
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 03 August 2023
Sec. Renal Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1215292
This article is part of the Research TopicAssociation between Diabetic Nephropathy and Diabetic Retinopathy or Non-diabetic NephropathyView all 8 articles
 Luxin Li1,2†
Luxin Li1,2† Yucen Dai1†
Yucen Dai1† Dan Ke1†
Dan Ke1† Jieting Liu1,2
Jieting Liu1,2 Peijian Chen1,2
Peijian Chen1,2 Dong Wei3
Dong Wei3 Tongtong Wang4
Tongtong Wang4 Yanjie Teng1,2
Yanjie Teng1,2 Xiaohuan Yuan1,2*
Xiaohuan Yuan1,2* Zhen Zhang2,5*
Zhen Zhang2,5*Diabetic nephropathy (DN) and diabetic retinopathy (DR) are the most serious and common diabetes-associated complications. DN and DR are all highly prevalent and dangerous global diseases, but the underlying mechanism remains to be elucidated. Ferroptosis, a relatively recently described type of cell death, has been confirmed to be involved in the occurrence and development of various diabetic complications. The disturbance of cellular iron metabolism directly triggers ferroptosis, and abnormal iron metabolism is closely related to diabetes. However, the molecular mechanism underlying the role of ferroptosis in DN and DR is still unclear, and needs further study. In this review article, we summarize and evaluate the mechanism of ferroptosis and its role and progress in DN and DR, it provides new ideas for the diagnosis and treatment of DN and DR.
Diabetes is the most common metabolic disorder in the world, and its incidence rate has risen sharply. The International Diabetes Federation estimates that 537 million people worldwide had diabetesin 2021, a number that is expected to rise to 783 million by 2045 (1). Diabetic vascular complications are one of the main causes of death and disability (2). The microvascular damage due to hyperglycemia can lead to diabetic microvascular complications, including diabetic nephropathy (DN) and diabetic retinopathy (DR). DN is the leading cause of chronic kidney failure and end-stage kidney disease. Oxidative stress induced by chronic hyperglycemia is considered a key mechanistic factor in the development of DN. DN is characterized by persistent albuminuria, elevated arterial blood pressure, and decreased glomerular filtration rate (3). Renal function in patients with diabetes frequently worsens in severity over time, and eventually to renal failure. The pathological changes are similar since both DN and DR have microvascular changes. Hyperglycaemia induces retinal oxidative stress, which plays an important role in DR (4). DR is a leading cause of progressive vision loss and blindness (5). The current research hotspots mainly focus on the exploration of the pathological mechanism of DN and DR.
Ferroptosis, an iron-dependent lipid peroxidation-driven new form of regulated cell death, is different from other types of cell death such as apoptosis, necrosis, autophagy, and pyroptosis (Table 1) (6, 7). Iron deposition leads to the formation of reactive oxygen species (ROS) and oxidative stress through the Fenton reaction, further promoting lipid peroxidation, a key factor in ferroptosis (8). Emerging evidence suggests a strong link between diabetic complications and ferroptosis. Hyperglycemia leads to the overproduction of ROS and induces oxidative stress in various organs of diabetic patients (9). High glucose could cause an overload of iron, and iron dysregulation induces the production of ROS and promotes oxidative stress, which leads to ferroptosis finally (10). Studies have shown that high ROS induces oxidative stress in kidney and retinal cells resulting in cell death (11, 12). This means that targeting ferroptosis may be an effective strategy to treat related DN and DR. This review summarized the mechanisms of ferroptosis and discussed the role of ferroptosis in DN and DR. In addition, we elaborated recent data about the novel actors of some “conventional” drugs or natural compounds as ferroptosis inducers or inhibitors to find common molecular mechanisms and therapeutic targets in DN and DR. It is of great significance to clarify the pathological mechanism of diabetic microvascular complications.
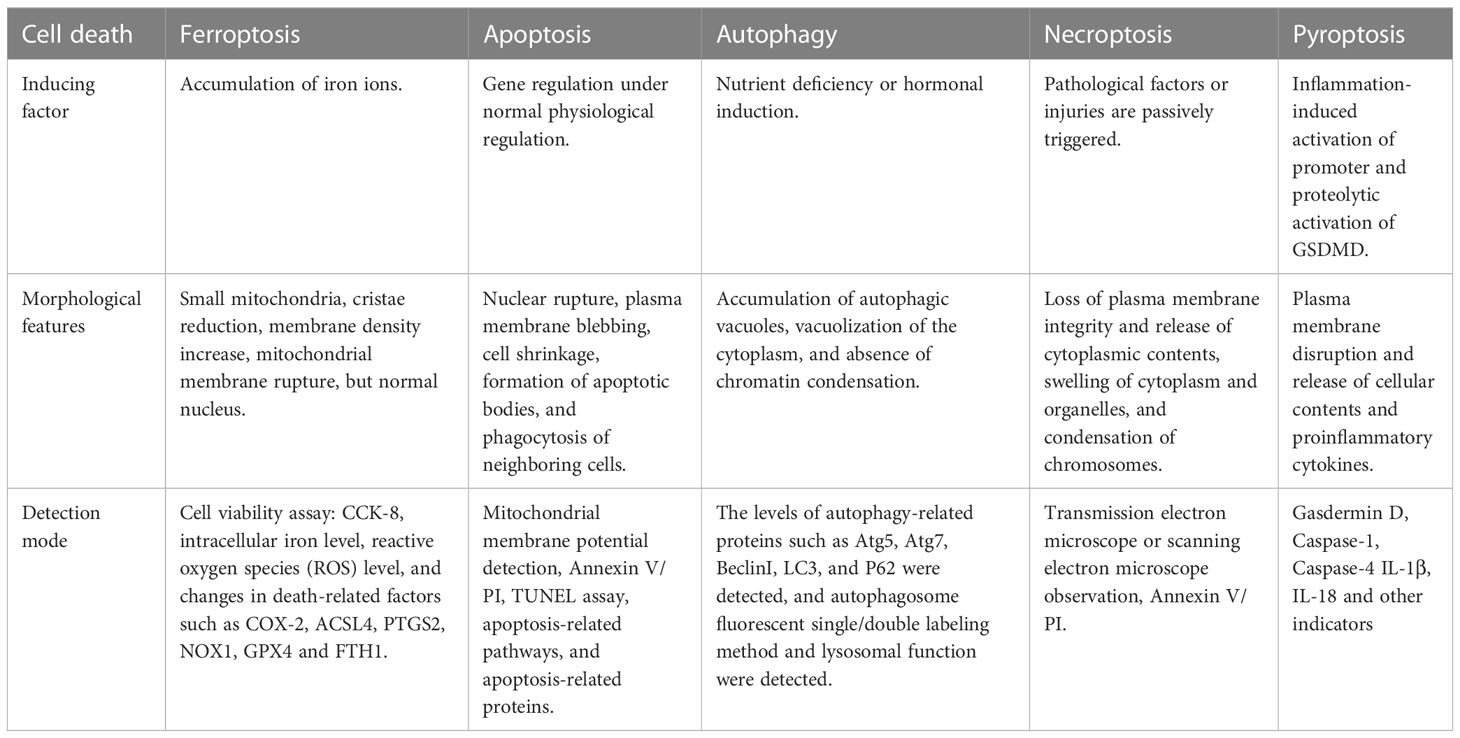
Table 1 Comparison of several common types of cell death.
Ferroptosis is a newly described type of iron-dependent programmed cell death. The basic principle is that divalent iron or ester oxygenase catalyzes lipid peroxidation of highly expressed unsaturated fatty acids on the cell membrane, which induces cell death (13). In addition to increased levels of peroxidation, it also reduces the decrease of glutathione peroxidase 4 (GPX4), the core enzyme regulating the antioxidant system (glutathione system, GSH system) (14). Ferroptosis is regulated by multiple signaling pathways (Figure 1) by the following mechanisms.
