 Shengnan Wang1†
Shengnan Wang1† Renyi Peng
Renyi Peng
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 21 August 2023
Sec. Diabetes: Molecular Mechanisms
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1230168
As important organelles of energetic and metabolism, changes in the dynamic state of mitochondria affect the homeostasis of cellular metabolism. Mitochondrial dynamics include mitochondrial fusion and mitochondrial fission. The former is coordinated by mitofusin-1 (Mfn1), mitofusin-2 (Mfn2), and optic atrophy 1 (Opa1), and the latter is mediated by dynamin related protein 1 (Drp1), mitochondrial fission 1 (Fis1) and mitochondrial fission factor (MFF). Mitochondrial fusion and fission are generally in dynamic balance and this balance is important to preserve the proper mitochondrial morphology, function and distribution. Diabetic conditions lead to disturbances in mitochondrial dynamics, which in return causes a series of abnormalities in metabolism, including decreased bioenergy production, excessive production of reactive oxygen species (ROS), defective mitophagy and apoptosis, which are ultimately closely linked to multiple chronic complications of diabetes. Multiple researches have shown that the incidence of diabetic complications is connected with increased mitochondrial fission, for example, there is an excessive mitochondrial fission and impaired mitochondrial fusion in diabetic cardiomyocytes, and that the development of cardiac dysfunction induced by diabetes can be attenuated by inhibiting mitochondrial fission. Therefore, targeting the restoration of mitochondrial dynamics would be a promising therapeutic target within type II diabetes (T2D) and its complications. The molecular approaches to mitochondrial dynamics, their impairment in the context of T2D and its complications, and pharmacological approaches targeting mitochondrial dynamics are discussed in this review and promise benefits for the therapy of T2D and its comorbidities.
Mitochondria represent one of the major resources of reactive oxygen species (ROS) and the main part of ATP production (1, 2). In the persistent hyperglycemic state of diabetes, mitochondria will increase the output of ROS, which leads to oxidative stress (OS) and tissue damage (3–5). Hyperglycemia can lead to a relative increase in ROS in three ways (causing respiratory chain electron transport blockage, burst production of ROS, and damage to the antioxidant system), mediating mitochondrial OS, damaging mitochondria and causing mitochondrial dysfunction (4, 6, 7). The normal function of mitochondria relies in their efficient quality of control with a highly plasticity of their dynamic structure, that makes them continually changeable through fusion and fission processes, namely mitochondrial dynamics (8, 9).
Mitochondrial fusion consists of outer membrane (OMM) and inner membrane (IMM) fusion, with OMM fusion are modulated by mitofusin-1 (Mfn1) and mitofusin-2 (Mfn2), whereas IMM fusion are modulated by optic atrophy 1 (Opa1) (10, 11). Dynamin related protein 1 (Drp1) is a crucial protein in mediating mitochondrial division, and other proteins involved in fission include mitochondrial fission factor (MFF) and mitochondrial fission 1 (Fis1) (11, 12). Regulatory mitochondrial dynamics is a complicated process in which mitochondria are coregulated by the above GTPases to maintain the homeostasis of their fission and fusion, which is fundamental to cellular functions including mitochondrial DNA distribution, mitochondrial functions, cell survival and signaling (8, 13, 14). Alterations in this balance may result in OS, mitochondrial impairment and metabolic modification, finally contributing to the occurrence of mitochondrial-related diseases (8, 15, 16).
Mitochondrial dynamics are markedly changed in type II diabetes (T2D) patients and are involved in the progression of insulin resistance, and not only that, mitochondrial dynamics are also closely correlated with the evolution of diabetic complications (6, 17–19). Therefore, it is significant to investigate the association between alterations in mitochondrial dynamics and T2D and its complications, and this article reviews existing research progress in this area.
Mitochondrial dynamics means that the dynamic equilibrium in which mitochondria are in a constant fusion and fission process. The mitochondrial fusion and fission processes are synergistic, highly conserved processes, and the two are generally in dynamic equilibrium, a balance that is essential for sustaining the proper mitochondrial morphology, distribution and function (8). Both abnormal fusion and fission will affect the normal function of mitochondria, for example, abnormal fission will result in small and broken mitochondrial morphology and abnormal fusion will result in extended mitochondria (6, 20).
The morphology of mitochondria can be changed by mitochondrial fusion, which refers to the phenomenon of two adjoining mitochondria merging their contents into one mitochondria with fibrous extensions and a network structure, and is present in all cells with mitochondria (10, 11, 21). Mitochondrial fusion is considered to be a key factor in maintaining a healthy mitochondrial network. The underlying principle is to achieve dilution of toxic substances in mitochondria and repolarization of membrane potential through fusion for the purpose of maintaining genetic and biochemical homogeneity (14). This function results in the selective fusion of healthy and less healthy mitochondria, while depolarized unhealthy mitochondria are rapidly become targets for decomposition (22, 23). Thus, in contrast to the active selection of fusion partners by mitochondria, the cytoplasmic environment is perhaps the pivotal determinant of fusion events, and a change in any one substance would affect this process (11, 14).
The structure of the Mfn molecule mediating outer mitochondrial membrane (OMM) fusion contains an GTPase domain, two hydrophobic heptapeptide repeat helix regions (HR1 and HR2), with two transmembrane domains (TM) (24, 25). The GTPase domain is essential for fusion activity and the TM is required for OMM insertion (24, 26). Mfn mediated OMM involves three steps, tethering, docking and merger (10). The two mitochondria must be in close contact to begin fusion, and once close contact is established, the OMM of the two mitochondria form trans-homologous or heterologous complexes via the HR2 and GTPase domains of Mfn, mediating OMM fusion (26–29). Mfn undergoes local and global conformational changes during GTP hydrolysis and forms a dimer through the GTPase domain to complete the OMM tether. As the cycle of GTP hydrolysis process continues, the mitochondria are continuously drawn closer together, eventually achieving OMM fusion (Figure 1A) (10, 29). Functionally, Mfn1 and Mfn2 are considered essential for mitochondrial fusion, and both have not only a degree of similarity as well as substantial differences (30). The tether efficiency of Mfn1 on mitochondrial membrane is higher than Mfn2, and the GTPase activity of purified Mfn1 is eight times more than the one of Mfn2 (29). In addition, defects in either Mfn1 or Mfn2 will result in abnormal mitochondrial morphology and reduced fusion rates. In Mfn2 deficient fibroblasts mitochondria appeared spherical or round with swelling, whereas Mfn1 deficient mitochondria were significantly more brokenness (31).
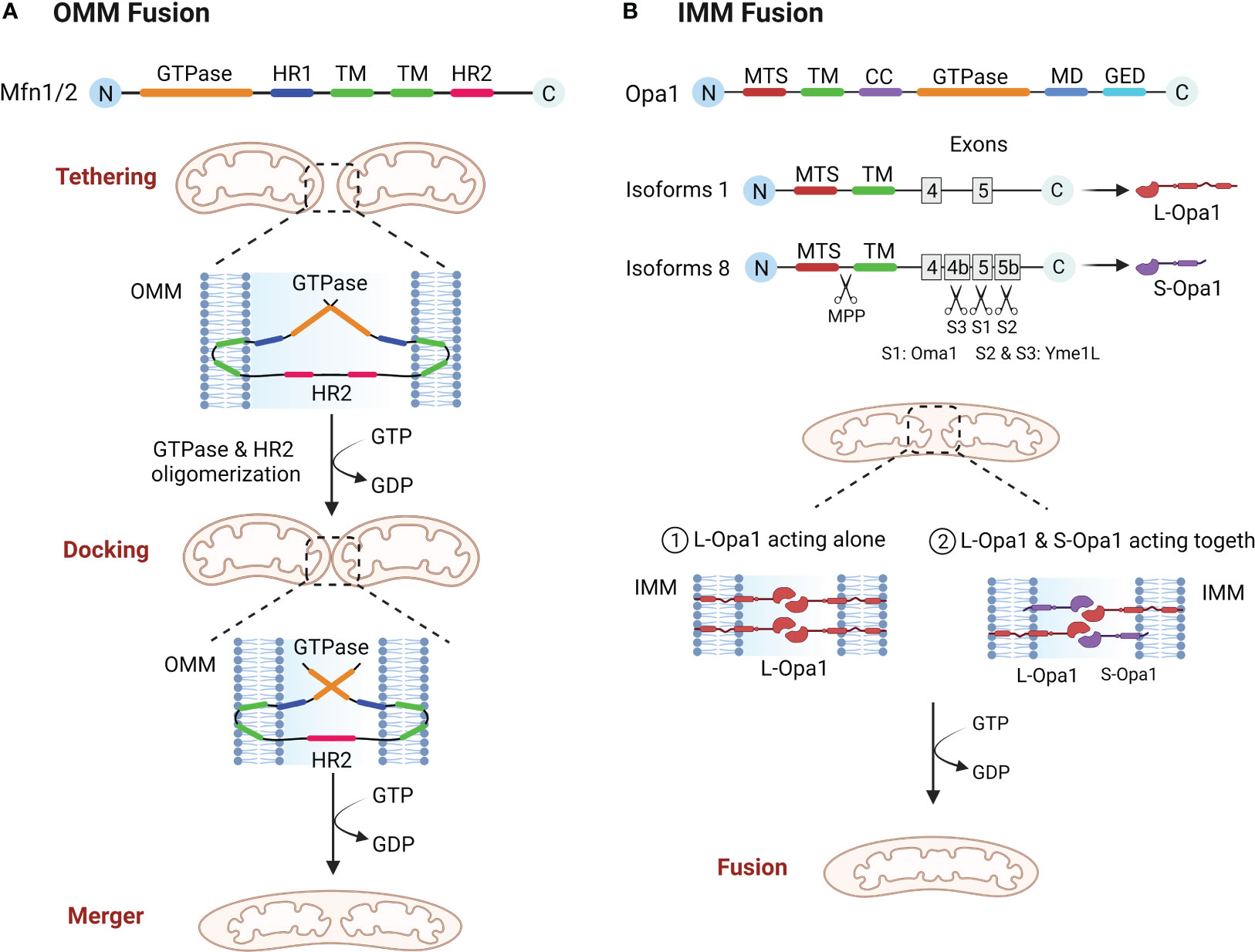
Figure 1 (A) OMM fusion. Mfn1 and Mfn2 tether mitochondria through oligomerization of HR2 and GTPase domain, and as GTP is hydrolyzed, mitochondria move closer together, eventually leading to OMM fusion. (B) IMM fusion. Opa1 needs to be hydrolyzed to L-Opa1 and S-Opa1 to promote IMM fusion. The figure shows two isoforms of Opa1, the mitochondrial targeting sequence (MTS) is first excised by MPP, and because isoform 1 lacks exons 4b and 5b it will not be cut, resulting in L-Opa1. While isoform 8 is cleaved in the S1 site by Oma1 and in the S2 and S3 sites by Yme1L, giving rise to S-Opa1. L-Opa1 then mediates IMM fusion either alone or in conjunction with S-Opa1.
After the OMM fusion, the IMM fusion must be followed up quickly and efficiently to completion (27). The structural composition of OPA1 is an mitochondrial targeting sequence (MTS), a subsequent TM, a coiled coil domain (CC), a highly conserved GTPase domain, an middle domain (MD) and a GTPase effector domain (GED) (10, 32). IMM fusions are more complex than OMM fusions in that the Opa1 gene coding has eight distinct isoforms and three cleavage sites (S1, S2, S3) and requires partial protein hydrolysis into two forms, L-Opa1 and S-Opa1, to successfully complete the fusion reaction (33, 34). Some of these researches suggest that L-Opa1 is integral to IMM and promotes IMM fusion, whereas S-Opa1 does not mediate fusion (35). However, Song and Ge et al. demonstrated that IMM fusion requires the joint collaboration of S-Opa1 and L-Opa1 (36, 37). Therefore, the exact mechanism of IMM fusion will have to be confirmed by further research. Analogous with Mfn proteins, Opa1 also constitutes oligomeric structures that drive IMM fusion through conformational changes driven by GTP hydrolysis (Figure 1B) (38). In addition, Opa1 is involved in cristae shaping and electron tomography has shown that Opa1 modulates the morphology of mitochondrial cristae, especially during apoptosis (39).
Mitochondria can divide to produce one or more daughter mitochondria, mainly mediated by Drp1, which has four different structural domains, including the GTPase domain, MD, variable domain and GED (11, 12). Drp1 in the cytoplasm need to be translocate to the mitochondria carry out its divisive function (40). The mitochondrial fission process undergoes three critical phases, marking of the fission site, oligomerization of Drp1 at the fission site and assembly into a helical superstructure, and contraction of the Drp1 and severing of the mitochondria (14). The preliminary step of mitochondrial fission is the initial contraction of the OMM, which is produced by the endoplasmic reticulum (ER) and actin filaments (41, 42). ER contacts mitochondria to form mitochondria-associated ER membranes (MAMs) to mediate the forming of mitochondrial fission sites, reducing the average mitochondrial diameter through the polymerization of ER-associated inverted formin 2 (INF2), myosin II, and Spire1C at the MAMs, which contributes to the formation of a Drp1 oligomerization loop at the fission site (41, 43, 44). Then there is the Drp1 recruitment, which is mainly accounted for by a class of small molecule proteins that are mosaic on the OMM (45). These include Fis1, MFF, 49 kDa and 51 kDa of mitochondrial dynamics protein (MiD49 and MiD51), the deletion of each of these molecules leads to significant stretching of the mitochondria (45). Drp1 is translocated to the fission site of the OMM in the cytoplasm and oligomerized, then assembles to a helical superstructure (46). Finally, the mitochondrial conformation is altered by hydrolysis of GTP, which continues to enhance mitochondrial contraction until two mitochondria are created (Figure 2) (11, 47). Following mitochondrial fission, Drp1 can be returned to the cytoplasm for reuse (11).
