
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 20 September 2023
Sec. Cancer Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1217875
This article is part of the Research Topic Impact of Type-2 Diabetes and Poor Metabolic Health on Aggressive Breast Cancer Biology View all 4 articles
 Barbara Mensah Sankofi1
Barbara Mensah Sankofi1 Estefania Valencia-Rincón1
Estefania Valencia-Rincón1 Malika Sekhri1
Malika Sekhri1 Adriana L. Ponton-Almodovar2,3,4
Adriana L. Ponton-Almodovar2,3,4 Jamie J. Bernard2,3,4
Jamie J. Bernard2,3,4 Elizabeth A. Wellberg1*
Elizabeth A. Wellberg1*Obesity and type 2 diabetes are chronic metabolic diseases that impact tens to hundreds of millions of adults, especially in developed countries. Each condition is associated with an elevated risk of breast cancer and with a poor prognosis after treatment. The mechanisms connecting poor metabolic health to breast cancer are numerous and include hyperinsulinemia, inflammation, excess nutrient availability, and adipose tissue dysfunction. Here, we focus on adipose tissue, highlighting important roles for both adipocytes and fibroblasts in breast cancer progression. One potentially important mediator of adipose tissue effects on breast cancer is the fibroblast growth factor receptor (FGFR) signaling network. Among the many roles of FGFR signaling, we postulate that key mechanisms driving aggressive breast cancer include epithelial-to-mesenchymal transition and cellular metabolic reprogramming. We also pose existing questions that may help better understand breast cancer biology in people with obesity, type 2 diabetes, and poor metabolic health.
Breast cancer is the second most commonly diagnosed malignancy in women (1), with almost 300,000 new cases and 43,000 deaths expected in 2023 (2). Heritable mutations account for some breast cancer diagnoses; a well-known example of which are the BRCA1/2 genes. However, most breast cancers arise from environmental or lifestyle-associated exposures. Two major contributors to breast cancer risk and mortality, although not necessarily separable, are obesity and type 2 diabetes (T2D). Approximately 1.9 billion adults worldwide have a BMI over 25 kg/m2, and 650 million (13%) of these have obesity (3). In the US, 42.4% of adults have obesity (43% of men and 41.9% of women). Globally, approximately 10.5% of adults have diabetes (4) and up to 90% of these cases are T2D (5). Excess body weight is a prominent risk factor for T2D; approximately 90% of people with T2D have overweight or obesity (6, 7). Importantly, according to the Centers for Disease Control and Prevention, more than 1 in 3 Americans have prediabetes and more than 80% of these people do not know they have it. Some strategies that promote weight loss or improve metabolic function, such as bariatric surgery, calorie restriction, or metformin, are known to associate with reduced breast cancer risk, suggesting that impairments in whole body metabolism are reversible (8–10). Once tumors form, poor metabolic health is likely to influence breast cancer risk and progression through multiple mechanisms.
At diagnosis, breast tumors are histologically defined based on the presence of specific receptors. These categories include hormone receptor-positive (HR+), which express the estrogen or progesterone receptors (ER/PR) and are collectively referred to here as estrogen receptor-positive (ER+), human epidermal growth factor receptor positive (HER2+), or triple-negative breast cancer (TNBC), which lacks all of these receptors. Each subtype has distinct treatment and mortality profiles that depend in part on the stage of tumor at diagnosis (localized versus distant) and on the expression of HR or HER2. The disease-free and disease-specific survival rates for ER+, HER2+, and TNBC (2) illustrate the unique biology of these tumors. Aside from the histological subtypes, there are also molecular subtypes that are reflected in tumor gene expression profiles including luminal A, luminal B, HER2-enriched, basal-like, and normal breast-like (11, 12) that each have unique survival patterns (13). TNBC or basal breast cancer cases occur frequently in women under 40 and people of African ancestry. These cases are characterized by greater early relapse and advanced disease stage at diagnosis. While the TNBC subtype is often more aggressive than ER+ breast cancer, approximately 40% of ER+ tumors may be resistant to endocrine therapy. Out of these, 15-20% demonstrate intrinsic and early resistance to treatments (14–16). Intrinsic or de novo resistance to endocrine therapies can occur with low ER expression (17), while acquired resistance occurs through several mechanisms such as mutations in the ESR1 gene, loss of ER expression, alteration of transcriptional co-regulatory proteins, growth factor receptor activation, and metabolic reprogramming (18, 19). Nearly 50% of patients with TNBC develop resistance to chemotherapy, attributable in part to poor cellular differentiation and the presence of cancer stem cells that frequently self-renew to drive chemoresistance (20). Recurrent ER+ tumors are insensitive to endocrine therapy, similar to TNBC, suggesting that endocrine therapy-resistant ER+ breast cancer may be functionally similar to TNBC, with potentially overlapping risk factors and drivers.
In this review, we describe some common features of aggressive breast cancers such as TNBC and endocrine-resistant ER+ tumors. We discuss factors that characterize poor metabolic health and the influence these have on breast tumor biology, emphasizing adipose tissue. While there are many reported mechanisms through which obesity and T2D impact cancer, our work has identified the fibroblast growth factor signaling network (FGF/FGFR) as one common pathway connecting metabolic dysfunction to both TNBC and endocrine-resistant ER+ breast tumors and we describe those studies (21–26). We also examine the various ways tumor FGFR signaling may be altered and potential downstream effects, including epithelial to mesenchymal transition and altered tumor metabolism, that contribute to the hallmarks of aggressive breast cancer.
Postmenopausal breast cancer is one of 13 obesity-associated cancers (27). However, obesity is associated with a lower risk for premenopausal breast cancer (28). This seems counterintuitive, and many scientists are still searching for an explanation for this phenomenon (29). In addition to menopausal status, the impact of obesity on breast cancer risk depends on the tumor subtype. Prior to menopause, a high BMI is associated with a lower risk for ER+ breast cancer (30–33), which may explain the overall lower incidence of breast cancer associated with obesity in young women. As mentioned above, ER+ tumors make up the majority of diagnosed cases and likely drive the statistics that link obesity to breast cancer as a whole. The risk for TNBC prior to menopause is elevated with obesity (33, 34). Obesity is defined as a BMI30kg/m2 by the World Health Organization; however, one large study indicated that the elevated risk for breast cancer diagnosis is apparent in women with a BMI>25kg/m2, which includes those considered “overweight” (35). The risk for contralateral breast cancer diagnosis in women who received conserving surgical treatment is significantly elevated in women with overweight or obesity (36).
Some studies have shown a link between obesity and a higher risk of TNBC (34, 37) and premenopausal ER-negative breast cancer (32, 38). In the Carolina Breast Cancer Study, obesity was associated with increased incidence of TNBC in both premenopausal and postmenopausal women of African descent (39). Similarly, the Women’s Circle Health Study showed that high waist-to-hip ratio was associated with an increased risk of premenopausal breast cancer in African Americans after adjusting for BMI (40). Another study also reported that obese premenopausal women had an 82% increased risk of TNBC compared with women with high BMI (34). Also, a meta-analysis by Pierobon and colleagues showed that premenopausal obese women have a 42% higher risk of developing TNBC compared with women with normal BMI (41)
Obesity is associated with a shorter overall survival in women with breast cancer and many but not all studies report a significant negative effect of obesity on recurrence-free or breast cancer-specific survival (29, 42–47). The relationship between obesity and breast cancer specific survival may depend on a variety of factors, including tumor subtype, lymph node involvement, tumor stage, and years since menopause Importantly, obesity is associated with other morbidities such as CVD, which may influence survival more strongly than breast cancer progression. Consequently, the epidemiological data describing the effects of obesity, metabolic function, and their associated sequelae on breast cancer mortality are not yet as consistent as those for breast cancer risk. This may be due, in part, to underpowered studies that cannot assess each variable and its impact on breast cancer-specific survival, or due to the complex physiology associated with metabolic health in humans. Early studies with extensive follow-up may not have taken into consideration the complexity of breast cancer subtypes or the driving mechanisms that underlie the BMI-breast cancer link.
Multiple studies have investigated the effect of diabetes on breast cancer risk and progression (48, 49). According to one meta-analysis, women with diabetes have a 23% higher risk of developing breast cancer than those without diabetes (50). Furthermore, a greater percentage of women with diabetes have a more advanced stage of breast cancer than their non-diabetic counterparts (48). A different study reported that women with diabetes had a 27% greater risk for breast cancer, but this was decreased to 16% when studies were considered that controlled for BMI, indicating the importance of obesity in the link between T2D and breast cancer (51). Fasting insulin and glucose, which precede diabetes diagnosis, have been associated with a greater risk for breast cancer in women with a BMI over 26 kg/m2 (52). A similar impact of diabetes is seen on all-cause or overall mortality and on breast cancer-specific mortality. In most cases, the relationship between diabetes and all-cause mortality is stronger than that for cancer-specific mortality since diabetes is associated with other chronic conditions that can shorten lifespan. One meta-analysis found that diabetes is linked with a 37% and 17% increased risk of all-cause mortality and breast cancer-specific mortality, respectively (53). Another study revealed that compared to those without diabetes, breast cancer patients with pre-existing diabetes had a 51% shorter overall survival time and a 28% shorter disease-free survival (54). The Cancer Prevention Cohort Study II which recruited one million US adults reported a 16% increase in breast cancer-specific mortality and a 2-fold increased risk of all-cause mortality (55). Another retrospective study reported a two-fold greater breast cancer-specific mortality in women with T2D (56). Diabetes also significantly affects the treatment of choice of breast cancer patients. For example, younger women with both diabetes and breast cancer were more likely to undergo surgical resection than their non-diabetic counterparts (57). Women with insulin-treated diabetes were less likely to undergo axillary lymph node dissection relative to their non-diabetic counterparts. Older patients (<65 years) were less likely to receive radiotherapy than their non-diabetic patients of the same age bracket (58). T2D is diagnosed based on hemoglobin A1c levels, which become elevated after insulin resistance is established. The environment of hyperinsulinemia, often referred to as prediabetes, may occur long before pancreatic beta-cell failure that characterizes T2D; however, it is difficult to capture the link between this environment and breast cancer risk without screening criteria to define the prediabetic state.
Beyond BMI, other host variables may better predict breast cancer risk than the amount of body fat and may indicate the importance of developing tools to routinely assess metabolic health before diabetes develops (25, 59). Adipose distribution often changes after menopause, when women experience more visceral adiposity or central obesity, reflected by a greater waist-to-hip ratio. Central obesity is associated with elevated risk of both ER+ and TNBC subtypes (32, 46). Adult weight gain (i.e. adipose expansion) is associated with elevated breast cancer risk in multiple studies (60–63). Metabolic health, defined by a variety of criteria, may be an independent predictor of breast cancer risk in some people. In several studies, postmenopausal women that were considered “metabolically unhealthy” using different approaches (e.g. high fasting insulin, high HOMA-IR, hepatic steatosis), had elevated breast cancer risk irrespective of BMI (64–66). However, a very recent study indicated no difference in postmenopausal breast cancer risk in women classified as metabolically healthy overweight/obese or as metabolically unhealthy lean using C-peptide measures (67). Deteriorating metabolic function may indicate the presence of prediabetes, which is characterized by sustained hyperinsulinemia. Insulin, a potent mitogenic and anabolic hormone, may support breast cancer progression through the activation of MAPK and PI3K pathways that lead to many pro-tumorigenic cellular changes. Elevated insulin is a feature of the “metabolically unhealthy” lean or obese phenotype, is often associated with visceral adiposity, and is linked to adult weight gain. According to the Women’s Health Initiative Study, higher levels of Insulin resistance in post-menopausal women are associated with higher breast cancer incidence and higher all-cause mortality after breast cancer. The role of insulin in regulating cancer growth has been recently reviewed (68). Currently, it remains to be established whether experimentally preventing hyperinsulinemia impacts breast cancer risk or progression.