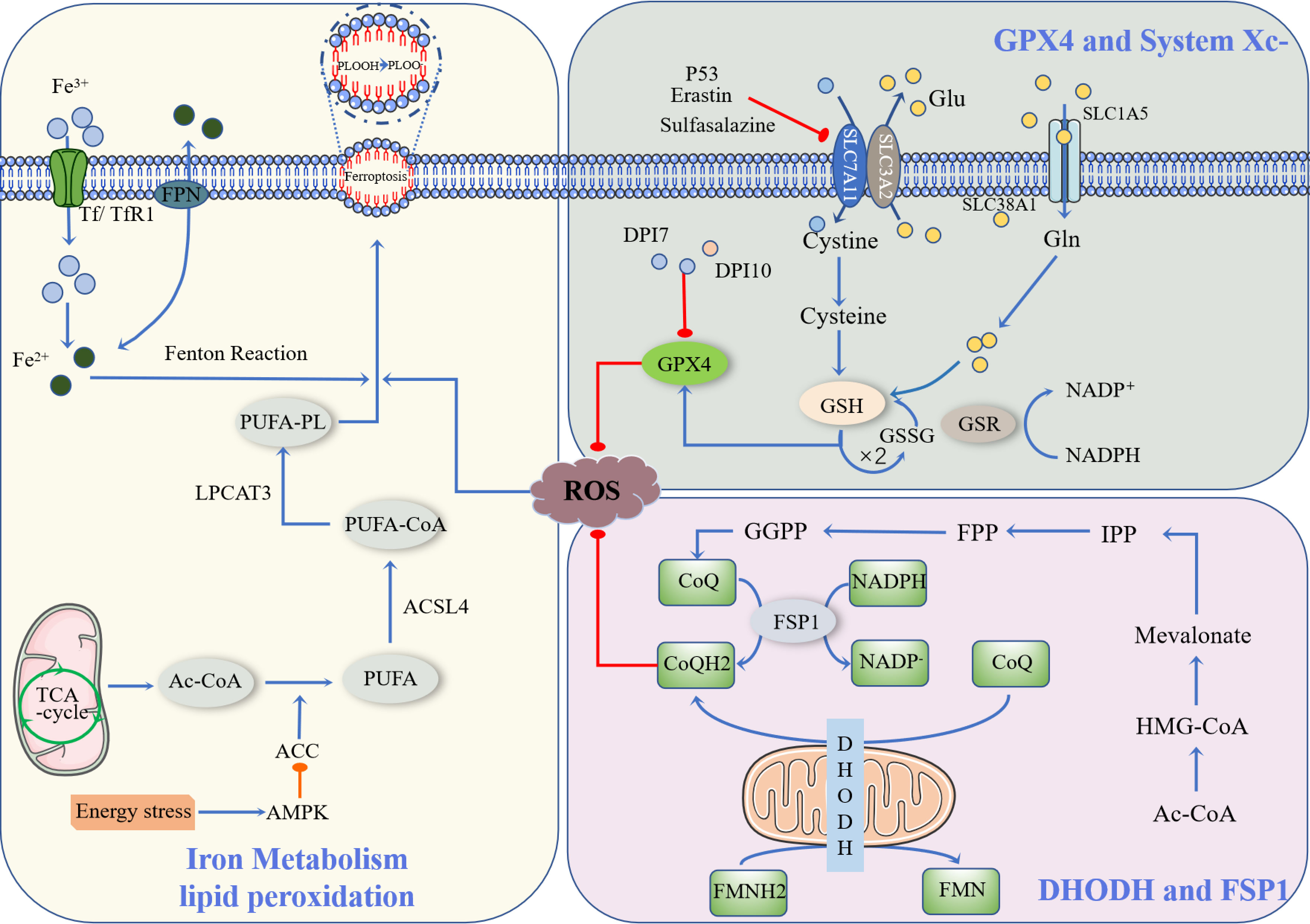
Figure 1 Occurrence and protective factors of Ferroptosis. Cystine and Glu are retroactively transported via the SLC7A11/SLC3A2 complex (p53, Erastin, Sulfasalazine, etc., can inhibit the complex). The cystine that enters the cell is transformed into cysteine by GSH biosynthesis using the electrons supplied by NADPH/H+, and oxidized glutathione (GSSG) is reduced by glutathione disulfide reductase (GSR). They finally enhance the activity of glutathione peroxidase 4 (GPX4). DHODH and FSP1 inhibit the occurrence of ferroptosis through CoQ/CoQH2, and Ac-CoA generates CoQ through a series of biosynthesis reactions. NADPH, as a hydrogen donor, assists FSP1 in reducing CoQ to CoQH2, while DHODH performs this process via FMNH2. CoQH2, in a reduced state, inhibits ferroptosis. Ac-CoA in mitochondria is catalyzed by ACC to produce PUFA, and PUFA is further catalyzed by ACSL4 to form PUFA-CoA, which is then activated by LPCAT3 as PUFA-PL, affecting the transmembrane properties of polyunsaturated fatty acids and thus promoting ferroptosis. Ferric ions are obtained by cells mainly through Tf/TfR1 protein transport and are acidified by endocytic spheroids and released from FPN into the cytoplasm as ferric ions. Excess iron further produces ROS through the Fenton reaction, resulting in an iron drop.
Currently, the known upstream pathways of ferroptosis ultimately point directly or indirectly to glutathione peroxides (GPXs), and therefore, affect the activity of GPXs by regulating GPX4, which is one of the most important ferroptosis defense pathways available (15). Among them, the GSH/GPX4 pathway is the main regulatory mechanism of ferroptosis, such as in the regulation of GPX4, inhibition of the cystine/glutamate reverse transporter system (System Xc-), glutamine metabolic pathway, and p53 regulatory axis (16).
GPX4 is an inhibitor of lipid peroxidation that degrades small molecule peroxides and relatively complex lipid peroxides (17). There are many members of the GPXs, including GPX1-GPX8, of which GPX4 plays an important role in ferroptosis. Yang et al. found that a decrease in GSH led to a decrease in GPX activity (18). GPX catalyzes hydrogen peroxide and degradation of hydrogen peroxide and inhibits the production of lipid reactive oxygen species, for which glutathione is an essential cofactor. The classical ferroptosis control axis requires cystine reduction to cysteine, GSH and/or thioredoxin reductase 1(TXNRD1)-dependent reduction of cystine to cysteine, GSH biosynthesis, and GPX4-mediated reduction of phospholipid hydroperoxide (PL-OOH) to its corresponding alcohol phospholipid hydroxide (PL-OH) using electrons provided by NADPH/H+, oxidized glutathione (GSSG) is recovered by glutathione disulfide reductase (GSR), GSH synthesis is inseparable from glutamine (Gln) and intracellular glutamine uptake is mainly dependent on the involvement of SLC1A5/SLC38A1 (19).
System Xc- is a heterodimer composed of SLC7A11 and SLC3A2, of which SLC7A11 is the main subunit that exerts its function (20). Downregulation of SLC7A11 can indirectly inhibit GPX4 activity by inhibiting the cysteine metabolic pathway, leading to reduced intracellular cystine levels and depletion of GSH biosynthesis, which in turn leads to lipid peroxide accumulation and ultimately induces cell ferroptosis (21). In addition, Erastin, a classical ferroptosis promoter, can target and inhibit SLC7A11 to induce ferroptosis (22). Related studies have shown that the ferroptosis process induced by sulfasalazine is also associated with inhibition of SLC7A11 (23). In addition, SLC7A11 is also associated with the p53 gene: p53 downregulates SLC7A11 expression and inhibits Systemic Xc- uptake of cysteine, leading to decreased cysteine-dependent glutathione peroxidase activity, reduced cellular antioxidant capacity, and increased lipid reactive oxygen species, resulting in cellular iron sagging (16). Inhibition of Systemic Xc- would lead to compensatory transcriptional upregulation of SLC7A11, preventing cystine uptake and thus leading to ferroptosis (24).
