 Congcong Guo1,2,3,4
Congcong Guo1,2,3,4 Yuying Cui1,2,3,5
Yuying Cui1,2,3,5 Mingwen Jiao6
Mingwen Jiao6 Jinming Yao1,2,3
Jinming Yao1,2,3 Junyu Zhao1,2,3
Junyu Zhao1,2,3 Yutian Tian1,2,3
Yutian Tian1,2,3 Jianjun Dong7*
Jianjun Dong7* Lin Liao1,2,3,5*
Lin Liao1,2,3,5*- 1Department of Endocrinology and Metabology, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
- 2Shandong Key Laboratory of Rheumatic Disease and Translational Medicine, the First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
- 3Shandong Institute of Nephrology, the First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
- 4College of Pharmacy, Shandong University of Traditional Chinese Medicine, Jinan, Shandong, China
- 5First Clinical Medical College, Shandong University of Traditional Chinese Medicin, Jinan, Shandong, China
- 6Department of General Surgery, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Jinan, Shandong, China
- 7Department of Endocrinology, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong, China
The energy needs of tubular epithelial components, especially proximal tubular epithelial cells (PTECs), are high and they heavily depend on aerobic metabolism. As a result, they are particularly vulnerable to various injuries caused by factors such as ischemia, proteinuria, toxins, and elevated glucose levels. Initial metabolic and phenotypic changes in PTECs after injury are likely an attempt at survival and repair. Nevertheless, in cases of recurrent or prolonged injury, PTECs have the potential to undergo a transition to a secretory state, leading to the generation and discharge of diverse bioactive substances, including transforming growth factor-β, Wnt ligands, hepatocyte growth factor, interleukin (IL)-1β, lactic acid, exosomes, and extracellular vesicles. By promoting fibroblast activation, macrophage recruitment, and endothelial cell loss, these bioactive compounds stimulate communication between epithelial cells and other interstitial cells, ultimately worsening renal damage. This review provides a summary of the latest findings on bioactive compounds that facilitate the communication between these cellular categories, ultimately leading to the advancement of tubulointerstitial fibrosis (TIF).
1 Introduction
Approximately 8% to 15% of the global population is impacted by chronic kidney disease (CKD), which has the potential to progress to end-stage renal failure. In such cases, the sole choices for survival are either dialysis or renal transplantation (1). CKD is not just a condition affecting the kidneys, but it is also linked to a higher chance of mortality from any cause and cardiovascular issues (2). Tubulointerstitial fibrosis (TIF), glomerulosclerosis, and vascular sclerosis are common features of progressive CKD, regardless of its initial cause, leading to the development of renal fibrosis. Studies have consistently demonstrated that the increase in TIF is the most reliable histological indicator of renal functional decline and a poorer prognosis (3–6). Despite decades of research, knowledge of TIF remains limited, and therapeutic advances are a daunting challenge (7). Fibrosis progression is contributed to by various cell types found in the tubulointerstitium, such as tubular epithelial cells, fibroblasts, endothelial cells, and inflammatory cells. According to a prior investigation, regions with active interstitial fibrosis were primarily distinguished by their peritubular distribution rather than perivascular distribution (5). The results suggested that tubular epithelial cells can transmit fibrogenic signals, thereby facilitating the advancement of progressive TIF. Additional investigation revealed that proximal tubular epithelial cells (PTECs) were capable of reproducing the histologic alterations in CKD and frequently occurred before the participation of other cellular components. Whether recovery or fibrosis occurs depends on the extent and frequency of PTECs damage. Restoration of normal kidney function is possible if the injury is mild and temporary. Nevertheless, in the case of a severe or persistent injury, the damaged epithelial cells may experience maladaptive healing and produce multiple active substances to trigger secondary responses through paracrine signaling (8). The paracrine signals promote the activation of fibroblasts, the recruitment of inflammatory cells, and the loss of endothelial cells. Hence, the interaction between these different cell types is crucial for the exchange of signals in the tubulointerstitium (9, 10). In the progression of TIF, this review will provide the advanced knowledge regarding the interaction between epithelial cells and other interstitial cells, known as crosstalk.
2 Metabolic changes in PTECs associated with survival after injury
Physiologically, the proximal tubule (PT), is in charge of preserving 99% of the water and solutes in the glomerular filtrate, while also ensuring the maintenance of acid/base equilibrium (11). Transcellular Na+ reabsorption through basolateral Na+-K+-ATPase is the primary driving force for reabsorption function, as depicted in Figure 1 (12). The sodium-dependent glucose transporters (SGLT) on the apical surface of PTECs recovers approximately 180 grams of glucose that has been filtered daily. However, little glucose is metabolized as an energetic substrate in PTECs (13). To ensure the energy supply of reabsorption, PTECs use fatty acid oxidation (FAO) as their preferred metabolic pathway, which generates 106 adenosine triphosphate (ATP) units compared to 36 APT units produced by glucose metabolism (14).
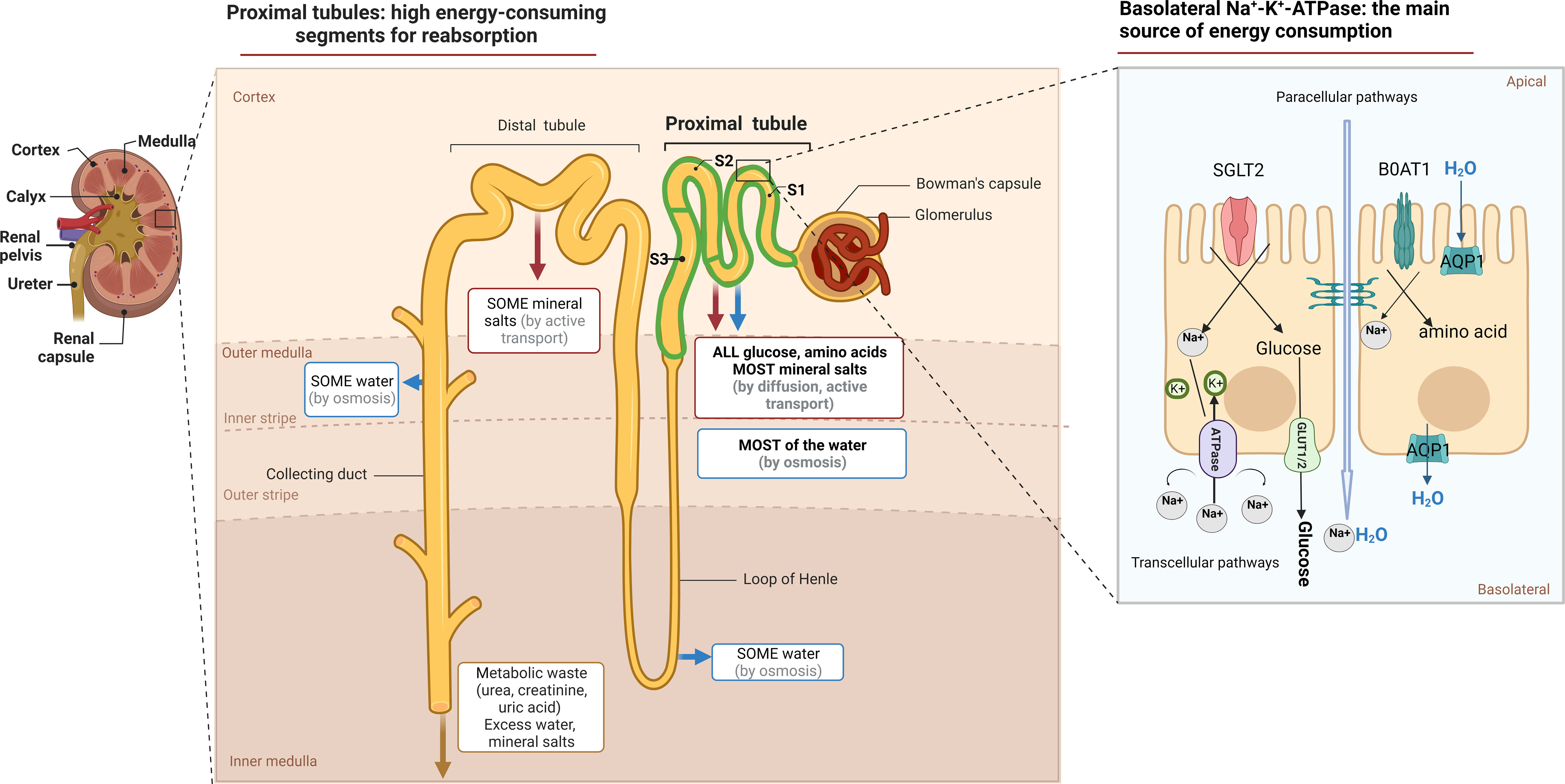
Figure 1 The function and metabolic characteristic of proximal tubule. Anatomically, the proximal tubule refers to the nephron that begins at the urinary pole of the glomerulus and ends at the Henle ‘s loop. Measuring approximately14 mm in length in humans, it constitutes three subtly distinct segments, S1, S2, S3, respectively. The tubular lumen of PTECs is accompanied by a brush border composed of cilia, which not only act as a mechanical sensor during reabsorption, but also increase the cell surface area, maintain normal nephron structure and play a reabsorption function. The reabsorption in the PTs occurs via both paracellular or transcellular pathways. SGLT2 located on the apical surface of the PTECs couples the transport of sodium and glucose in a 1:1 ratio and reabsorbs up to 90% of filtered glucose. The GLUT family located on the basolateral membrane provides an exit pathway for glucose back into the circulation. Various amino acids, similar to that of glucose, are actively transported across the luminal brush border membrane into PTECs, most of which by cotransport. While sodium–glucose or sodium–amino acids linked transport across the apical membrane of the PTECs is not of itself an energy-requiring process, their continuing activity is dependent on the maintenance of the electrochemical gradient for Na+, generated by Na+/K+ ATPase activity.
PTECs, known for their high energy consumption, possess abundant mitochondria, peroxisomes, and other metabolic organelles (15). According to reports, peroxisomes are harmed prior to the onset of mitochondrial dysfunction, making them a probable cause of the observed oxidative stress (16). Acute and chronic injury typically begins at the initial sites of the S2 and S3 proximal segments, likely due to their abundance of peroxisomes (17–19). Furthermore, mitochondrial dysfunction in injured PTECs has been widely reported in both acute kidney injury (AKI) and CKD (20). The process of anaerobic glycolysis can produce 2 ATP molecules without requiring functional mitochondria, resulting in the production of lactic acid. For survival, injured PTECs undergo metabolic reprogramming, which forces a shift from FAO to aerobic glycolysis (Figure 2A) (21, 22). In addition, some injured PTECs may undergo apoptosis and necrosis in AKI and CKD (23, 24). Thus, residual PTECs support compensatory changes in reabsorption, along with increased energy expenditure in the short term (25). Abnormal energy supply followed by metabolic rearrangement and increased energy consumption further exacerbate the damage to residual PTECs. FAO downregulation has been described in many AKI and CKD models (14, 26). The ischemia/reperfusion injury (IRI) -induced AKI model (27) and CKD model (28) have both confirmed the beneficial impact of increasing FAO in terms of protection. In recent times, the renoprotective properties of sodium-dependent glucose transporters-2 (SGLT-2) inhibitors on CKD have been shown (29), indicating that the kidney benefits of the drug may be related to decrease energy needs of PTECs and partially depend on reducing glycolysis (30).
