 Ziyuan Wang
Ziyuan Wang Zhuoran Cao
Zhuoran Cao Zhiying Yue
Zhiying Yue Zhengfeng Yang
Zhengfeng Yang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 04 September 2023
Sec. Clinical Diabetes
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1216907
This article is part of the Research Topic Non-insulin Hypoglycemic Drugs in Type 2 Diabetes Remission View all 7 articles
Diabetic Mellitus (DM), a chronic metabolic disorder disease characterized by hyperglycemia, is mainly caused by the absolute or relative deficiency of insulin secretion or decreased insulin sensitivity in target tissue cells. Dihydromyricetin (DMY) is a flavonoid compound of dihydroflavonol that widely exists in Ampelopsis grossedentata. This review aims to summarize the research progress of DMY in the treatment of DM. A detailed summary of related signaling induced by DMY are discussed. Increasing evidence implicates that DMY display hypoglycemic effects in DM via improving glucose and lipid metabolism, attenuating inflammatory responses, and reducing oxidative stress, with the signal transduction pathways underlying the regulation of AMPK or mTOR/autophagy, and relevant downstream cascades, including PGC-1α/SIRT3, MEK/ERK, and PI3K/Akt signal pathways. Hence, the mechanisms underlying the therapeutic implications of DMY in DM are still obscure. In this review, following with a brief introduction of the absorption, metabolism, distribution, and excretion characteristics of DMY, we summarized the current pharmacological developments of DMY as well as possible molecular mechanisms in the treatment of DM, aiming to push the understanding about the protective role of DMY as well as its preclinical assessment of novel application.
Diabetes Mellitus (DM) is a metabolic disease characterized by disordered lipid and carbohydrate metabolism. The prevalence of diabetes reported by the International Diabetes Federation shows that the number of adults aged 20-70 with diabetes worldwide is estimated to be 10.5%(536.6 million people)and is expected to rise to 12.2% (783.2 million) by 2045 (1, 2). DM patients will suffer pain from various DM complications, including kidney disease, diabetic foot, peripheral neuropathy, retinopathy and so on (3).
Diabetes, caused by a long-term combination of genetic and environmental factors, is mainly due to insulin deficiency or insufficient insulin secretion, leading to function disorders in glucose, lipid and protein metabolism (4). DM is largely classified into type I diabetes mellitus and type II diabetes mellitus (T2DM). T2DM has become the third most common non-communicable disease in the world. T2DM is characterized by insulin deficiency due to insulin secretion disorder and insulin resistance(IR) (5). The pathogenesis of T2DM may be caused by the massive generation of free radicals in response to high blood glucose and high free fatty acids, which leads to oxidative stress and inflammatory responses. The long-term inflammatory responses and the activation of oxidative stress-associated signaling pathways contribute to IR and impaired insulin secretion, thereby leading to the clinical symptoms of DM (6, 7). Until now, the etiology and pathogenesis for DM have not been fully clarified.
Dihydromyricetin (DMY), a 2,3-dihydroflavonol compound (Figure 1), is the main bioactive component isolated from the tender stem and leaves of the Chinese medicinal plant Ampelopsis grossedentata (A. grossedentata), which exert various biological effects, including anti-alcohol intoxication, anti-inflammatory, antibacterial, antioxidant, anti-tumor properties, as well as lipid and blood glucose-regulating effects (8, 9).
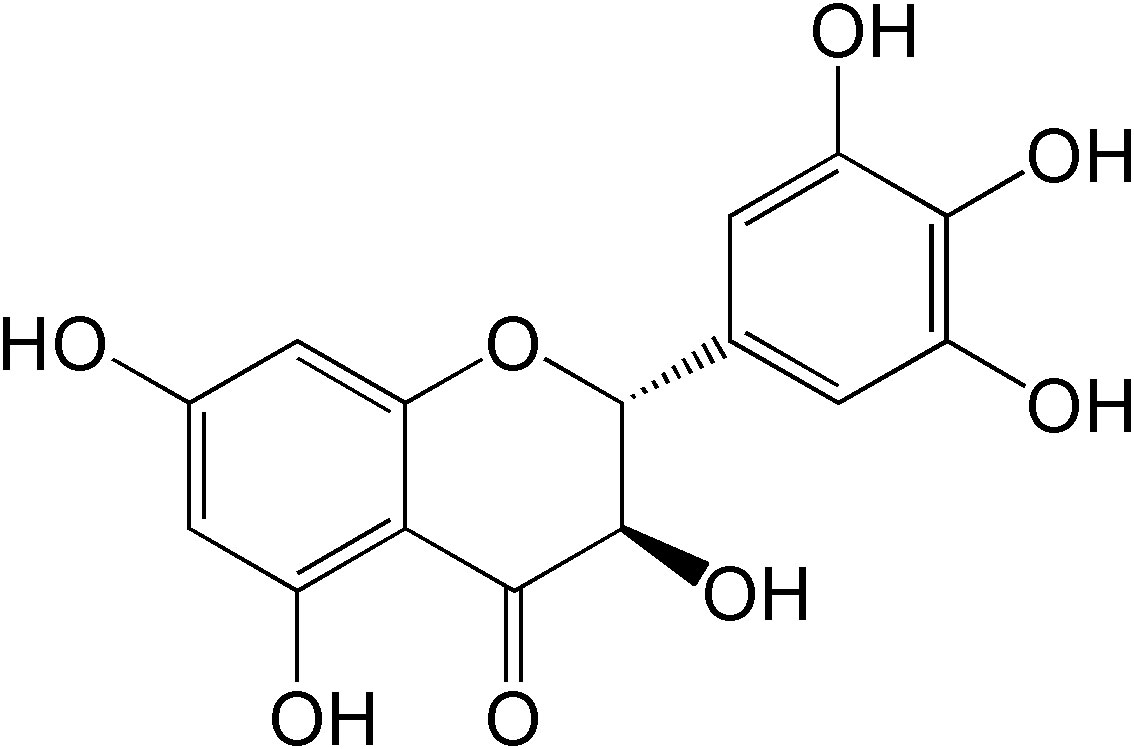
Figure 1 The chemical structure of DMY.
Pharmacokinetic studies have revealed that the half-life of DMY is approximately 3.70 ± 0.99 hours and the total time DMY remained in the body is around 5.98 ± 0.58 hours when DMY is orally administered at a dose of 20 mg/kg (10). Furthermore, once DMY is absorbed into the bloodstream, it would be easily metabolized and decomposed through methylation, sulfation, or reduction reaction. Fan et al. reported that after oral administration, unmetabolized DMY quickly distributes throughout various tissues, especially in the gastrointestinal tract. Based on in vitro metabolism studies, they also discovered that most of the original compounds and metabolites are excreted through feces, with only a small amount of original compounds being absorbed through the small intestine. These absorbed compounds enter the bloodstream through the portal vein, and reach the liver. In the liver, they undergo further processing through glucuronidation, sulfation, and methylation, resulting in phase II metabolites that are either absorbed by the body or excreted via urine (11). Due to extensive biotransformation occurring in the gastrointestinal tract and bloodstream, the oral bioavailability of DMY is relatively low (12). In short, they detected eight metabolites and proposed five metabolic pathways, including reduction, dehydroxylation, methylation, glucuronidation and sulfation.
Recently, both clinical and experimental studies have illustrated that DMY possesses preventive and therapeutic effects on DM and its complications (13). Mechanistically, DMY has been reported to be involved in improving glucose and lipid metabolism, alleviating oxidative stress as well as attenuating inflammatory response, however, the exact molecular mechanism of DMY in DM is still not fully elucidated. Previous studies have shown that DMY was able to activate AMP-activated protein kinase (AMPK) signal pathway to perform a potential anti-diabetic effect, with mechanism related to AMPK/mTOR/PGC-1α and MEK/ERK signaling pathways. The review aims to summarize the role and molecular mechanisms of DMY in the treatment of DM and its complication and all the effects are listed in Figure 2 and simplified in Table 1.