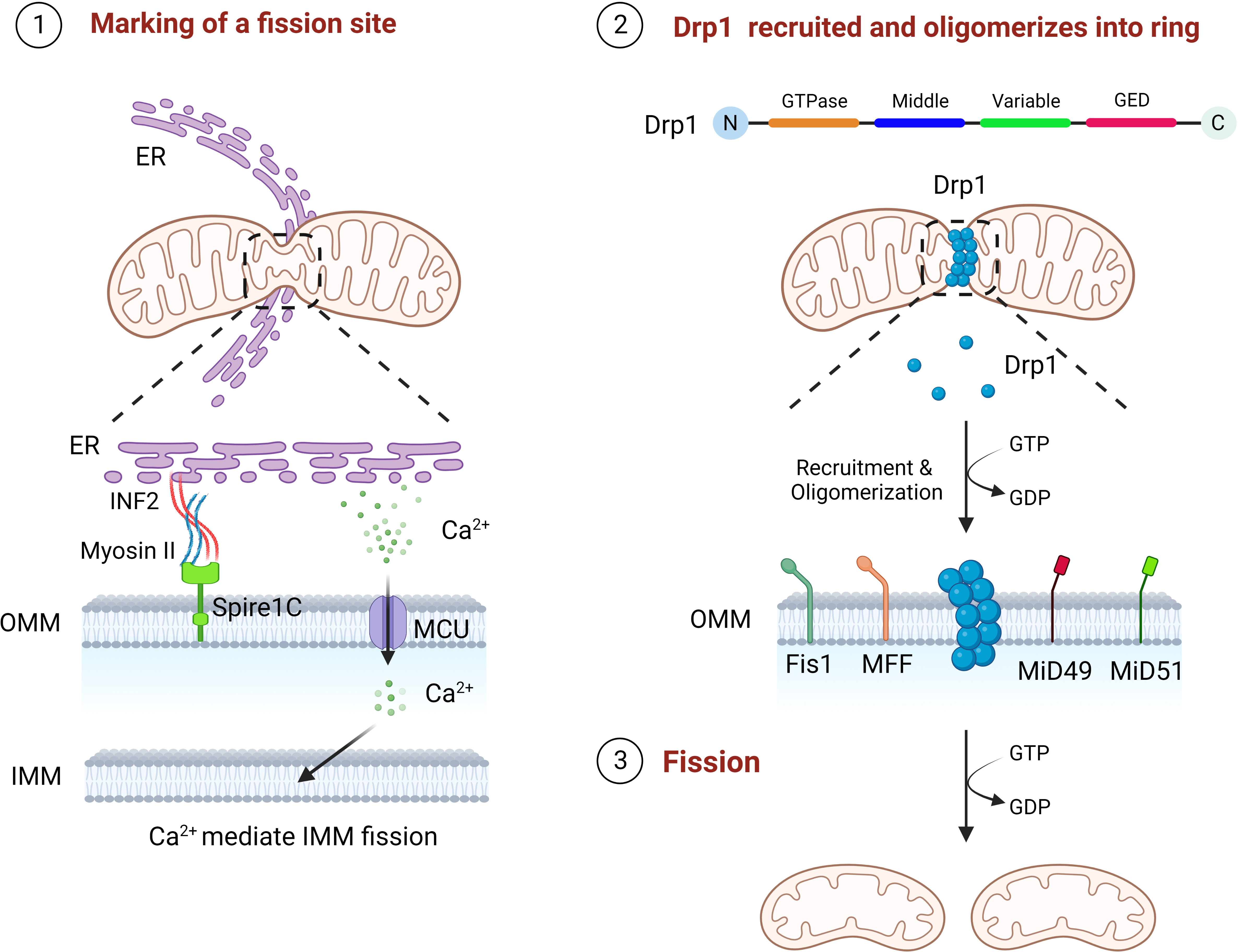
Figure 2 The three steps of mitochondrial fission. First, ER contacts mitochondria, polymerizes with Spire1C on mitochondria to form a fission site through INF2 and myosin II, and releases Ca2+ into mitochondria to mediate IMM fission. Drp1 is then recruited by Fis1, MFF, MiD49 and MiD50 to the mitochondrial fission site and oligomerizes into a loop. Finally, through GTP hydrolysis, Drp1 continuously contracts mitochondria until fission.
In addition, INF2 mediated actin polymerization facilitates the Ca2+ liberation from the ER at the MAMs, followed by Ca2+ passes through the mitochondrial calcium unidirectional transporter protein (MCU) into the mitochondria leading to IMM contraction (48). As the S637 residue of Drp1 can be dephosphorylated by Ca2+ dependent calcineurin, persistent elevated cytoplasmic Ca2+ levels further promotes Drp1 aggregation in mitochondria, suggesting that IMM fission is independent of, and precedes, OMM fission (48–50). More interestingly, S-Opa1 produced by Opa1 hydrolysis has been found to act in mitochondrial fission, where stress induced OMA1 hydrolyzes all of Opa1 into a short isomers and S-Opa1 is able to partly co-position with the MAMs, thereby inhibiting fusion and triggering mitochondrial breakage (51).
Mitochondrial fusion and fission are engaged directly or indirectly in mitochondrial maintenance, bioenergy production and cell death, which is essential in the development of diabetes mellitus and its complications (12, 13).
ATP produced by mitochondria through respiration and oxidative phosphorylation (OXPHOS) is the main source of energy in cell and along with the production of ROS (52, 53). Impaired OXPHOS leads to overproduction of ROS and gradually inhibit or deplete the body’s antioxidant system, eventually lead to the disorder of the body’s redox balance and result in OS and tissue damage (6, 54). As a vital mechanism for the homeostatic modulation of mitochondria, mitochondrial dynamics can be participated in the modification of OXPHOS processes (5). It is suggest that mitochondrial morphology may determine the level of intracellular ATP supply and the mode of respiratory metabolism (9, 55). Mitochondria in the fused state are rich in internal cristae structure, with close connected with the electron transport chain and high efficiency of OXPHOS. This is attributed to the fact that Opa1 and Mfn1/2 dependent mitochondrial fusion preserves mtDNA mass to maintain the electronic respiratory chain (ETC) complex and overall OXPHOS capacity (56, 57). In addition, Mfn2 could bind to a glycolysis rate-limiting enzyme (pyruvate kinase) to enhance mitochondrial OXPHOS and attenuate glycolysis (58). When the level of Drp1 mediated mitochondrial fission is elevated, OXPHOS capacity is reduced and the bioenergetic state is shifted to glycolysis, with a concomitant decrease in mitochondrial membrane potential and an increase in ROS production. By blocking Drp1 phosphorylation, mitochondrial fusion can be redirected and OXPHOS can be activated (56, 57). However, excessive mitochondrial fusion equally damages OXPHOS (57). Thus, coordinated mitochondrial dynamics homeostasis is essential for maintaining OXPHOS function and induces mitochondrial OS (Figure 3A).
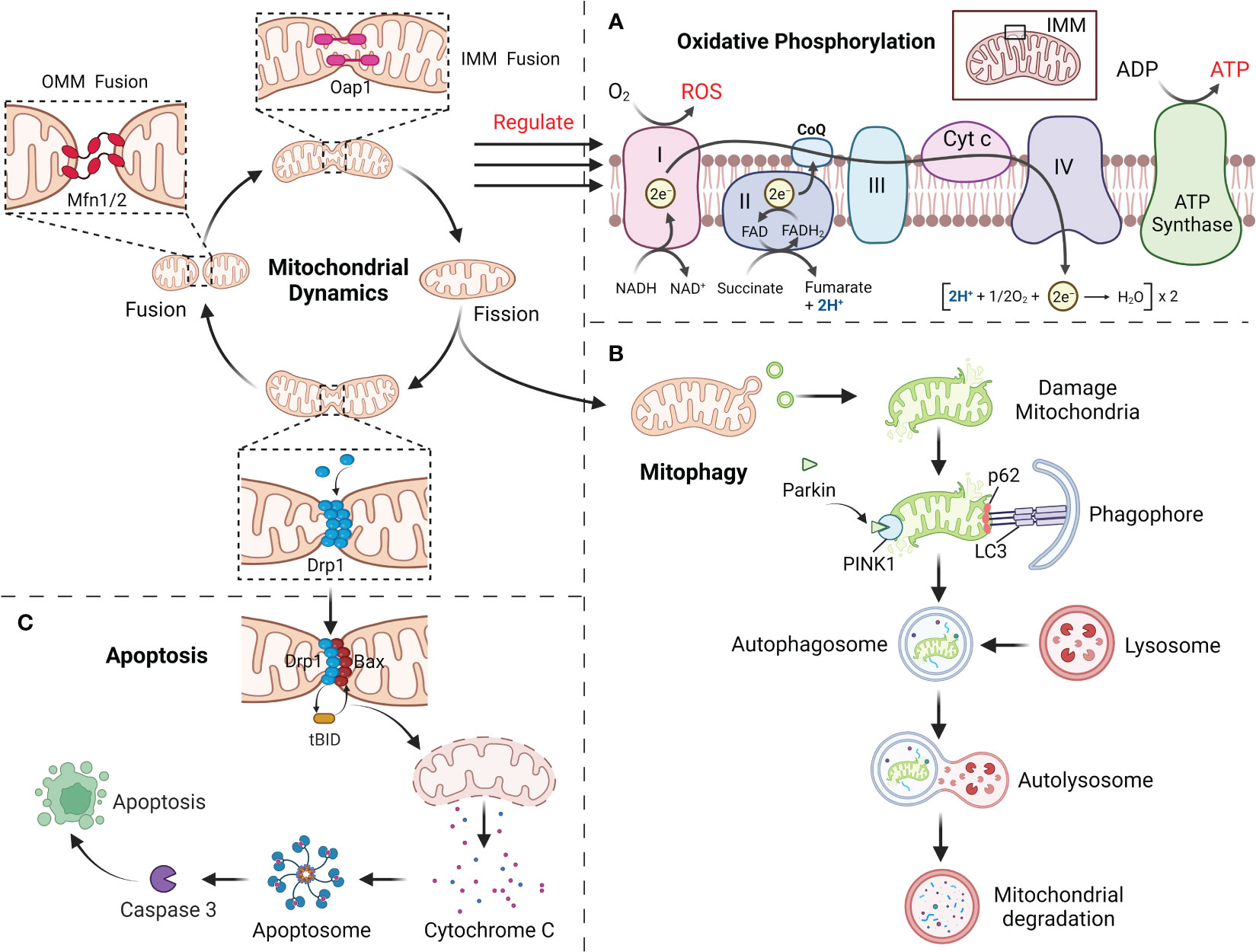
Figure 3 (A) Mitochondrial dynamics regulate the OXPHOS process, with fusion promoting OXPHOS and fission inhibiting it. (B) The damaged mitochondria first divide the damaged site through mitochondrial fission, and then degraded by mitophagy. (C) Mitochondrial fission mediates apoptosis through the release of Cyt c.
Mitochondria can maintain their homeostatic function by selectively degrading excess or damaged mitochondria within the cell, and this mechanism is called mitophagy (23, 59). Before mitophagy, the damaged mitochondria require Drp1 for mitochondrial fragments of appropriate size by mitochondrial fission (59). Moreover, Mfn1/2 and Opa1 in damaged mitochondria are degraded to prevent greater damage caused by the fusion of damaged mitochondria with healthy ones (23, 60). Mitophagy mainly involves PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin (59). When mitochondria have been injured, PINK1 accumulates in the OMM via outer membrane translocases, activating and recruiting Parkin, followed by ubiquitination of the proteins VDAC1 and Mfn1/2 in the mitochondrial outer by Parkin (61, 62). Subsequently, p62 accumulates on mitochondria, combines with ubiquitinated mitochondria together with LC3 and is phagocytosed by autophagosomes, which merge with lysosomes to form autophagic lysosomes that degrade the contained mitochondria (Figure 3B) (63). In addition, several mitochondrial LC3 receptors have been described that are independent of PINK1 in ubiquitin-independent autophagy, including BNIP3, Nix, FUNDC1 and BCL2L13, which include LC3-interacting regions that bind directly to LC3 and assemble damaged mitochondria to the autophagosome (64, 65). Mitochondrial dynamics are inextricably linked to mitophagy. FUNDC1 interacts with Drp1 and Opa1, and under normal conditions FUNDC1 can bind to Opa1 to promote mitochondrial fusion, whereas under stress conditions FUNDC1 detaches from Opa1, enhancing the recruitment of Drp1 by FUNDC1 at the MAMs and promoting mitochondrial fission as well as mitochondrial autophagy (66, 67). When mitophagy is impaired, mitochondrial dynamics will likewise be affected. PHB2 has been proved to be required for PINK1-mediated mitophagy, and PHB2 deletion causes selective absence of the long isoform of Opa1, which results in abnormal cristae morphogenesis and mitochondrial fragmentation (68, 69). Abolishing mitophagy induced by BNIP3L and FUNDC1 during cardiac progenitor cell differentiation will also result in consistent mitochondrial fission and donut-like formation of damaged mitochondria (70). This shows a strong association with mitochondrial dynamics and mitophagy.