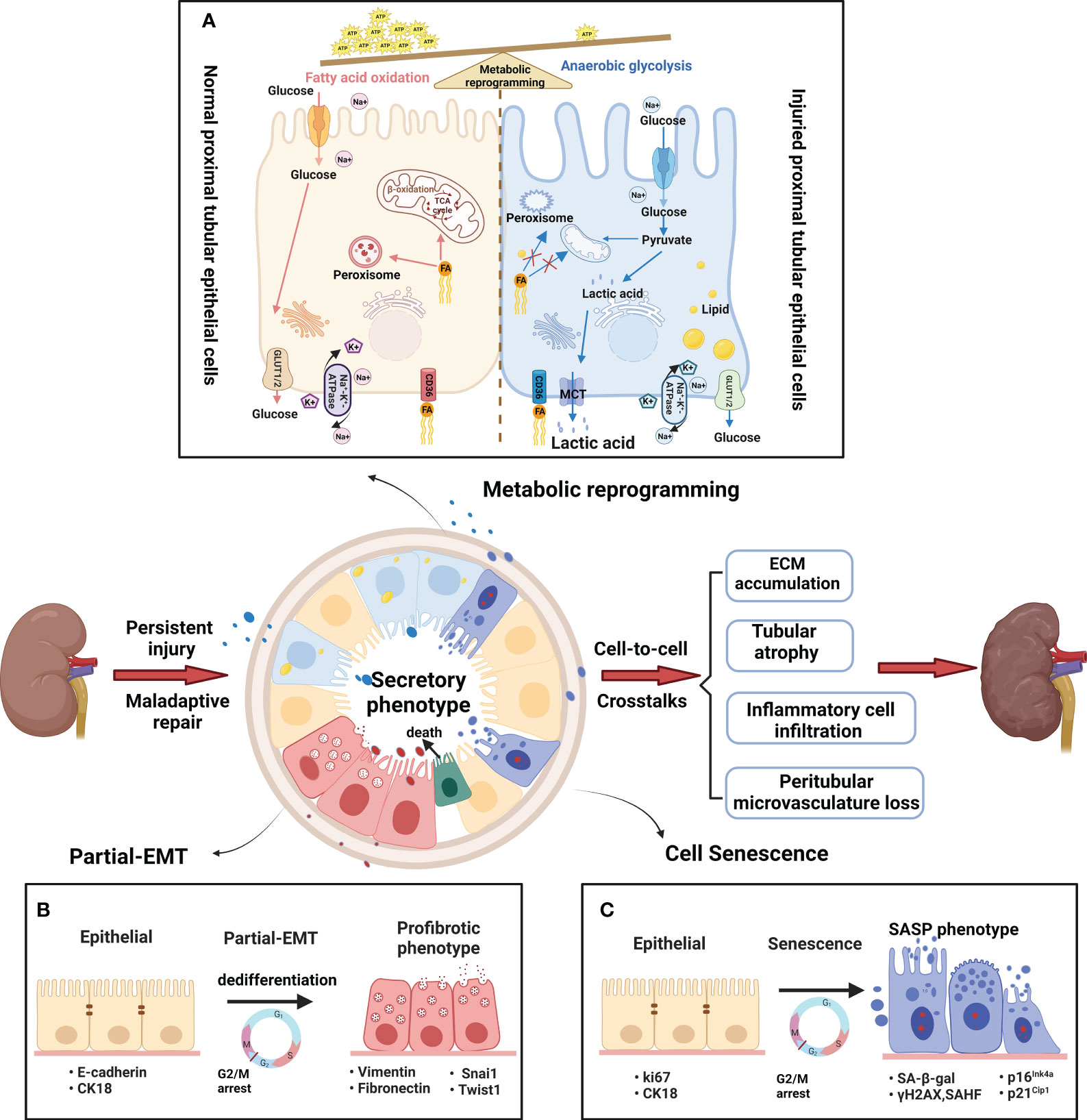
Figure 2 Metabolic and phenotypic changes in proximal tubular epithelial cells. (A) Metabolic rearrangement. The normal PTs rely mostly on fatty acid oxidation (FAO) to help meet energetic demands. The fatty acids are absorbed by PTECs through CD36 transporters, and then transported into mitochondria or peroxisomes to generate massive amounts of ATP. A large amount of glucose enters the PTs, but little glucose is metabolized in the normal proximal tubules. By contrast, the injured PTs rely mostly on anaerobic glycolysis to help meet energetic demands. Glycolysis is the process of converting reabsorbed glucose into pyruvate and generating small amounts of ATP. FAO decline due to mitochondrial dysfunction and peroxisome disruption in injured PTs. Therefore, anaerobic glycolysis is used here to the indicate metabolism of glucose to pyruvate and then lactic acid as contrasted with glucose oxidation (pyruvate which then enters the TCA cycle) (B) Partial epithelial–mesenchymal transition(pEMT). The pEMT is considered a “dedifferentiation- redifferentiation” process by which PTEC is characterized by the loss of epithelial markers (such as E-cadherin and cytokeratin) and gain of mesenchymal phenotype (vimentin) that is associated with increased production of profibrotic factors (fibronectin). During the process, the pEMT status of PTECs do not convert into interstitial fibroblasts but remain inside the tubule. Snai1 or Twist1 is a key factor in the process. (C) Cellular senescence. Cellular senescence is defined as a stable cell cycle arrest coupled with altered transcriptome and secretome. The senescent cells show typical changes with enlarged and flattened cell bodies, increased activity of senescence-associated β-galactosidase (SA-β-gal), and upregulation of cyclin-dependent kinase (CDK) inhibitors, such as p21Cip1 and p16Ink4a. Furthermore, senescent cells acquire senescence-associated secretory phenotype (SASP) to produce a host of proinflammatory and profibrotic cytokines. After persist and repeated injury, the metabolism and phenotype shift result in abnormal amplification of profibrogenic factors via cell-cell crosstalk. Finally, the peritubular microenvironment led to maladaptive repair with tubular atrophy, ECM accumulation, inflammatory cell infiltration and vascular rarefaction.
3 Phenotypic changes in PTECs associated with survival after injury
The partial epithelial-mesenchymal transition (pEMT) alterations and senescence are the main phenotypic changes experienced by PTECs. The introduction of the term “pEMT” was in response to the absence of proof indicating that tubular epithelial cells actively migrate across the basement membrane towards the renal interstitium (31). Residual PTECs within the tubule, as depicted in Figure 2B, experience dedifferentiation and enter epithelial G2/M arrest in order to facilitate the replacement of cells that have been lost due to apoptosis/necrosis. Initially, pEMT alterations are adaptive, allowing time for DNA repair and preventing genetic mutations. However, persistent pEMT alterations can promote TIF progression by modifying the epithelial secretome profile (32). The occurrence of accelerated senescence has been noticed in individuals with advanced clinical lesions and even in proteinuric patients who have a normal glomerular filtration rate (GFR), indicating that cellular senescence is an early occurrence in CKD (33). After kidney injury, the PT is typically the primary location for senescent cells (34). Senescence is an intricate and evolutionarily preserved process that has both advantageous and disadvantageous effects, which vary depending on whether the senescent condition is temporary or enduring (35). According to Figure 2C, when the senescent state persists, the harmful impacts of senescent cells are strongly linked to the development of a senescence-associated secretory phenotype (SASP). This phenotype generates numerous proinflammatory and profibrotic cytokines, creating an unfavorable microenvironment that contributes to chronic inflammation, adverse tissue remodeling, and fibrosis (36).
The pEMT and cell senescence, both of which involve dedifferentiation, cell cycle changes, and secretory phenotype alteration, have much in common. In fact, pEMT emphasizes phenotypic changes, and senescence emphasizes cell cycle changes. Recent research has affirmed that the inhibition of pEMT is observed when Snai1 or Twist1 is conditionally deleted in PTECs, as indicated by recent studies (32, 37). Silencing p16INK4a and p21Cip1 reduces PTEC senescence, improves tubular regeneration, and attenuates renal fibrosis in AKI and CDK models (38, 39).
4 Crosstalk between PTECs and other interstitial cells
Recent data strongly indicate that inhibiting FAO results in a phenotypic change, whereas reversing FAO reduces the impact of this change (40), suggesting that metabolic alterations may occur before phenotypic shifts in PTECs. Initially, the metabolic and phenotypic changes in PTECs in response to sublethal injury are to promote survival and escape cell death. However, this evolutionary adaptation in PTECs cannot be sustained and results in abnormal amplification of profibrogenic factors (32, 35, 41). The paracrine factors can be categorized into three groups based on the substances and size of the particles: microvesicles, exosomes, and paracrine mediators (42). Next, this review focuses on how increasing active substance crosstalk with interstitial cells promotes progressive TIF.
4.1 Crosstalk between PTECs and fibroblasts
It is difficult to find myofibroblasts in normal kidneys, however, a significant number of myofibroblasts positive for alpha smooth muscle actin (α-SMA) appear during TIF. Fibroblasts activation into myofibroblasts is considered the primary cells that are responsible for the production of extracellular matrix (ECM). During TIF, PTECs are initial reactors to injury, and myofibroblasts act as final executors. When fibroblasts cannot withstand persistent pathological damage, their roles change from “rescuer” to “destroyer.” The origins of myofibroblasts remain a subject of ongoing investigation. Although myofibroblasts have different origins, there is a widely held belief that resident fibroblasts serve as the precursors for myofibroblasts (43). However, recent studies have shown that marrow-derived fibroblasts contribute to 30% - 60% of myofibroblasts population in the fibrotic kidney, which contribute significantly to the development of renal fibrosis (44, 45). At present, only a few studies have suggested that the recruitment of marrow-derived fibroblasts into the kidney is dependent on the chemokine CXCL16 in tubular epithelial cells (46). However, the activation of marrow-derived fibroblasts may be modulated by T cells. Th2 cells secrete profibrotic factors IL-13 and IL-4 via activating transcription 6 (STAT6) signaling to stimulate marrow-derived fibroblast activation (47). Here, this review focuses on the direct experimental data of the paracrine pathways that mediate communication between PTECs and resident fibroblasts (Table 1).
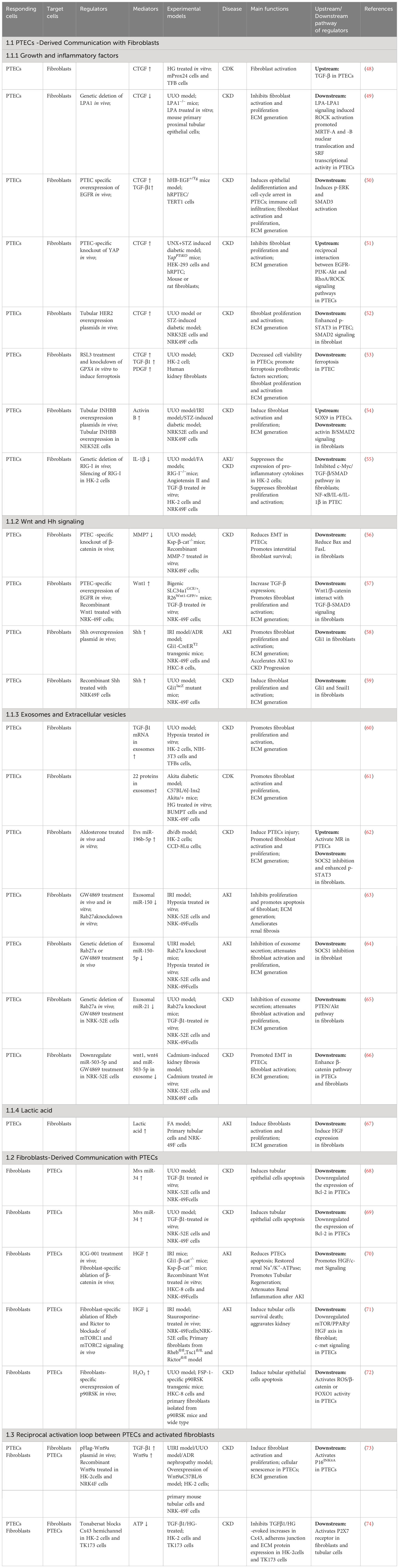
Table 1 Summary of mediators that mediate communication between proximal tubular epithelial cells and fibroblasts during kidney injury.