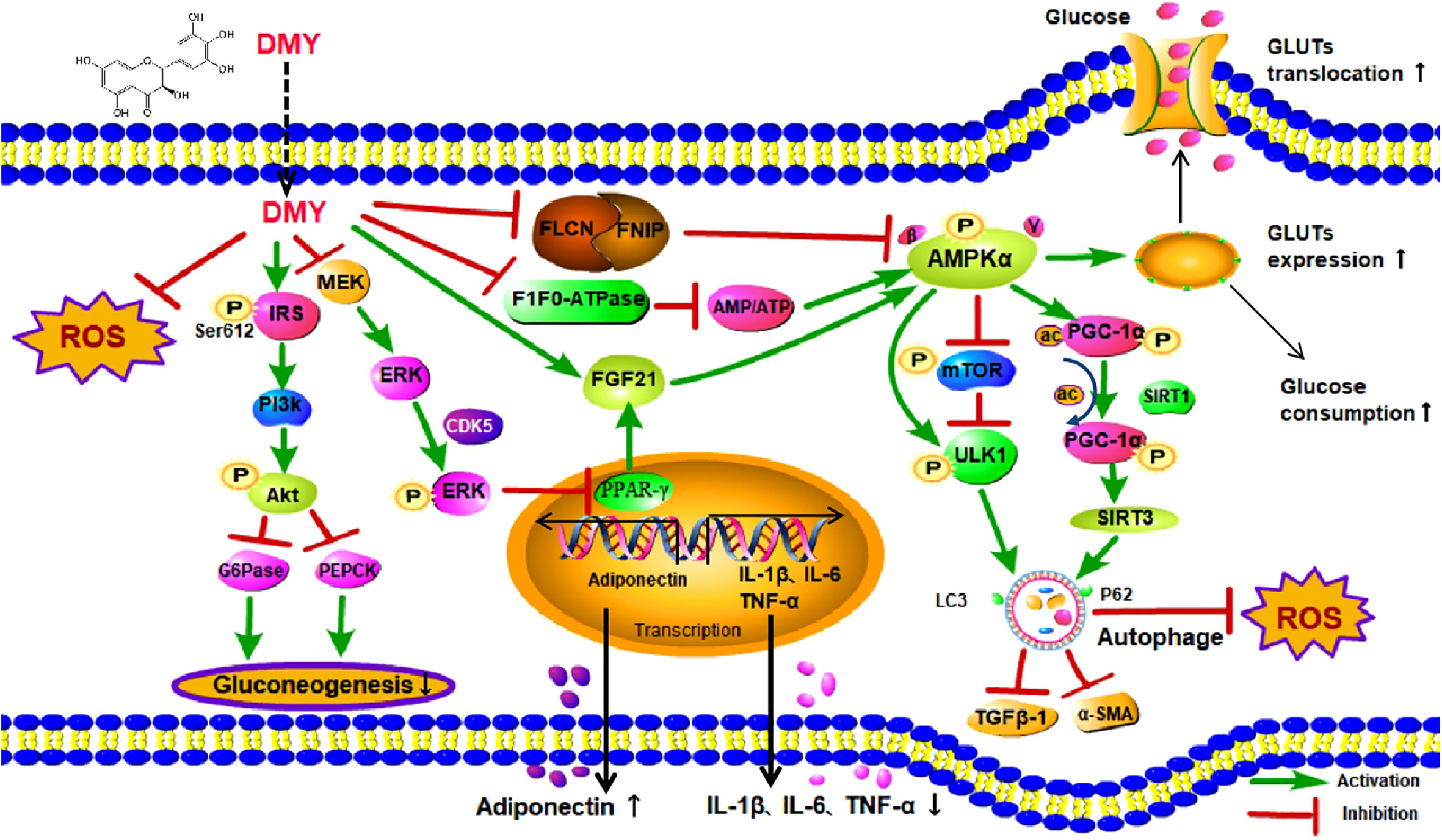
Figure 2 The effects and mechanisms on diabetes mellitus. DMY, Dihydromyricetin; ROS, Reactive oxygen species; IRS,insulin receptor substrate; MEK, mitogen-activated protein kinase kinase; ERK, extracellular regulated protein kinases; PI3K, phosphatidylinositol 3 kinase; Akt, protein kinase B; G6Pase, glucose-6-phosphatase; PEPCK, phosphoenolpyruvate carboxykina se;CDK5, cyclin dependent kinase-5; FLCN, Folliculin; FNIP1,Folliculin Interacting Protein 1; AMP, Cyclic Adenosine monophosphate; ATP,adenosine-triphosphate; FGF21, Fibroblast Growth Factor-21; PPARγ, peroxisome proliferator-activated receptor γ; IL-1β, Interleukin-1β; IL-6, Interleukin-6; TNF-α, tumor necrosis factor-α; AMPKα, Adenosine 5′-monophosphate activated protein α; mTOR, mammalian target of rapamycin; ULK1,unc-51-like kinase 1; TGFβ-1, transforming growth factor-β 1; α-SMA,α-Smooth muscle actin;PGC-1α, Peroxisome proliferator activated receptor co-activator-1α; SIRT1, Sirtuin1; SIRT3, Sirtuin3; P62, sequestosome 1; Gluts, Glucose transporters.
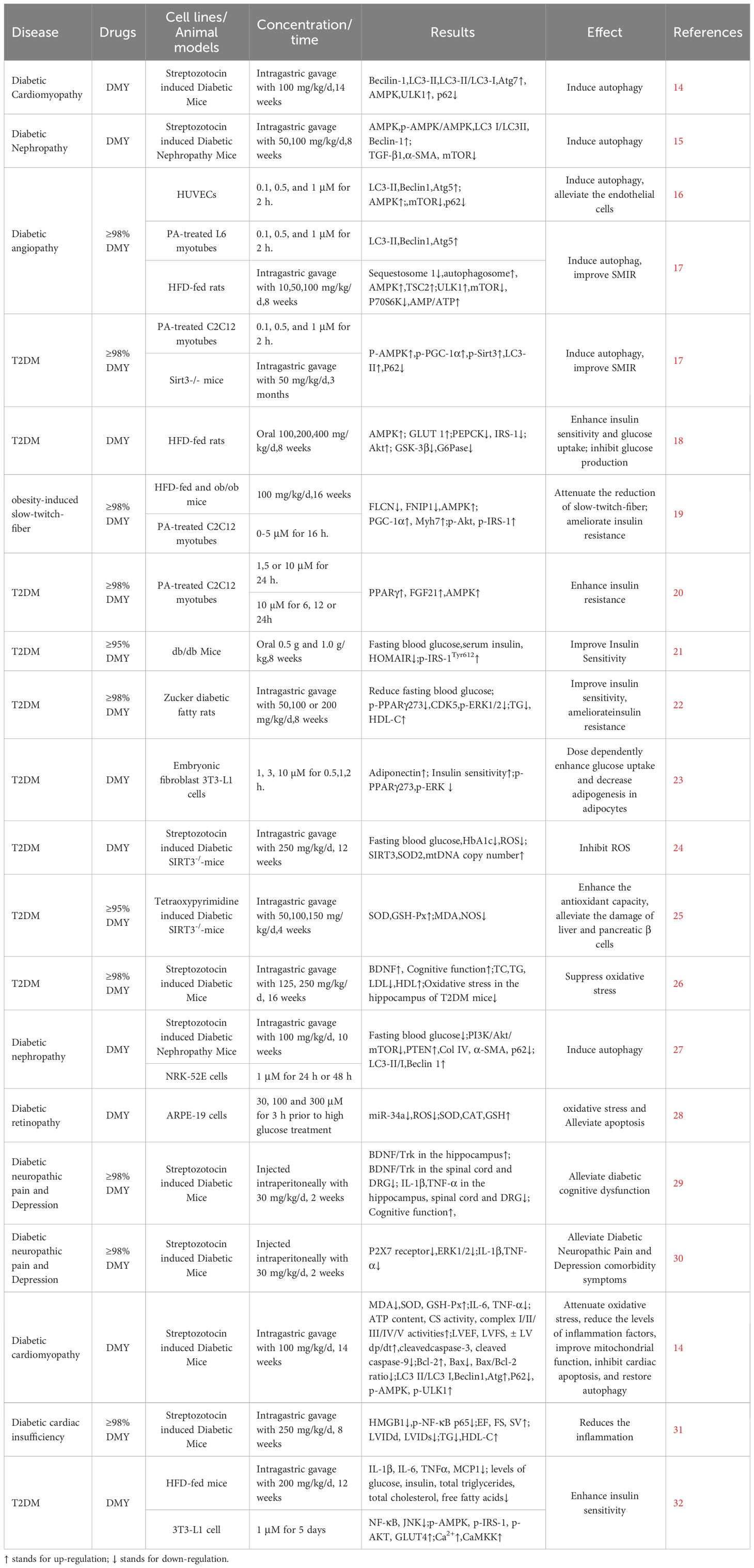
Table 1 Studies on the effects of DMY on DM.
