 J. Wesley Pike
J. Wesley Pike Seong Min Lee
Seong Min Lee Mark B. Meyer
Mark B. Meyer- 1Department of Biochemistry, University of Wisconsin-Madison, Madison, WI, United States
- 2Department of Nutritional Sciences, University of Wisconsin-Madison, Madison, WI, United States
The regulation of mineral homeostasis involves the three mineralotropic hormones PTH, FGF23 and 1,25-dihydroxyvitamin D3 (1,25(OH)2D3). Early research efforts focused on PTH and 1,25(OH)2D3 and more recently on FGF23 have revealed that each of these hormones regulates the expression of the other two. Despite early suggestions of transcriptional processes, it has been only recently that research effort have begun to delineate the genomic mechanisms underpinning this regulation for 1,25(OH)2D3 and FGF23; the regulation of PTH by 1,25(OH)2D3, however, remains obscure. We review here our molecular understanding of how PTH induces Cyp27b1 expression, the gene encoding the enzyme responsible for the synthesis of 1,25(OH)2D3. FGF23 and 1,25(OH)2D3, on the other hand, function by suppressing production of 1,25(OH)2D3. PTH stimulates the PKA-induced recruitment of CREB and its coactivator CBP at CREB occupied sites within the kidney-specific regulatory regions of Cyp27b1. PKA activation also promotes the nuclear translocation of SIK bound coactivators such as CRTC2, where it similarly interacts with CREB occupied Cyp27b1 sites. The negative actions of both FGF23 and 1,25(OH)2D3 appear to suppress Cyp27b1 expression by opposing the recruitment of CREB coactivators at this gene. Reciprocal gene actions are seen at Cyp24a1, the gene encoding the enzyme that degrades 1,25(OH)2D3, thereby contributing to the overall regulation of blood levels of 1,25(OH)2D3. Relative to PTH regulation, we summarize what is known of how 1,25(OH)2D3 regulates PTH suppression. These studies suggest that it is not 1,25(OH)2D3 that controls PTH levels in healthy subjects, but rather calcium itself. Finally, we describe current progress using an in vivo approach that furthers our understanding of the regulation of Fgf23 expression by PTH and 1,25(OH)2D3 and provide the first evidence that P may act to induce Fgf23 expression via a complex transcriptional mechanism in bone. It is clear, however, that additional advances will need to be made to further our understanding of the inter-regulation of each of these hormonal genes.
Introduction
The maintenance of mineral metabolism is regulated predominantly by 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), PTH and FGF23 (1, 2). While the effective concentrations of each of these hormones in the circulation is regulated through multiple mechanisms, all three are fundamentally derived in a tissue-specific manner via transcription from their respective genes. Interestingly, while early physiological observations led to the realization that each of these hormones also contributed significantly to the abundance of the other two, it has been only recently that research efforts have begun to identify the underlying molecular mechanisms responsible for altering the expression of these hormonal genes (2). The consequences of this inter-regulation, although frequently impacted by downstream non-transcriptional events as well, is the integration of the activities of these three different hormones with sometimes overlapping and often inhibitory actions at certain mineral regulating tissues including kidney and bone (Figure 1). There are, of course, frequent novel actions of each of these hormones at numerous tissues that are not involved in the active regulation of mineral homeostasis that further complicate the picture. In this paper, we consider advances that we have made at the genomic level that underlie the production of 1,25(OH)2D3 by PTH, FGF23 and 1,25(OH)2D3 through their actions on the Cyp27b1 and Cyp24a1 genes expressed in renal proximal tubules. We also review existing support for the mechanisms through which 1,25(OH)2D3 has been proposed in early studies to regulate PTH transcription, although advances here are limited. Finally, we discuss the results of our recent studies of the regulation of Fgf23 transcription using an approach involving gene modification via CRISPR-Cas9 gene editing followed by loss of Fgf23 regulation studies in vivo. These efforts attempt to identify the genomic control regions within the Fgf23 gene locus that mediate the regulatory actions of 1,25(OH)2D3, PTH and phosphate (P) at this gene and ultimately to identify unique factors involved in these transcriptional mechanisms. While our focus is on hormonal inter-regulation, it is worth noting that calcium (Ca) and P do not appear to directly control the expression of the renal Cyp27b1 and Cyp24a1 genes, but rather that Ca directly regulates PTH expression whereas P controls FGF23 levels; these in turn then control the production of 1,25(OH)2D3. For this reason, we also include a brief discussion of Ca and a direct consideration of P, with a particular emphasis on P regulation of Fgf23 expression. It will also be evident that it has been the application of several new technologies that helped advance our understanding of this inter-regulation, particularly that regarding regulation of PTH and FGF23. This paper does not represent a thorough review of these subjects, but rather a summary of the recent work that we have done aimed at understanding the molecular mechanisms of this regulation.
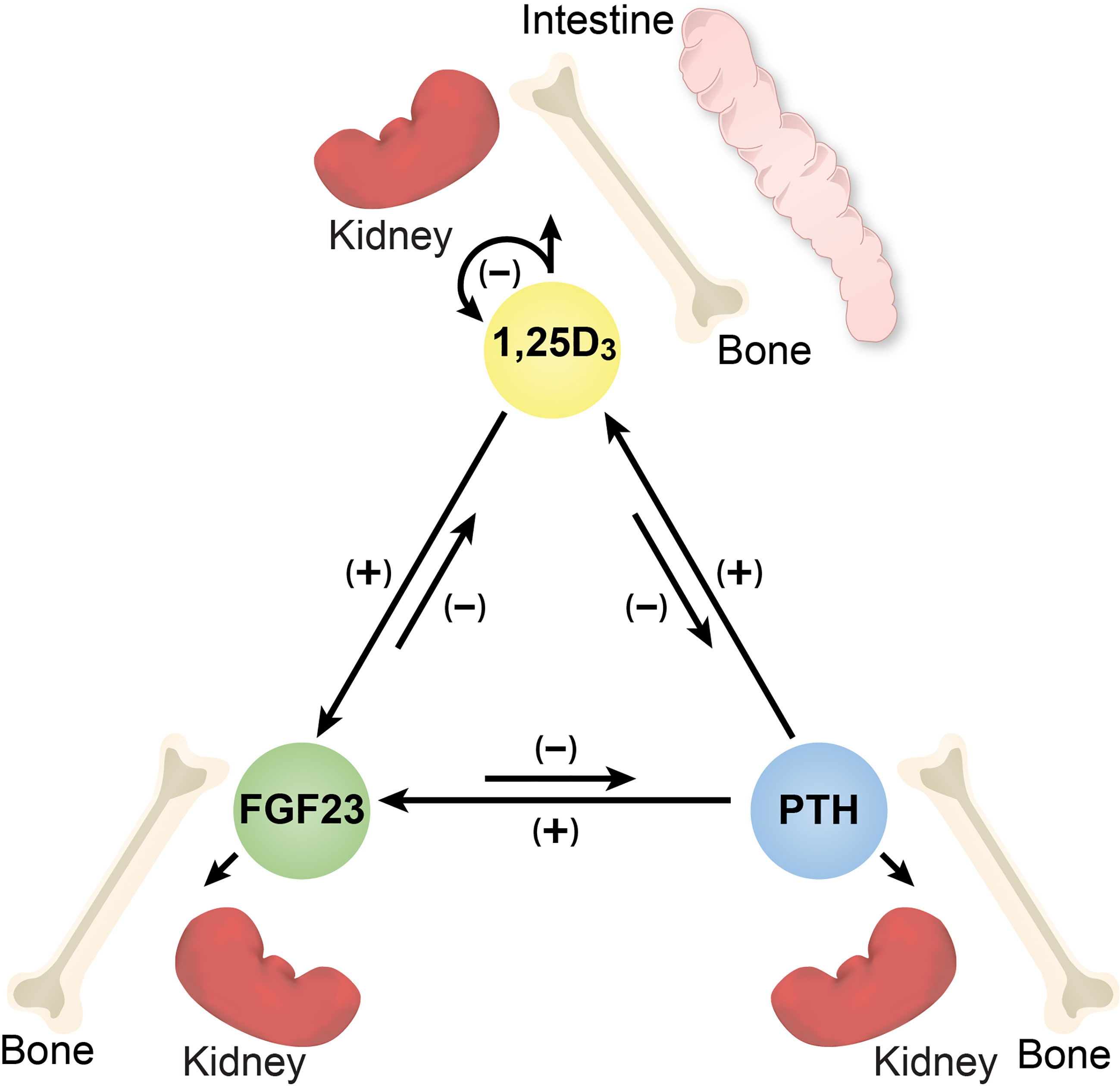
Figure 1 Diagram of the inter-regulation of PTH, FGF23 and 1,25(OH)2D3 and the target tissues relevant to each hormone. Arrows together with (+) or (-) signs indicate the direction and influence of each hormone.
New insights into transcriptional activation of genes
Before we address the regulation of renal 1,25(OH)2D3 which represents the primary endocrine source of this hormone, followed by parathyroid gland-produced PTH and bone-derived FGF23, we begin this paper by discussing the new approaches that we have taken to identify the mechanisms involved in gene regulation. Indeed, the development of chromatin-immunoprecipitation (ChIP) and other genomic procedures coupled to next-generation DNA sequencing technics (ChIP-seq analysis) and their applications have fully changed the way in which gene regulation is explored (3, 4). ChIP-seq analysis has seen widespread application in part to identify and characterize ligand-induced transcription factor DNA binding activity, to examine the recruitment of coregulators to these DNA bound factors at specific genomic sites, and to assess actions downstream of this binding, such as the recruitment of additional protein participants in the transcriptional pathways such as RNA polymerase II in an unbiased, genome-wide fashion. These studies have revealed a multitude of new details inherent to transcriptional processes at single genes and on a genome-wide scale; some of these principles confirm what has been previously determined while others identify modifications to those already known to exist. Others have advanced our understanding, revealing entirely new principles of gene regulation that must be now considered in mechanistic studies. Perhaps as important, they have also revealed how inherent technical bias drawn from over three decades of analyses using in vitro molecular biological methods involving cell-based transfection studies have led to numerous faulty conclusions regarding transcriptional mechanisms.