Mitochondria as a vital regulator of apoptosis, which play a vital role in the progression of apoptosis by liberating cytochrome c (Cyt c) and other pro-apoptotic factors (71, 72). Drp1, an important mitochondrial fission protein, not only affects mitochondrial morphology but also participates in the regulation of apoptosis (73). Mitochondrial fission itself does not lead to apoptosis, but mainly pro-apoptotic proteins (such as Bax) from the Bcl-2 family members are co-expressed with Drp1 at the fission site responsible for Cyt c release, an important early event in caspase 3 activation that ultimately induces apoptosis (74, 75). During apoptosis, recruitment of Drp1 is enhanced and Drp1 stimulates tBid-triggered Bax oligomerization by promoting hemifusion of cardiolipin-containing membranes and then initiates apoptosis by releasing Cyt c through a membrane hemifusion intermediate formed during mitochondrial fission (Figure 3C) (74, 75). Even in the absence of apoptotic triggers, Drp1 can directly regulate the Bax pore to affect the permeability activity of Bax to promote apoptosis. This is because the N-terminal structural domain of Bax interacts specifically and directly with Drp1 to form a homodimeric complex until cell death, and this interaction is enhanced during apoptosis (74).Downregulation of Drp1 caused by RNAi not only retards mitochondrial fission, but also inhibits Cyt c release and cell death (76). Fis1 was also involved in the regulation of apoptosis, and downregulation of Fis1 inhibited apoptosis to a significantly greater extent than downregulation of Drp1 (77). However, during cell differentiation, reduced levels of mitophagy induced by decreased Drp1 activity promote the early stages of apoptosis, whereas its overexpression prevents apoptosis, which provides another important link between apoptosis and mitochondrial dynamics (78). Furthermore, silencing of Mfn1 or Mfn2 will lead to increased mitochondrial fragmentation and sensitivity to apoptotic stimuli (79). Opa1 blocks the tBid-induced increase in Cyt c release from the cristae into the membrane gap, and Opa1 deficiency will also induce mitochondrial cristae abnormalities as well as spontaneous cell apoptosis (79). Restoration of mitochondrial fusion delays Bax activation and Cyt c release and reduces cell damage/apoptosis.
T2D is mainly distinguished by mitochondrial dysfunction, increased ROS content and decreased ATP levels (6). In T2D, hyperglycemia induces excessive mitochondria division and fragmentation in different cell types, with excessive production of ROS, reduced mitochondrial fusion and increased mitochondrial fission eventually resulting in mitochondrial dysfunction (80, 81). Under normal physiological conditions, damaged mitochondria are eliminated by mitophagy before they lead to excessive ROS production (81). In contrast, hyperglycemia in T2D inhibits the expression of mitophagy-related genes (NIZ, PINK1, Parkin), leading to impaired mitophagy (82). Reduced ATP levels due to mitochondrial dysfunction can be compensated by mitochondrial biogenesis, however, downregulation of a gene involved in mitochondrial biogenesis (PGC-1α) was observed in T2D (83). Abnormal mitochondrial biogenesis leads to energy dysregulation and accelerates ROS production, thereby aggravating the pathological pathway of diabetes development (84). Thus, abnormal mitochondrial dynamics induced by hyperglycemia may be the pathogenesis and common basis for the genesis of chronic complications of diabetes mellitus (Figure 4).
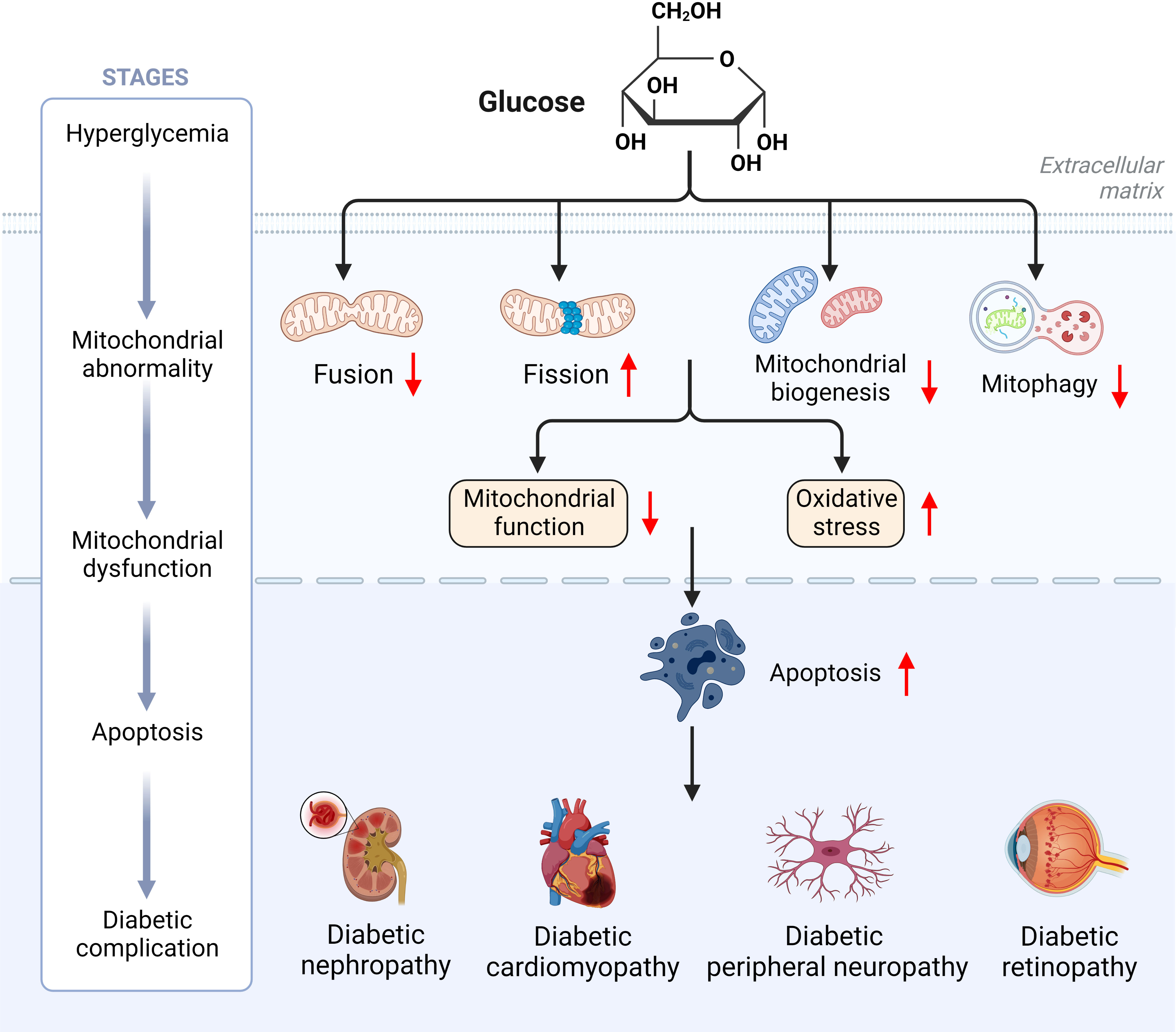
Figure 4 Hyperglycemia induces mitochondrial dysfunction ultimately leading to diabetic complications.
The kidney is a highly metabolic, energy-consuming and mitochondria-rich organ second only to the myocardium. In the diabetic state, the impairment of mitochondrial dynamics resulting in reduced mitochondrial quantity and capacity, and the accumulation of ROS are also considered to be a critical link the pathogenesis and evolution of DN, leading to a large number of cytokines and inflammatory mediators are released and downstream signaling pathways such as protein kinase C are activated, which further destroys the intrinsic cells of the kidney (podocytes, glomerular endothelial cells, thylakoid cells and tubular epithelial cells) and promotes the genesis and evolution of DN (15, 85–87).
Studies of renal cells in the diabetic setting showed altered renal mitochondrial dynamics, with increased mitochondrial Drp1 expression and decreased Mfn2 expression, suggesting that mitochondria tend to undergo fission and that the fusion process is inhibited (88). Disturbances in renal mitochondrial dynamics lead to histological and renal parameter abnormalities, Drp1 overexpression can lead to membrane matrix expansion, basement membrane thickening, podocyte damage and albuminuria (89). The associated proteins participating in mitochondrial fission and fusion are altered during the various stages of DN, and genetic modification of them can alleviate the symptoms of DN (88, 90). For example, DN mice specifically overexpressing Mfn2 or deletion of Drp1 can significantly improve kidney injury and mitochondrial ROS accumulation (91). Ultrastructural analysis showed that diabetic mice specifically deficient in Drp1 had reduced proteinuria and significantly improved membrane matrix expansion and foot cell morphology as well as mitochondrial structure compared with wild-type diabetic mice (91). Additionally, in Rho-associated kinase 1 (ROCK1) transgenic mice, it was observed that hyperglycemia phosphorylated Drp1 serine residues via ROCK1, thereby promoting the recruitment of Drp1 into mitochondria and ultimately inducing mitochondrial fission, while knockdown of ROCK1 reduced glomerular apoptosis and mitochondrial ROS production, decreased proteinuria and reduced podocyte shedding (92). Administration of Mdivi1, a pharmacological inhibitor of Drp1, significantly attenuated mitochondrial fission in mice and salvaged critical pathological characteristics of DN (93).
Asiatic acid was recently found to reduce tubular injury in DN by decreasing the expression of Drp1 and increasing the expression of Mfn1/2 via the Nrf-2 pathway (94). Existing studies have identified a variety of drugs that can modulate mitochondrial dynamics to alleviate the symptoms of DN, as shown in Table 1. In addition to alleviating the symptoms of DN, the modulation of mitochondrial dynamics may further exacerbate the damage of DN. When diabetic rats are administered Crocodile Oil, it will further increase Drp1 expression and decrease Mfn2 levels (95). Crocodile Oil use exacerbates diabetic kidney injury with a significant increase in mitochondrial ROS production and a decrease in mitochondrial membrane potential (MMP) compared to diabetic rats that do not receive Crocodile Oil (101). Therefore, by targeting and regulating mitochondrial fusion division-associated protein expression will influence the progression of DN. In addition, mitophagy, which is closely related to mitochondrial dynamics, is also expected to serve as a new target for the treatment of DN. PACS-2 promotes mitophagy and plays an important role in ameliorating tubular injury in diabetes (95).
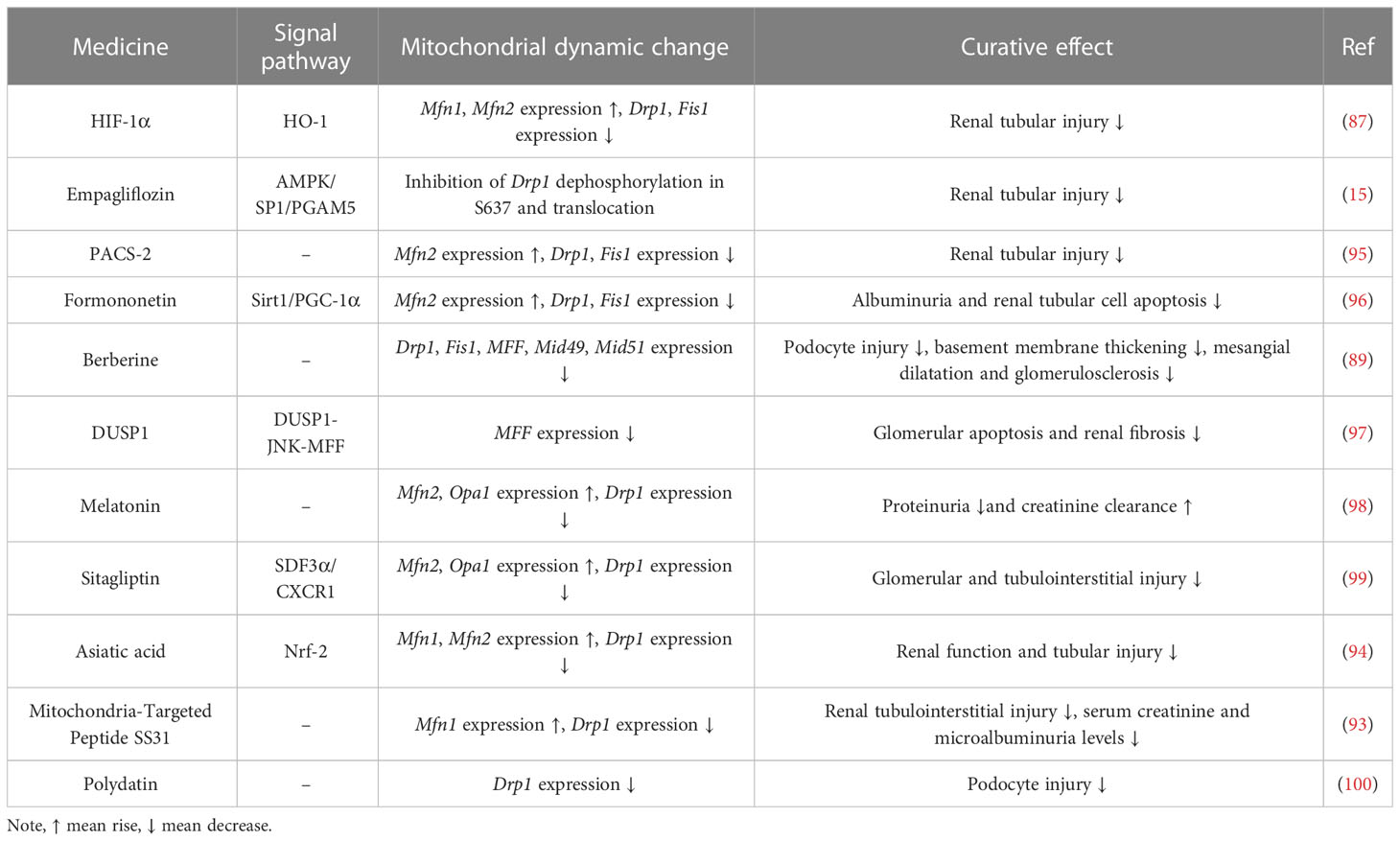
Table 1 Drugs targeting mitochondrial dynamics in the treatment of DN.
