 Yehan Bian
Yehan Bian Heidi Hahn
Heidi Hahn Anja Uhmann
Anja Uhmann- Institute of Human Genetics, Molecular Developmental Genetics, University Medical Center Göttingen, Göttingen, Germany
Hedgehog signaling plays pivotal roles in embryonic development, adult homeostasis and tumorigenesis. However, its engagement in the pituitary gland has been long underestimated although Hedgehog signaling and pituitary embryogenic development are closely linked. Thus, deregulation of this signaling pathway during pituitary development results in malformation of the gland. Research of the last years further implicates a regulatory role of Hedgehog signaling in the function of the adult pituitary, because its activity is also interlinked with homeostasis, hormone production, and most likely also formation of neoplasms of the gland. The fact that this pathway can be efficiently targeted by validated therapeutic strategies makes it a promising candidate for treating pituitary diseases. We here summarize the current knowledge about the importance of Hedgehog signaling during pituitary development and review recent data that highlight the impact of Hedgehog signaling in the healthy and the diseased adult pituitary gland.
1 Introduction
More than 40 years ago, the two Nobel laureates Nüsslein-Volhard and Wieschaus reported about lethal Drosophila melanogaster (D. melanogaster) variants caused by gene mutations that altered segmental pattering of the larvae. One mutant was almost entirely covered with denticles. Due to its ‘hedgehog-like’ appearance, the responsible gene was named hedgehog (hh). Simultaneously, the patch locus was identified, which later turned out to contain the gene encoding for the Hh receptor patched (Ptch) (1). Subsequent studies in D. melanogaster identified further genes encoding components of the Hh signaling pathway including suppressor of fused (sufu) and smoothened (smo) (2, 3). Today, it is known that the HH signaling pathway is highly conserved between D. melanogaster and vertebrates and that it plays a fundamental role in embryonic patterning and development of most tissues and organs. This is recapitulated in the adult, where the pathway regulates tissue stem cells necessary for organ repair and maintenance. Thus, its dysregulation results in developmental defects and in a variety of cancers in vertebrates (reviewed in (4)).
The pituitary is one of the organs that depend on HH signaling (5–8). This so called ‘Master Gland’ represents one of the most important endocrine glands of the human body and acts as a mediator between the hypothalamus and peripheral organs by regulating the activity of most hormone glands. Thus, the pituitary controls essential vital body functions, e.g. growth, metabolism, reproduction, childbirth, lactation, stress responses and water/salt balance (9). Numerous studies revealed that HH signaling is essential to pituitary development by promoting patterning, cell proliferation and cell type determination in coordination with other signaling pathways (5–8, 10). Several recent studies indicate the involvement of HH signaling in pituitary hormone regulation as well as in the development of pituitary diseases (11–16). However, the role of HH signaling in the adult pituitary has remained largely unexplored. In this review, we summarize and discuss the impact of HH signaling in development and homoeostasis of the pituitary as well as in pituitary disorders (e.g. hypopituitarism, hyperplasia of the gland, neoplasms, hormone deficiencies or overproduction). Targeting HH signaling is an attractive and eligible therapeutic strategy, which is used for the treatment of a wide range of diseases (reviewed in (17)). It remains to be seen, whether this pathway also provides a target in the treatment of pituitary diseases.
2 The HH signaling pathway
2.1 Canonical HH signaling
In vertebrates, three secreted HH ligands exist: sonic HH (SHH), indian HH and desert HH. Of these secreted factors, SHH is the most important and best-characterized one. Major components of the HH signaling pathway in vertebrates comprise the secreted HH ligands (e.g. SHH), the HH receptor and 12-transmembrane protein PTCH1, the seven-transmembrane G protein-coupled receptor and interaction partner of PTCH1 SMO, the suppressor of fused homolog (SUFUH) as a negative regulator of HH signaling as well as the glioma-associated oncogene (GLI) family of transcription factors (e.g. GLI1, GLI2, GLI3) (reviewed in (18–20)) (Figure 1).
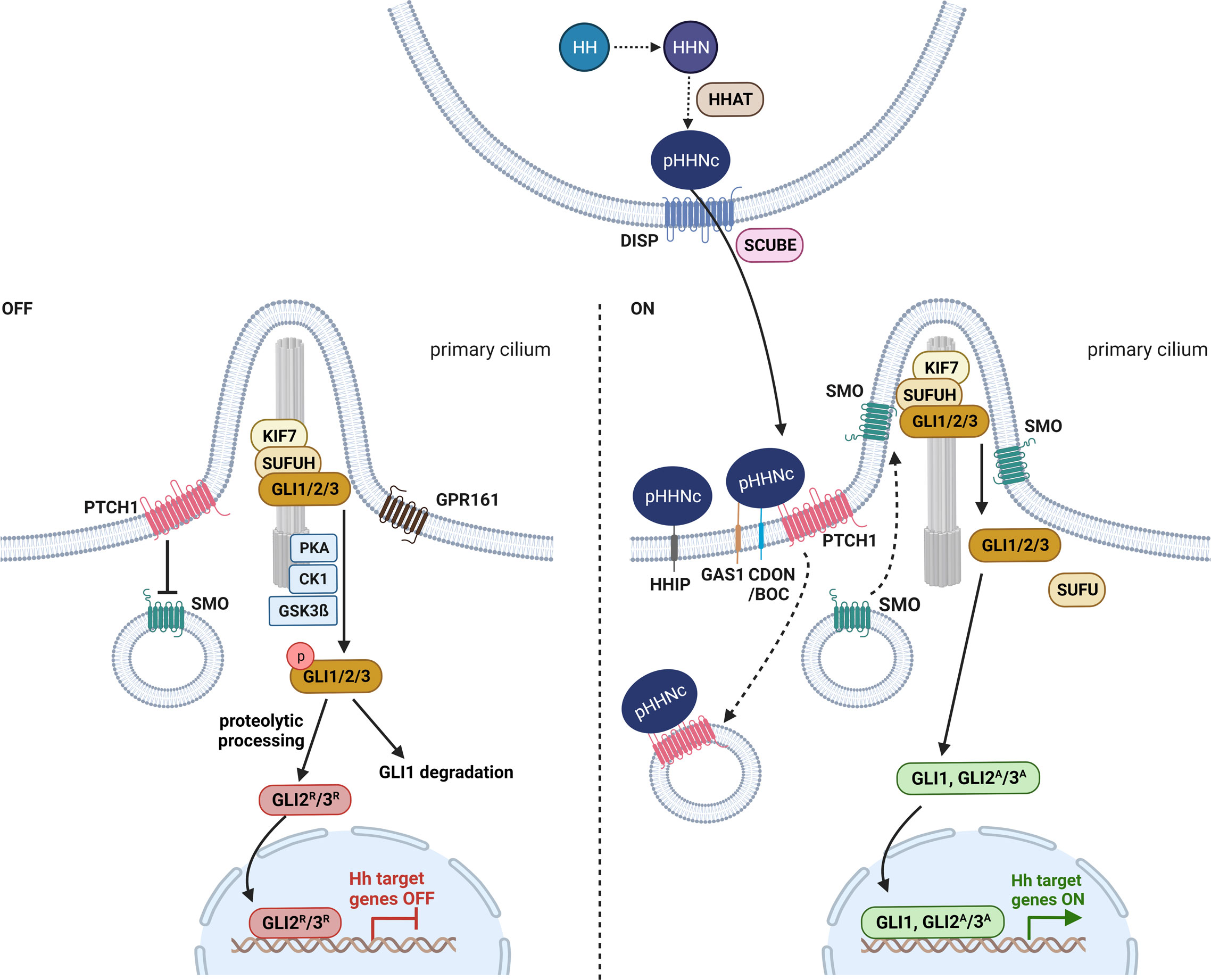
Figure 1 Hedgehog signaling in vertebrates. Before secretion, HH (hedgehog) is autoproteolytic cleaved into its N-terminal active form HHN and modified with cholesterol and HHAT (hedgehog acyltransferase)-mediated palmitoylation (pHHNc) (21, 22). pHHNc monomers are released from the membrane by DISP (dispatched) and SCUBE (signal peptide CUB-EGF-like domain-containing protein) (23). In the absence of pHHNc, PTCH1 (patched 1) accumulates in the primary cilium and inhibits SMO (smoothened) activity. GLI (transcription factor glioma associated oncogene) transcription factors are sequestered by KIF7 (kinesin-like protein 7) and SUFUH (suppressor of fused homolog) and phosphorylated by PKA (protein kinase A), GPR161 (G-protein coupled receptor 161), CK1 (casein kinase 1) and GSK3β (glycogen synthase kinase 3 β) kinases at the base of cilia, resulting in the degradation of GLI1 and the generation of GLI2/3 repressor (GLI2R/3R) forms. When pHHNc is present, it binds to its receptor PTCH1 and GAS1 (co-receptors growth arrest-specific protein 1), CDON (cell adhesion molecule-related/down-regulated by oncogenes) and BOC (brother of CDON), leading to the degradation of PTCH1 and the activation of SMO. Consequently, GLI dissociates from the SUFUH complex and acquires its active form (GLI1, GLI2A/3A) that promotes transcription of downstream target genes (19). Additionally, HHIP (hedgehog interacting protein) sequesters secreted pHHNc and negatively regulates HH signaling (24). The figure is based on (19, 20).
Before secretion, the HH precursor protein is autoproteolytic cleaved to its N-terminal active form (HHN). Concomitantly, the HHN is modified with a cholesterol moiety at its C-terminus (HHNc) that allows tethering of HHNc to the outer cell membrane and further a palmitoyl group at its N-terminus (pHHNc) by HH acyltransferase (HHAT) (21, 22) (Figure 1). The latter lipidation is required for interaction with the HH receptor PTCH1 at the signal-receiving cell surface (22). Finally, the 12-transmembrane protein dispatched (DISP) and the cholesterol-binding protein SCUBE (CUB-EGF-like domain-containing protein) release pHHNc from the signaling cell (23) (Figure 1). Both occurrence and delivery of monomeric SHH via specialized filopodia (cytoneme) are regulated by a complex of SHH, DISP, CDON (cell adhesion molecule-related, down-regulated by oncogenes) and BOC (brother of CDON) (25) (reviewed in (26)). BOC, CDON and the membrane-bound protein GAS1 (growth arrest-specific gene 1) are also required for activation of HH signaling in the target cells (25, 27–29) (Figure 1). However, monomeric pHHNc can also self-associate to soluble multimers and get released from the membrane (30, 31). Furthermore, oligomeric pHHNc can interact with heparan sulphate chains of glypicans and form lipoprotein particles (32, 33). Alternatively, pHHNc can be released via exovesicles (34).
The released pHHNc molecules are then either captured by the HH interacting protein (HHIP), resulting in sequestration of the ligand, or by its receptor PTCH1 (24) that forms complexes with its coreceptors CDON, BOC or GAS1 on the surface of receiving cells (25, 27–29, 35, 36) (Figure 1). The binding of pHHNc to PTCH1 abolishes the inhibitory function of PTCH1 on HH signaling activity and leads to downstream pathway activation mediated by GLI transcription factors (24).
In vertebrates, both HH signal reception and intracellular HH signal transmission require the primary or sensory cilium, which is a specialized antenna-like organelle that functions in signal sensing and transduction. The primary cilium is a singular protrusion from the cellular surface that can be found on almost every vertebrate cell. It consists of nine microtubule doublets that extend from the membrane-docked mature centriole (basal body) and form the microtubule-based cytoskeletal core of the cilium, the so called axoneme. The axoneme is surrounded by a ciliary membrane, which is a specialized and distinct compartment of the plasma membrane. Molecules and proteins essential for ciliogenesis, maintenance and signaling of the cilium are transported within the cilium bi-directionally along the microtubules by intraflagellar transport. With respect to HH signaling, initial signal reception, pathway activation and transcription factor processing are linked to the primary cilium (reviewed in (37)).
In the absence of HH ligands, PTCH1 concentrates in the ciliary membrane and inhibits the entry of SMO into the cilia and thus blocks its activity (18, 38) (Figure 1). It is proposed that PTCH1 does not physically interact with SMO, but rather regulates SMO by restricting its access to cholesterol in the ciliary membrane (39). Upon inhibition of SMO, cytoplasmatic SUFUH and kinesin-like protein 7 (KIF7) restrain GLI transcription factor activity, and protein kinases (e.g. casein kinase 1, CK1; protein kinase A, PKA; glycogen synthase kinase 3 β, GSK3β; G-protein coupled receptor 161, GPR161) induce phosphorylation of the GLI proteins (Figure 1). Afterwards, GLI proteins are either completely degraded (GLI1) or marked by ubiquitination for partial cleavage into truncated GLI repressor forms (e.g. GLI2R, GLI3R) that translocate to the nucleus and repress the transcription of HH signaling target genes (Figure 1). This posttranslational proteolytic processing regulates the availability of GLI activator and repressor forms (reviewed in (19)). Since the proteolytic processing of GLI2 is not as efficient as that of GLI3, GLI2 is typically found in its full-length activator form and only occasionally occurs in a weak truncated repressor form (40). In contrast, GLI3 is efficiently processed and acts as a strong transcriptional repressor (40–42).
In the presence of HH ligands, pHHNc binds to PTCH1 and coreceptors CDON, BOC or GAS1. The pHHNc-PTCH1 complex is internalized and subjected to degradation by the lysosome (Figure 1). In turn, SMO molecules spread over the ciliary membrane and promote the dissociation of GLI from the cytoplasmatic SUFUH kinase complex by an unknown mechanism (19, 38). Subsequently, the GLI transcription factors are stabilized in their full-length activator forms (e.g. GLI1, GLI2A/3A) and translocate into the nucleus, where they promote HH signaling target gene expression (e.g. HHIP, PTCH1, GLI1) (reviewed in (19, 37)) (Figure 1). In turn, the main targets can regulate HH signaling activity in negative (e.g. HHIP and PTCH1) or positive feedback loops (e.g. GLI1, GLI2) (reviewed in (19, 43)).
2.2 Non-canonical HH signaling
Besides the canonical pathway, the HH signaling pathway is also activated non-canonically by mechanisms that are either SMO-independent or SMO-dependent but without GLI involvement. Shortly, the SMO-independent mechanisms are related to the potential of PTCH1 to interact with phosphorylated cyclin B1 and to recruit caspase-3 that leads to caspase-9 activation and apoptosis. Both interactions are independent of SMO and can be prevented by HH ligand binding. More complex are SMO-dependent, GLI-independent mechanisms. For example, SMO can use heterotrimeric G-proteins to promote cellular Ca2+ influx that affects the cytoskeleton, angiogenesis, cellular differentiation, proliferation, apoptosis and migration. For further details, please refer to some excellent reviews on this topic (reviewed in (19, 43–47)).
Finally, a plethora of other signaling pathways activates or inactivates the signaling pathway independently of the HH ligand. For example, active RAS/RAF/MEK/ERK (also known as MAPK pathway) as well as PI3K/AKT signaling are able to stabilize GLI proteins and to stimulate their activity via mitogen-activated protein kinase (e.g. MEK or MAP2K) (48–52). Furthermore, oncogenic RAS can – dependent on the cancer entity – also inhibit GLI via MEK and/or ERK (extracellular signal-regulated kinase) (53, 54). Additionally, WNT signaling is able to interfere with GLI signal transduction. β-catenin can stabilize GLI1 protein or induce GLI1 transcription with the help of CRD-BP (coding region determinant-binding) (55) or sulfatase 2 (56), respectively. β-catenin can also physically interact with GLI1, which results in GLI1 degradation and inhibition of HH signaling (57). Vice versa, WNT signaling is also regulated by GLI proteins. Whereas active GLI2 can enhance β-catenin-dependent transcriptional activation (58), GLI3R – in the absence of HH ligands – inhibits WNT signaling via interactions with β-catenin (59). Additionally, crosstalks between the HH/GLI and WNT/β-catenin signaling pathways on the level of SUFUH, CK1 and GSK3β have been described (60, 61). GLI transcription factors are also regulated by several other factors such as TP53 and NUMB, NOTCH signaling or microRNAs (reviewed in (43, 45)).
3 HH signaling in the pituitary gland
In human adults, the pituitary is a pea-sized organ and lies in the pocket-shaped sella turcica at the cranial base of the sphenoid bone. It is covered by a flat piece of dura mater, the diaphragma sellae. The pituitary borders laterally on the wall of the cavernous sinus and antero-inferiorly on the posterior wall of the sphenoid sinus. Antero-superiorly, it lies in close proximity to the optic chiasm (62, 63).
Anatomically and functionally, the pituitary gland is composed of two distinct lobes: the adenohypophysis (anterior lobe) and the neurohypophysis (posterior lobe). The pituitary gland is connected with the superordinate hypothalamus via the infundibulum (infundibular stalk, pituitary stalk) and the hypophyseal portal system. The boundary between the adeno- and the neurohypophysis is called pars intermedia (intermediate lobe). In most animals, the pars intermedia is distinct from the fleshy, glandular adenohypophysis and the neural composition of the neurohypophysis. However, in humans, the intermediate lobe is only few cell layers thick and normally either very small or entirely absent in adulthood. As a result, it is often considered as a part of the adenohypophysis (reviewed in (9, 62, 63)) (Figure 2).
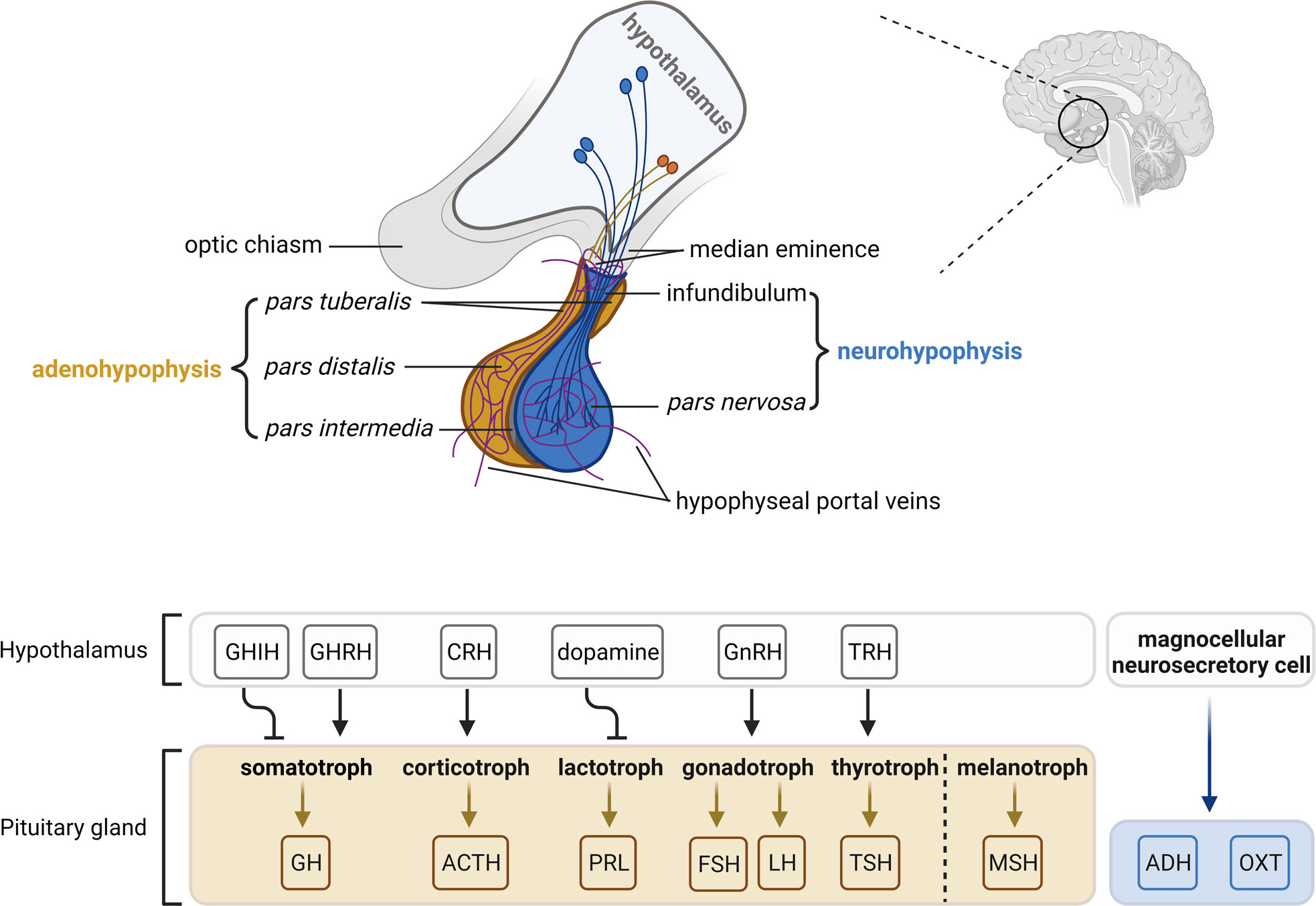
Figure 2 The hypothalamic-pituitary axis. The pituitary gland lies at the bottom of the hypothalamus and is composed of two functional components: the adenohypophysis consisting of the pars tuberalis, pars distalis and pars intermedia, and the neurohypophysis consisting of the infundibulum and pars nervosa. The pars distalis contains five endocrine cell types, which are somatotrophs, corticotrophs, lactotrophs, gonadotrophs and thyrotrophs that secrete growth hormone (GH), adrenocorticotropic hormone (ACTH), prolactin (PRL), follicle-stimulating hormone/luteinizing hormone (FSH/LH) and thyroid-stimulating hormone (TSH), respectively. The pars intermedia contains melanotrophs that secret melanocyte-stimulating hormone (MSH). Neurosecretory cells in the ventral nuclei of the hypothalamus secrete releasing/inhibitory hormones (growth hormone-inhibiting hormone, GHIH; growth hormone-releasing hormone, GHRH; corticotropin-releasing hormone, CRH; dopamine; gonadotropin-releasing hormone, GnRH; thyrotropin-releasing hormone, TRH) into hypophyseal veins at the median eminence and the infundibulum, which reach the adenohypophysis and regulate hormone activities. Magnocellular neurosecretory cells terminate in the pars nervosa and release oxytocin (OXT) and antidiuretic hormone (ADH) into the hypophyseal portal system. The figure is based on (9, 63).