Regarding the molecular actions of vitamin D, genome-wide studies using ChIP-seq analysis, as summarized in Table 1, have revealed many findings that are relevant not only to vitamin D actions but to the general actions of transcription factors that mediate the activities of numerous hormones and other regulatory components. As can be seen, these analyses in both human and mouse cultured cells as well as tissues in vivo indicate several thousand binding sites for the vitamin D receptor (VDR), a number consistent with other regulatory factors; the number and location of these sites for transcription factors in general are, however, dependent upon the factor involved, and are affected by species, the type of target cell studied, and nutritional status, among other variables (5). In the case of the VDR, most of these sites, but not all, are highly dependent upon 1,25(OH)2D3 pretreatment, indicating that like many nuclear receptors (NRs), the VDR represents a ligand-dependent DNA binding factor (6). The regulation of DNA binding activity of other factors is highly diverse but is a hallmark of gene regulation. VDR DNA binding is facilitated by the retinoid X receptor (RXR) which acts as a heterodimeric partner (7, 8). De novo sequence analysis across many VDR sites confirms the presence of at least one and often multiple regulatory DNA sequence motifs, each comprised of a typical vitamin D response element (VDRE) of two hexameric DNA half-sites separated by three base pairs. This DNA structural organization represents a typical VDRE now observed at hundreds of genes and in all species examined. Interestingly, however, VDR binding at sites known to function in repression are more diverse, and often contain novel DNA motifs, suggesting that the VDR may be bound to these sites indirectly via an association with other DNA or non-DNA binding proteins serving additional regulatory functions. These observations at genome-wide levels both confirm and extend our understanding of several key features of vitamin D action via the VDR at target genes and are generally consistent with features of other transcription factors of this type. Perhaps the most important discovery arising from unbiased genome-wide analyses of not only VDR/RXR binding sites, but other transcription factor regulatory sites termed enhancers is the observation that these regulatory sequences are not located exclusively near target gene promoters as previously suspected. Rather, they can be found at sites that are commonly distributed within intergenic regions both upstream and downstream of the genes they regulate, often with unregulated genes interspersed, and frequently within the introns of the target gene itself or in unregulated genes located adjacent to the regulated gene or both (9–14). These sites can be located 10’s if not 100’s of kilobases distal to a gene’s promoter, the locations of which may have an impact on their function. Perhaps even more interestingly, this regulatory capacity is frequently comprised of multiple interacting components, often 2 to 10 or more of these functional regulatory enhancer regions. Finally, each of these individual components is frequently modular in nature, capable of binding multiple interacting factors, that can span a kilobase or less to more complex segments that can span many kilobases in size; super-enhancers and other large regulatory regions to which the VDR often binds appear to represent consolidated collections of multiple enhancers that interact with numerous regulatory proteins and extend to over 20 kilobases.

Table 1 Overarching principles of vitamin D action in target cells.
Since multiple enhancers may control a single gene and the localization of transcription factors at sites on the genome do not identify the gene or genes they regulate, enhancer locations near a gene cannot be used to identify an enhancer’s target gene. Accordingly, the genome-wide number of transcription factor binding sites in a cell, for example, cannot be used to determine the number or identity of genes that are regulated by a given regulatory component. It would also appear that although the activation of some induced genes is temporally consistent with the direct regulatory actions of a transcription factor, other examples occur where this activity is not direct, but rather occurs following activation of cellular components that bind in a secondary fashion to a particular target gene. In the case of vitamin D, we will describe the regulation of FGF23 by 1,25(OH)2D3 and its receptor which may represent such an example. Mechanistically, it is also worth noting that although linear arrangements of binding sites at a gene locus are often schematically displayed, the actual arrangement in vivo is almost certainly a 3D complex of all or subsets or these regulatory units via looping. In addition, active enhancer regions near target genes that are characterized by open epigenetic chromatin structures provide access for regulatory factors that control the transcriptional output of the gene itself. This complexity also provides one example of why earlier studies using cell lines transfected with biased gene promoter plasmid constructs has frequently resulted in erroneous conclusions. These features will be observed in our studies of the renal genes for Cyp27b1 and Cyp24a1 and the genes for parathyroid gland produced PTH and skeletal produced FGF23.
Current advances in transcriptional regulation in vivo
The technological advances as above have led to new methods to characterize the details of gene regulation both in vitro and particularly in vivo, the latter at molecular levels equivalent to those heretofore achievable only in cultured cells. Details now extend to the coupling of changing epigenetic architecture in response to genetic and epigenetic action at regulatory loci to the recruitment of additional transcription factors that play direct roles in promoting or inhibiting transcriptional output (3). These include detection of epigenetic readers that affect the distribution and release of RNA polymerase II across the transcription unit to those that influence the downstream mechanisms essential for transcription of the gene per se that results in the production of RNA transcripts. Of additional advantage is the fact that in vivo studies can frequently benefit from an assessment of the overarching functional roles of specific enhancers using CRISPR/Cas9 methods to induce mutations into the genome of both cells in culture and in animals in vivo. While many of the methods to be discussed herein were developed initially in cell lines, much of what we will be discussed below regarding the regulation of the genes involving 1,25(OH)2D3 production by the kidney and FGF23 production in bone were conducted in wildtype and mutant mice in vivo.
Control of renal CYP27B1 AND CYP24A1 expression by PTH - raising the levels of endocrine 1,25(OH)2D3
Induction of Cyp27b1 expression by PTH in the kidney
We focus first on the regulation of 1,25(OH)2D3 production by the kidney. This hormone plays a central role in the regulation of mineral metabolism by virtue of its unique function to induce Ca and to a lesser extent P absorption from the diet via the intestinal enterocyte (15). Although this hormone also acts in bone and kidney as well, 1,25(OH)2D3 action in the intestine is novel. The primary regulator of 1,25(OH)2D3 production in the kidney is PTH which induces the expression of the Cyp27b1 gene whose enzymatic product 25(OH)-1α-vitamin D3 hydroxylase (CYP27B1) is responsible for the synthesis of 1,25(OH)2D3 (16). As important, however, is the ability of PTH to reciprocally suppress the expression of Cyp24a1, whose product 1,25(OH)2-24-vitamin D3 hydroxylase (CYP24A1) is responsible for the initial degradation of 1,25(OH)2D3 (17). Accordingly, Cyp24a1 is suppressed by PTH thereby reducing the degradation rate of 1,25(OH)2D3 and raising the levels of 1,25(OH)2D3. That the regulation of Cyp24a1 is transcriptional in nature has only recently been defined in molecular terms, as will be discussed herein. Together, these two enzymes regulate the level of 1,25(OH)2D3, which is secreted into the blood where it functions as an endocrine hormone. Thus, the coordinated regulation of both Cyp27b1 and Cyp24a1 expression in the kidney by PTH is essential to both the production as well as the maintenance of appropriate levels of circulating hormonal 1,25(OH)2D3. Since the elaboration of PTH from the parathyroid glands is controlled by decreasing blood levels of Ca, an inverse relationship emerges between blood Ca and PTH, and thus control of circulating 1,25(OH)2D3 (18). As will be discussed in the next section, Cyp27b1 is suppressed by the reciprocal actions of FGF23 and 1,25(OH)2D3, while Cyp24a1 is coordinately induced by each of these hormones.
While studies several decades ago revealed the essential role for PTH in the control of renal production of 1,25(OH)2D3, it has been only recently that the mechanisms associated with the induction of Cyp27b1 and suppression of Cyp24a1 by this hormone have emerged. These findings were facilitated by the technics discussed above, including both ChIP-seq analysis and CRISPR-Cas9 mediated gene editing. To further the relevance of their application as well as our results, we applied these techniques to mice and conducted each of these analyses in vivo. As PTH is known to stimulate cAMP, we explored using ChIP-seq analysis the possibility that PTH might induce CREB at specific sites across the Cyp27b1 locus in tissue derived from the kidney cortex of normal wildtype mice (19). Following in vivo treatment with exogenous PTH, we used antibodies to phosphorylated S133 CREB (pCREB) and discovered that pCREB could be detected at several sites upstream of the Cyp27b1 gene, one located within an intron of the adjacent Mettl1 gene and three located further upstream in an intron of the Mettl21b (Eef1akmt3) gene (Figure 2). None were found near the Cyp27b1 gene promoter region nor immediately upstream of the promoter in an intergenic region that had been the focus of efforts by many others. While pCREB levels were not induced initially by PTH at these sites, later time course experiments revealed that the actions of PTH were more rapid than anticipated, resulting in a striking and transient upregulation of pCREB followed by a return to baseline as tested in the initial experiment (20). PTH also exerted a profound increase in histone acetylation (H3K9ac and H3K27ac) across each of these pCREB-containing sites even at earliest times, suggesting that PTH likely exerted an increase in DNA accessibility around these factor-containing sites. Each of these activities with PTH, however, preceded an increase in the production of PTH-induced Cyp27b1 RNA transcripts. Most importantly, deletion of the pCREB containing site in the Mettl1 intron (M1) resulted in a profound mouse phenotype insensitive to exogenous PTH and consistent with a previously prepared mouse containing a mutation in Cyp27b1 that prevented the global expression of the active CYP27B1 enzyme (19, 21). The latter resulted in a total absence of circulating 1,25(OH)2D3 in these mice. Additional phenotypic features in both this unique Cyp27b1 mutant mouse and the M1 deleted mouse included hypocalcemia, hypophosphatemia, and striking hypocalcemic bone disease. Deletion of the three collective sites in the Mettl21b intron (M21) in the genome of mice resulted in a much more modest phenotype comprised of only slightly reduced sensitivity to PTH, and normal levels of Ca, P, and 1,25(OH)2D3 (19). These and additional studies, together with the profound epigenetic observations induced by PTH, suggest that each of these pCREB sites mediate PTH action, but that the pCREB site in M1 is likely a dominant requirement for physiologically impaired PTH action (20). Importantly, a double mutant containing deletions of the pCREB site in M1 and the multiple sites in M21 resulted in a decreased basal level, a profound resistance to PTH, strikingly reduced circulating levels of 1,25(OH)2D3 and an extreme phenotype similar to that of the Cyp27b1 null mouse; it was also slightly more severe than that seen in the M1 enhancer deleted mouse (21). Perhaps as interesting, a genomic analysis using ENCODE-produced DNase hypersensitivity (DHS) experiment revealed that all sites containing PTH-regulated pCREB exhibited an open chromatin profile present only in normal mouse kidney and absent in all other tissues examined (19, 22). More recent studies in isolated cells using ATAC-seq analysis revealed that this Cyp27b1 enhancer profile could be seen only in renal proximal tubules and not in other renal cell types nor in a variety of other tissue cell types as well (2). Finally, that these enhancers were active was confirmed most recently through single cell RNA expression studies that revealed that Cyp27b1 expression was limited to the renal proximal tubule segments S1 as well as S2 and S3 (23). While Cyp27b1 was expressed at very low levels in many non-renal tissues (NRTCs), it was not responsive to PTH in any of these tissues (19). Thus, we conclude that the kidney module responsible for PTH mediated upregulation of Cyp27b1 is unique to the kidney and responsible for the selective upregulation of Cyp27b1 by PTH.