The crosstalk between obesity, T2D, and breast cancer is intricate and mediated by multiple mechanisms including, inflammation, adipose tissue dysfunction, metabolic alterations, insulin, and hypoxia. Many of these mediators have been thoroughly reviewed elsewhere (68, 69). There are 13 obesity-associated cancers for which elevated risk is associated with a high BMI. Almost all of them, including liver, pancreas, endometrial, and certainly breast cancer, occur adjacent to or within adipose depots. The breast is composed of 90% adipose tissue, which contains many cell types including adipocytes, fibroblasts, vascular cells, and immune cells. Obesity and T2D have been implicated in adipose tissue dysfunction, which alters the cellular interactions within the breast that often support cancer therapy resistance. Generally, patients with obesity and/or T2D have poor responses to breast cancer therapies and greater mortality, as described above. As an endocrine organ, adipose tissue produces pro-inflammatory cytokines, adipokines, growth factors, and also aromatizes androgens into estrogens. These factors can alter the tumor microenvironment and promote aggressive breast cancer. Adipose tissue dysfunction leads to excessive production of adipokines such as leptin and adiponectin by the adipocytes (70). Low serum adiponectin levels were associated with a higher risk for metastasis, angiogenesis, and endocrine therapy resistance (71, 72). Besides adiponectin, obese individuals have a higher expression of leptin and its receptor which has been implicated in increasing the metastatic potential in the tumor microenvironment. Also, leptin has been shown to activate ERα signaling and increase aromatase activity leading to excessive proliferation and migration of tumor cells (73, 74). Knockdown of leptin in adipose-derived stromal cells co-cultured with ER+ breast cancer cells led to a reduction in tumor growth and expression of metastasis-related genes (75). Overall, leptin and adiponectin have opposing effects on breast tumorigenesis and their ratio may be modulated particularly in people with high BMI as it significantly increases breast cancer risk and metastasis.
After menopause, adipose tissue is a major source of estrogen in women, and women with obesity have elevated circulating and local levels of estrogens. The elevated levels of estrogen (estrone and estradiol) in women with obesity undoubtedly contribute to the greater breast cancer risk associated with a high BMI. Indeed, a recent study showed that, in women with BRCA mutations and a high BMI, estrogen biosynthesis was elevated compared to women with a low BMI, indicating a role for estrogen in DNA damage that contributes to breast cancer risk (76). However, aromatase inhibitors such as letrozole and anastrazole effectively reduce circulating estrogen levels by up to 98% in postmenopausal women, irrespective of BMI (77). Clinical studies testing higher aromatase inhibitor doses have found no added benefit to lowering estrogens in women with obesity (78) or to breast cancer outcomes in general (79, 80), suggesting that the mechanisms of obesity-associated breast cancer progression and mortality are estrogen-independent.
Both obesity and T2D influence pro-inflammatory immune cells implicated in breast cancer development and progression. Obesity is characterized by hypertrophic adipocytes that recruit macrophages and sustain chronic low-grade inflammation. The breast environment in women with obesity has been documented to have crown-like structures, which are named by the histological appearance of macrophages around dying adipocytes. These inflammatory foci are also present in women without obesity, but who are classified as metabolically unhealthy (81, 82), illustrating the potential limitation of using BMI to evaluate breast cancer risk on an individual level. Obesity promotes features of immune cell exhaustion in mice and humans (83, 84), but obesity is also associated with a better response to immune therapy, for example in retrospective studies of melanoma (85). Similar links are seen with T2D, where immune cells may be senescent and low-grade inflammation is present (86). In a small study of people with a genetic predisposition to diabetes (i.e. first-degree relatives of people with T2D) matched with controls for BMI, gender, and age, subcutaneous adipocyte hypertrophy was evident even prior to the development of obesity or overt T2D (87). Greater adipocyte size was associated with elevated fasting insulin, higher HOMA-IR, and proinflammatory cytokines such as IL1β and IL6 (87). Studies like these studies highlight the potentially tumor-promotional changes that could take place in the breast adipose microenvironment even before obesity or T2D develops. The intricate relationship surrounding obesity, metabolic dysfunction, and inflammation as it relates to breast cancer has been comprehensively reviewed by others (68, 81, 88).
Within the breast tumor environment, growth factors such as EGF, VEGF, IGFs, and FGFs are produced by fibroblasts, adipocytes, and by cancer cells themselves. Each of these stimulates aggressive features of breast tumors and can drive resistance to breast cancer therapies. Deregulated growth factor signaling influences several cancer hallmarks by supporting cell proliferation, rendering cancer cells resistant to apoptosis, promoting vascularization, and stimulating invasion and metastasis.
Fibroblast growth factor receptor (FGFR) signaling is crucial for breast development, tissue homeostasis, malignant transformation, and metastasis. There are 22 FGF ligands (18 of which signal through receptors) and 4 FGFR genes (89). Alternative splicing generates multiple isoforms of FGFRs 1-3, while FGFR4 has only one isoform (89). The ligands have paracrine or endocrine functions, with multiple ligand/receptor interactions possible. Dysregulated FGF/FGFR production or activation has been implicated in breast cancer progression and aggressive breast cancer phenotypes. FGF signaling regulates normal mammary stem cells and gland development, illustrated by the reduction in mammary outgrowth in mice lacking FGFR1 and FGFR2 (90). Conversely, activation of FGFR1 in mammary epithelium causes alveolar proliferation and early features of transformation (91). FGF and FGFR DNA alterations have been described in a variety of cancers, including breast cancer. For example, FGFR1 is frequently amplified and overexpressed in luminal breast tumors (up to 27% of cases), while FGFR2 is amplified in TNBC, although less frequently. Overexpression of FGFs and FGFRs in breast cancer cells can lead to aberrant pathway activation, which is why clinical trials have focused on targeting FGFRs in patients (92). The downstream effects of FGF signaling include activation of PI3K/AKT, MAPK, PLC, and JAK/STAT networks. Each of these pathways elicits a variety of responses that encompass the hallmarks of cancer such as proliferation, survival, migration, invasion, and changes in gene expression profiles and cell differentiation, including EMT. FGF signaling can promote growth, metastasis, and stemness in a variety of breast cancer cell subtypes and preclinical models (reviewed in (92).
FGFs are produced by breast cancer cells and by cells in the breast TME, including adipocytes and cancer-associated fibroblasts (CAFs). Obesity can impact each of the cells in the TME (25, 93). For example, one study found that adipose-derived fibroblasts from donors with obesity expressed elevated CAF markers compared to those from lean individuals. Co-culture of ER+ MCF7 cells with these obesity-associated fibroblasts facilitated proliferation and invasion more than those cultured with lean fibroblasts (94). Exposure of human fibroblasts to sera from women with obesity promoted pro-inflammatory changes that may impact signaling in breast cancer cells (95). In a mouse model of obesity, FGF1 expression was elevated in mammary adipose tissue after estrogen withdrawal-induced weight gain (23). Hypertrophic adipocytes secrete FGF1 as a proliferative signal to undifferentiated preadipocytes during weight gain (96). This normal signaling mechanism ensures healthy adipose tissue expansion through hyperplasia and new adipocyte formation, but can also support the proliferation of nearby breast cancer cells. ER+ tumors that continued to grow after estrogen loss had elevated levels of phosphorylated FGFR1, and inhibition of FGFR restored sensitivity of these tumors to estrogen deprivation (23). In subcutaneous adipose tissue biopsies taken from people with obesity before and after intentional weight gain, FGF1 expression increased significantly but only in those people who were classified as metabolically unhealthy. In human breast adipose, FGF1 expression positively correlated with BMI and with adipocyte size. In humans, high tumor levels of phosphorylated FGFR1 associated with a shorted disease-free survival after tamoxifen treatment and with an elevated BMI. An additional study found that circulating FGF2 was directly correlated with BMI in patients with breast cancer, and FGF2 treatment of obese tumor-bearing mice promoted resistance to anti-VEGF therapy (97). In TNBC (MDA-231), CAFs can stimulate FGFR1 signaling (98), and FGF2 from CAFs promotes cell migration and tumor growth through cancer cell FGFR1 (99). FGF2 produced by visceral adipose tissue stimulated growth of non-cancerous human skin cells and MCF10A human breast epithelial cells in soft agar and in low-attachment sphere cultures, which is a hallmark of malignant transformation (21, 22, 24). A pilot study using human serum in soft-agar MCF10A cultures showed that FGF2 signaling through FGFR1 may be necessary but not sufficient to promote sphere formation (100). There is compelling evidence demonstrating the role of FGF1 and FGF2 as potent angiogenic factors in mediating increased breast cancer risk and progression (92, 101, 102). One study showed that in ovariectomized or tamoxifen-treated mice, MCF7 cells overexpressing FGF1 displayed increased vascularization and enhanced metastatic potential (103). FGF2 was linked to obesity and elevated resistance to anti-VEGF therapy in a preclinical study. Its inhibition reduced vascular density and restored tumor susceptibility to anti-VEGF therapy in obese mice (97). In addition, crosstalk between FGF2 and VEGF synergistically amplifies breast tumor angiogenesis and metastasis (97, 104). These studies show that adipose-derived factors associated with obesity may not need to originate in the local tumor environment but can come from dysfunctional adipose around the body (25). Future research will fully define the FGF-mediated mechanisms that contribute to malignant transformation of ER-negative breast cells.
Like other growth factor receptors, FGFR can stimulate ER activation independently of the classical steroid hormone ligands (reviewed in (105); Figure 1). Obesity and T2D may promote features of aggressiveness in ER+ breast cancer, such as elevated proliferation, E2F activation, and growth factor signaling (106); pathways associated with a lack of response to endocrine therapy. In luminal breast tumors and cell lines, evidence shows that activated growth factor receptors can phosphorylate ER and its co-regulatory proteins via hormone-independent mechanisms (107). This ligand-independent activation of ER is often mediated through MAPK and PI3K/AKT signaling pathways that phosphorylate ER on several sites; the most commonly evaluated being S118, a target of MAPK, and S167, a target of AKT (Figure 1). Phosphorylation of ER by any pathway can contribute to the transcription of estrogen-responsive genes even in the presence of anti-estrogen therapies (108–110). CAF-derived FGF7 was shown to promote ER phosphorylation and breast cancer cell growth through FGFR2, which ultimately reduced the efficacy of endocrine therapies (111). FGF10/FGFR2 signaling can interfere with classical ER activation to facilitate resistance to endocrine therapies (112). FGF10 was also shown to promote EMT, migration, and colony formation in HR+ and TNBC cells (113). In multiple ER+ breast cancer cell lines, FGF1 and FGF2 can induce membrane ruffling, which often accompanies metastatic invasion (114). A different study showed that nuclear FGFR1 can interact with ER to promote cell proliferation and estrogen-independent transcription of ER target genes (115), showing that some of the effects of FGFR do not rely on FGF ligands. In isolated ER+ breast cancer cells, estrogen treatment was shown to maintain cancer stem cell populations through paracrine FGF9/FGFR3 signaling (116). Likewise, FGFR2 activation maintained mammary tumor-initiating populations in MMTV-PyMT mice, an aggressive model of luminal breast cancer (117). FGFR4, which can mediate signaling from FGF1, FGF4, and FGF8 subfamilies, may facilitate acquired and de novo resistance to endocrine therapies in breast cancer (118). Recently, FGF1 was shown to associate with greater phosphorylate of ER at S118, particularly in endocrine-resistant cells that grow in obese mice even after estrogen loss. Elevated pS118 ER associated with shorter recurrence free survival in patients with ER+ tumors (119). Altogether, the effects of FGF and other growth factors on breast cancer cells are broad and involve many different pathways. One potential target of FGFR signaling includes ER activation independently of estrogen ligands. Since the first line therapy for many breast cancer patients involves depleting peripheral estrogen production, it may be worth reconsidering how the tumor environment maintains ER activation through non-classical mechanisms, and whether this occurs more frequently in the context of poor metabolic health.