AMPK, a key energy stress sensor, is activated in response to an increased AMP/ATP ratio during various stress conditions, such as hypoxia, hypertrophy, hypoglycemia, low blood glucose levels, and other stressors (33). This crucial signaling pathway plays a role in distinguishing the distinct or similar regulation of cellular responses across different tissues and cell types.
Recent studies have revealed AMPK’s positive regulation of autophagy, which involves inhibiting the activity of the mammalian target of rapamycin (mTOR) complex and activating phosphorylated unc-51-like kinase 1 (ULK1) (34). Autophagy is essential for maintaining heart structure, function, and cardiomyocyte integrity. Additionally, AMPK has been linked to autophagy in other contexts, such as diabetic cardiomyopathy, where it may protect cardiomyocytes in diabetes through Bcl-2 dissociation from Beclin1 when activated (35). The alteration of AMPK activity in diabetic mice influences cardiac autophagy, as indicated by changes in the LC3II/LC3I ratio, Beclin1 expression, and autophagy-related gene 7 (Atg7) levels, all of which were reversed by DMY treatment (14).
In the context of diabetic complications, DMY has shown potential in various tissues. In renal tissues of diabetic rats, DMY increased AMPK protein expression levels, leading to improved glomerular extracellular matrix deposition and renal fibrosis, possibly through promoting autophagy and inhibiting the expression of transforming growth factor-beta (TGFβ-1) and Alpha-smooth muscle actin (α-SMA) (15). In human umbilical vascular endothelial cells (HUVEC) exposed to high glucose-induced injury, DMY up-regulated autophagy through the AMPK/mTOR pathway (16). Additionally, in skeletal muscle cells, DMY enhanced insulin sensitivity by activating the AMPK/mTOR/ULK1 pathway, which induced autophagy (36). Overall, the AMPK/mTOR signaling pathway offers promising therapeutic avenues for diabetes treatment by targeting autophagy processes. Nevertheless, further research is needed to fully elucidate the distinct and common mechanisms through which AMPK regulates autophagy across different tissues and cell types.
AMPK is a crucial intracellular serine/threonine-protein kinase, functioning as an energy stress sensor. It becomes activated by an increased AMP/ATP ratio during various stress conditions, such as hypoxia, hypertrophy, hypoglycemia, low blood glucose levels, and other stressors (33). Once activated, AMPK phosphorylates CREB (cAMP response element-binding protein), leading to the activation of peroxisome proliferator-activated receptor (PPARγ) coactivator-1α (PGC-1α) and downstream target Sirtuin 3 (SIRT3). PGC-1α serves as a transcriptional co-activator closely associated with the body’s energy metabolism, while SIRT3 induces autophagy through mitochondrial protein deacetylation, specifically mitochondrial autophagy, playing critical roles in cellular functions (37–39).
In a study conducted by Shi et al., DMY (a specific compound) was found to increase AMPK phosphorylation in skeletal muscle, subsequently upregulating PGC-1α expression and activating SIRT3. To validate the pathway’s role, the researchers used PGC-1α or AMPK siRNA transfection, resulting in reduced DMY-induced SIRT3 expression and suppressed autophagy in C2C12 myotubes. Consistent results were observed in skeletal muscle tissues of DMY-fed Stirt3-/- mice, confirming the involvement of the AMPK-PGC-1α-SIRT3 pathway (17).
These findings imply that DMY promotes autophagy, partially via activating the AMPK-PGC-1α-SIRT3 signaling pathway, thereby enhancing skeletal muscle insulin sensitivity in both in vitro and in vivo.
In conclusion, this study highlights the significance of the AMPK/PGC-1α/SIRT3 signaling pathway as a potential multi-targeted therapeutic approach for diabetes mellitus (DM) treatment. Furthermore, it provides valuable insights into the mechanisms underlying the anti-diabetic effects of DMY, emphasizing the essential role of AMPK-related signaling pathways in promoting autophagy in skeletal muscle cells.
Glucose transporter (GLUTs) is the major carrier of glucose transport in mammalian cells. AMPK serves as a crucial energy sensor to regulate lipid, cholesterol and glucose metabolism, from which AMPK activation leads to an increase in lipid and glucose catabolism (40). While a decrease in glucose production has advantageous effects on glucose homeostasis and peripheral insulin sensitivity (41). The overexpression of constitutively active AMPK in skeletal muscle cells leads to increased glucose uptake, accompanied by the trans-localization of GLUT1 and GLUT4 to the plasma membrane. This enhancement in glucose uptake and utilization further benefits skeletal muscle function. (42). Previous reports have confirmed the translocation of GLUT2 to the plasma membrane via the AMPK pathway in the liver (43). There have been studies showing that the activation of AMPK is correlated with the enhancement of glucose transport mediated by plasma membrane localized Glut1 activation (44). In another study, it was shown that DMY improves insulin resistance in rats fed with a high-fat diet (HFD) by facilitating the translocation of GLUT1 from the cytosol to the membrane. This effect is achieved through the activation of AMPK at Ser172 and AS160 signaling, which leads to an increase in glucose uptake (18).
Insulin primarily exerts its action through the insulin receptor substrate (IRS)-inositol phosphate kinase (PI3K)-protein kinase B (Akt) pathway (45). Liang Le et al. discovered that DMY downregulates the expression of G6Pase and PEPCK through the IRS/PI3K/Akt pathway, inhibiting gluconeogenesis and reducing glucose production (18, 46).
In studies involving fatty db/db mice, oral administration of DMY reduced fasting blood glucose, serum insulin levels, and insulin resistance index levels. The mechanism behind this effect appears to involve DMY inducing intracellular transduction of insulin signaling by up-regulating IRS-1 (Y612) tyrosine phosphorylation, mitigating insulin resistance and pancreatic fibrosis in the db/db mice (21).
Furthermore, various investigations have consistently indicated a direct correlation between insulin-stimulated glucose disposal rate and a decrease in the proportion of slow-twitch (type I) fibers, leading to reduced oxidative metabolism in skeletal muscles (46).
Folliculin (FLCN), a protein associated with kidney cancer, has been recently linked to AMPK regulation through the FLCN/Folliculin Interacting Protein 1 (FNIP1) complex and its interacting protein FNIP1. This complex acts as a negative regulator of AMPK activity, impacting the expression of PGC-1α (47).
In a study by Zhou et al., palmitic acid-induced C2C12 muscular tube exhibited a decrease in the expression of slow-twitch fiber-specific Myh7, along with increased levels of FLCN and FNIP1, AMPK inactivation, and reduced PGC-1α expression. However, DMY treatment reversed this phenomenon, significantly reducing palmitic acid-induced insulin resistance. This effect was evident by increased levels of p-Akt and p-IRS-1, as well as enhanced glucose uptake (19). These findings suggest that DMY may counteract the obesity-induced decrease of slow-twitch fibers by regulating the FLCN-FNIP1-AMPK signaling pathway, ultimately improving skeletal muscle insulin resistance in obese patients.
PPARγ, a member of the type II nuclear hormone receptor superfamily, plays a key role in the regulation of glucose and lipid metabolism. PPARγ is considered a clinically proven therapeutic target for T2DM (48). PPARγ agonists induce the expression of Fibroblast Growth Factor 21 (FGF21), further enhancing PPARγ activity in adipocytes (49). Additionally, FGF21 activates AMPK and enhances mitochondrial function and oxidative capacity, ultimately improving glucose tolerance (50). Zhou et al. reported that DMY activates PPARγ, leading to increased glucose uptake capacity in palmitic acid-induced insulin-resistant L6 myotube cells in a dose-dependent and time-dependent manner. Furthermore, DMY markedly up-regulated the expressions of p-IRS-1, p-Akt, p-AMPK, and FGF21 (20). These findings suggest that DMY may act as a possible agonist of PPARγ and enhance insulin sensitization by activating PPARγ and subsequently regulating the FGF21-AMPK signaling pathway.