3.1 Embryonal pituitary development
The pituitary gland originates from two germinal layers. The adenohypophysis including the pars intermedia originates from an evagination of the oral ectoderm at the roof of the developing oral cavity (Rathke’s pouch; RP) and thus is of epithelial origin. The neurohypophysis develops from descendants of the neural ectoderm, precisely from the ventral hypothalamic floor of the ventral diencephalon (reviewed in (62, 64)). The development of the human pituitary starts approximately at the 4th week of intrauterine development with the formation of the RP. During further growth of the pouch, the ventral diencephalon extends downwards to form the infundibulum. By the 5th week of the gestation, RP comes into contact with the infundibulum. Around the 6th to 8th week, a constriction forms at the base of the RP and closes completely, thus separating it from the oral epithelium and standing in close contact with the infundibulum. Upon proliferation, cells of the anterior wall of the RP form the anterior lobe and later on, a small part of the anterior lobe subsequently grows up along the infundibulum, covers it and forms the pars tuberalis. Cells of the posterior wall of the RP develop to the pars intermedia. The lumen of the pouch obliterates and cells of the infundibulum form the pituitary stalk and the neurohypophysis. Finally, cell patterning and terminal differentiation occur within the adenohypophysis to form five principal and specialized endocrine cell types (62) (see details in the forthcoming chapter, Figure 2).
As in humans, the adeno- and neurohypophysis in mice stem from the surface ectoderm and the neural ectoderm, respectively. The first sign of pituitary development occurs at 7.5 days post coitum (dpc) during murine embryogenesis. At this stage, a thickening of the ectoderm in the midline of the anterior neural ridge forms the hypophyseal placode that further develops into the RP between 9 to 10.5 dpc. By 10.5 dpc, when the definite RP has been formed, the neural ectoderm evaginates to form the infundibulum. The close and continuous contact of the neural ectoderm and the RP is essential for pituitary organogenesis. At 12.5 dpc, the pouch is completely separated from the underlying oral ectoderm. By 17 dpc, all parts of the murine pituitary have been developed. However, the lumen of the pouch persists as the pituitary cleft, separating the anterior and intermediate lobes in the mature gland (64) (Figure 3A).
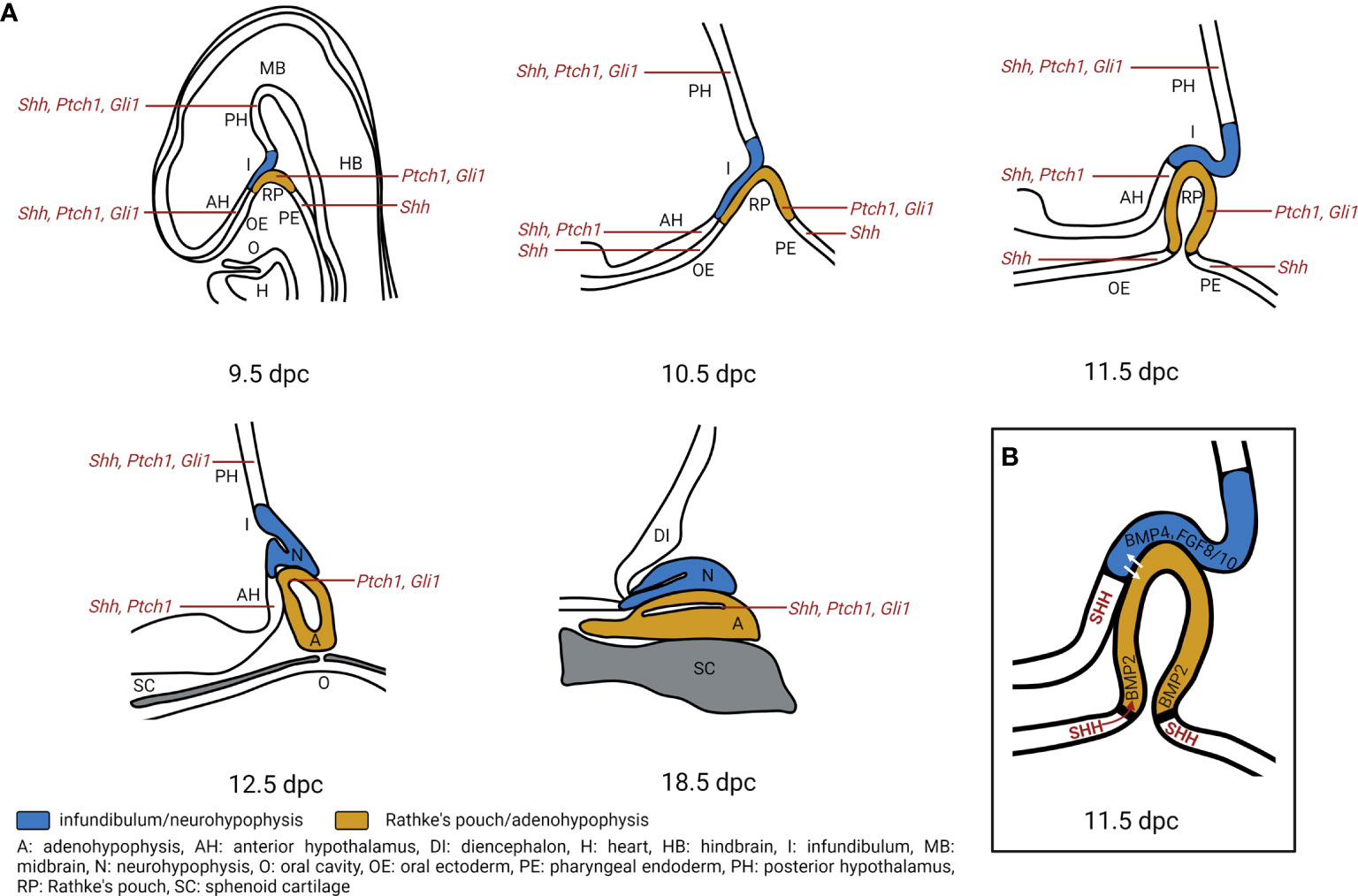
Figure 3 Hedgehog signaling regulates pituitary gland development. (A) Expression of Shh, Ptch1, Gli1 transcripts during pituitary gland development. During murine embryogenesis Shh is expressed in the anterior hypothalamus (AH), posterior hypothalamus (PH), oral ectoderm (OE) and pharyngeal endoderm (PE) that is attached to the developing Rathke’s pouch (RP) from 10 dpc to 12.5 dpc (6, 7). Ptch1 is expressed in the AH, PH, RP while Gli1 is expressed in PH and RP from 10.5 dpc to 14.5 dpc. By 18.5 dpc, Shh is detected in sporadic single cells within the anterior pituitary, while Ptch1, Gli1 are detected in the marginal zone of the pituitary (7). A, adenohypophysis; DI, diencephalon; H, heart; HB, hindbrain; I, infundibulum; MB, midbrain; N, neurohypophysis; O, oral cavity; SC, sphenoid cartilage. (B) Model of Hedgehog signaling regulation in pituitary gland development. SHH protein is expressed in the AH, together with BMP4. FGF8 and FGF10 that are expressed in the PH create an expression boundary within the hypothalamus that is essential for pituitary morphogenesis (10). Moreover, Shh expression marks a boundary between OE and RP which acts as an organizing center for ventral gene induction by inducing Bmp2 expression in the ventral RP. The ventrodorsal SHH/BMP2 signaling and dorsoventral FGF8/FGF10 signaling putatively induce expression of temporally and spatially specific transcription factors and promote cell fate determination (5, 6). Additionally, hypothalamic SHH expression is required for cell specification and proliferation of LHX3+/LHX4+ progenitors in the RP (7).
3.1.1 Signaling pathways and factors involved in pituitary development
Our current knowledge about embryonic development of the pituitary and the underlying mechanisms is mainly derived from mouse studies and phenotypes associated with human disorders that often share aspects with mouse models of defective pituitary development. Based on the respective observations, three general developmental steps can be distinguished in pituitary development: Firstly, initiation of pituitary organogenesis and formation of the RP, which are regulated by SIX homeodomain proteins (e.g. SIX1-6), paired-like homeobox proteins (e.g. HESX homeobox 1, HESX1; orthodenticle homeobox 2, OTX2; paired-like homeodomain transcription factor 1, PITX1; PITX2; PITX3), LIM homeodomain transcription factors (e.g. ISL LIM homeobox 1, ISL1; LIM homeobox 3, LHX3, LHX4) and by SOX2 (SRY-box transcription factor 2), β-catenin, NOTCH and SHH. Secondly, the proliferation and invagination of RP cells, which are controlled by bone morphogenic proteins (e.g. BMP2, BMP4), fibroblast growth factors (e.g. FGF8, FGF10, FGF18) and SHH. Thirdly, the lineage determination and cellular differentiation into endocrine cells, which is mediated by the stem cell marker PROP1 (homeobox protein prophet of PIT-1), PIT1 (or POU1F1, POU domain, class 1, transcription factor 1), GATA2 (GATA binding protein 2), NR5A1 (or SF1, steroidogenic factor 1) and TPIT (or TBX19, T-box transcription factor 19) (62, 64–66). Besides, additional factors expressed by the ventral diencephalon are implicated in regulation of pituitary formation. HESX1, SOX2, SOX3 and BMP4 expressed in the ventral diencephalon are regulators of RP progenitors, and OTX2, SIX6, SF1 (splicing factor 1), WNT5A, FGF8, FGF10 and FGF18 control further RP development (reviewed in (64, 67)). A summary of these transcription factors and signaling pathways as well as their roles in pituitary organogenesis are given in several excellent reviews (see (62, 64–67)). Here, we will focus on the role of HH signaling in the pituitary.
3.1.2 Expression of HH signaling components in pituitary development
Early studies show an essential role of HH signaling in pituitary development. Shh is expressed in the ventral diencephalon and the lateral oral ectoderm at 10 dpc in mice, before the formation of the RP (5–8). However, as soon as the pouch appears, Shh expression is specifically excluded from the RP, resulting in a Shh-expressing and non-expressing boundary. The oral epithelium and the ventral diencephalon lose their Shh expression at 12 dpc or 14 dpc, respectively (6). Recent data furthermore show that Shh transcripts are expressed in the developing hypothalamus from 9.5 to 12.5 dpc and become restricted to an area of the ventricular zone of the 3rd ventricle by 18.5 dpc (7) (Figure 3A). At 18.5 dpc Shh transcripts and Shh protein expression are also detectable in sporadic single cells within the adenohypophysis (7) (Figure 3A). Gli1 and Ptch1, main target genes of HH signaling, are expressed throughout pituitary development. Gli1 transcripts are expressed in the hypothalamus and in the RP between 9.5 and 12.5 dpc. Gli1 expression is mainly detected in the dorsal region of the periluminal epithelium of the RP by 14.5 dpc and is restricted to the marginal zone of the adenohypophysis by 18.5 dpc (Figure 3A). Ptch1 expression is detected in the hypothalamus except for the infundibulum from 9.5 to 14.5 dpc and is later restricted to the ventricular zone of the 3rd ventricle by 18.5 dpc. In the RP, Ptch1 is expressed between 9.5 dpc to 11.5 dpc. At 12.5 dpc, only a small region of the periluminal epithelium and by 18.5 dpc, only cells of the marginal zone express Ptch1 (7) (Figure 3A). Similarly, in human fetuses, SHH/SHH transcript and protein have been identified in the anterior hypothalamus and the pharyngeal endoderm but not in the developing RP at Carnegie stage 15 (estimated postfertilization age of 36 days, 5th week). GLI1 expression is detectable throughout the developing hypothalamus and the RP (7). Altogether, the expression of HH signaling components in pituitary and adjacent regions indicates that the developing pituitary is competent to receive and respond to HH signaling. Most likely, SHH protein, that is produced in SHH transcript-positive regions adjacent to the developing RP (e.g. developing hypothalamus and pharyngeal endoderm), activates HH signaling in regions containing undifferentiated precursors/stem cells (e.g. RP, periluminal epithelium, marginal zone) (7).
3.2 The adult pituitary gland
3.2.1 The hypothalamic-pituitary axis
The neurohypophysis is connected to the hypothalamus by the infundibulum. Magnocellular neurosecretory cells from the supraoptic and paraventricular nuclei of the hypothalamus project their axons to the neurohypophysis through the infundibulum (Figure 2). Magnocellular neurosecretory cells produce either oxytocin (OXT) and neurophysin I (NEU1) or vasopressin (antidiuretic hormone, ADH) and neurophysin II (NEU2) (Figure 2). OXT and ADH together with their respective carrier neurophysin proteins are stored and transported in neurosecretory granules of the axons called Herring bodies. Upon physiological stimuli like uterine stimulation during labor and suckling or increased osmolality of the plasma (e.g. hyponatremia), OXT-NEU1 or ADH-NEU2-containing vesicles are released from the axon terminals in the neurohypophysis into the systemic circulation via neurohypophyseal capillaries. In the bloodstream, OXT stimulates uterine contraction during labor and milk ejection from the breast, whereas ADH increases water reabsorption in the kidney and acts as a vasoconstrictor. Besides hypothalamic axons, the neurohypophysis also contains pituicytes that represent a subtype of non-neuronal glia cells assisting in storage and release of OXT and ADH (reviewed in (63, 68)).
Furthermore, the adenohypophysis is regulated by the hypothalamus via the hypophyseal portal system, which is a system of blood vessels. Neurosecretory axons in the hypothalamus terminate at the median eminence and release hypothalamic releasing and inhibitory hormones (e.g. corticotropin-releasing hormone, CRH; dopamine or prolactin release-inhibiting-hormone (PIH), growth hormone-releasing hormone, GHRH; gonadotropin-releasing hormone, GnRH; growth hormone-inhibiting hormone, GHIH; thyrotropin-releasing hormone, TRH). These releasing or inhibitory hormones reach the adenohypophysis via microcirculation and stimulate or inhibit the hormone secretion of their target endocrine cells, respectively (reviewed in (63, 68)) Figure 2).
As described in the afore-mentioned chapter, HH signaling is involved in the development of the hypothalamus-pituitary axis in that hypothalamic SHH expression promotes RP and pituitary formation (7, 10). However, little is known about the function of this pathway in the hypothalamus-pituitary axis in adulthood. Antonellis et al. reported expression of Shh, Ptch1, Smo, Gli1, and Gpr161 in differentiated neurons of the adult mouse hypothalamus (69). In respond to fasting, mice show increased HH signaling activity in the ventromedial hypothalamus and the paraventricular nucleus of the hypothalamus involved in the regulation of feeding behavior and to a lesser extent in the cortex (69). Since the hypothalamic-pituitary axis participates in appetite, metabolism and feeding behavior (70), it is tempting to speculate that regulation of HH signaling in feeding behavior is carried out via this axis. Yet, further investigations are needed to illustrate the role of HH signaling in the adult hypothalamic-pituitary axis. Several sporadic and congenital diseases of the hypothalamic-pituitary axis are closely associated with a deregulated HH signaling pathway (see below, see Tables 1, 2), making this research area particularly interesting.

Table 1 Human diseases associated with pituitary hypoplasia or deficiency.
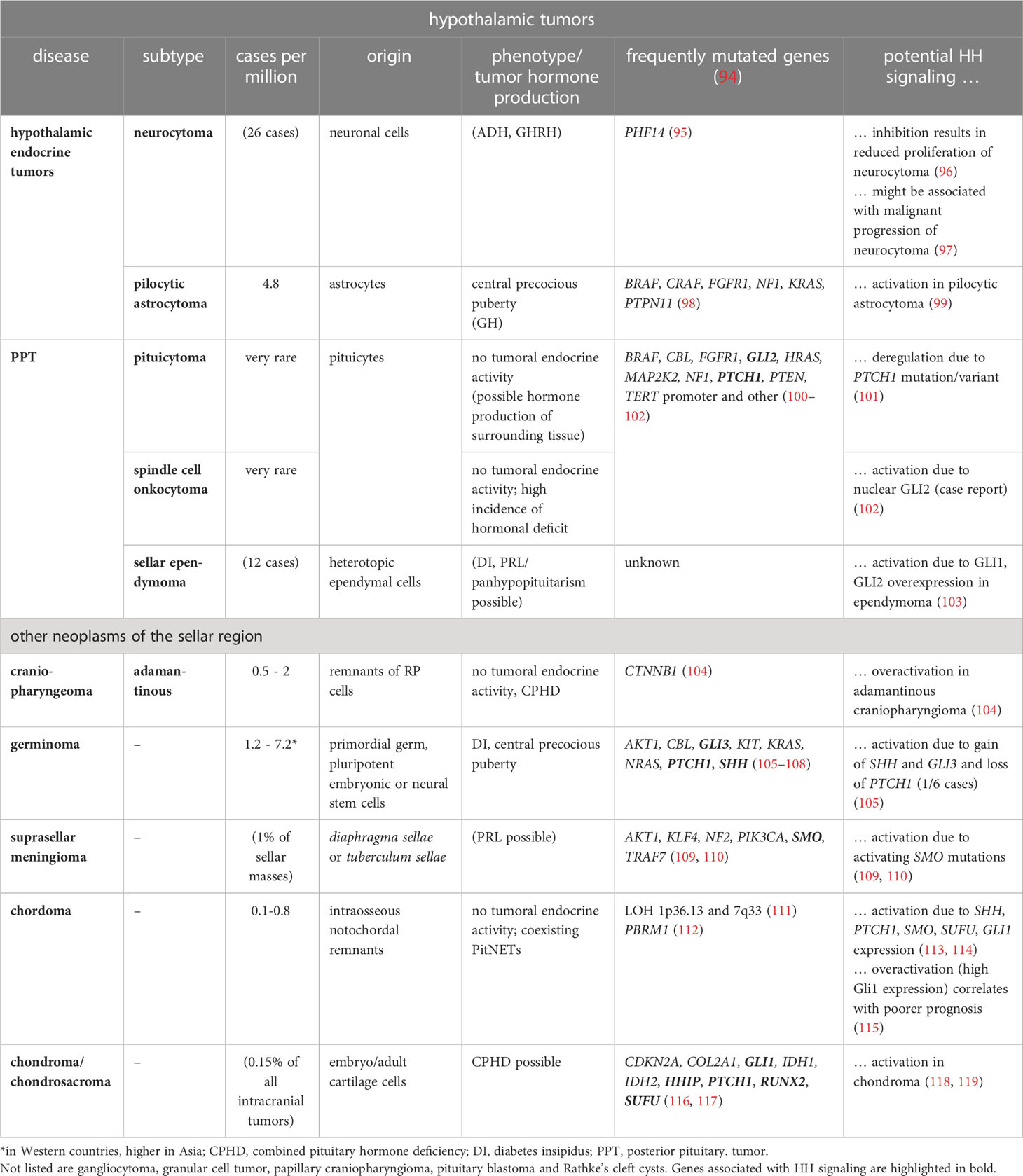
Table 2 HH signaling and other sporadic tumors of the sellar region.