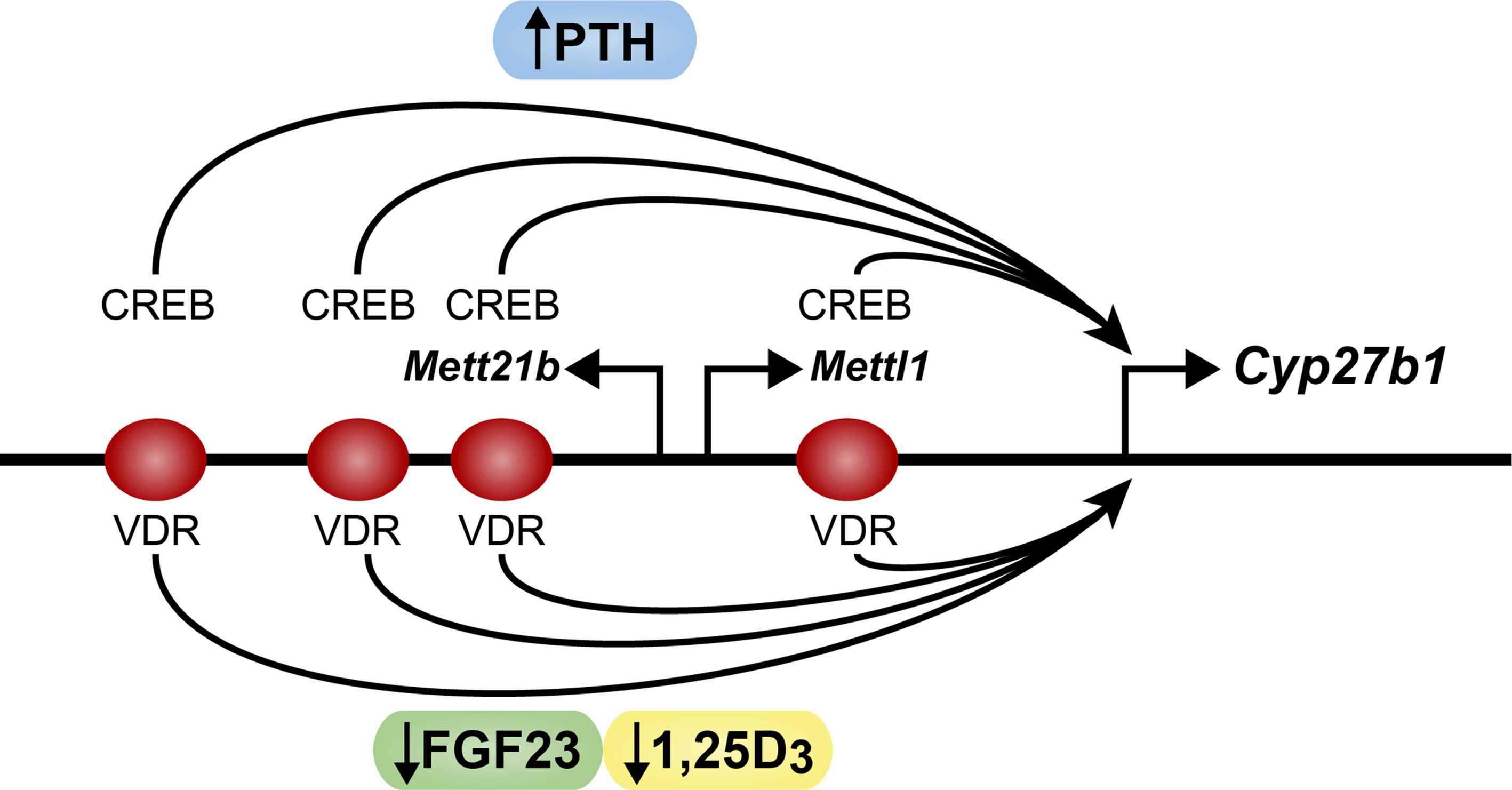
Figure 2 Schematic diagram of the mouse Cyp2b1 gene. The Cyp27b1, Mettl1 and Mettl21b genes are indicated. PTH (blue) sites of action at CREB are noted. FGF23 (green) and 1,25D3 (yellow) sites of action at the VDR are noted. Arrow indicates direction of transcription.
Our most recent studies using ChIP-seq analysis have revealed that while the levels of pCREB are not strongly induced by PTH, the mechanism through which the transcriptional activity of PKA induced pCREB is manifested is via the promoted recruitment of CBP, a CREB coactivator that exhibits histone H3K27 acetylation activity and is perhaps responsible for the ability of PTH to induce H3K27ac across the M1 and M21 sites (20, 23). Interestingly, additional chemical and genetic studies revealed that PTH via PKA also inhibits the salt-induced kinases (SIK) that result in the release of the cytoplasmic CREB regulated transcriptional coactivator (CRTC) class of CREB coactivators such as CRTC2 that also translocate to the nucleus where they bind to the CREB modules in M1 and M21 to facilitate Cyp27b1 activation. Thus, activation of Cyp27b1 in this case is mediated largely via the PTH mediated upregulation of two known coactivators of DNA-bound pCREB. The distinct role of each of these coactivators remains to be determined, however. Although widely expressed CBP retains multiple functions, one of which may be in the acetylation of histone H3K27; a second hypothetical function in the case of Cyp27b1 may be to participate in the 3D repositioning of the four distal CREB enhancer sites at or near the Cyp27b1 promoter (24). Perhaps as important in linking PTH to the upregulation of Cyp27b1 is our observation that treatment with PTH results in the recruitment of BRD4 at each of the M1 and M21 pCREB sites (20). This histone H3 lysine acetylase reader participates in the initial phases of the recruitment of RNA polymerase II to Cyp271b. Accordingly, we observed that PTH also induced a striking increase in RNA polymerase II and an increase in enrichment of the epigenetic histone methylation mark H3K36me3 across the Cyp27b1 gene body. The appearance of both factors reflects an increase in Cyp27b1 transcription and provides direct links to the PTH-induced Cyp27b1 RNA transcripts that are elevated in response to PTH treatment.
Suppression of Cyp24a1 expression in the kidney by PTH coordinately reduces the degradation of 1,25(OH)2D3
While PTH induces Cyp27b1 expression in the kidney, this hormone also imposes a reciprocal negative action on renal Cyp24a1 levels (19). This reduction in Cyp24a1 expression leads to reduced degradation of 1,25(OH)2D3 thereby facilitating the overall upregulation of renal 1,25(OH)2D3 that raises the hormones levels in the blood. While it has long been known that PTH suppresses Cyp24a1 production, the mechanism of this decrease has evaded discovery; indeed, studies during the past decade suggested a post-transcriptional mechanism involving Cyp24a1 transcript stabilization (25, 26). Interestingly, our recent ChIP-seq analyses of the renal Cyp24a1 gene revealed that PTH suppressed Cyp24a1 expression via a transcriptional mechanism, and that this downregulation was mediated via a large DNA segment that contains at least three widely spaced pCREB sites that are located immediately downstream of the Cyp24a1 gene body (Figure 3) (27). Like Cyp27b1, an open chromatin feature also characterized the DNA at each of these pCREB sites along this downstream segment, but only in the kidney proximal tubules (2). Importantly, however, deletion of this large DNA segment (DS1) containing these pCREB sites in the mouse using CRISPR/CAS9 gene editing revealed a loss of PTH mediated Cyp24a1 suppression, indicating that the pCREB features of this segment likely participated in renal Cyp24a1 downregulation. Mechanistically, however, recent ChIP-seq studies revealed only modest suppression of small amounts of residual pCREB together with an apparent blockade of CBP and CRTC2 binding at these pCREB sites as well as a site at the Cyp24a1 promoter in response to PTH (20). These limited changes in coregulator levels likely lead to the accompanying suppression of residual histone H3K9ac and H3K27ac and the absence of BRD4 observed at these sites, and therefore an inability of this gene to recruit RNA polymerase II. The corresponding absence of the transcriptional mark H3K36me3 indicated a lack of transcription. These features support the idea that PTH reduces Cyp24a1 expression in the kidney at the genetic level, consistent with our observed suppression of Cyp24a1 RNA transcripts, and does so by opposing the ability of the pCREB modules to interact with CREB coregulators essential for Cyp24a1 expression. Further studies will be necessary to identify the specific sites of action of PTH at one or more of these pCREB modules and to delineate the potential involvement of additional factors that may play a role in this suppression. Interestingly, as with pCREB components in the Cyp27b1 gene, the pCREB modules in the Cyp24a1 gene are uniquely absent in non-renal cells (NRTCs), supporting the concept that PTH is only capable of downregulating Cyp24a1 in the kidney (27). Thus, as with Cyp27b1, the Cyp24a1 gene retains regulatory features unique to the kidney. This reciprocal regulatory mechanism, together with the identified presence of both Cyp27b1 and Cyp24a1 in the same renal proximal tubules, support the coordinated involvement of both genes in the control of circulating 1,25(OH)2D3.
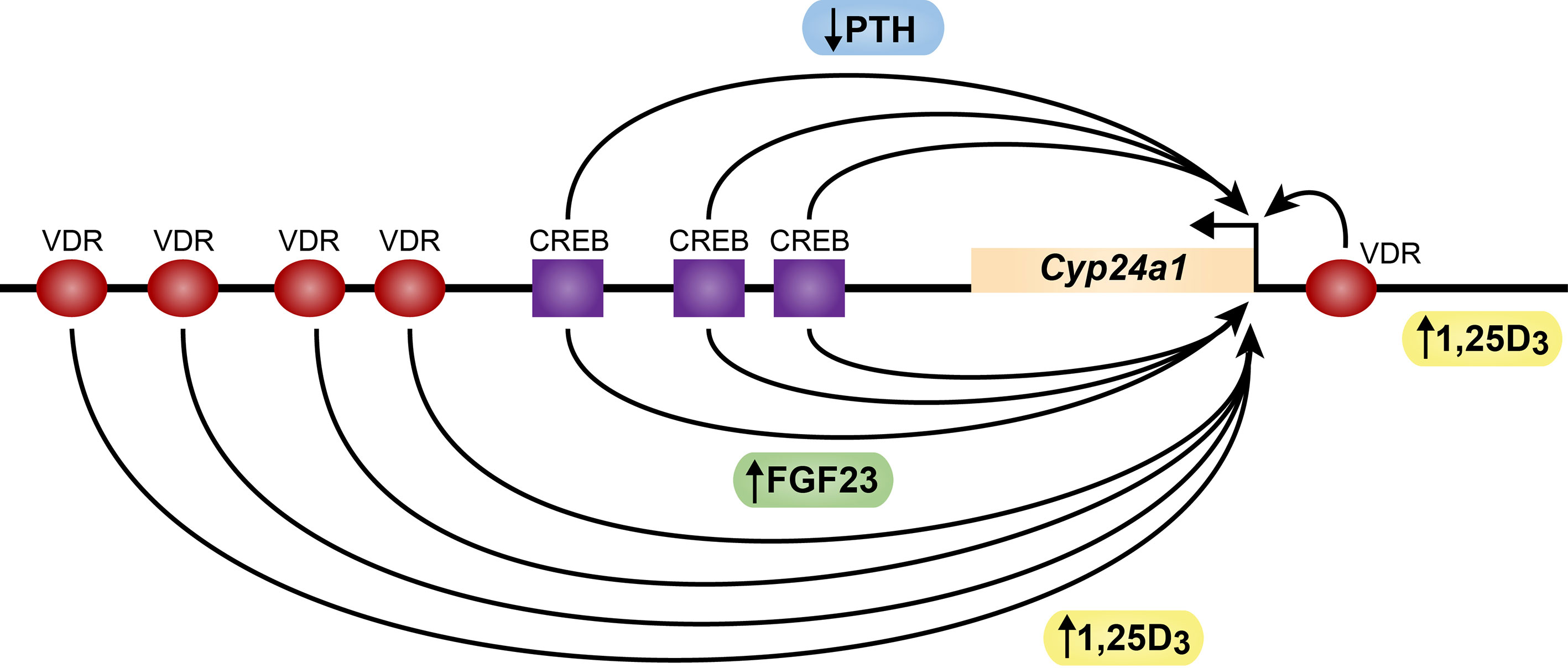
Figure 3 Schematic diagram of the mouse Cyp24a1 gene. The Cyp24a1 gene and downstream direction of transcription is noted. CREB sites (purple squares) and VDR sites (red ovals) are indicated. PTH (blue) sites of action negative action at CREB are noted. FGF23 (green) and 1,25D3 (yellow) sites of positive action at the VDR are noted.