Adiponectin is a crucial hormone predominantly synthesized and secreted by mature adipocytes, known for its antidiabetic, antiatherogenic, and anti-inflammatory functions. It exerts its effects by binding to adiponectin receptors (adipoR1 and adipoR2), leading to increased fatty acid oxidation, reduced hepatic glucose production, and improved insulin sensitivity (51). However, in the context of obesity, adipocytes become hypertrophic, which results in reduced secretion of adiponectin into the plasma and an imbalance in glucose homeostasis.
Moreover, the increased expression of adipose inflammatory cytokines leads to the impairment of insulin signaling pathways and reduced insulin-dependent glucose uptake, which further highlights the crucial role of adiponectin in insulin resistance (IR), type 2 diabetes mellitus (T2DM), and metabolic syndrome (52).
In their study, Lei et al. explored the effects of DMY on glucose uptake and adiponectin secretion in adipocytes. They observed that DMY treatment enhanced glucose uptake capacity in Zucker diabetic fatty rats and stimulated adipocytes to secrete adiponectin FGF21. This secreted adiponectin FGF21 demonstrated anti-diabetic efficacy by inhibiting extracellular signal-regulated kinase 1/2 (ERK)/cyclin-dependent kinase-5 (CDK5)-mediated PPARγ phosphorylation at Ser273 in adipose tissues (22). Consequently, DMY effectively delayed the onset of hyperglycemia and improved obesity-associated insulin resistance in diabetic rats without inducing weight gain.
Moreover, Liu et al. investigated whether DMY mitigates insulin resistance by inhibiting PPARγ phosphorylation at Ser273 via the MEK/ERK pathway in mouse fibroblast cells using the 3T3-L1 cell line. Their results showed that dexamethasone-treated adipocytes supplemented with DMY exhibited reduced insulin sensitivity for glucose uptake and decreased adipogenesis in a dose and time-dependent manner. Notably, pre-treatment of the PPARγ inhibitor, GW9662, effectively blocked the improvement of insulin resistance by DMY in differentiated adipocytes. Furthermore, both DMY and the mitogen-activated protein kinase (MEK) inhibitor, PD98059, significantly enhanced glucose uptake and adiponectin secretion in adipocytes (23). These findings suggest that DMY enhances glucose uptake in adipocytes by inhibiting the MEK/ERK pathway, which consequently down-regulates PPARγ phosphorylation at Ser273.
In summary, these studies demonstrate that DMY may have potent effects in reducing blood glucose levels, improving insulin sensitivity, and ameliorating lipid profiles. Agents that promote adiponectin secretion, such as DMY, could potentially be valuable candidates for treating metabolic diseases, including T2DM.
Oxidative stress refers to the critical imbalance between intracellular reactive oxygen species (ROS) production and scavenging in the body (53). For diabetic patients, oxidative stress and lipid peroxidation, caused by elevated levels of oxygen free radicals, play a significant role. In islet β cells, antioxidant enzymes function to reduce oxidative damage directly by decreasing the production of ROS and free radicals, protecting the cells from harm (54, 55). In a recent report, alloxan and/or STZ, extensively used in establishing T2DM animal models, mainly increase the production of ROS and inhibit free radical defense system, making ROS directly injure islet β cells, finally resulting in necrosis of pancreatic β cells, thus leading to hyperglycemia and inducing the occurrence of diabetes (56). In a study by Hua and his colleagues, male C57BL/6 mice, wild-type mice and SIRT3-/- mice were injected with STZ for 5 consecutive days (60 mg/kg/day. After a fortnight, DMY was given 250 mg/kg by gavage once a day for 12 weeks. They found that DMY treatment decreased fasting blood glucose and glycosylated hemoglobin level, improved endothelium-dependent relaxation of diabetic mice thoracic aorta, inhibited oxidative activity and ROS production, and enhanced SIRT3 and superoxide dismutase 2 protein expression (24). The results suggested that DMY improved profound endothelial dysfunction in diabetic mice via inhibiting oxidative stress and the production of ROS in a SIRT3-dependent manner.
Furthermore, high activities of superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), and catalase (CAT) contribute significantly to reducing the accumulation of intracellular ROS (57). Recently, Xiao et al. reported that DMY markedly increased the activities of antioxidative enzymes SOD and GSH-Px in the serum, liver, and pancreas of diabetic mice. Additionally, DMY decreased the levels of MDA and nitric oxide synthase, suggesting that it enhanced the antioxidant capacity of diabetic mice. This effect led to alleviation of liver and pancreatic β cell damage and promotion of insulin synthesis (25).
Moreover, Ling and colleagues demonstrated that DMY significantly decreased the production of MDA, increased the anti-oxidant defense system, including SOD, GSH-Px, and CAT, and inhibited the production of ROS in the hippocampus of T2DM mice. These effects resulted in reduced glucose and lipid metabolism, improved mitochondrial function, and ultimately ameliorated cognitive impairment (26). Guo et al. measured blood glucose, serum insulin and renal superoxide compound levels after DMY treatment in each mouse. They found that DMY significantly reduced MDA content and increased the activity of SOD in the kidney, DMY promoted autophagy and improved renal interstitial fibrosis in diabetic nephropathy (DN) by regulating the miR-155-5p/phosphatase and tensin homolog deleted on chromosome ten (PTEN) signaling pathway and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling pathway, suggesting that DMY had a protective effect in early kidney injury of diabetic nephropathy, which may be associated with improvement of anti-oxidative capability in the kidney (27). Recent studies have found that the ROS content in high-glucose stimulated ARPE-19 cells is significantly decreased, while the activities of SOD and CAT antioxidant enzymes, the concentration of GSH are both enhanced. Hence DMY has a protective effect on HG-induced oxidative stress of ARPE-19 cells, providing a promising therapeutic drug for the treatment of diabetic retinopathy (28).
Consistent with the above observations, oxidative stress plays a significant role in the development and progression of diabetes mellitus (DM) and its complications. Mitochondrial dysfunction is often associated with abnormal reactive oxygen species (ROS) production, and elevated ROS levels are linked to insulin resistance (IR) and islet dysfunction in type 2 diabetes mellitus (T2DM).Accumulating evidence suggests that DMY possesses a strong antioxidant effect with relatively good efficacy, indicating that natural antioxidant DMY can be clinically used as an adjunctive or second-line agent for preventing and treating diabetes and its complications.
Patients with type 2 diabetes mellitus (T2DM) often exhibit a mild chronic inflammatory state, characterized by elevated levels of cytokines and adipokines, as well as activation of pro-inflammatory pathways (58). Concurrently, DMY can directly target peripheral tissues to improve diabetic complications’ symptoms. Numerous reports have demonstrated that the upregulation of pro-inflammatory cytokine expression plays a crucial role in the development of T2DM complications, such as nephropathy, neuropathy, retinopathy, and various cardiovascular diseases. Among these complications, pro-inflammatory cytokines like interleukin-18 (IL-18), interleukin-1β, and tumor necrosis factor α (TNF-α) have been implicated in promoting insulin resistance (IR), impairing β cell function, and inducing cell apoptosis (59). Diabetic neuropathic pain (DNP) and depression are the most common and intractable chronic complications of DM, affecting nearly 25% of diabetic patients (60). Studies have found that DMY could inhibit brain-derived Neurotrophic Factor levels and the expression of Tyrosine Kinase receptor B in the nervous system, and thus improved DNP and depression by reducing the levels of IL-1β and TNF-α in the spinal cord (29). It prevents the inflammation induced tissueinjury by targeting P2X7 receptor (61) and BDNF/TrkB pathway, to reduce levels of pro-inflammatory factors (29). Guan et al. have also found that DMY could directly reduce the expression of P2X7 receptor, down regulate the activation of ERK1/2 pathway, and reduce the release of the inflammatory factor TNF and IL-1β in rats with DNP and depression (30). In addition, one group showed that DMY exerted an anti-inflammatory effect by reducing the levels of IL-6 and TNF-α, and also improved mitochondrial dysfunction through significant enhancement of cellular ATP content, citrate synthase activity and complex I/II/III/IV/V level, thus preventing from experimentally diabetic cardiomyopathy in STZ-induced diabetic mice (14). Another clinical trial found that DMY significantly improved IR in non-alcoholic fatty liver disease (NAFLD), regulated glucose and lipid metabolism, slowed the progression of hepatic steatosis, reduced blood levels of TNF-α, cytokeratin 18 and fibroblast growth factor 21. DMY also improved glucose and lipid metabolism and various biochemical indexes in patients with NAFLD, thus improving blood glucose levels (62).
