 Gonzalo-Martín Pérez-Arana1,2*Alfredo Díaz-Gómez3José Bancalero-de los Reyes4Manuel Gracia-Romero1Antonio Ribelles-García1
Gonzalo-Martín Pérez-Arana1,2*Alfredo Díaz-Gómez3José Bancalero-de los Reyes4Manuel Gracia-Romero1Antonio Ribelles-García1 Francisco Visiedo1,2
Francisco Visiedo1,2 Álvaro González-Domínguez2
Álvaro González-Domínguez2 David Almorza-Gomar2,5
David Almorza-Gomar2,5 José-Arturo Prada-Oliveira1,2*
José-Arturo Prada-Oliveira1,2*- 1Department of Human Anatomy and Embryology, University of Cadiz, Cádiz, Spain
- 2Institute for Biomedical Science Research and Innovation (INIBICA), University of Cadiz, Cádiz, Spain
- 3San Carlos Hospital, Andalusian Health System, Cádiz, Spain
- 4Complejo Hospitalario de Badajoz, Servicio Extremeño de Salud, Cádiz, Spain
- 5Operative Statistic and Research Department, University of Cádiz, Cádiz, Spain
The biological activity of glucagon has recently been proposed to both stimulate hepatic glucose production and also include a paradoxical insulinotropic effect, which could suggest a new role of glucagon in the pathophysiology type 2 diabetes mellitus (T2DM). An insulinotropic role of glucagon has been observed after bariatric/metabolic surgery that is mediated through the GLP-1 receptor on pancreatic beta cells. This effect appears to be modulated by other members of the proglucagon family, playing a key role in the beneficial effects and complications of bariatric/metabolic surgery. Glucagon serves a dual role after sleeve gastrectomy (SG) and Roux-en-Y gastric bypass (RYGB). In addition to maintaining blood glucose levels, glucagon exhibits an insulinotropic effect, suggesting that glucagon has a more complex function than simply an “anti-insulin hormone”.
1 Introduction
Glucagon, which was first identified in 1922, is a 29-amino acid peptide hormone that is produced and secreted by the alpha cell population within the pancreatic islets (1). In conjunction with insulin, glucagon serves to maintain glucose homeostasis. Glucagon stimulates hepatic glycogen breakdown via glycogen phosphorylase and gluconeogenesis via fructose 1,6-bisphosphatase activation; both mechanisms lead to elevated blood glucose (2). Glucagon also acts on lipid metabolism, stimulating beta-oxidation of fatty acids and facilitating the transport of fatty acids to the mitochondria by increasing the activity of carnitine acyltransferase (3). Diabetic hyperglycemia is often described as the result of impaired insulin secretion, but diabetic hyperglycemia also occurs due to inadequate glucagon release. This relationship has been reinforced by recent findings that glucagon/GLP-1 receptor dual agonists lead to improved blood glucose in type 2 diabetes mellitus (T2DM) patients.
Bariatric surgery (BS)/metabolic surgery (MS) has become the most powerful tool against T2DM (4). The study of the mechanisms by which BS/MS improves T2DM has included the investigation of a group of peptides called incretins. Among them are glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), as well as other peptides, which although we cannot consider incretins, are related such as peptide tyrosine-tyrosine (PYY) and ghrelin. The effects of incretins on the production, secretion, and action of insulin have been widely studied in animal models and humans. We focus on their effects on glucagon secretion and actions related to the two most performed variants of this surgery, sleeve gastrectomy (SG) and Roux-en-Y gastric bypass (RYGB).
2 Methods
To achieve our goal, we performed a selective search of scientific articles in many databases according to the PRISMA protocol (Figure 1). The protocol and results were registered at the RODIN-University of Cádiz repository (http://hdl.handle.net/10498/27147). The search was limited to standard scientific databases. The Boolean operators used were AND, OR and NOT, and the key words were Glucagon, GLP-1, Incretin, Sleeve Gastrectomy, Roux-en-Y Gastric Bypass, Glucose metabolism, Glycolysis and alpha cell. A total of 160 reports were selected for the preparation of this manuscript. We reduced the number to 50 main references according to editorial determination.
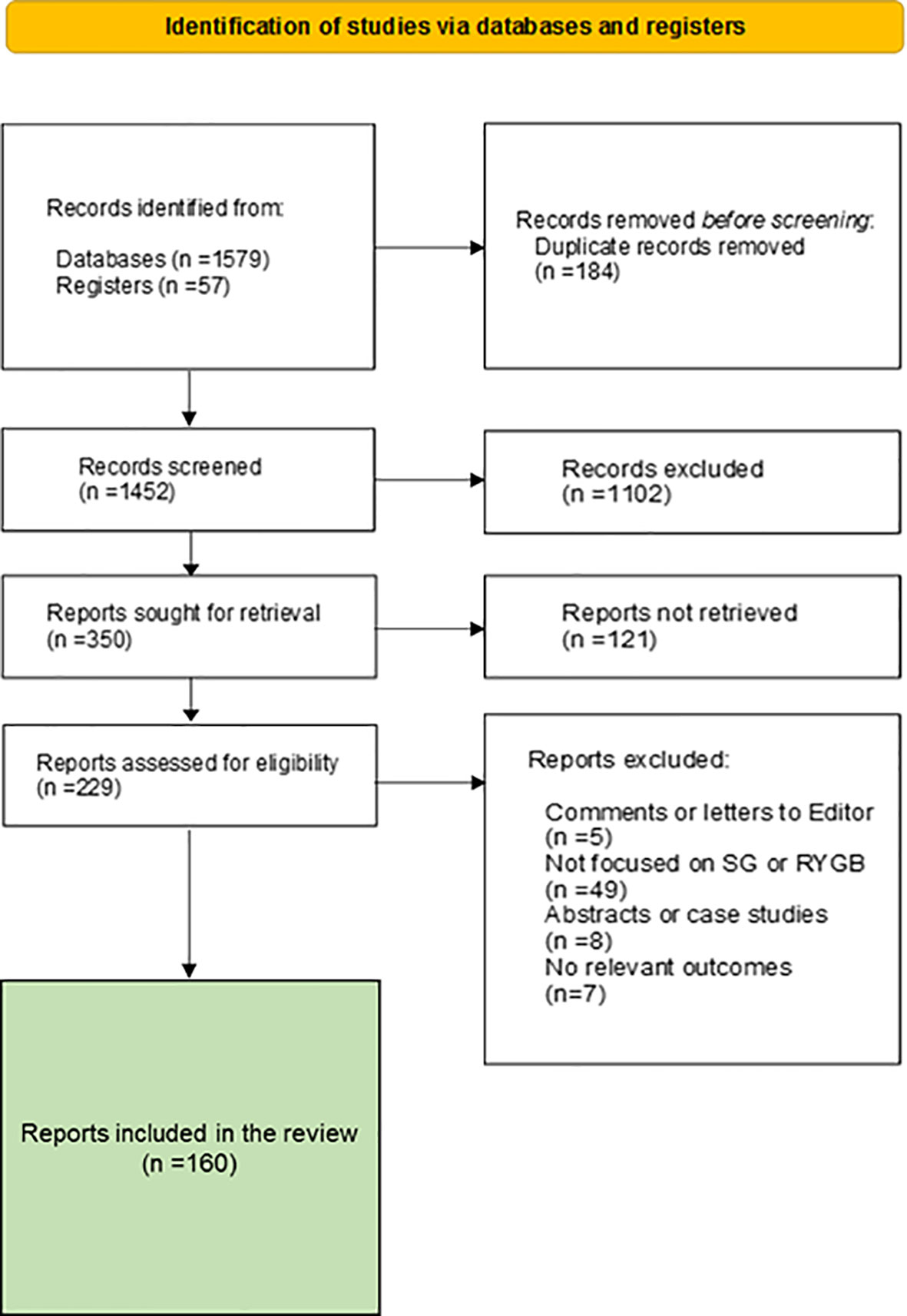
Figure 1 PRISMA flow diagram of the study.