3.2.2 Structure and function of the adenohypophysis
The adenohypophysis is the major organ of the endocrine system that hormonally regulates physiological processes like stress response, growth, reproduction, and lactation. It is composed of three regions: the pars distalis, the pars tuberalis (infundibularis) and the pars intermedia (reviewed in (63)) (Figure 2).
The pars distalis comprises the major part of the adenohypophysis and represents the region where most of the pituitary hormone production occurs. There are five types of endocrine cells: somatotrophs, corticotrophs, lactotrophs, gonadotrophs and thyrotrophs that secrete growth hormone (GH), adrenocorticotropic hormone (ACTH), prolactin (PRL), luteinizing hormone (LH) or follicle-stimulating hormone (FSH), and thyroid-stimulating hormone (TSH), respectively (Figure 2). The distribution of these endocrine cells in humans follows a certain pattern with somatotrophs and lactotrophs clustering in the lateral region, corticotrophs in the central regions, thyrotrophs in the anterior regions and gonadotroph scattered throughout the pars distalis (reviewed in (62)). Together with the non-endocrine folliculostellate (FS) cells, these endocrine cells form a 3D network that allows homotypic and heterotypic interactions between the cells, at least in rodents and fishes (120–125) (reviewed in (126)). Additionally, the pars distalis and the marginal zone – a single layered epithelium between the pars distalis and the pars intermedia – contain SOX2+ non-endocrine cells that exhibit stem cell properties since they self-renew and can differentiate into all five endocrine cell types. They contribute to organ homeostasis and probably are also in tumorigenesis due to their potential to differentiate into all mature hormone-producing lineages and their auto- and paracrine signaling (reviewed in (127, 128)). The adenohypophyseal hormone production and release are tightly controlled by hormones of the hypothalamus and from other endocrine organs in negative and positive feedback loops (e.g. liver, adrenal gland, thyroid gland etc.). For example, the release of GH is controlled by the hypothalamic hormones GHRH and GHIH, but also by the liver-born insulin-like growth factor-1 (IGF-1), GHIH of delta cells of the digestive system and by thyroid hormones (tri-iodothyronine (T3) and thyroxine (T4)) (reviewed in (62, 63)) (Figure 2).
The pars tuberalis represents a part of the sheath extending up from the pars distalis. It consists of gonadotrophs, thyrotrophs and FS cells that form an incomplete collar around the infundibulum/pituitary stalk of the neurohypophysis. Research of the last decade strongly indicates that the pars tuberalis is a major player in the regulation of seasonal reproduction and photoperiodicity, since it lies in direct proximity to the suprachiasmatic nuclei and expresses melatonin receptors and specific clock genes (reviewed in (62, 63, 129)).
The pars intermedia, as described above, represents the thin cell layers that mark the boundary between the adeno- and the neurohypophysis (Figure 2). In humans, it consists of colloid-filled cysts as well as of melanotrophs, corticotrophs, gonadotrophs and thyrotrophs (reviewed in (62)).
Somatotrophs constitute about 30-40% of all adenohypophyseal cells. They produce and release GH that exerts its effects in almost all tissues. GH promotes linear growth by stimulating protein synthesis and transport of amino acid through the cells. In the liver, GH induces the production of the somatomedins IGF-1 (somatomedin C), IGF-2 (somatomedin A) and vitronectin (VTN, with a somatomedin B-domain), which stimulate cell division and cellular proliferation. For example, somatomedins enhance T-cell proliferation, increase the absorption of calcium from the intestine and reduce calcium loss through urination, stimulate hepatic glycogenolysis to raise levels of glucose in the blood, increase the growth of soft and skeletal tissues and increase the uptake of non-esterified fatty acids by the muscle. The release of GH from somatotrophs in the adenohypophysis is positively controlled by hypothalamic GHRH. Negative feedback loops that inhibit GH secretion are mediated by high circulating GH concentrations and by liver-born IGF-1 that stimulates hypothalamic GHIH secretion (reviewed in (63, 130)).
Corticotropic cells and melanotrophs express pro-opiomelanocortin (POMC) which is the precursor protein for ACTH and β-lipotropin (β-LPH) in corticotrophs and for melanocyte-stimulating hormones (α-MSH, β-MSH, γ-MSH) in melanotrophs. In response to hypothalamic CRH stimulation, corticotrophs secrete ACTH that stimulates the adrenal cortex to produce cortisol. Via a negative feedback, cortisol inhibits the release of hypothalamic CRH and thus hypophyseal ACTH secretion. A functional role of β-LPH has not been proved so far. However, β-LPH is cleaved into β-endorphin and γ-LPH that is the precursor of β-MSH. In fact, corticotrophs produce and secrete β-endorphin acting as endogenous opioid neuropeptide that reduces stress and maintains homeostasis. Conversely, melanotrophs of the pars intermedia produce and secrete α-MSH, β-MSH and γ-MSH. These proteins are derived from different regions of POMC and bind with different preferences to different melanocortin receptors. Circulating α-MSH induces the release of melanin pigment in skin melanocytes during human fetal life and upon sun exposure in adulthood. In lower vertebrates, such as fish or amphibians, Msh from the pars intermedia induces skin darkening in response to changes in background color, whereas Msh released from hypothalamic neurons controls feeding and energy expenditure (reviewed in (63, 130)).
Lactotrophs comprise about 20% of all cells in the adenohypophysis. They produce and release PRL, which induces growth and development of the breast and maintains lactation in women. The production and secretion of PRL is primarily controlled by the hypothalamus that releases dopamine, which predominantly inhibits PRL release. Upon nipple stimulation of lactating women, sensory neurons are activated causing an inhibition of the dopamine release and thus an unopposed release of PRL. During pregnancy, the pituitary gland may increase by approximately 30% due to lactotroph hyperplasia (reviewed in (63, 130)).
Gonadotrophs release either FSH or LH. In females, FSH stimulates growth and development of follicles in the ovary and secretion of estrogens by the mature follicle, while LH triggers ovulation and stimulates the secretion of progesterone by the corpus luteum. In males, LH stimulates Leydig cells in the testes to secrete testosterone, whereas FSH stimulates Sertoli cells to secrete androgen-binding proteins that initiate spermatogenesis. In both males and females, these gonadotropic hormones help to stimulate the maturation of primordial germ cells. LH, FSH as well as human chorionic gonadotropin (hCG) and TSH (see below) are glycoprotein hormones. All these hormones are heterodimers consisting of non-covalently associated alpha (glycoprotein hormones, alpha polypeptide encoded by the CGA gene) and beta subunits (β-FSH, β-LH, β-hCG, β-TSH). The production and release of FSH and LH from the adenohypophysis are controlled by GnRH produced by the hypothalamus. Additionally, steroid sex hormones (e.g. estrogen) inhibit FSH and LH secretion via negative feedback loops (reviewed in (63, 130)).
Thyrotrophs compose around 5% of the adenohypophysis. They produce and release the glycoprotein hormone TSH that is controlled by the hypothalamic TRH. TSH functions as a stimulator of the thyroid gland to produce and release the hormones T3 and T4. In a negative feedback, T4 inhibits TSH secretion (reviewed in (63, 130)).
FS cells are non-endocrine cells of the adenohypophysis. They have a stellate shaped, follicular morphology with long cytoplasmic processes (123). Their lysosome content and their large number of microvilli on the apical side suggest that FS cells are phagocytic (131). Additionally, the FS cell mesh forms a 3D network, in which FS cells and endocrine cells reside and communicate via gap junction-mediated calcium wave propagation (123). Moreover, FS cells control adenohypophyseal cell function via cytokines and growth factors and modulate inflammatory response feedbacks by secreting inflammatory cytokines (reviewed in (132, 133)). Due to the lack of specific marker protein expression, the identification of FS cells remains challenging. So far, studies show that S100b and GFAP (glial fibrillary acidic protein) are strongly expressed in early, newly formed FS cells (134). Besides, other papers report that cytokeratin, vimentin, fibronectin (135) or SOX2 (16) are expressed in FS cells. However, whereas GFAP (136) and vimentin expression (137) suggest a neuroectodermal origin of FS cells, keratin or SOX2 expression rather implies epithelial-like (138) or stem cells-like characteristics. In addition, FS cells show high annexin A1 (ANXA1) levels, which is comparable to non-endocrine cells of the hypothalamus (139). Since glucocorticoids increase ANXA1 expression in FS cells, it is hypothesized that ANXA1+ FS cells subsequently regulate corticotrophs, which express ANXA1-associated G protein coupled receptors, via a paracrine mechanism (139–141). In the pars tuberalis, FS cells are localized in close association with GnRH-containing axon terminals, which regulate seasonal reproduction and photoperiodicity. Thus, FS cells may also be involved in transmission of photoperiodic stimuli to the reproductive system (63). Interestingly, the stem cell marker SOX2 is co-expressed with S100 protein in FS cells of adult mouse pituitaries (142) and in GH-producing pituitary neuroendocrine tumors (PitNETs) (143). Taking together, a bulk of data support the idea that FS cells function as sustentacular (mechanical/chemical) supporters for pituitary endocrine cells (144, 145). However, because these cells also show stem cell-like characteristics, the exact role of FS cells still remains mysterious.
3.2.3 HH Signaling in the healthy adult adenohypophysis
Vila et al. were the first scientists, who evaluated the potential role of HH signaling in the adult human adenohypophysis (13, 14). Their studies revealed that SHH and GLI1 are exclusively expressed in corticotrophs. Besides, they reported that PTCH1 is expressed in gonadotrophs and thyrotrophs, while PTCH2 (a PTCH1 paralog) is expressed in corticotrophs, somatotrophs and some lactotrophs (13, 14). Our recent data confirmed the SHH expression in corticotrophs of the human adenohypophysis. However, we additionally found SHH expression in somatotrophs and lactotrophs (11). In the adenohypophyseal tumor cell lines AtT-20 (corticotroph) and GH3 (lactotroph/somatotroph), expression of all four HH signaling components (SHH, PTCH1, PTCH2 and Gli1) is detected simultaneously (13, 14). This contrasts the data from human adenohypophysis samples that show no PTCH1 expression in corticotrophs and no GLI1 and PTCH2 expression in lactotrophs/somatotrophs (11, 13, 14). In addition, lineage-tracing experiments showed that approximately 2/3 of all somatotrophs and 2/3 of all FS cells descend from GLI1-expressing cells in the murine adenohypophysis (12), indicating a role of HH signaling in somatotrophs and FS cells. Together, these data are somewhat inconclusive. However, recent single cell RNA sequencing data revealed the expression of GLI1, GLI3 and GAS1 in human SOX2+ pituitary cells (146) and of Ptch1, Gli1/2/3 in FS cells and of Shh in gonadotrophs in chicken pituitary glands (147). Additionally, we re-analysed data from Cheung et al. (148) who sequenced single cells of adult male mouse pituitary glands and identified stem cells and hormone-producing cell types. Using this data set, we found that a subpopulation of murine somatotrophs expresses Gli1 while corticotrophs, gonadotrophs and lactotrophs cells do not. Together, these data support the conclusion that SOX2+ stem cells and/or FS cells as well as somatotrophs (at least in mice) express markers of active HH signaling. Therefore, HH signaling may be involved in pituitary homeostasis and/or tissue regeneration.
Furthermore, functional studies revealed that activation of HH signaling with recombinant SHH protein in AtT-20, GH3 and cultured rat pituitary cells results in increased ACTH, GH and/or PRL secretion, respectively (13, 14). A simultaneous treatment with SHH and CRH even increased ACTH production of AtT-20 and rat pituitary cells, whereas GLI1 inhibition reduces the CRH-induced stimulation of Pomc transcription (13). These data support the idea that active HH signaling directly regulates ACTH, PRL and GH secretion, and CRH signal transduction in the adult pituitary. Compared to these studies, our data rather hint towards an indirect hormone regulation by HH signaling in the adult adenohypophysis. We found that HH signaling activation in murine pituitary explants induces Acth, Gh, Prl and Cga expression (11), whereas a corticotroph-specific Ptch1 or Smo depletion in vivo does not (12). Moreover, neither corticotrophs nor lactotrophs show active HH signaling in adult mice (12). Interestingly, the FS cell line TtT/GF is responsive to HH signaling activation. Treatment with the SMO-aginist SAG of the FS cell line TtT/GF results in SMO localization to primary cilia and an enhanced expression of Gli1 and Gli2 (12). Remarkably, the supernatant from SAG-stimulated TtT/GF cells induces GH production and release in GH3 cells, which are mediated by the neuropeptide vasoactive intestinal protein (VIP) (12). The facts that GLI1 expression overlaps with the stem cell and FS cell marker SOX2 (11, 16) and that HH signaling activation induces the proliferation of SOX2+ cells in the pars distalis (11) further substantiate the assumption that HH signaling regulates pituitary hormone production indirectly via FS cells. This hypothesis is also supported by conditional Ptch1 depletion and concomitant activation of HH signaling in FS and CD45+ hematopoietic cells, which has a functional impact on multiple pituitary endocrine cells, including gonadotrophs and thyrotrophs (15). Nevertheless, future studies are needed to examine whether this concept is transferable to the human adenohypophysis and potentially also to corticotrophs and lactotrophs.
4 HH signaling in the diseased pituitary
Given that major components of HH signaling pathway are expressed during hypothalamic-pituitary development and in the adult pituitary, it is not surprising that the deregulation of this pathway is closely related to developmental defects or diseases of the hypothalamus and/or pituitary gland.
4.1 Hypothalamus and pituitary developmental defects caused by deregulated HH signaling
4.1.1 Lessons from animal models
The earliest hint for HH signaling involvement in development of pituitary diseases came from studies of Shh null mouse embryos that die just before or at birth and show forebrain malformations. These developmental defects result in a phenotype resembling holoprosencephaly in humans and in an agenesis of the pituitary gland (149). Similarly, Gas1 (27, 150) and Cdon null embryos (27, 151, 152) display microforms of holoprosencephaly and agenesis of the pituitary. Craniofacial developmental defects are also detected in Boc null mutants (153). These results support an essential role of HH signaling in pituitary development (see chapter ‘Expression of HH Signaling Components in Pituitary Development’ and Figures 3A, B). Hypothalamus-specific SHH depletion during murine embryogenesis results in pituitary hypoplasia and absence of the optic disc, which are key features of a rare developmental disorder in humans called Septo-optic dysplasia (SOD; please see below for detailed description) (10). These embryos develop a highly dysmorphic infundibulum that fails to protrude correctly from the diencephalon and is shifted anteriorly within the brain. Additionally, the formation of the anterior pituitary in these embryos is affected. The RP fails to pinch off the oral ectoderm and also shifts anteriorly into the brain (10). Likewise, a decrease in the SHH regulators SOX2 and SOX3 reduces SHH levels, expands FGF10 and BMP4 expression in the ventral diencephalon and thus may be the cause of SOD development (10). When SHH is depleted in a more restricted area of the anterior hypothalamus, RP development is completely arrested and an infundibulum fails to form. This results in complete pituitary agenesis, severe craniofacial defects, and telencephalic and hypothalamic abnormalities (7).
Moreover, HH signaling most likely promotes cell proliferation and cell-type determination in pituitary development. The before-mentioned hypothalamus-specific SHH depletion in mice results in a significant reduction of somatotrophs, corticotrophs and thyrotrophs (10). When SHH is depleted in the mouse anterior hypothalamus, Lhx3/Lhx4 expression is lost in the RP epithelium, which is accompanied by a progressive cell fate transformation of the epithelium. Besides, most of the hypothalamic factors controlling pituitary function (e.g. GHRH, GHIH, OXT, POMC) are lost or severely reduced in these mice, suggesting a loss of neuronal differentiation due to interrupted HH signaling (7). Similarly, a HHIP-mediated inhibition of HH signaling during very early stages in the oral ectoderm (prior to and during RP formation) results in a cystic rudiment of the pituitary gland that lacks the expression of ventral cell type markers (Bmp2, Gata2, Pou3f4) and of the terminal differentiation markers POMC and β-TSH (6). The study also implicates that Shh expression marks a boundary between the Shh-expressing oral ectoderm and the non-expressing RP. This boundary functions as an organizing center for ventral gene induction by inducing Bmp2 expression in the ventral RP and thus directing patterning and proliferation of the gland. Treier et al. furthermore hypothesized that ventrodorsal SHH/BMP signaling and dorsoventral FGF8/FGF10 activity lead to the induction of temporally and spatially restricted transcription factors expression and promote cell-fate determination (6) (Figure 3B). However, HH signaling inhibition (pharmacologically via the SMO-inhibitor cyclopamine or in smu/smu mutant embryos) during different zebrafish developmental stages revealed that HH signaling may also play a role even before invagination of the oral ectoderm and during early patterning of the gland (154). In contrast, HH signaling inactivation via a Gli2 mutation results in a variable loss of the adeno- and neurohypophysis in mice (155, 156). The Gli2 loss does not affect the overall patterning of the pituitary even not when Gli2 is specifically depleted before RP closure in cells of the anterior neural ridge and the early telencephalon (156). Its loss rather impacts on diencephalic expression of BMP4 and FGF8 and on proliferation of pituitary progenitor cells in the RP, which finally results in defective development of the neurohypophysis and of hormone-producing cells of the murine adenohypophysis (156). Interestingly, and in contrast to GLI2, neither GLI1 nor GLI3 depleted pituitaries show any abnormality. However, excessive production of GLI3, as in Shh null and Hhip-overexpressing embryos, may be responsible for severe pituitary phenotypes such as pituitary hypoplasia (6, 7, 10, 156). Together, these results show that inactivation of HH signaling results in a hypoplastic or missing pituitary.
Vice versa, an activation of the pathway during hypothalamic-pituitary formation results in a hyperplastic gland and/or hyperpituitarism in mice. Thus, ectopic SHH overexpression under control of the CGA-promoter in RP cells leads to an increased organ volume and elevated cell numbers of thyrotrophs and of LH-producing gonadotrophs (6). Ptch1 depletion in Hesx1 cell lineage, which marks the anterior neural plate including the prospective telencephalon, anterior hypothalamus and RP epithelium, results in HH signaling activation in RP cells from 10.5 dpc. This leads to increased proliferation of RP progenitors and development of a hyperplastic adenohypophysis due to an enlarged SOX2+ stem cell compartment (7). However, cell commitment (at 14.5 dpc) and differentiation of hormone-producing cells (at 18.5 dpc) in these embryos are unchanged (7). Similarly, overactive HH signaling in the ventral diencephalon due to a Tbx3 (T-box transcription factor 3) depletion results in reduced Fgf10 and increased Ptch1 expression in the infundibulum at 10.5 dpc. This goes along with hyperproliferation of the ventral diencephalon, hypoplasia of the adenohypophysis and lack of evagination of the infundibulum and thus of the neurohypophysis (157). Indeed, FGF10+ progenitors give rise to the neurohypophysis, whereas HH signaling orchestrates the development of the infundibulum and the RP and promotes the growth and differentiation of progenitors of the adenohypophysis, at least in chicks (158). Finally, pharmacological stimulation of HH signaling of a three-dimensional, ES-cell derived adenohypophysis tissue results in self-formation of RP-like structures in vitro. Moreover, these RP-like aggregates develop into functional corticotrophs or somatotrophs and lactotrophs upon manipulation of other signaling pathways (159).
Altogether, animal studies suggest that HH signaling plays a crucial role in pituitary development via regulation of patterning, cell proliferation and cell-fate determination. Moreover, HH signaling in both RP cells (6) and the developing hypothalamus (7, 157, 158) seems to be important for pituitary formation.
4.1.2 Human diseases
Similar to the above-mentioned animal models, mutations in the HH signaling pathway in humans result in defects of the pituitary. In fact, inactivating mutations in HH signaling components have been identified in subsets of patients suffering from SOD, Pallister-Hall syndrome (PHS), holoprosencephaly (HPE), isolated or combined pituitary hormone deficiency (CPHD), congenital hypopituitarism without HPE, or pituitary stalk interruption syndrome (PSIS) (reviewed in (75, 76, 160)) (Table 1).