Liu et al. found that the release of proinflammatory factor HMGB1 in plasma of diabetic rats was increased, and the expression of HMGB1 and phosphorylated NF-κB p65 protein in myocardial tissue was up-regulated, while the expression and release of HMGB1 and phosphorylated NF-κB p65 protein were significantly inhibited in DMY treated diabetic rats. It is reported that DMY may improve cardiomyopathy in diabetic rats by inhibiting the phosphorylation of HMGB1 and NF-κB p65 to reduce inflammatory responses (10). Researchers have shown that DMY reduced HFD-induced inflammatory cytokines (IL-1β, IL-6, TNFα, and MCP1) in the obese mice and could activate the PLC-CaMKK-AMPK signal pathway against inflammation-induced insulin resistance by targeting phospholipase C in peripheral tissue (32).
In recent years, ample research has indicated that the pathogenesis of T2DM is also closely linked to systemic low-grade chronic inflammation. Many inflammatory factors, such as TNF-α, IL-6 and C-reactive protein are not only directly involved in IR, but also closely associated with diabetic macro-vascular complications. The latest study indicated that DMY suppressed the expression of inflammatory factors, indicating that DMY would attenuate inflammatory responses and thus improve diabetes and relevant complications. Previous study have found that liver is one of the target organ of DMY, and it alleviated nonalcoholic fatty liver disease (NAFLD) via regulating lipid/glucose metabolism, probably due to its anti-inflammatory or sirtuins-dependent mechanism (32, 63) DMY was mainly absorbed in gastrointestinal tract, to ameliorate the liver inflammatory activity induced by DM, latest study has found that a long-circling DMY encapsulated liposome with good bioavailability that could target the liver in the long term and sustain DMY delivery. The primary finding of this study was that DMY encapsulated liposomes significantly reduced the liver inflammation induced by exhaustive exercise by promoting macrophage polarization from the M1 to M2 subtype by activating the SIRT3/HF-1α signaling pathway. Hence DMY may provide a novel insight into the research of the anti-diabetic mechanism from anti-inflammatory in clinical practice and lays a foundation for the further development of active components of Chinese herb for treating diabetes.
Diabetes mellitus (DM) comprises a group of chronic metabolic diseases characterized by chronic hyperglycemia resulting from defects in insulin secretion, insulin response, or both. DMY (dihydromyricetin), on the other hand, exerts protective or preventive effects in various systems, including the cardiovascular system, liver, musculoskeletal system, nervous system, and metabolic and endocrine diseases like DM. These beneficial effects are achieved through antioxidant, anti-inflammatory, anti-apoptotic, and other pathways. The underlying mechanism is related to the AMPK/mTOR/PGC-1α, MEK/ERK, etc. signaling pathway. Meanwhile, DMY can also alleviate DM and its complications such as diabetic foot, diabetic nephropathy, diabetic retinopathy and so forth by reducing inflammation, enhancing autophagy, and alleviating ROS via mainly targeting AMPK.
AMPK is an enzyme that plays a crucial role in cellular energy homeostasis, which exists as a heterotrimeric protein consisting of a catalytic α-subunit, regulatory β- subunits and γ-subunits. The α-subunit is the main catalytic subunit of AMPK. Under stress conditions, AMPK was activated by an increased intracellular AMP/ATP ratio. AMP binds to the AMPKγ subunit, causing conformational changes in the protein and promoting phosphorylation of AMPK on threonine residues (Thr172) (64).
In present studies, AMPK, as a key regulator of DMY regulating systemic energy balance, including regulation of glucose and lipid metabolism, attenuating inflammatory responses, and reducing oxidative stress, etc., has become a research hotspot in the field of T2DM (65). In addition to present signaling pathway discussed in the manuscript, another research further demonstrated that DMY could activate AMPK by inhibiting mitochondrial F1F0-ATPase activity, and then induce autophagy to improve skeletal muscle IR and treat diabetes. DMY could activate the AMPK/mTOR signaling pathway, trigger autophagy and act as the antagonism for high glucose-induced oxidative damage of endothelial cells, making it be a hopeful drug for the treatment of T2DM. Similarly, DMY could also mediate the AMPK-PGC-1α-SIRT3 signaling pathway to improve skeletal muscle insulin sensitivity and promote autophagy by activating the AMPK/mTOR signaling pathway, which can alleviate diabetic kidney injury and even retard the progression of diabetic nephropathy. Thus, these studies suggest that DMY is also a natural AMPK activator, and indicate that AMPK may be a clinical target of DMY in the treatment of DM. A recent clinical study showed that DMY can improve glucose and lipid metabolism in patients with T2DM (66). The major challenge for the translational research of DMY is its small molecular weight and poor solubility, which result in its low bioavailability. Currently, organic polymer materials are incorporated to facilitate the biological utilization of DMY, and to develop a more effective drug for clinical treatment of DM. the latest research have found that adipose tissue is the key organ for DMY to function, adipose tissue is multifunctional and plays an important role in whole-body energy homeostasis in DM, Lipid metabolism disorder is one of the pathological processes of DM (67, 68), it is reported that DMY had the capacity to decrease body weight, improve metabolic disorders of glucose and lipids and increase the consumption of energy by inducing WAT browning and offered new insight into the therapeutic action of this small molecule compound in obesity(69). Although lipid metabolism disorder is one of the pathological processes of diabetes, DMY may also had ability to improve metabolic disorders of glucose and lipids in DM.
DMY not only plays an important role in diabetes by targeting its activator AMPK but also exerts significant effects on its targets in other diseases. Sun et al. used the AD model of rats to verify whether DMY can exert the regulatory roles in AD via up-regulation of AMPK/SIRT1 signaling pathway to inhibit inflammatory responses, hippocampal neuronal apoptosis and ameliorate cognitive function, thus providing novel targets for the clinical treatment of AD (P. 70). Moreover, DMY has been shown to increase SIRT3 expression by activating the AMPK-PGC1α/ERRα signaling pathway, and promote the expression of mtDNA-encoded genes, restore the enzymatic activity of MRCs and increase the mtROS scavenging ability through the SIRT3-dependent mechanism, thus improving the mitochondrial respiratory capacity and redox homeostasis of hepatocyte to prevent NAFLD (57). In osteosarcoma cells, Zhao et al. found that DMY executes anti-tumor effects through the induction of apoptosis in osteosarcoma cells and the potential mechanism may be due to the activation of p38MAPK and AMPKα/GSK-3β/Sox2 signaling pathway (71). It has been reported that DMY ameliorated apoptosis and glucometabolic disorders through the regulation of AMPK/GLUT4 signaling pathway in PC12 cells. Specifically, DMY protected PC12 cells against MG-induced apoptosis and glycometabolic disorders, at least in part by restraining the hyperactivation of p-AMPK activity and normalizing the translocation of GLUT4 from the intracellular compartment, resulting in a balance in glucose uptake (72).