Control of renal CYP27B1 AND CYP24A1 expression and regulation by FGF23 and 1,25(OH)2D3 - decreasing the levels of 1,25(OH)2D3
FGF23 and 1,25(OH)2D3 decrease the expression of Cyp27b1 and oppose PTH induction via pCREB sites at regulatory enhancers
Returning to our investigation of renal Cyp27b1 regulation, we also explored the role of FGF23 and 1,25(OH)2D3 itself in renal Cyp27b1 expression. Interestingly, while PTH speaks to the unique and direct upregulatory control of renal 1,25(OH)2D3 production in response to an increased need to elevate blood Ca levels, both FGF23 and 1,25(OH)2D3 itself are negative regulators that seek to downregulate the production of 1,25(OH)2D3 in response to unique physiologic/metabolic determinants (28–30). Despite this, while both suppress Cyp27b1 expression, they may do so for different reasons, and as will be seen, by different mechanisms. Importantly, this suppression of Cyp27b1is also potentiated by the ability of FGF23 and 1,25(OH)2D3 to reciprocally induce Cyp24a1, thus collectively reducing blood 1,25(OH)2D3, as discussed in the next section (31). It is also worth noting that there is likely a distinction in the nature of the two signals in that while FGF23 represent a distal feedback mechanism prompted by circulating levels of P, 1,25(OH)2D3 and perhaps other regulatory components in the blood that control FGF23 expression from bone, identify a distal bone-kidney feedback axis. 1,25(OH)2D3 likely exerts its effects exclusively in the kidney via a short negative feedback mechanism. Nevertheless, despite an early understanding of the actions of 1,25(OH)2D3 and more recently of FGF23 to reduce 1,25(OH)2D3 production in the kidney via a downregulation of Cyp27b1, a delineation of the mechanisms involved have generally resisted research efforts. Importantly, while both FGF23 and 1,25(OH)2D3 suppress Cyp27b1 expression in wildtype mice, deletion of either the M1 or M21enhancer regions using CRISPR/Cas9 gene editing in vivo reduced but did not eliminate the ability of either to suppress Cyp27b1 expression (19). Deletion of both the M1 and M21 enhancers, on the other hand, fully abrogated the ability of both hormones to suppress renal Cyp27b1 expression, suggesting that both hormones may act at similar sites (Figure 2) (21). Unfortunately, while we confirmed the presence of 1,25(OH)2D3-inducible VDR and RXR at each of these sites in M1 and M21 using ChIP-seq analysis, the lack of identification of a factor responsible for FGF23 suppression prevented unequivocal confirmation that these sites were the precise targets of FGF23. Moreover, since there were no striking differences between FGF23 and 1,25(OH)2D3 as measured by changes in residual pCREB abundance, or through striking suppression in the levels of residual CBP or CRTC2 at the pCREB sites within these M1 or M21enhancers, or in the ability of FGF23 to confer a differential profile of histone acetylation, these studies did not advance our understanding (20). Finally, as suppressors neither hormone was able to alter BRD4 binding at each of the pCREB sites, to influence residual levels of RNA polymerase II or to affect H3K36me3 levels across the gene body (32). We also observed that both FGF23 and 1,25(OH)2D3 oppose the upregulation of Cyp27b1 in response to simultaneous treatment with PTH, although 1,25(OH)2D3 is more effective (20). These results suggest that renal Cyp27b1 is upregulated by PTH and that both FGF23 and 1,25(OH)2D3 appear to prevent both residual actions as well as the upregulation of Cyp27b1 by PTH. These results provide novel mechanistic evidence for how these three hormones dynamically regulate the expression of renal Cyp27b1 and therefore the production of circulating 1,25(OH)2D3.
FGF23 and 1,25(OH)2D3 induce the expression of renal Cyp24a1 and oppose PTH action via a dual FGF23/PTH sensitive pCREB containing region at the Cyp24a1 gene promoter and at multiple sites downstream of the gene
Cyp24a1 expression has long been known to be positively regulated by 1,25(OH)2D3 in tissues that include not only the kidney but in most VDR containing NRTCs as well. In contrast, while the more recent discovery that FGF23 regulates Cyp24a1 has suggested that this may occur in several tissue types, the bulk of evidence suggests that FGF23 regulation of Cyp24a1 is restricted to the kidney where FGF23 is biologically active (27). While FGF23 is known to mediate its extracellular actions via a coupling with Klotho that facilitates direct high affinity interaction with FGFR1C and FGFR3, the identity of the intracellular transcription factor and the mechanism whereby FGF23 induces Cyp24a1 and certain other genes as stated earlier remain to be clarified. This may contrast with the suggestion that FGF23 activates the ERK1/ERK2 pathway that induces the transcription factor EGR1 (33–36). Importantly, however, the deletion of EGR1 in the mouse led to only a minor suppression in renal Cyp27b1 expression while effects on Cyp24a1 were not reported (35). And not surprisingly, many other immediate early genes are induced by numerous cellular regulators such as FGF23. Regardless, our efforts to identify the general genomic site(s) of action of FGF23 at the Cyp24a1 gene suggest that FGF23 may act at open chromatin sites within the large DNA segment DS1 located downstream of the Cyp24a1 gene that contains multiple pCREB binding sites. We speculate that the localization of FGF23 action in this region may facilitate in future studies the identification of the FGF23 transcription factor itself. On the other hand, 1,25(OH)2D3 has long been known to induce Cyp24a1 expression via a specific couplet of VDREs located immediately upstream of the Cyp24a1 promoter (37–40). These sites bind the VDR and its heterodimer partner RXR and appear to function in all tissues that express Cyp24a1. Our more recent studies using ChIP-seq analysis and novel cell-based and in vivo assays, however, have now revealed an additional large intergenic region located even further downstream of the Cyp24a1 gene that contains numerous additional sites that bind the VDR/RXR heterodimer (Figure 3) (41). This discovery led to additional research that showed that many of these specific VDREs were functionally active in transfection assays. To functionally corroborate these earlier studies in vivo, we separately deleted the pCREB containing DS1 region downstream of Cyp24a1 as well as the downstream region containing VDR binding sites (DS2) using CRISPR/Cas9 gene editing in the mouse genome and assessed the ability of FGF23 and 1,25(OH)2D3 to induce Cyp24a1 following hormonal injection (27). Deletion of the DS1 segment resulted in the loss of FGF23 induction of Cyp24a1 in the kidney, as seen similarly for PTH suppression, but resulted in only a modest effect on the ability of 1,25(OH)2D3 to induce Cyp24a1. Loss of the DS2 region also resulted in a modest decrease in 1,25(OH)2D3 induction in the kidney and other tissues as well. While these results may appear to conflict, it is worth noting that retention of the apparently dominant VDRE located near the promoter remained active in vivo and thus likely underlined the limited loss of 1,25(OH)2D3 response seen in the DS2-deleted mouse. This complexity suggests that additional experiments are needed to dissect the contributions of the promoter proximal VDREs in the Cyp24a1 gene. However, we conclude that the sites of action of FGF23 and 1,25(OH)2D3 across this gene locus are likely distinct, with FGF23 sites located within the DS1 region and the 1,25(OH)2D3 inducible sites located within the DS2 region as well as at the promoter. Importantly, however, further analysis using ChIP-seq analysis revealed additional relevant information. Although both FGF23 and 1,25(OH)2D3 are strong agonists at the Cyp24a1 gene, 1,25(OH)2D3 is a much more robust inducer of Cyp24a1 than FGF23 (20). Thus, while each displayed common as well as distinct mechanistic effects on the gene, certain features appear to correlate directly with the more robust nature of 1,25(OH)2D3. For example, while neither affected the levels of pCREB present at the gene, both appeared to modestly induce CBP both across the downstream region as well as at the gene promoter, although 1,25(OH)2D3 exhibited more activity at DS2 and at the promoter proximal region. Interestingly, we noted that while both hormones exerted only nominal action on CRTC2, 1,25(OH)2D3 was much more effective at the Cyp24a1 promoter. This surprising finding suggests that in addition to PTH, 1,25(OH)2D3 may also be able to influence CRTC2 migration to the nucleus. While the actions of FGF23 and 1,25(OH)2D3 are not entirely clear at the levels of pCREB downstream factors, the two hormones’ actions are largely similar at the level of epigenetic modification and the consequences that lead to RNA upregulation. In short, both hormones induce H3K27ac across similar regions of the gene, although 1,25(OH)2D3 is unique in that it also strongly induces H3K9ac. Finally, both hormones induce the recruitment of BRD4, although FGF23 is most active at DS1 whereas 1,25(OH)2D3 is most active at DS2, yet both are active at the promoter. Finally, RNA polymerase II is strongly recruited to the Cyp24a1 gene under both hormone treatments, and this is reflected in the appearance of H3K36me3 across the gene body itself. Thus, there are both common as well as unique properties of both hormones, although it is clear 1,25(OH)2D3 is much more active at the Cyp24a1 gene than FGF23, a feature that can be seen in the relative ability of the two hormones to induce Cyp24a1 RNA transcripts. These data provide significant insight into the mechanisms through which FGF23 and 1,25(OH)2D3 regulate Cyp24a1, although additional work will be required to place the two hormones in perspective, not the least of which will be the identification of the transcription factor that mediates FGF23 action.
Summary of the regulation of 1,25(OH)2D3 levels in the blood by PTH, FGF23 and 1,25(OH)2D3
The above discussion defines the reciprocal co-regulation of Cyp27b1 and Cyp24a1 expression in the kidney that is central to the production and maintenance of endocrine 1,25(OH)2D3 by PTH, FGF23 and 1,25(OH)2D3 itself. These events are summarized in Figure 4. This importance is stressed at the in vivo level in mice when the M1 and M21 enhancers are deleted in the mouse, preventing the production of 1,25(OH)2D3 from being synthesized by CYP27B1. Under these conditions, the circulating levels of 1,25(OH)2D3 are strikingly reduced, but not eliminated. This level is insufficient to maintain the skeleton due to high PTH levels and reveals a striking Cyp27b1 null-like phenotype (21). The residual blood levels of 1,25(OH)2D3 are due, however, to the substantial co-reduction in Cyp24a1 expression that permits the small amounts of 1,25(OH)2D3 synthesized in the kidney to be sustained. We ultimately provide evidence of this hypothesis by rescuing this M1/M21-deleted mouse with a diet rich in Ca and P, which brings down PTH and raises FGF23 to normal levels thereby increasing renal Cyp24a1 expression. Under these conditions, the levels of endocrine 1,25(OH)2D3 in the blood are reduced to undetectable concentrations, indicating the importance of the regulation of both renal Cyp27b1 and Cyp24a1 levels in the maintenance of circulating 1,25(OH)2D3.