Cystine is an indispensable component necessary for the biosynthesis of GSH, a process catalyzed by glutamate-cysteine ligase and glutathione synthase, and a decrease in cellular antioxidant capacity (25). Glutathione is an essential cofactor for the function of GPXs, which results in a decrease in GPX activity. During glutamate-induced excitotoxic cell death, glutamate can initiate two pathways, one for calcium influx and the other for inhibition of Systemic Xc–dependent cysteine uptake pathway (24). Calcium chelators have no inhibitory effect on Erastin-induced ferroptosis, whereas excessive glutamate levels impede the function of Systemic Xc- and induce ferroptosis. Reduction of Systemic Xc- activity decreases cystine uptake; therefore, reduced cystine uptake leads to reduced GSH and eventually GPX4 activity, decreased cellular resistance to peroxidation, accumulation of lipid reactive oxygen species, and oxidative cell death (26).
p53 is an important tumor suppressor gene that mediates cell cycle inhibition, senescence, and apoptosis and plays an important role in tumorigenesis and progression (27). Recently, it has been shown that activated p53 can bind to the promoter region of the SLC7A11 gene, which inhibits the transcriptional activity of the SLC7A11 gene and affects the synthesis of GSH, which can induce the occurrence of ferroptosis (28). Jiang et al. also found that the messenger RNA and protein expression of SLC7A11 decreased significantly after upregulation of p53 gene expression, which confirmed that SLC7A11 is a new target of the p53 gene (29). p53 inhibits Systemic Xc- uptake of cystine by downregulating the expression of System Xc–component SLC7A11, leading to a decrease in cystine-dependent glutathione peroxidase activity, a decrease in cellular antioxidant capacity, and an increase in lipid reactive oxygen species, resulting in cellular ferroptosis (16).
In addition to the above-mentioned indirect action of acting on GSH that activates the GPX4 enzyme, it is also possible to eliminate GPX4 directly, such as Diphenyleneiodonium chloride-7(DPI-7), Diphenyleneiodonium chloride-10 (DPI-10), GPX4 inhibitors, squalene synthase, HMG-CoA reductase, etc (30). In summary, whether by direct or indirect means, the ultimate goal is to regulate the core enzyme GPX4, which can inhibit ferroptosis by eliminating lipid peroxides using reduced GSH.
Recent studies have identified ferroptosis suppressor protein 1 (FSP1) and the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH) as GPX4-independent ferroptosis defense pathways, both of which inhibit ferroptosis by using the reduced state of coenzyme Q10 (CoQ10) to prevent cell membrane lipid peroxidation and thus inhibit the occurrence of ferroptosis (31).
FSP1, formerly also known as apoptosis-inducing factor mitochondria-associated (AIFM2), was renamed FSP1 to avoid confusion due to the recent discovery of its role in inhibiting cellular ferroptosis (32). The inhibition of ferroptosis by FSP1 is mediated by CoQ10, which reduces ubiquinone (CoQ) to dihydro ubiquinone (CoQH2) in the cell membrane, and CoQH2 acts as an antioxidant blocking lipid peroxidation inhibiting ferroptosis from occurring (33).
Mitochondria are organelles wrapped by two membranes, the inner and outer membrane, and are the main site of aerobic respiration, with large amounts of ROS generated by the electron transfer process in the inner membrane (34). In addition to the synthesis of pyrimidine nucleotides, DHODH was found to inhibit ferroptosis in mitochondria by generating CoQH2 in the inner mitochondrial membrane, because CoQH2 can act as a radical trapping antioxidant to prevent lipid peroxidation and thus inhibit ferroptosis (35).
Acetyl coactivators in mitochondria produce the polyunsaturated fatty acids (PUFA) catalyzed by acetyl-CoA carboxylase (ACC) (36). However, the presence of PUFA does not directly lead to ferroptosis. This requires further catalysis by members of the long-chain acyl-coenzyme A synthases (ACSL) family, especially ACSL4, to form PUFA-CoA, which is then activated by LPCAT3 to PUFA-PL, affecting the transmembrane properties of polyunsaturated fatty acids and thus promoting ferroptosis (37). Activation of AMP-activated protein kinas (AMPK) by energy stress directly inhibits ACC, which limits PUFA synthesis and thus inhibits iron phagocytosis (38).
Cellular uptake of iron is mainly through the Tf/TfR1 protein transport pathway to obtain trivalent iron ions, but these trivalent irons still need to be released from FPN as divalent iron by acidification of endocytic cells (39). The divalent iron then enters the cytoplasm through the endosomes. Excess iron further generates ROS through the Fenton reaction, which leads to ferroptosis (40).
DN is the most common complication of diabetes mellitus and the leading cause of end-stage renal disease, which is a “global medical disaster” because of the difficulty of diagnosis and management (41, 42). However, the pathogenesis and pathophysiology of DN are complex. Therefore, further studies on its molecular mechanisms are needed to develop new therapeutic approaches.
Ferroptosis is a newly discovered form of regulated cell death that is different from apoptosis, necrosis, autophagy, and other forms and depends on iron and ROS. It is regulated by a variety of cellular metabolic pathways and various signaling pathways related to diseases (19). Ferroptosis can produce a large amount of ROS, and the kidney is sensitive to oxidative stress due to its rich mitochondrial structure, suggesting that ferroptosis may be related to DN. The persistent hyperglycemic environment promotes ROS production and increases basal levels of several renal cell types, including mesangial cells, podocytes, and tubular epithelial cells (43). The identification of related genes also reveals the involvement of ferroptosis in the pathogenesis of DN (44). Recently, it was shown that inhibition of ferroptosis may slow the progression of DN in a variety of cellular and animal models.
Mesangial cells are a special type of smooth muscle cells distributed between the capillary rings of glomerular capillaries, and their damage is the basic pathological change of DN (45). Sustained hyperglycemia induces hyperplasia of the glomerular mesangium, leading to thickening of the basement membrane, sclerosis of the glomerular capillary wall, and ultimately proteinuria (46). High mobility group box 1 (HMGB1) is known to act as a pro-inflammatory cytokine that is involved in many diabetic complications (47). Related experiments have shown that high glucose (HG)-induced ferroptosis of mesangial cells can be prevented by inhibiting HMGB1 (48). Ferroptosis is also associated with podocyte injury in diabetic patients. Podocytes are an important component of the glomerular filtration barrier (GFB), and recently, it was revealed that podocytes regulate the GFB through endocytosis, and podocyte injury is considered one of the main mechanisms leading to GFB damage (49, 50). GPX4 and SLC7A11 are considered key proteins in the prevention of ferroptosis, and the loss or inhibition of GPX4/SLC7A11 can induce ferroptosis (51). In vitro studies demonstrate that Sp1-mediated upregulation of Prdx6 expression inhibits iron accumulation and increases the expression of SLC7A11 and GPX4, preventing podocyte injury in DN (52). Another study also revealed that Ginkgolide B may improve DN by protecting the kidney from ferroptosis and oxidative stress damage by inhibiting the ubiquitination of GPX4 (53).
Renal tubular damage is the key factor of DN. The formation of advanced glycation end products (AGEs) associated with hyperglycemia plays a central role in the pathogenesis of diabetic nephropathy. High glucose induces oxidative stress by increasing the production of ROS, excessive iron in cells damages cell function by producing ROS, and renal tubular cells are more sensitive to oxidative stress and lipid peroxidation (54, 55). Wang et al. (56) establish a DN model using streptozotocin (STZ) and db/db mice and found that the expression levels of ferroptosis-related markers, ACSL4 and GPX4, were increased in kidney tissue, especially in the renal tubules. Rosiglitazone, an ACSL4 inhibitor, has a protective effect on DN mice by attenuating ferroptosis, which provides a possibility for the treatment of DN with ferroptosis. In addition, some drugs for diabetes, such as dapagliflozin, improve renal tubular injury by inhibiting ferroptosis (51). Recently, some researchers have found that empagliflozin prevents the progression of ferroptosis by promoting AMPK-mediated (the transcription factor nuclear factor red line related factor 2) Nrf2 activation pathway in vivo and vitro (57). Chinese medicine’s active ingredients, such as licorice flavone, calycosin, and 7-hydroxycoumarin, also inhibit ferroptosis and reduce renal tubular damage induced by diabetes to achieve the effect of DN treatment (58–60). These results suggest that ferroptosis mediates renal tubular injury in DN, and inhibiting ferroptosis in renal tubular cells may play a role in the treatment of DN.