No matter whether DMY functions in diabetes by improving glucose and lipid homeostasis in the liver, adipose tissue, and skeletal muscle, or assists in reducing NAFLD through AMPK/PGCl-α/ERR-α or facilitated tumor cell apoptosis through AMPK/GSK3/SOX2 and regulated the AMPK/GLUT4 signaling pathway in the treatment of diabetic encephalopathy, AMPK has been recognized as the most upstream of these cascades, making AMPK is a key target for DMY in the treatment of these diseases. However, how DMY activates AMPK and whether it also mediates other related signaling pathways such as AMPK-NLRP3 to treat diabetes still requires to be elucidated. In the future, molecular docking and molecular dynamics simulations utilized to verify the association between DMY and its potential targets is full of great significance. Furthermore, exploration of the specific binding sites where DMY activates AMPK, such as Thr172, will provide compelling evidence for AMPK to become an attractive potential target for DMY to treat DM. Despite majority of studies focused on the mechanism and potential functions of DMY in treating DM, further study is needed to determine whether DMY combined with first-line diabetes drugs would exhibit better efficacy.
This review provides a systematic summary and in-depth discussion of DMY in the context of signal transduction pathways that regulate AMPK or mTOR/autophagy, as well as relevant downstream cascades, such as PGC-1α/SIRT3, MEK/ERK, and PI3K/Akt signaling pathways. Indeed, from a pharmacological standpoint, DMY holds promise as a valuable flavonoid compound with potential to prevent the initiation and progression of several diseases. While hypothesized mechanisms of action have been reported, further comprehensive mechanistic and toxicological studies are essential to advance our understanding of DMY’s effects. According to ClinicalTrials.gov, there are only four clinical studies published on DMY aganist human diseases up to date (Table 2), and we find that clinical studies of DMY are very limited, as the low bioavailability of DMY is a challenge. Another reason is the "dose-time-toxic-function" of DMY, which is still unclear. Conducting such studies will be crucial in expediting experimental research and preclinical investigations, ultimately paving the way for DMY to potentially be developed into commercial drugs in the near future.
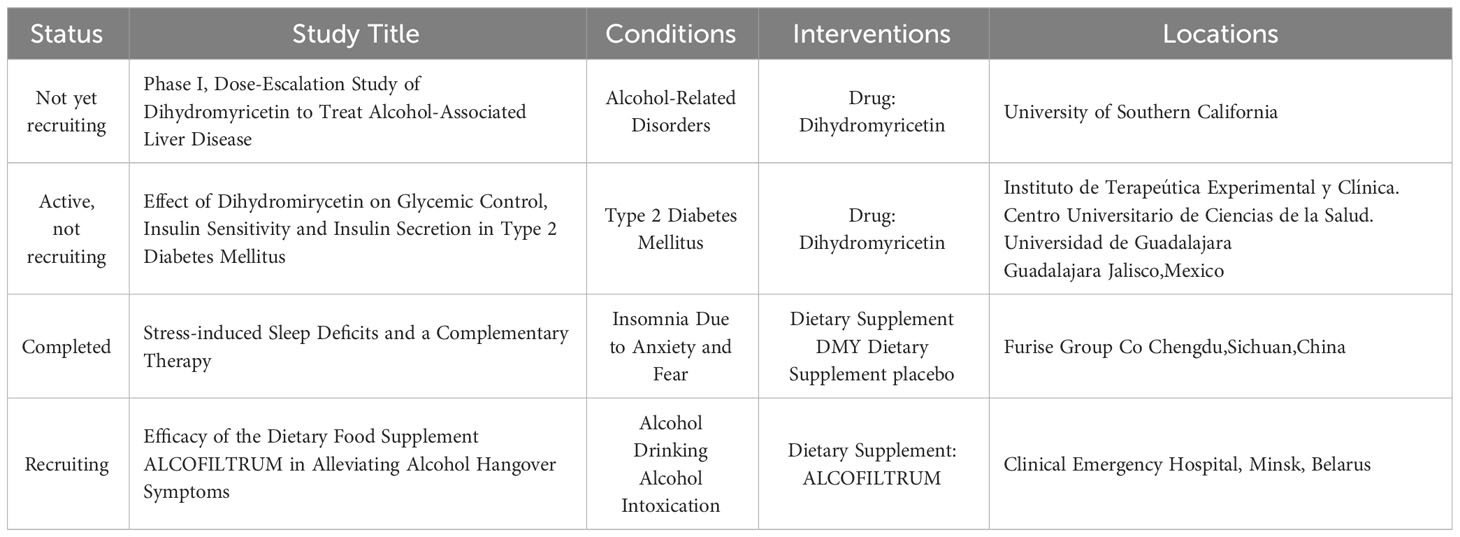
Table 2 Clinical trials for Dihydromyricetin.
ZW was in charge of searching all the relative papers and writing this manuscript. ZC was in charge of drawing the picture. ZFY and ZYY gave their valuable and professional suggestions, guided in organizing and drafting this manuscript and provided funding. ZFY reviewed the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the National Natural Science Foundation of China (82003170 to ZYY), the National Natural Science Foundation of China (82272645 & 81971555 to ZFY); The Shanghai Rising-Star Program (23QA1408100 to ZYY); the Natural Science Foundation of Shanghai (23ZR1451800 to ZYY); the Shanghai Municipal Health Commission (2022YQ065 to ZYY).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Park JJ. Epidemiology, pathophysiology, diagnosis and treatment of heart failure in diabetes. Diabetes Metab J (2021) 45(2):146–57. doi: 10.4093/dmj.2020.0282
2. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
3. Cole JB, Florez JC. Genetics of diabetes mellitus and diabetes complications. Nat Rev Nephrol (2020) 16(7):377–90. doi: 10.1038/s41581-020-0278-5
4. Mahajan A, Spracklen CN, Zhang W, Ng MCY, Petty LE, Kitajima H. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat Genet (2022) 54(5):560–72. doi: 10.1038/s41588-022-01058-3
5. Rahman MM, Islam MR, Rabbi F, Islam MT, Sultana S, Ahmed M, et al. Bioactive Compounds and Diabetes Mellitus: Prospects and Future Challenges. Curr Pharm Des. (2022) 28(16):1304–1320. doi: 10.2174/1381612828666220412090808
6. Fox ME, Lobo MK. The molecular and cellular mechanisms of depression: a focus on reward circuitry. Mol Psychiatry (2019) 24(12):1798–815. doi: 10.1038/s41380-019-0415-3
7. Halim M, Halim A. The effects of inflammation, aging and oxidative stress on the pathogenesis of diabetes mellitus (type 2 diabetes). Diabetes Metab Syndrome: Clin Res Rev (2019) 13(2):1165–72. doi: 10.1016/j.dsx.2019.01.040
8. Liu D, Mao Y, Ding L, Zeng XA. Dihydromyricetin: A review on identification and quantification methods, biological activities, chemical stability, metabolism and approaches to enhance its bioavailability. Trends Food Sci Technol (2019) 91:586–97. doi: 10.1016/j.tifs.2019.07.038
9. Tong H, Zhang X, Tan L, Jin R, Huang S, Li X. Multitarget and promising role of dihydromyricetin in the treatment of metabolic diseases. Eur J Pharmacol (2020) 870:172888. doi: 10.1016/j.ejphar.2019.172888
10. Liu S, Liu Q, Peng Q, Zhang Y, Wang J. [Dihydromyricetin improves cardiac insufficiency by inhibiting HMGB1 in diabetic rats]. Nan Fang Yi Ke Da Xue Xue Bao (2022) 42(5):641–8. doi: 10.12122/j.issn.1673-4254.2022.05.03
11. Fan L, Tong Q, Dong WW. Tissue distribution, excretion, and metabolic profile of dihydromyricetin, a flavonoid from vine tea (Ampelopsis grossedentata) after oral administration in rats. J Agric Food Chem (2017) 65(23):4597–604. doi: 10.1021/acs.jafc.7b01155
12. Fan L, Zhao X, Tong Q, Zhou X, Chen J, Xiong W, et al. Interactions of Dihydromyricetin, a Flavonoid from Vine Tea (Ampelopsis grossedentata) with Gut Microbiota. J Food Sci (2018) 83(5):1444–53. doi: 10.1111/1750-3841.14128
13. Zhang J, Chen Y, Luo H, Sun L, Xu M, Yu J, et al. Recent update on the pharmacological effects and mechanisms of dihydromyricetin. Front Pharmacol (2018) 9:1204. doi: 10.3389/fphar.2018.01204
14. Wu B, Lin J, Luo J, Han D, Fan M, Guo T, et al. Dihydromyricetin protects against diabetic cardiomyopathy in streptozotocin-induced diabetic mice. BioMed Res Int (2017) 3(21):1–13. doi: 10.1155/2017/3764370
15. Chen H, Gong XH, Chen LJ, Xia S, Zhu JD, Nutrition DO. Dihydromyricetin Prevents against Diabetic Nephropathy through the Activation of AMPK Pathway. Progress in Modern Biomedicine (2017) 17(34):6613–9. doi: 10.13241/j.cnki.pmb.2017.34.003
16. Liang X, Zhang T, Shi L, Kang C, Wan J, Zhou Y, et al. Ampelopsin protects endothelial cells from hyperglycemia-induced oxidative damage by inducing autophagy via the AMPK signaling pathway. Biofactors (2016) 41(6):463–75. doi: 10.1002/biof.1248