SOD is a rare developmental disorder and is characterized by optic nerve hypoplasia combined with midline abnormalities of the brain and pituitary hypoplasia/hypopituitarism (71). Thus, SOD patients can suffer from isolated GH deficiency (GHD), CPHD and/or diabetes insipidus. Whereas the most common disease-causing mutations are found in HESX1, SOX2 and other genes, recently a causative variant in SHH in a SOD family has been identified (74). Mice with a conditional Shh deletion in the hypothalamus recapitulate this SOD phenotype (see above) (10). Moreover, the impact of HH signaling in SOD is substantiated by the fact that SOX2 induces SHH expression (10, 91) and directly regulates HESX1/Hesx1 transcription (161).
Similarly, the rare PHS is related to mutational inactivation of HH signaling during embryonal development. This autosomal dominant inherited disease is caused by frameshift/nonsense or splicing mutations in the second third of the GLI3 gene (from nucleotides 1998-3481) that lead to an exclusive expression of the GLI3R (162, 163). This results in developmental defects like mesoaxial polydactyly, hypothalamic hamartoma, and sometimes in GHD and panhypopituitarism (reviewed in (164)).
Furthermore, variants in PTCH1 (77), CDON (35), GLI2 (78), DISP1 (79), and GAS1 (80) have been detected in patients with HPE. Interestingly, approximately 17% of familial HPE are associated with inactivating SHH mutations or SHH haploinsufficiency (76, 81–84). The phenotypes of HPE range from most severe forms that include cyclopia and loss of the pituitary gland to less severe forms with milder symptoms such as hypotelorism, a single midline incisor, and hypopituitarism (165–167). Thus, HPE patients often suffer from several endocrine problems. These include GHD or ACTH deficiency (165, 168), which result in short stature or adrenal hypoplasia, respectively. In addition, loss-of-function mutations in GLI2 are also within the HPE spectrum, but are associated with a distinctive phenotype. The primary features of this phenotype are defective anterior pituitary formation and pan-hypopituitarism (78). This is similar to the developmental defects found in Gli2 mutant mice (155, 156) (see above). However, more recent reports rather associate GLI2 variants with polydactyly, pituitary deficiency and subtle facial phenotypes (169–173). In addition, GLI2 mutations are apparently also responsible for congenital hypopituitarism without HPE or for CPHD (i.e. impaired GH production combined with an impairment of at least one other anterior pituitary hormone) (85, 170, 174, 175). The mutational spectrum of these phenotypes includes GLI2 variants that result in loss of GLI2 function, or at least has a negative impact on the proteins function (174, 175). CPHD patients with GLI2 loss can suffer from deficiencies in somatotrophs, thyrotrophs and gonadotrophs, maldescended neurohypophysis or ADH deficiency (170, 174, 176). However, GLI2 mutations have also been detected in some patients with isolated GHD (see below) (75, 85).
Finally, mutations in PTCH1, PTCH2 (177), GPR161, HHAT and CDON have been found in PSIS patients (86, 178, 179). PSIS is characterized by a combination of three specific findings on magnetic resonance imaging, which are an interrupted pituitary stalk, an absent or ectopic posterior pituitary and anterior pituitary hypoplasia. Besides these characteristics, PSIS is also associated with other midline and ophthalmic abnormalities and variable endocrine disorders such as hypoglycemia, growth failure, or CPHD (86, 178, 179).
It also should be mentioned that hypopituitarism is a symptom of other syndromes that are caused by variants in genes that may regulate HH signaling activity. These syndromes include Prader-Willi (75), Williams (180), Kabuki (181), Axenfeld-Rieger (182, 183), Johanson-Blizzard (184) or Oliver-McFarlane syndromes (185). However, the description of the mechanisms by which the respective genes may regulate HH signaling activity in pituitary development is unknown or out of the scope of this review. In addition, several isolated hormone deficiencies are linked to deregulated HH signaling. For example, some patients with isolated GHD show GLI2 mutations (75, 85). Similarly, the development of central hypogonadism in Kallmann or CHARGE syndromes has been linked to HH signaling. Thus, some Kallmann syndrome patients show mutations in WDR11 (WD repeat domain 11) that modulate HH signaling activity by regulating GLI3 processing (88, 89). Likewise, 65% of CHARGE syndrome patients show variants of CHD7 (chromodomain helicase DNA binding protein 7) that physically interacts with SOX2 to regulate a set of common target genes including GLI3 (91). Additionally, some forms of central diabetes insipidus may be linked to deregulated HH signaling. For example, WFS1 (Wolframin ER transmembrane glycoprotein) mutations, which are found in 95% of Wolfram syndrome patients, lead to GLI1 degradation in in vitro analyses (93) (Table 1).
4.2 HH signaling in sellar neoplasm
It is well known that mutational activation of HH signaling results in a broad range of tumor entities (44, 177–179). Currently three basic mechanisms for mutational activation of the pathway in cancers are proposed. The first mechanism is mutation-driven and ligand-independent and was detected in patients suffering from the inherited Nevoid basal-cell carcinoma syndrome (NBCCS) (86, 180). This disease is caused by heterozygous PTCH1 mutations resulting in aberrant HH signaling activity in the absence of the HH ligand (86, 180). Besides developmental defects, NBCCS patients show an increased risk for a variety of tumors (e.g. medulloblastoma, basal cell carcinoma). Thus, the detection of PTCH1 mutations in this syndrome was the first link between HH signaling and cancer. Besides PTCH1 mutations, the mutation-driven subtype of HH signaling activation also includes activating mutations in SMO, GLI1 or GLI2 or inactivating mutations in GAS1, CDON or GLI3. The second type of activation is ligand-dependent and autocrine. This mechanism is characterized by an overexpression and oversecretion of HH ligands by the respective tumor, which results in an autocrine stimulation of tumor cell growth. Similarly, the third mechanism is characterized by an overexpression and oversecretion of HH ligands by the tumor cells. However, in this case the ligand-dependent activation is paracrine, in that the tumor-born HH ligands activate HH signaling in tumor and/or in stromal cells (reviewed in (181)). HH-associated malignancies are usually characterized by high expression levels of downstream targets like GLI1 and/or GLI2 as indicators for a pathologically overactive pathway. However, whereas the role of HH signaling in classical HH-associated tumors such as medulloblastoma and basal cell carcinoma is well known, that of neoplasms of the pituitary or the sellar region is far from clear.
4.2.1 Neoplasm of the sellar region
Neoplastic lesions in the sellar region can be classified as PitNETs (182), hypothalamic (endocrine) tumors, tumors of the neurohypophysis (posterior pituitary tumors, PPTs) and other tumors. The development of tumors in the sellar region is either sporadic, phenotypic for congenital disorders like hypothalamic hamartoma or associated with syndromes that predispose to neuroendocrine tumors (NETs). These syndromes encompass neurofibromatosis type 1 (NF1), Lynch syndrome, multiple endocrine neoplasia 1 (MEN type 1 and 2), Von Hippel-Lindau (VHL) syndrome, McCune-Albright syndrome, DICER1 syndrome, Carney complex (CNC), Succinate dehydrogenase deficiency, familial isolated pituitary adenoma (FIPA) and familial infantile gigantism (X-LAG syndrome) (reviewed in (147, 184))
PitNETs comprise approximately 15% of all primary sporadic brain tumors and are the most common pituitary disease (186). Most PitNETs are slow-growing and benign. A metastatic behavior of PitNETs is exceptionally rare (< 0.5% of PitNETs) (187). PitNETs are frequently hormone-secreting, which indicates an origin from hormone-secreting cells of the adenohypophysis. They are classified based on their histological expression pattern of certain transcription factors, such as TPIT (or TBX19), PIT1 (or POU1F1) or SF1 (NR5A1). TPIT+ PitNETs account for approximately 15% of PitNETs. Because these tumors secrete ACTH (corticotroph), which results in excess of ACTH, the phenotype of the patients resembles that of Cushing disease. PIT1+ PitNETs are subdivided into tumors, which release GH and/or CGA (somatotroph), PRL (lactotroph), TSH (thyrotroph) or GH together with PRL and/or TSH (mammasomatotroph, mature plurihormonal, immature PIT1-lineage). Over 33% of resected PitNETs are SF1+ and secrete FSH and LH (gonadotroph) (reviewed in (188–190)). PitNETs that express TPIT, PIT1 or SF1 on protein level and one or more hormones at clinically irrelevant levels, are further classified as silent PitNETs (191). Together with null cell adenoma (see below), they were historically categorized into the group of non-functioning pituitary adenomas (NFPAs). However, according to the newest WHO classification guideline, null cell adenoma together with unclassified plurihormonal tumors now account for the fourth class of PitNETs, the undefined PitNETs. Undefined PitNETs are negative for TPIT, PIT1 and SF1, but whereas unclassified plurihormonal tumors secrete ACTH, GH, PRL, TSH, FSH and/or LH, null cell adenomas do not (reviewed in (188–190, 192)). However, silent and null cell PitNETs can also lead to hormonal deficiencies due to compression of the surrounding pituitary areas (Table 3).
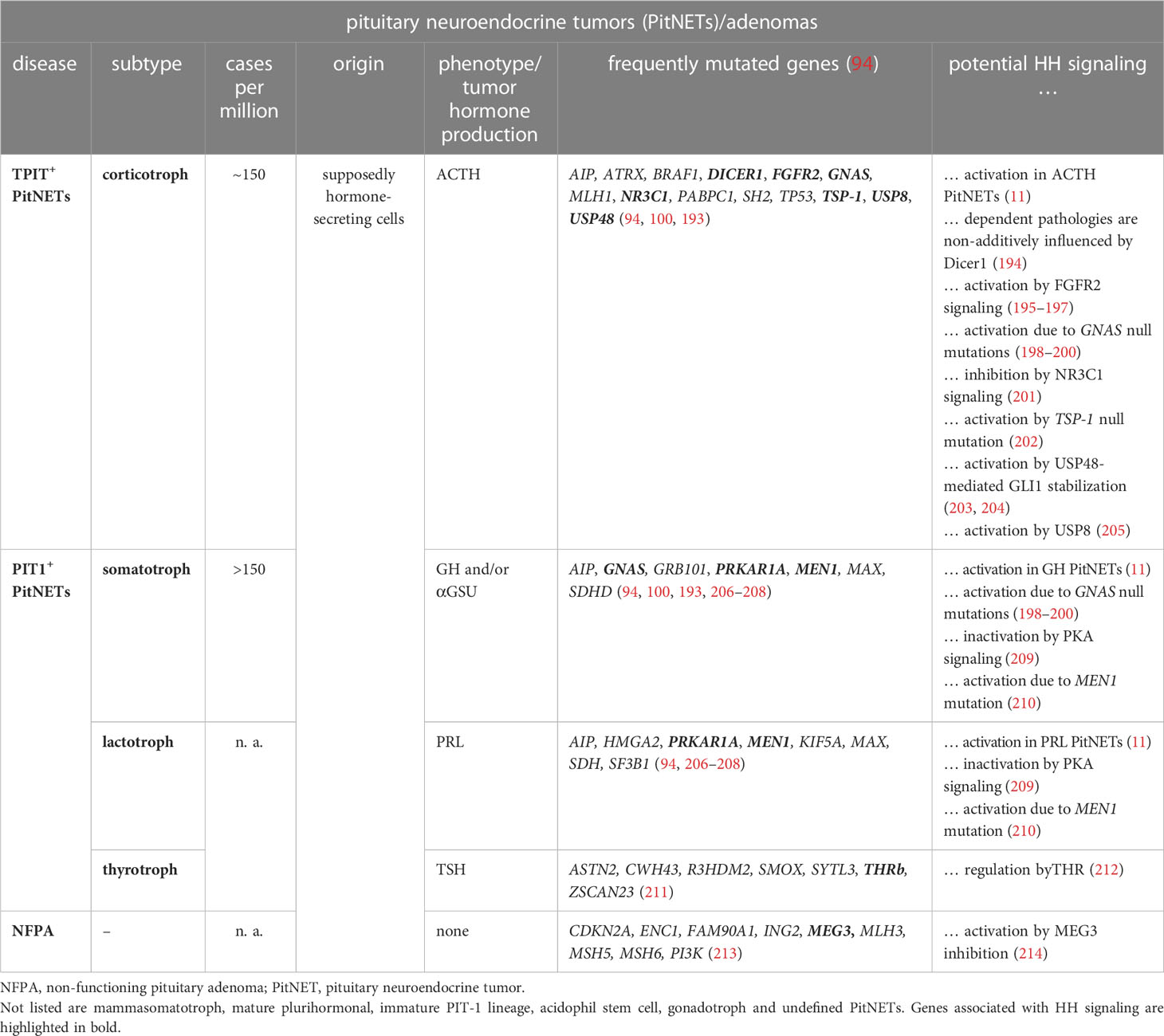
Table 3 HH signaling and PitNETs.
Sporadic PitNETs frequently show epigenetic changes (215–217), whereas common mutations are quite rare (reviewed in (218)). Nevertheless, subsets of sporadic corticotroph, somatotroph and lactotroph PitNETs can harbor activating mutations in GNAS (guanine nucleotide binding protein, alpha stimulating), which result in constitutive activation of cyclic AMP signaling (reviewed in (193)). The tumors can also show AIP (aryl hydrocarbon receptor interacting protein) mutations, whose role in tumorigenesis is still unclear (reviewed in (219)). Furthermore, some GH- or PRL-producing PitNETs carry mutations in MEN1, CDKN1B (cyclin-dependent kinase inhibitor 1B), MAX (MYC associated factor X) or PRKAR1A (protein kinase CAMP-dependent type I regulatory subunit alpha) and ACTH-secreting tumors can harbor USP8 (ubiquitin specific peptidase 8), USP48, DICER1, MLH1 (MutL homolog 1) or MSH2 (MutS homolog 2) mutations. In addition, pituitary stalk hemangioblastomas can harbor VHL (Von Hippel-Lindau tumor suppressor) mutations (reviewed in (193)) (Table 3).
A small subset of PitNETs is also caused by germline mutations associated with familial endocrine syndromes, whose disease-causing loci are identical to those found in sporadic PitNETs (reviewed in (218)) (Table 4). For example, the autosomal dominant inherited MEN types 1, 4 and 5 are caused by heterozygous inactivating mutations in MEN1, CDKN1B and MAX, respectively (reviewed in (193)). Depending on the mutated gene, MEN1, MEN4 and MEN5 patients develop PitNETs in 30%, 42% and 80% of cases, respectively, which are mostly PRL- or GH-releasing tumors. Postzygotic activating mutations in GNAS cause McCune-Albright syndrome that leads to somatotroph/corticotroph PitNETs and pituitary hyperplasia in 6% and 14% of patients, respectively (235). GNAS mutation are also found in GH-secreting PitNETs of patients suffering from the autosomal dominant inherited NF1 syndrome (231). 2% of all PitNETs are caused by the autosomal dominant FIPA syndrome that predisposes to functioning/non-functioning PitNETs (236). 20% of FIPA patients have heterozygote inactivating AIP mutations, whereas the genetic cause in the remaining patients is unknown (193). Patients suffering from the autosomal dominant inherited Carney complex CNC develop pituitary hyperplasia, which results in the formation of lactotroph or somatotroph PitNETs in 20% of the cases (237, 238). The molecular cause is heterozygous inactivating mutations of PRKAR1A or PRKAR1B genes (232) (reviewed in (193)). Moreover, congenital genetic variants of the DICER1 gene cause the extremely rare DICER1 syndrome that predisposes to the formation of pituitary blastoma, ACTH-dependent hypercortisolism and corticotroph PitNETs (233, 234). Furthermore, germline mutations in VHL in the Von-Hippel-Lindau syndrome result in various benign and malignant tumors of the central nervous system including pituitary stalk hemangioblastomas (227). Finally, MLH1 and MSH2 mutations are responsible for Lynch syndrome, which leads to the formation of aggressive ACTH-secreting PitNETs (222, 223).
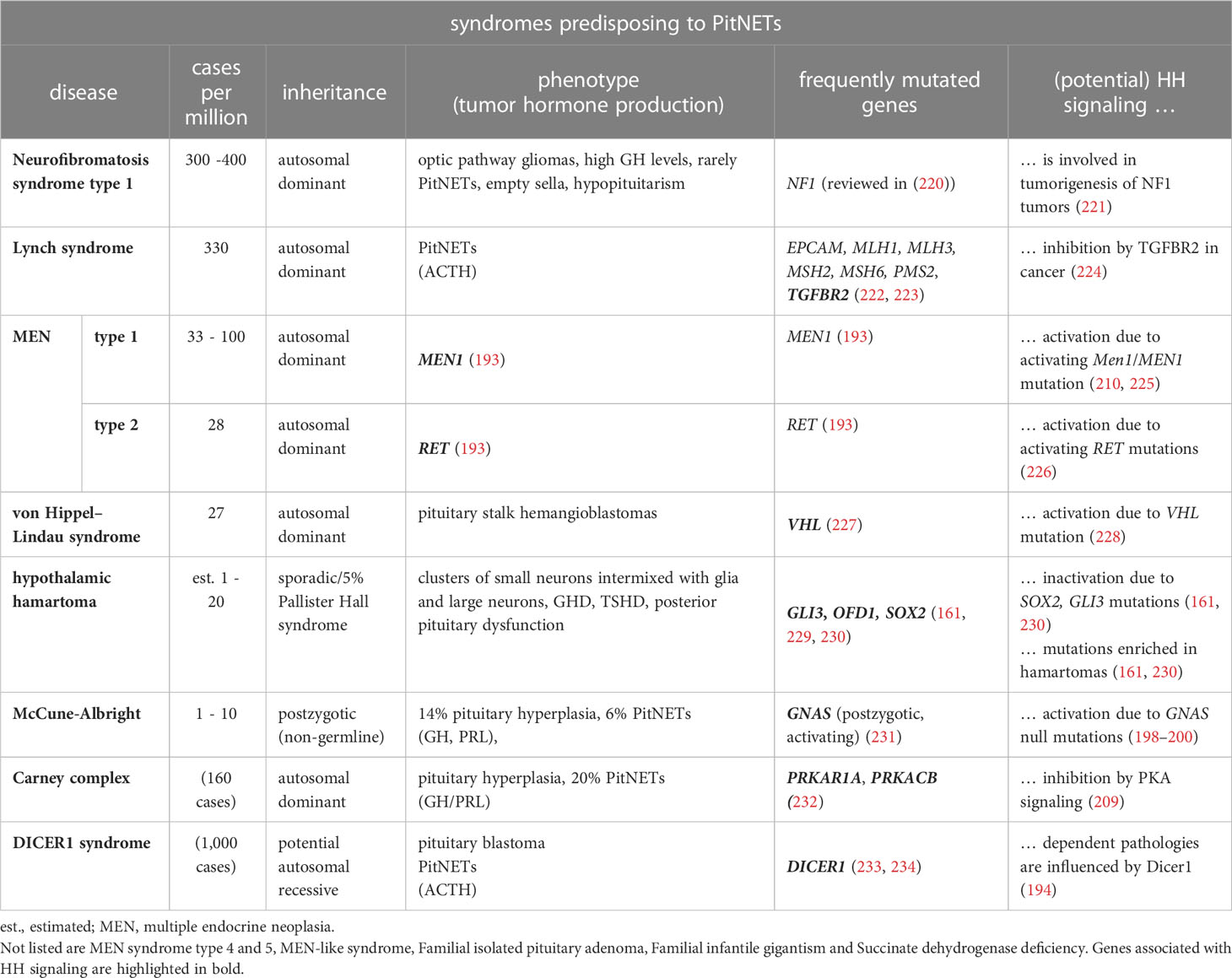
Table 4 Human syndromes predisposing for PitNETs.
Hypothalamic tumors are subdivided into gangliocytoma, neurocytoma and pilocytic astrocytoma (low-grade optic/hypothalamic glioma), whereas PPTs are categorized into pituicytoma, granular cell tumors (GCT), spindle cell oncocytoma (SCO), sellar ependymoma (SE). Other tumors of the sellar region are craniopharyngiomas, germinomas, suprasellar meningiomas, chordoma, chondroma, Rathke’s cleft cysts (RCC) and hamartoma. Except for PitNETs, tumors of the sellar region have different cellular origins such as the craniopharyngeal duct (craniopharyngiomas), or arachnoid cells (meningiomas) or are primordial germ cell remnants (germinomas). Nevertheless, pituicytomas, GCT, SCO and SE, that together account for only 0.5% of all sellar tumors, probably represent a spectrum of a unique neuropathological entity that most likely originate from pituicytes of the neurohypophysis (183). Whereas hypothalamic endocrine tumors can lead to acromegaly, Cushing disease or precocious puberty due to excess GHRH, CRH or GnRH/PRL production, respectively (reviewed in (239)), PPTs as well as all other tumors of the sellar region are hormonally inactive. With exception of the malignant chordoma and chondroma, hypothalamic tumor as well as PTTs are benign neoplasms (Table 2).