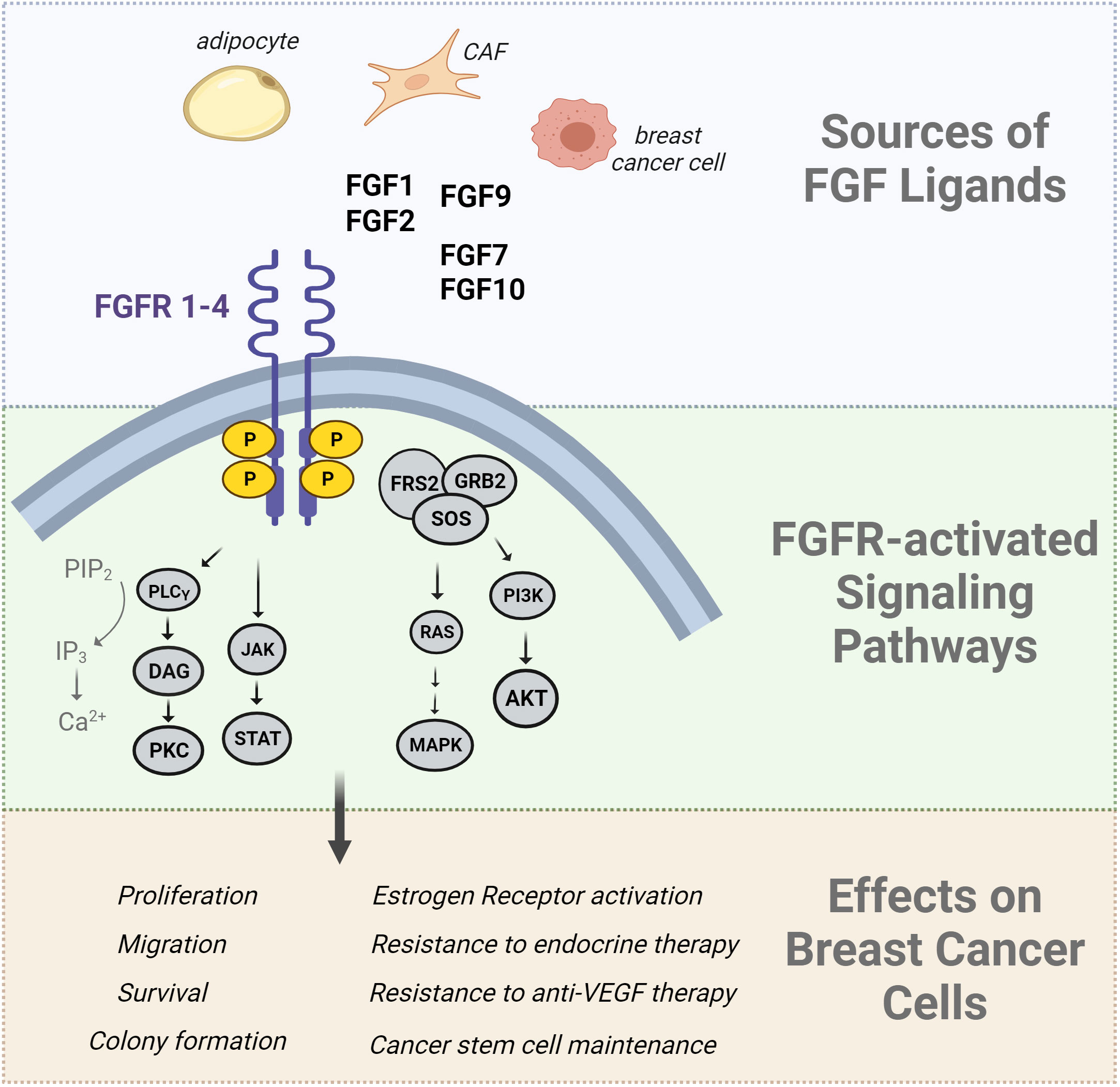
Figure 1 Summary of FGF effects in breast cancer cells. The Fibroblast Growth Factor (FGF) ligands are produced by a variety of cells including adipocytes, cancer associated fibroblasts (CAFs), and breast cancer cells. FGFs bind to FGF receptors, inducing receptor dimerization and transphosphorylation of its intracellular kinase domain. The FGFR signaling network activates downstream signal transduction pathways such as the RAS/MAPK pathway, PI3K/AKT pathway, PLCγ pathway, and JAK/STAT pathways. The downstream effects of these pathways include an increase in cell proliferation, migration, angiogenesis, and survival. Downstream-activated effector molecules of FGFR signaling can also activate the estrogen receptor in a ligand-independent manner which is implicated in endocrine therapy resistance.
One mechanism by which obesity and T2D support breast cancer growth and progression may be through metabolic reprogramming that involves alterations in glycolytic and mitochondrial metabolism. As an important hallmark of cancer, metabolic reprogramming allows cancer cells to adapt their increasing energy demands to available resources for growth, motility, proliferation, and function. Aerobic glycolysis has been widely and consistently observed in many cancer types. The rapid ability of cells to convert glucose into pyruvate and lactate can lead to substantial ATP production but can also support biomass generation. Mitochondrial metabolism also generates precursors for fatty acids, nucleotides, and amino acids (120). Several studies have demonstrated that metabolic reprogramming is a prominent feature in therapy resistance and aggressiveness in breast cancer cells. The metabolic milieu that characterizes T2D and often obesity provides a favorable microenvironment to support breast cancer growth, including sustained hyperglycemia and dyslipidemia (121, 122).
Elevated glycolytic metabolism is a feature of aggressive breast cancer, including TNBC and endocrine-resistant ER-positive cancer cells. High levels of tumor glucose uptake (FDG-PET) predicted a shorter progression-free survival interval in patients on endocrine therapy (123). Tamoxifen-resistant human breast cancer cells (LCC2 and LCC9) have greater glycolytic activity than the MCF7 cells from which they were derived and are susceptible to glycolytic inhibition (124). Multiple studies on specific glycolytic enzymes and transporters show the importance of this metabolic pathway in aggressive breast cancer cell behavior. For example, overexpression of the glucose transporter GLUT1 in TNBC cells supports the invasion, migration, and metastatic potential of these cells (125, 126). HK2 expression is associated with tamoxifen resistance in ER+ breast cancer cells (127), and with proliferation of TNBC cells (128). In a recent study, Zhu et al. demonstrated that the transcription factor ETV4 regulates breast cancer stemness and glycolic metabolism by modulating HK activity in both TNBC and ER+ breast cancer cells (129). ENO1, which catalyzes the reversible conversion of 2-phosphoglycerate and phosphoenolpyruvate, has been implicated in tamoxifen resistance by activation of ER and inhibition of apoptosis (130, 131). Lactate production by lactate dehydrogenase (LDH) enzymes is a feature of enhanced glycolytic activity in cancer cells. The LDH enzymes are significantly elevated in TNBC tumors and associate with shorter overall and disease-free survival (132) and with brain metastasis (133). LDH activity is also elevated in cancer-associated adipocytes, an important component of the tumor microenvironment, suggesting a crosstalk that can be promoted by obesity (134). Overexpression of MCT1 and MCT4, monocarboxylate transporters that shuttle lactate across the plasma membrane, strongly associates with worse survival in TNBC and endocrine-resistant breast cancer cells (133, 135). Treatment of tamoxifen-resistant MCF7 cells with FGF1 led to elevated expression of ETV4 and glycolytic genes, including ENO1, LDH, and SLC2A1. Functional evaluation of metabolism after FGF1 treatment demonstrated enhanced glycolysis in endocrine-resistant cells but not oxygen consumption, consistent with metabolic reprogramming and aggressive phenotype (119). Acidification of the tumor microenvironment is suggested to provide multiple advantages including increased angiogenesis, genomic instability, and potentially selecting for apoptosis-resistant tumor clones (136). Overall, altered cancer cell metabolism, particularly a shift to rapid glycolysis and lactate production, is a feature of aggressive breast tumors, whether TNBC or endocrine therapy resistant ER+. Tumors that benefit from or rely on elevated glucose metabolism may thrive in an environment of poor metabolic health in which glucose, as well as other nutrients are readily available, and that also produces growth-promoting signals from multiple cell types in the breast.
Many potential mechanisms of obesity- or diabetes-associated signaling pathways could stimulate or maintain aggressive cancer cell behavior. One such mechanism may be epithelial to mesenchymal transition (EMT); a feature of aggressive cancer cells that undergo invasion and metastasis. EMT has been linked to altered cancer cell metabolism and to poor metabolic health. The epithelial phenotype is often associated with luminal breast cancers, regardless of ER expression (137). Cells of the basal subtype or TNBC tumors have greater expression of mesenchymal genes, such as SNAI1 (Snail), SNAI2 (Slug), VIM (vimentin), and various MMPs. Multiple studies have suggested that metabolic reprogramming both supports and is caused by the EMT process and related genes (136, 138), and cancer metastasis has been hypothesized to be under metabolic control (139). Several glycolytic enzymes can induce an EMT phenotype, including aldolase A, LDHA, and pyruvate dehydrogenase kinase-1 (136). On the other hand, EMT related proteins may influence metabolic activity in cancer cells (136). One example is a study showing that the Snail transcription factor can inhibit mitochondrial cytochrome C oxidase activity, and may facilitate the loss of fructose-1,6-bisphosphatase 1 in basal cancer cells, which enhances glucose uptake (140). A comparison of metabolic activity between breast cancer cells representing the luminal subtype (MCF7, T47D) and the basal/mesenchymal subtype (MDA-231, MDA-435) revealed distinct metabolic signatures that were influenced by EMT proteins (141). Compared to luminal cells, the basal cells had impaired, but not completely defective mitochondrial function, were less dependent on mitochondrial ATP production, and displayed elevated glycolysis and lactate production. Knockdown of E-cadherin or β-catenin in luminal cells phenocopied the basal cell metabolic phenotype, decreasing mitochondrial respiration and increasing lactate production. Low expression of mitochondrial oxidative genes is associated with metastatic potential and poor clinical outcomes across multiple cancer types (142). In cervical cancer cells, Porporato et al. showed that mitochondria must be present, but defective, in cells undergoing metastasis, based on increased cell migration that accompanied partial electron transport chain inhibition (139). The investigators suggested that metastatic progenitor cells are characterized by mitochondrial superoxide production, both from overloaded TCA cycling and from defective electron transport chain activity (139). Recently, the concept of mitochondrial overload resurfaced with the suggestion that glycolytic production of lactate occurs when cancer cell mitochondria cannot oxidize NADH as fast as glycolysis can produce it (120). It is currently not well understood if aggressive or metastatic breast cancer cells prefer glycolysis because of specific defects in mitochondrial electron transport subunits. A thorough investigation of how mitochondrial function may be altered during breast cancer progression, or in specific populations such as cancer stem cells, may help better target therapies to patients.