3 Glucagon secretion and modulating agents: the role of incretins
Preproglucagon is encoded by the Gcg gene, which is expressed in pancreatic alpha cells, L cells from the gut and many neurons from the nucleus of the solitary tract in the brain (5). Different products are produced in these cell types depending on preproglucagon processing by different prohormone convertase family members (PCs) (6). PC2 is exclusively responsible for the production of glucagon in pancreatic alpha cells, whereas PC1/3 is responsible for the formation of glucagon-like peptide-1 (GLP-1) in the same cells (7). In addition, PC1/3 is present in other cell types, such as neurons and L-cells producing GLP-1, GLP-2, glicentin and oxyntomodulin (8).
Glucagon is primarily produced in alpha cells and its secretion is stimulated by low blood glucose levels. When low blood glucose levels are detected, circulating glucose is taken up by alpha cells via glucose transporter 1 (GLUT1), which exhibits high glucose affinity. The low intracellular ATP/ADP ratio in the alpha cell leads to ATP-sensitive potassium (KATP) channel closure. Depolarization of the plasma membrane and, ultimately, voltage-dependent calcium channel activation allow the flow of calcium into cells and glucagon exocytosis mediated by the SNARE complex of exocytosis proteins (9).
However, low glucose is not the only important stimulus for glucagon release. Amino acids (AAs) have long been known to stimulate glucagon release. The most powerful stimulators of glucagon release include arginine, alanine and glutamine. Recently, glutamine-stimulated glucagon secretion has been linked to high interstitial glutamate concentrations and glutamate-induced changes in plasma membrane polarization (7). Thus, the presence of high plasma levels of AAs induces a strong glucagonotropic effect, preventing potential hypoglycemia due to AA-induced insulin secretion (8). Moreover, high glucagon plasma levels induce hepatic uptake of AAs and activation of the urea cycle in the liver, leading to enhanced hepatic AA turnover and hypoaminoacidemia. Thus, a feedback relationship exists between the liver and the alpha cell population (10).
Glucagon secretion can be modulated by a number of factors, including insulin secretion itself or the release of somatostatin by pancreatic delta cells, limiting glucagon and insulin release (11) (Table 1). In this context, free fatty acids (FFAs), among many other factors, seem to play a key role in the upregulation of glucagon and insulin secretion through FFAR4/GPR120 receptor binding in delta cells and decreasing somatostatin release by pancreatic delta cells. Additionally, infusion of amylin, a 37-amino-acid peptide produced by beta cells at a ratio of 1:100 to insulin, limits arginine-stimulated glucagon secretion in rats (12). Pancreatic polypeptide (PP), a member of the neuropeptide Y family released by PP cells within the islets, acts on alpha and delta cells to inhibit glucagon and somatostatin secretion as well as binding to PPYR1, the Y4 subtype of the G-protein coupled receptor (GPCR) family.
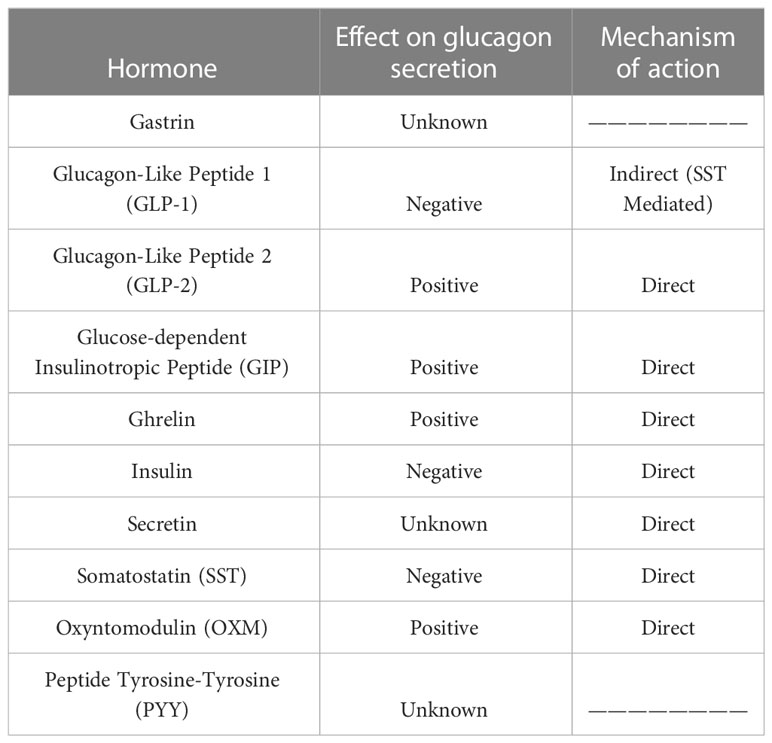
Table 1 Effects of gastrointestinal hormones on glucagon secretion.
3.1 GLP-1
Among the glucagon-secretion modulators, a few gastrointestinal hormones deserve special mention. The first is GLP-1, a peptide of its own family primarily produced in intestinal L cells. A very small portion of GLP-1 is also produced and locally released within the pancreatic islets by alpha cells (13). The effect of GLP-1 on glucagon secretion is controversial; the presence of GLP-1 on isolated alpha cell cultures leads to enhanced glucagon secretion; however, many authors have proposed an inhibitory effect in isolated pancreas and GLP-1-infused patients. This finding suggests an indirect mechanism mediated by an intermediate factor. Supporting relationship, the GLP-1 receptor (GLP-1R) has been detected on the surface of pancreatic delta cells, and perfusion of somatostatin-14 (SST-14) in isolated rat pancreas led to diminished glucagon release by alpha cells (14).
3.2 GIP
GIP is another gastrointestinal incretin hormone produced by K cells, primarily in the duodenum. GIP is able to enhance glucagon secretion of alpha cells in rats, healthy people or patients with T2DM both in the hyperglycemic and euglycemic state. This effect appears to be direct and due to the binding of GIP to its receptor (GIPR) on the alpha cell surface (15) and resulting increases in intracellular calcium concentrations, leading to glucagon vesicle release (16). Some authors propose that loss of beta cell GIPR function due to cell stress in T2DM patients may reduce the inhibitory effect of insulin on glucagon secretion, leading to elevated GIP-stimulated glucagon secretion even during hyperglycemia (17), but this hypothesis is controversial.
3.3 Ghrelin
The gastrointestinal peptides that act on glucagon secretion also include ghrelin, a 28-amino-acid peptide primarily produced by X/A-like cells in the oxyntic glands of the gastric fundus. A small amount of ghrelin is also produced in the pancreatic islets by epsilon cells (18). Ghrelin receptor (GHSR) has been detected in alpha cell populations, and a direct stimulating effect of acyl ghrelin on glucagon secretion was observed in a pancreatic alpha cell line clone in isolated islets (19). The exact mechanism of ghrelin-enhanced glucagon secretion remains unknown.
3.4 GLP-2
GLP-2 is another enterohormone encoded by the Gcg gene and processed by PC1/3 in intestinal L cells. GLP-2 has also been described inside the pancreatic islets, and a glucagonotropic effect occurs after intravenous administration of GLP-2 to patients (20). Thus, GLP-2 may act directly by binding to its receptors on alpha cells, leading to glucagon release. However, cross-activation of GLP-1R by other members of the preproglucagon family has also been described. Therefore, we cannot rule out activation of GLP-2R on the surface of alpha cells by GLP-1 or by the released glucagon, thereby serving as a mechanism to amplify glucagon secretion.