DCM is a nonischemic and nonhypertensive cardiomyopathy caused by diabetes-related metabolic disorders and represents one of the common diabetic complications (102). Deficiency of myocardial energy is highly associated with the development and evolution of various cardiac pathologies (103). Under normal physiological conditions, mitochondrial OXPHOS accounts for the vast majority of cardiac ATP requirements; however, in the presence with insulin resistance, myocardial use of insulin-stimulated glucose uptake and glucose utilization is reduced and fatty acid uptake and oxidation rates are increased, and this altered substrate preference plays an essential role in the pathophysiology of DCM (103–105). When fatty acid intake in the heart increases, it can cause mitochondrial structural remodeling, significantly reduce the minimum diameter, and regulate Opa1 and Drp1 post-translational modifications, thereby promoting mitochondrial fission (106).
DCM is initially marked by myocardial fibrosis, left ventricular hypertrophy and diastolic dysfunction, which later manifests as systolic dysfunction and eventually clinical heart failure (102, 104, 107). Heart failure is distinguished by altered redox regulation of the myocardium, mainly in the form of OS, and increased ROS have been described in animal models of heart failure (107–109). We have previously described the association between mitochondrial dynamics and OS, and alterations in mitochondrial dynamics are observed in all pathological changes in DCM, as shown in Table 2. Disturbed mitochondrial dynamics in cardiomyocytes, followed by mitochondrial OXPHOS and mitochondrial respiratory chain dysfunction, lead to reduced ATP production and excessive ROS production, ultimately leading to cardiomyocyte death (112). Hu’s study indicated that the reduced expression of Mfn2 in DCM could be partly attributed to the result of reduced expression of peroxisome proliferator-activated receptor α (PPARα) (112). Mfn2 deficiency not only leads to impaired glucose tolerance, the progression of hyperinsulinemia and insulin resistance, but also affects the normal development of cardiac muscle cells (117). Mitophagy has been shown to be critical in protecting cardiac function during DCM, and Mfn2 may have a pivotal role in cardiac autophagy by facilitating fusion between autophagic vesicles and lysosomes (118, 119). Parkin-mediated mitophagy protects against high-fat diet-induced cardiac invasion of hypertrophy, lipid accumulation and diastolic dysfunction in the heart, and when mitophagy is impaired, mitochondrial dysfunction and lipid accumulation are induced, thereby exacerbating DCM (118).
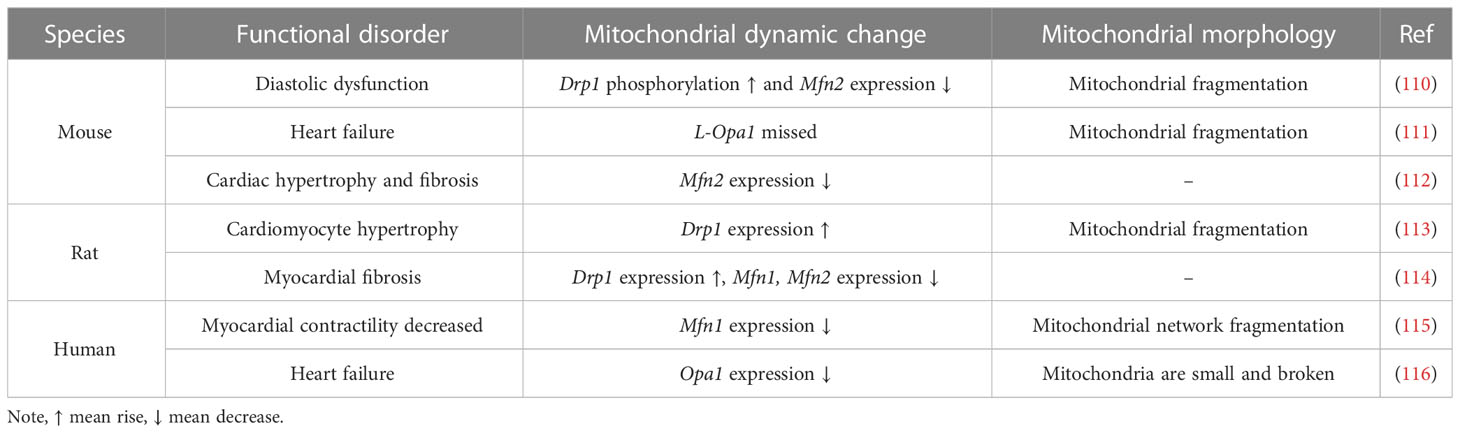
Table 2 Mitochondrial dynamics and morphology under DCM in different species.
Calcineurin (CaN) is an important regulator of cardiac hypertrophy and heart failure, and CaN is activated in response to increased calcium concentrations in cells (120, 121). High glucose stimulation induces upregulation of Orai1 (Ca2+ release-activated calcium channel protein 1) expression, and Orai1 is able to mediate calcium inward flow to bind CaN and calmodulin (CaM). CaM then binds to CaN to form an active phosphatase that catalyzes subunit A (CnA) and phosphorylates- extracellular signal-regulated kinase (p-ERK1/2) as targets to suppress Drp1 phosphorylation at S637 and induce Drp1 phosphorylation at S616, respectively, facilitating mitochondrial fission and speeding up cardiac hypertrophy induced by high glucose (113, 122). Suppression of the Orai1-Ca2+-CnA or ERK-Drp1 prevents high glucose induced cardiomyocyte hypertrophy (113). Excessive mitochondrial fission in DCM patients and administration of the mitochondrial fusion promoter M1 dramatically enhanced mitochondrial fusion and increased Opa1 expression levels in diabetic hearts, improving myocardial fibrosis and OS in cardiomyocytes (123). Melatonin supplementation also inhibited Drp1 mediated fission further reducing OS and promoting cardiomyocyte survival during hyperglycemic stress (124). Furthermore, by regulating the upstream factors of mitochondrial dynamics might be a new pathway for the treatment of DCM. Paeonol and Brain natriuretic peptide promotes Opa1-mediated mitochondrial fusion in the DCM to maintain cardiac mitochondrial function through activation of the CK2α-Stat3 pathway and PKG-STAT1 pathway, respectively (125, 126). Nicotinamide riboside promotes Mfn2-mediated mitochondrial fusion via the SIRT2-PGC1α-PPARα pathway, reduces diabetes-induced cardiomyocyte apoptosis, and prevents diabetes-induced cardiac insufficiency (127). Aldehyde dehydrogenase 2 (ALDH2) regulates mitochondrial fusion and fission through the PI3K/AKT/mTOR pathway in DCM patients and attenuates ischemia and reperfusion injury (128). Similarly, secreted frizzled related-protein 2 (SFRP2) and andrographolide have been successively reported to alleviate DCM in recent years by regulating the balance of mitochondrial dynamics (129, 130). Therefore, maintaining the myocardial mitochondrial fusion and fission balance is key to maintaining myocardial mitochondrial function, and targeting mitochondrial dynamics is a target for potentially intervening in metabolic disorder-related myocardial diseases such as diabetic cardiomyopathy and obesity cardiomyopathy.
DPN is a frequent neurological complication of diabetes, manifested by sensory, motor or autonomic disorders (131). Hyperglycemia and hyperlipidemia in diabetic patients facilitate the pathogenesis of neuropathy (132). The increase in long-chain saturated fatty acids due to dyslipidemia will induce mitochondrial depolarization and impair axonal mitochondrial transport, thereby impairing the mitochondrial dynamics of sensory neurons and ultimately ATP loss and neuronal apoptosis (132, 133). The excess glucose associated with hyperglycemia triggers nutrient overload in neurons, overloading glycolysis and the tricarboxylic acid cycle, leading to impaired OXPHOS processes, depolarization of MPP, inhibition of the rate of electron transfer and reduced ATP production (132, 134). These alterations in neuronal bioenergetics are accompanied by an increase in intracellular OS (135). To compensate for this reduction in bioenergy, neurons increase mitochondrial mass in response to OS through the division of existing mitochondria (136). However, excess glucose enhances the expression of the pro-apoptotic proteins Bim and Bax as well as Drp1, activating and localizing and thus promoting pro-apoptotic fission in mitochondria (137).
Painful diabetic neuropathy (PDN) is a clinical manifestation of DPN that includes small fiber degeneration and neuropathic pain, manifested by pathological pain and overexcitation of dorsal root ganglion (DRG) neurons (138). In a high fat diet induced PDN mouse model, mitochondria in DRG injurious neurons have a broken morphology after 2 weeks, prior to episodes of mechanically abnormal pain and small fiber degeneration (139). This indicates that overly fission and fragmented mitochondrial morphology may be the basis for PDN axon degeneration. DRG injured neurons also show elevated calcium levels, which may be the result of mitochondrial dysfunction (139). Decreased ATP levels reduce Na+-K+ ATPase function, elevate intracellular Na+, and reverse Na+-Ca2+ exchange, leading to elevated calcium levels in neuronal cell axons and mediating increased calcium-dependent mitochondrial fission (140). Persistent elevation of Ca2+ has been shown to be a critical ingredient of the signaling pathway causing axonal degeneration in the central and peripheral nervous system (141, 142). Through the specific removal of mitochondrial calcium transporters to prevent calcium from entering the mitochondria, normal mitochondrial morphology can be restored and axonal degeneration can be prevented (139). Furthermore, sensory neurons with downregulated Mfn2 expression showed delayed transport of mitochondria to distal axons and reduced frequency of mitochondrial motility (143). This transport defect interrupts the correct positioning of mitochondria, disrupts axonal and synaptic dysfunction, and may eventually lead to axonal degeneration (144, 145).
Tang Luo Ning, a traditional Chinese compound prescription, significantly reduced the expression of mitochondrial fission protein in sciatic nerve Schwann cells, significantly increased the expression levels of Mfn1/2 and Opa1, and ameliorated nerve sheath disease in DPN rats (146). Isoliquiritigenin promotes mitochondrial biogenesis by mediating SIRT1 activation and regulates mitochondrial dynamics homeostasis to attenuate damage under diabetic neuropathy (147). Currently, studies targeting mitochondrial dynamics for the treatment of DPN are in their preliminary stages, it is evident that in diabetes, abnormalities in mitochondrial dynamics can lead to mitochondrial dysfunction resulting in neuronal damage and loss, and ultimately diabetic neuropathy. Therefore, by targeting mitochondrial dynamics possibly a promising approach for disease treatment in patients suffering from diabetic neuropathy.
DR is one of the typical complications of diabetes and diabetic optic neurodegeneration is an early stage of DR pathogenesis and may be associated with the development of microvascular abnormalities (148, 149). Microvascular abnormalities within the retina include retinal vascular leakage and decellularized capillaries (149). In the pathogenesis of DR, accelerated capillary apoptosis by mitochondrial dysfunction is prior to the development of the histopathological features of DR (150, 151).
Mitochondrial disruption within diabetic retinal capillaries is partly related to the decreased expression of Opa1 induced by diabetes (152). The research has demonstrated that Opa1 expression were remarkably inhibited in the retinas of diabetic mice, where the proportion of S-Opa1 to L-Opa1 was also reduced (153). Opa1 reduction results in mitochondrial fragmentation through the development of mitochondrial swelling and local mitochondrial contraction (154). In addition, increased Bax expression and Cyt c release promote apoptosis of retinal endothelial cells (REC) and lead to increased acellular capillaries (AC) and pericyte loss (PL) which are features of DR pathogenesis (152, 153). These phenomena were more severe in diabetic mice with Opa1 gene deletion (153).
Reduced expression of Mfn2 and overexpression of Drp1 and Fis1 were also found in the retinal choroidal system of animals and humans with DR, while Mfn1 levels remained unchanged (155, 156). Decreased Mfn2 expression may be due to DNA hypermethylation of its promoter in a hyperglycemic environment, leading to reduced binding of transcription factors and consequent inhibition of gene transcription (157). Regulation of Mfn2, Drp1 and Fis1 expression may protect mitochondrial homeostasis and suppress diabetic retinopathy. For example, reducing hyperglycemia-induced aberrant overexpression of Drp1 and Fis1 by combined siRNA approaches can effectively prevent mitochondrial breakage, improve mitochondrial respiration function and inhibit the apoptosis of RECs (156, 158, 159).
In summary, the both Mfn2 and Opa1 were downregulated under high glucose conditions is coincident with the upregulation of Drp1 and Fis1. However, even if hyperglycemia is reversed to normal blood sugar levels, mitochondrial dynamics will still be impaired (160). Reduced fusion and increased fission of mitochondria eventually stimulate the discharge of Cyt c, inducing apoptosis in retinal cells like RECs and Müller cells (151). Thus, maintaining mitochondrial quality control and interfering with metabolic memory phenomena by directly regulating mitochondrial dynamics prevents further progression of DR (161). In recent years drugs that would prevent excessive mitochondrial fission have received much attention in the treatment of DR. Melatonin can guard the blood-retinal barrier by maintaining mitochondrial homeostasis. The mechanism is to upregulate the expression of genes linked to mitochondrial biogenesis (e.g., PGC-1α, NRF2 and PPAR-γ) and downregulate the expression of genes linked to mitochondrial fission (e.g., Drp1 and Fis1) (162). Drp1 can be de-SUMO-ized by SUMO-specific proteinase 3 (SENP3). Activation of de-SUMO-ized Drp1 disrupts mitochondrial dynamics, and increasing SUMO-ized Drp1 levels in retinal microvascular endothelial cells by inhibiting SENP3 expression reduces hyperglycemia-induced mitochondrial damage and apoptosis, thereby attenuating retinal permeability and increasing DR (163). Metformin prevents retinal ischaemia/reperfusion injury by increasing Mfn2 and Opa1-mediated mitochondrial fusion via AMPK (164). Current drugs and approaches that can be used to treat DR by targeting mitochondrial dynamics are shown in Table 3.