Many of the genes encoding glycolytic enzymes are regulated by the HIF1α transcription factor that is upregulated or stabilized in hypoxic environments (143). The consequences to cancer cells include EMT, invasion, metastasis, therapy resistance, and metabolic reprogramming (143). The obese or diabetic breast tissue is often characterized by reduced vascularization, which leads to hypoxia and inflammation. This, coupled with the excess available nutrients is the prime environment to support tumor progression. Sustained hyperinsulinemia in a mouse model of T2D promoted ER+ breast cancer cell growth associated with stabilization of HIF1α (144). A recent study reported that obesity associated with greater DNA damage in the breast epithelium of women with BRCA mutations (76). In several models, DNA damage was shown to be enhanced by estrogen and insulin signaling; two hormones that likely mediate many adverse effects of obesity on breast cancer. The top pathway enriched in isolated breast epithelial organoids from women with obesity and BRCA mutations was HIF1α signaling (76). These two reports clearly link a poor metabolic environment to changes that could facilitate glycolytic metabolism in tumors. Future studies are needed to define the metabolic changes in cancer cells in these contexts, and whether they can be targeted to prevent disease progression.
Within the breast, CAFs and cancer cells have intricate interactions where each can reprogram its metabolism to benefit the other (145). A recent study using murine models of breast cancer found that cancer-associated adipocytes, which are present at the invasive front of tumors, underwent dedifferentiation to a myofibroblast-like cell type that expressed markers of immune cells and ECM remodeling (146). These de-differentiated adipocytes had enhanced glycolytic metabolism and oxygen consumption compared to CAFs that did not arise from adipocytes (146). Preventing adipocyte dedifferentiation reduced tumor growth under chow and high-fat diet conditions (146), indicating the tumor promotional role of lipids when they are present in excess. Another recent study evaluated breast adipose tissue fibroblasts isolated from tissue immediately adjacent to tumors (CAFs) and distant from tumors in the same individuals, classified as either lean (BMI<25kg/m2) or obese (BMI>35kg/m2) (147). The CAFs from women with obesity expressed higher levels of myoepithelial markers such as CD29, alpha-smooth muscle actin (ACTA2), and connective tissue growth factor (CTGF). In contrast, the CAFs from lean individuals expressed markers associated with inflammation such as IL1β and CXCL10. Notably, FGF2 and to a lesser extent, FGF1, were each more highly expressed in CAFs compared to distant fibroblasts, but the distant fibroblasts from women with obesity had greater FGF2 expression than the lean group. This could indicate a more widespread impact of the tumor on the breast in obesity, or it could reflect a tumor-promotional milieu present throughout the obese breast tissue. In several different assays, the most prominent effect of fibroblast conditioned media on cancer cells, including proliferation and motility, appeared to be from the site of origin, with CAFs imparting a more aggressive cancer cell phenotype than distant fibroblasts. However, compared to all other groups (lean CAFs, lean or obese distant fibroblasts), obese CAFs promoted greater growth of luminal breast cancer spheroids in direct co-culture assays. Interestingly, the pathways altered in ER+/HER2+ (BT474) cancer cells cultured with lean CAFs included cell migration and EMT, while those altered by obese CAFs included cell cycle progression and cellular metabolism. In TNBC (MDA-231), the obese CAFs induced a cancer stem cell-like phenotype, while in ER+/HER2+ cells the impact was greater for EMT (147). Reprogramming of CAF metabolism towards glycolysis and lactate production can promote the growth of TNBC in vivo (148). A different study found that extracellular vesicles (EVs) from adipocytes induced genes associated with EMT and cancer stem cells in ER+ and TNBC cell lines (149). Conditioned media from breast adipocytes collected from women with obesity stimulated migration and invasion of breast cancer cells, but this phenotype was attenuated when EVs were depleted (149). TNBC cells grafted in a murine model of streptozotocin-induced diabetes showed greater tumor growth and had elevated expression of EMT markers and glycolytic enzymes compared to cells grown in non-diabetic mice (150). Diet-induced obesity accelerated growth and elevated cancer stem cell and EMT markers in MMTV-Wnt1 mice, a model of TNBC (151). The effects of obesity in this study were mediated by circulating leptin, produced by adipose tissue.
FGFR signaling has been linked to EMT in various cancer types, such as lung, pancreas, and prostate; an effect that may be FGFR isoform-specific (152). In endometrial cancer, FGFR2 mutations were associated with features of EMT, such as high vimentin staining, low E-cadherin, and tumor budding (153). FGFR1 was identified as a potential mediator of EMT-induced drug resistance in EGFR mutant non-small cell lung cancer (154). In HER2-transformed breast cancer cells, FGF2/FGFR1 were identified to be highly expressed in Lapatinib-resistant populations (155). FGF signaling, through the MAPK pathway, stabilized the Twist transcription factor to help maintain the mesenchymal, drug-resistant cell population (155). A separate study showed that, in chemotherapy-resistant MCF7 and MDA-231 cells, glycolytic genes were elevated compared to parental lines, and FGFR4 inhibition reduced glycolytic flux (156). Overexpression of N-cadherin in MMTV-Neu mice, a model of HER2+ breast cancer, did not alter primary tumor latency or growth, but significantly augmented lung metastasis (157). In cell lines derived from these primary tumors, N-cadherin overexpression was associated with greater levels and phosphorylation of FGFR1 and with greater relative levels of the mesenchymal IIIc isoform of FGFR2 compared to the epithelial IIIb isoform. The expression of EMT markers Snail and Slug was elevated in N-cadherin overexpressing cells and was dependent upon FGFR.
Metabolism of other nutrients besides glucose, such as amino acids and lipids, is often altered in aggressive breast tumors. Breast cancer cells are often dependent on glutamine for the production of carbon and nitrogen needed for proliferation, invasion, and metastasis (158–160). Serum glutamine levels are elevated in individuals with obesity and T2D, as well as in breast cancer patients. The amino acid transporters SLC6A14 and SLC1A5 are upregulated in breast cancer cells and implicated in therapy resistance (161). TNBC displays increased glutamine uptake and glutamine-related enzymes (160) and reduction of glutamine transporters reduces the proliferation and migration of TNBC (159). Circulating free fatty acids were correlated with increased proliferation and aggressiveness of ER+ breast cancer via activation of ER and mTOR pathways and reprogrammed cancer cell metabolism (162). In a preclinical model, the induction of hypercholesterolemia in mice resulted in greater breast cancer growth (163). Tamoxifen resistance in ER+ breast cancer cells was found to be linked to deregulation of cholesterol pathways and altered lysosomal integrity (164). ER-dependent breast cancer cells develop resistance to aromatase inhibition through epigenetic and transcriptomic activation of cholesterol biosynthesis that contributes to aggressive breast phenotypes (165). Key enzymes and mediators of fatty acid metabolic pathways have been shown to drive metabolic reprogramming of endocrine-resistant invasive lobular carcinoma cells (166). As more and more investigators incorporate aspects of obesity and metabolic disease into preclinical models and in the analysis of clinical specimens, our understanding of how these environments associate with and influence aggressive tumor cell metabolism will greatly improve.
Obesity and T2D are closely linked chronic diseases that impact millions of adults worldwide. The proportion of children with obesity has increased in recent decades, foretelling a future where most adults may be impacted by poor metabolic health. The relationship surrounding obesity, T2D, and breast cancer is incredibly complex, but scientists have uncovered many interesting mechanisms. Growth factor signaling from the tumor environment, specifically FGF/FGFR pathways, may be one important link between poor metabolic health and aggressive breast cancer. Cancer cells themselves may benefit from a complex relationship between EMT and metabolic reprogramming that is influenced by host metabolic health through growth factor production and excess nutrient availability (Figure 2). People who are metabolically healthy and who do not have obesity do develop and die from breast cancer, but statistically, it is less likely compared to those with poor metabolic health. It is not known if aggressive breast cancers are unique between these two groups of individuals, and currently, BMI and metabolic health is not factored into treatment decisions for breast cancer. Based on the studies reviewed here, some specific knowledge gaps remain. For example, several preclinical studies show a link between FGFR and ER activation. The loss of classical ER target gene expression signature is associated with endocrine therapy resistance, but it isn’t clear if growth factor receptor activation alters the ER-dependent transcriptome resulting in a distinct estrogen-independent ER transcriptome. Is there a gene expression signature that could indicate what is activating ER in endocrine-resistant breast cancers? In some people with obesity or T2D, a strategy to directly target ER may be more effective than targeting estrogen synthesis. Another area for investigation involves the similarities and differences between TNBC and endocrine-resistant ER+ breast cancers. In addition to estrogen-independent ER+ tumor growth, FGFs, and FGFR have been linked to malignant transformation of ER-negative breast cells, potentially accelerating TNBC formation. Are there common driving mechanisms that can be targeted in these tumors specifically? Further, what are the unique molecular responses of endocrine-sensitive and endocrine-resistant breast cancer cells to FGF treatment? From the host perspective, FGF signaling is a normal, necessary growth-promoting pathway, so is it feasible to target these factors directly for prevention or treatment of breast cancer. In summary, the prevalent incidence of obesity, T2D, and poor metabolic health, which may remain undefined, illustrates the importance of paying attention to BMI, adiposity, and individual metabolic state when evaluating breast cancer risk or prognosis. Future research will help illuminate the role of whole-body metabolism in breast cancer incidence and progression.