Recent studies have shown that ferroptosis is involved in renal fibrosis of DN, which is the terminal pathological change of DN (56). Heme oxygenase-1 (HO-1) plays an important role in anti-oxidative stress injury, and hypoxia-inducible factor-1 (HIF-1) is a key molecule involved in alleviating hypoxia-induced injury. Excessive HO-1 degradation of oxidized heme leads to iron overload, which results in oxidative stress and lipid peroxidation (61). Feng et al. (62) demonstrated that diabetes enhanced renal tubular damage accelerated tubular iron overload, and aggravated the accumulation of ROS in mouse kidneys through the HIF-1α/HO-1 pathway. In contrast, the ferroptosis inhibitor, Ferrostatin-1, inhibited renal tubular iron overload, renal oxidative stress, and lipid peroxidation in diabetic mice. Renal tubular damage in diabetes may be considered a major cause of renal fibrosis. A large number of studies have shown that the TGF-β/Smad signaling pathway plays a crucial role in renal fibrosis (63). Transforming growth factor-β1 (TGF-β1) is an important factor leading to renal fibrosis, and relevant studies have reported that TGF-β1-induced renal tubular cell death is related to DN (64). Nrf2 is an important regulator of the antioxidant system and realizes redox reactions with multiple downstream targets. Salusin-β participates in HG-induced ferroptosis in HK-2 cells in an Nrf2-dependent manner (65). Fenofibrate inhibits ferroptosis and delays the progression of diabetic nephropathy by up-regulating Nrf2 and 7-hydroxycoumarin by activating the Nrf-2/HO-1 pathway (60, 66). Notoginsenoside R1 (NGR1) is a novel saponin derived from Panax notoginseng. NGR1 promotes Nrf2-mediated HO-1 expression to prevent DN, eliminates ROS that induces apoptosis and TGF-β signaling, and plays a renoprotective role in DN by inhibiting oxidative stress-induced apoptosis and renal fibrosis (67).
In addition, several researchers reviewed the latest findings and emerging trends in ferroptosis research and highlighted that the tumor suppressor p53 has a dual role in ferroptosis. On the one hand, p53 enhances ferroptosis by inhibiting SLC7A11 expression or promoting SAT1 and GLS2 expression. On the other hand, p53 inhibits ferroptosis by inhibiting DPP4 activity or inducing CDKN1A/p21 expression (68). Our previous studies also confirmed that hyperglycemia regulates pathogenic processes in DN through a miR-23b/G3BP2 feedback circuit involving p38MAPK and p53 (69). This suggests that p53 may be an attractive therapeutic target for regulating ferroptosis against DN. Some researchers have analyzed the mechanisms, pathways, and genes related to ferroptosis in DN through bioinformatics, revealing Hub genes related to ferroptosis in DN that mainly include FPR3, C3AR1, CD14, ITGB2, RAC2, and ITGAM. Moreover, there are non-coding genes that interact with the Hub genes and these mainly include has-miR-572, has-miR-29a-3p, has-miR-29b-3p, has-miR-208a-3p, has-miR-153-3p, has-miR-29c-3p, etc. The transcription factors related to DN mainly include HIF1α, KLF4, KLF5, RUNX1, SP1, VDR, and WT1. A KEGG pathway enrichment analysis showed that the MAPK signaling pathway was significantly enriched (70–72). These observations also serve as a starting point for future mechanistic studies. In conclusion, ferroptosis plays a crucial role in the development of DN, and ferroptosis may be the future direction of DN treatment.
DR is a common and specific microvascular complication of diabetes mellitus. Blindness caused by DR has a great impact on the life of patients (73). The disease is negatively affected by diabetes mellitus, which alters normal cellular interactions, leading to severe vascular abnormalities, the loss of the blood-retinal barrier, and impaired neuronal function. Conventional therapies have not responded well to recent treatments, and a new treatment is needed (74). With evidence supporting the interaction between iron metabolism and diabetes, the molecular mechanism of ferroptosis in the pathogenesis of DR has also attracted attention, which provides a new therapeutic target for the treatment of DR (75).
Retinal pericyte loss is one of the earliest changes associated with DR. Although the pathophysiological mechanisms of DR are complex, vascular endothelial damage, increased vascular permeability, and neovascularization are the most common phenomena (76). Vascular endothelial growth factor (VEGF) plays a leading role in the occurrence and progression of DR, and berberine inhibits insulin-induced retinal endothelial cell activation through the Akt/mTOR/HIF-1α/VEGF pathway to improve insulin-induced DR progression (77). Increased vascular permeability in the early stage of DR leads to macular edema in the later stage, which is related to the release of proinflammatory cytokines (74). TRIM46 promotes HG-induced ferroptosis in human retinal capillary endothelial cells (HRCECs) by regulating the ubiquitination and degradation of GPX4 (76). A recent study showed that TRIM46 aggravates HG-induced hyperpermeability and inflammatory response of HRCECs by promoting IκBα ubiquitination (78). In addition, ferroptosis is characterized by the accumulation of lipid ROS, particularly the loss of the activity of lipid hydroperoxides and the lipid repair enzyme GPX4 (54). Fan et al. (79) found that BMS309403, an inhibitor of FABP4, promoted peroxisome proliferator-activated receptor γ (PPARγ), thereby regulating PPARγ-mediated ferroptosis to alleviate lipid peroxidation and oxidative stress in DR.
Studies on non-coding RNAs and related signaling pathways demonstrate a link between DR and ferroptosis. Previous research by our group showed that miR-200b was closely associated with diabetes and its complications (80–82), and miR-200b regulated VEGF-mediated alterations in DR (80). Zhu et al. (83) found that the knockdown of circ-PSEN1 reduced ferroptosis in ARPE19 cells induced by high glucose through the miR-200b-3p/CFL2 axis. Zhou et al. (84) also found that HG-induced ferroptosis in retinal epithelial cells could be inhibited by blocking the miR-338-3p/SLC1A5 axis. In addition, Astragaloside-IV may inhibit DR by reducing miR-138-5p expression and subsequently increasing Sirt1/Nrf2 activity and cellular antioxidant capacity to alleviate ferroptosis, resulting in reduced cell death (85). In addition, Liu et al. (86) expanded the understanding of the relationship between autophagy and ferroptosis. They found that the GMFB antibody, lysosomal activator NKH477, CMA activator QX77, and ferroptosis inhibitor lipstatin-1 effectively prevented early DR, which had a strong clinical application value. In summary, ferroptosis plays a crucial role in the development of DR and may provide a new therapeutic strategy for the treatment of DR.
This article reviews the role and potential mechanisms of ferroptosis in DN and DR. Although diabetes has been investigated for decades, the effective prevention and treatment of diabetic complications is a challenging clinical issue. Long-term hyperglycemia and genetic susceptibility increase the risk of microvascular complications in patients with diabetes (87, 88). The microvascular complications of diabetes are related to the long-term damage and dysfunction of various organs and systems, including the kidney and retina, which may lead to end-stage renal disease and blindness, leading to a significant increase in incidence rate and mortality (89, 90). DN and DR are interrelated through a common pathophysiological mechanism. The main biological mechanism of DN and DR can be linked through the excessive production of ROS, downstream intracellular signal pathways their regulators, which can be used as therapeutic targets for the treatment of diabetes complications (91, 92).
Ferroptosis is a new form of cell death characterized by iron-dependent accumulation of lipid peroxides to lethal levels (93). Iron is an essential trace metal element involved in many physiological processes of the human body. However, excess iron can generate oxidative stress and cause tissue damage (94). Given the association between diabetes and ferroptosis, starting from the key target of ferroptosis may improve diabetes. In recent years, there has been increasing research on ferroptosis in DN and DR. The pathophysiology of DN and DR is complex, which involves many types of cells. Multiple signaling pathways are involved in the ferroptosis process of DN and DR, including Nrf2, HIF-1, TGF-β1, VEGF, etc (Figure 2). Some ferroptosis inhibitors and iron chelators have shown good regulatory effects in animal and cellular experiments related to DN and DR. Many antiferroptotic natural products and drugs also provide new ideas and targets for the treatment of DN and DR (Table 2). Although selective inhibition of ferroptosis has been proven to substantially improve kidney function and play a retinal protected role in various animal models and cell models, clinical trials have not yet been performed with ferroptosis-specific inhibitors to treat DN and DR. The exact mechanism of ferroptosis needs to be explored through further study. Monitoring and controlling ferroptosis related factors may be a promising measure for early diagnosis and treatment of diabetes. However, there have been no established specific markers to demonstrate the presence of ferroptosis in vivo. Identifying key biomarkers of ferroptosis will also facilitate our understanding of its role and progress in diabetic complications. To sum up, although its biological function and molecular mechanism have not been thoroughly elucidated, ferroptosis has become a hot spot in diabetic complications research.