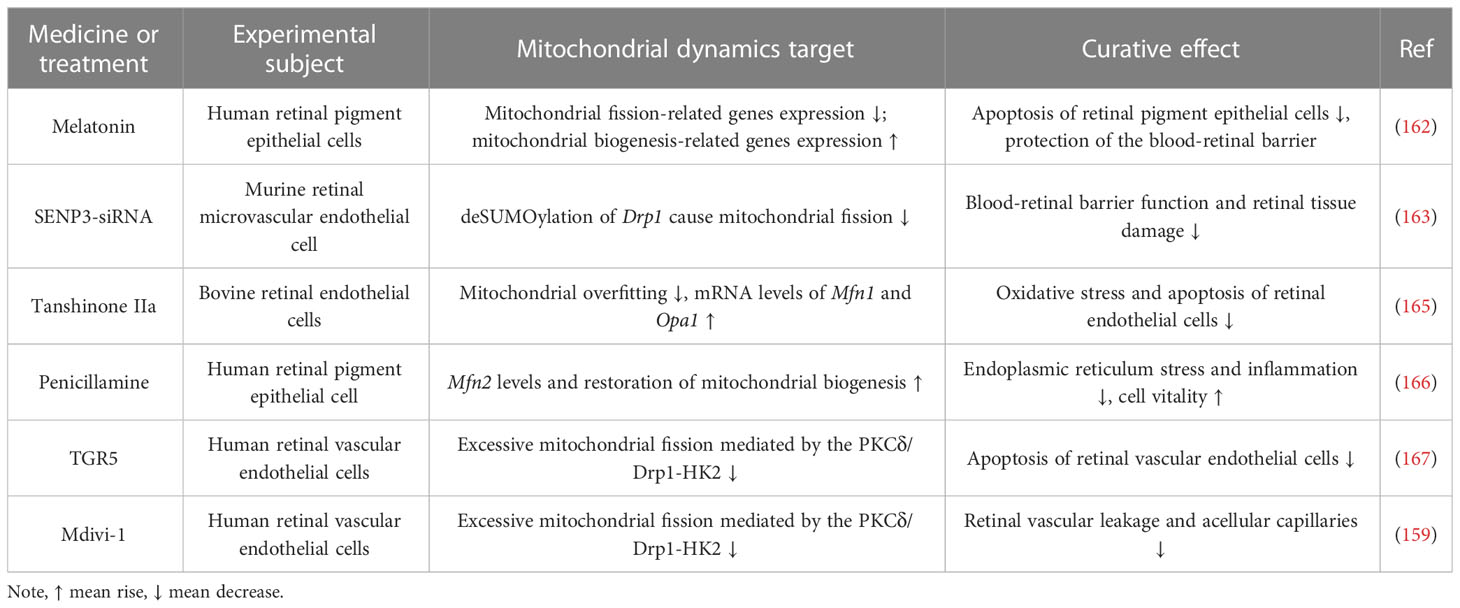
Table 3 Drugs and methods for targeting mitochondrial dynamics in the treatment of DR.
In addition, retinal cells could preserve themselves in a high glucose environment through mitophagy. Thioredoxin-interacting protein (TXNIP) is upregulated in a high glucose environment, inducing nitroso-modification of Drp1 and subsequently promoting TXNIP translocation to the mitochondria, mediating autophagy in a variety of pathways to maintain healthy mitochondria (168). The degree of hyperglycemia determines the level of mitochondrial autophagy; when the glucose concentration reaches 50 mM, mitophagy is inhibited, causing excessive mitochondrial fission and a tendency toward apoptosis (169, 170). Mitophagy has the potential to be a new therapeutic approach for DR.
During the previous years, research on the exploitation of drugs or methods for mitochondrial fusion and fission has increased each year. One of the more thoroughly investigated drugs for excessive mitochondrial fission under hyperglycemic conditions is mdivi-1. In addition to repressing Drp1 activity in cells, mdivi-1 reduces OS and inflammation and increase insulin sensitivity in diabetic mice under insulin-resistant conditions (171, 172). However, when administered for more than 24 hours mdivi-1 will reduce the number of mitochondria and induce apoptosis (173). SGLT-2 inhibitors such as Dapagliflozin and Empagliflozin can reduce glucose by lowering the glucose uptake threshold and promoting the excretion of glucose from the urine (174). Not only that, SGLT-2 inhibitors also prevent mitochondrial swelling and enhance mitochondrial restoration and regeneration, and can improve mitochondrial dysfunction by inhibiting abnormal mitochondrial fission through AMPK (15, 175, 176). In addition, the use of SGLT-2 inhibitor drugs increases the risk of diabetic ketoacidosis (177).
In addition to drugs that broadly modulate mitochondrial dynamics, drugs applied to mitochondria in specific conditions have also been reported successively. For example, HIF-1α is a hypoxia-inducible factor that is expressed primarily in renal tubular cells and predisposes renal tubules to hypoxia (171, 172). It has been suggested that proximal renal tubule cells are the initiator and key therapy target of DN. HIF-1α can improve mitochondrial dynamics and limit mitochondria-dependent apoptosis in DN renal tubular cells through the HO-1 pathway (87, 172). This change may have occurred through the HO-1/CO pathway (87). The HO-1/CO pathway has been well characterized for its antioxidant and anti-inflammatory effects in in vivo and in vitro stress models, but its potential in regulating altered mitochondrial dynamics needs further investigation (178–180). While the potential benefits of these drugs with the ability to modulate mitochondrial dynamics are well documented, the adverse effects resulting from long-term use cannot be ignored (6).
In addition, MAMs is a new target for the treatment of diabetes and is involved in various physiological processes such as mitochondrial dynamics, mitochondrial autophagy, Ca2+ signaling and lipid metabolism (181, 182). In diabetes, hyperglycemia promotes excessive formation of MAMs in the body, leading to a range of mitochondrial dysfunctions (183). Fundc1 is a key molecule involved in MAMs formation, and by inhibiting its expression it can reduce the excessive formation of MAMs and effectively ameliorate diabetes and its complications, such as TRPV1 and SIRT3 (183–185). However, several other studies have contradicted this, indicating that mitochondrial dysfunction, apoptosis, and tissue damage in the diabetic setting are associated with disruption of MAMs integrity and a reduction in its formation (95, 186, 187). Further in-depth studies are still needed for the use of MAMs in diabetes and its complications.
Mitochondria are organelles with high dynamic changes in eukaryotic cells, and are one of the most important organelles in maintaining homeostasis in the body. It adapts to changes in the external environment, maintaining the function of tissues and organs through mitochondrial dynamics, and participating in various physiological procedures such as intracellular energy metabolism, autophagy and apoptosis, during which mis-regulation can lead to disease states, making their function vital to life. In the diabetic state, mitochondrial morphology is fragmented, and imbalance of mitochondrial dynamics causes disturbance in cellular energy metabolism and damage to pancreatic β-cells and peripheral tissues and organs, thus promoting the progression of diabetic complications. Targeting mitochondrial dynamic balance can effectively improve the progression of diabetes and its complications (Figure 5). The contribution of mitochondrial dynamics in chronic complications such as DN and DCM is gradually being explored, but a holistic picture is lacking and the specific molecular mechanisms and pathways of action are yet to be elucidated. There have been significant breakthroughs in recent years in the therapy of type II diabetes and its complications through targeted modulation of mitochondrial dynamics. The development of new therapeutic drugs based on the idea of adjusting the homeostasis of mitochondrial dynamics is expected to bring new light to the treatment of T2D and its comorbidities.
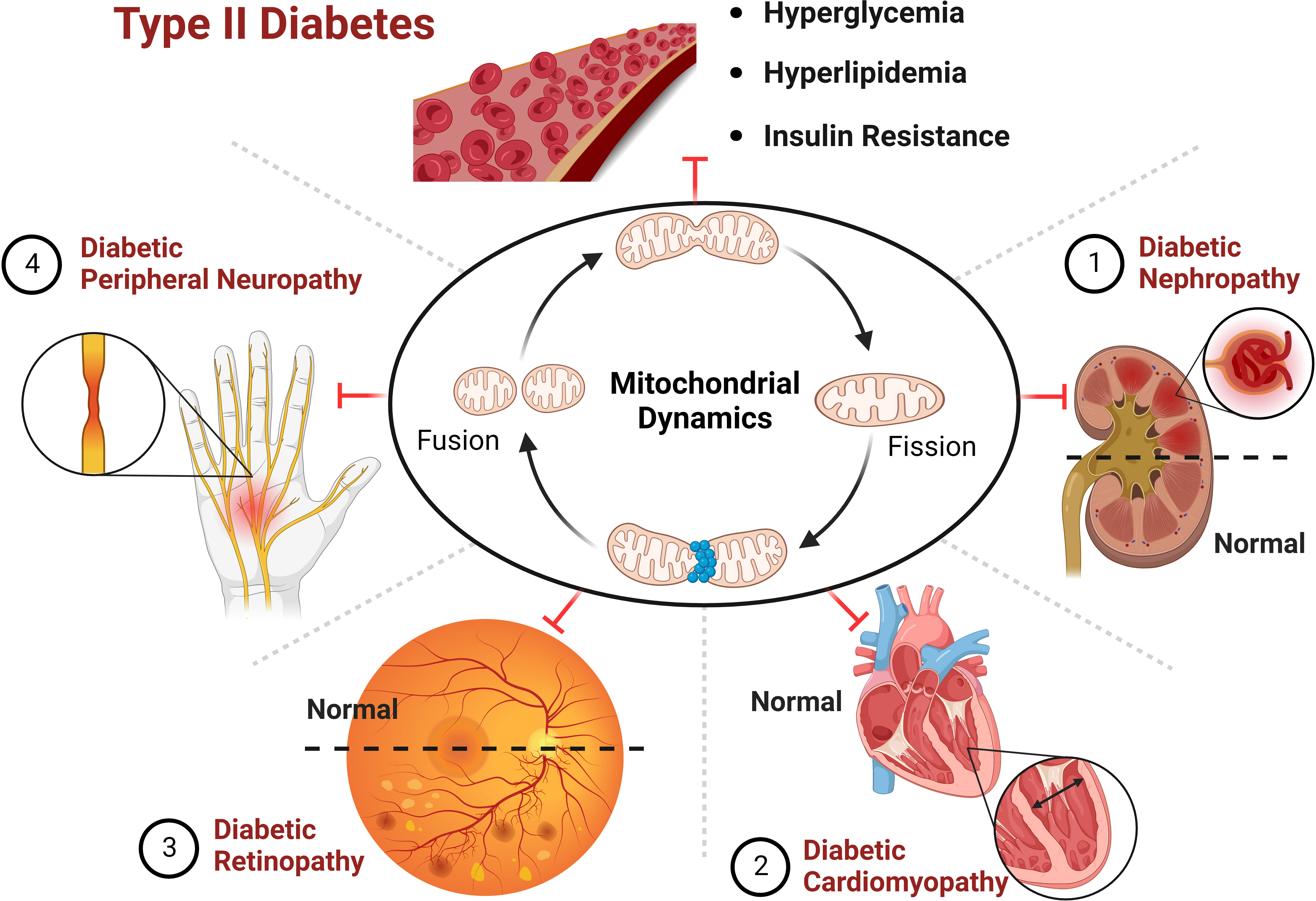
Figure 5 Improving the progression of diabetes and its complications by targeting mitochondrial dynamics.
SW and HZ contributed equally. SW and HZ, Investigation, data curation, writing - original draft, writing - review & editing. SL, YaL and YuL, Software, Supervision, Validation. YiL, RP and HJ, Conceptualization, Writing- Reviewing and Editing. All authors contributed to the article and approved the submitted version.
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1230168/full#supplementary-material
ROS, reactive oxygen species; OS, oxidative stress; Mfn1, mitofusin-1; Mfn2, mitofusin-2; Opa1, optic atrophy 1; Drp1, dynamin related protein 1; Fis1, mitochondrial fission 1; MFF, mitochondrial fission factor; T2D, type II diabetes; OMM, outer mitochondrial membrane; HR1, helix regions 1; HR2, helix regions 2; TM, transmembrane domains; IMM, inner mitochondrial membrane; MTS, mitochondrial targeting sequence; CC, coiled coil domain; MD, middle domain; GED, GTPase effector domain; ER, endoplasmic reticulum; INF2, inverted formin 2; MCU, mitochondrial calcium unidirectional; OXPHOS, oxidative phosphorylation; PINK1, PTEN-induced putative kinase 1; Cyt c, cytochrome c; DN, diabetic nephropathy; ROCK1, rho-associated kinase 1; DCM, diabetic cardiomyopathy; PPARα, peroxisome proliferator-activated receptor α; CaN, calcineurin; CaM, calmodulin; CnA, catalyzes subunit A; p-ERK1/2, phosphorylates- extracellular signal-regulated kinase; DPN, diabetic peripheral neuropathy; PDN, painful diabetic neuropathy; DRG, dorsal root ganglion; DR, diabetic retinopathy; REC, retinal endothelial cell; AC, acellular capillaries; PL, pericyte loss; TXNIP, thioredoxin-interacting protein.