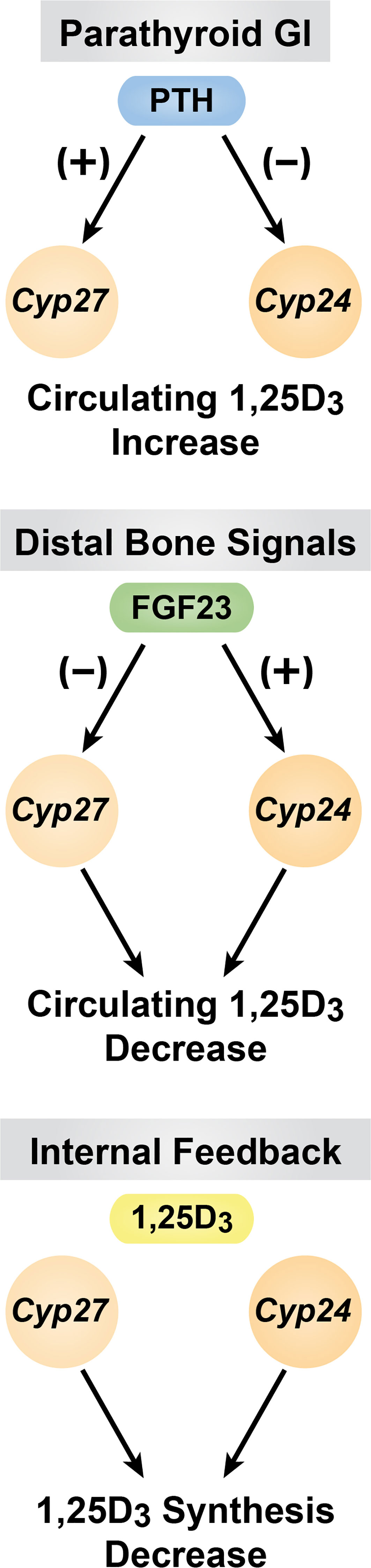
Figure 4 Summary of the of PTH, FGF23 and 1,25(OH)2D3 are diagrammed.
Control of PTH gene expression by 1,25(OH)2D3 and FGF23
As PTH represents the primary stimulator of the 1,25(OH)2D3 synthesis by renal Cyp27b1 and likewise retards the hormone’s degradation via suppression of Cyp24a1, it should not be surprising that circulating 1,25(OH)2D3 might be a primary feedback suppressor of PTH expression. In fact, early studies suggested that was indeed the case. Accordingly, both cells in culture and rodents treated with exogenous 1,25(OH)2D3 exhibited a striking downregulation at the level of PTH mRNA in response to the hormone. Response was uncharacteristically slow in both, however, requiring 48 hours to achieve baseline levels suggesting additional mechanisms (42–44). Many of these mechanisms of PTH processing have been reported (45). One of the primary effects of 1,25(OH)2D3 and indeed most effective agonists of this hormone is to suppress PTH levels in various disease states that include chronic kidney disease (CKD) and perhaps other diseases characterized by inflammation (46–48). Zemplar represents one such analog of 1,25(OH)2D3 that has been effective in PTH suppression, although it is worth noting that this analog is considerably less effective in promoting calcium absorption than 1,25(OH)2D3 (49, 50).
The mechanism whereby 1,25(OH)2D3 might suppress PTH gene expression was explored several decades ago, when it was revealed that the VDR suppressed an artificial construct comprised of a reporter linked to the proximal region of the PTH gene. Accordingly, the VDR was found to bind to a sequence described as a negative VDRE located immediately upstream of the PTH start site (51). This mechanism was advanced based upon the use of traditional cell-based transfection assays of the era. Presently, the validity of this approach has been called into question, as it fails in general to account for numerous features of gene expression which were identified earlier in this manuscript. This result also remains in question as the ensuing years have not been able to substantiate the concept of a negative VDRE sequence. Indeed, the ability of hormones to suppress gene expression is quite complex, and generally involves the interaction of a suppressor with additional elements within an enhancer. Interestingly, recent studies focusing on the PTH gene using ChIP-seq analysis of thyroparathyroid tissue revealed that 1,25(OH)2D3 may induce VDR binding to a site located immediately downstream of the PTH gene (Figure 5). While preliminary, these data do not confirm VDR binding activity at a site upstream of the PTH gene.
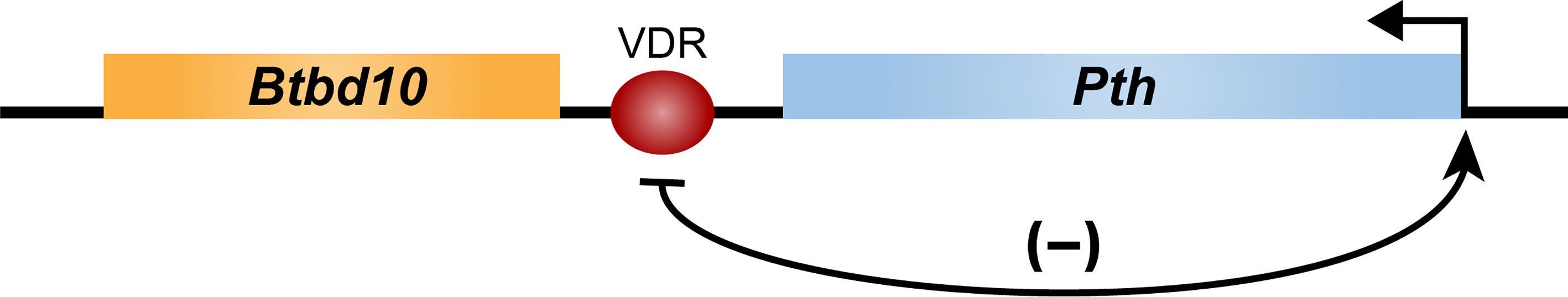
Figure 5 Schematic diagram of the mouse Pth gene located in blue. Arrow indicates direction of transcription. Location of a single VDR binding site (red oval) indicated at the 3’ end of the Pth gene, as assessed by ChIP-seq analysis of thyroparathyroid tissue.
Perhaps most interesting are recent studies in which a mouse model was examined wherein the VDR was selectively deleted from the parathyroid glands using the PTH gene promoter driving CRE (52). Surprisingly, loss VDR expression in the parathyroid glands had no effect on basal levels of PTH in these mice that could account for changes in blood PTH levels. There was also no effect of VDR deletion on the degree of parathyroid hypertrophy. These studies suggest that the effects of 1,25(OH)2D3 on PTH may be secondary to a hormone induced rise in blood Ca which likely reduces PTH levels.
Over the past decades, the discovery of the G protein-based calcium sensing receptor that modulates PTH regulation via Ca sensitivity has been the focus of research on PTH regulation (53). The literature on this subject is vast and has led to the development of therapeutically relevant compounds that are in use currently.
Regulation of PTH by FGF23
The literature is replete with studies suggesting that FGF23 regulates PTH. These studies support both positive as well as negative effects and are almost always conditional (54). Given that lack of insight into the impact of environment on FGF23 action on PTH, a definitive impact remains to be established mechanistically.
Summary of PTH gene regulation
This short summary outlines what is largely known historically of PTH gene regulation. It is not surprising that key details remain to be explored, as useful parathyroid cells are generally not available thereby restricting the use of current techniques described earlier in this manuscript.
Induction of Fgf23 gene expression in bone by PTH and 1,25(OH)2D3
FGF23 represents a long sought after bone-derived peptide hormone that regulates P homeostasis. FGF23 binds to the fibroblast growth factor receptors (FGFRs) FGFR1c and FGFR3 in the kidney, decreasing P reabsorption in the proximal tubules via the positional regulation in the brush border of Na,-P transporters NAPI2A and NAPI2C (29, 30, 55). This repositioning results in the loss of P recovery by the kidney and ensuing hypophosphatemia. FGF23 also downregulates renal Cyp27b1 and induces Cyp24a1 gene expression as detailed earlier, thereby reducing the synthesis of 1,25(OH)2D3 and thus the potential intestinal uptake of both Ca and P. The mechanism of action of FGF23 is dependent upon the exceptionally high affinity of this hormone for the renal FGF receptors by virtue of their interaction with α-klotho, a renal co-receptor that functions to elevate the subsequent binding affinity of FGF23 for the appropriate FGFRs (33). Activation of the FGFRs is believed to result in a triggering of the ERK1/ERK2 signaling pathway that is known to activate the transcription factors EGR1 and perhaps other immediate early genes. While these pathways are known to be involved in the regulation of certain genes by FGF23, the loss of EGR1 via mutation in mice, as stated earlier, had little to no effect on Cyp27b1 expression, suggesting that other unidentified transcription factors may be targets of this pathway (35).
FGF23 is regulated by a myriad of physiologic modulators that represent not only transcriptional inducers, but factors that are involved in FGF23 processing as well. This complexity emerged as a result of a number of genetic P related disease states in humans that not only facilitated the discovery of FGF23 but identified many of the key features of additional components essential to P related control and post transcriptional regulation of FGF23 as well. Significant interest has been sustained over several years now in understanding the transcriptional regulation of Fgf23 by not only the primary mineral regulating hormones such as PTH and 1,25(OH)2D3, but factors that participate in this regulation of Fgf23 at the genomic level. Indeed, early studies suggested the locations of VDRE-like sequences within the upstream region of the Fgf23 gene using traditional transfection assays (56–58). These were suggested to mediate a secondary activity of the VDR/RXR heterodimer; others suggested that the actions of PTH action occurred via the NURR1 transcription factor (59). Perhaps as important as the above hormones, however, is role and mechanism of action of P itself. Due to the temporal features of virtually all dietary P studies, it remained unclear whether P action at the Fgf23 gene was transcriptional or post-transcriptional in nature (60). In some cases, factors involved in the transcriptional regulation of Fgf23 by immune activators, iron and other inducers have been observed, given previous knowledge of their pathways. In view of some of the uncertainties that have emerged, we took an exclusively in vivo approach in which to identify the sites of action of PTH, 1,25(OH)2D3 and P at the Fgf23 gene, with the supposition that the identification of the precise genomic regions that mediate such regulation might provide clues as to the nature of transcription factors involved. While certain factors seem likely to participate in the induction of Fgf23 by PTH and 1,25(OH)2D3, for example CREB and the VDR, there do not appear to be likely candidates for a pathway(s) that might mediate P regulation, necessitating an unbiased search. As will be seen in the following description of our current efforts, the presumption of involvement and the mechanisms of such involvement of anticipated specific targets in these early studies has been highly misleading. Indeed, for example, while it is known that the VDR mediates the actions of 1,25(OH)2D3 at numerous target genes, it appears that the pathway whereby Fgf23 is induced by 1,25(OH)2D3 may be indirect and involve the activity of secondary factors. CREB activation represents a natural mediator of PTH. However, although many of the upstream components of PKA induction by PTH are now well known, the molecular mechanisms through which this pathway triggers controls of SIK inhibition and regulates CREB activity remain to be clarified, as highlighted in our discussion of PTH regulation of coregulators of CREB at the Cyp27b1 gene (20, 23). Furthermore, PTH activates additional signaling pathways and many of the factors that mediate gene regulation via these pathways also remain unclear as well. P on the other hand, requires first the initial determination of whether a transcriptional mechanism of action may be involved prior to the search for mediators, including not only the pathways and the transcription factors involved but the identity and potential role for cellular P sensitizers analogous to that for Ca in the parathyroid glands as well (61–64). Accordingly, we approached our search in an unbiased fashion using several of the techniques outlined in the earlier section on gene regulation, together with additional methods that have been mostly in vivo in the mouse.