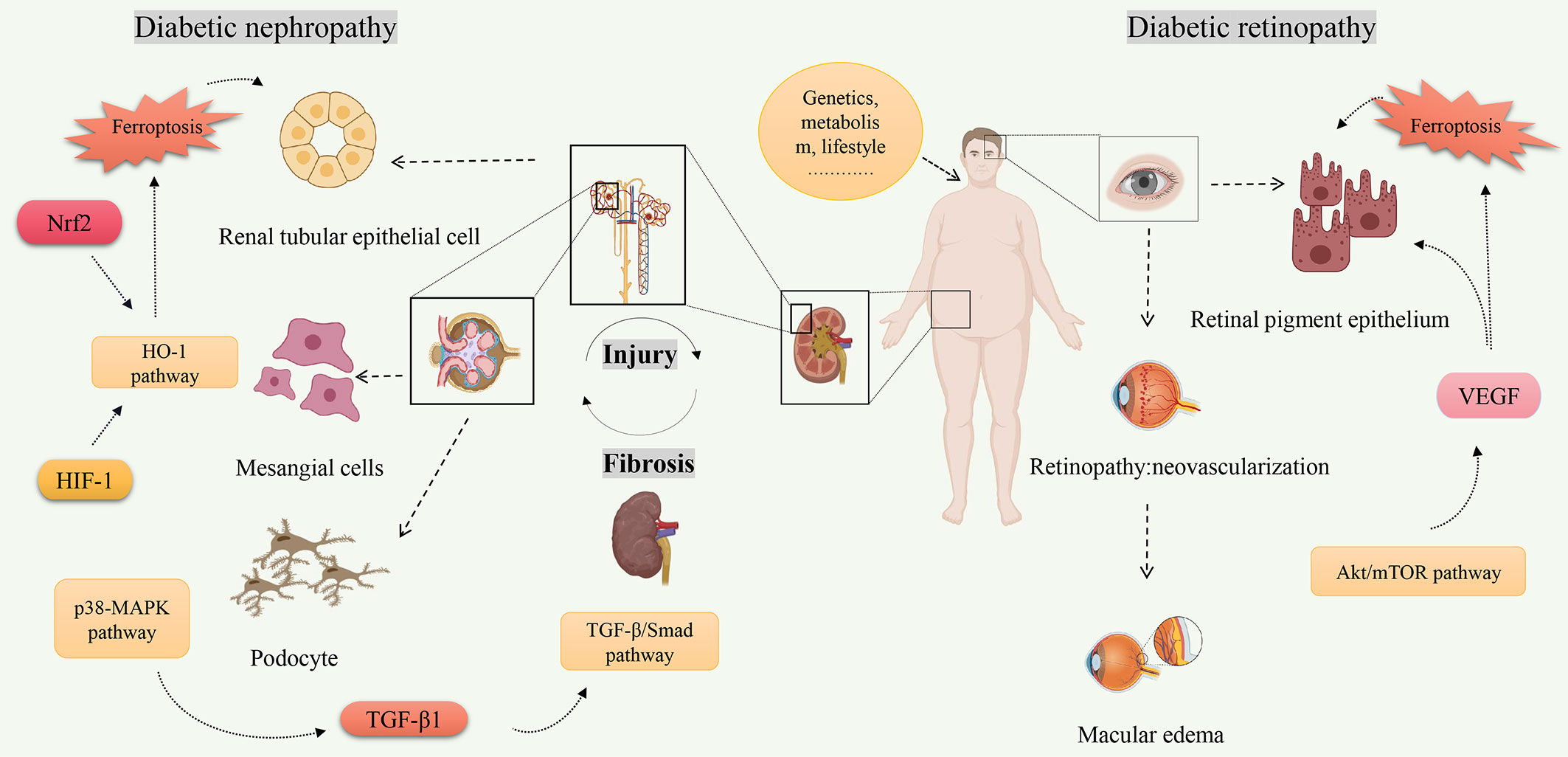
Figure 2 The related signaling pathways on ferroptosis of diabetic nephropathy and diabetic retinopathy. Created by Biorender.com. High-risk factors for diabetes (increasing age, overweight or obesity, etc.) are closely related to the occurrence of diabetes. Hyperglycemia is one of the main clinical manifestations of diabetes, and also one of the high-risk factors leading to diabetes complications. Molecules (Nrf2, HIF-1, TGF-β1, VEGF, etc.) regulate ferroptosis through multiple signaling pathways (TGF-β/Smad pathway, Akt/mTOR pathway, HO-1 pathway, etc.) and play a role in pathological progression of diabetic nephropathy and diabetic retinopathy.
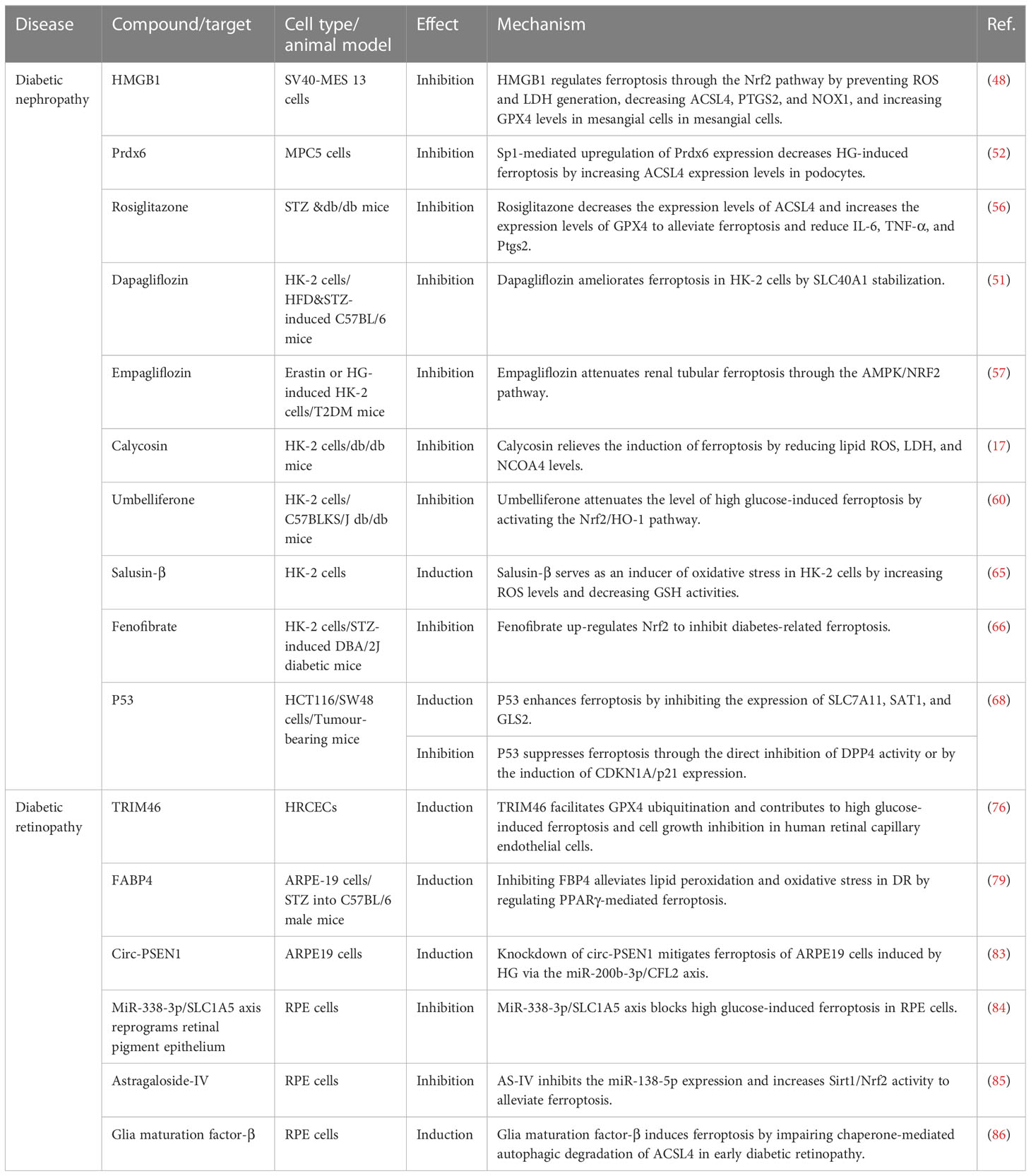
Table 2 Drugs or targets against ferroptosis in diabetic nephropathy and retinopathy.