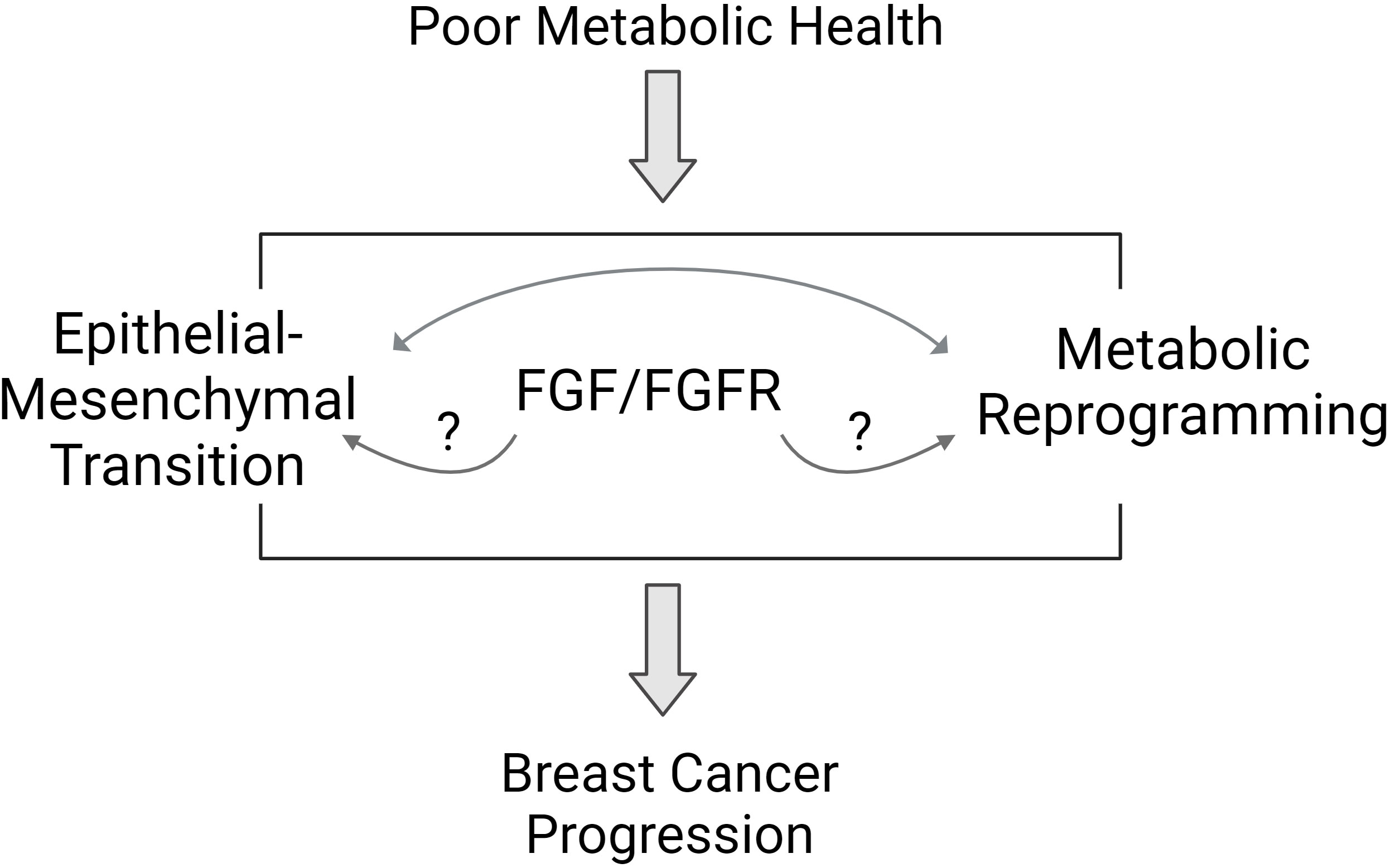
Figure 2 Proposed pathways mediating the link between poor metabolic health and breast cancer biology. Poor metabolic health influences breast cancer progression through a variety of mechanisms. Studies suggest that the FGFR signaling pathway may mediate some of the effects of whole-body metabolism on breast tumor biology. Features of aggressive breast cancer include epithelial to mesenchymal transition (EMT) and metabolic reprogramming. FGFR signaling may influence each of these areas to promote aggressive breast cancer behavior.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by NIH R01 CA241156 (EW) and R01 ES030695 (JB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; EMT, epithelial to mesenchymal transition; ER, estrogen receptor; HER2, human epidermal growth factor receptor; TNBC, triple-negative breast cancer; T2D, type 2 diabetes; BMI, body mass index.
2. Seer N. Cancer stat facts: female breast cancer subtypes. (2016-2020). Available at: https://seer.cancer.gov/statfacts/html/breast-subtypes.html
4. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
5. Mobasseri M, Shirmohammadi M, Amiri T, Vahed N, Hosseini Fard H, Ghojazadeh M. Prevalence and incidence of type 1 diabetes in the world: a systematic review and meta-analysis. Health Promot Perspect (2020) 10:98–115. doi: 10.34172/hpp.2020.18
6. Bhupathiraju SN, Hu FB. Epidemiology of obesity and diabetes and their cardiovascular complications. Circ Res (2016) 118:1723–35. doi: 10.1161/CIRCRESAHA.115.306825
7. Leitner DR, Fruhbeck G, Yumuk V, Schindler K, Micic D, Woodward E, et al. Obesity and type 2 diabetes: two diseases with a need for combined treatment strategies - EASO can lead the way. Obes Facts (2017) 10:483–92. doi: 10.1159/000480525
8. Goodwin PJ, Stambolic V. Obesity and insulin resistance in breast cancer–chemoprevention strategies with a focus on metformin. Breast (2011) 20 Suppl 3:S31–35. doi: 10.1016/S0960-9776(11)70291-0
9. Chlebowski RT, Luo J, Anderson GL, Barrington W, Reding K, Simon MS, et al. Weight loss and breast cancer incidence in postmenopausal women. Cancer (2019) 125:205–12. doi: 10.1002/cncr.31687
10. Lovrics O, Butt J, Lee Y, Lovrics P, Boudreau V, Anvari M, et al. The effect of bariatric surgery on breast cancer incidence and characteristics: A meta-analysis and systematic review. Am J Surg (2021) 222:715–22. doi: 10.1016/j.amjsurg.2021.03.016
11. Perou CM, Sørlie T, Eisen MB, Van De Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature (2000) 406:747–52. doi: 10.1038/35021093
12. Sorlie T, Perou CM, TibshIrani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A (2001) 98:10869–74. doi: 10.1073/pnas.191367098
13. Sorlie T. Molecular portraits of breast cancer: tumour subtypes as distinct disease entities. Eur J Cancer (2004) 40:2667–75. doi: 10.1016/j.ejca.2004.08.021
14. Anurag M, Ellis MJ, Haricharan S. DNA damage repair defects as a new class of endocrine treatment resistance driver. Oncotarget (2018) 9:36252–3. doi: 10.18632/oncotarget.26363
15. Anurag M, Punturi N, Hoog J, Bainbridge MN, Ellis MJ, Haricharan S. Comprehensive profiling of DNA repair defects in breast cancer identifies a novel class of endocrine therapy resistance drivers. Clin Cancer Res (2018) 24:4887–99. doi: 10.1158/1078-0432.CCR-17-3702
16. Lei JT, Anurag M, Haricharan S, Gou X, Ellis MJ. Endocrine therapy resistance: new insights. Breast (2019) 48 Suppl 1:S26–s30. doi: 10.1016/S0960-9776(19)31118-X
17. Rani A, Stebbing J, Giamas G, Murphy J. Endocrine resistance in hormone receptor positive breast cancer-from mechanism to therapy. Front Endocrinol (Lausanne) (2019) 10:245. doi: 10.3389/fendo.2019.00245
18. Haque MM, Desai KV. Pathways to endocrine therapy resistance in breast cancer. Front Endocrinol (2019) 10. doi: 10.3389/fendo.2019.00573
19. Belachew EB, Sewasew DT. Molecular mechanisms of endocrine resistance in estrogen-receptor-positive breast cancer. Front Endocrinol (2021) 12:599586. doi: 10.3389/fendo.2021.599586
20. Bai X, Ni J, Beretov J, Graham P, Li Y. Triple-negative breast cancer therapeutic resistance: Where is the Achilles' heel? Cancer Lett (2021) 497:100–11. doi: 10.1016/j.canlet.2020.10.016
21. Chakraborty D, Benham V, Bullard B, Kearney T, Hsia HC, Gibbon D, et al. Fibroblast growth factor receptor is a mechanistic link between visceral adiposity and cancer. Oncogene (2017) 36:6668–79. doi: 10.1038/onc.2017.278
22. Benham V, Chakraborty D, Bullard B, Bernard JJ. A role for FGF2 in visceral adiposity-associated mammary epithelial transformation. Adipocyte (2018) 7:113–20. doi: 10.1080/21623945.2018.1445889
23. Wellberg EA, Kabos P, Gillen AE, Jacobsen BM, Brechbuhl HM, Johnson SJ, et al. FGFR1 underlies obesity-associated progression of estrogen receptor-positive breast cancer after estrogen deprivation. JCI Insight (2018) 3. doi: 10.1172/jci.insight.120594
24. Boothby-Shoemaker W, Benham V, Paithankar S, Shankar R, Chen B, Bernard JJ. The relationship between leptin, the leptin receptor and FGFR1 in primary human breast tumors. Cells (2020) 9. doi: 10.3390/cells9102224
25. Bernard JJ, Wellberg EA. The tumor promotional role of adipocytes in the breast cancer micro- and macro-environment. Am J Pathol (2021) 191:1342–52. doi: 10.1016/j.ajpath.2021.02.006
26. Wellberg EA, Corleto KA, Checkley LA, Jindal S, Johnson G, Higgins JA, et al. Preventing ovariectomy-induced weight gain decreases tumor burden in rodent models of obesity and postmenopausal breast cancer. Breast Cancer Res (2022) 24:42. doi: 10.1186/s13058-022-01535-x
27. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, et al. Body fatness and cancer–viewpoint of the IARC working group. N Engl J Med (2016) 375:794–8. doi: 10.1056/NEJMsr1606602
28. Matthews SB, Thompson HJ. The obesity-breast cancer conundrum: an analysis of the issues. Int J Mol Sci (2016) 17. doi: 10.3390/ijms17060989
29. Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D, et al. Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ (2007) 335:1134. doi: 10.1136/bmj.39367.495995.AE
30. Ritte R, Lukanova A, Berrino F, Dossus L, Tjonneland A, Olsen A, et al. Adiposity, hormone replacement therapy use and breast cancer risk by age and hormone receptor status: a large prospective cohort study. Breast Cancer Res (2012) 14:R76. doi: 10.1186/bcr3186
31. Munsell MF, Sprague BL, Berry DA, Chisholm G, Trentham-Dietz A. Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. Epidemiol Rev (2014) 36:114–36. doi: 10.1093/epirev/mxt010
32. Nagrani R, Mhatre S, Rajaraman P, Soerjomataram I, Boffetta P, Gupta S, et al. Central obesity increases risk of breast cancer irrespective of menopausal and hormonal receptor status in women of South Asian Ethnicity. Eur J Cancer (2016) 66:153–61. doi: 10.1016/j.ejca.2016.07.022
33. Picon-Ruiz M, Morata-Tarifa C, Valle-Goffin JJ, Friedman ER, Slingerland JM. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J Clin (2017) 67:378–97. doi: 10.3322/caac.21405
34. Chen L, Cook LS, Tang MT, Porter PL, Hill DA, Wiggins CL, et al. Body mass index and risk of luminal, HER2-overexpressing, and triple negative breast cancer. Breast Cancer Res Treat (2016) 157:545–54. doi: 10.1007/s10549-016-3825-9
35. Majed B, Dozol A, Ribassin-Majed L, Senouci K, Asselain B. Increased risk of contralateral breast cancers among overweight and obese women: a time-dependent association. Breast Cancer Res Treat (2011) 126:729–38. doi: 10.1007/s10549-010-1153-z
36. Majed B, Moreau T, Asselain B, Curie Institute Breast Cancer G. Overweight, obesity and breast cancer prognosis: optimal body size indicator cut-points. Breast Cancer Res Treat (2009) 115:193–203. doi: 10.1007/s10549-008-0065-7
37. Dolle JM, Daling JR, White E, Brinton LA, Doody DR, Porter PL, et al. Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomarkers Prev (2009) 18:1157–66. doi: 10.1158/1055-9965.EPI-08-1005
38. Kawai M, Malone KE, Tang MT, Li CI. Height, body mass index (BMI), BMI change, and the risk of estrogen receptor-positive, HER2-positive, and triple-negative breast cancer among women ages 20 to 44 years. Cancer (2014) 120:1548–56. doi: 10.1002/cncr.28601
39. Millikan RC, Newman B, Tse CK, Moorman PG, Conway K, Dressler LG, et al. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat (2008) 109:123–39. doi: 10.1007/s10549-007-9632-6
40. Mcgee SA, Durham DD, Tse CK, Millikan RC. Determinants of breast cancer treatment delay differ for African American and White women. Cancer Epidemiol Biomarkers Prev (2013) 22:1227–38. doi: 10.1158/1055-9965.EPI-12-1432
41. Pierobon M, Frankenfeld CL. Obesity as a risk factor for triple-negative breast cancers: a systematic review and meta-analysis. Breast Cancer Res Treat (2013) 137:307–14. doi: 10.1007/s10549-012-2339-3
42. Senie RT, Rosen PP, Rhodes P, Lesser ML, Kinne DW. Obesity at diagnosis of breast carcinoma influences duration of disease-free survival. Ann Intern Med (1992) 116:26–32. doi: 10.7326/0003-4819-116-1-26
43. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med (2003) 348:1625–38. doi: 10.1056/NEJMoa021423
44. Ewertz M, Gray KP, Regan MM, Ejlertsen B, Price KN, Thurlimann B, et al. Obesity and risk of recurrence or death after adjuvant endocrine therapy with letrozole or tamoxifen in the breast international group 1-98 trial. J Clin Oncol (2012) 30:3967–75. doi: 10.1200/JCO.2011.40.8666
45. Petrelli F, Cortellini A, Indini A, Tomasello G, Ghidini M, Nigro O, et al. Association of obesity with survival outcomes in patients with cancer: A systematic review and meta-analysis. JAMA Netw Open (2021) 4:e213520. doi: 10.1001/jamanetworkopen.2021.3520
46. Bonet C, Crous-Bou M, Tsilidis KK, Gunter MJ, Kaaks R, Schulze MB, et al. The association between body fatness and mortality among breast cancer survivors: results from a prospective cohort study. Eur J Epidemiol (2023) 38:545–57. doi: 10.1007/s10654-023-00979-5
47. Chan DSM, Vieira R, Abar L, Aune D, Balducci K, Cariolou M, et al. Postdiagnosis body fatness, weight change and breast cancer prognosis: Global Cancer Update Program (CUP global) systematic literature review and meta-analysis. Int J Cancer (2023) 152:572–99. doi: 10.1002/ijc.34322
48. Srokowski TP, Fang S, Hortobagyi GN, Giordano SH. Impact of diabetes mellitus on complications and outcomes of adjuvant chemotherapy in older patients with breast cancer. J Clin Oncol (2009) 27:2170–6. doi: 10.1200/JCO.2008.17.5935
49. Peairs KS, Barone BB, Snyder CF, Yeh HC, Stein KB, Derr RL, et al. Diabetes mellitus and breast cancer outcomes: a systematic review and meta-analysis. J Clin Oncol (2011) 29:40–6. doi: 10.1200/JCO.2009.27.3011
50. De Bruijn KM, Arends LR, Hansen BE, Leeflang S, Ruiter R, Van Eijck CH. Systematic review and meta-analysis of the association between diabetes mellitus and incidence and mortality in breast and colorectal cancer. Br J Surg (2013) 100:1421–9. doi: 10.1002/bjs.9229
51. Boyle P, Boniol M, Koechlin A, Robertson C, Valentini F, Coppens K, et al. Diabetes and breast cancer risk: a meta-analysis. Br J Cancer (2012) 107:1608–17. doi: 10.1038/bjc.2012.414
52. Muti P, Quattrin T, Grant BJ, Krogh V, Micheli A, Schunemann HJ, et al. Fasting glucose is a risk factor for breast cancer: a prospective study. Cancer Epidemiol Biomarkers Prev (2002) 11:1361–8.