PTH upregulates Fgf23 expression in bone in vivo via a potential CREB factor
Early exploration of the Fgf23 gene using ChIP-seq analysis revealed four regions across the Fgf23 gene locus in osteocytes that were enriched for H3K27ac, an epigenetic mark that has been associated with an open chromatin state critical for enhancer function (Figure 6) (65). Two sites were located upstream of Fgf23 gene at -16 kb and -38 kb, one near the promoter region, and one in a downstream intron at +7kb. This discovery prompted the CRISPR mediated deletion of each region and a search for loss of functional activation by PTH. While PTH induction of Fgf23 mRNA in bone was lost in the distal -16kb (DE16K) deleted mouse, no effect was seen in any of the other mouse mutants. Further ChIP-seq analysis of osteocytes revealed the presence of pCREB exclusively at this distal enhancer. Interestingly, recent studies suggest that several SIK inhibitors that promote the recruitment of the CREB receptor transcriptional coactivator 2, (CRTC2) to the Cyp27b1 gene in mouse kidney also induces Fgf23 in the bones of those same mice (20). These results suggest that the mediator of PTH action may indeed be pCREB seen bound at DE16K. While further work will be necessary to establish the precise role for pCREB at the Fgf23 gene, its location 16kb upstream does not contain a NURR1 site. Additional ChIP-seq analyses also fail to identify the presence of NURR1 across the entire Fgf23 binding motif while revealing binding at other additional known sites for NURR1 including the osteocalcin gene in response to PTH. Further studies of NURR1 activation by PTH and 1,25(OH)2D3 in rat UMR bone cells revealed that the pattern of temporal induction of NURR1, as well as NURR2 and NURR3 and other immediate early genes such as EGR1, 2 and 3 are also upregulated (Pike, unpublished)). These studies allow us to conclude that it is unlikely that the mediator of PTH action at Fgf23 in bone is NURR1. In mice, the rapid actions of exogenous PTH resulted in PTH induction of Fgf23 bone mRNA, and prevented the delayed but coordinated detection of upregulated iFGF23 in the blood, as will be seen for 1,25(OH)2D3 and P.
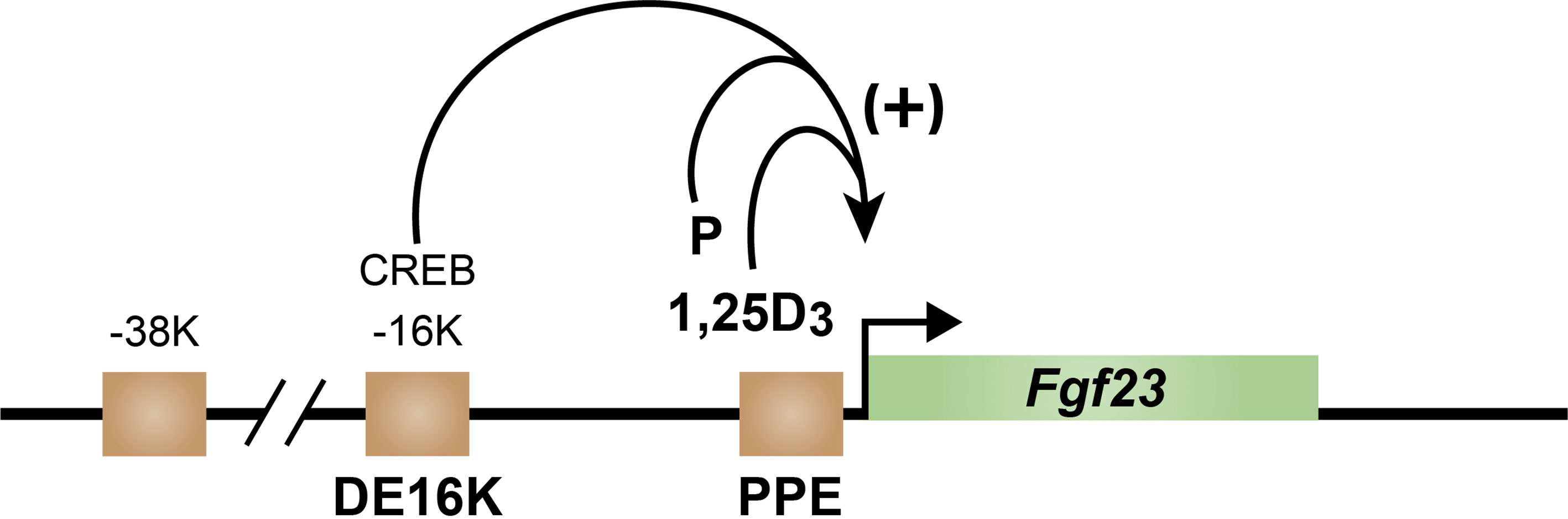
Figure 6 Schematic diagram of the mouse fgf23 gene located in green with arrow indicting direction of transcription. Brown squares indicate regions of increased epigenetic acetylation. Positive regulation by PTH occurs from the CREB site in the DE16K enhancer (DE16K) and by 1,25D3 and P at the promoter-proximal enhancer (PPE).
1,25(OH)2D3 upregulation of Fgf23 expression in bone in vivo may involve a secondary process
1,25(OH)2D3 upregulates Fgf23 expression at the transcriptional level and is known to require the presence of VDR in both mice and in cells in culture. In cells, this induction is rapid and reminiscent of most other genes that are induced by this hormone (57). Nevertheless, our earliest studies of Fgf23 in the osteocyte revealed that despite this classic temporal regulation of Fgf23 by 1,25(OH)2D3, we were unable to identify the presence of the VDR at any sites across the Fgf23 gene locus, despite its presence at numerous known vitamin D target genes in these bone cells (66). Our most recent attempts to identify VDR as well as RXR binding sites at Fgf23 using ChIP-seq analysis in other bone cell lines were also unable to identify the presence of these two receptors, again despite the presence of this heterodimer at all known genes we inspected. These data suggested that the induction of Fgf23 by 1,25(OH)2D3 might be indirect, and mediated by the ability of this hormone and its receptor to rapidly induce one or more secondary transcription factors that might interact directly with the Fgf23 gene. This result prompted further exploration of the Fgf23 gene and revealed that deletion of only the promoter proximal (PPE) but not the DE16K region of Fgf23 in mice using CRISPR/Cas9 gene editing was essential for loss of 1,25(OH)2D3 induction of Fgf23 (67). The unusual nature of this activity of 1,25(OH)2D3 dictated a further unbiased dissection of the PPE by creating a series of mouse mutants containing unique genomic deletions upstream of the Fgf23 start site. Numerous internal mutations and specific deletions were also created as well. These series of mutant mice were then explored for loss of function at the Fgf23 gene as above to map in an unbiased manner response to not only 1,25(OH)2D3 but to P (see below) and other activators as well. Accompanying the quantification of bone Fgf23 mRNA, we also determined whether any changes in the level of mRNA resulted in a change in the circulating levels of intact (i) FGF23 (iFGF23); a change in both was necessary for us to conclude that the deletion resulted in a loss of function. The loss of response to 1,25(OH)2D3 occurred in mutant mice in which deletions were created downstream of the VDRE-like sequence proposed by Liu et al. (58) and upstream of a site suggested to contain a proposed NURR1-like site termed the NURR1/VDRE like sequence as discussed above which was believed to be a potential target for PTH as well as 1,25(OH)2D3 (57, 59). Internal base mutations of this NURR1/VDRE sequence alone as well as small deletions both upstream and downstream of this site had no effect on the ability of 1,25(OH)2D3 to induce bone Fgf23 mRNA or to upregulate blood levels of iFGF23. These data suggest that neither the upstream VDRE-like sequences proposed earlier nor that of the NURR1/VDRE sequence proposed more recently, nor sites more proximal to this latter proposed site mediates the actions of 1,25(OH)2D3. A recent alternative evaluation of this region appears to concur, speculating in detail that this site and transcription factors other than NURR1 may be involved (15). Perhaps more importantly, the DNA segment located between these two hypothetical mediators of 1,25(OH)2D3 does not contain a potential VDRE-like DNA motif capable of directly binding the VDR, supporting our earlier suggestion that the VDR does not bind directly within the Fgf23 gene locus. Thus, we are currently exploring this mapped region of 1,25(OH)2D3 sensitive genomic space that appears to be functionally relevant in vivo in an unbiased search for potential transcription factor candidates that are amenable to technical confirmation as mediators of 1,25(OH)2D3 action. Importantly, these data suggest a novel, yet transcriptionally consistent, genomic mechanism whereby 1,25(OH)2D3 induces the expression of an important gene involved in mineral homeostasis.
P upregulates Fgf23 expression in bone in vivo via distinct acute and chronic mechanisms
P induces an upregulation of Fgf23, even in mice devoid of the VDR (31). The mechanism by which this mineral functions to induce Fgf23 as well as other genes is not known. This appears to be largely because the study of this mineral over many decades in vivo has involved dietary P loading experiments of a generally prolonged nature, wherein lower or elevated P levels in the diet were delivered to mice. This delivery system results in a lengthy delay (days) in the measurement of P response, including the upregulation of Fgf23 and prevented exploration of mechanisms whereby P might induce Fgf23. To overcome this issue, we developed a method whereby elevated dietary P could be rapidly delivered to mice and the upregulation of Fgf23 mRNA evaluated within several hours as described previously; importantly, both the upregulation of Fgf23 mRNA as well as blood iFGF23 could be detected within that time frame (60). Subsequent studies revealed an elevated response to high P as early as 1 h. The rapidity of this response suggested that the mechanisms might be transcriptional in nature. More definitively, however, we also discovered that deletion of the PPE but not the DE16K of the FGF23 gene resulted in a loss of this rapid induction by high P, providing strong support for the idea that this mechanism likely involved transcriptional regulation. Surprisingly, however, despite this loss of acute induction, P appeared to upregulate Fgf23 mRNA and iFGF23 through an additional chronic mechanism apparent in this PPE mouse mutant beginning within 24 h that resulted in normal levels of Fgf23 mRNA and blood iFGF23 by 48 h. It is worth noting here that these results did not appear to arise through an unusual underlying feeding mechanism inherent to the approach, as diet without P was used as feeding controls. Thus, high P appears to induce Fgf23 by two different processes that differ temporally and mechanistically.