3.5 Oxyntomodulin
Another interesting peptide is oxyntomodulin (OXM), a peptide related to GLP-1 and GLP-2 obtained from proglucagon processing by PC1/3 in L cells along the intestine but also at the caudal part of the nucleus of the solitary tract in the Central Nervous System (CNS) (21). OXM stimulates glucagon secretion by binding to glucagon receptors (GCGR) with low affinity, as has been demonstrated in vitro and in animal models (22). In practice, OXM is rapidly degraded by the DPP-4 enzyme, leading to a small amount of circulating OXM and negligible activity in carbohydrate metabolism.
3.6 PYY
PYY is a molecule co-secreted with GLP-1 by intestinal L cells. Its mechanism of action on alpha cells remains unknown, but PYY has been shown to stimulate glucagon production in cultured islets from diabetic GK rats. More recently, PYY expression inside pancreatic islets from rodents and humans and GLP-1–mediated paracrine activity in islets have been documented. However, the sensitivity of PYY to degradation by DPP-4 activity and the limited quantities in which it is expressed makes studying PYY difficult (23).
4 Glucagon and glucose metabolism
4.1 Physiological effects of glucagon on glucose metabolism
The regulation exerted by the insulin–glucagon pair on carbohydrate metabolism is well known. After a meal, high blood glucose levels stimulate insulin release by pancreatic beta cells, leading to glucose uptake and glycogen production in the liver and muscles in a process called glycogenesis, and insulin also inhibits glucagon release.
Glucagon is secreted by the pancreatic alpha cell population in situations of hypoglycemia or stress, reaching blood concentrations of up to 1 ng/mL in such situations. When glucagon reaches the target tissues, it binds to glucagon receptor (GCGR), a G-protein-coupled receptor with seven transmembrane domains on the cell surface.
The effects of glucagon in the liver also suggest paradoxical situations, such as those related to protein synthesis in fasting periods through transient protein kinase mechanistic target of rapamycin 1/2 (mTORC1/2) activation thanks to the GTPase activity of RAP-1 aided by exchange protein activated by cAMP (EPAC). The increase in hepatic sensitivity to insulin induced by glucagon due to phosphorylation of AKT at Ser 473 is similar to the effect of insulin on mTORC2, which has been observed in primary cultures of hepatocytes and is of special therapeutic interest (24).
After a meal, high blood glucose levels stimulate insulin release from the pancreatic beta cell population to the bloodstream, inhibit the pancreatic secretion of glucagon and enhance glucose uptake and glycogen storage, primarily in the liver and muscles. These effects lead to decreased blood glucose levels, facilitating euglycemia. In contrast, during fasting periods of 2 to 12 hours, blood glucose level depletion is prevented by the action of glucagon secreted by the pancreatic alpha cell population. As mentioned above, this initially induces glycogenolysis in peripheral tissues and the liver, but when glycogen reserves are depleted, glucagon also induces hepatic gluconeogenesis, ensuring maintenance of proper blood glucose levels. However, the role of glucagon in hepatic gluconeogenesis is complex; a recent study proposed that the mechanism of gluconeogenesis is more dependent on the availability of gluconeogenic amino acids than on the action of glucagon itself (24). Glucagon increases the availability of gluconeogenic amino acids. In this sense, glucagon regulates hepatic glucose metabolism more by the glycogenolysis pathway than by gluconeogenesis. This relationship is supported by the poor hepatic glucose production after glucagon administration in patients with low hepatic glycogen reserves subjected to a low carbohydrate diet. Therefore, glucagon may lead to immediate regulation mediated by glycogenolysis, or medium-term regulation mediated by gluconeogenesis (Figure 2).
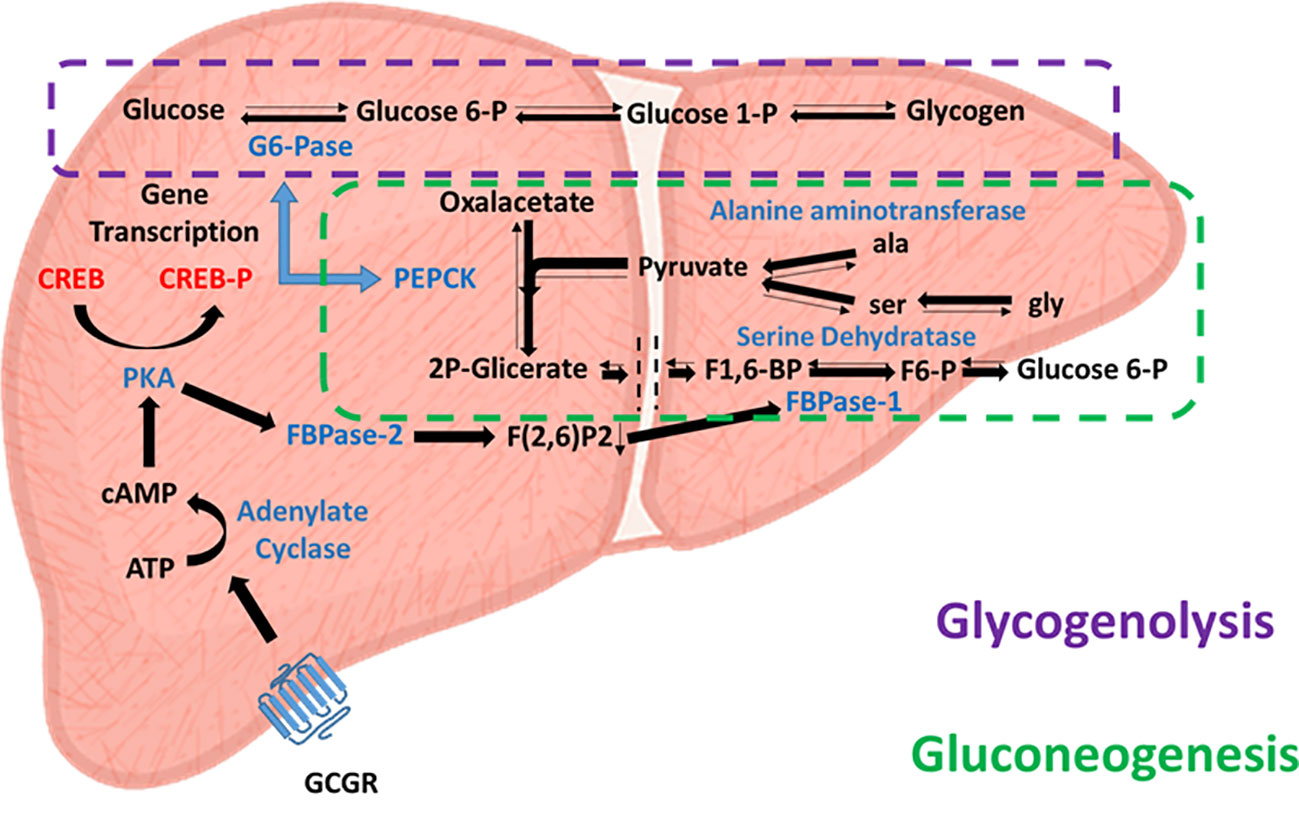
Figure 2 Glucagon-activated glycogenolytic and gluconeogenic pathways. Effects of glucagon receptor (Cgcr) activation on hepatic glycolysis (dashed purple line) and gluconeogenesis (dashed green line) pathways. Enzymes involved are in blue, substrates are in black, and transcription factors are in red.