4.1.1 PTECs-derived communication with fibroblasts
4.1.1.1 Growth and inflammatory factors
The advancement of TIF has been linked to the involvement of transforming growth factor (TGF-β), connective tissue growth factor (CTGF), and platelet-derived growth factor (PDGF) (10). Furthermore, apart from alterations in metabolism and phenotype, ferroptosis-mediated PTEC death triggers the stimulation of neighboring fibroblasts through the enhancement of the paracrine function of pro-fibrotic substances (TGF-β1, CTGF, and PDGF) within PTECs (53). TGF-β1, out of the three TGF-β isoforms (β1, β2, β3), is regarded as a dominant initiator because it can trigger the transformation of fibroblasts into myofibroblasts and promote the production of ECM (75, 76). It is now well accepted that both canonical (SMAD-dependent) and non-canonical (SMAD-independent) signaling pathways serve as the two major downstream pathways that promote TGF-β1-mediated myofibroblast activation (77). Latency-associated peptide (LAP) forms complexes with TGF-βs when they are secreted, rendering them inactive. Integrin αvβ6 binds to TGF-β1 LAP. The hidden TGF-β1 complexes attach to αvβ6, enabling fully developed TGF-β1 to reach TGF-β receptors (TβRII) and initiate the traditional TGF-β signaling (78). Furthermore, empirical proof indicated that the transmission of TGF-β mRNA into fibroblasts through exosomes was an alternative method to trigger TGF-β activity (60). Despite the strong association between renal fibrosis and TGF-β1, attempts to treat diabetic nephropathy using clinical trials focused on TGF-β1 or neutralizing antibodies against it have not yielded any positive outcomes (79). Possible explanations for these results are that other growth factors may have negated its benefit. For example, apart from TGF-β1, activin B derived from tubules (which is a part of the TGF-β family) is involved in the development of renal fibrosis by stimulating neighboring fibroblasts through paracrine signaling in a unilateral ureteral obstruction (UUO) model (54). CTGF (also referred to as CCN2) is another growth factor that has a significant impact on the reaction to long-term kidney damage. Several signaling pathways in damaged PTECs can mediate CTGF expression in CKD. Persistent activation of YAP (yes-associated protein)) (51), HER2 (human epidermal growth factor receptor 2) (52), EGFR (epidermal growth factor receptor) (50) and the LPA-LPA1 signaling pathway (49) in tubular cells is required for activating fibroblasts by inducing CTGF.CTGF serves as a partner for TGF-β.CTGF does not possess a specific receptor, however, it seems to engage with TβRII in fibroblasts to facilitate the development of a pro-fibrotic atmosphere. CTGF has the ability to attach to lipoprotein receptor-related proteins 6 (LRP6), which results in the activation of β-catenin and the promotion of TIF (80). Patients with diabetic nephropathy exhibit increased levels of CTGF in both their plasma and urine (81, 82). TGF-β by not PDGF mediates high glucose induction of CTGF expression in PTECs. Moreover, damaged PTECs within fibrotic tissue exhibited heightened TGF-β functionality, indicated by the phosphorylation of downstream Smad2, alongside elevated CTGF levels (83). The findings from these studies suggest that several growth factors are crucial in facilitating the progression of CKD, and focusing solely on one growth factor is inadequate to modify the trajectory of TIF.
Besides the growth factors, damaged PTECs can trigger the inflammatory response by inducing various inflammatory factors such as interleukin (IL)-1β and IL-6. Multiple studies have shown that retinoic acid-inducible gene-I (RIG-I), implicated in numerous chronic inflammatory conditions (84), plays a crucial role in a proinflammatory pathway (85). In unilateral UUO and folic acid renal fibrosis models, retinoic acid-inducible gene-I (RIG-I) functions as a promoter of NF-κB (nuclear factor-kappa B) signaling, leading to the activation of NF-κB signaling and facilitating the production and secretion of IL-1β and IL-6 in PTECs. The release of proinflammatory cytokines by PTECs greatly increases the expression of c-Myc in fibroblasts, leading to the activation of TGF-β/Smad signaling-mediated fibroblast activation and the synthesis of ECM components (55).
4.1.1.2 Wnt and Hh signaling
Wnt/β-catenin signaling remains inactive in normal adults, however, it gets reactivated in the kidneys following both acute and chronic damage. The Wnt ligands consist of a group of proteins that are secreted to transmit signals. Humans have a total of 19 members belonging to the WNT family, including WNT1, WNT2, WNT2B, WNT3, WNT3A, WNT4, WNT5A, WNT5B, WNT6, WNT7A, WNT7B, WNT8A, WNT8B, WNT9A, WNT9B, WNT10A, WNT10B, WNT11, and WNT16. Each one has the ability to attach to the Frizzled and LRP5/6 (lipoprotein receptor-related proteins 5 and 6) receptors, resulting in the activation of canonical signaling pathway via β-catenin nuclear translocation and the increase in expression of Wnt target genes. Numerous researches have indicated that the timely and suitable initiation of Wnt/β-catenin signaling is commonly believed to have a protective effect (86). For example, in AKI models like IRI and folic acid nephropathy, the elimination of β-catenin specifically in the tubules has demonstrated the worsening of kidney damage through the elevation of PTEC apoptosis (87). In stark opposition to the detrimental impact of tubule-specific elimination of β-catenin, the elimination of β-catenin in fibroblasts protects kidneys from tubular apoptosis and hinders renal inflammation in AKI (70). In the CKD model, the depletion of β-catenin in the tubules does not have a significant impact on the apoptosis of tubular cells, which contradicts its function in AKI (56). There was no variation in the severity or extent of renal fibrosis after UUO when β-catenin depletion occurred, however, it enhanced the survival of interstitial fibroblasts through the MMP-7 (matrix metalloproteinases-7)/FasL (Fas ligand) pathway (56). The difference might be associated with the unique characteristics of the injuries in AKI compared to CKD. Unlike the beneficial transient activity in acute injury, continuous stimulation of Wnt signaling in PTECs is harmful and triggers epithelial pEMT, along with unregulated fibroblast activation and excessive accumulation of ECM in long-term harm (57, 88). Collectively, these findings indicate that the function of Wnt/β-catenin activation is influenced by both the stage of the disease (AKI versus CKD) and the specific tissue components (renal tubules versus fibroblasts).
Similar to Wnt/β-catenin signaling, Hedgehog (Hh) signaling controls tissue arrangement, differentiation, and cellular proliferation. Shh, one of the three Hh ligands (including Dhh and Ihh), is the most extensively studied. Shh binds to the Patched1 receptor on the membrane leads to the release of the protein smoothened (Smo), which in turn promotes the transcription of Gli1/2 effectors. The immunohistochemical staining revealed that the Shh protein was primarily found in the tubular epithelial cells, while the interstitial cells showed mostly no signs of Shh staining. Fascinatingly, only fibroblasts in the UUO model expressed the receptor Patched1 and the Gli transcriptional targets (59). In different animal models of CKD, PTECs are primarily responsible for the induction of Shh, which specifically stimulates fibroblast proliferation and worsens fibrotic kidney lesions (58). Hence, the signaling pathway of Shh/Gli1 plays a vital function in facilitating communication between epithelial cells and fibroblasts in the process of renal fibrogenesis. In the treatment of various fibrotic kidney disorders, Targeting Shh/Gli1 signaling could be considered as a new strategy.
4.1.1.3 Exosomes and extracellular vesicles
Exosomes and extracellular vesicles (EVs) have the ability to effectively transport bioactive substances, such as miRNAs, proteins, and mRNA, to facilitate the exchange of genetic information between cells (89). According to a recent investigation, twenty-two proteins discovered in tubular exosomes have the ability to facilitate tubulointerstitial communication and control the advancement of renal fibrosis in DKD (61). Exosomes can transport hypoxia-induced TGF-β mRNA, which then stimulates fibroblasts, thereby playing a role in the progression of renal fibrosis (60). Small noncoding RNAs called miRNAs control gene expression by binding target mRNAs, leading to degradation or translational repression. In addition to directly controlling the expression of target genes in target cells, miRNAs can also regulate the expression of target genes in different cells by delivering them through exosomes. After an ischemia‒reperfusion incident, exosomes derived from PTECs stimulate fibroblast activation and proliferation through the action of miR-150-5p and miR-150 (63, 64). MiR-21, extensively researched, is known to be involved in the control of fibroblast proliferation (90). In vitro, the activation of fibroblasts was expedited by miR-21-containing exosomes derived from epithelial cells, while in vivo, the presence of miR-21-deficient exosomes alleviated renal fibrosis after UUO by acting on the PTEN/Akt pathway (65). Rab27a, a member of the RAB GTPase family, plays a significant role in the production and release of exosomes (91). Multiple studies suggest that Rab27a knockout or GW4869 (exosome secretion inhibition) treatment can ameliorate renal fibrosis in AKI and CKD models (64, 65). In addition to profibrotic miRNAs, PTECs-derived exosomal miR-503-5p protects against cadmium nephrotoxicity and is capable of suppressing pEMT and fibroblast activation (66). Novel mediators of cellular crosstalk are emerging in the form of PTEC-derived EVs, which have a larger volume compared to exosomes (92). In the study conducted by Renzhi Hu, it was found that miR-196b-5p (EVs) facilitate communication between proximal tubular epithelial cells (PTECs) and fibroblasts in the development of renal fibrosis caused by aldosterone in individuals with diabetes. This communication is achieved by suppressing the expression of SOCS2 and promoting the phosphorylation of Stat3, as stated in the research (62).
4.1.1.4 Lactic acid
Lactic acid, the final product of glycolysis, has been traditionally considered a metabolic byproduct. Recently, the role of lactic acid in cell proliferation has gained significant attention (93). Fibroblasts take up lactate excreted by tubules via monocarboxylate transporter-1 (MCT1). Tubule-derived lactic acid is required for fibroblast proliferation, which is considered to facilitate repair in the early phase of AKI (67). This protective effect of lactic acid could potentially be linked to instructing fibroblasts in generating hepatocyte growth factor (HGF) (67, 94). However, in a CKD model, inhibiting aerobic glycolysis in PTECs suppressed fibroblasts from transforming into matrix-producing myofibroblasts (95). These results suggest the differential roles of lactic acid in different stages of disease progression.
4.1.2 Fibroblasts-derived communication with PTECs
When faced with injury, certain PTECs undergo apoptotic death, resulting in the destruction of the kidney parenchyma. Initially, fibroblasts create a conducive atmosphere to facilitate the regeneration of PTECs in response to temporary damage. HGF, a protein with renoprotective properties, is synthesized by fibroblasts that binds to the c-met receptor found on both PTECs and fibroblasts. The downstream effects of HGF (hepatocyte growth factor)/c-met (cellular-mesenchymal epithelial transition factor) signal transduction include cell survival, proliferation, differentiation, and migration (96). In an AKI model, activation of mTORC1 (mammalian target of rapamycin complex 1) and mTORC2 (mammalian target of rapamycin complex 2) signaling in fibroblasts safeguards against ischemia/reperfusion-induced PTECs death by inducing HGF. β-Catenin is an upstream negative regulator of HGF. In AKI, the reduction of PTECs apoptosis and the promotion of regeneration are achieved by increasing HGF expression through the specific elimination of β-catenin in fibroblasts (70, 71). After kidney injury, tubules serve as the primary origin of Wnt ligands (97). As mentioned previously, overexpressed Wnt1 in PTECs could specifically target fibroblasts, resulting in the activation of β-catenin (57). Consequently, it suppresses the expression of HGF in fibroblasts and lead to PTECs apoptosis. The findings suggest that the interaction between Wnt and HGF signaling facilitates the bidirectional communication between PTECs and fibroblasts, playing a crucial role in promoting TIF progression.
The UUO model demonstrates that the stimulation of 90 kDa ribosomal protein S6 kinase 1 (p90RSK) triggers kidney fibrosis by causing fibroblast-induced PTECs apoptosis through the activation of the β-catenin/FOXO1 signaling pathway via ROS (98). The secretion of detrimental molecules through microvesicles was previously reported to be a self-defense strategy in cells (99). In the CKD model, miR-34a was reported to promote cell apoptosis by inhibiting the anti-apoptotic Bcl-2, which was upregulated in the fibroblasts. The injured fibroblasts secrete miR-34a via microvesicles to avoid apoptosis. Nonetheless, the miR-34a carried in microvesicles subsequently disrupts the tubular basement membrane and triggers PTECs apoptosis by specifically targeting Bcl-2 (B-cell lymphoma-2) (68, 69). These findings suggest that persistent harmful triggers in CKD can lead to extensive activation of signaling agents and disrupt the defensive impact of fibroblasts, leading to progressive tissue damage and functional decline.
4.1.3 Reciprocal activation between PTECs and fibroblasts
The pro-fibrotic microenvironment is facilitated by a reciprocal activation loop between senescent/pEMT epithelial cells and activated fibroblasts. Recently, experimental data further confirmed that Wnt9a is involved in regulating communication via paracrine or autocrine mechanisms. The induction of senescent tubular cells by Wnt9a resulted in the secretion of TGF-β1, a component of the SASP. This secretion subsequently caused the proliferation of interstitial fibroblasts and the transformation into myofibroblasts. Subsequently, TGF-β1 stimulated the expression of Wnt9a in fibroblasts, resulting in the establishment of a mutual activation cycle between senescent tubular cells and activated fibroblasts (73). Connexin 43 (Cx43) is a connexin (Cx) that constitutes gap junctions. Adjacent cells can directly exchange small signaling molecules. In vitro, Cx43 and P2X7R are critical factors contributing to epithelial-fibroblast communication circuits. Blocking Cx43-mediated hemichannel ATP release can decrease P2X7 receptor (P2X7R) activation in PTECs and fibroblasts, thus preventing diabetic nephropathy by blocking epithelial cells phenotype transformation and fibroblast activation (74).