53. Zhou Y, Zhang X, Gu C, Xia J. Influence of diabetes mellitus on mortality in breast cancer patients. ANZ J Surg (2015) 85:972–8. doi: 10.1111/ans.12877
54. Zhao XB, Ren GS. Diabetes mellitus and prognosis in women with breast cancer: A systematic review and meta-analysis. Med (Baltimore) (2016) 95:e5602. doi: 10.1097/MD.0000000000005602
55. Campbell PT, Newton CC, Patel AV, Jacobs EJ, Gapstur SM. Diabetes and cause-specific mortality in a prospective cohort of one million U.S. adults. Diabetes Care (2012) 35:1835–44. doi: 10.2337/dc12-0002
56. Lee KN, Torres MA, Troeschel AN, He J, Gogineni K, Mccullough LE. Type 2 diabetes, breast cancer specific and overall mortality: Associations by metformin use and modification by race, body mass, and estrogen receptor status. PLoS One (2020) 15:e0232581. doi: 10.1371/journal.pone.0232581
57. Van De Poll-Franse LV, Houterman S, Janssen-Heijnen ML, Dercksen MW, Coebergh JW, Haak HR. Less aggressive treatment and worse overall survival in cancer patients with diabetes: a large population based analysis. Int J Cancer (2007) 120:1986–92. doi: 10.1002/ijc.22532
58. Yancik R, Wesley MN, Ries LA, Havlik RJ, Edwards BK, Yates JW. Effect of age and comorbidity in postmenopausal breast cancer patients aged 55 years and older. JAMA (2001) 285:885–92. doi: 10.1001/jama.285.7.885
59. Raychaudhuri S, Dieli-Conwright CM, Cheng RK, Barac A, Reding KW, Vasbinder A, et al. A review of research on the intersection between breast cancer and cardiovascular research in the Women's Health Initiative (WHI). Front Oncol (2022) 12:1039246. doi: 10.3389/fonc.2022.1039246
60. Suzuki R, Orsini N, Saji S, Key TJ, Wolk A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status–a meta-analysis. Int J cancer J Int du Cancer (2009) 124:698–712. doi: 10.1002/ijc.23943
61. Vrieling A, Buck K, Kaaks R, Chang-Claude J. Adult weight gain in relation to breast cancer risk by estrogen and progesterone receptor status: a meta-analysis. Breast Cancer Res Treat (2010) 123:641–9. doi: 10.1007/s10549-010-1116-4
62. Gathirua-Mwangi WG, Zollinger TW, Murage MJ, Pradhan KR, Champion VL. Adult BMI change and risk of Breast Cancer: National Health and Nutrition Examination Survey (NHANES) 2005-2010. Breast Cancer (2015) 22:648–56. doi: 10.1007/s12282-015-0638-3
63. Keum N, Greenwood DC, Lee DH, Kim R, Aune D, Ju W, et al. Adult weight gain and adiposity-related cancers: a dose-response meta-analysis of prospective observational studies. J Natl Cancer Inst (2015) 107. doi: 10.1093/jnci/djv088
64. Gunter MJ, Xie X, Xue X, Kabat GC, Rohan TE, Wassertheil-Smoller S, et al. Breast cancer risk in metabolically healthy but overweight postmenopausal women. Cancer Res (2015) 75:270–4. doi: 10.1158/0008-5472.CAN-14-2317
65. Kabat GC, Kim MY, Lee JS, Ho GY, Going SB, Beebe-Dimmer J, et al. Metabolic obesity phenotypes and risk of breast cancer in postmenopausal women. Cancer Epidemiol Biomarkers Prev (2017) 26:1730–5. doi: 10.1158/1055-9965.EPI-17-0495
66. Park YM, White AJ, Nichols HB, O'brien KM, Weinberg CR, Sandler DP. The association between metabolic health, obesity phenotype and the risk of breast cancer. Int J Cancer (2017) 140:2657–66. doi: 10.1002/ijc.30684
67. Mahamat-Saleh Y, Rinaldi S, Kaaks R, Biessy C, Gonzalez-Gil EM, Murphy N, et al. Metabolically defined body size and body shape phenotypes and risk of postmenopausal breast cancer in the European Prospective Investigation into Cancer and Nutrition. Cancer Med (2023) 12:12668–82. doi: 10.1002/cam4.5896
68. Zhang AMY, Wellberg EA, Kopp JL, Johnson JD. Hyperinsulinemia in obesity, inflammation, and cancer. Diabetes Metab J (2021) 45:285–311. doi: 10.4093/dmj.2021.0131
69. Kim DS, Scherer PE. Obesity, diabetes, and increased cancer progression. Diabetes Metab J (2021) 45:799–812. doi: 10.4093/dmj.2021.0077
70. Barone I, Caruso A, Gelsomino L, Giordano C, Bonofiglio D, Catalano S, et al. Obesity and endocrine therapy resistance in breast cancer: Mechanistic insights and perspectives. Obes Rev (2022) 23. doi: 10.1111/obr.13358
71. Mantzoros C, Petridou E, Dessypris N, Chavelas C, Dalamaga M, Alexe DM, et al. Adiponectin and breast cancer risk. J Clin Endocrinol Metab (2004) 89:1102–7. doi: 10.1210/jc.2003-031804
72. Chu DT, Phuong TNT, Tien NLB, Tran DK, Nguyen TT, Thanh VV, et al. The effects of adipocytes on the regulation of breast cancer in the tumor microenvironment: an update. Cells (2019) 8. doi: 10.3390/cells8080857
73. Catalano S, Mauro L, Marsico S, Giordano C, Rizza P, Rago V, et al. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J Biol Chem (2004) 279:19908–15. doi: 10.1074/jbc.M313191200
74. Geisler J, Haynes B, Ekse D, Dowsett M, Lønning PE. Total body aromatization in postmenopausal breast cancer patients is strongly correlated to plasma leptin levels. J Steroid Biochem Mol Biol (2007) 104:27–34. doi: 10.1016/j.jsbmb.2006.09.040
75. Strong AL, Ohlstein JF, Biagas BA, Rhodes LV, Pei DT, Tucker HA, et al. Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res (2015) 17:112. doi: 10.1186/s13058-015-0622-z
76. Bhardwaj P, Iyengar NM, Zahid H, Carter KM, Byun DJ, Choi MH, et al. Obesity promotes breast epithelium DNA damage in women carrying a germline mutation in BRCA1 or BRCA2. Sci Transl Med (2023) 15:eade1857. doi: 10.1126/scitranslmed.ade1857
77. Folkerd EJ, Dixon JM, Renshaw L, A'hern RP, Dowsett M. Suppression of plasma estrogen levels by letrozole and anastrozole is related to body mass index in patients with breast cancer. J Clin Oncol (2012) 30:2977–80. doi: 10.1200/JCO.2012.42.0273
78. Elliott MJ, Ennis M, Pritchard KI, Townsley C, Warr D, Elser C, et al. Association between BMI, vitamin D, and estrogen levels in postmenopausal women using adjuvant letrozole: a prospective study. NPJ Breast Cancer (2020) 6:22. doi: 10.1038/s41523-020-0166-y
79. Jonat W, Howell A, Blomqvist C, Eiermann W, Winblad G, Tyrrell C, et al. A randomised trial comparing two doses of the new selective aromatase inhibitor anastrozole (Arimidex) with megestrol acetate in postmenopausal patients with advanced breast cancer. Eur J Cancer (1996) 32A:404–12. doi: 10.1016/0959-8049(95)00014-3
80. Buzdar AU, Jones SE, Vogel CL, Wolter J, Plourde P, Webster A. A phase III trial comparing anastrozole (1 and 10 milligrams), a potent and selective aromatase inhibitor, with megestrol acetate in postmenopausal women with advanced breast carcinoma. Arimidex Study Group. Cancer (1997) 79:730–9. doi: 10.1002/(SICI)1097-0142(19970215)79:4<730::AID-CNCR10>3.0.CO;2-0
81. Iyengar NM, Gucalp A, Dannenberg AJ, Hudis CA. Obesity and cancer mechanisms: tumor microenvironment and inflammation. J Clin Oncol (2016) 34:4270–6. doi: 10.1200/JCO.2016.67.4283
82. Brown KA, Iyengar NM, Zhou XK, Gucalp A, Subbaramaiah K, Wang H, et al. Menopause is a determinant of breast aromatase expression and its associations with BMI, inflammation, and systemic markers. J Clin Endocrinol Metab (2017) 102:1692–701. doi: 10.1210/jc.2016-3606
83. Wang Z, Aguilar EG, Luna JI, Dunai C, Khuat LT, Le CT, et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat Med (2019) 25:141–51. doi: 10.1038/s41591-018-0221-5
84. Porsche CE, Delproposto JB, Geletka L, O'rourke R, Lumeng CN. Obesity results in adipose tissue T cell exhaustion. JCI Insight (2021) 6. doi: 10.1172/jci.insight.139793
85. Mcquade JL, Daniel CR, Hess KR, Mak C, Wang DY, Rai RR, et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: a retrospective, multicohort analysis. Lancet Oncol (2018) 19:310–22. doi: 10.1016/S1470-2045(18)30078-0
86. Prattichizzo F, De Nigris V, La Sala L, Procopio AD, Olivieri F, Ceriello A. "Inflammaging" as a druggable target: A senescence-associated secretory phenotype-centered view of type 2 diabetes. Oxid Med Cell Longev (2016) 2016:1810327. doi: 10.1155/2016/1810327
87. Henninger AM, Eliasson B, Jenndahl LE, Hammarstedt A. Adipocyte hypertrophy, inflammation and fibrosis characterize subcutaneous adipose tissue of healthy, non-obese subjects predisposed to type 2 diabetes. PLoS One (2014) 9:e105262. doi: 10.1371/journal.pone.0105262
89. Fearon AE, Grose RP. The FGFR receptor family. In: Wheeler D, Yarden Y, editors. Receptor Tyrosine Kinases: Family and Subfamilies. Cham: Springer (2015).