4.2 Glucagon and type 2 diabetes mellitus
Patients with type 2 diabetes mellitus (T2DM) exhibit high plasma glucagon levels in the fasting periods and lack the ability to inhibit its secretion after a meal. Historically, steps have been taken to ameliorate the disease. Diabetic patients were administered somatostatin, which limited hyperglycemia in most patients (25). Later, glucagon receptor (Gcgr -/-) knockout mice treated with streptozotocin also exhibited euglycemia without hypoglycemic episodes in an interesting study (26). However, in this model, worse glycemic control may develop if GCGR ablation is induced after the onset of STZ-induced diabetes. Moreover, this partial control of hyperglycemia was lost when the animals were treated with a GLP-1R antagonist. These findings lead us to consider the establishment of an alternative pathway to GCGR after its ablation, the role of GLP-1R in this alternative pathway and the need for time to establish the pathway. Since that time, several studies have tested the GCGR antagonist effect on glucose metabolism in animals and humans with T2DM, confirming beneficial antidiabetogenic effects (27). However, the use of GCGR antagonists as pharmaceuticals has drawbacks, such as a notable increase in hepatic transaminases and LDL cholesterol (28). Other ways of limiting the effect of glucagon on glucose metabolism that have been studied include the use of anti-GCGR antibodies, which is efficient but also exhibited many problems in preclinical trials, such as alpha cell hyperplasia in treated animals, a common problem for any agents that disrupts glucagon receptor signaling (29). Similar beneficial effects on glucose metabolism have been demonstrated by Gcgr-antisense oligonucleotide administration, which leads to inhibited Gcgr expression; only mild and local side effects have been observed when these oligonucleotides are used (30).
Paradoxically, glucagon stimulates dose-dependent insulin release via GLP-1R in beta cells in rodents (31). Considering the therapeutic role of GLP-1R agonists such as exenatide, liraglutide, or lixisenatide on glucose tolerance (32), it is easy to understand why the combined effect of glucagon/GLP-1 dual co-agonists on glucose and lipid metabolism is being widely studied (33) and why they might exhibit a superior and more complete effect than single agonists. Therefore, several glucagon/GLP-1R co-agonists have been developed with different affinities for the two receptors and varying effects on glucose tolerance in rats and mice, likely due to different glucagon receptor tissue distributions in each receptor. Despite this difficulty, a handful of these co-agonists, as LY2944876/TT-401, LY3305677 (Eli Lilly), SAR425899 (Sanofi-Aventis), Efinopegdutide (Merk), BI 456906 (Boehringer Ingelheim), Efocipegtrutide (Hanmi Pharmaceutical), JNJ-54728518 (Jannsen) or cotadutide (AstraZeneca), have moved to different phases of clinical trials (33–36) oriented toward the treatment of obesity rather than diabetes. Finally, a triple agonist of GIPR, GLP1R and GCGR called Retratutide has also recently been tested in a phase 2 trial. This showed a great ability to substantially reduce body weight. However, it also presented a series of adverse gastrointestinal events, although limited to patients treated with high doses (37).
5 Glucagon and metabolic surgery
Today, metabolic surgery is one of the most effective weapons in the therapeutic arsenal against T2DM, and since its appearance, many surgical variants have been developed with varying degrees of success (4). Two approaches stand out for their safety and efficiency: Roux-en-y-gastric bypass (RYGB) and Sleeve gastrectomy (SG). RYGB is a surgery involving mixed malabsorptive and restrictive components that limit the volume of the stomach and the absorption length of the small intestine, whereas SG is a restrictive surgery that only affects the gastric volume (Figure 3). Most studies on the pathophysiological mechanisms underlying the improvement of glucose metabolism after these surgeries focus on insulin production and secretion (38), and few focus on the effect on glucagon, often resulting in an incomplete view of the phenomena that lead to changes in glucose metabolism after surgery. We have compiled and summarized what is known about the role of glucagon after these two surgeries.
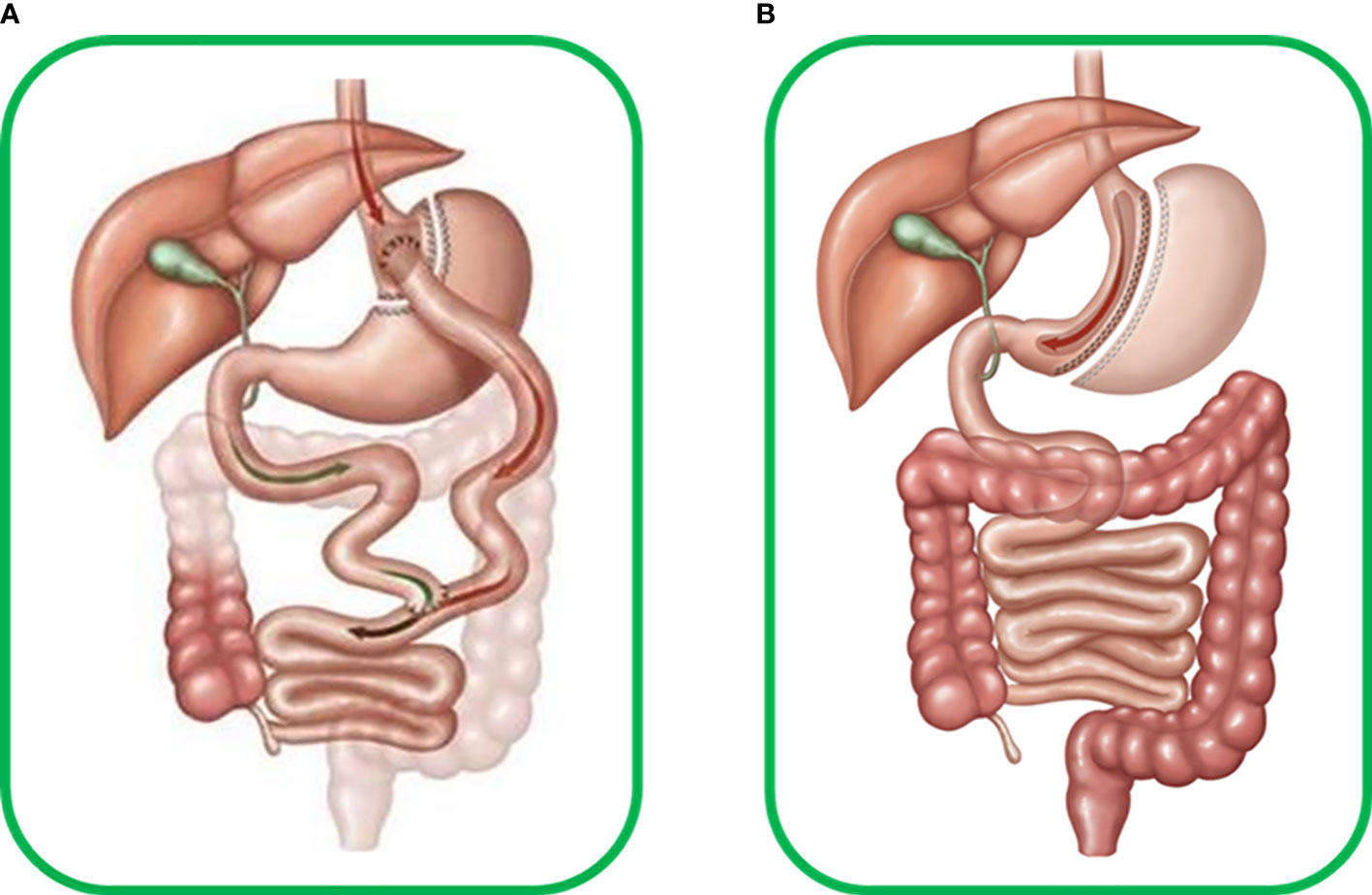
Figure 3 (A) Human Roux-en-Y gastric bypass surgery (RYGB) includes transverse section of the stomach from the great to the minor curvature, producing a new gastric pouch with limited volume that continues to the alimentary limb connecting to the medium portion of the jejunum. The antrum, duodenum, and initial portion of jejunum form the biliopancreatic limb, which excludes food flux. Thus RYGB becomes a mixed malabsorptive and restrictive technique. (B) Human sleeve gastrectomy (SG) surgery, which involves important reduction of around 80-95% of total gastric volume due to resection of most of great gastric curvature with ablation of most of the antrum, stomach corpus, and gastric fundus, forming a cylindrical pouch and preserving the pylorus and minor gastric curvature, thus becoming a restrictive technique.