4.2.2 HH signaling in PitNETs
Several of the genetic variants in sporadic and syndrome-associated PitNETs are linked to HH signaling (Tables 3, 4). GLI1 is known to inhibit the mismatch repair (MMR) pathway (reviewed in (240)) whose members MLH1 and MSH2 are mutated in sporadic corticotroph PitNETs (193) and also are the reason for Lynch syndrome that is associated with aggressive ACTH-secreting PitNETs (222, 223). Moreover, USP8 (205, 241) and USP48, which are mutated in corticotroph PitNETs (193), are positive regulators of HH signaling activity (203, 204). USP8 prevents SMO ubiquitylation and increases HH signaling activity by promoting SHH-induced cell surface accumulation of SMO (205, 242), whereas USP48 interacts with GLI1 in the nucleus and thereby protects GLI1 from proteasome-dependent degradation. The resulting GLI1 protein stabilization enhances the proliferation and colony formation ability of glioma cells in vitro. Additionally, active HH signaling induces USP48 expression via a positive feedback loop by GLI1 binding to the USP48 promoter (203).
In contrast, GNAS, MEN1, PRKAR1A and DICER1, which are mutated in sporadic PitNETs (Table 3) and in familiar diseases predisposing for PitNETs (Table 4), are known HH signaling repressors (198–200, 209, 210, 225). Thus, both GNAS and DICER1 act as tumor suppressors in HH-dependent medulloblastoma formation in mice (194, 200). Similarly, VHL, which is mutated in pituitary stalk hemangioblastoma of VHL patients, decreases GLI1-mediated promoter transactivation and thus inhibits expression of HH signaling target genes (228).
Together, these data suggest a functional role of HH signaling in PitNETs. However, the observation that USP8 and USP48 rather positively regulate HH signaling, whereas GNAS, MEN1, PRKAR1A, DICER1 and VHL rather negatively regulate this signaling pathway in PitNETs, does not allow a definitive conclusion, whether it is active or inactive HH signaling that is involved in formation of these tumors. In fact, the first report about the HH signaling activation status in PitNETs showed that ACTH-secreting Cushing tumors produced low SHH protein, whereas its receptors PTCH1 and PTCH2 were detectable at variable levels (14). Subsequent in vitro analyses revealed that SHH stimulation of corticotroph, somatotroph or lactotroph PitNETs inhibits cell proliferation and elevates secretion of ACTH, GH or PRL, respectively. Thus, it was hypothesized that active HH signaling is necessary for hormone production, whereas loss of HH signaling activity results in tumor formation in the adult pituitary (14). However, if this hypothesis turns out to be true, PitNETs would be one of the rare tumor entities that depend on an inactive HH signaling cascade for their development or at least for their maintenance, as reported for colorectal cancer (243). In addition, this hypothesis contradicts two observations: firstly, it interferes with the growth-activating role of HH signaling during pituitary development (78, 156, 170, 244, 245); secondly, this assumption would imply that PitNETs need active HH signaling for hormone production. However, this is obviously not the case since Cushing tumors are generally SHH negative (14). In fact, a growth-activating role of HH signaling in PitNETs is underlined by a study by Lampichler et al., which demonstrates that GLI1, as a major indicator for active HH signaling, is present in 87% of all PitNETs and significantly correlates with the expression of the stem cell marker SOX2 and the proliferation marker MKI67 (246). Moreover, purmorphamine-induced HH signaling activation increases cell viability of AtT-20 cells, whereas inhibition of GLI1 results in cell death (246). Since GLI1/GLI1 expression was found in almost all human pituitary tumors studied, whereas SHH mRNA was present in few tumors samples (246) (see also (14)), the authors hypothesized that GLI1 expression in pituitary tumors is stimulated in a ligand-independent way and plays an important role in tumorigenesis, while the reduction in SHH expression is likely a compensatory mechanism.
Contrarily, we found that almost all PitNETs express SHH/SHH, which strongly and significantly correlates with GLI1 mRNA expression levels. Remarkably, this is identical with classical HH-associated malignancies, which include the highly aggressive medulloblastoma and the benign basal cell carcinoma (see above). Since ACTH-, GH- and PRL-immunopositive PitNETs show the highest SHH and GLI1 levels, we propose that HH signaling activity may play a role in formation and/or maintenance of these pituitary tumor subtypes (11) as it is known for classical HH-associated tumors. Similarily, Yavropoulou et al. found higher PTCH1, PTCH2, GLI1 and GLI3 levels in somatotroph PitNETs compared to normal pituitary samples. However, they did not observe any hint for HH signaling activity in PRL-secreting PitNETs and NFPA, which even showed lower PTCH1, PTCH2, GLI1, GLI3 and HHIP levels than control tissue (247). All these studies do not answer whether the expression level of HH-related genes is pathogenetically associated with the development of the repective PitNET or simply reflects a compensation mechanism for tumor growth initiated by other factors (247). Nevertheless, several recent publications support the assumption that active HH signaling plays a central role in PitNETs. For example, functional analysis of differentially expressed genes between PitNETs and normal pituitaries identified the HH signaling pathway as one of the three pathways significantly enriched in the tumors (248). In addition, PitNETs with a proliferation MKI76-index above 3% express significantly higher GLI1/GLI1 levels than tumors with a proliferation index below 3% or normal pituitaries (249). In line with this observation, activation of HH signaling also induces the proliferation of in vitro cultured PitNETs cells, whereas inhibition of the pathway inhibits it (249). Moreover, recent single cell transcriptomes of 21 PitNETs compared to 3 normal pituitary glands show that the SOX2+ stem-like cell compartment of PitNETs (tumor stem cells) is enriched for the expression of genes involved in HH signaling and expresses higher GLI1 and GLI2 levels (250).
Together, these functional analyses of recent years strongly reinforce a tumor-supportive role of HH signaling in PitNETs. However, additional studies are needed to substantiate this assumption and to test therapeutic possibilities by targeting the HH signaling pathway in this tumor entity.
4.2.3 HH signaling in hypothalamic endocrine tumors and PPTs
In terms of the genetic landscape of sporadic hypothalamic tumors and PPTs, the majority of the tumors show activating mutations in the RAS/RAF/MEK/ERK signaling pathway. For example, gangliocytoma show mutations in BRAF, KRAS, RAF1, NF1 (neurofibromin 1) and CDKN2A (cyclin dependent kinase inhibitor 2A) (251) and tumors of the glia in FGFR, HRAS, BRAF and NF1 (100). Interestingly, hypothalamic tumors or PPTs also harbor mutations in HH signaling components. Recently, a PTCH1 variant has been described in pituicytoma (101). Loss of PTCH1 together with copy gains of SHH, SMO and GLI3 have been reported in 1/6 germinomas (105). Additionally, some chordoma show activating mutations in HH signaling components (115). These studies indicate that HH signaling may also play a role in tumorigenesis in some of these tumor entities. Indeed, histological examination of hypothalamic tumors or PPTs also reveal HH signaling activity in several of these neoplasms. GAB1 (GRB2 associated binding protein 1) that can serve as a histological read-out for active HH signaling (252), is upregulated in malignant neurocytoma (97). Moreover, PTCH1 and GLI1 expression are elevated in 45% of analyzed pilocytic astrocytoma (99) and a strong nuclear GLI2 signal, indicative for active HH signaling, has been detected in a case of SCO (102). Similarly, active HH signaling may be present in SE, since ependymoma in general overexpress GLI1 and GLI2 (103). Active HH signaling has also been detected in chordoma and adamantinomatous craniopharyngioma (ACP) (104, 113, 115, 118, 119, 253–256). Surprisingly, HH signaling apparently has anti-tumorigenic properties in the latter tumor entity. Thus, inhibition of HH signaling increases the proliferation of human APC cells and decreases the median survival of APC-bearing mice due to premature development of highly proliferative and vascularized undifferentiated tumors (257). Similarly, hypothalamic hamartomas, that are clusters of small neurons intermixed with glia and large neurons, often are associated with PHS caused by exclusive GLI3R expression. They also contain SOX2 mutations (161, 229, 258, 259) that lead to inefficient HH signaling (91). This indicates that HH signaling inactivation may not necessarily contribute to the formation this tumor entity (Table 2).
Together, these data suggest that HH signaling in APC and hypothalamic hamartomas has a rather tumor-suppressive function, whereas in hypothalamic endocrine tumors and PPTs it may have an oncogenic function. However, HH signaling activity in gangliocytoma, pilocytic astrocytoma (low-grade optic/hypothalamic glioma), GCT, suprasellar meningioma or RCC has not been reported so far. It will be highly interesting to see whether HH signaling also plays a functional role in these tumor entities.
5 Conclusions and future directions
Research of the last twenty years has greatly improved our knowledge about the impact of HH signaling in the embryonic and adult pituitary. Animal experiments show that cells in the developing pituitary are competent to receive and respond to HH signaling, and that HH signaling promotes patterning, cell proliferation and terminal differentiation during pituitary organogenesis. In line with these observations, aberrant HH signaling activity is frequently found in human developmental disorders associated with pituitary malformations or hormone deficiencies, indicating that the HH signaling cascade plays an important role during pituitary development in humans as well. Very recent data further suggest that HH signaling is also active in adulthood and regulates pituitary homeostasis and hormone production. In addition, HH signaling seems to have a tumor-supportive role for PitNETs and PPTs. However, additional studies are needed to support these findings and to test whether HH signaling may be an intervention target for treatment of pituitary diseases.
So far, a large number of HH signaling pathway inhibitors have been developed and have entered clinical trials as anti-cancer agents. To date, two SMO inhibitors (i.e. vismodegib, sonidegib) have been approved by the US Food and Drug Administration (FDA) and by the European Medicines Agency (EMA) for treating locally advanced basal cell carcinoma. Both drugs are currently tested in various clinical trials for other tumor entities either as a single agent or in combination with other anticancer drugs (reviewed in (17)). Thus, vismodegib or sonidegib are currently tested in combination with either the focal adhesion kinase inhibitor GSK2256098, the AKT-inhibitor capivasertib or the CDK4/6-inhibitor abemaciclib in the treatment of meningioma (NCT02523014). They are also used in combination with gemcitabine, the CDK4/6-inhibitor ribociclib, the BRAF-inhibitor trametinib or the cytokine filgrastim for treatment of brain tumors (like astrocytoma, meningioma, ependymoma) in children and young adults (NCT03434262). However, to date treatments of pituitary neoplasms with HH signaling inhibitors have been only reported in mouse models. For example, Ding et al. showed that inhibition of HH signaling by vismodegib blocks tumor growth of xenotransplanted sterol carrier protein 2-overexpressing GH3 cells (249). Currently, dependent on their tumor size and hormone production, PitNETs are subjected to surgery and/or radiation therapy. Medications can help to block excess hormone secretion. Thus, corticotroph PitNETs that cause Cushing syndrome are treated with drugs controlling cortisol production in the adrenal gland such as ketoconazole, mitotane and metyrapone. Somatotroph or lactotroph PitNETs are treated with somatostatin analogs or with the dopamine agonists cabergoline and bromocriptine, respectively (reviewed in (260)). Although most of pituitary tumors are benign, some of them show an aggressive behavior and are either locally invasive, rapidly growing or even metastasizing (189, 261). In addition, some PitNETs recidivate (262–264). For these tumors, the therapy is rather experimental and definitely needs improvement (260). Thus, there is hope that a better understanding of the role of HH signaling in the pituitary will contribute to better treatment options in case of disease.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
The work was funded by financial support of the University Medical Center Göttingen, Göttingen, Germany and the Deutsche Forschungsgemeinschaft (UH228/6-3).
Acknowledgments
The authors acknowledge BioRender, which was used for creating the Figures 1–3 and the support by the Open Access Publication Funds of the Göttingen University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in drosophila. Nature (1980) 287(5785):795–801. doi: 10.1038/287795a0
2. Hooper JE. Distinct pathways for autocrine and paracrine wingless signalling in drosophila embryos. Nature (1994) 372(6505):461–4. doi: 10.1038/372461a0
3. Preat T. Characterization of suppressor of fused, a complete suppressor of the fused segment polarity gene of drosophila melanogaster. Genetics (1992) 132(3):725–36. doi: 10.1093/genetics/132.3.725
4. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell (2008) 15(6):801–12. doi: 10.1016/j.devcel.2008.11.010
5. Treier M, Gleiberman AS, O'Connell SM, Szeto DP, McMahon JA, McMahon AP, et al. Multistep signaling requirements for pituitary organogenesis in vivo. Genes Dev (1998) 12(11):1691–704. doi: 10.1101/gad.12.11.1691
6. Treier M, O'Connell S, Gleiberman A, Price J, Szeto DP, Burgess R, et al. Hedgehog signaling is required for pituitary gland development. Development (2001) 128(3):377–86. doi: 10.1242/dev.128.3.377
7. Carreno G, Apps JR, Lodge EJ, Panousopoulos L, Haston S, Gonzalez-Meljem JM, et al. Hypothalamic sonic hedgehog is required for cell specification and proliferation of Lhx3/Lhx4 pituitary embryonic precursors. Development (2017) 144(18):3289–302. doi: 10.1242/dev.153387
8. Scully KM, Rosenfeld MG. Pituitary development: regulatory codes in mammalian organogenesis. Science (2002) 295(5563):2231–5. doi: 10.1126/science.1062736
9. Perez-Castro C, Renner U, Haedo MR, Stalla GK, Arzt E. Cellular and molecular specificity of pituitary gland physiology. Physiol Rev (2012) 92(1):1–38. doi: 10.1152/physrev.00003.2011
10. Zhao L, Zevallos SE, Rizzoti K, Jeong Y, Lovell-Badge R, Epstein DJ. Disruption of Soxb1-dependent sonic hedgehog expression in the hypothalamus causes septo-optic dysplasia. Dev Cell (2012) 22(3):585–96. doi: 10.1016/j.devcel.2011.12.023
11. Pyczek J, Buslei R, Schult D, Holsken A, Buchfelder M, Hess I, et al. Hedgehog signaling activation induces stem cell proliferation and hormone release in the adult pituitary gland. Sci Rep (2016) 6:24928. doi: 10.1038/srep24928
12. Botermann DS, Brandes N, Frommhold A, Hess I, Wolff A, Zibat A, et al. Hedgehog signaling in endocrine and folliculo-stellate cells of the adult pituitary. J Endocrinol (2021) 248(3):303–16. doi: 10.1530/JOE-20-0388
13. Vila G, Papazoglou M, Stalla J, Theodoropoulou M, Stalla GK, Holsboer F, et al. Sonic hedgehog regulates crh signal transduction in the adult pituitary. FASEB J (2005) 19(2):281–3. doi: 10.1096/fj.04-2138fje
14. Vila G, Theodoropoulou M, Stalla J, Tonn JC, Losa M, Renner U, et al. Expression and function of sonic hedgehog pathway components in pituitary adenomas: evidence for a direct role in hormone secretion and cell proliferation. J Clin Endocrinol Metab (2005) 90(12):6687–94. doi: 10.1210/jc.2005-1014
15. Ren YA, Monkkonen T, Lewis MT, Bernard DJ, Christian HC, Jorgez CJ, et al. S100a4-Cre-Mediated deletion of Patched1 causes hypogonadotropic hypogonadism: role of pituitary hematopoietic cells in endocrine regulation. JCI Insight (2019) 5(14). doi: 10.1172/jci.insight.126325
16. Andoniadou CL, Matsushima D, Mousavy Gharavy SN, Signore M, Mackintosh AI, Schaeffer M, et al. Sox2(+) Stem/Progenitor cells in the adult mouse pituitary support organ homeostasis and have tumor-inducing potential. Cell Stem Cell (2013) 13(4):433–45. doi: 10.1016/j.stem.2013.07.004
17. Xin M, Ji X, de la Cruz LK, Thareja S, Wang B. Strategies to target the hedgehog signaling pathway for cancer therapy. Med Res Rev (2018) 38(3):870–913. doi: 10.1002/med.21482
18. Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science (2007) 317(5836):372–6. doi: 10.1126/science.1139740
19. Kaushal JB, Batra SK, Rachagani S. Hedgehog signaling and its molecular perspective with cholesterol: a comprehensive review. Cell Mol Life Sci (2022) 79(5):266. doi: 10.1007/s00018-022-04233-1
20. Briscoe J, Therond PP. The mechanisms of hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol (2013) 14(7):416–29. doi: 10.1038/nrm3598
