoreactivity and inhibitory function in the hippocampus. Endocrinology (2007) 148(1):92–102. doi: 10.1210/en.2006-0164
49. Shiraki A, Akane H, Ohishi T, Wang L, Morita R, Suzuki K, et al. Similar distribution changes of GABAergic interneuron subpopulations in contrast to the different impact on neurogenesis between developmental and adult-stage hypothyroidism in the hippocampal dentate gyrus in rats. Arch Toxicol (2012) 86(10):1559–69. doi: 10.1007/s00204-012-0846-y
50. Sawano E, Takahashi M, Negishi T, Tashiro T. Thyroid hormone-dependent development of the GABAergic pre- and post-synaptic components in the rat hippocampus. Int J Dev Neurosci (2013) 31(8):751–61. doi: 10.1016/j.ijdevneu.2013.09.007
51. Cunha Menezes E, Rabelo Santos P, Costa Goes T, Barboza Carvalho VC, Teixeira-Silva F, Stevens HE, et al. Effects of a rat model of gestational hypothyroidism on forebrain dopaminergic, GABAergic, and serotonergic systems and related behaviors. Behav Brain Res (2019) 366:77–87. doi: 10.1016/j.bbr.2019.03.027
52. Ramhøj L, Guyot R, Svingen T, Kortenkamp A, Flamant F, Axelstad M. Is periventricular heterotopia a useful endpoint for developmental thyroid hormone system disruption in mouse toxicity studies? Regul Toxicol Pharmacol (2023), 105445. doi: 10.1016/j.yrtph.2023.105445
53. Goodman JH, Gilbert ME. Modest thyroid hormone insufficiency during development induces a cellular malformation in the corpus callosum: a model of cortical dysplasia. Endocrinology (2007) 148(6):2593–7. doi: 10.1210/en.2006-1276
54. Friauf E, Wenz M, Oberhofer M, Nothwang HG, Balakrishnan V, Knipper M, et al. Hypothyroidism impairs chloride homeostasis and onset of inhibitory neurotransmission in developing auditory brainstem and hippocampal neurons. Eur J Neurosci (2008) 28(12):2371–80. doi: 10.1111/j.1460-9568.2008.06528.x
55. Quignodon L, Vincent S, Winter H, Samarut J, Flamant F. A point mutation in the activation function 2 domain of thyroid hormone receptor alpha1 expressed after CRE-mediated recombination partially recapitulates hypothyroidism. Mol Endocrinol (2007) 21(10):2350–60. doi: 10.1210/me.2007-0176
56. Tinnikov A, Nordström K, Thorén P, Kindblom JM, Malin S, Rozell B, et al. Retardation of post-natal development caused by a negatively acting thyroid hormone receptor α1. EMBO J (2002) 21(19):5079–87. doi: 10.1093/emboj/cdf523
57. Flamant FDR, Poguet A-L, Plateroti M, Chassande O, Gauthier K, Streichenberger N, et al. Congenital hypothyroid Pax8–/– mutant mice can be rescued by inactivating the TRα Gene. Mol Endocrinol (2002) 16(1):24–32. doi: 10.1210/mend.16.1.0766
58. Hashimoto K, Curty FH, Borges PP, Lee CE, Abel ED, Elmquist JK, et al. An unliganded thyroid hormone receptor causes severe neurological dysfunction. Proc Natl Acad Sci (2001) 98(7):3998–4003. doi: 10.1073/pnas.051454698
59. Portella AC, Carvalho F, Faustino L, Wondisford FE, Ortiga-Carvalho TM, Gomes FC. Thyroid hormone receptor beta mutation causes severe impairment of cerebellar development. Mol Cell Neurosci (2010) 44(1):68–77. doi: 10.1016/j.mcn.2010.02.004
60. Forrest D, Erway LC, Ng L, Altschuler R, Curran T. Thyroid hormone receptor β is essential for development of auditory function. Nat Genet (1996) 13:354–7. doi: 10.1038/ng0796-354
61. Sandhofer C, Schwartz HL, Mariash CN, Forrest D, Oppenheimer JH. Beta isoforms are not essential for thyroid hormone-dependent acceleration of PCP-2 and myelin basic protein gene expression in the developing brains of neonatal mice. Mol Cell Endocrinol (1998) 137:109–15. doi: 10.1016/S0303-7207(98)00005-7