90. Pond AC, Bin X, Batts T, Roarty K, Hilsenbeck S, Rosen JM. Fibroblast growth factor receptor signaling is essential for normal mammary gland development and stem cell function. Stem Cells (2013) 31:178–89. doi: 10.1002/stem.1266
91. Welm BE, Freeman KW, Chen M, Contreras A, Spencer DM, Rosen JM. Inducible dimerization of FGFR1: development of a mouse model to analyze progressive transformation of the mammary gland. J Cell Biol (2002) 157:703–14. doi: 10.1083/jcb.200107119
92. Francavilla C, O'brien CS. Fibroblast growth factor receptor signalling dysregulation and targeting in breast cancer. Open Biol (2022) 12:210373. doi: 10.1098/rsob.210373
93. Hillers-Ziemer LE, Kuziel G, Williams AE, Moore BN, Arendt LM. Breast cancer microenvironment and obesity: challenges for therapy. Cancer Metastasis Rev (2022) 41:627–47. doi: 10.1007/s10555-022-10031-9
94. Strong AL, Pei DT, Hurst CG, Gimble JM, Burow ME, Bunnell BA. Obesity enhances the conversion of adipose-derived stromal/stem cells into carcinoma-associated fibroblast leading to cancer cell proliferation and progression to an invasive phenotype. Stem Cells Int (2017) 2017:9216502. doi: 10.1155/2017/9216502
95. Degraffenried L, Harlow B, Davalos A, Brenner A, Jolly C, Hursting S. Obese conditions induce changes in stromal fibroblast phenotype. Cancer Res (2018) 78.
96. Wang S, Cao S, Arhatte M, Li D, Shi Y, Kurz S, et al. Adipocyte Piezo1 mediates obesogenic adipogenesis through the FGF1/FGFR1 signaling pathway in mice. Nat Commun (2020) 11:2303. doi: 10.1038/s41467-020-16026-w
97. Incio J, Ligibel JA, Mcmanus DT, Suboj P, Jung K, Kawaguchi K, et al. Obesity promotes resistance to anti-VEGF therapy in breast cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med (2018) 10. doi: 10.1126/scitranslmed.aag0945
98. Wu X, Zahari MS, Renuse S, Sahasrabuddhe NA, Chaerkady R, Kim MS, et al. Quantitative phosphoproteomic analysis reveals reciprocal activation of receptor tyrosine kinases between cancer epithelial cells and stromal fibroblasts. Clin Proteomics (2018) 15:21. doi: 10.1186/s12014-018-9197-x
99. Suh J, Kim DH, Lee YH, Jang JH, Surh YJ. Fibroblast growth factor-2, derived from cancer-associated fibroblasts, stimulates growth and progression of human breast cancer cells via FGFR1 signaling. Mol Carcinog (2020) 59:1028–40. doi: 10.1002/mc.23233
100. Holowatyj AN, Gigic B, Warby CA, Ose J, Lin T, Schrotz-King P, et al. The use of human serum samples to study Malignant transformation: A pilot study. Cells (2021) 10. doi: 10.3390/cells10102670
101. Chen J, Zhou Z, Yao Y, Dai J, Zhou D, Wang L, et al. Dipalmitoylphosphatidic acid inhibits breast cancer growth by suppressing angiogenesis via inhibition of the CUX1/FGF1/HGF signalling pathway. J Cell Mol Med (2018) 22:4760–70. doi: 10.1111/jcmm.13727
102. Francavilla C, O'brien CS. Fibroblast growth factor receptor signalling dysregulation and targeting in breast cancer. Open Biol (2022) 12. doi: 10.1098/rsob.210373
103. Zhang L, Kharbanda S, Chen D, Bullocks J, Miller DL, Ding IY, et al. MCF-7 breast carcinoma cells overexpressing FGF-1 form vascularized, metastatic tumors in ovariectomized or tamoxifen-treated nude mice. Oncogene (1997) 15:2093–108. doi: 10.1038/sj.onc.1201386
104. Madu CO, Wang S, Madu CO, Lu Y. Angiogenesis in breast cancer progression, diagnosis, and treatment. J Cancer (2020) 11:4474–94. doi: 10.7150/jca.44313
105. Piasecka D, Braun M, Kitowska K, Mieczkowski K, Kordek R, Sadej R, et al. FGFs/FGFRs-dependent signalling in regulation of steroid hormone receptors - implications for therapy of luminal breast cancer. J Exp Clin Cancer Res (2019) 38:230. doi: 10.1186/s13046-019-1236-6
106. Gao Q, Lopez-Knowles E, Cheang MCU, Morden J, Ribas R, Sidhu K, et al. Impact of aromatase inhibitor treatment on global gene expression and its association with antiproliferative response in ER+ breast cancer in postmenopausal patients. Breast Cancer Res (2019) 22:2. doi: 10.1186/s13058-019-1223-z
107. Alfakeeh A, Brezden-Masley C. Overcoming endocrine resistance in hormone receptor-positive breast cancer. Curr Oncol (2018) 25:S18–27. doi: 10.3747/co.25.3752
108. Nagira K, Sasaoka T, Wada T, Fukui K, Ikubo M, Hori S, et al. Altered subcellular distribution of estrogen receptor alpha is implicated in estradiol-induced dual regulation of insulin signaling in 3T3-L1 adipocytes. Endocrinology (2006) 147:1020–8. doi: 10.1210/en.2005-0825
109. De Leeuw R, Neefjes J, Michalides R. A role for estrogen receptor phosphorylation in the resistance to tamoxifen. Int J Breast Cancer (2011) 2011:232435. doi: 10.4061/2011/232435
110. Anbalagan M, Rowan BG. Estrogen receptor alpha phosphorylation and its functional impact in human breast cancer. Mol Cell Endocrinol (2015) 418 Pt 3:264–72. doi: 10.1016/j.mce.2015.01.016
111. Turczyk L, Kitowska K, Mieszkowska M, Mieczkowski K, Czaplinska D, Piasecka D, et al. FGFR2-driven signaling counteracts tamoxifen effect on ERalpha-positive breast cancer cells. Neoplasia (2017) 19:791–804. doi: 10.1016/j.neo.2017.07.006
112. Campbell TM, Castro M, De Oliveira KG, Ponder B, Meyer KB. ERalpha binding by transcription factors NFIB and YBX1 enables FGFR2 signaling to modulate estrogen responsiveness in breast cancer. Cancer Res (2018) 78:410–21. doi: 10.1158/0008-5472.CAN-17-1153
113. Abolhassani A, Riazi GH, Azizi E, Amanpour S, Muhammadnejad S, Haddadi M, et al. FGF10: type III epithelial mesenchymal transition and invasion in breast cancer cell lines. J Cancer (2014) 5:537–47. doi: 10.7150/jca.7797
114. Johnston CL, Cox HC, Gomm JJ, Coombes RC. bFGF and aFGF induce membrane ruffling in breast cancer cells but not in normal breast epithelial cells: FGFR-4 involvement. Biochem J (1995) 306(Pt 2):609–16. doi: 10.1042/bj3060609
115. Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, et al. Association of FGFR1 with ERalpha maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER+ Breast cancer. Clin Cancer Res (2017) 23:6138–50. doi: 10.1158/1078-0432.CCR-17-1232
116. Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U S A (2010) 107:21737–42. doi: 10.1073/pnas.1007863107
117. Kim S, Dubrovska A, Salamone RJ, Walker JR, Grandinetti KB, Bonamy GM, et al. FGFR2 promotes breast tumorigenicity through maintenance of breast tumor-initiating cells. PLoS One (2013) 8:e51671. doi: 10.1371/journal.pone.0051671
118. Levine KM, Ding K, Chen L, Oesterreich S. FGFR4: A promising therapeutic target for breast cancer and other solid tumors. Pharmacol Ther (2020) 214:107590. doi: 10.1016/j.pharmthera.2020.107590
119. Castillo-Castrejon M, Sankofi BM, Murguia SJ, Udeme A-A, Cen HH, Xia YH, et al. FGF1 supports glycolytic metabolism through the estrogen receptor in endocrine-resistant and obesity-associated breast cancer. Breast Cancer Res (2023) 25:99. doi: 10.1186/s13058-023-01699-0
120. Wang Y, Patti GJ. The Warburg effect: a signature of mitochondrial overload. Trends Cell Biol (2023). doi: 10.1016/j.tcb.2023.03.013
121. Intlekofer AM, Finley LWS. Metabolic signatures of cancer cells and stem cells. Nat Metab (2019) 1:177–88. doi: 10.1038/s42255-019-0032-0
122. Lv L, Yang S, Zhu Y, Zhai X, Li S, Tao X, et al. Relationship between metabolic reprogramming and drug resistance in breast cancer. Front Oncol (2022) 12:942064. doi: 10.3389/fonc.2022.942064
123. Kurland BF, Peterson LM, Lee JH, Schubert EK, Currin ER, Link JM, et al. Estrogen receptor binding (18F-FES PET) and glycolytic activity (18F-FDG PET) predict progression-free survival on endocrine therapy in patients with ER+ Breast cancer. Clin Cancer Res (2017) 23:407–15. doi: 10.1158/1078-0432.CCR-16-0362
124. Woo YM, Shin Y, Lee EJ, Lee S, Jeong SH, Kong HK, et al. Inhibition of aerobic glycolysis represses akt/mTOR/HIF-1alpha axis and restores tamoxifen sensitivity in antiestrogen-resistant breast cancer cells. PLoS One (2015) 10:e0132285. doi: 10.1371/journal.pone.0132285
125. Hussein YR, Bandyopadhyay S, Semaan A, Ahmed Q, Albashiti B, Jazaerly T, et al. Glut-1 expression correlates with basal-like breast cancer. Transl Oncol (2011) 4:321–7. doi: 10.1593/tlo.11256
126. Oh S, Kim H, Nam K, Shin I. Glut1 promotes cell proliferation, migration and invasion by regulating epidermal growth factor receptor and integrin signaling in triple-negative breast cancer cells. BMB Rep (2017) 50:132–7. doi: 10.5483/BMBRep.2017.50.3.189
127. Liu X, Miao W, Huang M, Li L, Dai X, Wang Y. Elevated hexokinase II expression confers acquired resistance to 4-hydroxytamoxifen in breast cancer cells. Mol Cell Proteomics (2019) 18:2273–84. doi: 10.1074/mcp.RA119.001576
128. Wang J, Tao M, Wang T, Wang Z, Xiao J, Ding S, et al. [Knockdown of hexokinase 2 (HK2) inhibits breast cancer cell proliferation and reduces their resistance to fluorouracil]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi (2021) 37:722–7.