17. Shi L, Zhang T, Zhou Y, Zeng X, Ran L, Zhang Q, et al. Dihydromyricetin improves skeletal muscle insulin sensitivity by inducing autophagy via the AMPK-PGC-1α-Sirt3 signaling pathway. Endocrine (2015) 50(2):378–89. doi: 10.1007/s12020-015-0599-5
18. Le L, Jiang B, Wan W, Zhai W, Xiao P. Metabolomics reveals the protective of Dihydromyricetin on glucose homeostasis by enhancing insulin sensitivity OPEN. Sci Rep (2016) 6:36184. doi: 10.1038/srep36184
19. Zhou Q, Gu Y, Lang H, Wang X, Chen K, Gong X, et al. Dihydromyricetin prevents obesity-induced slow-twitch-fiber reduction partially via FLCN/FNIP1/AMPK pathway. Biochim Biophys Acta Mol Basis Dis (2017) 1863(6):1282–91. doi: 10.1016/j.bbadis.2017.03.019
20. Zhou Y, Wu Y, Qin Y, Liu L, Wan J, Zou L, et al. Ampelopsin improves insulin resistance by activating PPARγ and subsequently up-regulating FGF21-AMPK signaling pathway. PloS One (2016) 11(7):e0159191. doi: 10.1371/journal.pone.0159191
21. He J, Zhang J, Dong L, Dang X, Wang L, Cheng L, et al. Dihydromyricetin attenuates metabolic syndrome and improves insulin sensitivity by upregulating insulin receptor substrate-1 (Y612) tyrosine phosphorylation in db/db mice. Diabetes Metab syndrome Obes Targets Ther (2019) 12:2237–49. doi: 10.2147/DMSO.S218487
22. Liu L, Wan J, Lang H, Si M, Zhu J, Zhou Y, et al. Dihydromyricetin delays the onset of hyperglycemia and ameliorates insulin resistance without excessive weight gain in Zucker diabetic fatty rats. Mol Cell Endocrinol (2017) 439(5):105–15. doi: 10.1016/j.mce.2016.10.028
23. Liu L, Zhou M, Lang H, Zhou Y, Mi M. Dihydromyricetin enhances glucose uptake by inhibition of MEK/ERK pathway and consequent down-regulation of phosphorylation of PPARγ in 3T3-L1 cells. J Cell Mol Med (2018) 22(2):1247–56. doi: 10.1111/jcmm.13403
24. Hua Y, Zhang Y, Gong W, Ding Y, Shen J, Li H, et al. Dihydromyricetin improves endothelial dysfunction in diabetic mice via oxidative stress inhibition in a SIRT3-dependent manner. Int J Mol Sci (2020) 21(18):1–15. doi: 10.3390/ijms21186699
25. Lu F, Wang E, Qin T, Yu J, University G. Hypoglycemic effect of dihydromyricetin from Ampelopsis grossedentata on diabetic mice. Pharmacol Clinics Chin Materia Medica. (2016) 03):45–8. doi: 10.13412/j.cnki.zyyl.2016.03.012
26. Ling H, Zhu Z, Yang J, He J, Yang S, Wu D, et al. Dihydromyricetin improves type 2 diabetes-induced cognitive impairment via suppressing oxidative stress and enhancing brain-derived neurotrophic factor-mediated neuroprotection in mice. Acta Biochim Biophys Sin (2018) 50(3):298–306. doi: 10.1093/abbs/gmy003
27. Guo L, Tan K, Luo Q, Bai X. Dihydromyricetin promotes autophagy and attenuates renal interstitial fibrosis by regulating miR-155-5p/PTEN signaling in diabetic nephropathy. Bosn J Basic Med Sci (2020) 20(3):372–80. doi: 10.17305/bjbms.2019.4410
28. Li W, Xiao H. Dihydromyricetin alleviates high glucose-induced oxidative stress and apoptosis in human retinal pigment epithelial cells by downregulating miR-34a expression. Diabetes Metab Syndr Obes (2021) 14:387–97. doi: 10.2147/DMSO.S290633
29. Ge H, Guan S, Shen Y, Sun M, Gao Y. Dihydromyricetin affects BDNF levels in the nervous system in rats with comorbid diabetic neuropathic pain and depression. Sci Rep (2019) 9(1):1–12. doi: 10.1038/s41598-019-51124-w
30. Guan S, Shen Y, Ge H, Xiong W, He L, Liu L, et al. Dihydromyricetin alleviates diabetic neuropathic pain and depression comorbidity symptoms by inhibiting P2X(7) receptor. Front Psychiatry (2019) 10:770. doi: 10.3389/fpsyt.2019.00770
31. Liu L, Yin X, Wang X, Li X. Determination of dihydromyricetin in rat plasma by LC-MS/MS and its application to a pharmacokinetic study. Pharm Biol (2017) 55(1):657–62. doi: 10.1080/13880209.2016.1266669
32. Hou L, Jiang F, Huang B, Zheng W, Jiang Y, Cai G, et al. Dihydromyricetin ameliorates inflammation-induced insulin resistance via phospholipase C-caMKK-AMPK signal pathway. Oxid Med Cell Longev (2021) 2021:8542809. doi: 10.1155/2021/8542809
33. Rodríguez C, Muñoz M, Contreras C, Prieto D. AMPK, metabolism, and vascular function. FEBS J (2021) 288(12):3746–71. doi: 10.1111/febs.15863
34. González A, Hall MN, Lin SC, Hardie DG. AMPK and TOR: the yin and yang of cellular nutrient sensing and growth control. Cell Metab (2020) 31(3):472–92. doi: 10.1016/j.cmet.2020.01.015
35. He C, Zhu H, Li H, Zou M, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes (2013) 62(4):1270–81. doi: 10.2337/db12-0533
36. Shi L, Zhang T, Liang X, Hu Q, Huang J, Zhou Y, et al. Dihydromyricetin improves skeletal muscle insulin resistance by inducing autophagy via the AMPK signaling pathway. Mol Cell Endocrinol (2015) 409:92–102. doi: 10.1016/j.mce.2015.03.009
37. Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol (2009) 20(2):98–105. doi: 10.1097/MOL.0b013e328328d0a4
38. Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, et al. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PloS One (2010) 5(7):e11707. doi: 10.1371/journal.pone.0011707
39. Wang D, Cao L, Zhou X, Wang G, Ma Y, Hao X, et al. Mitigation of honokiol on fluoride-induced mitochondrial oxidative stress, mitochondrial dysfunction, and cognitive deficits through activating AMPK/PGC-1α/Sirt3. J Hazard Mater (2022) 437:129381. doi: 10.1016/j.jhazmat.2022.129381
40. Chang YC, Chan MH, Yang YF, Li CH, Hsiao M. Glucose transporter 4: Insulin response mastermind, glycolysis catalyst and treatment direction for cancer progression. Cancer Lett (2023) 563:216179. doi: 10.1016/j.canlet.2023.216179
41. Entezari M, Hashemi D, Taheriazam A, Zabolian A, Mohammadi S, Fakhri F, et al. AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: A pre-clinical and clinical investigation. BioMed Pharmacother (2022) 146:112563. doi: 10.1016/j.biopha.2021.112563
42. Fryer LG, Foufelle F, Barnes K, Baldwin SA, Woods A, Carling D. Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem J (2002) 363(Pt 1):167–74. doi: 10.1042/bj3630167
43. Luo Z, Zhang Y, Li F, He J, Ding H, Yan L, et al. Resistin induces insulin resistance by both AMPK-dependent and AMPK-independent mechanisms in HepG2 cells. Endocrine (2009) 36(1):60–9. doi: 10.1007/s12020-009-9198-7
44. Abbud W, Habinowski S, Zhang JZ, Kendrew J, Elkairi FS, Kemp BE, et al. Stimulation of AMP-activated protein kinase (AMPK) is associated with enhancement of glut1-mediated glucose transport. Arch Biochem Biophysics (2000) 380(2):347–52. doi: 10.1006/abbi.2000.1935