To further explore the site of acute action of P at the Fgf23 gene, we mapped the loss of P induced response of Fgf23 mRNA and iFGF23 in each of the mutant mice developed for our studies described above with 1,25(OH)2D3. As with the steroid hormone, a loss of P response was identified both upstream of the NURR1/VDRE-like site. Deletion of the NURR1/VDR-like site also had no effect on loss of Fgf23 mRNA or Fgf23 induction. Thus, a single upstream DNA segment identical to that for 1,25(OH)2D3 induction was apparent (67). It is worth noting, however, that this segment was relatively lengthy. In addition, the time course for P was much more rapid for P than for 1,25(OH)2D3. Therefore, it cannot be concluded that both P and 1,25(OH)2D3 target the same DNA sequence(s) or utilize the same transcription factor(s). Studies are in progress with additional mutant mice to refine the sites of action of P at the Fgf23 gene and to identify functional motifs useful for further exploration of the mechanism of acute P action.
In order to gain additional support for the identity of possible targets of both 1,25)OH)2D3 and P, we also explored the general response in bone via RNA-seq analysis Our working hypothesis is that a genome wide response as determined by an RNA-seq analysis and subsequent analysis of regulated RNAs might provide information regarding not only a relationship between the two inducers, but might reveal additional useful pathways for P in particular, and possible reveal novel transcription factor candidates as well. Of course, the actions of each of these inducers may not involve the induction of regulators via transcription but rather the activation of other signaling pathways as well. One pathway that is common to both 1,25(OH)2D3 and P is an upregulation of Wnt/β-catenin signaling, observed via an increase in the expression of numerous Wnts and the downregulation of numerous Wnt inhibitors (68). However, this overlap is not broadly displayed and many of the genes that are regulated by 1,25(OH)2D3 do not overlap those regulated by P. Future studies will be necessary to determine the relevance of these data with those derived from our mechanistic studies of Fgf23 regulation in the mutant mice.
Concluding remarks
The regulation of mineral metabolism is highly complex and involves three dominant hormones that includes PTH, 1,25(OH)2D3, and FGF23. While these hormones coordinate the regulation of both Ca and P in multiple organs including the intestine, bone, and kidney, they also exert additional hormone-dependent actions in additional tissues that are unrelated to the control of mineral metabolism. Relative to mineral metabolism, numerous diseases are associated with the actions of these hormones; altered mineral metabolism also emerges due to a wide variety of human disease states. Importantly, each of these hormones coordinate actions that include the regulation of the other two. While this has been widely known, the molecular mechanisms responsible for this inter-regulation are only now emerging, due in part to the development of new technologies that have enable the studies described herein.
Author contributions
JP drafted and edited the manuscript; MM edited the manuscript; SL edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants DK-117475, AR-064424, and AR-118174 from the National Institutes of Health to JP. This work was also supported by a Cancer Center Support Grant P30 CA014520.
Acknowledgments
We thank our collaborators for their contributions to the research summarized in this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. DeLuca HF. Overview of general physiologic features and functions of vitamin d. Am J Clin Nutr (2004) 80:1689S–96S. doi: 10.1093/ajcn/80.6.1689S
2. Pike JW, Lee SM, Benkusky NA, Meyer MB. Genomic mechanisms governing mineral homeostasis and the regulation and maintenance of vitamin d metabolism. JBMR Plus (2021) 5:e10433. doi: 10.1002/jbm4.10433
3. Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature (2012) 489:75–82. doi: 10.1038/nature11232
4. Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature (2012) 489:83–90. doi: 10.1038/nature11212
5. Meyer MB, Goetsch PD, Pike JW. Genome-wide analysis of the VDR/RXR cistrome in osteoblast cells provides new mechanistic insight into the actions of the vitamin d hormone. J Steroid Biochem Mol Biol (2010) 121:136–41. doi: 10.1016/j.jsbmb.2010.02.011
6. Pike JW, Meyer MB. Fundamentals of vitamin d hormone-regulated gene expression. J Steroid Biochem Mol Biol (2014) 144PA:5–11. doi: 10.1016/j.jsbmb.2013.11.004
7. Liao J, Ozono K, Sone T, McDonnell D, Pike J. Vitamin d receptor interaction with specific DNA requires a nuclear protein and 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U.S.A. (1990) 87:9751–5. doi: 10.1073/pnas.87.24.9751
8. Kliewer SA, Umesono K, Mangelsdorf DJ, Evans RM. Retinoid X receptor interacts with nuclear receptors in retinoic acid, thyroid hormone and vitamin D3 signalling. Nature (1992) 355:446–9. doi: 10.1038/355446a0
9. Meyer MB, Benkusky NA, Lee CH, Pike JW. Genomic determinants of gene regulation by 1,25-dihydroxyvitamin D3 during osteoblast-lineage cell differentiation. J Biol Chem (2014) 289:19539–54. doi: 10.1074/jbc.M114.578104
10. Meyer MB, Benkusky NA, Pike JW. 1,25-dihydroxyvitamin D3 induced histone profiles guide discovery of VDR action sites. J Steroid Biochem Mol Biol (2014) 144 Pt A:19–21. doi: 10.1016/j.jsbmb.2013.09.005
11. Kim S, Yamazaki M, Zella LA, Meyer MB, Fretz JA, Shevde NK, et al. Multiple enhancer regions located at significant distances upstream of the transcriptional start site mediate RANKL gene expression in response to 1,25-dihydroxyvitamin D3. J Steroid Biochem Mol Biol (2007) 103:430–4. doi: 10.1016/j.jsbmb.2006.12.020
12. Onal M, Bishop KA, St John HC, Danielson AL, Riley EM, Piemontese M, et al. A DNA segment spanning the mouse Tnfsf11 transcription unit and its upstream regulatory domain rescues the pleiotropic biologic phenotype of the RANKL null mouse. J Bone Miner Res (2015) 30:855–68. doi: 10.1002/jbmr.2417
13. Onal M, Galli C, Fu Q, Xiong J, Weinstein RS, Manolagas SC, et al. The RANKL distal control region is required for the increase in RANKL expression, but not the bone loss, associated with hyperparathyroidism or lactation in adult mice. Mol Endocrinol (2012) 26:341–8. doi: 10.1210/me.2011-1149
14. Meyer MB, Benkusky NA, Onal M, Pike JW. Selective regulation of Mmp13 by 1,25(OH). J Steroid Biochem Mol Biol (2016) 164:258–64. doi: 10.1016/j.jsbmb.2015.09.001
15. Haussler MR, Livingston S, Sabir ZL, Haussler CA, Jurutka PW. Vitamin d receptor mediates a myriad of biological actions dependent on its 1,25-dihydroxyvitamin d ligand: distinct regulatory themes revealed by induction of klotho and fibroblast growth factor-23. JBMR Plus (2021) 5:e10432. doi: 10.1002/jbm4.10432
16. Jones G, Prosser DE, Kaufmann M. Cytochrome P450-mediated metabolism of vitamin d. J Lipid Res (2014) 55:13–31. doi: 10.1194/jlr.R031534
17. Jones G, Prosser DE, Kaufmann M. 25-hydroxyvitamin d-24-hydroxylase (CYP24A1): its important role in the degradation of vitamin d. Arch Biochem Biophys (2012) 523:9–18. doi: 10.1016/j.abb.2011.11.003
18. Quinn SJ, Thomsen AR, Egbuna O, Pang J, Baxi K, Goltzman D, et al. CaSR-mediated interactions between calcium and magnesium homeostasis in mice. Am J Physiol Endocrinol Metab (2013) 304:E724–733. doi: 10.1152/ajpendo.00557.2012
19. Meyer MB, Benkusky NA, Kaufmann M, Lee SM, Onal M, Jones G, et al. A kidney-specific genetic control module in mice governs endocrine regulation of the cytochrome P450 gene Cyp27b1 essential for vitamin D3 activation. J Biol Chem (2017) 292:17541–58. doi: 10.1074/jbc.M117.806901
20. Meyer MB, Benkusky NA, Lee SM, Yoon SH, Mannstadt M, Wein MN, et al. Rapid genomic changes by mineralotropic hormones and kinase SIK inhibition drive coordinated renal Cyp27b1 and Cyp24a1 expression via CREB modules. J Biol Chem (2022) 298(11):102559. doi: 10.1016/j.jbc.2022.102559
21. Meyer MB, Benkusky NA, Kaufmann M, Lee SM, Redfield RR, Jones G, et al. Targeted genomic deletions identify diverse enhancer functions and generate a kidney-specific, endocrine-deficient Cyp27b1 pseudo-null mouse. J Biol Chem (2019) 294:9518–35. doi: 10.1074/jbc.RA119.008760
22. Stamatoyannopoulos JA, Snyder M, Hardison R, Ren B, Gingeras T, Gilbert DM, et al. An encyclopedia of mouse DNA elements (Mouse ENCODE). Genome Biol (2012) 13:418. doi: 10.1186/gb-2012-13-8-418
23. Yoon SH, Meyer MB, Arevalo Rivas C, Tekguc M, Zhang C, Wang JS, et al. A parathyroid hormone/salt-inducible kinase signaling axis controls renal vitamin d activation and organismal calcium homeostasis. J Clin Invest (2023) 133(9):e163627. doi: 10.1172/JCI163627
24. Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev (2000) 14:1553–77. doi: 10.1101/gad.14.13.1553
25. Zierold C, Mings JA, DeLuca HF. Parathyroid hormone regulates 25-hydroxyvitamin D(3)-24-hydroxylase mRNA by altering its stability. Proc Natl Acad Sci U.S.A. (2001) 98:13572–6. doi: 10.1073/pnas.241516798
26. Zierold C, Reinholz GG, Mings JA, Prahl JM, DeLuca HF. Regulation of the procine 1,25-dihydroxyvitamin D3-24-hydroxylase (CYP24) by 1,25-dihydroxyvitamin D3 and parathyroid hormone in AOK-B50 cells. Arch Biochem Biophys (2000) 381:323–7. doi: 10.1006/abbi.2000.1964
27. Meyer MB, Lee SM, Carlson AH, Benkusky NA, Kaufmann M, Jones G, et al. A chromatin-based mechanism controls differential regulation of the cytochrome P450 gene Cyp24a1 in renal and nonrenal tissues. J Biol Chem (2019) 294:14467–81. doi: 10.1074/jbc.RA119.010173
28. Tanaka Y, DeLuca H. Rat renal 25-hydroxyvitamin D3 1- and 24-hydroxylases: their in vivo regulation. Am J Physiol (1984) 246:E168–173. doi: 10.1152/ajpendo.1984.246.2.E168
29. Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, et al. FGF-23 is a potent regulator of vitamin d metabolism and phosphate homeostasis. J Bone Miner Res (2004) 19:429–35. doi: 10.1359/JBMR.0301264
30. Shimada T, Kakitani M, Yamazaki Y, Hasegawa H, Takeuchi Y, Fujita T, et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin d metabolism. J Clin Invest (2004) 113:561–8. doi: 10.1172/JCI19081
31. Shimada T, Yamazaki Y, Takahashi M, Hasegawa H, Urakawa I, Oshima T, et al. Vitamin d receptor-independent FGF23 actions in regulating phosphate and vitamin d metabolism. Am J Physiol Renal Physiol (2005) 289:F1088–1095. doi: 10.1152/ajprenal.00474.2004
32. Van de Velde S, Wiater E, Tran M, Hwang Y, Cole PA, Montminy M. CREB promotes beta cell gene expression by targeting its coactivators to tissue-specific enhancers. Mol Cell Biol (2019) 39(17):e00200–19. doi: 10.1128/MCB.00200-19
33. Quarles LD. Fibroblast growth factor 23 and α-klotho co-dependent and independent functions. Curr Opin Nephrol Hypertens (2019) 28:16–25. doi: 10.1097/MNH.0000000000000467
34. Kovesdy CP, Quarles LD. Fibroblast growth factor-23: what we know, what we don't know, and what we need to know. Nephrol Dial Transplant (2013) 28:2228–36. doi: 10.1093/ndt/gft065
35. Portale AA, Zhang MY, David V, Martin A, Jiao Y, Gu W, et al. Characterization of FGF23-dependent egr-1 cistrome in the mouse renal proximal tubule. PloS One (2015) 10:e0142924. doi: 10.1371/journal.pone.0142924
36. Chanakul A, Zhang MY, Louw A, Armbrecht HJ, Miller WL, Portale AA, et al. FGF-23 regulates CYP27B1 transcription in the kidney and in extra-renal tissues. PloS One (2013) 8:e72816. doi: 10.1371/journal.pone.0072816
37. Ozono K, Yamagata M, Ohyama Y, Nakajima S. Direct repeat 3-type element lacking the ability to bind to the vitamin d receptor enhances the function of a vitamin d-responsive element. J Steroid Biochem Mol Biol (1998) 66:263–9. doi: 10.1016/S0960-0760(98)00055-7
38. Ohyama Y, Ozono K, Uchida M, Yoshimura M, Shinki T, Suda T, et al. Functional assessment of two vitamin d-responsive elements in the rat 25-hydroxyvitamin D3 24-hydroxylase gene. J Biol Chem (1996) 271:30381–5. doi: 10.1074/jbc.271.48.30381
39. Zierold C, Darwish H, DeLuca H. Identification of a vitamin d-response element in the rat calcidiol (25-hydroxyvitamin D3) 24-hydroxylase gene. Proc Natl Acad Sci U.S.A. (1994) 91:900–2. doi: 10.1073/pnas.91.3.900
40. Zierold C, Darwish H, DeLuca H. Two vitamin d response elements function in the rat 1,25-dihydroxyvitamin d 24-hydroxylase promoter. J Biol Chem (1995) 270:1675–8. doi: 10.1074/jbc.270.4.1675
41. Meyer MB, Goetsch PD, Pike JW. A downstream intergenic cluster of regulatory enhancers contributes to the induction of CYP24A1 expression by 1alpha,25-dihydroxyvitamin D3. J Biol Chem (2010) 285:15599–610. doi: 10.1074/jbc.M110.119958
42. Russell J, Silver J, Sherwood LM. The effects of calcium and vitamin d metabolites on cytoplasmic mRNA coding for pre-proparathyroid hormone in isolated parathyroid cells. Trans Assoc Am Physicians (1984) 97:296–303.