4.2 Crosstalk between PTECs and macrophages
Renal fibrosis encompasses different types of inflammatory cells, with macrophages being the primary immune cell involved in renal tissue, ultimately leading to the development of renal fibrosis (100). Macrophages not only detect different inflammatory stimuli but are also affected by diverse cell populations, resulting in a change in the proinflammatory characteristics and the secretion of inflammatory agents (101). Several research studies have indicated that macrophages possess the ability to release pro-fibrotic substances and have a multifaceted impact on the development of renal fibrosis (102). Initially, in response to injury, PTECs release different chemokines and cytokines in the vicinity of peritubular compartments to lure and guide the entry of macrophages into the tubulointerstitial area. Afterward, macrophages become active and generate a blend of soluble substances, which consist of inflammatory cytokines, fibrotic cytokines, and MMPs. In this section, we provide a summary of empirical evidence regarding the paracrine mechanisms that facilitate communication between epithelial cells and macrophages (Table 2).
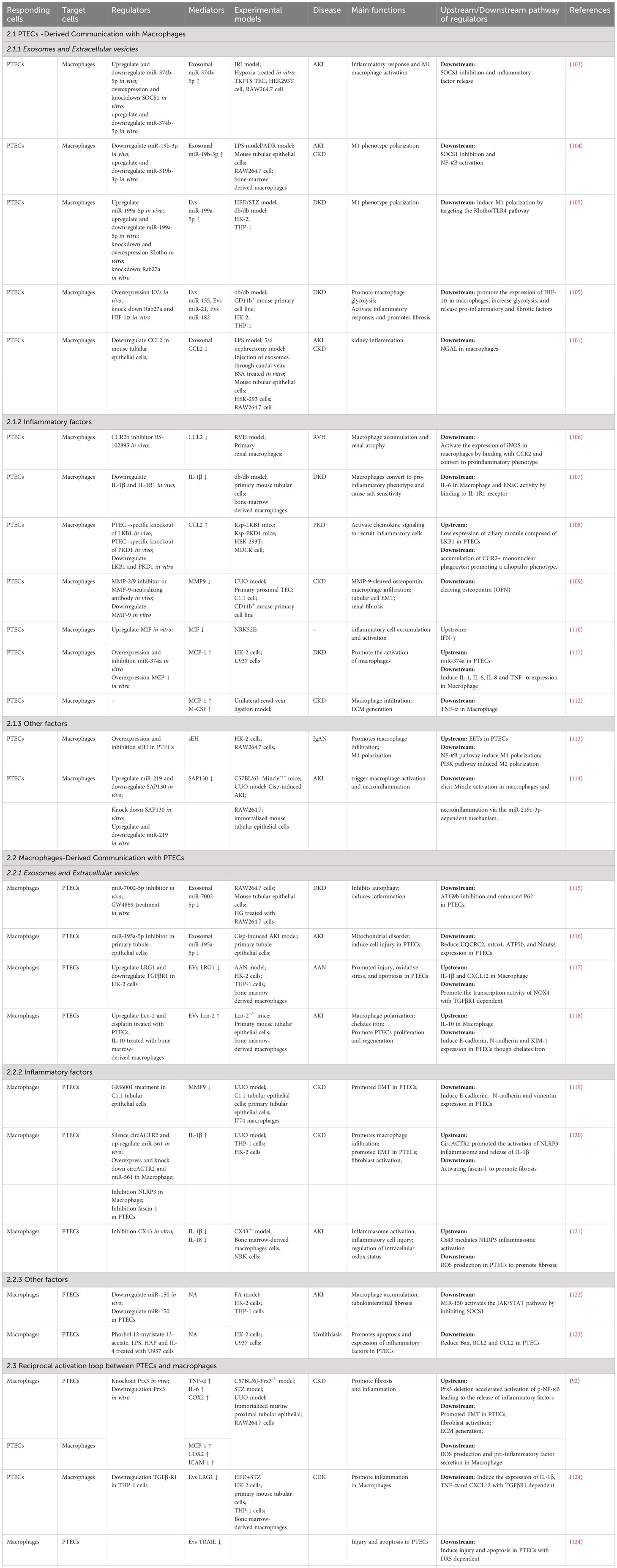
Table 2 Summary of mediators that mediate communication between proximal tubular epithelial cells and macrophages during kidney injury.
4.2.1 PTECs-derived communication with macrophages
4.2.1.1 Exosomes and Extracellular Vesicles
MiRNAs are the predominant intracellular communication molecules among the exosomes and EVs derived from PTECs, which induce phenotypic changes in macrophages. Exosomes derived from PTECs have demonstrated the ability to suppress the expression of suppressor of cytokine signaling-1 (SOCS1) in macrophages, resulting in the conversion of macrophages to the M1 phenotype (103, 104). Likewise, miR-199a-5p in EVs originating from human serum albumin-induced PTECs triggered M1 polarization and expedited the advancement of DKD by targeting the Klotho/TLR4 (toll like receptor 4) pathway (92). It has also been shown that PTECs-derived EVs (containing miRNA-155, miRNA-21 and miRNA-182) induce glycolysis in renal macrophages by inducing the overexpressing HIF-1α (105). Macrophages can release factors and glycolytic products that directly contribute to tissue fibrosis when there is an elevated glycolytic flux (105). These studies indicate that PTECs-derived EVs can not only affect the phenotype of macrophages but also affect the metabolic status of macrophages.
In addition to miRNAs, EVs can carry proteins for cell-to-cell information transfer. Chemokine 2 (CCL2), also referred to as monocyte chemoattractant protein or monocyte chemoattractant protein-1 (MCP-1), is an inflammatory factor that has demonstrated a crucial function in the infiltration of macrophages, resulting in tissue damage (125). CCL2 is produced by injured PTECs and transported to macrophages in the form of EVs, which plays a key role in albumin-induced tubulointerstitial inflammation (101). CCL2 is not only transmitted as EVs between PTECs and macrophages but also mediates signal transmission through receptor-ligand binding. Chemokine receptor 2 (CCR2) is the primary receptor for CCL2 and is found in renal macrophages (106). Research has indicated that CCL2/CCR2 signaling enhances the pro-inflammatory characteristics of immune cells in different experimental models of CKD (106, 108).
4.2.1.2 Inflammatory factors
In addition to secreting numerous inflammatory factors, macrophages can also be influenced by inflammatory factors like IL-1β, matrix metalloprotein 9 (MMP9), migration inhibitory factor (MIF), and monocyte chemoattractant protein-1 (MCP-1). IL-1β released from PTECs polarizes macrophages to a pro-inflammatory phenotype, and these cells release IL-6 through an IL-1β/IL-1βR binding mode (107). Notably, this research demonstrated that PTECs rather than macrophages serve as the primary origin of IL-1β. Furthermore, the buildup of IL-6 can result in renal inflammation and salt sensitivity, which exhibits a strong correlation with the development of hypertension. The cellular sources of MMP-9 are associated with different stages of nephropathy. During the initial phase, PTECs serve as the primary source. With the progression of TIF, PTECs, macrophages and myofibroblasts can all produce MMP-9 (109). Additional macrophages may be recruited by MMP-9 derived from macrophages and PTECs macrophages through osteopontin cleavage (126). Inhibition of MMP-9 in the early phase of renal fibrosis and the late phase of nephropathy could decrease tubular cell pEMT (109). Macrophage migration inhibitory factor (MIF) plays a crucial role in both the innate and adaptive immune systems by activating macrophages (127). MIF is enzymatically active, setting it apart from other pro-inflammatory cytokines (128), and has the ability to control cell proliferation and differentiation (129). PTECs have demonstrated the ability of interferon-γ (110) to enhance the production and release of MIF. Monocyte chemoattractant protein (MCP)-1 is a chemokine responsible for attracting macrophages to inflammation areas. Damaged PTECs can also secrete MCP-1, which is negatively regulated by miR-374a (111), leading to increased macrophage infiltration and recruitment in the kidney (112).
4.2.1.3 Other factors
Soluble epoxide hydrolase (sEH) metabolizes epoxyeicosatrienoic acids (EETs) into dihydroxyeicosatrienoic acids (DHETs). It is believed that the function of sEH is the primary factor affecting the availability of EET (130). EET has various biological functions, such as vasodilation and inhibiting inflammation (113). The excessive expression of sEH in PTECs triggers the NF-κB pathway in macrophages, resulting in the activation of M1 macrophages. Conversely, suppression of sEH expression promotes M2 polarization via the PI3K pathway, in the context of IgA nephropathy (113). According to recent research, endogenous damage-associated molecular patterns (DAMPs) have the potential to activate the innate immune system. Endogenous molecules released after cell injury can activate inflammatory pathways during aseptic inflammation (131). Damaged cells release these molecules which then stimulate pattern recognition receptors (PRRs) found on immune cells (132, 133). Mincle, a crucial receptor for recognizing patterns, detects cell death and triggers the release of inflammatory cytokines, leading to the influx of inflammatory cells into injured tissues (134). Sin3A associated protein 130 (SAP130) belongs to the small ribose protein group and exits from injured PTECs. It acts as a recognized natural binder of Mincle in macrophages (135). SAP130 binding to Mincle can drive necroinflammation in AKI (136).
4.2.2 Macrophages-derived communication with PTECs
4.2.2.1 Exosomes and extracellular vesicles
Paracrine communication also occurs during macrophage-derived communication with PTECs. Single-stranded miRNAs are the most extensively studied factors (137). Macrophages can encapsulate miR-7002-5p and miR-195a-5p in exosomes. Specifically, exosomes carry excess miR-7002-5p and inhibit autophagy by targeting Atg9b in PTECs in the presence of high glucose, which subsequently induces tubular dysfunction and inflammation (115). miR-195a-5p is upregulated in a model of cisplatin-induced AKI. Macrophages transport miR-195a-5p through exosomes, resulting in tubular mitochondrial dysfunction and cell damage (116).
Proteins are also important paracrine mediators (138). LRG1 (leucine-rich-alpha-2-glycoprotein 1), also known as leucine-rich alpha-2-glycoprotein 1, has the ability to directly interact with TGF-β receptor 1 (TGF-βR1) and modify TGF-β signaling to produce its effects. Macrophages release extracellular vesicles (EVs) abundant in LRG1, which have been demonstrated to cause injury and apoptosis in PTECs via TGF-β1-dependent mechanisms (117). Following renal injury, macrophages communicate unidirectionally with PTECs, resulting in negative effects. However, a 2020 study revealed that macrophages with an anti-inflammatory phenotype can release macrophage-secreted lipocalin-2 (Lcn-2) EV2.Lcn-2 promotes PTEC regeneration after injury by chelating iron (118). The discovery implies that the type of macrophage affects the inflammatory surroundings and has a crucial part in deciding the course of PTEC restoration (139).
4.2.2.2 Inflammatory factors
Macrophages can release a variety of inflammatory factors, but only a few factors have been reported to act on PTECs. Activated macrophage-conditioned medium is enriched in MMP-9 and can induce EMT phenotypic transformation in PTECs (119). In macrophages, circACTR2 has the ability to attach to miRNAs, stimulate the activation of NLRP3 (nOD-like receptor thermal protein domain associated protein 3) inflammasome, and trigger the secretion of IL-1β. Additionally, in PTECs, the upregulation of fascin-1 expression caused by IL-1β leads to renal fibrosis (120). Disruption of the redox equilibrium in macrophages can partially trigger the activation of the NLRP3 inflammasome, resulting in the release of IL-1β and IL-18 (120). In addition, Cx43 can activate the NLRP3 inflammasome, producing the same effect as CircACTR2 (121).
4.2.2.3 Other factors
In kidney disease, although cellular communication has been extensively studied, many mechanisms remain unknown. MiR-150 was significantly upregulated in PTECs after coculture with macrophages. The presence of this element has the ability to inhibit the SOCS1/JAK (janus kinase)/STAT(signal transducer and activator of transcription) pathway, resulting in a reduction in the infiltration of macrophages (122). PTECs can aggravate apoptosis and proinflammatory factor expression in response to stimulation with conditioned medium derived from macrophages (123). The intermediate conditions in which these phenomena occur require further exploration.