62. Peeters RP, Hernandez A, Ng L, Ma M, Sharlin DS, Pandey M, et al. Cerebellar abnormalities in mice lacking type 3 deiodinase and partial reversal of phenotype by deletion of thyroid hormone receptor alpha1. Endocrinology (2013) 154(1):550–61. doi: 10.1210/en.2012-1738
63. Wallis K, Sjögren M, van Hogerlinden M, Silberberg G, Fisahn A, Nordström K, et al. Locomotor deficiencies and aberrant development of subtype-specific GABAergic interneurons caused by an unliganded thyroid hormone receptor α1. J Neurosci (2008) 28(8):1904–15. doi: 10.1523/JNEUROSCI.5163-07.2008
64. Markossian S, Guyot R, Richard S, Teixeira M, Aguilera N, Bouchet M, et al. CRISPR/cas9 editing of the mouse Thra gene produces models with variable resistance to thyroid hormone. Thyroid (2018) 28(1):139–50. doi: 10.1089/thy.2017.0389
65. Mayerl S, Muller J, Bauer R, Richert S, Kassmann CM, Darras VM, et al. Transporters MCT8 and OATP1C1 maintain murine brain thyroid hormone homeostasis. J Clin Invest (2014) 124(5):1987–99. doi: 10.1172/JCI70324
66. Amano I, Takatsuru Y, Toya S, Aijima A, Iwasaki T, Grasberger H, et al. Aberrant cerebellar development in mice lacking dual oxidase maturation factors. Thyroid (2016) 26(5):741–52. doi: 10.1089/thy.2015.0034
67. Barez-Lopez S, Grijota-Martinez C, Auso E, Fernandez-de Frutos M, Montero-Pedrazuela A, Guadano-Ferraz A. Adult mice lacking Mct8 and Dio2 proteins present alterations in peripheral thyroid hormone levels and severe brain and motor skill impairments. Thyroid (2019) 29(11):1669–82. doi: 10.1089/thy.2019.0068
68. Valcárcel-Hernández V, Guillén-Yunta M, Bueno-Arribas M, Montero-Pedrazuela A, Grijota-Martínez C, Markossian S, et al. A CRISPR/Cas9-engineered avatar mouse model of monocarboxylate transporter 8 deficiency displays distinct neurological alterations. Neurobiol Dis (2022) 174:105896. doi: 10.1016/j.nbd.2022.105896
69. Guadaño-Ferraz A, Benavides-Piccione R, Venero C, Lancha C, Vennström B, Sandi C, et al. Lack of thyroid hormone receptor alpha1 is associated with selective alterations in behavior and hippocampal circuits. Mol Psychiatry (2003) 8(1):30–8. doi: 10.1038/sj.mp.4001196
70. Hu H, Gan J, Jonas P. Interneurons. Fast-spiking, parvalbumin(+) GABAergic interneurons: from cellular design to microcircuit function. Science (2014) 345(6196):1255263. doi: 10.1126/science.1255263
71. Kriener B, Hu H, Vervaeke K. Parvalbumin interneuron dendrites enhance gamma oscillations. Cell Rep (2022) 39(11):110948. doi: 10.1016/j.celrep.2022.110948
72. Hadjab-Lallemend S, Wallis K, van Hogerlinden M, Dudazy S, Nordstrom K, Vennstrom B, et al. A mutant thyroid hormone receptor alpha1 alters hippocampal circuitry and reduces seizure susceptibility in mice. Neuropharmacology (2010) 58(7):1130–9. doi: 10.1016/j.neuropharm.2010.02.005
73. Ben-Ari Y. The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience (2014) 279:187–219. doi: 10.1016/j.neuroscience.2014.08.001
74. Wang Y, Fisahn A, Sinha I, Nguyen DP, Sterzenbach U, Lallemend F, et al. Hippocampal transcriptome profile of persistent memory rescue in a mouse model of THRA1 mutation-mediated resistance to thyroid hormone. Sci Rep (2016) 6:18617. doi: 10.1038/srep18617
75. Mittag J, Lyons DJ, Sallstrom J, Vujovic M, Dudazy-Gralla S, Warner A, et al. Thyroid hormone is required for hypothalamic neurons regulating cardiovascular functions. J Clin Invest (2013) 123(1):509–16. doi: 10.1172/JCI65252
76. Harder L, Dudazy-Gralla S, Muller-Fielitz H, Hjerling Leffler J, Vennstrom B, Heuer H, et al. Maternal thyroid hormone is required for parvalbumin neurone development in the anterior hypothalamic area. J Neuroendocrinol (2018) 30(3):e12573. doi: 10.1111/jne.12573