Conceptualization, LL and ZZ; original draft preparation, LL, YD and DK; collected the literatures, YD, DW and TW; review and editing, XY and ZZ; supervision, YT, JL, and PC. All authors contributed to the article and approved the submitted version.
This work was supported by the Local Colleges and Universities Talent Development Funding from Heilongjiang Provincial Department of Finance (No. 2020GSP09), the Natural Science Foundation of Heilongjiang Province (No. LH2022H099), the Basic Scientific Research Project of University belongs to Heilongjiang (No. 2020-KYYWF-0771), the Traditional Chinese Medicine Scientific Research Fund Project of Heilongjiang Province (No. ZYW2022-090) and the Project of Young Innovative Talents Training Program of Regular Undergraduate Colleges and Universities in Heilongjiang Province (No. UNPYSCT-2020064).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Yang Y, Qiu W, Meng Q, Liu M, Lin W, Yang H, et al. GRB10 rs1800504 polymorphism is associated with the risk of coronary heart disease in patients with type 2 diabetes mellitus. Front Cardiovasc Med (2021) 8:728976. doi: 10.3389/fcvm.2021.728976
3. Jung J, Park WY, Kim YJ, Kim M, Choe M, Jin K, et al. 3-hydroxybutyrate ameliorates the progression of diabetic nephropathy. Antioxid (Basel) (2022) 11(2):381. doi: 10.3390/antiox11020381
4. Fu Z, Wang Z, Liu CH, Gong Y, Cakir B, Liegl R, et al. Fibroblast growth factor 21 protects photoreceptor function in type 1 diabetic mice. Diabetes (2018) 67(5):974–85. doi: 10.2337/db17-0830
5. Alam NM, Mills WC 4th, Wong AA, Douglas RM, Szeto HH, Prusky GT. A mitochondrial therapeutic reverses visual decline in mouse models of diabetes. Dis Model Mech (2015) 8(7):701–10. doi: 10.1242/dmm.020248
6. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
7. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol (2019) 12(1):34. doi: 10.1186/s13045-019-0720-y
8. Li Y, Sun M, Cao F, Chen Y, Zhang L, Li H, et al. The ferroptosis inhibitor liproxstatin-1 ameliorates LPS-induced cognitive impairment in mice. Nutrients (2022) 14(21):4599. doi: 10.3390/nu14214599
9. Park JE, Lee H, Kim SY, Lim Y. Lespedeza bicolor extract ameliorated renal inflammation by regulation of NLRP3 inflammasome-associated hyperinflammation in type 2 diabetic mice. Antioxid (Basel) (2020) 9(2):148. doi: 10.3390/antiox9020148
10. Deng Q, Zhu Y, Zhang M, Fei A, Liang J, Zheng J, et al. Ferroptosis as a potential new therapeutic target for diabetes and its complications. Endocr Connect (2023) 12(3):e220419. doi: 10.1530/EC-22-0419
11. Coughlan MT, Sharma K. Challenging the dogma of mitochondrial reactive oxygen species overproduction in diabetic kidney disease. Kidney Int (2016) 90(2):272–9. doi: 10.1016/j.kint.2016.02.043
12. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
13. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
14. Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, et al. Lipid peroxidation drives gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe (2018) 24(1):97–108.e4. doi: 10.1016/j.chom.2018.05.009
15. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature (2019) 572(7769):402–6. doi: 10.1038/s41586-019-1426-6
16. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
17. Yang S, Xie Z, Pei T, Zeng Y, Xiong Q, Wei H, et al. Salidroside attenuates neuronal ferroptosis by activating the Nrf2/HO1 signaling pathway in Aβ(1-42)-induced Alzheimer's disease mice and glutamate-injured HT22 cells. Chin Med (2022) 17(1):82. doi: 10.1016/j.cell.2017.09.021
18. Feng Q, Yu X, Qiao Y, Pan S, Wang R, Zheng B, et al. Ferroptosis and acute kidney injury (AKI): Molecular mechanisms and therapeutic potentials. Front Pharmacol (2022) 13:858676. doi: 10.3389/fphar.2022.858676
19. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol (2021) 22(4):266–82. doi: 10.1038/s41580-020-00324-8
20. Liu Y, Zhou L, Xu Y, Li K, Zhao Y, Qiao H, et al. Heat shock proteins and ferroptosis. Front Cell Dev Biol (2022) 10:864635. doi: 10.3389/fcell.2022.864635
21. Huang W, Chen K, Lu Y, Zhang D, Cheng Y, Li L, et al. ABCC5 facilitates the acquired resistance of sorafenib through the inhibition of SLC7A11-induced ferroptosis in hepatocellular carcinoma. Neoplasia (2021) 23(12):1227–39. doi: 10.1016/j.neo.2021.11.002
22. Duan C, Jiao D, Wang H, Wu Q, Men W, Yan H, et al. Activation of the PPARγ Prevents ferroptosis-induced neuronal loss in response to intracerebral hemorrhage through synergistic actions with the nrf2. Front Pharmacol (2022) 13:869300. doi: 10.3389/fphar.2022.869300
23. Hong T, Lei G, Chen X, Li H, Zhang X, Wu N, et al. PARP inhibition promotes ferroptosis via repressing SLC7A11 and synergizes with ferroptosis inducers in BRCA-proficient ovarian cancer. Redox Biol (2021) 42:101928. doi: 10.1016/j.redox.2021.101928
24. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
25. Xl L, Gy Z, Guo R, Cui N. Ferroptosis in sepsis: The mechanism, the role and the therapeutic potential. Front Immunol (2022) 13:956361. doi: 10.3389/fimmu.2022.956361
26. Kinowaki Y, Taguchi T, Onishi I, Kirimura S, Kitagawa M, Yamamoto K. Overview of ferroptosis and synthetic lethality strategies. Int J Mol Sci (2021) 22(17):9271. doi: 10.3390/ijms22179271
27. Xiao C, Wang Y, Zheng M, Chen J, Song G, Zhou Z, et al. RBBP6 increases radioresistance and serves as a therapeutic target for preoperative radiotherapy in colorectal cancer. Cancer Sci (2018) 109(4):1075–87. doi: 10.1111/cas.13516
28. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology (2017) 66(2):449–65. doi: 10.1002/hep.29117
29. Ou M, Jiang Y, Ji Y, Zhou Q, Du Z, Zhu H, et al. Role and mechanism of ferroptosis in neurological diseases. Mol Metab (2022) 61:101502. doi: 10.1016/j.molmet.2022.101502
30. Pandrangi SL, Chittineedi P, Chalumuri SS, Meena AS, Neira Mosquera JA, Sánchez SN, et al. Role of intracellular iron in switching apoptosis to ferroptosis to target therapy-resistant cancer stem cells. Molecules (2022) 27(9):3011. doi: 10.3390/molecules27093011
31. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol (2023) 20(1):7–23. doi: 10.1038/s41569-022-00735-4
32. Tonnus W, Belavgeni A, Beuschlein F, Eisenhofer G, Fassnacht M, Kroiss M, et al. The role of regulated necrosis in endocrine diseases. Nat Rev Endocrinol (2021) 17(8):497–510. doi: 10.1038/s41574-021-00499-w
33. Mao C, Lei G, Zhuang L, Gan B. Phospholipase iPLA2β acts as a guardian against ferroptosis. Cancer Commun (Lond) (2021) 41(11):1082–5. doi: 10.1002/cac2.12231
34. Sreekumar PG, Kannan R. Mechanisms of protection of retinal pigment epithelial cells from oxidant injury by humanin and other mitochondrial-derived peptides: Implications for age-related macular degeneration. Redox Biol (2020) 37:101663. doi: 10.1016/j.redox.2020.101663
35. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature (2021) 593(7860):586–90. doi: 10.1038/s41586-021-03539-7