43. Silver J, Russell J, Sherwood LM. Regulation by vitamin d metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc Natl Acad Sci U.S.A. (1985) 82:4270–3. doi: 10.1073/pnas.82.12.4270
44. Silver J, Naveh-Many T, Mayer H, Schmelzer HJ, Popovtzer MM. Regulation by vitamin d metabolites of parathyroid hormone gene transcription in vivo in the rat. J Clin Invest (1986) 78:1296–301. doi: 10.1172/JCI112714
45. Bell O, Silver J, Naveh-Many T. Identification and characterization of cis-acting elements in the human and bovine PTH mRNA 3'-untranslated region. J Bone Miner Res (2005) 20:858–66. doi: 10.1359/JBMR.041227
46. Plum LA, DeLuca HF. Vitamin d, disease and therapeutic opportunities. Nat Rev Drug Discov (2010) 9:941–55. doi: 10.1038/nrd3318
47. Plum LA, Fitzpatrick LA, Ma X, Binkley NC, Zella JB, Clagett-Dame M, et al. 2MD, a new anabolic agent for osteoporosis treatment. Osteoporos Int (2006) 17:704–15. doi: 10.1007/s00198-005-0036-3
48. Brown AJ, Dusso AS, Slatopolsky E. Vitamin d analogues for secondary hyperparathyroidism. Nephrol Dial Transplant (2002) 17 Suppl;10:10–9. doi: 10.1093/ndt/17.suppl_10.10
49. Ahmed OM, Ali TM, Abdel Gaid MA, Elberry AA. Effects of enalapril and paricalcitol treatment on diabetic nephropathy and renal expressions of TNF-α, p53, caspase-3 and bcl-2 in STZ-induced diabetic rats. PloS One (2019) 14:e0214349. doi: 10.1371/journal.pone.0214349
50. Schuster A, Al-Makki A, Shepler B. Use of paricalcitol as adjunctive therapy to renin-Angiotensin-Aldosterone system inhibition for diabetic nephropathy: a systematic review of the literature. Clin Ther (2019) 41:2416–23. doi: 10.1016/j.clinthera.2019.09.009
51. Demay MB, Kiernan MS, DeLuca HF, Kronenberg HM. Sequences in the human parathyroid hormone gene that bind the 1,25-dihydroxyvitamin D3 receptor and mediate transcriptional repression in response to 1,25-dihydroxyvitamin D3. Proc Natl Acad Sci U.S.A. (1992) 89:8097–101. doi: 10.1073/pnas.89.17.8097
52. Meir T, Levi R, Lieben L, Libutti S, Carmeliet G, Bouillon R, et al. Deletion of the vitamin d receptor specifically in the parathyroid demonstrates a limited role for the receptor in parathyroid physiology. Am J Physiol Renal Physiol (2009) 297:F1192–1198. doi: 10.1152/ajprenal.00360.2009
53. Brown EM, MacLeod RJ. Extracellular calcium sensing and extracellular calcium signaling. Physiol Rev (2001) 81:239–97. doi: 10.1152/physrev.2001.81.1.239
54. Lavi-Moshayoff V, Wasserman G, Meir T, Silver J, Naveh-Many T. PTH increases FGF23 gene expression and mediates the high-FGF23 levels of experimental kidney failure: a bone parathyroid feedback loop. Am J Physiol Renal Physiol (2010) 299:F882–889. doi: 10.1152/ajprenal.00360.2010
55. Shimada T, Mizutani S, Muto T, Yoneya T, Hino R, Takeda S, et al. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc Natl Acad Sci U.S.A. (2001) 98:6500–5. doi: 10.1073/pnas.101545198
56. Kolek OI, Hines ER, Jones MD, LeSueur LK, Lipko MA, Kiela PR, et al. 1alpha,25-dihydroxyvitamin D3 upregulates FGF23 gene expression in bone: the final link in a renal-gastrointestinal-skeletal axis that controls phosphate transport. Am J Physiol Gastrointest Liver Physiol (2005) 289:G1036–1042. doi: 10.1152/ajpgi.00243.2005
57. Kaneko I, Saini RK, Griffin KP, Whitfield GK, Haussler MR, Jurutka PW. FGF23 gene regulation by 1,25-dihydroxyvitamin d: opposing effects in adipocytes and osteocytes. J Endocrinol (2015) 226:155–66. doi: 10.1530/JOE-15-0225
58. Liu S, Tang W, Zhou J, Stubbs JR, Luo Q, Pi M, et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin d. J Am Soc Nephrol (2006) 17:1305–15. doi: 10.1681/ASN.2005111185
59. Meir T, Durlacher K, Pan Z, Amir G, Richards WG, Silver J, et al. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int (2014) 86:1106–15. doi: 10.1038/ki.2014.215
60. Lee SM, Carlson AH, Onal M, Benkusky NA, Meyer MB, Pike JW. A control region near the fibroblast growth factor 23 gene mediates response to phosphate, 1,25(OH)2D3, and LPS In vivo. Endocrinology (2019) 160:2877–91. doi: 10.1210/en.2019-00622
61. Simic P, Kim W, Zhou W, Pierce KA, Chang W, Sykes DB, et al. Glycerol-3-phosphate is an FGF23 regulator derived from the injured kidney. J Clin Invest (2020) 130:1513–26. doi: 10.1172/JCI131190
62. Zhou W, Simic P, Zhou IY, Caravan P, Vela Parada X, Wen D, et al. Kidney glycolysis serves as a mammalian phosphate sensor that maintains phosphate homeostasis. J Clin Invest (2023) 133(8):e164610. doi: 10.1172/JCI164610
63. Couasnay G, Bon N, Devignes CS, Sourice S, Bianchi A, Véziers J, et al. PiT1/Slc20a1 is required for endoplasmic reticulum homeostasis, chondrocyte survival, and skeletal development. J Bone Miner Res (2019) 34:387–98. doi: 10.1002/jbmr.3609
64. Bon N, Frangi G, Sourice S, Guicheux J, Beck-Cormier S, Beck L. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol Metab (2018) 11:197–204. doi: 10.1016/j.molmet.2018.02.007
65. Onal M, Carlson AH, Thostenson JD, Benkusky NA, Meyer MB, Lee SM, et al. A novel distal enhancer mediates inflammation-, PTH-, and early onset murine kidney disease-induced expression of the mouse Fgf23 gene. JBMR Plus (2018) 2:32–47. doi: 10.1002/jbm4.10023
66. St John HC, Bishop KA, Meyer MB, Benkusky NA, Leng N, Kendziorski C, et al. The osteoblast to osteocyte transition: epigenetic changes and response to the vitamin D3 hormone. Mol Endocrinol (2014) 28:1150–65. doi: 10.1210/me.2014-1091
67. Pike JW, Meyer MB, St John HC, Benkusky NA. Epigenetic histone modifications and master regulators as determinants of context dependent nuclear receptor activity in bone cells. Bone (2015) 81:757–64. doi: 10.1016/j.bone.2015.03.012
Keywords: mineral regulating hormones, transcription, ChIP-seq analysis, CRISPR/Cas9, mutant mice, Cyp27b1/Cyp24a1 genes, PTH gene, FGF23 gene
Citation: Pike JW, Lee SM and Meyer MB (2023) Molecular insights into mineralotropic hormone inter-regulation. Front. Endocrinol. 14:1213361. doi: 10.3389/fendo.2023.1213361
Received: 27 April 2023; Accepted: 13 June 2023;
Published: 27 June 2023.
Edited by:
Rik Mencke, University Medical Center Groningen, NetherlandsReviewed by:
Sylvia Christakos, Rutgers, The State University of New Jersey, United StatesAdriana Dusso, Washington University in St. Louis, United States
Copyright © 2023 Pike, Lee and Meyer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: J. Wesley Pike, anBpa2VAd2lzYy5lZHU=