129. Zhu T, Zheng J, Zhuo W, Pan P, Li M, Zhang W, et al. ETV4 promotes breast cancer cell stemness by activating glycolysis and CXCR4-mediated sonic Hedgehog signaling. Cell Death Discovery (2021) 7:126. doi: 10.1038/s41420-021-00508-x
130. Tu SH, Chang CC, Chen CS, Tam KW, Wang YJ, Lee CH, et al. Increased expression of enolase alpha in human breast cancer confers tamoxifen resistance in human breast cancer cells. Breast Cancer Res Treat (2010) 121:539–53. doi: 10.1007/s10549-009-0492-0
131. Cancemi P, Buttacavoli M, Roz E, Feo S. Expression of alpha-enolase (ENO1), myc promoter-binding protein-1 (MBP-1) and matrix metalloproteinases (MMP-2 and MMP-9) reflect the nature and aggressiveness of breast tumors. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20163952
132. Huang X, Li X, Xie X, Ye F, Chen B, Song C, et al. High expressions of LDHA and AMPK as prognostic biomarkers for breast cancer. Breast (2016) 30:39–46. doi: 10.1016/j.breast.2016.08.014
133. Dong T, Liu Z, Xuan Q, Wang Z, Ma W, Zhang Q. Tumor LDH-A expression and serum LDH status are two metabolic predictors for triple negative breast cancer brain metastasis. Sci Rep (2017) 7:6069. doi: 10.1038/s41598-017-06378-7
134. Kalezic A, Udicki M, Srdic Galic B, Aleksic M, Korac A, Jankovic A, et al. Lactate metabolism in breast cancer microenvironment: contribution focused on associated adipose tissue and obesity. Int J Mol Sci (2020) 21. doi: 10.3390/ijms21249676
135. San-Millán I, Julian CG, Matarazzo C, Martinez J, Brooks GA. Is lactate an oncometabolite? Evidence supporting a role for lactate in the regulation of transcriptional activity of cancer-related genes in MCF7 breast cancer cells. Front Oncol (2019) 9:1536. doi: 10.3389/fonc.2019.01536
136. Huang R, Zong X. Aberrant cancer metabolism in epithelial-mesenchymal transition and cancer metastasis: Mechanisms in cancer progression. Crit Rev Oncol Hematol (2017) 115:13–22. doi: 10.1016/j.critrevonc.2017.04.005
137. Pfefferle AD, Herschkowitz JI, Usary J, Harrell JC, Spike BT, Adams JR, et al. Transcriptomic classification of genetically engineered mouse models of breast cancer identifies human subtype counterparts. Genome Biol (2013) 14:R125. doi: 10.1186/gb-2013-14-11-r125
138. Sciacovelli M, Frezza C. Metabolic reprogramming and epithelial-to-mesenchymal transition in cancer. FEBS J (2017) 284:3132–44. doi: 10.1111/febs.14090
139. Porporato PE, Payen VL, Perez-Escuredo J, De Saedeleer CJ, Danhier P, Copetti T, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep (2014) 8:754–66. doi: 10.1016/j.celrep.2014.06.043
140. Dong C, Yuan T, Wu Y, Wang Y, Fan TW, Miriyala S, et al. Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell (2013) 23:316–31. doi: 10.1016/j.ccr.2013.01.022
141. Lunetti P, Di Giacomo M, Vergara D, De Domenico S, Maffia M, Zara V, et al. Metabolic reprogramming in breast cancer results in distinct mitochondrial bioenergetics between luminal and basal subtypes. FEBS J (2019) 286:688–709. doi: 10.1111/febs.14756
142. Gaude E, Frezza C. Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun (2016) 7:13041. doi: 10.1038/ncomms13041
143. Gentric G, Mechta-Grigoriou F. Tumor cells and cancer-associated fibroblasts: an updated metabolic perspective. Cancers (Basel) (2021) 13. doi: 10.3390/cancers13030399
144. Wang L, Fan J, Yan CY, Ling R, Yun J. Activation of hypoxia-inducible factor-1alpha by prolonged in vivo hyperinsulinemia treatment potentiates cancerous progression in estrogen receptor-positive breast cancer cells. Biochem Biophys Res Commun (2017) 491:545–51. doi: 10.1016/j.bbrc.2017.03.128
145. Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin Cancer Biol (2014) 25:47–60. doi: 10.1016/j.semcancer.2014.01.005
146. Zhu Q, Zhu Y, Hepler C, Zhang Q, Park J, Gliniak C, et al. Adipocyte mesenchymal transition contributes to mammary tumor progression. Cell Rep (2022) 40:111362. doi: 10.1016/j.celrep.2022.111362
147. Ritter A, Kreis NN, Roth S, Friemel A, Safdar BK, Hoock SC, et al. Cancer-educated mammary adipose tissue-derived stromal/stem cells in obesity and breast cancer: spatial regulation and function. J Exp Clin Cancer Res (2023) 42:35. doi: 10.1186/s13046-022-02592-y
148. Becker LM, O'connell JT, Vo AP, Cain MP, Tampe D, Bizarro L, et al. Epigenetic reprogramming of cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast cancer. Cell Rep (2020) 31:107701. doi: 10.1016/j.celrep.2020.107701
149. La Camera G, Gelsomino L, Malivindi R, Barone I, Panza S, De Rose D, et al. Adipocyte-derived extracellular vesicles promote breast cancer cell Malignancy through HIF-1alpha activity. Cancer Lett (2021) 521:155–68. doi: 10.1016/j.canlet.2021.08.021
150. Viedma-Rodriguez R, Martinez-Hernandez MG, Martinez-Torres DI, Baiza-Gutman LA. Epithelial mesenchymal transition and progression of breast cancer promoted by diabetes mellitus in mice are associated with increased expression of glycolytic and proteolytic enzymes. Horm Cancer (2020) 11:170–81. doi: 10.1007/s12672-020-00389-z
151. Bowers LW, Rossi EL, Mcdonell SB, Doerstling SS, Khatib SA, Lineberger CG, et al. Leptin signaling mediates obesity-associated CSC enrichment and EMT in preclinical TNBC models. Mol Cancer Res (2018) 16:869–79. doi: 10.1158/1541-7786.MCR-17-0508
152. Szymczyk J, Sluzalska KD, Materla I, Opalinski L, Otlewski J, Zakrzewska M. FGF/FGFR-dependent molecular mechanisms underlying anti-cancer drug resistance. Cancers (Basel) (2021) 13. doi: 10.3390/cancers13225796
153. Adamczyk-Gruszka O, Horecka-Lewitowicz A, Gruszka J, Wawszczak-Kasza M, Strzelecka A, Lewitowicz P. FGFR-2 and epithelial-mesenchymal transition in endometrial cancer. J Clin Med (2022) 11. doi: 10.3390/jcm11185416
154. Raoof S, Mulford IJ, Frisco-Cabanos H, Nangia V, Timonina D, Labrot E, et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene (2019) 38:6399–413. doi: 10.1038/s41388-019-0887-2
155. Brown WS, Akhand SS, Wendt MK. FGFR signaling maintains a drug persistent cell population following epithelial-mesenchymal transition. Oncotarget (2016) 7:83424–36. doi: 10.18632/oncotarget.13117
156. Xu M, Chen S, Yang W, Cheng X, Ye Y, Mao J, et al. FGFR4 links glucose metabolism and chemotherapy resistance in breast cancer. Cell Physiol Biochem (2018) 47:151–60. doi: 10.1159/000489759
157. Qian X, Anzovino A, Kim S, Suyama K, Yao J, Hulit J, et al. N-cadherin/FGFR promotes metastasis through epithelial-to-mesenchymal transition and stem/progenitor cell-like properties. Oncogene (2014) 33:3411–21. doi: 10.1038/onc.2013.310
158. Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer (2016) 16:619–34. doi: 10.1038/nrc.2016.71
159. Van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene (2016) 35:3201–8. doi: 10.1038/onc.2015.381
160. Lampa M, Arlt H, He T, Ospina B, Reeves J, Zhang B, et al. Glutaminase is essential for the growth of triple-negative breast cancer cells with a deregulated glutamine metabolism pathway and its suppression synergizes with mTOR inhibition. PLoS One (2017) 12:e0185092. doi: 10.1371/journal.pone.0185092
161. Karunakaran S, Ramachandran S, Coothankandaswamy V, Elangovan S, Babu E, Periyasamy-Thandavan S, et al. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J Biol Chem (2011) 286:31830–8. doi: 10.1074/jbc.M111.229518
162. Madak-Erdogan Z, Band S, Zhao YC, Smith BP, Kulkoyluoglu-Cotul E, Zuo Q, et al. Free Fatty Acids Rewire Cancer Metabolism in Obesity-Associated Breast Cancer via Estrogen Receptor and mTOR Signaling. Cancer Res (2019) 79:2494–510. doi: 10.1158/0008-5472.CAN-18-2849
163. Alikhani N, Ferguson RD, Novosyadlyy R, Gallagher EJ, Scheinman EJ, Yakar S, et al. Mammary tumor growth and pulmonary metastasis are enhanced in a hyperlipidemic mouse model. Oncogene (2013) 32:961–7. doi: 10.1038/onc.2012.113
164. Hultsch S, Kankainen M, Paavolainen L, Kovanen RM, Ikonen E, Kangaspeska S, et al. Association of tamoxifen resistance and lipid reprogramming in breast cancer. BMC Cancer (2018) 18:850. doi: 10.1186/s12885-018-4757-z
165. Nguyen VT, Barozzi I, Faronato M, Lombardo Y, Steel JH, Patel N, et al. Differential epigenetic reprogramming in response to specific endocrine therapies promotes cholesterol biosynthesis and cellular invasion. Nat Commun (2015) 6:10044. doi: 10.1038/ncomms10044
Keywords: breast cancer, adipose, fibroblast growth factor, obesity, diabetes
Citation: Sankofi BM, Valencia-Rincón E, Sekhri M, Ponton-Almodovar AL, Bernard JJ and Wellberg EA (2023) The impact of poor metabolic health on aggressive breast cancer: adipose tissue and tumor metabolism. Front. Endocrinol. 14:1217875. doi: 10.3389/fendo.2023.1217875
Received: 06 May 2023; Accepted: 30 August 2023;
Published: 20 September 2023.
Edited by:
Victoria Seewaldt, City of Hope, United StatesReviewed by:
Cinzia Giordano, University of Calabria, ItalyCopyright © 2023 Sankofi, Valencia-Rincón, Sekhri, Ponton-Almodovar, Bernard and Wellberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth A. Wellberg, ZWxpemFiZXRoLXdlbGxiZXJnQG91aHNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.