77. Kim KH, Lee J, Ahn CH, Yu HW, Choi JY, Lee HY, et al. Association between thyroid function and heart rate monitored by wearable devices in patients with hypothyroidism. Endocrinol Metab (Seoul) (2021) 36(5):1121–30. doi: 10.3803/EnM.2021.1216
78. Laing BT, Anderson MS, Bonaventura J, Jayan A, Sarsfield S, Gajendiran A, et al. Anterior hypothalamic parvalbumin neurons are glutamatergic and promote escape behavior. Curr Biol (2023) 33(15):3215–28 e7. doi: 10.1016/j.cub.2023.06.070
79. Fauquier T, Chatonnet F, Picou F, Richard S, Fossat N, Aguilera N, et al. Purkinje cells and Bergmann glia are primary targets of the TRalpha1 thyroid hormone receptor during mouse cerebellum postnatal development. Development (2014) 141(1):166–75. doi: 10.1242/dev.103226
80. Fauquier T, Romero E, Picou F, Chatonnet F, Nguyen X-N, Quignodon L, et al. Severe impairment of cerebellum development in mice expressing a dominant-negative mutation inactivating thyroid hormone receptor alpha1 isoform. Dev Biol (2011) 356(2):350–8. doi: 10.1016/j.ydbio.2011.05.657
81. Picou F, Fauquier T, Chatonnet F, Richard S, Flamant F. Deciphering direct and indirect influence of thyroid hormone with mouse genetics. Mol Endocrinol (2014) 28(4):429–41. doi: 10.1210/me.2013-1414
82. Picou F, Fauquier T, Chatonnet F, Flamant F. A bimodal influence of thyroid hormone on cerebellum oligodendrocyte differentiation. Mol Endocrinol (2012) 26(4):608–18. doi: 10.1210/me.2011-1316
83. Yu L, Shimokawa N, Iwasaki T, Xu M, Lesmana R, Koibuchi N, et al. Aberrant cerebellar development of transgenic mice expressing dominant-negative thyroid hormone receptor in cerebellar Purkinje cells. Endocrinology (2015) 156(4):1565–76. doi: 10.1210/en.2014-1079
84. Ninomiya A, Amano I, Kokubo M, Takatsuru Y, Ishii S, Hirai H, et al. Long-term depression-inductive stimulation causes long-term potentiation in mouse Purkinje cells with a mutant thyroid hormone receptor. Proc Natl Acad Sci U S A (2022) 119(45):e2210645119. doi: 10.1073/pnas.2210645119
85. Mayerl S, Chen J, Salveridou E, Boelen A, Darras VM, Heuer H. Thyroid hormone transporter deficiency in mice impacts multiple stages of GABAergic interneuron development. Cereb Cortex (2022) 32(2):329–41. doi: 10.1093/cercor/bhab211
86. Xu Q, Guo L, Moore H, Waclaw RR, Campbell K, Anderson SA. Sonic hedgehog signaling confers ventral telencephalic progenitors with distinct cortical interneuron fates. Neuron (2010) 65(3):328–40. doi: 10.1016/j.neuron.2010.01.004
87. Chatonnet F, Flamant F, Morte B. A temporary compendium of thyroid hormone target genes in brain. Biochim Biophys Acta (2015) 1849(2):122–9. doi: 10.1016/j.bbagrm.2014.05.023
88. Thompson CC. Thyroid hormone-responsive genes in developing cerebellum include a novel synaptotagmin and a hairless homolog. J Neurosci (1996) 16(24):7832–40. doi: 10.1523/JNEUROSCI.16-24-07832.1996
89. Denver RJ, Ouellet L, Furling D, Kobayashi A, Fujii-Kuriyama Y, Puymirat J. Basic transcription element-binding protein (BTEB) is a thyroid hormone-regulated gene in the developing central nervous system. Evidence for a role in neurite outgrowth. J Biol Chem (1999) 274(33):23128–34. doi: 10.1074/jbc.274.33.23128
90. Diez D, Grijota-Martinez C, Morreale de Escobar G, Bernal J, Morte B, Pinchera A, et al. Thyroid hormone action in the adult brain: gene expression profiling of the effects of single and multiple doses of Triiodo-l-thyronine in the rat striatum. Endocrinology (2008) 149(8):3989–4000. doi: 10.1210/en.2008-0350