5.1 The role of glucagon after RYGB
The induction of postprandial hyperglucagonemia in patients undergoing RYGB has long been observed (39). However, other bariatric techniques that involve duodenal exclusion, such as the duodenal-endoluminal sleeve (DES), seem to stimulate hyperglucagonemia (40). This is a paradoxical situation given that RYGB also stimulates insulin secretion and limits high glucose levels. However, supporting these observations, long-term increased alpha cell mass has been observed in Wistar rodent models after RYGB (41), and elevated plasma glucagon levels have been observed after the oral glucose tolerance test (OGTT) but not after intravenous glucose administration in T2DM patients after RYGB (39). Furthermore, glucagon gene expression and glucagon expression are increased in patients after RYGB, as determined by small intestine biopsy, indicating new sources of alternative glucagon secretion. This finding may be explained by modification of the intestinal mucosa and glucagon gene processing by L cells due to early contact with food derived from RYGB surgery. However, the number of patients studied is small, and recent studies combining liquid chromatography and mass spectrometry techniques did not observe increased fasting or postprandial glucagon levels in T2DM patients one year after RYGB (42). These findings therefore raise suspicions of cross-detection of other proglucagon-like molecules, such as glicentin or OXM (43). It is necessary to establish the sensitivity and specificity of these techniques and determine which is the most suitable for this type of study. In this sense, a recent study has established a close correlation between glucagon levels measured using sandwich enzyme-linked immunosorbent assay (ELISA) in pancreatectomized patients and glucagon-related hormone levels such as OXM or glicentin. These findings lead us to consider a cross-reaction between molecules of the same family. Therefore, it is necessary to confirm the hyperglucagonemia detected after RYGB using other techniques, preferably LC-MS. Meanwhile, all data referring to the presence and amount of glucagon announced in the studies should be taken with great caution.
The important role that glucagon could play after RYGB is also not clear. Episodes of severe postprandial hypoglycemia are one of the most important and frequent complications following RYGB surgery. Some authors attribute this phenomenon to an excessive and inappropriate insulin response after the meal. However, the published data are inconclusive. . In addition, there is a C-terminal 19-29 mini-glucagon from the fragmentation of glucagon during its processing and released together with it. Mini-glucagon 19-29 has a potent limiting effect on insulin secretion (44, 45) and it could happen that RYGB modifies the released ratios of glucagon/mini-glucagon 19-29, which could explain the episodes of postprandial hypoglycemia.
At the same time, many authors have proposed that the high postprandial plasma glucagon level is the key to avoiding severe hypoglycemic episodes after meals in RYGB patients (46). However, Tharakan et al. proposed that the combination of early elevated plasma glucagon and GLP-1 levels after meals may imply an insulinotropic effect triggering postprandial hypoglycemia (47), building on previous studies in which these hypoglycemic states were limited using GLP-1R antagonists. Therefore, some GLP-1R antagonists, such as exendin (9–39), may be of special therapeutic interest. Thus, postprandial glucagon has a dual role. It acts first as a GLP-1 agonist, leading to an insulinotropic effect through GLP-1R, and subsequently stimulates the liver to avoid hypoglycemia as a safety mechanism. In this sense, some experiments performed in diabetic rodents suggest that the response to GLP-1R agonists prior to surgery may act as a predictor of the future effect of RYGB on glucose metabolism and are of significant therapeutic interest. Thus, the response to GLP-1 R prior to surgery would indicate the degree of sensitivity of the patient´s GLP-1R to GLP-1 and other similar molecules such as glucagon. Giving us an idea about the efficiency of surgery to improve glucose metabolism but also about the possibilities of postprandial hypoglycemia episodes after surgery (48).
Another possible explanation for postprandial hypoglycemia is glucagon resistance. Hepatic steatosis and certain diets have been related to decreased hepatic GCGR expression in animals, probably due to GCGR internalization (49). However, given the presence of liver fat in animals and the consumption of a high-fat diet by patients after RYGB, this situation does not persist and postprandial hypoglycemia can be seen even years after bariatric/metabolic surgery. RYGB also appears to increase circulating levels of GIP in diabetic-GK rats. This is of great interest considering the possible synergistic insulinotropic effects established with GLP-1 in cultured diabetic and nondiabetic human islets (50) and its demonstrated insulinotropic effect in T2DM obese patients. However, the role of GIP on glucagon secretion is not entirely clear. Many authors propose a GIP-stimulated glucagon secretion even during hyperglycemia, but this hypothesis is controversial since in Type 1 diabetes patients without insulin secretion, GIP infusion cannot stimulate glucagon secretion during hyperglycemia (51). Therefore, more research is needed in this field to clarify these events.
Previous studies by our group have demonstrated the fundamental role of PYY as a regulatory molecule for the increased secretion of both GLP-1 and GIP after RYGB in diabetic-GK rats when PYY exhibits secretion patterns prior to those of GLP-1 and GIP after ingestion (52).
Within the glucagon family of molecules, GLP-2 also appears to be increased after RYGB (53), which is consistent with high postprandial glucagon levels given the glucagonotropic effect of GLP-2. This relationship suggests a dual role for GLP-2 in maintaining glucose homeostasis indirectly by stimulating glucagon secretion and facilitating the growth and adaptive response of the intestinal absorptive loop (54).
5.2 The role of glucagon after SG
Today, SG is the most common restrictive surgical procedure performed in the USA, with undeniable postsurgical benefits for glucose metabolism. Many studies have been conducted to elucidate how SG improves T2DM, implying modified bile acid circulation, elevated amino acid levels, and modified incretin expression (55). We will focus on those that relate changes in glucose metabolism after surgery with glucagon.
Elevated glucagon: insulin ratios have been reported in diabetic-GK rats after SG, and an early increase in serum glucagon levels has been reported in patients. These findings are consistent with what was observed in previous studies by our group reporting a long-term increase in pancreatic alpha cell mass in healthy rodents after SG with potent glucagon secretion capable of buffering exogenous insulin administration (56). This finding could explain the improved insulin secretion reported after SG because beta cell-expressing GCGR is activated by glucagon in rodents. However, beta cell-specific Gcgr knockout animals exhibit normoglycemia, suggesting that the insulinotropic activity of glucagon is mediated mainly through GLP-1R present in beta cell populations (57). Therefore, the elevated plasma GLP-1 levels observed after SG suggest a joint effect of both on insulin release after SG. However, T2DM patients’ serum glucagon levels decrease approximately 6 months after surgery, but the improved glucose metabolism persists (58). Could this phenomenon be explained by the paracrine activity of these hormones in the pancreas rather than by their circulating levels? Indeed, SG has been shown to modify the cellular composition and activity of pancreatic islets, even increasing the residual pancreatic ghrelin cell-producing population (59). Furthermore, a ghrelin-positive effect on glucagon secretion has been reported using cultured mouse islets and alphaTC1 and InR1G9 alpha cell lines. However, ghrelin Knockout and Ghsr(-/-) Knockout mice exhibit no changes in plasma glucose patterns after OGTT or IPTT, suggesting a limited paracrine effect of ghrelin on pancreatic islets. It is possible that this paracrine effect also occurs in the case of glucagon secretion. Nonetheless, it is difficult to investigate these results in humans, and further efforts are needed.