4.2.3 Reciprocal activation between PTECs and macrophages
Communication between PTECs and macrophages is not unidirectional, and feedback can occur. Damaged tubular epithelial cells trigger the activation of macrophages, which subsequently secrete pro-inflammatory cytokines, leading to the induction of apoptosis in the injured cells. Peroxide reductases (Prxs) belong to a group of alcohol peroxidases that function as cellular thiol peroxidases. Among the six members, Prx3 stands out as a distinctive antioxidant (140). It specifically localizes to mitochondria and has a crucial function in preserving mitochondrial reactive oxygen species (ROS) (141), while oxidative stress caused by ROS serves as a significant mediator of CKD. The acceleration of CKD through positive feedback between macrophages and PTECs has been demonstrated by the removal of Prx3 (92). The deletion of Prx3 in PTECs accelerates the development of fibrosis and inflammation, which are accompanied by increased oxidative stress in the mitochondria. Furthermore, the deletion of Prx3 triggers the activation of macrophages, resulting in the increase of pro-inflammatory cytokines and the expedited release of pro-inflammatory and profibrotic cytokines by PTECs. Similarly, there is a negative feedback loop between PTECs and macrophages. Several research studies have indicated that EVs derived from PTECs stimulate the inflammatory macrophage phenotype and trigger the secretion of EVs derived from macrophages (101). Moreover, EVs trigger programmed cell death in PTECs. Notably, EVs enriched with leucine-rich alpha-2-glycoprotein 1 (LRG1) stimulate macrophages through TGF-βR1-dependent mechanisms, while EVs enriched with tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) trigger apoptosis in injured PTECs through death receptor 5 (DR5)-dependent mechanisms (124). To sum up, the reciprocal communication between PTECs and macrophages plays a crucial role in the development and upkeep of homeostasis in mammals, and it has the potential to expedite the advancement of kidney disease.
4.3 Crosstalk between PTECs and endothelial cells
Most nutrient exchange processes occur in the adjacent tubular epithelium, which is tightened by the peritubular capillary network in the renal cortex. Substances and water are absorbed again from the PT, absorbed by the vascular system, and returned to the circulation, while the substance is also secreted into the PT. Nephrogenesis and the assembly of microvessels are closely linked in the developing kidney (142). In chronic kidney disease, the separation of endothelial cells, which are thin cells positioned amidst the vessel wall and the flow of blood, results in a scarcity of blood vessels and inadequate blood provision, thereby exacerbating renal fibrosis. The complex communication networks between epithelial cells and endothelial cells have been demonstrated to impact angiogenesis, regulation of solutes, and the high expression of certain genes (143).
The gene family of vascular endothelial growth factor (VEGF) is composed of VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor (PlGF). Originally identified as a factor that increases the permeability of blood vessels (144), VEGF-A plays a crucial part in the development of blood vessels by stimulating angiogenesis. VEGF-A is found on various types of cells, such as PTECs, and its receptor (Kdr/Vegfr2) is believed to be primarily situated on peritubular capillary endothelial cells in the vasculature behind the glomerulus (145). The demonstration of VEGF’s involvement in preserving elevated permeability and widespread angiogenesis in various tumors and endocrine glands has been established (146). EVs secretion by upregulating COX-2 expression endothelial cells have been shown to increase HIF-1α expression and VEGF-A production in PTECs (147). PTECs induce VEGF-dependent capillary vessel formation in cocultured endothelial cells, and hypoxia further increases angiogenesis. Further studies showed that knockdown of the VEGF-A gene in PTECs resulted in selective shedding of peritubular microvessels. The specific mechanism of VEGF-A in renal PTECs is achieved by mediating crosstalk in VEGFR2-expressing endothelial cells. This form of communication is also linked to a significant rise in Epo production within the kidney, resulting in significant erythrocytosis (145).
TNF-α receptors (148) have been observed in the S2 and S3 sections of proximal tubules, indicating the presence of PTECs. When cocultured with endothelial cells (HGECs), epithelial cells may produce inflammatory mediators by sensing TNF-α released from HGECs (149). Furthermore, transcriptomic analysis showed that differentially expressed genes in PTECs were mainly enriched in extracellular matrix (e.g., lysyl oxidase), intercellular communication (e.g., IL-6), and transport pathways (e.g., neutrophil gelatinase-associated lipocalin 2) in coculture systems involving endothelial cells. On the other hand, the genes that were expressed differently in endothelial cells mainly acted as regulators of epithelial cell function, such as proteins in the extracellular matrix (e.g., collagen I, III, IV and V, laminin IV) and cytokines/growth factors (e.g., HGF, VEGF-C, etc.) (150). The discovery reveals an intricate communication system between microvascular endothelial cells and epithelial cells that ultimately affects PTECs function.
5 Conclusion
CKD is most commonly attributed to diabetes, hypertension and AKI.As the early CKD caused by these diseases are often asymptomatic, the Kidney Disease Improving Global Outcomes (KDIGO) conference emphasized the importance of early identification and early treatments of CKD in order to delay the advancement of the disease (151). Hence, it is imperative to ascertain the initial cellular occurrences that trigger renal fibrosis and discover novel targets for therapeutic intervention. The high energy consumption and metabolic characteristics of PTECs established their pivotal role as an initial cell affected in the early phase of renal injury. To survive, PTECs undergo shifts toward glycolysis enhancement, pEMT and cellular senescence. These transitions can be either positive or negative depending on their timing and duration. Except for HGF and MIF, the majority of active compounds facilitate this harmful communication between the damaged epithelial cells and interstitial cells in CKD (Figure 3). Therefore, understanding the timing of targeted PTECs phenotypic shifts and clarifying the mechanism of the intricate system of communication may be the key to preventing and treating nephropathy in the future.
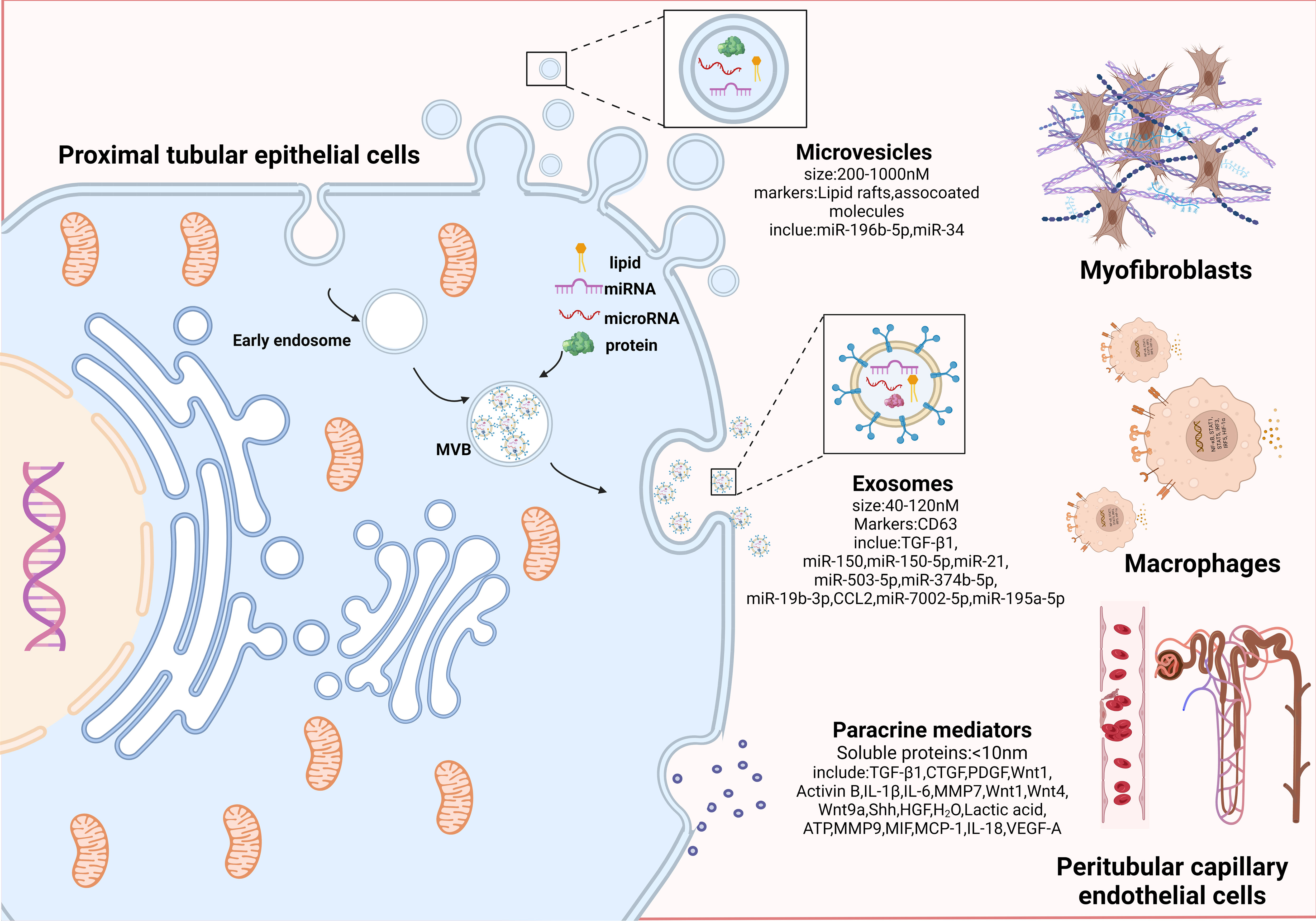
Figure 3 Proximal tubular epithelial cells crosstalk with other interstitial cells to promote progressive tubulointerstitial fibrosis. An overview of paracrine manner. Paracrine signals can be divided into three categories according to effector substances and secretion particle size: microvesicles, exosomes, and paracrine mediators. Microvesicles (200-1000nM in size; markers: Lipid rafts and associated molecules) are shed directly from the plasma membrane by outward budding; exosomes are small membrane particles (40 to 120nM in size; markers: CD63) released by fusion of multivesicular bodies with the plasma membrane, which encapsulate miRNAs and proteins internally; paracrine mediators are the secretion of various paracrine factors composed of soluble proteins by fusion of paracrine granules with the plasma membrane. Among them, microvesicles and exosomes are collectively referred to as extracellular vesicles (EVs), because they can efficiently carry bioactive molecules including mRNA, proteins, and microRNAs to facilitate transfer of genetic information between different cells. PTECs affect myofibroblasts activation and ECM production through bioactive substances (miR-196b-5p, TGF-β1, etc.). PTECs mediates macrophage activation and accumulation by secreting inflammatory mediators (CCL2, MMP9, IL-18, etc.). Communication between PTECs and epithelial occurs via paracrine secretion of VEGF-A. Combined with the above intercellular communication medias centered on PTECs, they jointly lead to the progression of TIF.
Author contributions
CG: Conceptualization, Data curation, Formal analysis, Funding acquisition, Writing – original draft. YC: Data curation, Formal analysis, Visualization, Writing – original draft. MJ: Software, Writing – review & editing. JY: Writing – review & editing, Investigation. JZ: Data curation, Writing – original draft. YT: Data curation, Visualization, Writing – review & editing. JD: Supervision, Writing – review & editing. LL: Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded by National Natural Science Foundation of China Grants (NO.82170847,82105044), key project of Shandong Provincial Natural Science Foundation of China Grants (NO. ZR2020KH004); Shandong Provincial Natural Science Foundation of China Grants (No. ZR 2020QH310).