91. Morte B, Ceballos A, Diez D, Grijota-Martínez C, Dumitrescu AM, Di Cosmo C, et al. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology (2010) 151(5):2381–7. doi: 10.1210/en.2009-0944
92. Poguet AL, Legrand C, Feng X, Yen PM, Meltzer P, Samarut J, et al. Microarray analysis of knockout mice identifies cyclin D2 as a possible mediator for the action of thyroid hormone during the postnatal development of the cerebellum. Dev Biol (2003) 254(2):188–99. doi: 10.1016/S0012-1606(02)00039-8
93. Fang Y, Dang P, Liang Y, Zhao D, Wang R, Xi Y, et al. Histological, functional and transcriptomic alterations in the juvenile hippocampus in a mouse model of thyroid hormone resistance. Eur Thyroid J (2022) 11(2). doi: 10.1530/ETJ-21-0097
94. Gil-Ibañez P, García-García F, Dopazo J, Bernal J, Morte B. Global transcriptome analysis of primary cerebrocortical cells: identification of genes regulated by triiodothyronine in specific cell types. Cereb Cortex (2017) 27(1):706–17. doi: 10.1093/cercor/bhv273
95. Chatonnet F, Guyot R, Picou F, Bondesson M, Flamant F. Genome-wide search reveals the existence of a limited number of thyroid hormone receptor alpha target genes in cerebellar neurons. PloS One (2012) 7(5):e30703. doi: 10.1371/journal.pone.0030703
96. Neveu I, Arenas E. Neurotrophins promote the survival and development of neurons of the cerebellum of hypothyroid rats in vivo. J Cell Biol (1996) 133(3):631–46. doi: 10.1083/jcb.133.3.631
97. Aramaki M, Wu X, Liu H, Liu Y, Cho YW, Song M, et al. Transcriptional control of cone photoreceptor diversity by a thyroid hormone receptor. Proc Natl Acad Sci U S A (2022) 119(49):e2209884119. doi: 10.1073/pnas.2209884119
98. Ariyani W, Miyazaki W, Amano I, Koibuchi N. Involvement of integrin alphavbeta3 in thyroid hormone-induced dendritogenesis. Front Endocrinol (Lausanne) (2022) 13:938596. doi: 10.3389/fendo.2022.938596
99. Avci HX, Lebrun C, Wehrle R, Doulazmi M, Chatonnet F, Morel MP, et al. Thyroid hormone triggers the developmental loss of axonal regenerative capacity via thyroid hormone receptor alpha1 and kruppel-like factor 9 in Purkinje cells. Proc Natl Acad Sci U S A (2012) 109(35):14206–11. doi: 10.1073/pnas.1119853109
100. Royland JE, Parker JS, Gilbert ME. A genomic analysis of subclinical hypothyroidism in hippocampus and neocortex of the developing rat brain. J Neuroendocrinol (2008) 20(12):1319–38. doi: 10.1111/j.1365-2826.2008.01793.x
101. Bernal J, Morte B, Diez D. Thyroid hormone regulators in human cerebral cortex development. J Endocrinol (2022) 255(3):R27–36. doi: 10.1530/JOE-22-0189
102. Wirth EK, Roth S, Blechschmidt C, Holter SM, Becker L, Racz I, et al. Neuronal 3’,3,5-triiodothyronine (T3) uptake and behavioral phenotype of mice deficient in Mct8, the neuronal T3 transporter mutated in Allan-Herndon-Dudley syndrome. J Neurosci (2009) 29(30):9439–49. doi: 10.1523/JNEUROSCI.6055-08.2009
103. Diez D, Morte B, Bernal J. Single-cell transcriptome profiling of thyroid hormone effectors in the human fetal neocortex: expression of SLCO1C1, DIO2, and THRB in specific cell types. Thyroid (2021) 31(10):1577–88. doi: 10.1089/thy.2021.0057
104. Ren J, Flamant F. Thyroid hormone as a temporal switch in mouse development. Eur Thyroid J (2023) 12(2). doi: 10.1530/ETJ-22-0225
105. Lear BP, Moore DL. Moving CNS axon growth and regeneration research into human model systems. Front Neurosci (2023) 17:1198041. doi: 10.3389/fnins.2023.1198041
106. Batista G, Hensch TK. Critical period regulation by thyroid hormones: potential mechanisms and sex-specific aspects. Front Mol Neurosci (2019) 12:77. doi: 10.3389/fnmol.2019.00077