Finally, SG has also been correlated to increases in the circulating levels of GIP in diabetic rodents (60). However, this relationship has not been confirmed in patients. The effect can be understood based on the idea that the anatomical rearrangements of the digestive tube due to SG surgery do not affect the duodenum. Other authors have reported increases in GIP in the immediate postoperative period. However, these levels normalized 12 months later (61). This change indicates a probable adaptive phenomenon in the digestive tube. Together, these data rule out the possible insulinotropic synergistic role of GIP as proposed in the case of RYGB.
6 Outlook, limitations and future prospects
In this review, we briefly discussed the physiology of glucagon, its implication in T2DM, and its role in glucose improvement after bariatric/metabolic surgery. Several insights have been gained from studies on the role of glucagon in improved glucose metabolism after metabolic and bariatric surgery, and one of the most important is that glucagon is not an anti-insulin hormone due to its reported insulinotropic actions and postsurgical high plasma levels, exhibiting a dual role in the regulation of glucose homeostasis. Another important aspect is the possible modulation of glucagon effects by other hormones whose expression is modified by surgery, such as GLP-1, ghrelin, or GIP. Many studies have been performed to elucidate these complex interactions, but many of them occur in the paracrine environment, and it is very difficult to translate this research from animal models to patients. Finally, metabolic/bariatric surgery has allowed the exploration of modifications and alternative sources of glucagon as parts of a compensatory mechanism of the anatomical and functional rearrangements derived from surgery.
However, many issues remain to be elucidated, which represent promising areas for future research. In this sense, the possible use of glucagon agonists to treat T2DM based on their insulinotropic effects is intriguing. Furthermore, the findings of the endocrine pancreas and functional assays performed in animals that have undergone metabolic/bariatric surgery support the study of glucagon and its phenomena of dumping and relapse in T2DM after surgery seems to be important. Thus, there remains significant areas to explore to understand the role of glucagon.
Author contributions
The author's contributions were equal in the whole manuscript. All authors co-participate in references selection, reading, analysis and manuscript writing. The initial version of the manuscript was written by G-MP-A and J-AP-O. All authors contributed to the article and approved the submitted version.
Funding
The authors declare the research project was supported by University of Cadiz and Institute for Biomedical Science Research and Innovation (INIBICA).
Acknowledgments
We appreciate the helpful assistance of Tugiana-Peal of Becerro Asociation. The scientific articles consulted for the preparation of this review were obtained from the following databases: PubMed, Science Direct, Google Scholar.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Banting FG, Best CH, Collip JB, Campbell WR, Fletcher AA. Pancreatic extracts in the treatment of diabetes mellitus. Diabetes (1956) 5(1):69–71. doi: 10.2337/diab.5.1.69
2. Quesada I, Tudurí E, Ripoll C, Nadal A. Physiology of the pancreatic alpha-cell and glucagon secretion: role in glucose homeostasis and diabetes. J Endocrinol (2008) 199:5–19. doi: 10.1677/joe-08-0290
3. Müller TD, Finan B, Clemmensen C, DiMarchi RD, Tschöp MH. The new biology and pharmacology of glucagon. Physiol Rev (2017) 97:721–66. doi: 10.1152/physrev.00025.2016
4. Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Aminian A, Brethauer SA, et al. Bariatric surgery versus intensive medical therapy for diabetes 5-year outcomes. N Engl J Med (2017) 376:641–51. doi: 10.1056/NEJMoa1600869
5. Kilimnik G, Kim A, Steiner DF, Friedman TC, Hara M. Intraislet production of GLP-1 by activation of prohormone convertase 1/3 in pancreatic α-cells in mouse models of ß-cell regeneration. Islets (2010) 2:149–55. doi: 10.4161/isl.2.3.11396
6. Holt MK, Richards JE, Cook DR, Brierley DI, Williams DL, Reimann F, et al. Preproglucagon neurons in the nucleus of the solitary tract are the main source of brain glp-1, mediate stress-induced hypophagia, and limit unusually large intakes of food. Diabetes (2019) 68:21–33. doi: 10.2337/db18-0729
7. Zmazek J, Grubelnik V, Markovič R, Marhl M. Modeling the amino acid effect on glucagon secretion from pancreatic alpha cells. Metabolites (2022) 4:348. doi: 10.3390/metabo12040348
8. Ang T, Bruce CR, Kowalski GM. Postprandial aminogenic insulin and glucagon secretion can stimulate glucose flux in humans. Diabetes (2019) 5:939–46. doi: 10.2337/db18-1138
9. Rorsman P, Salehi SA, Abdulkader F, Braun M, MacDonald PE. K(ATP)-channels and glucose-regulated glucagon secretion. Trends Endocrinol Metab (2008) 19(8):277–84. doi: 10.1016/j.tem.2008.07.003
10. Wewer-Albrechtsen NJ. Glucagon receptor signaling in metabolic diseases. Peptides (2018) 100:42–7. doi: 10.1016/j.peptides.2017.11.016
11. Vergari E, Knudsen JG, Ramracheya R, Salehi A, Zhang Q, Adam J, et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat Commun (2019) 10:139. doi: 10.1038/s41467-018-08193-8
12. Silvestre RA, Rodríguez-Gallardo J, Jodka C, Parkes DG, Pittner RA, Young AA, et al. Selective amylin inhibition of the glucagon response to arginine is extrinsic to the pancreas. Am J Physiol Endocrinol Metab (2001) 280:E443–9. doi: 10.1152/ajpendo.2001.280.3.e443
13. Whalley NM, Pritchard LE, Smith DM, White A. Processing of proglucagon to GLP-1 in pancreatic α-cells: Is this a paracrine mechanism enabling GLP-1 to act on β-cells? J Endocrinol (2011) 211:99–106. doi: 10.1530/JOE-11-0094
14. Xu SFS, Andersen DB, Izarzugaza JMG, Kuhre RE, Holst JJ. In the rat pancreas, somatostatin tonically inhibits glucagon secretion and is required for glucose-induced inhibition of glucagon secretion. Acta Physiol (Oxf) (2020) 229:e13464. doi: 10.1111/apha.13464