Acknowledgments
Schematics were created with BioRender.com. (https://app.biorender.com/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Global, regional, and national burden of chronic kidney disease 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet (London England) (2020) 395:709–33. doi: 10.1016/S0140-6736(20)30045-3
2. Kuwahara M, Takehara E, Sasaki Y, Azetsu H, Kusaka K, Shikuma S, et al. Effects of cardiovascular events on end-stage renal disease and mortality in patients with chronic kidney disease before dialysis. Ther Apher Dial (2016) 20:12–9. doi: 10.1111/1744-9987.12332
3. Hewitson TD. Renal tubulointerstitial fibrosis: common but never simple. Am J Physiol Renal Physiol (2009) 296:F1239–44. doi: 10.1152/ajprenal.90521.2008
4. Rodriguez-Iturbe B, Garcia Garcia G. The role of tubulointerstitial inflammation in the progression of chronic renal failure. Nephron Clin Pract (2010) 116:c81–8. doi: 10.1159/000314656
5. Phillips AO, Steadman R. Diabetic nephropathy: the central role of renal proximal tubular cells in tubulointerstitial injury. Histol Histopathology (2002) 17:247–52. doi: 10.14670/HH-17.247
6. Ruilope LM, Agarwal R, Anker SD, Bakris GL, Filippatos G, Nowack C, et al. Design and baseline characteristics of the finerenone in reducing cardiovascular mortality and morbidity in diabetic kidney disease trial. Am J Nephrol (2019) 50:345–56. doi: 10.1159/000503712
7. Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol (2020) 16:269–88. doi: 10.1038/s41581-019-0248-y
8. Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, et al. Severity and frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol (2016) 27:2393–406. doi: 10.1681/ASN.2015060647
9. Tan RJ, Zhou D, Liu Y. Signaling crosstalk between tubular epithelial cells and interstitial fibroblasts after kidney injury. Kidney Dis (Basel) (2016) 2:136–44. doi: 10.1159/000446336
10. Gewin L, Zent R, Pozzi A. Progression of chronic kidney disease: too much cellular talk causes damage. Kidney Int (2017) 91:552–60. doi: 10.1016/j.kint.2016.08.025
11. Stormark TA, Strommen K, Iversen BM, Matre K. Three-dimensional ultrasonography can detect the modulation of kidney volume in two-kidney, one-clip hypertensive rats. Ultrasound Med Biol (2007) 33:1882–8. doi: 10.1016/j.ultrasmedbio.2007.06.010
12. Yu ASL. Paracellular transport and energy utilization in the renal tubule. Curr Opin Nephrol Hypertens (2017) 26:398–404. doi: 10.1097/MNH.0000000000000348
13. Uchida S, Endou H. Substrate specificity to maintain cellular ATP along the mouse nephron. Am J Physiol (1988) 255:F977–83. doi: 10.1152/ajprenal.1988.255.5.F977
14. Gewin LS. Sugar or fat? Renal tubular metabolism reviewed in health and disease. Nutrients (2021) 13:1580. doi: 10.3390/nu13051580
15. Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol (2017) 13:629–46. doi: 10.1038/nrneph.2017.107
16. Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, et al. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol (2011) 22:1505–16. doi: 10.1681/ASN.2011020203
17. Xu C, Zhou X, Xie T, Zhou Y, Zhang Q, Jiang S, et al. Renal tubular Bim mediates the tubule-podocyte crosstalk via NFAT2 to induce podocyte cytoskeletal dysfunction. Theranostics (2020) 10:6806–24. doi: 10.7150/thno.43145
18. Liu BC, Tang TT, Lv LL, Lan HY. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int (2018) 93:568–79. doi: 10.1016/j.kint.2017.09.033
19. El-Achkar TM, Dagher PC. Tubular cross talk in acute kidney injury: a story of sense and sensibility. Am J Physiol Renal Physiol (2015) 308:F1317–23. doi: 10.1152/ajprenal.00030.2015
20. Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol (2014) 306:F367–78. doi: 10.1152/ajprenal.00571.2013
21. Lan R, Geng H, Singha PK, Saikumar P, Bottinger EP, Weinberg JM, et al. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J Am Soc Nephrol (2016) 27:3356–67. doi: 10.1681/ASN.2015020177
22. Kwan B, Fuhrer T, Zhang J, Darshi M, Van Espen B, Montemayor D, et al. Metabolomic markers of kidney function decline in patients with diabetes: evidence from the chronic renal insufficiency cohort (CRIC) study. Am J Kidney Dis (2020) 76:511–20. doi: 10.1053/j.ajkd.2020.01.019
23. Maremonti F, Meyer C, Linkermann A. Mechanisms and models of kidney tubular necrosis and nephron loss. J Am Soc Nephrol (2022) 33:472–86. doi: 10.1681/ASN.2021101293
24. Zhou X, Xu C, Dong J, Liao L. Role of renal tubular programed cell death in diabetic kidney disease. Diabetes Metab Res Rev (2023) 39:e3596. doi: 10.1002/dmrr.3596
25. Harris DC, Chan L, Schrier RW. Remnant kidney hypermetabolism and progression of chronic renal failure. Am J Physiol (1988) 254:F267–76. doi: 10.1152/ajprenal.1988.254.2.F267
26. Simon N, Hertig A. Alteration of fatty acid oxidation in tubular epithelial cells: from acute kidney injury to renal fibrogenesis. Front Med (2015) 2:52. doi: 10.3389/fmed.2015.00052
27. Idrovo JP, Yang WL, Nicastro J, Coppa GF, Wang P. Stimulation of carnitine palmitoyltransferase 1 improves renal function and attenuates tissue damage after ischemia/reperfusion. J Surg Res (2012) 177:157–64. doi: 10.1016/j.jss.2012.05.053
28. Miguel V, Tituana J, Herrero JI, Herrero L, Serra D, Cuevas P, et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest (2021) 131:e140695. doi: 10.1172/JCI140695
29. Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Committees and investigators, dapagliflozin in patients with chronic kidney disease. N Engl J Med (2020) 383:1436–46. doi: 10.1056/NEJMoa2024816
30. Li J, Liu H, Takagi S, Nitta K, Kitada M, Srivastava SP, et al. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight (2020) 5:e129034. doi: 10.1172/jci.insight.129034
31. Lovisa S, Zeisberg M, Kalluri R. Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol Metab (2016) 27:681–95. doi: 10.1016/j.tem.2016.06.004
32. Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med (2015) 21:998–1009. doi: 10.1038/nm.3902
33. Verzola D, Gandolfo MT, Gaetani G, Ferraris A, Mangerini R, Ferrario F, et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol (2008) 295:F1563–73. doi: 10.1152/ajprenal.90302.2008
34. Xu J, Zhou L, Liu Y. Cellular senescence in kidney fibrosis: pathologic significance and therapeutic strategies. Front Pharmacol (2020) 11:601325. doi: 10.3389/fphar.2020.601325
35. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol (2021) 22:75–95. doi: 10.1038/s41580-020-00314-w
36. Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest (2013) 123:966–72. doi: 10.1172/JCI64098
37. Grande MT, Sanchez-Laorden B, Lopez-Blau C, De Frutos CA, Boutet A, Arevalo M, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med (2015) 21:989–97. doi: 10.1038/nm.3901
38. Lee DH, Wolstein JM, Pudasaini B, Plotkin M. INK4a deletion results in improved kidney regeneration and decreased capillary rarefaction after ischemia-reperfusion injury. Am J Physiol Renal Physiol (2012) 302:F183–91. doi: 10.1152/ajprenal.00407.2011
39. Jin J, Tao J, Gu X, Yu Z, Wang R, Zuo G, et al. P16 (INK4a) deletion ameliorated renal tubulointerstitial injury in a stress-induced premature senescence model of bmi-1 deficiency. Sci Rep (2017) 7:7502. doi: 10.1038/s41598-017-06868-8
40. Kang HM, Ahn SH, Choi P, Ko YA, Han SH, Chinga F, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med (2015) 21:37–46. doi: 10.1038/nm.3762
41. Yin XN, Wang J, Cui LF, Fan WX. Enhanced glycolysis in the process of renal fibrosis aggravated the development of chronic kidney disease. Eur Rev Med Pharmacol Sci (2018) 22:4243–51. doi: 10.26355/eurrev_201807_15419
42. Kusuma GD, Carthew J, Lim R, Frith JE. Effect of the microenvironment on mesenchymal stem cell paracrine signaling: opportunities to engineer the therapeutic effect. Stem Cells Dev (2017) 26:617–31. doi: 10.1089/scd.2016.0349
43. Mack M, Yanagita M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int (2015) 87:297–307. doi: 10.1038/ki.2014.287
44. An C, Jia L, Wen J, Wang Y. Targeting bone marrow-derived fibroblasts for renal fibrosis. Adv Exp Med Biol (2019) 1165:305–22. doi: 10.1007/978-981-13-8871-2_14
45. An C, Jiao B, Du H, Tran M, Song B, Wang P, et al. Jumonji domain-containing protein-3 (JMJD3) promotes myeloid fibroblast activation and macrophage polarization in kidney fibrosis. Br J Pharmacol (2023) 180:2250–65. doi: 10.1111/bph.16096
46. Chen G, Lin SC, Chen J, He L, Dong F, Xu J, et al. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol JASN (2011) 22:1876–86. doi: 10.1681/ASN.2010080881
47. Jiao B, An C, Du H, Tran M, Wang P, Zhou D, et al. STAT6 deficiency attenuates myeloid fibroblast activation and macrophage polarization in experimental folic acid nephropathy. Cells (2021) 10:3057. doi: 10.3390/cells10113057
48. Kobayashi T, Inoue T, Okada H, Kikuta T, Kanno Y, Nishida T, et al. Connective tissue growth factor mediates the profibrotic effects of transforming growth factor-beta produced by tubular epithelial cells in response to high glucose. Clin Exp Nephrol (2005) 9:114–21. doi: 10.1007/s10157-005-0347-x
49. Sakai N, Chun J, Duffield JS, Lagares D, Wada T, Luster AD, et al. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int (2017) 91:628–41. doi: 10.1016/j.kint.2016.09.030
50. Overstreet JM, Wang Y, Wang X, Niu A, Gewin LS, Yao B, et al. Selective activation of epidermal growth factor receptor in renal proximal tubule induces tubulointerstitial fibrosis. FASEB J (2017) 31:4407–21. doi: 10.1096/fj.201601359RR
51. Chen J, Wang X, He Q, Bulus N, Fogo AB, Zhang MZ, et al. YAP activation in renal proximal tubule cells drives diabetic renal interstitial fibrogenesis. Diabetes (2020) 69:2446–57. doi: 10.2337/db20-0579
52. Li H, Shao F, Qian B, Sun Y, Huang Z, Ding Z, et al. Upregulation of HER2 in tubular epithelial cell drives fibroblast activation and renal fibrosis. Kidney Int (2019) 96:674–88. doi: 10.1016/j.kint.2019.04.012
53. Zhang B, Chen X, Ru F, Gan Y, Li B, Xia W, et al. Liproxstatin-1 attenuates unilateral ureteral obstruction-induced renal fibrosis by inhibiting renal tubular epithelial cells ferroptosis. Cell Death Dis (2021) 12:843. doi: 10.1038/s41419-021-04137-1
54. Sun Y, Cai H, Ge J, Shao F, Huang Z, Ding Z, et al. Tubule-derived INHBB promotes interstitial fibroblast activation and renal fibrosis. J Pathol (2022) 256:25–37. doi: 10.1002/path.5798
55. Zhou Z, Ni J, Li J, Huo C, Miao N, Yin F, et al. RIG-I aggravates interstitial fibrosis via c-Myc-mediated fibroblast activation in UUO mice. J Mol Med (Berl) (2020) 98:527–40. doi: 10.1007/s00109-020-01879-x
56. Zhou D, Tan RJ, Zhou L, Li Y, Liu Y. Kidney tubular beta-catenin signaling controls interstitial fibroblast fate via epithelial-mesenchymal communication. Sci Rep (2013) 3:1878. doi: 10.1038/srep01878
57. Maarouf OH, Aravamudhan A, Rangarajan D, Kusaba T, Zhang V, Welborn J, et al. Paracrine wnt1 drives interstitial fibrosis without inflammation by tubulointerstitial cross-talk. J Am Soc Nephrol (2016) 27:781–90. doi: 10.1681/ASN.2014121188
58. Zhou D, Li Y, Zhou L, Tan RJ, Xiao L, Liang M, et al. Sonic hedgehog is a novel tubule-derived growth factor for interstitial fibroblasts after kidney injury. J Am Soc Nephrol (2014) 25:2187–200. doi: 10.1681/ASN.2013080893
59. Ding H, Zhou D, Hao S, Zhou L, He W, Nie J, et al. Sonic hedgehog signaling mediates epithelial-mesenchymal communication and promotes renal fibrosis. J Am Soc Nephrol (2012) 23:801–13. doi: 10.1681/ASN.2011060614