21. Chamoun Z, Mann RK, Nellen D, von Kessler DP, Bellotto M, Beachy PA, et al. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science (2001) 293(5537):2080–4. doi: 10.1126/science.1064437
22. Coupland CE, Andrei SA, Ansell TB, Carrique L, Kumar P, Sefer L, et al. Structure, mechanism, and inhibition of hedgehog acyltransferase. Mol Cell (2021) 81(24):5025–38.e10. doi: 10.1016/j.molcel.2021.11.018
23. Tukachinsky H, Kuzmickas RP, Jao CY, Liu J, Salic A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep (2012) 2(2):308–20. doi: 10.1016/j.celrep.2012.07.010
24. Griffiths SC, Schwab RA, El Omari K, Bishop B, Iverson EJ, Malinauskas T, et al. Hedgehog-interacting protein is a multimodal antagonist of hedgehog signalling. Nat Commun (2021) 12(1):7171. doi: 10.1038/s41467-021-27475-2
25. Hall ET, Dillard ME, Stewart DP, Zhang Y, Wagner B, Levine RM, et al. Cytoneme delivery of sonic hedgehog from ligand-producing cells requires myosin 10 and a dispatched-Boc/Cdon Co-receptor complex. Elife (2021) 10. doi: 10.7554/eLife.61432
26. Daly CA, Hall ET, Ogden SK. Regulatory mechanisms of cytoneme-based morphogen transport. Cell Mol Life Sci (2022) 79(2):119. doi: 10.1007/s00018-022-04148-x
27. Allen BL, Tenzen T, McMahon AP. The hedgehog-binding proteins Gas1 and cdo cooperate to positively regulate shh signaling during mouse development. Genes Dev (2007) 21(10):1244–57. doi: 10.1101/gad.1543607
28. Lee CS, Buttitta L, Fan CM. Evidence that the wnt-inducible growth arrest-specific gene 1 encodes an antagonist of sonic hedgehog signaling in the somite. Proc Natl Acad Sci USA (2001) 98(20):11347–52. doi: 10.1073/pnas.201418298
29. Martinelli DC, Fan CM. Gas1 extends the range of hedgehog action by facilitating its signaling. Genes Dev (2007) 21(10):1231–43. doi: 10.1101/gad.1546307
30. Chen MH, Li YJ, Kawakami T, Xu SM, Chuang PT. Palmitoylation is required for the production of a soluble multimeric hedgehog protein complex and long-range signaling in vertebrates. Genes Dev (2004) 18(6):641–59. doi: 10.1101/gad.1185804
31. Gallet A, Ruel L, Staccini-Lavenant L, Therond PP. Cholesterol modification is necessary for controlled planar long-range activity of hedgehog in drosophila epithelia. Development (2006) 133(3):407–18. doi: 10.1242/dev.02212
32. Eugster C, Panakova D, Mahmoud A, Eaton S. Lipoprotein-heparan sulfate interactions in the hh pathway. Dev Cell (2007) 13(1):57–71. doi: 10.1016/j.devcel.2007.04.019
33. Panakova D, Sprong H, Marois E, Thiele C, Eaton S. Lipoprotein particles are required for hedgehog and wingless signalling. Nature (2005) 435(7038):58–65. doi: 10.1038/nature03504
34. Therond PP. Release and transportation of hedgehog molecules. Curr Opin Cell Biol (2012) 24(2):173–80. doi: 10.1016/j.ceb.2012.02.001
35. Bae GU, Domene S, Roessler E, Schachter K, Kang JS, Muenke M, et al. Mutations in cdon, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am J Hum Genet (2011) 89(2):231–40. doi: 10.1016/j.ajhg.2011.07.001
36. Izzi L, Levesque M, Morin S, Laniel D, Wilkes BC, Mille F, et al. Boc and Gas1 each form distinct shh receptor complexes with Ptch1 and are required for shh-mediated cell proliferation. Dev Cell (2011) 20(6):788–801. doi: 10.1016/j.devcel.2011.04.017
37. Ho EK, Stearns T. Hedgehog signaling and the primary cilium: implications for spatial and temporal constraints on signaling. Development (2021) 148(9). doi: 10.1242/dev.195552
38. Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate smoothened functions at the primary cilium. Nature (2005) 437(7061):1018–21. doi: 10.1038/nature04117
39. Kinnebrew M, Iverson EJ, Patel BB, Pusapati GV, Kong JH, Johnson KA, et al. Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. Elife (2019) 8. doi: 10.7554/eLife.50051
40. Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol (2006) 26(9):3365–77. doi: 10.1128/MCB.26.9.3365-3377.2006
41. Buttitta L, Mo R, Hui CC, Fan CM. Interplays of Gli2 and Gli3 and their requirement in mediating shh-dependent sclerotome induction. Development (2003) 130(25):6233–43. doi: 10.1242/dev.00851
42. Bai CB, Stephen D, Joyner AL. All mouse ventral spinal cord patterning by hedgehog is gli dependent and involves an activator function of Gli3. Dev Cell (2004) 6(1):103–15. doi: 10.1016/s1534-5807(03)00394-0
43. Sigafoos AN, Paradise BD, Fernandez-Zapico ME. Hedgehog/Gli signaling pathway: transduction, regulation, and implications for disease. Cancers (Basel) (2021) 13(14). doi: 10.3390/cancers13143410
44. Brennan D, Chen X, Cheng L, Mahoney M, Riobo NA. Noncanonical hedgehog signaling. Vitam Horm (2012) 88:55–72. doi: 10.1016/B978-0-12-394622-5.00003-1
45. Aberger F, Kern D, Greil R, Hartmann TN. Canonical and noncanonical Hedgehog/Gli signaling in hematological malignancies. Vitam Horm (2012) 88:25–54. doi: 10.1016/B978-0-12-394622-5.00002-X
46. Teperino R, Aberger F, Esterbauer H, Riobo N, Pospisilik JA. Canonical and non-canonical hedgehog signalling and the control of metabolism. Semin Cell Dev Biol (2014) 33:81–92. doi: 10.1016/j.semcdb.2014.05.007
47. Pietrobono S, Gagliardi S, Stecca B. Non-canonical hedgehog signaling pathway in cancer: activation of gli transcription factors beyond smoothened. Front Genet (2019) 10:556. doi: 10.3389/fgene.2019.00556
48. Riobo NA, Haines GM, Emerson CP Jr. Protein kinase c-delta and mitogen-activated Protein/Extracellular signal-regulated kinase-1 control gli activation in hedgehog signaling. Cancer Res (2006) 66(2):839–45. doi: 10.1158/0008-5472.CAN-05-2539
49. Riobo NA, Lu K, Ai X, Haines GM, Emerson CP Jr. Phosphoinositide 3-kinase and akt are essential for sonic hedgehog signaling. Proc Natl Acad Sci USA (2006) 103(12):4505–10. doi: 10.1073/pnas.0504337103
50. Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require hedgehog-gli signaling regulated by interactions between Gli1 and the ras-Mek/Akt pathways. Proc Natl Acad Sci USA (2007) 104(14):5895–900. doi: 10.1073/pnas.0700776104
51. Ji Z, Mei FC, Johnson BH, Thompson EB, Cheng X. Protein kinase a, not epac, suppresses hedgehog activity and regulates glucocorticoid sensitivity in acute lymphoblastic leukemia cells. J Biol Chem (2007) 282(52):37370–7. doi: 10.1074/jbc.M703697200
52. Ji Z, Mei FC, Xie J, Cheng X. Oncogenic kras activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem (2007) 282(19):14048–55. doi: 10.1074/jbc.M611089200
53. Lauth M, Bergstrom A, Shimokawa T, Tostar U, Jin Q, Fendrich V, et al. Dyrk1b-dependent autocrine-to-Paracrine shift of hedgehog signaling by mutant ras. Nat Struct Mol Biol (2010) 17(6):718–25. doi: 10.1038/nsmb.1833
54. Bauer J, Cuvelier N, Ragab N, Simon-Keller K, Nitzki F, Geyer N, et al. Context-dependent modulation of aggressiveness of pediatric tumors by individual oncogenic ras isoforms. Oncogene (2021) 40(31):4955–66. doi: 10.1038/s41388-021-01904-4
55. Noubissi FK, Goswami S, Sanek NA, Kawakami K, Minamoto T, Moser A, et al. Wnt signaling stimulates transcriptional outcome of the hedgehog pathway by stabilizing Gli1 mrna. Cancer Res (2009) 69(22):8572–8. doi: 10.1158/0008-5472.CAN-09-1500
56. Nakamura I, Fernandez-Barrena MG, Ortiz-Ruiz MC, Almada LL, Hu C, Elsawa SF, et al. Activation of the transcription factor Gli1 by wnt signaling underlies the role of sulfatase 2 as a regulator of tissue regeneration. J Biol Chem (2013) 288(29):21389–98. doi: 10.1074/jbc.M112.443440
57. Zinke J, Schneider FT, Harter PN, Thom S, Ziegler N, Toftgard R, et al. Beta-Catenin-Gli1 interaction regulates proliferation and tumor growth in medulloblastoma. Mol Cancer (2015) 14(1):17. doi: 10.1186/s12943-015-0294-4
58. Pantazi E, Gemenetzidis E, Teh MT, Reddy SV, Warnes G, Evagora C, et al. Gli2 is a regulator of beta-catenin and is associated with loss of e-cadherin, cell invasiveness, and long-term epidermal regeneration. J Invest Dermatol (2017) 137(8):1719–30. doi: 10.1016/j.jid.2016.11.046
59. Ulloa F, Itasaki N, Briscoe J. Inhibitory Gli3 activity negatively regulates Wnt/Beta-catenin signaling. Curr Biol (2007) 17(6):545–50. doi: 10.1016/j.cub.2007.01.062
60. Song L, Li ZY, Liu WP, Zhao MR. Crosstalk between Wnt/Beta-catenin and Hedgehog/Gli signaling pathways in colon cancer and implications for therapy. Cancer Biol Ther (2015) 16(1):1–7. doi: 10.4161/15384047.2014.972215
61. Meng X, Poon R, Zhang X, Cheah A, Ding Q, Hui CC, et al. Suppressor of fused negatively regulates beta-catenin signaling. J Biol Chem (2001) 276(43):40113–9. doi: 10.1074/jbc.M105317200
62. Larkin S, Ansorge O. Development and microscopic anatomy of the pituitary gland. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, editors. Endotext. South Dartmouth (MA): MDText.com, Inc. (2000).
63. Lechan RM, Toni R. Functional anatomy of the hypothalamus and pituitary. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, editors. Endotext. South Dartmouth (MA): MDText.com, Inc (2000).
64. Kelberman D, Rizzoti K, Lovell-Badge R, Robinson IC, Dattani MT. Genetic regulation of pituitary gland development in human and mouse. Endocr Rev (2009) 30(7):790–829. doi: 10.1210/er.2009-0008
65. Davis SW, Castinetti F, Carvalho LR, Ellsworth BS, Potok MA, Lyons RH, et al. Molecular mechanisms of pituitary organogenesis: in search of novel regulatory genes. Mol Cell Endocrinol (2010) 323(1):4–19. doi: 10.1016/j.mce.2009.12.012
66. Zhu X, Gleiberman AS, Rosenfeld MG. Molecular physiology of pituitary development: signaling and transcriptional networks. Physiol Rev (2007) 87(3):933–63. doi: 10.1152/physrev.00006.2006
67. Alatzoglou KS, Gregory LC, Dattani MT. Development of the pituitary gland. Compr Physiol (2020) 10(2):389–413. doi: 10.1002/cphy.c150043
68. Christ-Crain M, Ball S. The neurohypophysis: endocrinology of vasopressin and oxytocin. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, editors. Endotext. South Dartmouth (MA): MDText.com, Inc (2000).
69. Antonellis PJ, Engle SE, Brewer KM, Berbari NF. The hedgehog signaling pathway is expressed in the adult mouse hypothalamus and modulated by fasting. eNeuro (2021) 8(5). doi: 10.1523/ENEURO.0276-21.2021
70. Yoo ES, Yu J, Sohn JW. Neuroendocrine control of appetite and metabolism. Exp Mol Med (2021) 53(4):505–16. doi: 10.1038/s12276-021-00597-9
71. Gregory LC, Dattani MT. The molecular basis of congenital hypopituitarism and related disorders. J Clin Endocrinol Metab (2020) 105(6). doi: 10.1210/clinem/dgz184
72. McCabe MJ, Gaston-Massuet C, Tziaferi V, Gregory LC, Alatzoglou KS, Signore M, et al. Novel Fgf8 mutations associated with recessive holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J Clin Endocrinol Metab (2011) 96(10):E1709–18. doi: 10.1210/jc.2011-0454
73. Xatzipsalti M, Voutetakis A, Stamoyannou L, Chrousos GP, Kanaka-Gantenbein C. Congenital hypopituitarism: various genes, various phenotypes. Horm Metab Res (2019) 51(2):81–90. doi: 10.1055/a-0822-3637
74. Reis LM, Seese S, Maheshwari M, Basel D, Weik L, McCarrier J, et al. Novel genetic diagnoses in septo-optic dysplasia. Genes (Basel) (2022) 13(7). doi: 10.3390/genes13071165
75. Bosch IAL, Katugampola H, Dattani MT. Congenital hypopituitarism during the neonatal period: epidemiology, pathogenesis, therapeutic options, and outcome. Front Pediatr (2020) 8:600962. doi: 10.3389/fped.2020.600962
76. Muenke M, Cohen MM Jr. Genetic approaches to understanding brain development: holoprosencephaly as a model. Ment Retard Dev Disabil Res Rev (2000) 6(1):15–21. doi: 10.1002/(SICI)1098-2779(2000)6:1<15::AID-MRDD3>3.0.CO;2-8
77. Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, et al. Mutations in patched-1, the receptor for sonic hedgehog, are associated with holoprosencephaly. Hum Genet (2002) 110(4):297–301. doi: 10.1007/s00439-002-0695-5
78. Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, et al. Loss-of-Function mutations in the human Gli2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci USA (2003) 100(23):13424–9. doi: 10.1073/pnas.2235734100
79. Roessler E, Ma Y, Ouspenskaia MV, Lacbawan F, Bendavid C, Dubourg C, et al. Truncating loss-of-Function mutations of Disp1 contribute to holoprosencephaly-like microform features in humans. Hum Genet (2009) 125(4):393–400. doi: 10.1007/s00439-009-0628-7
80. Pineda-Alvarez DE, Roessler E, Hu P, Srivastava K, Solomon BD, Siple CE, et al. Missense substitutions in the Gas1 protein present in holoprosencephaly patients reduce the affinity for its ligand, shh. Hum Genet (2012) 131(2):301–10. doi: 10.1007/s00439-011-1078-6
81. Belloni E, Muenke M, Roessler E, Traverso G, Siegel-Bartelt J, Frumkin A, et al. Identification of sonic hedgehog as a candidate gene responsible for holoprosencephaly. Nat Genet (1996) 14(3):353–6. doi: 10.1038/ng1196-353
82. Roessler E, Belloni E, Gaudenz K, Vargas F, Scherer SW, Tsui LC, et al. Mutations in the c-terminal domain of sonic hedgehog cause holoprosencephaly. Hum Mol Genet (1997) 6(11):1847–53. doi: 10.1093/hmg/6.11.1847
83. Roessler E, Ward DE, Gaudenz K, Belloni E, Scherer SW, Donnai D, et al. Cytogenetic rearrangements involving the loss of the sonic hedgehog gene at 7q36 cause holoprosencephaly. Hum Genet (1997) 100(2):172–81. doi: 10.1007/s004390050486
84. Gurrieri F, Trask BJ, van den Engh G, Krauss CM, Schinzel A, Pettenati MJ, et al. Physical mapping of the holoprosencephaly critical region on chromosome 7q36. Nat Genet (1993) 3(3):247–51. doi: 10.1038/ng0393-247
85. Franca MM, Jorge AA, Carvalho LR, Costalonga EF, Otto AP, Correa FA, et al. Relatively high frequency of non-synonymous Gli2 variants in patients with congenital hypopituitarism without holoprosencephaly. Clin Endocrinol (Oxf) (2013) 78(4):551–7. doi: 10.1111/cen.12044
86. Fang X, Zhang Y, Cai J, Lu T, Hu J, Yuan F, et al. Identification of novel candidate pathogenic genes in pituitary stalk interruption syndrome by whole-exome sequencing. J Cell Mol Med (2020) 24(20):11703–17. doi: 10.1111/jcmm.15781
87. Kang S, Graham JM Jr., Olney AH, Biesecker LG. Gli3 frameshift mutations cause autosomal dominant pallister-hall syndrome. Nat Genet (1997) 15(3):266–8. doi: 10.1038/ng0397-266
88. Kim YJ, Osborn DP, Lee JY, Araki M, Araki K, Mohun T, et al. Wdr11-mediated hedgehog signalling defects underlie a new ciliopathy related to kallmann syndrome. EMBO Rep (2018) 19(2):269–89. doi: 10.15252/embr.201744632
89. Kim HG, Ahn JW, Kurth I, Ullmann R, Kim HT, Kulharya A, et al. Wdr11, a wd protein that interacts with transcription factor Emx1, is mutated in idiopathic hypogonadotropic hypogonadism and kallmann syndrome. Am J Hum Genet (2010) 87(4):465–79. doi: 10.1016/j.ajhg.2010.08.018
90. Vissers LE, van Ravenswaaij CM, Admiraal R, Hurst JA, de Vries BB, Janssen IM, et al. Mutations in a new member of the chromodomain gene family cause charge syndrome. Nat Genet (2004) 36(9):955–7. doi: 10.1038/ng1407
91. Engelen E, Akinci U, Bryne JC, Hou J, Gontan C, Moen M, et al. Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat Genet (2011) 43(6):607–11. doi: 10.1038/ng.825
92. Inoue H, Tanizawa Y, Wasson J, Behn P, Kalidas K, Bernal-Mizrachi E, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet (1998) 20(2):143–8. doi: 10.1038/2441
93. Zmyslowska A, Kuljanin M, Malachowska B, Stanczak M, Michalek D, Wlodarczyk A, et al. Multiomic analysis on human cell model of wolfram syndrome reveals changes in mitochondrial morphology and function. Cell Commun Signal (2021) 19(1):116. doi: 10.1186/s12964-021-00791-2
94. Srirangam Nadhamuni V, Korbonits M. Novel insights into pituitary tumorigenesis: genetic and epigenetic mechanisms. Endocrine Rev (2020) 41(6):821–46. doi: 10.1210/endrev/bnaa006
95. Zhang D, Yong WH, Movassaghi M, Rodriguez FJ, Yang I, McKeever P, et al. Whole exome sequencing identifies Phf14 mutations in neurocytoma and predicts responsivity to the pdgfr inhibitor sunitinib. Biomedicines (2022) 10(11):2842. doi: 10.3390/biomedicines10112842
96. Clement V, Sanchez P, de Tribolet N, Radovanovic I, Ruiz i Altaba A. Hedgehog-Gli1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr Biol (2007) 17(2):165–72. doi: 10.1016/j.cub.2006.11.033
97. Sugita Y, Furuta T, Komaki S, Ohshima K, Sakata K, Morioka M. Malignant progression of an extraventricular neurocytoma arising from the viiith cranial nerve: a case report and literature review. Neuropathology (2019) 39(2):120–6. doi: 10.1111/neup.12533
98. Ryall S, Tabori U, Hawkins C. Pediatric low-grade glioma in the era of molecular diagnostics. Acta Neuropathol Commun (2020) 8(1):30. doi: 10.1186/s40478-020-00902-z
99. Rush SZ, Abel TW, Valadez JG, Pearson M, Cooper MK. Activation of the hedgehog pathway in pilocytic astrocytomas. Neuro Oncol (2010) 12(8):790–8. doi: 10.1093/neuonc/noq026
100. Schmid S, Solomon DA, Perez E, Thieme A, Kleinschmidt-DeMasters BK, Giannini C, et al. Genetic and epigenetic characterization of posterior pituitary tumors. Acta Neuropathol (2021) 142(6):1025–43. doi: 10.1007/s00401-021-02377-1
101. Viaene AN, Lee EB, Rosenbaum JN, Nasrallah IM, Nasrallah MP. Histologic, immunohistochemical, and molecular features of pituicytomas and atypical pituicytomas. Acta Neuropathol Commun (2019) 7(1):69. doi: 10.1186/s40478-019-0722-6
102. Alexandrescu S, Brown RE, Tandon N, Bhattacharjee MB. Neuron precursor features of spindle cell oncocytoma of adenohypophysis. Ann Clin Lab Sci (2012) 42(2):123–9.