15. Chia CW, Odetunde JO, Kim W, Carlson OD, Ferrucci L, Egan JM. GIP contributes to islet trihormonal abnorMalities in type 2 diabetes. J Clin Endocrinol Metab (2014) 99:2477–85. doi: 10.1210/jc.2013-3994
16. Singh B, Khattab F, Gilon P. Glucose inhibits glucagon secretion by decreasing (Ca2+)c and by reducing the efficacy of Ca2+ on exocytosis via somatostatin-dependent and independent mechanisms. Mol Metab (2022) 61:101495. doi: 10.1016/j.molmet.2022.101495
17. Piteau S, Olver A, Kim SJ, Winter K, Pospisilik JA, Lynn F, et al. Reversal of islet GIP receptor down-regulation and resistance to GIP by reducing hyperglycemia in the Zucker rat. Biochem Biophys Res Commun (2007) 362:1007–12. doi: 10.1016/j.bbrc.2007.08.115
18. Wiedemann T, Bielohuby M, Müller TD, Bidlingmaier M, Pellegata NS. Obesity in MENX rats is accompanied by high circulating levels of ghrelin and improved insulin sensitivity. Diabetes (2016) 65:406–20. doi: 10.2337/db15-0374
19. Shen XX, Li HL, Pan L, Hong J, Xiao J, Hermansen K, et al. Glucotoxicity and α cell dysfunction: involvement of the PI3K/Akt pathway in glucose-induced insulin resistance in rat islets and clonal αTC1-6 cells. Endocr Res (2012) 1:12–24. doi: 10.3109/07435800.2011.610855
20. Hansen NL, Brønden A, Nexøe-Larsen CC, Christensen AS, Sonne DP, Rehfeld JF, et al. Glucagon-Like peptide 2 inhibits postprandial gallbladder emptying in man: a randomized, double-blinded, crossover study. Clin Transl Gastroenterol (2020) 11:e00257. doi: 10.14309/ctg.0000000000000257
21. Holst JJ, Albrechtsen NJW, Gabe MBN, Rosenkilde MM. Oxyntomodulin: Actions and role in diabetes. Peptides (2018) 100:48–53. doi: 10.1016/j.peptides.2017.09.018
22. Baggio LL, Huang Q, Brown TJ, Drucker DJ. Oxyntomodulin and glucagon-like peptide-1 differentially regulate murine food intake and energy expenditure. Gastroenterology (2004) 127:546–58. doi: 10.1053/j.gastro.2004.04.063
23. Guida C, McCulloch LJ, Godazgar M, Stephen SD, Baker C, Basco D, et al. Sitagliptin and Roux-en-Y gastric bypass modulate insulin secretion via regulation of intra-islet PYY. Diabetes Obes Metab (2018) 20(3):571–81. doi: 10.1111/dom.13113
24. Kim T, Holleman CL, Nason S, Arble DM, Ottaway N, Chabenne J, et al. Hepatic glucagon receptor signaling enhances insulin-stimulated glucose disposal in rodents. Diabetes (2018) 11:2157–66. doi: 10.2337/db18-0068
25. Gerich JE, Lorenzi M, Schneider V, Kwan CW, Karam JH, Guillemin R, et al. Inhibition of pancreatic glucagon responses to arginine by somatostatin in normal man and in insulin-dependent diabetics. Diabetes (1974) 23(11):876–80. doi: 10.2337/diab.23.11.876
26. Conarello SL, Jiang G, Mu J, Li Z, Woods J, Zycband E, et al. Glucagon receptor knockout mice are resistant to diet-induced obesity and streptozotocin-mediated beta cell loss and hyperglycaemia. Diabetologia (2007) 50:142–50. doi: 10.1007/s00125-006-0481-3
27. Kazda CM, Ding Y, Kelly RP, Garhyan P, Shi C, Lim CN, et al. Evaluation of efficacy and safety of the glucagon receptor antagonist ly2409021 in patients with type 2 Diabetes: 12- and 24-Week Phase 2 Studies. Diabetes Care (2016) 39:1241–9. doi: 10.2337/dc17-er06
28. Vajda EG, Logan D, Lasseter K, Armas D, Plotkin DJ, Pipkin JD, et al. Pharma-cokinetics and pharmacodynamics of single and multiple doses of the glucagon receptor antagonist LGD-6972 in healthy subjects and subjects with type 2 diabetes mellitus. Diabetes Obes Metab (2017) 19:24–32. doi: 10.1111/dom.12752
29. Longuet C, Robledo AM, Dean ED, Dai C, Ali S, McGuinness I, et al. Liver-specific disruption of the murine glucagon receptor produces α-cell hyperplasia: evidence for a circulating α-cell growth factor. Diabetes (2013) 62:1196–205. doi: 10.2337/db11-1605
30. Van Dongen MG, Geerts BF, Morgan ES, Brandt TA, De Kam ML, Romijn JA, et al. First proof of pharmacology in humans of a novel glucagon receptor antisense drug. J Clin Pharmacol (2015) 55:298–306. doi: 10.1002/jcph.396
31. Capozzi ME, Wait JB, Koech J, Gordon AN, Coch RW, Svendsen B, et al. Glucagon lowers glycemia when β-cells are active. JCI Insight (2019) 5(16):e129954. doi: 10.1172/jci.insight.129954
32. Nauck MA, Quast DR, Wefers J, Meier JJ. GLP-1 receptor agonists in the treatment of type 2 diabetes-state-of-the-art. Mol Metabol (2021) 46:101102. doi: 10.1016/j.molmet.2020.101102
33. Hope DCD, Vincent ML, Tan TMM. Striking the Balance: GLP-1/Glucagon co-agonism as a treatment strategy for obesity. Front Endocrinol (Lausanne) (2021) 12:735019. doi: 10.3389/fendo.2021.735019
34. Sánchez-Garrido MA, Brandt SJ, Clemmensen C, Müller TD, DiMarchi RD, Tschöp MH. GLP-1/glucagon receptor co-agonism for treatment of obesity. Diabetologia (2017) 10:1851–61. doi: 10.1007/s00125-017-4354-8
35. Ambery PD, Klammt S, Posch MG, Petrone M, Pu W, Rondinone L, et al. MEDI0382, a GLP-1/glucagon receptor dual agonist, meets safety and tolerability endpoints in a single-dose, healthy-subject, randomized, Phase 1 study. Br J Clin Pharmacol (2018) 10:2325–35. doi: 10.1111/bcp.13688
36. Parker VER, Hoang T, Schlichthaar H, Gibb FW, Wenzel B, Posch MG, et al. Efficacy and safety of cotadutide, a dual glucagon-like peptide-1 and glucagon receptor agonist, in a randomized phase 2a study of patients with type 2 diabetes and chronic kidney disease. Diabetes Obes Metab (2022) 24(7):1360–9. doi: 10.1111/dom.14712
37. Jastreboff AM, Kaplan LM, Frías JP, Wu Q, Du Y, Gurbuz S, et al. Retatrutide phase 2 obesity trial investigators. Triple-hormone-receptor agonist retatrutide for obesity - A phase 2 trial. N Engl J Med (2023). doi: 10.1056/NEJMoa2301972
38. Liu DF, Ma ZY, Zhang CS, Lin Q, Li MW, Su KZ, et al. The effects of bariatric surgery on dyslipidemia and insulin resistance in overweight patients with or without type 2 diabetes: a systematic review and network meta-analysis. Surg Obes Relat Dis (2021) 17:1655–72. doi: 10.1016/j.soard.2021.04.005
39. Jorsal T, Wewer Albrechtsen NJ, Christensen MM, Mortensen B, Wandall E, Langholz E, et al. Investigating intestinal glucagon after roux-en-Y gastric bypass surgery. J Clin Endocrinol Metab (2019) 104(12):6403–16. doi: 10.1210/jc.2019-00062
40. Habegger KM, Al-Massadi O, Heppner KM, Myronovych A, Holland J, Berger J, et al. Duodenal nutrient exclusion improves metabolic syndrome and stimulates villus hyperplasia. Gut (2014) 63(8):1238–46. doi: 10.1136/gutjnl-2013-304583