60. Borges FT, Melo SA, Ozdemir BC, Kato N, Revuelta I, Miller CA, et al. TGF-beta1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J Am Soc Nephrol (2013) 24:385–92. doi: 10.1681/ASN.2012101031
61. Wen J, Ma Z, Livingston MJ, Zhang W, Yuan Y, Guo C, et al. Decreased secretion and profibrotic activity of tubular exosomes in diabetic kidney disease. Am J Physiol Renal Physiol (2020) 319:F664–73. doi: 10.1152/ajprenal.00292.2020
62. Hu R, Li X, Peng C, Gao R, Ma L, Hu J, et al. miR-196b-5p-enriched extracellular vesicles from tubular epithelial cells mediated aldosterone-induced renal fibrosis in mice with diabetes. BMJ Open Diabetes Res Care (2020) 8:e001101. doi: 10.1136/bmjdrc-2019-001101
63. Guan H, Peng R, Mao L, Fang F, Xu B, Chen M. Injured tubular epithelial cells activate fibroblasts to promote kidney fibrosis through miR-150-containing exosomes. Exp Cell Res (2020) 392:112007. doi: 10.1016/j.yexcr.2020.112007
64. Zhou X, Zhao S, Li W, Ruan Y, Yuan R, Ning J, et al. Tubular cell-derived exosomal miR-150-5p contributes to renal fibrosis following unilateral ischemia-reperfusion injury by activating fibroblast in vitro and in vivo. Int J Biol Sci (2021) 17:4021–33. doi: 10.7150/ijbs.62478
65. Zhao S, Li W, Yu W, Rao T, Li H, Ruan Y, et al. Exosomal miR-21 from tubular cells contributes to renal fibrosis by activating fibroblasts via targeting PTEN in obstructed kidneys. Theranostics (2021) 11:8660–73. doi: 10.7150/thno.62820
66. Liang L, Huang K, Yuan W, Liu L, Zou F, Wang G. Dysregulations of miR-503-5p and Wnt/beta-catenin pathway coordinate in mediating cadmium-induced kidney fibrosis. Ecotoxicol Environ Saf (2021) 224:112667. doi: 10.1016/j.ecoenv.2021.112667
67. Shen Y, Jiang L, Wen P, Ye Y, Zhang Y, Ding H, et al. Tubule-derived lactate is required for fibroblast activation in acute kidney injury. Am J Physiol Renal Physiol (2020) 318:F689–701. doi: 10.1152/ajprenal.00229.2019
68. Zhou Y, Xiong M, Niu J, Sun Q, Su W, Zen K, et al. Secreted fibroblast-derived miR-34a induces tubular cell apoptosis in fibrotic kidney. J Cell Sci (2014) 127:4494–506. doi: 10.1242/jcs.155523
69. Li H, Xu Y, Zhang Q, Xu H, Xu Y, Ling K. Microvesicles containing miR-34a induce apoptosis of proximal tubular epithelial cells and participate in renal interstitial fibrosis. Exp Ther Med (2019) 17:2310–6. doi: 10.3892/etm.2019.7197
70. Zhou D, Fu H, Xiao L, Mo H, Zhuo H, Tian X, et al. Fibroblast-specific beta-catenin signaling dictates the outcome of AKI. J Am Soc Nephrol (2018) 29:1257–71. doi: 10.1681/ASN.2017080903
71. Gui Y, Lu Q, Gu M, Wang M, Liang Y, Zhu X, et al. Fibroblast mTOR/PPARgamma/HGF axis protects against tubular cell death and acute kidney injury. Cell Death Differ (2019) 26:2774–89. doi: 10.1038/s41418-019-0336-3
72. Lin L, White S, Hu K. A co-culture model to study the effect of kidney fibroblast-p90rsk on epithelial cell survival. Methods Mol Biol (2021) 2346:63–71. doi: 10.1007/7651_2020_292
73. Luo C, Zhou S, Zhou Z, Liu Y, Yang L, Liu J, et al. Wnt9a promotes renal fibrosis by accelerating cellular senescence in tubular epithelial cells. J Am Soc Nephrol (2018) 29:1238–56. doi: 10.1681/ASN.2017050574
74. Williams BM, Cliff CL, Demirel I, Squires PE, Hills CE. Blocking connexin 43 hemichannel-mediated ATP release reduces communication within and between tubular epithelial cells and medullary fibroblasts in a model of diabetic nephropathy. Diabetes Med (2022) 39:e14963. doi: 10.1111/dme.14963
75. Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol (1993) 122:103–11. doi: 10.1083/jcb.122.1.103
76. Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor-beta. J Biol Chem (1987) 262:6443–6. doi: 10.1016/S0021-9258(18)48258-0
77. Chang JF, Hsieh CY, Lu KC, Chen YW, Liang SS, Lin CC, et al. Therapeutic targeting of aristolochic acid induced uremic toxin retention, SMAD 2/3 and JNK/ERK pathways in tubulointerstitial fibrosis: nephroprotective role of propolis in chronic kidney disease. Toxins (2020) 12:364. doi: 10.3390/toxins12060364
78. Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell (1999) 96:319–28. doi: 10.1016/S0092-8674(00)80545-0
79. Voelker J, Berg PH, Sheetz M, Duffin K, Shen T, Moser B, et al. Anti-TGF-beta1 antibody therapy in patients with diabetic nephropathy. J Am Soc Nephrol (2017) 28:953–62. doi: 10.1681/ASN.2015111230
80. Phanish MK, Winn SK, Dockrell ME. Connective tissue growth factor-(CTGF, CCN2)–a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol (2010) 114:e83–92. doi: 10.1159/000262316
81. Nguyen TQ, Tarnow L, Jorsal A, Oliver N, Roestenberg P, Ito Y, et al. Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes Care (2008) 31:1177–82. doi: 10.2337/dc07-2469
82. Riser BL, Cortes P, DeNichilo M, Deshmukh PV, Chahal PS, Mohammed AK, et al. Urinary CCN2 (CTGF) as a possible predictor of diabetic nephropathy: preliminary report. Kidney Int (2003) 64:451–8. doi: 10.1046/j.1523-1755.2003.00130.x
83. Geng H, Lan R, Singha PK, Gilchrist A, Weinreb PH, Violette SM, et al. Lysophosphatidic acid increases proximal tubule cell secretion of profibrotic cytokines PDGF-B and CTGF through LPA2- and Galphaq-mediated Rho and alphavbeta6 integrin-dependent activation of TGF-beta. Am J Pathol (2012) 181:1236–49. doi: 10.1016/j.ajpath.2012.06.035
84. Liu F, Wu S, Ren H, Gu J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat Cell Biol (2011) 13:254–62. doi: 10.1038/ncb2167
85. Imaizumi T, Tanaka H, Tajima A, Tsuruga K, Oki E, Sashinami H, et al. Retinoic acid-inducible gene-I (RIG-I) is induced by IFN-gamma in human mesangial cells in culture: possible involvement of RIG-I in the inflammation in lupus nephritis. Lupus (2010) 19:830–6. doi: 10.1177/0961203309360540
86. Zhou D, Tan RJ, Fu H, Liu Y. Wnt/beta-catenin signaling in kidney injury and repair: a double-edged sword. Lab Invest (2016) 96:156–67. doi: 10.1038/labinvest.2015.153
87. Zhou D, Li Y, Lin L, Zhou L, Igarashi P, Liu Y. Tubule-specific ablation of endogenous beta-catenin aggravates acute kidney injury in mice. Kidney Int (2012) 82:537–47. doi: 10.1038/ki.2012.173
88. Hao S, He W, Li Y, Ding H, Hou Y, Nie J, et al. Targeted inhibition of beta-catenin/CBP signaling ameliorates renal interstitial fibrosis. J Am Soc Nephrol (2011) 22:1642–53. doi: 10.1681/ASN.2010101079
89. Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell (2016) 164:1226–32. doi: 10.1016/j.cell.2016.01.043
90. Xu H, Ling M, Xue J, Dai X, Sun Q, Chen C, et al. Exosomal microRNA-21 derived from bronchial epithelial cells is involved in aberrant epithelium-fibroblast cross-talk in COPD induced by cigarette smoking. Theranostics (2018) 8:5419–33. doi: 10.7150/thno.27876
91. Song L, Tang S, Han X, Jiang Z, Dong L, Liu C, et al. KIBRA controls exosome secretion via inhibiting the proteasomal degradation of Rab27a. Nat Commun (2019) 10:1639. doi: 10.1038/s41467-019-09720-x
92. Hwang I, Uddin MJ, Lee G, Jiang S, Pak ES, Ha H. Peroxiredoxin 3 deficiency accelerates chronic kidney injury in mice through interactions between macrophages and tubular epithelial cells. Free Radical Biol Med (2019) 131:162–72. doi: 10.1016/j.freeradbiomed.2018.12.002
93. Grist JT, Jarvis LB, Georgieva Z, Thompson S, Kaur Sandhu H, Burling K, et al. Extracellular lactate: A novel measure of T cell proliferation. J Immunol (2018) 200:1220–6. doi: 10.4049/jimmunol.1700886
94. Ghani QP, Wagner S, Becker HD, Hunt TK, Hussain MZ. Regulatory role of lactate in wound repair. Methods Enzymol (2004) 381:565–75. doi: 10.1016/S0076-6879(04)81036-X
95. Ding H, Jiang L, Xu J, Bai F, Zhou Y, Yuan Q, et al. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am J Physiol Renal Physiol (2017) 313:F561–75. doi: 10.1152/ajprenal.00036.2017
96. Liu Y. Hepatocyte growth factor in kidney fibrosis: therapeutic potential and mechanisms of action. Am J Physiol Renal Physiol (2004) 287:F7–16. doi: 10.1152/ajprenal.00451.2003
97. Zhou D, Fu H, Zhang L, Zhang K, Min Y, Xiao L, et al. Tubule-derived wnts are required for fibroblast activation and kidney fibrosis. J Am Soc Nephrol (2017) 28:2322–36. doi: 10.1681/ASN.2016080902
98. Lin L, Shi C, Sun Z, Le NT, Abe JI, Hu K. The Ser/Thr kinase p90RSK promotes kidney fibrosis by modulating fibroblast-epithelial crosstalk. J Biol Chem (2019) 294:9901–10. doi: 10.1074/jbc.RA119.007904
99. Pilzer D, Fishelson Z. Mortalin/GRP75 promotes release of membrane vesicles from immune attacked cells and protection from complement-mediated lysis. Int Immunol (2005) 17:1239–48. doi: 10.1093/intimm/dxh300
100. Noh H, Yu MR, Kim HJ, Lee JH, Park BW, Wu IH, et al. Beta 2-adrenergic receptor agonists are novel regulators of macrophage activation in diabetic renal and cardiovascular complications. Kidney Int (2017) 92:101–13. doi: 10.1016/j.kint.2017.02.013
101. Lv LL, Feng Y, Wen Y, Wu WJ, Ni HF, Li ZL, et al. Exosomal CCL2 from tubular epithelial cells is critical for albumin-induced tubulointerstitial inflammation. J Am Soc Nephrol JASN (2018) 29:919–35. doi: 10.1681/ASN.2017050523
102. Calle P, Hotter G. Macrophage phenotype and fibrosis in diabetic nephropathy. Int J Mol Sci (2020) 21:2806. doi: 10.3390/ijms21082806
103. Ding C, Zheng J, Wang B, Li Y, Xiang H, Dou M, et al. Exosomal microRNA-374b-5p from tubular epithelial cells promoted M1 macrophages activation and worsened renal ischemia/reperfusion injury. Front Cell Dev Biol (2020) 8:587693. doi: 10.3389/fcell.2020.587693
104. Lv LL, Feng Y, Wu M, Wang B, Li ZL, Zhong X, et al. Exosomal miRNA-19b-3p of tubular epithelial cells promotes M1 macrophage activation in kidney injury. Cell Death Differentiation (2020) 27:210–26. doi: 10.1038/s41418-019-0349-y
105. Jia Y, Chen J, Zheng Z, Tao Y, Zhang S, Zou M, et al. Tubular epithelial cell-derived extracellular vesicles induce macrophage glycolysis by stabilizing Hif-1α in diabetic kidney disease. Mol Med (2022) 28:95. doi: 10.1186/s10020-022-00525-1
106. Kashyap S, Warner GM, Hartono SP, Boyilla R, Knudsen BE, Zubair AS, et al. Blockade of CCR2 reduces macrophage influx and development of chronic renal damage in murine renovascular hypertension. Am J Physiol Renal Physiol (2016) 310:F372–84. doi: 10.1152/ajprenal.00131.2015
107. Veiras LC, Bernstein EA, Cao D, Okwan-Duodu D, Khan Z, Gibb DR, et al. Tubular IL-1β Induces salt sensitivity in diabetes by activating renal macrophages. Circ Res (2022) 131:59–73. doi: 10.1161/CIRCRESAHA.121.320239
108. Viau A, Bienaimé F, Lukas K, Todkar AP, Knoll M, Yakulov TA, et al. Cilia-localized LKB1 regulates chemokine signaling, macrophage recruitment, and tissue homeostasis in the kidney. EMBO J (2018) 37:e98615. doi: 10.15252/embj.201798615
109. Tan TK, Zheng G, Hsu TT, Lee SR, Zhang J, Zhao Y, et al. Matrix metalloproteinase-9 of tubular and macrophage origin contributes to the pathogenesis of renal fibrosis via macrophage recruitment through osteopontin cleavage. Lab Investigation; A J Tech Methods Pathol (2013) 93:434–49. doi: 10.1038/labinvest.2013.3
110. Rice EK, Nikolic-Paterson DJ, Hill PA, Metz CN, Bucala R, Atkins RC, et al. Interferon-gamma induces macrophage migration inhibitory factor synthesis and secretion by tubular epithelial cells. Nephrol (Carlton Vic.) (2003) 8:156–61. doi: 10.1046/j.1440-1797.2003.00152.x
111. Yang Z, Guo Z, Dong J, Sheng S, Wang Y, Yu L, et al. miR-374a regulates inflammatory response in diabetic nephropathy by targeting MCP-1 expression. Front Pharmacol (2018) 9:900. doi: 10.3389/fphar.2018.00900
112. Zhou W, Guo H. Relationshipbetweentubulointerstitial fibrosis andmononuclear-macrophage. J Peking Univ (2003) 35:503–5.