103. Modena P, Lualdi E, Facchinetti F, Veltman J, Reid JF, Minardi S, et al. Identification of tumor-specific molecular signatures in intracranial ependymoma and association with clinical characteristics. J Clin Oncol (2006) 24(33):5223–33. doi: 10.1200/JCO.2006.06.3701
104. Andoniadou CL, Gaston-Massuet C, Reddy R, Schneider RP, Blasco MA, Le Tissier P, et al. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol (2012) 124(2):259–71. doi: 10.1007/s00401-012-0957-9
105. Kuleszo D, Koczkowska M, Lipska-Zietkiewicz BS, Grajkowska W, Adamkiewicz-Drozynska E, Dembowska-Baginska B, et al. Comparative genomic analysis of intracranial germ cell tumors - the preliminary study focused on sonic hedgehog signaling pathway. Contemp Oncol (Pozn) (2017) 21(4):279–84. doi: 10.5114/wo.2017.72390
106. Fukushima S, Otsuka A, Suzuki T, Yanagisawa T, Mishima K, Mukasa A, et al. Mutually exclusive mutations of kit and ras are associated with kit mrna expression and chromosomal instability in primary intracranial pure germinomas. Acta Neuropathol (2014) 127(6):911–25. doi: 10.1007/s00401-014-1247-5
107. Wang L, Yamaguchi S, Burstein MD, Terashima K, Chang K, Ng HK, et al. Novel somatic and germline mutations in intracranial germ cell tumours. Nature (2014) 511(7508):241–5. doi: 10.1038/nature13296
108. Kubota Y, Seki M, Kawai T, Isobe T, Yoshida M, Sekiguchi M, et al. Comprehensive genetic analysis of pediatric germ cell tumors identifies potential drug targets. Commun Biol (2020) 3(1):544. doi: 10.1038/s42003-020-01267-8
109. Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol (2016) 33(4):237–47. doi: 10.1007/s10014-016-0271-7
110. Jungwirth G, Warta R, Beynon C, Sahm F, von Deimling A, Unterberg A, et al. Intraventricular meningiomas frequently harbor Nf2 mutations but lack common genetic alterations in Traf7, Akt1, smo, Klf4, Pik3ca, and tert. Acta Neuropathol Commun (2019) 7(1):140. doi: 10.1186/s40478-019-0793-4
111. Larizza L, Mortini P, Riva P. Update on the cytogenetics and molecular genetics of chordoma. Hereditary Cancer Clin Pract (2005) 3(1):29. doi: 10.1186/1897-4287-3-1-29
112. Bai J, Shi J, Li C, Wang S, Zhang T, Hua X, et al. Whole genome sequencing of skull-base chordoma reveals genomic alterations associated with recurrence and chordoma-specific survival. Nat Commun (2021) 12(1):757. doi: 10.1038/s41467-021-21026-5
113. Akhavan-Sigari R, Schulz-Schaeffer W, Angelika Harcej A, Rohde V. The importance of the hedgehog signaling pathway in tumorigenesis of spinal and cranial chordoma. J Clin Med (2019) 8(2). doi: 10.3390/jcm8020248
114. Bai J, Shi J, Zhang Y, Li C, Xiong Y, Koka H, et al. Gene expression profiling identifies two chordoma subtypes associated with distinct molecular mechanisms and clinical outcomes. Clin Cancer Res (2023) 29(1):261–70. doi: 10.1158/1078-0432.CCR-22-1865
115. Yang C, Yong L, Liang C, Li Y, Ma Y, Wei F, et al. Genetic landscape and ligand-dependent activation of sonic hedgehog-Gli1 signaling in chordomas: a novel therapeutic target. Oncogene (2020) 39(24):4711–27. doi: 10.1038/s41388-020-1324-2
116. Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, et al. Frequent mutation of the major cartilage collagen gene Col2a1 in chondrosarcoma. Nat Genet (2013) 45(8):923–6. doi: 10.1038/ng.2668
117. Nicolle R, Ayadi M, Gomez-Brouchet A, Armenoult L, Banneau G, Elarouci N, et al. Integrated molecular characterization of chondrosarcoma reveals critical determinants of disease progression. Nat Commun (2019) 10(1):4622. doi: 10.1038/s41467-019-12525-7
118. Tiet TD, Hopyan S, Nadesan P, Gokgoz N, Poon R, Lin AC, et al. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am J Pathol (2006) 168(1):321–30. doi: 10.2353/ajpath.2006.050001
119. Campbell VT, Nadesan P, Ali SA, Wang CY, Whetstone H, Poon R, et al. Hedgehog pathway inhibition in chondrosarcoma using the smoothened inhibitor ipi-926 directly inhibits sarcoma cell growth. Mol Cancer Ther (2014) 13(5):1259–69. doi: 10.1158/1535-7163.Mct-13-0731
120. Budry L, Lafont C, El Yandouzi T, Chauvet N, Conejero G, Drouin J, et al. Related pituitary cell lineages develop into interdigitated 3d cell networks. Proc Natl Acad Sci USA (2011) 108(30):12515–20. doi: 10.1073/pnas.1105929108
121. Liu NA, Ren M, Song J, Rios Y, Wawrowsky K, Ben-Shlomo A, et al. In vivo time-lapse imaging delineates the zebrafish pituitary proopiomelanocortin lineage boundary regulated by Fgf3 signal. Dev Biol (2008) 319(2):192–200. doi: 10.1016/j.ydbio.2008.03.039
122. Levavi-Sivan B, Bloch CL, Gutnick MJ, Fleidervish IA. Electrotonic coupling in the anterior pituitary of a teleost fish. Endocrinology (2005) 146(3):1048–52. doi: 10.1210/en.2004-1415
123. Fauquier T, Guerineau NC, McKinney RA, Bauer K, Mollard P. Folliculostellate cell network: a route for long-distance communication in the anterior pituitary. Proc Natl Acad Sci USA (2001) 98(15):8891–6. doi: 10.1073/pnas.151339598
124. Royan MR, Siddique K, Csucs G, Puchades MA, Nourizadeh-Lillabadi R, Bjaalie JG, et al. 3d atlas of the pituitary gland of the model fish medaka (Oryzias latipes). Front Endocrinol (Lausanne) (2021) 12:719843. doi: 10.3389/fendo.2021.719843
125. Yoshitomi M, Ohta K, Kanazawa T, Togo A, Hirashima S, Uemura KI, et al. Three-dimensional ultrastructural analyses of anterior pituitary gland expose spatial relationships between endocrine cell secretory granule localization and capillary distribution. Sci Rep (2016) 6:36019. doi: 10.1038/srep36019
126. Santiago-Andres Y, Golan M, Fiordelisio T. Functional pituitary networks in vertebrates. Front Endocrinol (Lausanne) (2020) 11:619352. doi: 10.3389/fendo.2020.619352
127. Even-Zohar N, Metin Armagan D, Melmed S. Chapter one - pituitary stem cells. In: Litwack G, editor. Vitamins and hormones. (Cambridge, Massachusetts: Academic Press) (2021) 116:1–19.
128. Willis TL, Lodge EJ, Andoniadou CL, Yianni V. Cellular interactions in the pituitary stem cell niche. Cell Mol Life Sci (2022) 79(12):612. doi: 10.1007/s00018-022-04612-8
129. Wood S, Loudon A. The pars tuberalis: the site of the circannual clock in mammals? Gen Comp Endocrinol (2018) 258:222–35. doi: 10.1016/j.ygcen.2017.06.029
130. Ilahi S, Ilahi TB. Adenohypophysis (Pars Anterior, Anterior Pituitary). 2022 Oct 3. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing. (2023).
131. Inoue K, Couch EF, Takano K, Ogawa S. The structure and function of folliculo-stellate cells in the anterior pituitary gland. Arch Histol Cytol (1999) 62(3):205–18. doi: 10.1679/aohc.62.205
132. Denef C. Paracrinicity: the story of 30 years of cellular pituitary crosstalk. J Neuroendocrinol (2008) 20(1):1–70. doi: 10.1111/j.1365-2826.2007.01616.x
133. Morris J, Christian H. Folliculo-stellate cells: paracrine communicators in the anterior pituitary. Open Neuroendocrinol J (2011) 4:77–89. doi: 10.2174/1876528901104010077
134. Horvath E, Kovacs K. Folliculo-stellate cells of the human pituitary: a type of adult stem cell? Ultrastruct Pathol (2002) 26(4):219–28. doi: 10.1080/01913120290104476
135. Liu YC, Tanaka S, Inoue K, Kurosumi K. Localization of fibronectin in the folliculo-stellate cells of the rat anterior pituitary by the double bridge peroxidase-antiperoxidase method. Histochemistry (1989) 92(1):43–5. doi: 10.1007/BF00495014
136. Velasco ME, Roessmann U, Gambetti P. The presence of glial fibrillary acidic protein in the human pituitary gland. J Neuropathol Exp Neurol (1982) 41(2):150–63. doi: 10.1097/00005072-198203000-00005
137. Marin F, Boya J, Lopez-Carbonell A. Immunocytochemical localization of vimentin in stellate cells (Folliculo-stellate cells) of the rat, cat and rabbit pituitary pars distalis. Anat Embryol (Berl) (1989) 179(5):491–5. doi: 10.1007/BF00319592
138. Shimada T. Immunohistochemical localization of keratin in bull, goat, and sheep anterior pituitary glands. Cell Tissue Res (1992) 267(2):251–60. doi: 10.1007/BF00302962
139. Devnath S, Inoue K. An insight to pituitary folliculo-stellate cells. J Neuroendocrinol (2008) 20(6):687–91. doi: 10.1111/j.1365-2826.2008.01716.x
140. Traverso V, Christian HC, Morris JF, Buckingham JC. Lipocortin 1 (Annexin 1): a candidate paracrine agent localized in pituitary folliculo-stellate cells. Endocrinology (1999) 140(9):4311–9. doi: 10.1210/endo.140.9.7008
141. John CD, Theogaraj E, Christian HC, Morris JF, Smith SF, Buckingham JC. Time-specific effects of perinatal glucocorticoid treatment on anterior pituitary morphology, annexin 1 expression and adrenocorticotrophic hormone secretion in the adult female rat. J Neuroendocrinol (2006) 18(12):949–59. doi: 10.1111/j.1365-2826.2006.01493.x
142. Fauquier T, Rizzoti K, Dattani M, Lovell-Badge R, Robinson IC. Sox2-expressing progenitor cells generate all of the major cell types in the adult mouse pituitary gland. Proc Natl Acad Sci USA (2008) 105(8):2907–12. doi: 10.1073/pnas.0707886105
143. Soukup J, Cesak T, Hornychova H, Michalova K, Michnova L, Netuka D, et al. Stem cell transcription factor Sox2 is expressed in a subset of folliculo-stellate cells of growth hormone-producing pituitary neuroendocrine tumours and its expression shows no association with tumour size or Igf1 levels: a clinicopathological study of 109 cases. Endocr Pathol (2020) 31(4):337–47. doi: 10.1007/s12022-020-09634-1
144. Inoue K, Mogi C, Ogawa S, Tomida M, Miyai S. Are folliculo-stellate cells in the anterior pituitary gland supportive cells or organ-specific stem cells? Arch Physiol Biochem (2002) 110(1-2):50–3. doi: 10.1076/apab.110.1.50.911
145. Le Tissier PR, Mollard P. Renewing an old interest: pituitary folliculostellate cells. J Neuroendocrinol (2021) 33(11):e13053. doi: 10.1111/jne.13053
146. Zhang S, Cui Y, Ma X, Yong J, Yan L, Yang M, et al. Single-cell transcriptomics identifies divergent developmental lineage trajectories during human pituitary development. Nat Commun (2020) 11(1):5275. doi: 10.1038/s41467-020-19012-4
147. Zhang J, Lv C, Mo C, Liu M, Wan Y, Li J, et al. Single-cell rna sequencing analysis of chicken anterior pituitary: a bird's-eye view on vertebrate pituitary. Front Physiol (2021) 12:562817. doi: 10.3389/fphys.2021.562817
148. Cheung LYM, George AS, McGee SR, Daly AZ, Brinkmeier ML, Ellsworth BS, et al. Single-cell rna sequencing reveals novel markers of Male pituitary stem cells and hormone-producing cell types. Endocrinology (2018) 159(12):3910–24. doi: 10.1210/en.2018-00750
149. Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, et al. Cyclopia and defective axial patterning in mice lacking sonic hedgehog gene function. Nature (1996) 383(6599):407–13. doi: 10.1038/383407a0
150. Seppala M, Depew MJ, Martinelli DC, Fan CM, Sharpe PT, Cobourne MT. Gas1 is a modifier for holoprosencephaly and genetically interacts with sonic hedgehog. J Clin Invest (2007) 117(6):1575–84. doi: 10.1172/JCI32032
151. Cole F, Krauss RS. Microform holoprosencephaly in mice that lack the ig superfamily member cdon. Curr Biol (2003) 13(5):411–5. doi: 10.1016/s0960-9822(03)00088-5
152. Zhang W, Kang JS, Cole F, Yi MJ, Krauss RS. Cdo functions at multiple points in the sonic hedgehog pathway, and cdo-deficient mice accurately model human holoprosencephaly. Dev Cell (2006) 10(5):657–65. doi: 10.1016/j.devcel.2006.04.005
153. Echevarria-Andino ML, Allen BL. The hedgehog Co-receptor boc differentially regulates shh signaling during craniofacial development. Development (2020) 147(23). doi: 10.1242/dev.189076
154. Sbrogna JL, Barresi MJ, Karlstrom RO. Multiple roles for hedgehog signaling in zebrafish pituitary development. Dev Biol (2003) 254(1):19–35. doi: 10.1016/s0012-1606(02)00027-1
155. Park HL, Bai C, Platt KA, Matise MP, Beeghly A, Hui CC, et al. Mouse Gli1 mutants are viable but have defects in shh signaling in combination with a Gli2 mutation. Development (2000) 127(8):1593–605. doi: 10.1242/dev.127.8.1593
156. Wang Y, Martin JF, Bai CB. Direct and indirect requirements of Shh/Gli signaling in early pituitary development. Dev Biol (2010) 348(2):199–209. doi: 10.1016/j.ydbio.2010.09.024
157. Trowe MO, Zhao L, Weiss AC, Christoffels V, Epstein DJ, Kispert A. Inhibition of Sox2-dependent activation of shh in the ventral diencephalon by Tbx3 is required for formation of the neurohypophysis. Development (2013) 140(11):2299–309. doi: 10.1242/dev.094524
158. Fu T, Towers M, Placzek MA. Fgf10(+) progenitors give rise to the chick hypothalamus by rostral and caudal growth and differentiation. Development (2017) 144(18):3278–88. doi: 10.1242/dev.153379
159. Suga H, Kadoshima T, Minaguchi M, Ohgushi M, Soen M, Nakano T, et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature (2011) 480(7375):57–62. doi: 10.1038/nature10637
160. Dubourg C, Bendavid C, Pasquier L, Henry C, Odent S, David V. Holoprosencephaly. Orphanet J Rare Dis (2007) 2:8. doi: 10.1186/1750-1172-2-8
161. Kelberman D, Rizzoti K, Avilion A, Bitner-Glindzicz M, Cianfarani S, Collins J, et al. Mutations within Sox2/Sox2 are associated with abnormalities in the hypothalamo-Pituitary-Gonadal axis in mice and humans. J Clin Invest (2006) 116(9):2442–55. doi: 10.1172/JCI28658
162. Johnston JJ, Olivos-Glander I, Killoran C, Elson E, Turner JT, Peters KF, et al. Molecular and clinical analyses of Greig cephalopolysyndactyly and pallister-hall syndromes: robust phenotype prediction from the type and position of Gli3 mutations. Am J Hum Genet (2005) 76(4):609–22. doi: 10.1086/429346
163. Demurger F, Ichkou A, Mougou-Zerelli S, Le Merrer M, Goudefroye G, Delezoide AL, et al. New insights into genotype-phenotype correlation for Gli3 mutations. Eur J Hum Genet (2015) 23(1):92–102. doi: 10.1038/ejhg.2014.62
164. Biesecker LG. Gli3-related pallister-hall syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, editors. Genereviews. Seattle (WA): University of Washington, Seattle (1993).
165. Cameron FJ, Khadilkar VV, Stanhope R. Pituitary dysfunction, morbidity and mortality with congenital midline malformation of the cerebrum. Eur J Pediatr (1999) 158(2):97–102. doi: 10.1007/s004310051026
166. Johnson VP. Holoprosencephaly: a developmental field defect. Am J Med Genet (1989) 34(2):258–64. doi: 10.1002/ajmg.1320340228
167. Kjaer I, Fischer-Hansen B. Human fetal pituitary gland in holoprosencephaly and anencephaly. J Craniofac Genet Dev Biol (1995) 15(4):222–9.
168. Rappaport R, Czernichow P, Prevost C, Jullien M. Delayed growth with raised circulating growth hormone and incapacity to produce somatomedin (Dwarfism of laron type). Ann Pediatr (Paris) (1977) 24(1):63–7.
169. Bear KA, Solomon BD, Antonini S, Arnhold IJ, Franca MM, Gerkes EH, et al. Pathogenic mutations in Gli2 cause a specific phenotype that is distinct from holoprosencephaly. J Med Genet (2014) 51(6):413–8. doi: 10.1136/jmedgenet-2013-102249
170. Franca MM, Jorge AA, Carvalho LR, Costalonga EF, Vasques GA, Leite CC, et al. Novel heterozygous nonsense Gli2 mutations in patients with hypopituitarism and ectopic posterior pituitary lobe without holoprosencephaly. J Clin Endocrinol Metab (2010) 95(11):E384–91. doi: 10.1210/jc.2010-1050
171. Rahimov F, Ribeiro LA, de Miranda E, Richieri-Costa A, Murray JC. Gli2 mutations in four Brazilian patients: how wide is the phenotypic spectrum? Am J Med Genet A (2006) 140(23):2571–6. doi: 10.1002/ajmg.a.31370
172. Valenza F, Cittaro D, Stupka E, Biancolini D, Patricelli MG, Bonanomi D, et al. A novel truncating variant of Gli2 associated with culler-Jones syndrome impairs hedgehog signalling. PloS One (2019) 14(1):e0210097. doi: 10.1371/journal.pone.0210097
173. Kordass U, Schroder C, Elbracht M, Soellner L, Eggermann T. A familial Gli2 deletion (2q14.2) not associated with the holoprosencephaly syndrome phenotype. Am J Med Genet A (2015) 167A(5):1121–4. doi: 10.1002/ajmg.a.36972
174. Flemming GM, Klammt J, Ambler G, Bao Y, Blum WF, Cowell C, et al. Functional characterization of a heterozygous Gli2 missense mutation in patients with multiple pituitary hormone deficiency. J Clin Endocrinol Metab (2013) 98(3):E567–75. doi: 10.1210/jc.2012-3224
175. Babu D, Fanelli A, Mellone S, Muniswamy R, Wasniewska M, Prodam F, et al. Novel Gli2 mutations identified in patients with combined pituitary hormone deficiency (Cphd): evidence for a pathogenic effect by functional characterization. Clin Endocrinol (Oxf) (2019) 90(3):449–56. doi: 10.1111/cen.13914
176. Gregory LC, Gaston-Massuet C, Andoniadou CL, Carreno G, Webb EA, Kelberman D, et al. The role of the sonic hedgehog signalling pathway in patients with midline defects and congenital hypopituitarism. Clin Endocrinol (Oxf) (2015) 82(5):728–38. doi: 10.1111/cen.12637
177. Smyth I, Narang MA, Evans T, Heimann C, Nakamura Y, Chenevix-Trench G, et al. Isolation and characterization of human patched 2 (Ptch2), a putative tumour suppressor gene inbasal cell carcinoma and medulloblastoma on chromosome 1p32. Hum Mol Genet (1999) 8(2):291–7. doi: 10.1093/hmg/8.2.291
178. Obara-Moszynska M, Budny B, Kaluzna M, Zawadzka K, Jamsheer A, Rohde A, et al. Cdon gene contributes to pituitary stalk interruption syndrome associated with unilateral facial and abducens nerve palsy. J Appl Genet (2021) 62(4):621–9. doi: 10.1007/s13353-021-00649-w
179. Bashamboo A, Bignon-Topalovic J, Rouba H, McElreavey K, Brauner R. A nonsense mutation in the hedgehog receptor cdon associated with pituitary stalk interruption syndrome. J Clin Endocrinol Metab (2016) 101(1):12–5. doi: 10.1210/jc.2015-2995
180. Wang YK, Samos CH, Peoples R, Pérez-Jurado LA, Nusse R, Francke U. A novel human homologue of the drosophila frizzled wnt receptor gene binds wingless protein and is in the williams syndrome deletion at 7q11.23. Hum Mol Genet (1997) 6(3):465–72. doi: 10.1093/hmg/6.3.465
181. Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, et al. Exome sequencing identifies Mll2 mutations as a cause of kabuki syndrome. Nat Genet (2010) 42(9):790–3. doi: 10.1038/ng.646
182. Nishimura DY, Swiderski RE, Alward WL, Searby CC, Patil SR, Bennet SR, et al. The forkhead transcription factor gene Fkhl7 is responsible for glaucoma phenotypes which map to 6p25. Nat Genet (1998) 19(2):140–7. doi: 10.1038/493
183. Semina EV, Reiter R, Leysens NJ, Alward WL, Small KW, Datson NA, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, rieg, involved in rieger syndrome. Nat Genet (1996) 14(4):392–9. doi: 10.1038/ng1296-392
184. Zenker M, Mayerle J, Lerch MM, Tagariello A, Zerres K, Durie PR, et al. Deficiency of Ubr1, a ubiquitin ligase of the n-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-blizzard syndrome). Nat Genet (2005) 37(12):1345–50. doi: 10.1038/ng1681
185. Patsi O, De Beaufort C, Kerschen P, Cardillo S, Soehn A, Rautenberg M, et al. A new Pnpla6 mutation presenting as Oliver mcfarlane syndrome. J Neurol Sci (2018) 392:1–2. doi: 10.1016/j.jns.2018.06.016
186. Melmed S. Pituitary-tumor endocrinopathies. N Engl J Med (2020) 382(10):937–50. doi: 10.1056/NEJMra1810772
187. Asa SL, Ezzat S. Pituitary carcinoma: reclassification and implications in the net schema. Endocrine Oncol (2022) 2(1):R14–23. doi: 10.1530/eo-22-0041
188. Mete O, Lopes MB. Overview of the 2017 who classification of pituitary tumors. Endocr Pathol (2017) 28(3):228–43. doi: 10.1007/s12022-017-9498-z
189. Asa SL, Casar-Borota O, Chanson P, Delgrange E, Earls P, Ezzat S, et al. From pituitary adenoma to pituitary neuroendocrine tumor (Pitnet): an international pituitary pathology club proposal. Endocr Relat Cancer (2017) 24(4):C5–8. doi: 10.1530/ERC-17-0004
190. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 who classification of pituitary tumors. Endocr Pathol (2022) 33(1):6–26. doi: 10.1007/s12022-022-09703-7
191. Nishioka H, Inoshita N, Mete O, Asa SL, Hayashi K, Takeshita A, et al. The complementary role of transcription factors in the accurate diagnosis of clinically nonfunctioning pituitary adenomas. Endocr Pathol (2015) 26(4):349–55. doi: 10.1007/s12022-015-9398-z
192. Drummond JB, Ribeiro-Oliveira A Jr., Soares BS. Non-functioning pituitary adenomas. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, editors. Endotext. South Dartmouth (MA): MDText.com, Inc. (2000).