1. Burke PJ. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer (2017) 3:857–70. doi: 10.1016/j.trecan.2017.10.006
2. Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu S-S. Calcium, ATP, and ROS, a mitochondrial love-hate triangle. Am J Physiol Cell Physiol (2004) 287:C817–833. doi: 10.1152/ajpcell.00139.2004
3. Singh A, Kukreti R, Saso L, Kukreti S. Mechanistic insight into oxidative stress-triggered signaling pathways and type 2 diabetes. Molecules (2022) 27:950. doi: 10.3390/molecules27030950
4. Rehman K, Akash MSH. Mechanism of generation of oxidative stress and pathophysiology of type 2 diabetes mellitus, how are they interlinked? J Cell Biochem (2017) 118:3577–85. doi: 10.1002/jcb.26097
5. Willems PHGM, Rossignol R, Dieteren CEJ, Murphy MP, Koopman WJH. Redox homeostasis and mitochondrial dynamics. Cell Metab (2015) 22:207–18. doi: 10.1016/j.cmet.2015.06.006
6. Rovira-Llopis S, Bañuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM. Mitochondrial dynamics in type 2 diabetes, Pathophysiological implications. Redox Biol (2017) 11:637–45. doi: 10.1016/j.redox.2017.01.013
7. Wu M-Y, Yiang G-T, Lai T-T, Li C-J. The oxidative stress and mitochondrial dysfunction during the pathogenesis of diabetic retinopathy. Oxid Med Cell Longev (2018) 2018:3420187. doi: 10.1155/2018/3420187
8. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
9. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab (2016) 27:105–17. doi: 10.1016/j.tem.2015.12.001
10. Gao S, Hu J. Mitochondrial fusion, the machineries in and out. Trends Cell Biol (2021) 31:62–74. doi: 10.1016/j.tcb.2020.09.008
11. van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol (2013) 5:a011072. doi: 10.1101/cshperspect.a011072
12. Adebayo M, Singh S, Singh AP, Dasgupta S. Mitochondrial fusion and fission, The fine-tune balance for cellular homeostasis. FASEB J (2021) 35:e21620. doi: 10.1096/fj.202100067R
13. Giacomello M, Pyakurel A, Glytsou C, Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol (2020) 21:204–24. doi: 10.1038/s41580-020-0210-7
14. Pernas L, Scorrano L. Mito-morphosis, mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol (2016) 78:505–31. doi: 10.1146/annurev-physiol-021115-105011
15. Liu X, Xu C, Xu L, Li X, Sun H, Xue M, et al. Empagliflozin improves diabetic renal tubular injury by alleviating mitochondrial fission via AMPK/SP1/PGAM5 pathway. Metabolism (2020) 111:154334. doi: 10.1016/j.metabol.2020.154334
16. Archer SL. Mitochondrial dynamics–mitochondrial fission and fusion in human diseases. N Engl J Med (2013) 369:2236–51. doi: 10.1056/NEJMra1215233
17. Lin H-Y, Weng S-W, Chang Y-H, Su Y-J, Chang C-M, Tsai C-J, et al. The causal role of mitochondrial dynamics in regulating insulin resistance in diabetes, link through mitochondrial reactive oxygen species. Oxid Med Cell Longev (2018) 2018:7514383. doi: 10.1155/2018/7514383
18. Dubé JJ, Collyer ML, Trant S, Toledo FGS, Goodpaster BH, Kershaw EE, et al. Decreased mitochondrial dynamics is associated with insulin resistance, metabolic rate, and fitness in African Americans. J Clin Endocrinol Metab (2020) 105:1210–20. doi: 10.1210/clinem/dgz272
19. Houzelle A, Jörgensen JA, Schaart G, Daemen S, van Polanen N, Fealy CE, et al. Human skeletal muscle mitochondrial dynamics in relation to oxidative capacity and insulin sensitivity. Diabetologia (2021) 64:424–36. doi: 10.1007/s00125-020-05335-w
20. Yapa NMB, Lisnyak V, Reljic B, Ryan MT. Mitochondrial dynamics in health and disease. FEBS Lett (2021) 595:1184–204. doi: 10.1002/1873-3468.14077
21. Silva Ramos E, Motori E, Brüser C, Kühl I, Yeroslaviz A, Ruzzenente B, et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PloS Genet (2019) 15:e1008085. doi: 10.1371/journal.pgen.1008085
22. Narendra DP, Youle RJ. Targeting mitochondrial dysfunction, role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal (2011) 14:1929–38. doi: 10.1089/ars.2010.3799
23. Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol (2011) 12:9–14. doi: 10.1038/nrm3028
24. Rojo M, Legros F, Chateau D, Lombès A. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mamMalian homologs of the transmembrane GTPase Fzo. J Cell Sci (2002) 115:1663–74. doi: 10.1242/jcs.115.8.1663
25. Chandhok G, Lazarou M, Neumann B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev Camb Philos Soc (2018) 93:933–49. doi: 10.1111/brv.12378
26. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science (2004) 305:858–62. doi: 10.1126/science.1099793
27. Meeusen S, McCaffery JM, Nunnari J. Mitochondrial fusion intermediates revealed. vitro Sci (2004) 305:1747–52. doi: 10.1126/science.1100612
28. Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell (2009) 20:3525–32. doi: 10.1091/mbc.e09-03-0252
29. Ishihara N, Eura Y, Mihara K. Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci (2004) 117:6535–46. doi: 10.1242/jcs.01565
30. Eura Y, Ishihara N, Yokota S, Mihara K. Two mitofusin proteins, mamMalian homologues of FZO, with distinct functions are both required for mitochondrial fusion. J Biochem (2003) 134:333–44. doi: 10.1093/jb/mvg150
31. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol (2003) 160:189–200. doi: 10.1083/jcb.200211046
32. Zhang D, Zhang Y, Ma J, Zhu C, Niu T, Chen W, et al. Cryo-EM structures of S-OPA1 reveal its interactions with membrane and changes upon nucleotide binding. Elife (2020) 9:e50294. doi: 10.7554/eLife.50294
33. Olichon A, Elachouri G, Baricault L, Delettre C, Belenguer P, Lenaers G. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ (2007) 14:682–92. doi: 10.1038/sj.cdd.4402048
34. Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, et al. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet (2001) 109:584–91. doi: 10.1007/s00439-001-0633-y
35. Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J (2006) 25:2966–77. doi: 10.1038/sj.emboj.7601184
36. Ge Y, Shi X, Boopathy S, McDonald J, Smith AW, Chao LH. Two forms of Opa1 cooperate to complete fusion of the mitochondrial inner-membrane. Elife (2020) 9:e50973. doi: 10.7554/eLife.50973
37. Song Z, Chen H, Fiket M, Alexander C, Chan DC. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J Cell Biol (2007) 178:749–55. doi: 10.1083/jcb.200704110
38. Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U.S.A. (2004) 101:15927–32. doi: 10.1073/pnas.0407043101
39. Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell (2006) 126:177–89. doi: 10.1016/j.cell.2006.06.025
40. Smirnova E, Griparic L, Shurland D-L, van der Bliek AM. Dynamin-related protein drp1 is required for mitochondrial division in mamMalian cells. MBoC (2001) 12:2245–56. doi: 10.1091/mbc.12.8.2245
41. Korobova F, Ramabhadran V, Higgs HN. An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science (2013) 339:464–7. doi: 10.1126/science.1228360
42. Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK. ER tubules mark sites of mitochondrial division. Science (2011) 334:358–62. doi: 10.1126/science.1207385
43. Manor U, Bartholomew S, Golani G, Christenson E, Kozlov M, Higgs H, et al. Lippincott-Schwartz J. A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife (2015) 4:e08828. doi: 10.7554/eLife.08828
44. Korobova F, Gauvin TJ, Higgs HN. A role for myosin II in mamMalian mitochondrial fission. Curr Biol (2014) 24:409–14. doi: 10.1016/j.cub.2013.12.032
45. Losón OC, Song Z, Chen H, Chan DC. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol Biol Cell (2013) 24:659–67. doi: 10.1091/mbc.E12-10-0721
46. Ji W, Hatch AL, Merrill RA, Strack S, Higgs HN. Actin filaments target the oligomeric maturation of the dynamin GTPase Drp1 to mitochondrial fission sites. eLife (2015) 4:e11553. doi: 10.7554/eLife.11553
47. Pagliuso A, Cossart P, Stavru F. The ever-growing complexity of the mitochondrial fission machinery. Cell Mol Life Sci (2018) 75:355–74. doi: 10.1007/s00018-017-2603-0
48. Chakrabarti R, Ji W-K, Stan RV, de Juan Sanz J, Ryan TA, Higgs HN. INF2-mediated actin polymerization at the ER stimulates mitochondrial calcium uptake, inner membrane constriction, and division. J Cell Biol (2018) 217:251–68. doi: 10.1083/jcb.201709111
49. Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, et al. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U.S.A. (2008) 105:15803–8. doi: 10.1073/pnas.0808249105
50. Cribbs JT, Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep (2007) 8:939–44. doi: 10.1038/sj.embor.7401062
51. Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E, et al. The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission. J Cell Biol (2014) 204:919–29. doi: 10.1083/jcb.201308006
52. Smeitink JA, Zeviani M, Turnbull DM, Jacobs HT. Mitochondrial medicine, a metabolic perspective on the pathology of oxidative phosphorylation disorders. Cell Metab (2006) 3:9–13. doi: 10.1016/j.cmet.2005.12.001
53. Rossmann MP, Dubois SM, Agarwal S, Zon LI. Mitochondrial function in development and disease. Dis Model Mech (2021) 14:dmm048912. doi: 10.1242/dmm.048912
54. Apel K, Hirt H. Reactive oxygen species, metabolism, oxidative stress, and signal transduction. Annu Rev Plant Biol (2004) 55:373–99. doi: 10.1146/annurev.arplant.55.031903.141701
55. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab (2013) 17:491–506. doi: 10.1016/j.cmet.2013.03.002
56. Yao C-H, Wang R, Wang Y, Kung C-P, Weber JD, Patti GJ. Mitochondrial fusion supports increased oxidative phosphorylation during cell proliferation. Elife (2019) 8:e41351. doi: 10.7554/eLife.41351
57. Punter KB, Chu C, Chan EYW. Mitochondrial dynamics and oxidative phosphorylation as critical targets in cancer. Endocr Relat Cancer (2023) 30:e220229. doi: 10.1530/ERC-22-0229
58. Li T, Han J, Jia L, Hu X, Chen L, Wang Y. PKM2 coordinates glycolysis with mitochondrial fusion and oxidative phosphorylation. Protein Cell (2019) 10:583–94. doi: 10.1007/s13238-019-0618-z
59. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ (2013) 20:31–42. doi: 10.1038/cdd.2012.81
60. Dorn GW, Kitsis RN. The mitochondrial dynamism-mitophagy-cell death interactome, multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res (2015) 116:167–82. doi: 10.1161/CIRCRESAHA.116.303554
61. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature (2015) 524:309–14. doi: 10.1038/nature14893
62. Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol (2016) 26:733–44. doi: 10.1016/j.tcb.2016.05.008
63. Ni H-M, Williams JA, Ding W-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol (2015) 4:6–13. doi: 10.1016/j.redox.2014.11.006
64. Yoo S-M, Jung Y-K. A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells (2018) 41:18–26. doi: 10.14348/molcells.2018.2277
65. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ (2009) 16:939–46. doi: 10.1038/cdd.2009.16
66. Chen M, Chen Z, Wang Y, Tan Z, Zhu C, Li Y, et al. Mitophagy receptor FUNDC1 regulates mitochondrial dynamics and mitophagy. Autophagy (2016) 12:689–702. doi: 10.1080/15548627.2016.1151580
67. Wu W, Li W, Chen H, Jiang L, Zhu R, Feng D. FUNDC1 is a novel mitochondrial-associated-membrane (MAM) protein required for hypoxia-induced mitochondrial fission and mitophagy. Autophagy (2016) 12:1675–6. doi: 10.1080/15548627.2016.1193656
68. Wei Y, Chiang W-C, Sumpter R, Mishra P, Levine B. Prohibitin 2 is an inner mitochondrial membrane mitophagy receptor. Cell (2017) 168:224–238.e10. doi: 10.1016/j.cell.2016.11.042
69. Merkwirth C, Dargazanli S, Tatsuta T, Geimer S, Löwer B, Wunderlich FT, et al. Prohibitins control cell proliferation and apoptosis by regulating OPA1-dependent cristae morphogenesis in mitochondria. Genes Dev (2008) 22:476–88. doi: 10.1101/gad.460708
70. Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, et al. BNIP3L/NIX and FUNDC1-mediated mitophagy is required for mitochondrial network remodeling during cardiac progenitor cell differentiation. Autophagy (2019) 15:1182–98. doi: 10.1080/15548627.2019.1580095
71. Estaquier J, Vallette F, Vayssiere J-L, Mignotte B. The mitochondrial pathways of apoptosis. Adv Exp Med Biol (2012) 942:157–83. doi: 10.1007/978-94-007-2869-1_7
72. Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol (2020) 21:85–100. doi: 10.1038/s41580-019-0173-8
73. Ugarte-Uribe B, García-Sáez AJ. Apoptotic foci at mitochondria, in and around Bax pores. Philos Trans R Soc Lond B Biol Sci (2017) 372:20160217. doi: 10.1098/rstb.2016.0217
74. Jenner A, Peña-Blanco A, Salvador-Gallego R, Ugarte-Uribe B, Zollo C, Ganief T, et al. DRP1 interacts directly with BAX to induce its activation and apoptosis. EMBO J (2022) 41:e108587. doi: 10.15252/embj.2021108587
75. Peña-Blanco A, García-Sáez AJ. Bax, Bak and beyond - mitochondrial performance in apoptosis. FEBS J (2018) 285:416–31. doi: 10.1111/febs.14186
76. Wu H, Li G, Chen W, Luo W, Yang Z, You Z, et al. Drp1 knockdown represses apoptosis of rat retinal endothelial cells by inhibiting mitophagy. Acta Histochem (2022) 124:151837. doi: 10.1016/j.acthis.2021.151837
77. Lee Y, Jeong S-Y, Karbowski M, Smith CL, Youle RJ. Roles of the mamMalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell (2004) 15:5001–11. doi: 10.1091/mbc.e04-04-0294
78. Pernaute B, Pérez-Montero S, Sánchez Nieto JM, Di Gregorio A, Lima A, Lawlor K, et al. DRP1 levels determine the apoptotic threshold during embryonic differentiation through a mitophagy-dependent mechanism. Dev Cell (2022) 57:1316–1330.e7. doi: 10.1016/j.devcel.2022.04.020
79. Suen D-F, Norris KL, Youle RJ. Mitochondrial dynamics and apoptosis. Genes Dev (2008) 22:1577–90. doi: 10.1101/gad.1658508
80. Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U.S.A. (2006) 103:2653–8. doi: 10.1073/pnas.0511154103
81. Shan Z, Fa WH, Tian CR, Yuan CS, Jie N. Mitophagy and mitochondrial dynamics in type 2 diabetes mellitus treatment. Aging (Albany NY) (2022) 14:2902–19. doi: 10.18632/aging.203969
82. Scheele C, Nielsen AR, Walden TB, Sewell DA, Fischer CP, Brogan RJ, et al. Altered regulation of the PINK1 locus, a link between type 2 diabetes and neurodegeneration? FASEB J (2007) 21:3653–65. doi: 10.1096/fj.07-8520com
83. Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet (2003) 34:267–73. doi: 10.1038/ng1180
84. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414:813–20. doi: 10.1038/414813a
85. Xiao L, Xu X, Zhang F, Wang M, Xu Y, Tang D, et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol (2017) 11:297–311. doi: 10.1016/j.redox.2016.12.022
86. Yao L, Liang X, Qiao Y, Chen B, Wang P, Liu Z. Mitochondrial dysfunction in diabetic tubulopathy. Metabolism (2022) 131:155195. doi: 10.1016/j.metabol.2022.155195
87. Jiang N, Zhao H, Han Y, Li L, Xiong S, Zeng L, et al. HIF-1α ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif (2020) 53:e12909. doi: 10.1111/cpr.12909
88. Narongkiatikhun P, Chattipakorn SC, Chattipakorn N. Mitochondrial dynamics and diabetic kidney disease, Missing pieces for the puzzle of therapeutic approaches. J Cell Mol Med (2022) 26:249–73. doi: 10.1111/jcmm.17116
89. Qin X, Zhao Y, Gong J, Huang W, Su H, Yuan F, et al. Berberine protects glomerular podocytes via inhibiting drp1-mediated mitochondrial fission and dysfunction. Theranostics (2019) 9:1698–713. doi: 10.7150/thno.30640
90. Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics, regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int (2013) 83:568–81. doi: 10.1038/ki.2012.441
91. Ayanga BA, Badal SS, Wang Y, Galvan DL, Chang BH, Schumacker PT, et al. Dynamin-related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy. J Am Soc Nephrol (2016) 27:2733–47. doi: 10.1681/ASN.2015101096
92. Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, et al. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab (2012) 15:186–200. doi: 10.1016/j.cmet.2012.01.009
93. Yang S-K, Li A-M, Han Y-C, Peng C-H, Song N, Yang M, et al. Mitochondria-targeted peptide SS31 attenuates renal tubulointerstitial injury via inhibiting mitochondrial fission in diabetic mice. Oxid Med Cell Longev (2019) 2019:2346580. doi: 10.1155/2019/2346580
94. Ji Y, Zhang X, Chen J, Song S, Fang S, Wang Z, et al. Asiatic acid attenuates tubular injury in diabetic kidney disease by regulating mitochondrial dynamics via the Nrf-2 pathway. Phytomedicine (2023) 109:154552. doi: 10.1016/j.phymed.2022.154552
95. Li C, Li L, Yang M, Yang J, Zhao C, Han Y, et al. PACS-2 ameliorates tubular injury by facilitating endoplasmic reticulum-mitochondria contact and mitophagy in diabetic nephropathy. Diabetes (2022) 71:1034–50. doi: 10.2337/db21-0983
96. Huang Q, Chen H, Yin K, Shen Y, Lin K, Guo X, et al. Formononetin attenuates renal tubular injury and mitochondrial damage in diabetic nephropathy partly via regulating sirt1/PGC-1α Pathway. Front Pharmacol (2022) 13:901234. doi: 10.3389/fphar.2022.901234
97. Sheng J, Li H, Dai Q, Lu C, Xu M, Zhang J, et al. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J Cell Physiol (2019) 234:3043–57. doi: 10.1002/jcp.27124
98. Agil A, Chayah M, Visiedo L, Navarro-Alarcon M, Rodríguez Ferrer JM, Tassi M, et al. Melatonin improves mitochondrial dynamics and function in the kidney of Zücker diabetic fatty rats. J Clin Med (2020) 9:2916. doi: 10.3390/jcm9092916
99. Zhang Q, He L, Dong Y, Fei Y, Wen J, Li X, et al. Sitagliptin ameliorates renal tubular injury in diabetic kidney disease via STAT3-dependent mitochondrial homeostasis through SDF-1α/CXCR4 pathway. FASEB J (2020) 34:7500–19. doi: 10.1096/fj.201903038R
100. Ni Z, Tao L, Xiaohui X, Zelin Z, Jiangang L, Zhao S, et al. Polydatin impairs mitochondria fitness and ameliorates podocyte injury by suppressing Drp1 expression. J Cell Physiol (2017) 232:2776–87. doi: 10.1002/jcp.25943
101. Wai Linn T, Kobroob A, Ngernjan M, Amornlerdpison D, Lailerd N, Wongmekiat O. Crocodile oil disrupts mitochondrial homeostasis and exacerbates diabetic kidney injury in spontaneously diabetic torii rats. Biomolecules (2022) 12:1068. doi: 10.3390/biom12081068
102. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy, an update of mechanisms contributing to this clinical entity. Circ Res (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
103. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia (2014) 57:660–71. doi: 10.1007/s00125-014-3171-6
104. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy, a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia (2018) 61:21–8. doi: 10.1007/s00125-017-4390-4
105. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
106. Tsushima K, Bugger H, Wende AR, Soto J, Jenson GA, Tor AR, et al. Mitochondrial reactive oxygen species in lipotoxic hearts induce post-translational modifications of AKAP121, DRP1, and OPA1 that promote mitochondrial fission. Circ Res (2018) 122:58–73. doi: 10.1161/CIRCRESAHA.117.311307
107. Mallat Z, Philip I, Lebret M, Chatel D, Maclouf J, Tedgui A. Elevated levels of 8-iso-prostaglandin F2alpha in pericardial fluid of patients with heart failure, a potential role for in vivo oxidant stress in ventricular dilatation and progression to heart failure. Circulation (1998) 97:1536–9. doi: 10.1161/01.cir.97.16.1536
108. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res (2021) 128:1487–513. doi: 10.1161/CIRCRESAHA.121.318241
109. Sugamura K, Keaney JF. Reactive oxygen species in cardiovascular disease. Free Radic Biol Med (2011) 51:978–92. doi: 10.1016/j.freeradbiomed.2011.05.004
110. Lozhkin A, Vendrov AE, Ramos-Mondragón R, Canugovi C, Stevenson MD, Herron TJ, et al. Mitochondrial oxidative stress contributes to diastolic dysfunction through impaired mitochondrial dynamics. Redox Biol (2022) 57:102474. doi: 10.1016/j.redox.2022.102474
111. Wai T, García-Prieto J, Baker MJ, Merkwirth C, Benit P, Rustin P, et al. Imbalanced OPA1 processing and mitochondrial fragmentation cause heart failure in mice. Science (2015) 350:aad0116. doi: 10.1126/science.aad0116
112. Hu L, Ding M, Tang D, Gao E, Li C, Wang K, et al. Targeting mitochondrial dynamics by regulating Mfn2 for therapeutic intervention in diabetic cardiomyopathy. Theranostics (2019) 9:3687–706. doi: 10.7150/thno.33684
113. Wu Q-R, Zheng D-L, Liu P-M, Yang H, Li L-A, Kuang S-J, et al. High glucose induces Drp1-mediated mitochondrial fission via the Orai1 calcium channel to participate in diabetic cardiomyocyte hypertrophy. Cell Death Dis (2021) 12:216. doi: 10.1038/s41419-021-03502-4
114. Liu M, Ai J, Shuai Z, Tang K, Li Z, Huang Y. Adropin alleviates myocardial fibrosis in diabetic cardiomyopathy rats, A preliminary study. Front Cardiovasc Med (2021) 8:688586. doi: 10.3389/fcvm.2021.688586
115. Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, et al. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation (2014) 130:554–64. doi: 10.1161/CIRCULATIONAHA.113.008476
116. Chen L, Gong Q, Stice JP, Knowlton AA. Mitochondrial OPA1, apoptosis, and heart failure. Cardiovasc Res (2009) 84:91–9. doi: 10.1093/cvr/cvp181
117. Kasahara A, Cipolat S, Chen Y, Dorn GW, Scorrano L. Mitochondrial fusion directs cardiomyocyte differentiation via calcineurin and Notch signaling. Science (2013) 342:734–7. doi: 10.1126/science.1241359
118. Tong M, Saito T, Zhai P, Oka S-I, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet-induced diabetic cardiomyopathy. Circ Res (2019) 124:1360–71. doi: 10.1161/CIRCRESAHA.118.314607
119. Zhao T, Huang X, Han L, Wang X, Cheng H, Zhao Y, et al. Central role of mitofusin 2 in autophagosome-lysosome fusion in cardiomyocytes. J Biol Chem (2012) 287:23615–25. doi: 10.1074/jbc.M112.379164
120. Vásquez-Trincado C, García-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol (2016) 594:509–25. doi: 10.1113/JP271301
122. Rusnak F, Mertz P. Calcineurin, form and function. Physiol Rev (2000) 80:1483–521. doi: 10.1152/physrev.2000.80.4.1483
123. Ding M, Liu C, Shi R, Yu M, Zeng K, Kang J, et al. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol (Oxf) (2020) 229:e13428. doi: 10.1111/apha.13428
124. Ding M, Feng N, Tang D, Feng J, Li Z, Jia M, et al. Melatonin prevents Drp1-mediated mitochondrial fission in diabetic hearts through SIRT1-PGC1α pathway. J Pineal Res (2018) 65:e12491. doi: 10.1111/jpi.12491
125. Liu C, Han Y, Gu X, Li M, Du Y, Feng N, et al. Paeonol promotes Opa1-mediated mitochondrial fusion via activating the CK2α-Stat3 pathway in diabetic cardiomyopathy. Redox Biol (2021) 46:102098. doi: 10.1016/j.redox.2021.102098
126. Chang P, Zhang X, Zhang J, Wang J, Wang X, Li M, et al. BNP protects against diabetic cardiomyopathy by promoting Opa1-mediated mitochondrial fusion via activating the PKG-STAT3 pathway. Redox Biol (2023) 62:102702. doi: 10.1016/j.redox.2023.102702
127. Hu L, Guo Y, Song L, Wen H, Sun N, Wang Y, et al. Nicotinamide riboside promotes Mfn2-mediated mitochondrial fusion in diabetic hearts through the SIRT1-PGC1α-PPARα pathway. Free Radic Biol Med (2022) 183:75–88. doi: 10.1016/j.freeradbiomed.2022.03.012
128. Tan X, Chen Y-F, Zou S-Y, Wang W-J, Zhang N-N, Sun Z-Y, et al. ALDH2 attenuates ischemia and reperfusion injury through regulation of mitochondrial fusion and fission by PI3K/AKT/mTOR pathway in diabetic cardiomyopathy. Free Radic Biol Med (2023) 195:219–30. doi: 10.1016/j.freeradbiomed.2022.12.097
129. Ma T, Huang X, Zheng H, Huang G, Li W, Liu X, et al. SFRP2 improves mitochondrial dynamics and mitochondrial biogenesis, oxidative stress, and apoptosis in diabetic cardiomyopathy. Oxid Med Cell Longevity (2021) 2021:9265016. doi: 10.1155/2021/9265016
130. Fu F, Liu C, Shi R, Li M, Zhang M, Du Y, et al. Punicalagin protects against diabetic cardiomyopathy by promoting opa1-mediated mitochondrial fusion via regulating PTP1B-stat3 pathway. Antioxid Redox Signal (2021) 35:618–41. doi: 10.1089/ars.2020.8248
131. Selvarajah D, Kar D, Khunti K, Davies MJ, Scott AR, Walker J, et al. Diabetic peripheral neuropathy, advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol (2019) 7:938–48. doi: 10.1016/S2213-8587(19)30081-6
132. Sajic M, Rumora AE, Kanhai AA, Dentoni G, Varatharajah S, Casey C, et al. High dietary fat consumption impairs axonal mitochondrial function. In Vivo J Neurosci (2021) 41:4321–34. doi: 10.1523/JNEUROSCI.1852-20.2021