36. Barrow AD, Edeling MA, Trifonov V, Luo J, Goyal P, Bohl B, et al. Natural killer cells control tumor growth by sensing a growth factor. Cell (2018) 172(3):534–548.e19. doi: 10.1016/j.cell.2017.11.037
37. Wu X, Li Y, Zhang S, Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics (2021) 11(7):3052–9. doi: 10.7150/thno.54113
38. Mu T, Hu H, Ma Y, Feng X, Zhang J, Gu Y. Regulation of key genes for milk fat synthesis in ruminants. Front Nutr (2021) 8:765147. doi: 10.3389/fnut.2021.765147
39. Wang YM, Gong FC, Qi X, Zheng YJ, Zheng XT, Chen Y, et al. Mucin 1 inhibits ferroptosis and sensitizes vitamin E to alleviate sepsis-induced acute lung injury through GSK3β/keap1-nrf2-GPX4 pathway. Oxid Med Cell Longev 2022 (2022) p:2405943. doi: 10.1155/2022/2405943
40. Tian R, Abarientos A, Hong J, Hashemi SH, Yan R, Dräger N, et al. Genome-wide CRISPRi/a screens in human neurons link lysosomal failure to ferroptosis. Nat Neurosci (2021) 24(7):1020–34. doi: 10.1038/s41593-021-00862-0
41. Fu H, Liu S, Bastacky SI, Wang X, Tian XJ, Zhou D. Diabetic kidney diseases revisited: A new perspective for a new era. Mol Metab (2019) 30:250–63. doi: 10.1016/j.molmet.2019.10.005
42. Bommer C, Heesemann E, Sagalova V, Manne-Goehler J, Atun R, Bärnighausen T, et al. The global economic burden of diabetes in adults aged 20-79 years: a cost-of-illness study. Lancet Diabetes Endocrinol (2017) 5(6):423–30. doi: 10.1016/S2213-8587(17)30097-9
43. Saleh H, Salama M, Hussein RM. Polyethylene glycol capped gold nanoparticles ameliorate renal ischemia-reperfusion injury in diabetic mice through AMPK-Nrf2 signaling pathway. Environ Sci Pollut Res Int (2022) 29(51):77884–907. doi: 10.1007/s11356-022-21235-5
44. Wang X, Jiang L, Liu XQ, Huang YB, Zhu W, Zeng HX, et al. Identification of genes reveals the mechanism of cell ferroptosis in diabetic nephropathy. Front Physiol (2022) 13:890566. doi: 10.3389/fphys.2022.890566
45. Yang YY, Deng RR, Chen Z, Yao LY, Yang XD, Xiang DX. Piperazine ferulate attenuates high glucose−induced mesangial cell injury via the regulation of p66(Shc). Mol Med Rep (2021) 23(5):374. doi: 10.3892/mmr.2021.12013
46. Hu Y, Shi R, Mo R, Hu F. Nomogram for the prediction of diabetic nephropathy risk among patients with type 2 diabetes mellitus based on a questionnaire and biochemical indicators: a retrospective study. Aging (Albany NY) (2020) 12(11):10317–36. doi: 10.18632/aging.103259
47. Sohn E, Kim J, Kim CS, Lee YM, Kim JS. Extract of polygonum cuspidatum attenuates diabetic retinopathy by inhibiting the high-mobility group box-1 (HMGB1) signaling pathway in streptozotocin-induced diabetic rats. Nutrients (2016) 8(3):140. doi: 10.3390/nu8030140
48. Wu Y, Zhao Y, Yang HZ, Wang YJ, Chen Y. HMGB1 regulates ferroptosis through Nrf2 pathway in mesangial cells in response to high glucose. Biosci Rep (2021) 41(2):BSR20202924. doi: 10.1042/BSR20202924
49. Daehn IS, Duffield JS. The glomerular filtration barrier: a structural target for novel kidney therapies. Nat Rev Drug Discovery (2021) 20(10):770–88. doi: 10.1038/s41573-021-00242-0
50. Tian X, Bunda P, Ishibe S. Podocyte endocytosis in regulating the glomerular filtration barrier. Front Med (Lausanne) (2022) 9:801837. doi: 10.3389/fmed.2022.801837
51. Huang B, Wen W, Ye S. Dapagliflozin ameliorates renal tubular ferroptosis in diabetes via SLC40A1 stabilization. Oxid Med Cell Longev (2022) 2022:9735555. doi: 10.1155/2022/9735555
52. Zhang Q, Hu Y, Hu JE, Ding Y, Shen Y, Xu H, et al. Sp1-mediated upregulation of Prdx6 expression prevents podocyte injury in diabetic nephropathy via mitigation of oxidative stress and ferroptosis. Life Sci (2021) 278:119529. doi: 10.1016/j.lfs.2021.119529
53. Chen J, Ou Z, Gao T, Yang Y, Shu A, Xu H, et al. Ginkgolide B alleviates oxidative stress and ferroptosis by inhibiting GPX4 ubiquitination to improve diabetic nephropathy. BioMed Pharmacother (2022) 156:113953. doi: 10.1016/j.biopha.2022.113953
54. Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell (2022) 185(14):2401–21. doi: 10.1016/j.cell.2022.06.003
55. Wu XQ, Zhang DD, Wang YN, Tan YQ, Yu XY, Zhao YY. AGE/RAGE in diabetic kidney disease and ageing kidney. Free Radic Biol Med (2021) 171:260–71. doi: 10.1016/j.freeradbiomed.2021.05.025
56. Wang Y, Bi R, Quan F, Cao Q, Lin Y, Yue C, et al. Ferroptosis involves in renal tubular cell death in diabetic nephropathy. Eur J Pharmacol (2020) 888:173574. doi: 10.1016/j.ejphar.2020.173574
57. Lu Q, Yang L, Xiao JJ, Liu Q, Ni L, Hu JW, et al. Empagliflozin attenuates the renal tubular ferroptosis in diabetic kidney disease through AMPK/NRF2 pathway. Free Radic Biol Med (2023) 195:89–102. doi: 10.1016/j.freeradbiomed.2022.12.088
58. Tan H, Chen J, Li Y, Li Y, Zhong Y, Li G, et al. Glabridin, a bioactive component of licorice, ameliorates diabetic nephropathy by regulating ferroptosis and the VEGF/Akt/ERK pathways. Mol Med (2022) 28(1):58. doi: 10.1186/s10020-022-00481-w
59. Huang D, Shen P, Wang C, Gao J, Ye C, Wu F. Calycosin plays a protective role in diabetic kidney disease through the regulation of ferroptosis. Pharm Biol (2022) 60(1):990–6. doi: 10.1080/13880209.2022.2067572
60. Jin T, Chen C. Umbelliferone delays the progression of diabetic nephropathy by inhibiting ferroptosis through activation of the Nrf-2/HO-1 pathway. Food Chem Toxicol (2022) 163:112892. doi: 10.1016/j.fct.2022.112892
61. Jiang N, Zhao H, Han Y, Li L, Xiong S, Zeng L, et al. HIF-1α ameliorates tubular injury in diabetic nephropathy via HO-1-mediated control of mitochondrial dynamics. Cell Prolif (2020) 53(11):e12909. doi: 10.1111/cpr.12909
62. Feng X, Wang S, Sun Z, Dong H, Yu H, Huang M, et al. Ferroptosis Enhanced Diabetic Renal Tubular Injury via HIF-1α/HO-1 Pathway in db/db Mice. Front Endocrinol (Lausanne) (2021) 12:626390. doi: 10.3389/fendo.2021.626390
63. Yu XY, Sun Q, Zhang YM, Zou L, Zhao YY. TGF-β/smad signaling pathway in tubulointerstitial fibrosis. Front Pharmacol (2022) 13:860588. doi: 10.3389/fphar.2022.860588
64. Zhao L, Zou Y, Liu F. Transforming growth factor-beta1 in diabetic kidney disease. Front Cell Dev Biol (2020) 8:187. doi: 10.3389/fcell.2020.00187
65. Wang WJ, Jiang X, Gao CC, Chen ZW. Salusin−β participates in high glucose−induced HK−2 cell ferroptosis in a Nrf−2−dependent manner. Mol Med Rep (2021) 24(3):674. doi: 10.3892/mmr.2021.12313
66. Li S, Zheng L, Zhang J, Liu X, Wu Z. Inhibition of ferroptosis by up-regulating Nrf2 delayed the progression of diabetic nephropathy. Free Radic Biol Med (2021) 162:435–49. doi: 10.1016/j.freeradbiomed.2020.10.323
67. Zhang B, Zhang X, Zhang C, Shen Q, Sun G, Sun X. Notoginsenoside R1 Protects db/db Mice against Diabetic Nephropathy via Upregulation of Nrf2-Mediated HO-1 Expression. Molecules (2019) 24(2):247. doi: 10.3390/molecules24020247
68. Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med (2019) 133:162–8. doi: 10.1016/j.freeradbiomed.2018.05.074
69. Zhao B, Li H, Liu J, Han P, Zhang C, Bai H, et al. MicroRNA-23b targets ras GTPase-activating protein SH3 domain-binding protein 2 to alleviate fibrosis and albuminuria in diabetic nephropathy. J Am Soc Nephrol (2016) 27(9):2597–608. doi: 10.1681/ASN.2015030300
70. Hu Y, Liu S, Liu W, Zhang Z, Liu Y, Sun D, et al. Bioinformatics analysis of genes related to iron death in diabetic nephropathy through network and pathway levels based approaches. PloS One (2021) 16(11):e0259436. doi: 10.1371/journal.pone.0259436
71. Zhou LT, Zhang ZJ, Cao JY, Chen H, Zhu YS, Wu X, et al. The unique molecular mechanism of diabetic nephropathy: a bioinformatics analysis of over 250 microarray datasets. Clin Kidney J (2021) 14(6):1626–38. doi: 10.1093/ckj/sfaa190
72. Liu D, Zhou W, Mao L, Cui Z, Jin S. Identification of ferroptosis-related genes and pathways in diabetic kidney disease using bioinformatics analysis. Sci Rep (2022) 12(1):22613. doi: 10.1038/s41598-022-26495-2
73. Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet (2010) 376(9735):124–36. doi: 10.1016/S0140-6736(09)62124-3
74. Antonetti DA, Silva PS, Stitt AW. Current understanding of the molecular and cellular pathology of diabetic retinopathy. Nat Rev Endocrinol (2021) 17(4):195–206. doi: 10.1038/s41574-020-00451-4
75. Chen YJ, Chen JT, Tai MC, Liang CM, Chen YY, Chen WL. Serum iron and risk of diabetic retinopathy. Nutrients (2020) 12(8):2297. doi: 10.3390/nu12082297
76. Zhang J, Qiu Q, Wang H, Chen C, Luo D. TRIM46 contributes to high glucose-induced ferroptosis and cell growth inhibition in human retinal capillary endothelial cells by facilitating GPX4 ubiquitination. Exp Cell Res (2021) 407(2):112800. doi: 10.1016/j.yexcr.2021.112800
77. Wang N, Zhang C, Xu Y, Tan HY, Chen H, Feng Y. Berberine improves insulin-induced diabetic retinopathy through exclusively suppressing Akt/mTOR-mediated HIF-1α/VEGF activation in retina endothelial cells. Int J Biol Sci (2021) 17(15):4316–26. doi: 10.7150/ijbs.62868
78. Shen H, Gong Q, Zhang J, Wang H, Qiu Q, Zhang J, et al. TRIM46 aggravated high glucose-induced hyper permeability and inflammatory response in human retinal capillary endothelial cells by promoting IκBα ubiquitination. Eye Vis (Lond) (2022) 9(1):35. doi: 10.1186/s40662-022-00305-2
79. Fan X, Xu M, Ren Q, Fan Y, Liu B, Chen J, et al. Downregulation of fatty acid binding protein 4 alleviates lipid peroxidation and oxidative stress in diabetic retinopathy by regulating peroxisome proliferator-activated receptor γ-mediated ferroptosis. Bioengineered (2022) 13(4):10540–51. doi: 10.1080/21655979.2022.2062533
80. McArthur K, Feng B, Wu Y, Chen S, Chakrabarti S. MicroRNA-200b regulates vascular endothelial growth factor-mediated alterations in diabetic retinopathy. Diabetes (2011) 60(4):1314–23. doi: 10.2337/db10-1557
81. Cao Y, Feng B, Chen S, Chu Y, Chakrabarti S. Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Invest Ophthalmol Vis Sci (2014) 55(11):7321–31. doi: 10.1167/iovs.14-15167
82. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes (2016) 65(3):768–79. doi: 10.2337/db15-1033
83. Zhu Z, Duan P, Song H, Zhou R, Chen T. Downregulation of Circular RNA PSEN1 ameliorates ferroptosis of the high glucose treated retinal pigment epithelial cells via miR-200b-3p/cofilin-2 axis. Bioengineered (2021) 12(2):12555–67. doi: 10.1080/21655979.2021.2010369
84. Zhou J, Sun C, Dong X, Wang H. A novel miR-338-3p/SLC1A5 axis reprograms retinal pigment epithelium to increases its resistance to high glucose-induced cell ferroptosis. J Mol Histol (2022) 53(3):561–71. doi: 10.1007/s10735-022-10070-0
85. Tang X, Li X, Zhang D, Han W. Astragaloside-IV alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of miR-138-5p/Sirt1/Nrf2. Bioengineered (2022) 13(4):8240–54. doi: 10.1080/21655979.2022.2049471
86. Liu C, Sun W, Zhu T, Shi S, Zhang J, Wang J, et al. Glia maturation factor-β induces ferroptosis by impairing chaperone-mediated autophagic degradation of ACSL4 in early diabetic retinopathy. Redox Biol (2022) 52:102292. doi: 10.1016/j.redox.2022.102292
87. Elshaer SL, Lemtalsi T, El-Remessy AB. High glucose-mediated tyrosine nitration of PI3-kinase: A molecular switch of survival and apoptosis in endothelial cells. Antioxid (Basel) (2018) 7(4):47. doi: 10.3390/antiox7040047
88. Sekhar RV, McKay SV, Patel SG, Guthikonda AP, Reddy VT, Balasubramanyam A, Jahoor F. Glutathione synthesis is diminished in patients with uncontrolled diabetes and restored by dietary supplementation with cysteine and glycine. Diabetes Care (2011) 34(1):162–7. doi: 10.2337/dc10-1006
89. Chen Z, Miao F, Braffett BH, Lachin JM, Zhang L, Wu X, et al. DNA methylation mediates development of HbA1c-associated complications in type 1 diabetes. Nat Metab (2020) 2(8):744–62. doi: 10.1038/s42255-020-0231-8
90. Yun KJ, Kim HJ, Kim MK, Kwon HS, Baek KH, Roh YJ, et al. Risk factors for the development and progression of diabetic kidney disease in patients with type 2 diabetes mellitus and advanced diabetic retinopathy. Diabetes Metab J (2016) 40(6):473–81. doi: 10.4093/dmj.2016.40.6.473
91. Huang C, Xue LF, Hu B, Liu HH, Huang SB, Khan S, et al. Calycosin-loaded nanoliposomes as potential nanoplatforms for treatment of diabetic nephropathy through regulation of mitochondrial respiratory function. J Nanobiotechnol (2021) 19(1):178. doi: 10.1186/s12951-021-00917-1
92. Rodriguez-Carrizalez AD, Castellanos-González JA, Martínez-Romero EC, Miller Arrevillaga G, Román-Pintos LM, Pacheco-Moisés FP, et al. The antioxidant effect of ubiquinone and combined therapy on mitochondrial function in blood cells in non-proliferative diabetic retinopathy: A randomized, double-blind, phase IIa, placebo-controlled study. Redox Rep (2016) 21(4):190–5. doi: 10.1179/1351000215Y.0000000032
93. Yu F, Zhang Q, Liu H, Liu J, Yang S, Luo X, et al. Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov (2022) 8(1):40. doi: 10.1038/s41421-022-00375-5
Keywords: diabetic nephropathy, diabetic retinopathy, ferroptosis, regulatory cell death, oxidative stress
Citation: Li L, Dai Y, Ke D, Liu J, Chen P, Wei D, Wang T, Teng Y, Yuan X and Zhang Z (2023) Ferroptosis: new insight into the mechanisms of diabetic nephropathy and retinopathy. Front. Endocrinol. 14:1215292. doi: 10.3389/fendo.2023.1215292
Received: 01 May 2023; Accepted: 19 July 2023;
Published: 03 August 2023.
Edited by:
Xiaoyong Yu, Shaanxi Provincial Hospital of Traditional Chinese Medicine, ChinaReviewed by:
Hangxing Yu, Chongqing Hospital of Traditional Chinese Medicine, ChinaCopyright © 2023 Li, Dai, Ke, Liu, Chen, Wei, Wang, Teng, Yuan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhen Zhang, emhhbmd6aGVuQG1kam11LmVkdS5jbg==; Xiaohuan Yuan, eXVhbnhpYW9odWFuQG1kam11LmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.