193. Chang M, Yang C, Bao X, Wang R. Genetic and epigenetic causes of pituitary adenomas. Front Endocrinol (Lausanne) MDText.com, Inc. (2020) 11:596554. doi: 10.3389/fendo.2020.596554
194. Constantin L, Constantin M, Wainwright BJ. Microrna biogenesis and hedgehog-patched signaling cooperate to regulate an important developmental transition in granule cell development. Genetics (2016) 202(3):1105–18. doi: 10.1534/genetics.115.184176
195. Mukhopadhyay A, Krishnaswami SR, Cowing-Zitron C, Hung NJ, Reilly-Rhoten H, Burns J, et al. Negative regulation of shh levels by kras and Fgfr2 during hair follicle development. Dev Biol (2013) 373(2):373–82. doi: 10.1016/j.ydbio.2012.10.024
196. Meuser M, Deuper L, Rudat C, Aydoğdu N, Thiesler H, Zarnovican P, et al. Fgfr2 signaling enhances the shh-Bmp4 signaling axis in early ureter development. Development (2022) 149(1). doi: 10.1242/dev.200021
197. Farreny MA, Agius E, Bel-Vialar S, Escalas N, Khouri-Farah N, Soukkarieh C, et al. Fgf signaling controls shh-dependent oligodendroglial fate specification in the ventral spinal cord. Neural Dev (2018) 13(1):3. doi: 10.1186/s13064-018-0100-2
198. Regard JB, Malhotra D, Gvozdenovic-Jeremic J, Josey M, Chen M, Weinstein LS, et al. Activation of hedgehog signaling by loss of gnas causes heterotopic ossification. Nat Med (2013) 19(11):1505–12. doi: 10.1038/nm.3314
199. Pusapati GV, Kong JH, Patel BB, Gouti M, Sagner A, Sircar R, et al. G Protein-coupled receptors control the sensitivity of cells to the morphogen sonic hedgehog. Sci Signal (2018) 11(516). doi: 10.1126/scisignal.aao5749
200. He X, Zhang L, Chen Y, Remke M, Shih D, Lu F, et al. The G protein A subunit gαs is a tumor suppressor in sonic hedgehog-driven medulloblastoma. Nat Med (2014) 20(9):1035–42. doi: 10.1038/nm.3666
201. Bongiovanni D, Tosello V, Saccomani V, Dalla Santa S, Amadori A, Zanovello P, et al. Crosstalk between hedgehog pathway and the glucocorticoid receptor pathway as a basis for combination therapy in T-cell acute lymphoblastic leukemia. Oncogene (2020) 39(42):6544–55. doi: 10.1038/s41388-020-01453-2
202. Wang JM, Isenberg JS, Billiar TR, Chen AF. Thrombospondin-1/Cd36 pathway contributes to bone marrow-derived angiogenic cell dysfunction in type 1 diabetes Via sonic hedgehog pathway suppression. Am J Physiol Endocrinol Metab (2013) 305(12):E1464–72. doi: 10.1152/ajpendo.00516.2013
203. Zhou A, Lin K, Zhang S, Ma L, Xue J, Morris SA, et al. Gli1-induced deubiquitinase Usp48 aids glioblastoma tumorigenesis by stabilizing Gli1. EMBO Rep (2017) 18(8):1318–30. doi: 10.15252/embr.201643124
204. Mallick S, Chakrabarti J, Eschbacher J, Moraitis AG, Greenstein AE, Churko J, et al. Genetically engineered human pituitary corticotroph tumor organoids exhibit divergent responses to glucocorticoid receptor modulators. Transl Res (2023) 256:56–72. doi: 10.1016/j.trsl.2023.01.002
205. Xia R, Jia H, Fan J, Liu Y, Jia J. Usp8 promotes smoothened signaling by preventing its ubiquitination and changing its subcellular localization. PloS Biol (2012) 10(1):e1001238. doi: 10.1371/journal.pbio.1001238
206. Xekouki P, Brennand A, Whitelaw B, Pacak K, Stratakis CA. The 3pas: an update on the association of pheochromocytomas, paragangliomas, and pituitary tumors. Horm Metab Res (2019) 51(7):419–36. doi: 10.1055/a-0661-0341
207. Xekouki P, Pacak K, Almeida M, Wassif CA, Rustin P, Nesterova M, et al. Succinate dehydrogenase (Sdh) d subunit (Sdhd) inactivation in a growth-Hormone-Producing pituitary tumor: a new association for sdh? J Clin Endocrinol Metab (2012) 97(3):E357–66. doi: 10.1210/jc.2011-1179
208. Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, et al. Germline aip mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab (2012) 97(4):E663–70. doi: 10.1210/jc.2011-2291
209. Arveseth CD, Happ JT, Hedeen DS, Zhu JF, Capener JL, Klatt Shaw D, et al. Smoothened transduces hedgehog signals Via activity-dependent sequestration of pka catalytic subunits. PloS Biol (2021) 19(4):e3001191. doi: 10.1371/journal.pbio.3001191
210. Gurung B, Feng Z, Iwamoto DV, Thiel A, Jin G, Fan CM, et al. Menin epigenetically represses hedgehog signaling in Men1 tumor syndrome. Cancer Res (2013) 73(8):2650–8. doi: 10.1158/0008-5472.CAN-12-3158
211. Sapkota S, Horiguchi K, Tosaka M, Yamada S, Yamada M. Whole-exome sequencing study of thyrotropin-secreting pituitary adenomas. J Clin Endocrinol Metab (2017) 102(2):566–75. doi: 10.1210/jc.2016-2261
212. Desouza LA, Sathanoori M, Kapoor R, Rajadhyaksha N, Gonzalez LE, Kottmann AH, et al. Thyroid hormone regulates the expression of the sonic hedgehog signaling pathway in the embryonic and adult mammalian brain. Endocrinology (2011) 152(5):1989–2000. doi: 10.1210/en.2010-1396
213. Salomon MP, Wang X, Marzese DM, Hsu SC, Nelson N, Zhang X, et al. The epigenomic landscape of pituitary adenomas reveals specific alterations and differentiates among acromegaly, cushing's disease and endocrine-inactive subtypes. Clin Cancer Res (2018) 24(17):4126–36. doi: 10.1158/1078-0432.Ccr-17-2206
214. Yu F, Geng W, Dong P, Huang Z, Zheng J. Lncrna-Meg3 inhibits activation of hepatic stellate cells through smo protein and mir-212. Cell Death Dis (2018) 9(10):1014. doi: 10.1038/s41419-018-1068-x
215. Ezzat S, Cheng S, Asa S. Epigenetics of pituitary tumors: pathogenetic and therapeutic implications. Mol Cell Endocrinol (2018) 469:70–6. doi: 10.1016/j.mce.2017.07.011
216. Asa S, Mete O, Ezzat S. Genomics and epigenomics of pituitary tumors: what do pathologists need to know? Endocrine Pathol (2021) 32:3–16. doi: 10.1007/s12022-021-09663-4
217. Bahreini F, Jabbari P, Gossing W, Aziziyan F, Frohme M, Rezaei N. The role of noncoding rnas in pituitary adenoma. Epigenomics (2021) 13:1421–37. doi: 10.2217/epi-2021-0165
218. Russ S, Anastasopoulou C, Shafiq I. Pituitary adenoma. In: Statpearls. Treasure Island (FL): StatPearls Publishing (2022).
219. Daly AF, Beckers A. The role of aip mutations in pituitary adenomas: 10 years on. Endocrine (2017) 55(2):333–5. doi: 10.1007/s12020-016-1194-0
220. Wang W, Wei CJ, Cui XW, Li YH, Gu YH, Gu B, et al. Impacts of Nf1 gene mutations and genetic modifiers in neurofibromatosis type 1. Front Neurol (2021) 12:704639. doi: 10.3389/fneur.2021.704639
221. Endo H, Utani A, Matsumoto F, Kuroki T, Yoshimoto S, Ichinose M, et al. A possible paracrine hedgehog signalling pathway in neurofibromas from patients with neurofibromatosis type 1. Br J Dermatol (2003) 148(2):337–41. doi: 10.1046/j.1365-2133.2003.05178.x
222. Bengtsson D, Joost P, Aravidis C, Askmalm Stenmark M, Backman AS, Melin B, et al. Corticotroph pituitary carcinoma in a patient with lynch syndrome (Ls) and pituitary tumors in a nationwide ls cohort. J Clin Endocrinol Metab (2017) 102(11):3928–32. doi: 10.1210/jc.2017-01401
223. Uraki S, Ariyasu H, Doi A, Furuta H, Nishi M, Sugano K, et al. Atypical pituitary adenoma with Men1 somatic mutation associated with abnormalities of DNA mismatch repair genes; Mlh1 germline mutation and Msh6 somatic mutation. Endocr J (2017) 64(9):895–906. doi: 10.1507/endocrj.EJ17-0036
224. Yuan J, Yi K, Yang L. Tgfbr2 regulates hedgehog pathway and cervical cancer cell proliferation and migration by mediating Smad4. J Proteome Res (2020) 19(8):3377–85. doi: 10.1021/acs.jproteome.0c00239
225. Duan S, Sawyer TW, Sontz RA, Wieland BA, Diaz AF, Merchant JL. Gfap-directed inactivation of Men1 exploits glial cell plasticity in favor of neuroendocrine reprogramming. Cell Mol Gastroenterol Hepatol (2022) 14(5):1025–51. doi: 10.1016/j.jcmgh.2022.06.009
226. Bohinc B, Michelotti G, Diehl AM. Hedgehog signaling in human medullary thyroid carcinoma: a novel signaling pathway. Thyroid (2013) 23(9):1119–26. doi: 10.1089/thy.2012.0474
227. Lonser RR, Butman JA, Kiringoda R, Song D, Oldfield EH. Pituitary stalk hemangioblastomas in Von hippel-lindau disease. J Neurosurg (2009) 110(2):350–3. doi: 10.3171/2008.4.17532
228. Cho HK, Kim SY, Kim KH, Kim HH, Cheong J. Tumor suppressor protein vhl inhibits hedgehog-gli activation through suppression of Gli1 nuclear localization. FEBS Lett (2013) 587(7):826–32. doi: 10.1016/j.febslet.2013.01.050
229. Graham JM Jr., Saunders R, Fratkin J, Spiegel P, Harris M, Klein RZ. A cluster of pallister-hall syndrome cases, (Congenital hypothalamic hamartoblastoma syndrome). Am J Med Genet Suppl (1986) 2:53–63. doi: 10.1002/ajmg.1320250609
230. Saitsu H, Sonoda M, Higashijima T, Shirozu H, Masuda H, Tohyama J, et al. Somatic mutations in Gli3 and Ofd1 involved in sonic hedgehog signaling cause hypothalamic hamartoma. Ann Clin Transl Neurol (2016) 3(5):356–65. doi: 10.1002/acn3.300
231. Hozumi K, Fukuoka H, Odake Y, Takeuchi T, Uehara T, Sato T, et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type 1. Endocr J (2019) 66(10):853–7. doi: 10.1507/endocrj.EJ19-0035
232. Forlino A, Vetro A, Garavelli L, Ciccone R, London E, Stratakis CA, et al. Prkacb and Carney complex. N Engl J Med (2014) 370(11):1065–7. doi: 10.1056/NEJMc1309730
233. Solarski M, Rotondo F, Foulkes WD, Priest JR, Syro LV, Butz H, et al. Dicer1 gene mutations in endocrine tumors. Endocr Relat Cancer (2018) 25(3):R197–r208. doi: 10.1530/erc-17-0509
234. de Kock L, Sabbaghian N, Plourde F, Srivastava A, Weber E, Bouron-Dal Soglio D, et al. Pituitary blastoma: a pathognomonic feature of germ-line Dicer1 mutations. Acta Neuropathol (2014) 128(1):111–22. doi: 10.1007/s00401-014-1285-z
235. Chen CJ, Liu JY, Cheng SN, Chang FW, Yuh YS. Mccune-albright syndrome associated with pituitary microadenoma: patient report. J Pediatr Endocrinol Metab (2004) 17(3):365–9. doi: 10.1515/jpem.2004.17.3.365
236. Beckers A, Aaltonen LA, Daly AF, Karhu A. Familial isolated pituitary adenomas (Fipa) and the pituitary adenoma predisposition due to mutations in the aryl hydrocarbon receptor interacting protein (Aip) gene. Endocr Rev (2013) 34(2):239–77. doi: 10.1210/er.2012-1013
237. Watson JC, Stratakis CA, Bryant-Greenwood PK, Koch CA, Kirschner LS, Nguyen T, et al. Neurosurgical implications of Carney complex. J Neurosurg (2000) 92(3):413–8. doi: 10.3171/jns.2000.92.3.0413
238. Irvine AD, Armstrong DK, Bingham EA, Hadden DR, Nevin NC, Hughes AE. Evidence for a second genetic locus in Carney complex. Br J Dermatol (1998) 139(4):572–6. doi: 10.1046/j.1365-2133.1998.02450.x
239. Asa SL, Mete O. Hypothalamic endocrine tumors: an update. J Clin Med (2019) 8(10). doi: 10.3390/jcm8101741
240. Meng E, Hanna A, Samant RS, Shevde LA. The impact of hedgehog signaling pathway on DNA repair mechanisms in human cancer. Cancers (Basel) (2015) 7(3):1333–48. doi: 10.3390/cancers7030839
241. Uzilov AV, Taik P, Cheesman KC, Javanmard P, Ying K, Roehnelt A, et al. Usp8 and Tp53 drivers are associated with cnv in a corticotroph adenoma cohort enriched for aggressive tumors. J Clin Endocrinol Metab (2021) 106(3):826–42. doi: 10.1210/clinem/dgaa853
242. Zhou Z, Yao X, Pang S, Chen P, Jiang W, Shan Z, et al. The deubiquitinase Uchl5/Uch37 positively regulates hedgehog signaling by deubiquitinating smoothened. J Mol Cell Biol (2018) 10(3):243–57. doi: 10.1093/jmcb/mjx036
243. Gerling M, Buller NV, Kirn LM, Joost S, Frings O, Englert B, et al. Stromal hedgehog signalling is downregulated in colon cancer and its restoration restrains tumour growth. Nat Commun (2016) 7:12321. doi: 10.1038/ncomms12321
244. Odent S, Atti-Bitach T, Blayau M, Mathieu M, Aug J, Delezo de AL, et al. Expression of the sonic hedgehog (Shh ) gene during early human development and phenotypic expression of new mutations causing holoprosencephaly. Hum Mol Genet (1999) 8(9):1683–9. doi: 10.1093/hmg/8.9.1683
245. Kimura T, Furudate S. Pituitary gh and prolactin deficiency and testis enlargement in hypothyroid rats caused by goitrogen methimazole. Exp Anim (1996) 45(4):369–75. doi: 10.1538/expanim.45.369
246. Lampichler K, Ferrer P, Vila G, Lutz MI, Wolf F, Knosp E, et al. The role of proto-oncogene Gli1 in pituitary adenoma formation and cell survival regulation. Endocr Relat Cancer (2015) 22(5):793–803. doi: 10.1530/ERC-15-0109
247. Yavropoulou MP, Maladaki A, Topouridou K, Kotoula V, Poulios C, Daskalaki E, et al. Expression pattern of the hedgehog signaling pathway in pituitary adenomas. Neurosci Lett (2016) 611:94–100. doi: 10.1016/j.neulet.2015.10.076
248. Zhou W, Ma CX, Xing YZ, Yan ZY. Identification of candidate target genes of pituitary adenomas based on the DNA microarray. Mol Med Rep (2016) 13(3):2182–6. doi: 10.3892/mmr.2016.4785
249. Ding X, Fan K, Hu J, Zang Z, Zhang S, Zhang Y, et al. Scp2-mediated cholesterol membrane trafficking promotes the growth of pituitary adenomas Via hedgehog signaling activation. J Exp Clin Cancer Res (2019) 38(1):404. doi: 10.1186/s13046-019-1411-9
250. Zhang Q, Yao B, Long X, Chen Z, He M, Wu Y, et al. Single-cell sequencing identifies differentiation-related markers for molecular classification and recurrence prediction of pitnet. Cell Rep Med (2023) 4(2):100934. doi: 10.1016/j.xcrm.2023.100934
251. Pekmezci M, Villanueva-Meyer JE, Goode B, Van Ziffle J, Onodera C, Grenert JP, et al. The genetic landscape of ganglioglioma. Acta Neuropathol Commun (2018) 6(1):47. doi: 10.1186/s40478-018-0551-z
252. Boetto J, Lerond J, Peyre M, Tran S, Marijon P, Kalamarides M, et al. Gab1 overexpression identifies hedgehog-activated anterior skull base meningiomas. Neuropathol Appl Neurobiol (2021) 47(6):748–55. doi: 10.1111/nan.12740
253. Coy S, Du Z, Sheu SH, Woo T, Rodriguez FJ, Kieran MW, et al. Distinct patterns of primary and motile cilia in rathke's cleft cysts and craniopharyngioma subtypes. Mod Pathol (2016) 29(12):1446–59. doi: 10.1038/modpathol.2016.153
254. Holsken A, Sill M, Merkle J, Schweizer L, Buchfelder M, Flitsch J, et al. Adamantinomatous and papillary craniopharyngiomas are characterized by distinct epigenomic as well as mutational and transcriptomic profiles. Acta Neuropathol Commun (2016) 4:20. doi: 10.1186/s40478-016-0287-6
255. Gump JM, Donson AM, Birks DK, Amani VM, Rao KK, Griesinger AM, et al. Identification of targets for rational pharmacological therapy in childhood craniopharyngioma. Acta Neuropathol Commun (2015) 3:30. doi: 10.1186/s40478-015-0211-5
256. Gomes DC, Jamra SA, Leal LF, Colli LM, Campanini ML, Oliveira RS, et al. Sonic hedgehog pathway is upregulated in adamantinomatous craniopharyngiomas. Eur J Endocrinol (2015) 172(5):603–8. doi: 10.1530/EJE-14-0934
257. Carreno G, Boult JKR, Apps J, Gonzalez-Meljem JM, Haston S, Guiho R, et al. Shh pathway inhibition is protumourigenic in adamantinomatous craniopharyngioma. Endocr Relat Cancer (2019) 26(3):355–66. doi: 10.1530/ERC-18-0538
258. Hildebrand MS, Griffin NG, Damiano JA, Cops EJ, Burgess R, Ozturk E, et al. Mutations of the sonic hedgehog pathway underlie hypothalamic hamartoma with gelastic epilepsy. Am J Hum Genet (2016) 99(2):423–9. doi: 10.1016/j.ajhg.2016.05.031
259. Ng YT, Kerrigan JF, Prenger EC, White WL, Rekate HL. Successful resection of a hypothalamic hamartoma and a rathke cleft cyst. case report. J Neurosurg (2005) 102(1 Suppl):78–80. doi: 10.3171/ped.2005.102.1.0078
260. Ilie MD, Lasolle H, Raverot G. Emerging and novel treatments for pituitary tumors. J Clin Med (2019) 8(8). doi: 10.3390/jcm8081107
261. Raverot G, Vasiljevic A, Jouanneau E, Lasolle H. Confirmation of a new therapeutic option for aggressive or dopamine agonist-resistant prolactin pituitary neuroendocrine tumors. Eur J Endocrinol (2019) 181(2):C1–3. doi: 10.1530/EJE-19-0359
262. Rickert CH, Dockhorn-Dworniczak B, Busch G, Moskopp D, Albert FK, Rama B, et al. Increased chromosomal imbalances in recurrent pituitary adenomas. Acta Neuropathol (2001) 102(6):615–20. doi: 10.1007/s004010100413
263. Rickert CH, Scheithauer BW, Paulus W. Chromosomal aberrations in pituitary carcinoma metastases. Acta Neuropathol (2001) 102(2):117–20. doi: 10.1007/s004010100430
Keywords: hedgehog signaling, pituitary development, hypothalamic-pituitary axis, hormone production, pituitary disorders, pituitary tumors
Citation: Bian Y, Hahn H and Uhmann A (2023) The hidden hedgehog of the pituitary: hedgehog signaling in development, adulthood and disease of the hypothalamic-pituitary axis. Front. Endocrinol. 14:1219018. doi: 10.3389/fendo.2023.1219018
Received: 08 May 2023; Accepted: 19 June 2023;
Published: 05 July 2023.
Edited by:
Hidenori Fukuoka, Kobe University, JapanReviewed by:
Cuiqi Zhou, Cedars Sinai Medical Center, United StatesKarine Rizzoti, The Francis Crick Institute, United Kingdom
Copyright © 2023 Bian, Hahn and Uhmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anja Uhmann, auhmann@gwdg.de