41. Pérez-Arana GM, Gómez-Diaz A, DeLosReyes-Bancalero J, Camacho-Ramírez A, Fernández-Vivero J, Ribelles-García A, et al. The long-term failure of RYGB surgery in improving T2D is related to hyperinsulinism. Ann Anat (2022) 240:151855. doi: 10.1016/j.aanat.2021.151855
42. Alexiadou K, Cuenco J, Howard J, Wewer Albrechtsen N,J, Ilesanmi I, Kamocka A, et al. Proglucagon peptide secretion profiles in type 2 diabetes before and after bariatric surgery: 1-year prospective study. BMJ Open Diabetes Res Care (2020) 8(1):e001076. doi: 10.1136/bmjdrc-2019-001076
43. Kobayashi M, Waki H, Nakayama H, Miyachi A, Mieno E, Hamajima H, et al. Pseudo-hyperglucagonemia was observed in pancreatectomized patients when measured by glucagon sandwich enzyme-linked immunosorbent assay. J Diabetes Investig (2021) 2:286–9. doi: 10.1111/jdi.13325
44. Bataille D, Fontés G, Costes S, Longuet C, Dalle S. The glucagon-miniglucagon interplay: a new level in the metabolic regulation. Ann N Y Acad Sci (2006) 1070:161–6. doi: 10.1196/annals.1317.005
45. Dalle S, Fontés G, Lajoix AD, LeBrigand L, Gross R, Ribes G, et al. Miniglucagon (glucagon 19-29): a novel regulator of the pancreatic islet physiology. Diabetes (2002) 51(2):406–12. doi: 10.2337/diabetes.51.2.406
46. Øhrstrøm CC, Hansen DL, Kielgast UL, Bergmann ML, Veedfald S, Holst JJ, et al. Counterregulatory responses to postprandial hypoglycemia after Roux-en-Y gastric bypass. Surg Obes Relat Dis (2021) 17:55–63. doi: 10.1016/j.soard.2020.08.037
47. Tharakan G, Behary P, Wewer-Albrechtsen NJ, Chahal H, Kenkre J, Miras AD, et al. Roles of increased glycaemic variability, GLP-1 and glucagon in hypoglycaemia after Roux-en-Y gastric bypass. Eur J Endocrinol (2017) 177:455–64. doi: 10.1530/EJE-17-0446
48. Habegger KM, Heppner KM, Amburgy SE, Ottaway N, Holland J, Raver C, et al. GLP-1R responsiveness predicts individual gastric bypass efficacy on glucose tolerance in rats. Diabetes (2014) 63(2):505–13. doi: 10.2337/db13-0511
49. Lizarraga-Mollinedo E, Fernández-Millán E, Martín JDT, Martínez-Honduvilla C, Escrivá F, Álvarez C. Early undernutrition induces glucagon resistance and insulin hypersensitivity in the liver of suckling rats. Am J Physiol Endocrinol Metab (2012) 302:E1070–7. doi: 10.1152/ajpendo.00495.2011
50. Lupi R, Del Guerra S, D'Aleo V, Boggi U, Filipponi F, Marchetti P. The direct effects of GLP-1 and GIP, alone or in combination, on human pancreatic islets. Regul Pept (2010) 2-3:129–32. doi: 10.1016/j.regpep.2010.04.009
51. Christensen M, Knop FK, Vilsbøll T, Aaboe K, Holst JJ, Madsbad S, et al. Glucagon-like peptide-2, but not glucose-dependent insulinotropic polypeptide, stimulates glucagon release in patients with type 1 diabetes. Regul Pept (2010) 163(1-3):96–101. doi: 10.1016/j.regpep.2010.05.004
52. Camacho-Ramírez A, Prada-Oliveira JA, Ribelles-García A, Almorza-Gomar D, Pérez-Arana GM. The leading role of peptide tyrosine tyrosine in glycemic control after Roux-en-Y gastric bypass in rats. Obes Surg (2020) 2:697–706. doi: 10.1007/s11695-019-04239-y
53. Pérez-Arana GM, Díaz-Gómez A, Camacho-Ramírez A, Ribelles-García A, Almorza-Gomar D, Gracia-Romero M, et al. Peptide tyrosine-tyrosine triggers GLP-2-mediated intestinal hypertrophy after roux-en-Y gastric bypass. Obes Surg (2022) 32(12):4023–32. doi: 10.1007/s11695-022-06328
54. Le Roux CW, Borg C, Wallis K, Vincent RP, Bueter M, Goodlad R, et al. Gut hypertrophy after gastric bypass is associated with increased glucagon-like peptide 2 and intestinal crypt cell proliferation. Ann Surg (2010) 1:50–6. doi: 10.1097/SLA.0b013e3181d3d21f
55. Gu L, Lin K, Du N, Ng DM, Lou D, Chen P. Differences in the effects of laparoscopic sleeve gastrectomy and laparoscopic Roux-en-Y gastric bypass on gut hormones: systematic and meta-analysis. Surg Obes Relat Dis. (2021) 17:444–55. doi: 10.1016/j.soard.2020.10.018
56. Bancalero-DelosReyes J, Camacho-Ramírez A, Fernández-Vivero J, Ribelles-García A, Macías-Rodríguez M, Almorza-Gomar D, et al. Glucagon-producing cell expansion in Wistar rats. Changes to islet architecture after sleeve gastrectomy. Obes Surg (2021) 31:2241–9. doi: 10.1007/s11695-021-05264-6
57. Tomas A, Jones B, Leech C. New insights into beta-cell GLP-1 receptor and cAMP signaling. J Molec Biol (2020) 432:1347–66. doi: 10.1016/j.jmb.2019.08.009
58. Yang C, Brecht J, Weiß C, Reissfelder C, Otto M, Buchwald JN, et al. Serum glucagon, bile acids, and FGF-19: metabolic behavior patterns after roux-en-Y gastric bypass and vertical sleeve gastrectomy. Obes Surg (2021) 31(11):4939–46. doi: 10.1007/s11695-021-05677-3
59. Gray SM, Niu J, Zhang A, Svendsen B, Campbell JE, D’Alessio DA, et al. Intraislet ghrelin signaling does not regulate insulin secretion from adult mice. Diabetes (2019) 68:1795–805. doi: 10.2337/db19-0079
60. Qiu NC, Li W, Liu ME, Cen XX, Shan CX, Zhang W, et al. Comparison of great curvature plication with duodenal-jejunal bypass (GCP-DJB) and Sleeve Gastrectomy (SG) on metabolic indices and gut hormones in type 2 diabetes mellitus rats. Obes Surg (2018) 12:4014–21. doi: 10.1007/s11695-018-3459-6
Keywords: glucagon, diabetes, sleeve gastrectomy, Roux-en-Y gastric bypass, incretin, glucose, alpha cell
Citation: Pérez-Arana G-M, Díaz-Gómez A, Bancalero-de los Reyes J, Gracia-Romero M, Ribelles-García A, Visiedo F, González-Domínguez Á, Almorza-Gomar D and Prada-Oliveira J-A (2023) The role of glucagon after bariatric/metabolic surgery: much more than an “anti-insulin” hormone. Front. Endocrinol. 14:1236103. doi: 10.3389/fendo.2023.1236103
Received: 07 June 2023; Accepted: 21 July 2023;
Published: 11 August 2023.
Edited by:
Undurti Narasimha Das, UND Life Sciences LLC, United StatesReviewed by:
Finbarr P. M. O'Harte, Ulster University, United KingdomTamer Coskun, Eli Lilly, United States
Copyright © 2023 Pérez-Arana, Díaz-Gómez, Bancalero-de los Reyes, Gracia-Romero, Ribelles-García, Visiedo, González-Domínguez, Almorza-Gomar and Prada-Oliveira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gonzalo-Martín Pérez-Arana, Z29uemFsby5wZXJlekB1Y2EuZXM=; José-Arturo Prada-Oliveira, YXJ0dXJvLnByYWRhQHVjYS5lcw==