133. Hinder LM, Figueroa-Romero C, Pacut C, Hong Y, Vivekanandan-Giri A, Pennathur S, et al. Long-chain acyl coenzyme A synthetase 1 overexpression in primary cultured Schwann cells prevents long chain fatty acid-induced oxidative stress and mitochondrial dysfunction. Antioxid Redox Signal (2014) 21:588–600. doi: 10.1089/ars.2013.5248
134. Liu Y-P, Shao S-J, Guo H-D. Schwann cells apoptosis is induced by high glucose in diabetic peripheral neuropathy. Life Sci (2020) 248:117459. doi: 10.1016/j.lfs.2020.117459
135. Feldman EL, Nave K-A, Jensen TS, Bennett DLH. New horizons in diabetic neuropathy, mechanisms, bioenergetics, and pain. Neuron (2017) 93:1296–313. doi: 10.1016/j.neuron.2017.02.005
136. Van Laar VS, Berman SB. The interplay of neuronal mitochondrial dynamics and bioenergetics, implications for Parkinson’s disease. Neurobiol Dis (2013) 51:43–55. doi: 10.1016/j.nbd.2012.05.015
137. Leinninger GM, Backus C, Sastry AM, Yi Y-B, Wang C-W, Feldman EL. Mitochondria in DRG neurons undergo hyperglycemic mediated injury through Bim, Bax and the fission protein Drp1. Neurobiol Dis (2006) 23:11–22. doi: 10.1016/j.nbd.2006.01.017
138. Feldman EL, Callaghan BC, Pop-Busui R, Zochodne DW, Wright DE, Bennett DL, et al. Diabetic neuropathy. Nat Rev Dis Primers (2019) 5:42. doi: 10.1038/s41572-019-0097-9
139. Ds G SH, Nd J DR, Sl E, Ca R, Re M, Am M, et al. Mitochondrial calcium uniporter deletion prevents painful diabetic neuropathy by restoring mitochondrial morphology and dynamics. Pain (2022) 163:560–78. doi: 10.1097/j.pain.0000000000002391
140. Waxman SG. Ions, energy and axonal injury, towards a molecular neurology of multiple sclerosis. Trends Mol Med (2006) 12:192–5. doi: 10.1016/j.molmed.2006.03.001
141. Ak P, Jgj H ME, Ja B, Sg W. Sodium channels, mitochondria, and axonal degeneration in peripheral neuropathy. Trends Mol Med (2016) 22:377–390. doi: 10.1016/j.molmed.2016.03.008
142. Jt W, Za M, Ba B. Axon degeneration, molecular mechanisms of a self-destruction pathway. J Cell Biol (2012) 196:7–18. doi: 10.1083/jcb.201108111
143. Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci (2007) 27:422–30. doi: 10.1523/JNEUROSCI.4798-06.2007
144. Baloh RH. Mitochondrial dynamics and peripheral neuropathy. Neuroscientist (2008) 14:12–8. doi: 10.1177/1073858407307354
145. Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci (2010) 30:4232–40. doi: 10.1523/JNEUROSCI.6248-09.2010
146. Zhu J, Yang X, Li X, Han S, Zhu Y, Xu L. Tang Luo Ning, a traditional Chinese compound prescription, ameliorates schwannopathy of diabetic peripheral neuropathy rats by regulating mitochondrial dynamics in vivo and in vitro. Front Pharmacol (2021) 12:650448. doi: 10.3389/fphar.2021.650448
147. Yerra VG, Kalvala AK, Kumar A. Isoliquiritigenin reduces oxidative damage and alleviates mitochondrial impairment by SIRT1 activation in experimental diabetic neuropathy. J Nutr Biochem (2017) 47:41–52. doi: 10.1016/j.jnutbio.2017.05.001
148. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet (2010) 376:124–36. doi: 10.1016/S0140-6736(09)62124-3
149. Kollias AN, Ulbig MW. Diabetic retinopathy, Early diagnosis and effective treatment. Dtsch Arztebl Int (2010) 107:75–83; quiz 84. doi: 10.3238/arztebl.2010.0075
150. Kang Q, Yang C. Oxidative stress and diabetic retinopathy, Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
151. Wu Y, Zou H. Research progress on mitochondrial dysfunction in diabetic retinopathy. Antioxidants (Basel) (2022) 11:2250. doi: 10.3390/antiox11112250
152. Kim D, Roy S. Effects of diabetes on mitochondrial morphology and its implications in diabetic retinopathy. Invest Ophthalmol Visual Sci (2020) 61:10. doi: 10.1167/iovs.61.10.10
153. Kim D, Votruba M, Roy S. Opa1 deficiency promotes development of retinal vascular lesions in diabetic retinopathy. Int J Mol Sci (2021) 22:5928. doi: 10.3390/ijms22115928
154. Griparic L, van der Wel NN, Orozco IJ, Peters PJ, van der Bliek AM. Loss of the intermembrane space protein Mgm1/OPA1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem (2004) 279:18792–8. doi: 10.1074/jbc.M400920200
155. Zhong Q, Kowluru RA. Diabetic retinopathy and damage to mitochondrial structure and transport machinery. Invest Ophthalmol Vis Sci (2011) 52:8739–46. doi: 10.1167/iovs.11-8045
156. Kim D, Sankaramoorthy A, Roy S. Downregulation of drp1 and fis1 inhibits mitochondrial fission and prevents high glucose-induced apoptosis in retinal endothelial cells. Cells (2020) 9:1662. doi: 10.3390/cells9071662
157. Duraisamy AJ, Mohammad G, Kowluru RA. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim Biophys Acta Mol basis Dis (2019) 1865:1617–26. doi: 10.1016/j.bbadis.2019.03.013
158. Kim D, Sesaki H, Roy S. Reduced levels of drp1 protect against development of retinal vascular lesions in diabetic retinopathy. Cells (2021) 10:1379. doi: 10.3390/cells10061379
159. Zhang M-Y, Zhu L, Bao X, Xie T-H, Cai J, Zou J, et al. Inhibition of Drp1 ameliorates diabetic retinopathy by regulating mitochondrial homeostasis. Exp Eye Res (2022) 220:109095. doi: 10.1016/j.exer.2022.109095
160. Mohammad G, Kowluru RA. Mitochondrial dynamics in the metabolic memory of diabetic retinopathy. J Diabetes Res (2022) 2022:3555889. doi: 10.1155/2022/3555889
161. Kowluru RA, Alka K. Mitochondrial quality control and metabolic memory phenomenon associated with continued progression of diabetic retinopathy. Int J Mol Sci (2023) 24:8076. doi: 10.3390/ijms24098076
162. Doğanlar ZB, Doğanlar O, Kurtdere K, Güçlü H, Chasan T, Turgut E. Melatonin prevents blood-retinal barrier breakdown and mitochondrial dysfunction in high glucose and hypoxia-induced in vitro diabetic macular edema model. Toxicol In Vitro (2021) 75:105191. doi: 10.1016/j.tiv.2021.105191
163. Chen M, Zhang Q, Wang S, Zheng F. Inhibition of diabetes-induced Drp1 deSUMOylation prevents retinal vascular lesions associated with diabetic retinopathy. Exp Eye Res (2023) 226:109334. doi: 10.1016/j.exer.2022.109334
164. Zhang K, Wang T, Sun G-F, Xiao J-X, Jiang L-P, Tou F-F, et al. Metformin protects against retinal ischemia/reperfusion injury through AMPK-mediated mitochondrial fusion. Free Radic Biol Med (2023) 205:47–61. doi: 10.1016/j.freeradbiomed.2023.05.019
165. Qian S, Qian Y, Huo D, Wang S, Qian Q. Tanshinone IIa protects retinal endothelial cells against mitochondrial fission induced by methylglyoxal through glyoxalase 1. Eur J Pharmacol (2019) 857:172419. doi: 10.1016/j.ejphar.2019.172419
166. Aloysius Dhivya M, Sulochana KN, Bharathi Devi SR. High glucose induced inflammation is inhibited by copper chelation via rescuing mitochondrial fusion protein 2 in retinal pigment epithelial cells. Cell Signal (2022) 92:110244. doi: 10.1016/j.cellsig.2022.110244
167. Zhang M-Y, Zhu L, Zheng X, Xie T-H, Wang W, Zou J, et al. TGR5 activation ameliorates mitochondrial homeostasis via regulating the PKCδ/drp1-HK2 signaling in diabetic retinopathy. Front Cell Dev Biol (2021) 9:759421. doi: 10.3389/fcell.2021.759421
168. Sharma I, Yadav KS, Mugale MN. Redoxisome and diabetic retinopathy, Pathophysiology and therapeutic interventions. Pharmacol Res (2022) 182:106292. doi: 10.1016/j.phrs.2022.106292
169. Zhang Y, Xi X, Mei Y, Zhao X, Zhou L, Ma M, et al. High-glucose induces retinal pigment epithelium mitochondrial pathways of apoptosis and inhibits mitophagy by regulating ROS/PINK1/Parkin signal pathway. BioMed Pharmacother (2019) 111:1315–25. doi: 10.1016/j.biopha.2019.01.034
170. Hombrebueno JR, Cairns L, Dutton LR, Lyons TJ, Brazil DP, Moynagh P, et al. Uncoupled turnover disrupts mitochondrial quality control in diabetic retinopathy. JCI Insight (2019) 4:e129760. doi: 10.1172/jci.insight.129760
171. Liu J, Wei Q, Guo C, Dong G, Liu Y, Tang C, et al. Hypoxia, HIF, and associated signaling networks in chronic kidney disease. Int J Mol Sci (2017) 18:950. doi: 10.3390/ijms18050950
172. Re G. Proximal tubulopathy, prime mover and key therapeutic target in diabetic kidney disease. Diabetes (2017) 66:791–800. doi: 10.2337/db16-0796
173. Salabei JK, Hill BG. Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol (2013) 1:542–51. doi: 10.1016/j.redox.2013.10.011
174. Garcia-Ropero A, Badimon JJ, Santos-Gallego CG. The pharmacokinetics and pharmacodynamics of SGLT2 inhibitors for type 2 diabetes mellitus, the latest developments. Expert Opin Drug Metab Toxicol (2018) 14:1287–302. doi: 10.1080/17425255.2018.1551877
175. Bode D, Semmler L, Wakula P, Hegemann N, Primessnig U, Beindorff N, et al. Dual SGLT-1 and SGLT-2 inhibition improves left atrial dysfunction in HFpEF. Cardiovasc Diabetol (2021) 20:7. doi: 10.1186/s12933-020-01208-z
176. Zannad F, Ferreira JP, Butler J, Filippatos G, Januzzi JL, Sumin M, et al. Effect of empagliflozin on circulating proteomics in heart failure, mechanistic insights into the EMPEROR programme. Eur Heart J (2022) 43:4991–5002. doi: 10.1093/eurheartj/ehac495
177. Taylor SI, Blau JE, Rother KI. SGLT2 inhibitors may predispose to ketoacidosis. J Clin Endocrinol Metab (2015) 100:2849–52. doi: 10.1210/jc.2015-1884
178. Wenzel P, Rossmann H, Müller C, Kossmann S, Oelze M, Schulz A, et al. Heme oxygenase-1 suppresses a pro-inflammatory phenotype in monocytes and determines endothelial function and arterial hypertension in mice and humans. Eur Heart J (2015) 36:3437–46. doi: 10.1093/eurheartj/ehv544
179. Jais A, Einwallner E, Sharif O, Gossens K, Lu TT-H, Soyal SM, et al. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell (2014) 158:25–40. doi: 10.1016/j.cell.2014.04.043
180. Shi J, Yu T, Song K, Du S, He S, Hu X, et al. Dexmedetomidine ameliorates endotoxin-induced acute lung injury in vivo and in vitro by preserving mitochondrial dynamic equilibrium through the HIF-1a/HO-1 signaling pathway. Redox Biol (2021) 41:101954. doi: 10.1016/j.redox.2021.101954
181. Barazzuol L, Giamogante F, Calì T. Mitochondria Associated Membranes (MAMs), Architecture and physiopathological role. Cell Calcium (2021) 94:102343. doi: 10.1016/j.ceca.2020.102343
182. Yang S, Zhou R, Zhang C, He S, Su Z. Mitochondria-associated endoplasmic reticulum membranes in the pathogenesis of type 2 diabetes mellitus. Front Cell Dev Biol (2020) 8:571554. doi: 10.3389/fcell.2020.571554
183. Wu S, Lu Q, Ding Y, Wu Y, Qiu Y, Wang P, et al. Hyperglycemia-driven inhibition of AMP-activated protein kinase α2 induces diabetic cardiomyopathy by promoting mitochondria-associated endoplasmic reticulum membranes. In Vivo Circ (2019) 139:1913–36. doi: 10.1161/CIRCULATIONAHA.118.033552
184. Wei X, Wei X, Lu Z, Li L, Hu Y, Sun F, et al. Activation of TRPV1 channel antagonizes diabetic nephropathy through inhibiting endoplasmic reticulum-mitochondria contact in podocytes. Metabolism (2020) 105:154182. doi: 10.1016/j.metabol.2020.154182
185. Chang Y, Wang C, Zhu J, Zheng S, Sun S, Wu Y, et al. SIRT3 ameliorates diabetes-associated cognitive dysfunction via regulating mitochondria-associated ER membranes. J Transl Med (2023) 21:494. doi: 10.1186/s12967-023-04246-9
186. Yang M, Han Y, Luo S, Xiong X, Zhu X, Zhao H, et al. MAMs protect against ectopic fat deposition and lipid-related kidney damage in DN patients. Front Endocrinol (Lausanne) (2021) 12:609580. doi: 10.3389/fendo.2021.609580
Keywords: mitochondrial dynamics, mitochondrial fusion, mitochondrial fission, type II diabetes, diabetic complications
Citation: Wang S, Zhao H, Lin S, Lv Y, Lin Y, Liu Y, Peng R and Jin H (2023) New therapeutic directions in type II diabetes and its complications: mitochondrial dynamics. Front. Endocrinol. 14:1230168. doi: 10.3389/fendo.2023.1230168
Received: 28 May 2023; Accepted: 07 August 2023;
Published: 21 August 2023.
Edited by:
Sangwon Kim, John Hopkins University, United StatesReviewed by:
Farhad R Danesh, University of Texas MD Anderson Cancer Center, United StatesCopyright © 2023 Wang, Zhao, Lin, Lv, Lin, Liu, Peng and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yinai Liu, MjE0NjEzMzgwMTJAc3R1Lnd6dS5lZHUuY24=; Renyi Peng, MjAxNzAwMzJAd3p1LmVkdS5jbg==; Huanzhi Jin, amluaHVhbnpoaUBzaW5hLmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.