113. Wang Q, Liang Y, Qiao Y, Zhao X, Yang Y, Yang S, et al. Expression of soluble epoxide hydrolase in renal tubular epithelial cells regulates macrophage infiltration and polarization in IgA nephropathy. Am J Physiol Renal Physiol (2018) 315:F915–f926. doi: 10.1152/ajprenal.00534.2017
114. Lv LL, Wang C, Li ZL, Cao JY, Zhong X, Feng Y, et al. SAP130 released by damaged tubule drives necroinflammation via mirna-219c/mincle signaling in acute kidney injury. Cell Death Dis (2021) 12:866. doi: 10.1038/s41419-021-04131-7.
115. Zhao J, Chen J, Zhu W, Qi XM, Wu YG. Exosomal miR-7002-5p derived from highglucose-induced macrophages suppresses autophagy in tubular epithelial cells by targeting Atg9b. FASEB J (2022) 36:e22501. doi: 10.1096/fj.202200550RR
116. Yuan L, Yang J, Liu F, Li L, Liu J, Chen Y, et al. Macrophage-derived exosomal miR-195a-5p impairs tubular epithelial cells mitochondria in acute kidney injury mice. FASEB J (2023) 37:e22691. doi: 10.1096/fj.202200644R
117. Jiang W, Xu C, Xu S, Su W, Du C, Dong J, et al. Macrophage-derived, LRG1-enriched extracellular vesicles exacerbate aristolochic acid nephropathy in a TGFβR1-dependent manner. Cell Biol Toxicol (2022) 38:629–48. doi: 10.1007/s10565-021-09666-1
118. Urbschat A, Thiemens AK, Mertens C, Rehwald C, Meier JK, Baer PC, et al. Macrophage-secreted lipocalin-2 promotes regeneration of injured primary murine renal tubular epithelial cells. Int J Mol Sci (2020) 21:2038. doi: 10.3390/ijms21062038
119. Tan TK, Zheng G, Hsu TT, Wang Y, Lee VW, Tian X, et al. Macrophage matrix metalloproteinase-9 mediates epithelial-mesenchymal transition in vitro in murine renal tubular cells. Am J Pathol (2010) 176:1256–70. doi: 10.2353/ajpath.2010.090188
120. Fu H, Gu YH, Tan J, Yang YN, Wang GH. CircACTR2 in macrophages promotes renal fibrosis by activating macrophage inflammation and epithelial-mesenchymal transition of renal tubular epithelial cells. Cell Mol Life Sci CMLS (2022) 79:253. doi: 10.1007/s00018-022-04247-9
121. Huang Y, Mao Z, Zhang Z, Obata F, Yang X, Zhang X, et al. Connexin43 contributes to inflammasome activation and lipopolysaccharide-initiated acute renal injury via modulation of intracellular oxidative status. Antioxidants Redox Signaling (2019) 31:1194–212. doi: 10.1089/ars.2018.7636
122. Luan J, Fu J, Wang D, Jiao C, Cui X, Chen C, et al. miR-150-based RNA interference attenuates tubulointerstitial fibrosis through the SOCS1/JAK/STAT pathway in vivo and in vitro. Mol Ther Nucleic Acids (2020) 22:871–84. doi: 10.1016/j.omtn.2020.10.008
123. Yu J, Deng Y, Tao Z, Liang W, Guan X, Wu J, et al. The effects of HAP and macrophage cells to the expression of inflammatory factors and apoptosis in HK-2 cells of vitro co-cultured system. Urolithiasis (2018) 46:429–43. doi: 10.1007/s00240-017-1032-8
124. Jiang WJ, Xu CT, Du CL, Dong JH, Xu SB, Hu BF, et al. Tubular epithelial cell-to-macrophage communication forms a negative feedback loop via extracellular vesicle transfer to promote renal inflammation and apoptosis in diabetic nephropathy. Theranostics (2022) 12:324–39. doi: 10.7150/thno.63735
125. Navarro-González JF, Mora-Fernández C, Muros de Fuentes M, García-Pérez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol (2011) 7:327–40. doi: 10.1038/nrneph.2011.51
126. Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arteriosclerosis Thrombosis Vasc Biol (2007) 27:2302–9. doi: 10.1161/ATVBAHA.107.144824
127. Bernhagen J, Calandra T, Mitchell RA, Martin SB, Tracey KJ, Voelter W, et al. MIF is a pituitary-derived cytokine that potentiates lethal endotoxaemia. Nature (1993) 365:756–9. doi: 10.1038/365756a0
128. Benigni F, Atsumi T, Calandra T, Metz C, Echtenacher B, Peng T, et al. The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest (2000) 106:1291–300. doi: 10.1172/JCI9900
129. Filbey KJ, Varyani F, Harcus Y, Hewitson JP, Smyth DJ, McSorley HJ, et al. Macrophage migration inhibitory factor (MIF) is essential for type 2 effector cell immunity to an intestinal helminth parasite. Front Immunol (2019) 10:2375. doi: 10.3389/fimmu.2019.02375
130. He J, Wang C, Zhu Y, Ai D. Soluble epoxide hydrolase: a potential target for metabolic diseases. J Diabetes (2016) 8:305–13. doi: 10.1111/1753-0407.12358
131. DeWolf SE, Kasimsetty SG, Hawkes AA, Stocks LM, Kurian SM, McKay DB. DAMPs released from injured renal tubular epithelial cells activate innate immune signals in healthy renal tubular epithelial cells. Transplantation (2022) 106:1589–99. doi: 10.1097/TP.0000000000004038
132. Dai H, Thomson AW, Rogers NM. Dendritic cells as sensors, mediators, and regulators of ischemic injury. Front Immunol (2019) 10:2418. doi: 10.3389/fimmu.2019.02418
133. Jang HR, Rabb H. Immune cells in experimental acute kidney injury. Nat Rev Nephrol (2015) 11:88–101. doi: 10.1038/nrneph.2014.180
134. Lu X, Nagata M, Yamasaki S. Mincle: 20 years of a versatile sensor of insults. Int Immunol (2018) 30:233–9. doi: 10.1093/intimm/dxy028
135. Yamasaki S, Ishikawa E, Sakuma M, Hara H, Ogata K, Saito T. Mincle is an ITAM-coupled activating receptor that senses damaged cells. Nat Immunol (2008) 9:1179–88. doi: 10.1038/ni.1651
136. Kim JW, Roh YS, Jeong H, Yi HK, Lee MH, Lim CW, et al. Spliceosome-associated protein 130 exacerbates alcohol-induced liver injury by inducing NLRP3 inflammasome-mediated IL-1β in mice. Am J Pathol (2018) 188:967–80. doi: 10.1016/j.ajpath.2017.12.010
137. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Sci (New York N.Y.) (2020) 367:eaau6977. doi: 10.1126/science.aau6977
138. Leipziger J, Praetorius H. Renal autocrine and paracrine signaling: A story of self-protection. Physiol Rev (2020) 100:1229–89. doi: 10.1152/physrev.00014.2019
139. Jung M, Sola A, Hughes J, Kluth DC, Vinuesa E, Viñas JL, et al. Infusion of IL-10-expressing cells protects against renal ischemia through induction of lipocalin-2. Kidney Int (2012) 81:969–82. doi: 10.1038/ki.2011.446
140. Cox AG, Winterbourn CC, Hampton MB. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem J (2009) 425:313–25. doi: 10.1042/BJ20091541
141. Hu W, Dang XB, Wang G, Li S, Zhang YL. Peroxiredoxin-3 attenuates traumatic neuronal injury through preservation of mitochondrial function. Neurochemistry Int (2018) 114:120–6. doi: 10.1016/j.neuint.2018.02.004
142. Abrahamson DR, Robert B, Hyink DP, St John PL, Daniel TO. Origins and formation of microvasculature in the developing kidney. Kidney Int (1998) Supplement 67:S7–11. doi: 10.1046/j.1523-1755.1998.06702.x
143. Tasnim F, Zink D. Cross talk between primary human renal tubular cells and endothelial cells in cocultures. Am J Physiol Renal Physiol (2012) 302:F1055–62. doi: 10.1152/ajprenal.00621.2011
144. Dvorak HF, Brown LF, Detmar M, Dvorak AM. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol (1995) 146:1029–39. doi: 10.1200/JCO.2002.10.088
145. Dimke H, Sparks MA, Thomson BR, Frische S, Coffman TM, Quaggin SE. Tubulovascular cross-talk by vascular endothelial growth factor a maintains peritubular microvasculature in kidney. J Am Soc Nephrol JASN (2015) 26:1027–38. doi: 10.1681/ASN.2014010060
146. LeCouter J, Kowalski J, Foster J, Hass P, Zhang Z, Dillard-Telm L, et al. Identification of an angiogenic mitogen selective for endocrine gland endothelium. Nature (2001) 412:877–84. doi: 10.1038/35091000
147. Fernandez-Martínez AB, Torija AV, Carracedo J, Ramirez R, de Lucio-Cazaña FJ. Microparticles released by vascular endothelial cells increase hypoxia inducible factor expression in human proximal tubular HK-2 cells. Int J Biochem Cell Biol (2014) 53:334–42. doi: 10.1016/j.biocel.2014.05.025
148. Nee LE, McMorrow T, Campbell E, Slattery C, Ryan MP. TNF-alpha and IL-1beta-mediated regulation of MMP-9 and TIMP-1 in renal proximal tubular cells. Kidney Int (2004) 66:1376–86. doi: 10.1111/j.1523-1755.2004.00900.x
149. Álvarez RS, Jancic C, Garimano N, Sacerdoti F, Paton AW, Paton JC, et al. Crosstalk between human microvascular endothelial cells and tubular epithelial cells modulates pro-inflammatory responses induced by shiga toxin type 2 and subtilase cytotoxin. Toxins (2019) 11:648. doi: 10.3390/toxins11110648
150. Aydin S, Signorelli S, Lechleitner T, Joannidis M, Pleban C, Perco P, et al. Influence of microvascular endothelial cells on transcriptional regulation of proximal tubular epithelial cells. Am J Physiol Cell Physiol (2008) 294:C543–54. doi: 10.1152/ajpcell.00307.2007
151. Shlipak MG, Tummalapalli SL, Boulware LE, Grams ME, Ix JH, Jha V, et al. The case for early identification and intervention of chronic kidney disease: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int (2021) 99:34–47. doi: 10.1016/j.kint.2020.10.012
Keywords: tubulointerstitial fibrosis, crosstalk, chronic kidney disease, proximal tubular epithelial cells, molecular mechanism
Citation: Guo C, Cui Y, Jiao M, Yao J, Zhao J, Tian Y, Dong J and Liao L (2024) Crosstalk between proximal tubular epithelial cells and other interstitial cells in tubulointerstitial fibrosis after renal injury. Front. Endocrinol. 14:1256375. doi: 10.3389/fendo.2023.1256375
Received: 10 July 2023; Accepted: 22 November 2023;
Published: 08 January 2024.
Edited by:
Yao-Wu Liu, Xuzhou Medical University, ChinaReviewed by:
Hao Du, UCONN Health, United StatesJia-Feng Chang, Taipei Veterans General Hospital, Taiwan
Copyright © 2024 Guo, Cui, Jiao, Yao, Zhao, Tian, Dong and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Liao, bGlhb2xpbkBzZGZtdS5lZHUuY24=; Jianjun Dong, ZG9uZ2ppYW5qdW5Ac2R1LmVkdS5jbg==