107. Reh RK, Dias BG, Nelson CA 3rd, Kaufer D, Werker JF, Kolb B, et al. Critical period regulation across multiple timescales. Proc Natl Acad Sci U S A (2020) 117(38):23242–51. doi: 10.1073/pnas.1820836117
108. Tao HW, Li YT, Zhang LI. Formation of excitation-inhibition balance: inhibition listens and changes its tune. Trends Neurosci (2014) 37(10):528–30. doi: 10.1016/j.tins.2014.09.001
109. Gogolla N, Leblanc JJ, Quast KB, Sudhof TC, Fagiolini M, Hensch TK. Common circuit defect of excitatory-inhibitory balance in mouse models of autism. J Neurodev Disord (2009) 1(2):172–81. doi: 10.1007/s11689-009-9023-x
110. Hashemi E, Ariza J, Rogers H, Noctor SC, Martinez-Cerdeno V. The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb Cortex (2017) 27(3):1931–43. doi: 10.1093/cercor/bhw021
111. Schwede M, Nagpal S, Gandal MJ, Parikshak NN, Mirnics K, Geschwind DH, et al. Strong correlation of downregulated genes related to synaptic transmission and mitochondria in post-mortem autism cerebral cortex. J Neurodev Disord (2018) 10(1):18. doi: 10.1186/s11689-018-9237-x
112. Berbel P, Navarro D, Roman GC. An evo-devo approach to thyroid hormones in cerebral and cerebellar cortical development: etiological implications for autism. Front Endocrinol (Lausanne) (2014) 5:146. doi: 10.3389/fendo.2014.00146
113. Navarro D, Alvarado M, Navarrete F, Giner M, Obregon MJ, Manzanares J, et al. Gestational and early postnatal hypothyroidism alters VGluT1 and VGAT bouton distribution in the neocortex and hippocampus, and behavior in rats. Front Neuroanat (2015) 9(9). doi: 10.3389/fnana.2015.00009
114. Jiang X, Lachance M, Rossignol E. Involvement of cortical fast-spiking parvalbumin-positive basket cells in epilepsy. Prog Brain Res (2016) 226:81–126. doi: 10.1016/bs.pbr.2016.04.012
115. Godoy LD, Prizon T, Rossignoli MT, Leite JP, Liberato JL. Parvalbumin role in epilepsy and psychiatric comorbidities: from mechanism to intervention. Front Integr Neurosci (2022) 16:765324. doi: 10.3389/fnint.2022.765324
116. Holbech H, Matthiessen P, Hansen M, Schuurmann G, Knapen D, Reuver M, et al. ERGO: breaking down the wall between human health and environmental testing of endocrine disrupters. Int J Mol Sci (2020) 21(8):2954. doi: 10.3390/ijms21082954
117. Gouwens NW, Sorensen SA, Baftizadeh F, Budzillo A, Lee BR, Jarsky T, et al. Integrated morphoelectric and transcriptomic classification of cortical GABAergic cells. Cell (2020) 183(4):935–53 e19. doi: 10.1016/j.cell.2020.09.057
118. Sreenivasan VKA, Dore R, Resch J, Maier J, Dietrich C, Henck J, et al. Single-cell RNA-based phenotyping reveals a pivotal role of thyroid hormone receptor alpha for hypothalamic development. Development (2023) 150(3). doi: 10.1242/dev.201228
Keywords: brain development, parvalbumin interneurons, thyroid hormone receptors, animal models, hypothyroidism, neurological disorders
Citation: Richard S, Ren J and Flamant F (2023) Thyroid hormone action during GABAergic neuron maturation: The quest for mechanisms. Front. Endocrinol. 14:1256877. doi: 10.3389/fendo.2023.1256877
Received: 11 July 2023; Accepted: 18 September 2023;
Published: 03 October 2023.
Edited by:
Laurent M. Sachs, Muséum National d’Histoire Naturelle, FranceReviewed by:
Heike Heuer, Essen University Hospital, GermanyCopyright © 2023 Richard, Ren and Flamant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frédéric Flamant, RnJlZGVyaWMuZmxhbWFudEBlbnMtbHlvbi5mcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.