45. Gao XK, Rao XS, Cong XX, Sheng ZK, Sun YT, Xu SB, et al. Phase separation of insulin receptor substrate 1 drives the formation of insulin/IGF-1 signalosomes. Cell Discovery (2022) 8(1):60. doi: 10.1038/s41421-022-00426-x
46. Xu D, Li C, Jiang Z, Wang L, Huang H, Li Z, et al. The hypoglycemic mechanism of catalpol involves increased AMPK-mediated mitochondrial biogenesis. Acta pharmacologica Sin (2020) 41(6):791–9. doi: 10.1038/s41401-019-0345-2
47. Reyes N, Banks G, Tsang M, Margineantu D, Gu H, Djukovic D, et al. Fnip1 regulates skeletal muscle fiber type specification, fatigue resistance, and susceptibility to muscular dystrophy. Proc Natl Acad Sci United States America (2015) 112(2):424–9. doi: 10.1073/pnas.1413021112
48. Frkic RL, Richter K, Bruning JB. The therapeutic potential of inhibiting PPARγ phosphorylation to treat type 2 diabetes. J Biol Chem (2021) 297(3):101030. doi: 10.1016/j.jbc.2021.101030
49. Yu Y, Du H, Wei S, Feng L, Li J, Fan Y, et al. Adipocyte-derived exosomal miR-27a induces insulin resistance in skeletal muscle through repression of PPARγ. Theranostics (2018) 8(8):2171–88. doi: 10.7150/thno.22565
50. Qiang L, Accili D. FGF21 and the second coming of PPARγ. Cell (2012) 148(3):397–8. doi: 10.1016/j.cell.2012.01.020
51. Yanai H, Yoshida H. Beneficial effects of adiponectin on glucose and lipid metabolism and atherosclerotic progression: mechanisms and perspectives. Int J Mol Sci (2019) 20(5):1190. doi: 10.3390/ijms20051190
52. Kita S, Maeda N, Shimomura I. Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest (2019) 129(10):4041–9. doi: 10.1172/JCI129193
53. Liguori I, Russo G, Curcio F, Bulli G, Aran L, Della-Morte D, et al. Oxidative stress, aging, and diseases. Clin Interv Aging (2018) 13:757–72. doi: 10.2147/CIA.S158513
54. Malik A, Morya RK, Saha S, Singh PK, Bhadada S, Rana SV. Oxidative stress and inflammatory markers in type 2 diabetic patients. Eur J Clin Invest (2020) 50(6):e13238. doi: 10.1111/eci.13238
55. Zhang P, Li T, Wu X, Nice EC, Huang C, Zhang Y. Oxidative stress and diabetes: antioxidative strategies. Front Med (2020) 14(5):583–600. doi: 10.1007/s11684-019-0729-1
56. Lenzen S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia (2008) 51(2):216–226. doi: 10.1007/s00125-007-0886-7
57. Zeng X, Yang J, Hu O, Huang J, Ran L, Chen M, et al. Dihydromyricetin ameliorates nonalcoholic fatty liver disease by improving mitochondrial respiratory capacity and redox homeostasis through modulation of SIRT3 signaling. Antioxidants Redox Signaling (2019) 30(2):163–83. doi: 10.1089/ars.2017.7172
58. Magalhães JP, Santos DA, Correia IR, Hetherington-Rauth M, Ribeiro R, Raposo JF, et al. Impact of combined training with different exercise intensities on inflammatory and lipid markers in type 2 diabetes: a secondary analysis from a 1-year randomized controlled trial. Cardiovasc Diabetol (2020) 19(1):169. doi: 10.1186/s12933-020-01136-y
59. Esser N, Paquot N, Scheen AJ. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin Investig Drugs (2015) 24(3):283–307. doi: 10.1517/13543784.2015.974804
60. Vas PRJ, Papanas N. Depression and diabetic peripheral neuropathy: birds of a feather, but when do they flock together? Exp Clin Endocrinol Diabetes (2020) 128(5):347–9. doi: 10.1055/a-0808-4269
61. Ge H, Sun M, Wei X, Zhang M, Tu H, Hao Y, et al. Protective effects of dihydromyricetin on primary hippocampal astrocytes from cytotoxicity induced by comorbid diabetic neuropathic pain and depression. Purinergic Signal. (2020) 16(4):585–599. doi: 10.1007/s11302-020-09752-9
62. Chen S, Zhao X, Wan J, Ran L, Qin Y, Wang X, et al. Dihydromyricetin improves glucose and lipid metabolism and exerts anti-inflammatory effects in nonalcoholic fatty liver disease: A randomized controlled trial. Pharmacol Res (2015) 99:74–81. doi: 10.1016/j.phrs.2015.05.009
63. Chen J, Wang X, Xia T, Bi Y, Liu B, Fu J, et al. Molecular mechanisms and therapeutic implications of dihydromyricetin in liver disease. Biomedicine pharmacotherapy (2021) 142:111927. doi: 10.1016/j.biopha.2021.111927
64. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol (2018) 19(2):121–35. doi: 10.1038/nrm.2017.95
65. Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy status. Cell Metab (2018) 27(2):299–313. doi: 10.1016/j.cmet.2017.10.009
66. Ran L, Wang X, Lang H, Xu J, Wang J, Liu H, et al. Ampelopsis grossedentata supplementation effectively ameliorates the glycemic control in patients with type 2 diabetes mellitus. Eur J Clin Nutr (2019) 73(5):776–82. doi: 10.1038/s41430-018-0282-z
67. Shan T, Xiong Y, Zhang P, Li Z, Jiang Q, Bi P, et al. Lkb1 controls brown adipose tissue growth and thermogenesis by regulating the intracellular localization of CRTC3. Nat Commun (2016) 7:12205. doi: 10.1038/ncomms12205
68. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature (2018) 560(7716):102–6. doi: 10.1038/s41586-018-0353-2
69. Leng Q, Zhou J, Li C, Xu Y, Liu L, Zhu Y, et al. Dihydromyricetin ameliorates diet-induced obesity and promotes browning of white adipose tissue by upregulating IRF4/PGC-1α. Nutr Metab (2022) 19(1):38. doi: 10.1186/s12986-022-00672-6
70. Sun P, Yin J, Liu L, Guo J, Wang S, Qu C, et al. Protective role of Dihydromyricetin in Alzheimer's disease rat model associated with activating AMPK/SIRT1 signaling pathway. Bioscience Rep (2019) 39(1):BSR20180902. doi: 10.1042/BSR20180902
71. Zhao Z, Yin J, Wu M, Song G, Xie X, Zou C, et al. Dihydromyricetin activates AMP-activated protein kinase and P38(MAPK) exerting antitumor potential in osteosarcoma. Cancer Prev Res (Philadelphia Pa.) (2014) 7(9):927–38. doi: 10.1158/1940-6207.CAPR-14-0067
Keywords: dihydromyricetin, diabetes, diabetes complications, AMPK, research progress
Citation: Wang Z, Cao Z, Yue Z and Yang Z (2023) Research progress of dihydromyricetin in the treatment of diabetes mellitus. Front. Endocrinol. 14:1216907. doi: 10.3389/fendo.2023.1216907
Received: 10 May 2023; Accepted: 18 August 2023;
Published: 04 September 2023.
Edited by:
Chunping Zeng, The Fifth Affiliated Hospital of Guangzhou Medical University, ChinaReviewed by:
Weinan Zhou, University of Illinois at Urbana-Champaign, United StatesCopyright © 2023 Wang, Cao, Yue and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengfeng Yang, emhlbmdmZW5nLnlhbmdAc2hnaC5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.