 Si-jin Zhang
Si-jin Zhang Xian-zheng Qin2†
Xian-zheng Qin2† Jie Zhou
Jie Zhou Bin-feng He
Bin-feng He Surendra Shrestha
Surendra Shrestha Jing Zhang
Jing Zhang Wei-ping Hu
Wei-ping Hu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 10 October 2023
Sec. Obesity
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1204744
This article is part of the Research Topic Obesity Paradox and Associated Complications in Chronic Obstructive Pulmonary Disease View all 4 articles
Background: Obesity and chronic obstructive pulmonary disease (COPD) are prevailing worldwide, bringing a heavy medical burden. Clinical and pathophysiological relationship between obesity and COPD is paradoxical and elusive. We aim to explore their inherent associations from clinical, genetic, and animal levels.
Methods: We performed literature review and cohort analysis of patients with COPD to compare lung function, symptom, and prognosis among different weight groups. After retrieving datasets of obesity and COPD in Gene Expression Omnibus (GEO) database, we carried out differentially expressed gene analysis, functional enrichment, protein–protein interactions network, and weighted gene co-expression network analysis. Then, we acquired paraffin-embedded lung tissues of fatty acid–binding protein 4–Cre-BMPR2fl/fl conditional knockout (CKO) mice that were characterized by adipocyte-specific knockout of bone morphogenetic protein receptor 2 (BMPR2) for staining and analysis.
Results: Our cohort study reports the effect of obesity on COPD is inconsistent with previous clinical studies. Lung function of overweight group was statistically superior to that of other groups. We also found that the inflammatory factors were significantly increased hub genes, and cytokine-associated pathways were enriched in white adipose tissue of patients with obesity. Similarly, injury repair–associated genes and pathways were further enhanced in the small airways of patients with COPD. CKO mice spontaneously developed lung injury, emphysema, and pulmonary vascular remodeling, along with increased infiltration of macrophages. BMPR2-defiecient adipocytes had dysregulated expression of adipocytokines.
Conclusion: Inflammation and abnormal repair might be potential mechanisms of the pathological association between obesity and COPD. BMPR2-associated adipocyte dysfunction promoted lung inflammation and aberrant repair, in which adipocytokines might play a role and thus could be a promising therapeutic target.
Characterized by chronic inflammation, remodeling of small airways, and emphysema (1), chronic obstructive pulmonary disease (COPD) has been a major public health problem. More than 380 million people are estimated to suffer from COPD worldwide (2). World Health Organization predicted that COPD would be the third leading cause of death and rank the fifth in the burden of disease (1). Poor prophylaxis and management of COPD also increase patient’s morbidity and mortality (3, 4).Thus, it is important to understand pathogenesis of COPD, which could guide us for better management of COPD.
With lifestyle changes, more people became overweight and obese rather than underweight. The 2014 Global Non-communicable Disease Survey showed that about 40% of adults were overweight and 10.8%–14.9% were obese (5). Body mass index (BMI) had been widely used to access prevalence of obesity. It has been well established that increasing BMI is a major risk factor for cardiovascular disease (6), osteoarthritis (7), diabetes mellitus (8), and choledocholithiasis (9). However, the relationship between obesity and clinical outcome of COPD seems to be more complex (10). Some studies report that both adiposity and underweight were risk factors for COPD (11). In contrast, other studies found overweight to significantly improved the survival of patients with COPD, which was defined as “obesity paradox” (12). From the perspective of respiratory physiology, patients with obesity with COPD may experience lower lung hyperinflation because of modified mechanical properties of chest wall, compared with their lean counterparts (13). Moreover, patients with overweight with COPD often present a mild degree of emphysema, probably accounting for decreased mortality (14).
It was hard to elucidate the sophisticated effects of the adipose tissue on lung (13). Regulation of adipose tissue function could be a promising therapeutic target, because it acts as a systemic regulator in response to the environmental changes, like inflammation (15). Similar to COPD, increased systemic inflammation was also associated with excessive fat mass, especially regarding tumor necrosis factor–α (TNFα), interleukin-6, C-reactive protein, and metabolic syndrome (12). There have been several classical meta-analyses on the relationship between BMI and prognosis or mortality of patients with COPD. For example, Evangelos et al. reported that BMI was one of the most commonly used index in the development of prognostic models of COPD (16). Xiong et al. reported that patients with COPD being overweight or obese had a protective effect against mortality (17). The phenomenon of “obesity paradox” also exists in COPD animal models. For cigarette smoke-exposed rodents, the obese mice presented more severe emphysema and pulmonary inflammation compared with lean group (18). For rodents who received lipopolysaccharide of E. coli, obese rats showed less deterioration of lung function, lower phagocytosis of monocytes in blood and macrophages in adipose tissue, as well as reduced migration of neutrophils in comparison with normal weight rats (19). Moreover, Wu et al. reported that obesity could alleviate ventilator-induced lung injury through Signal transducer and activator of transcription 3-Suppressor of cytokine signaling 3 (STAT3–SOCS3) signaling pathway (20).
Adipocytokines are a class of bioactive mediators secreted by adipocytes, including leptin, adiponectin, fibroblast growth factor 21, bone morphogenetic protein–4 (BMP4), and BMP7, with ability of regulating systemic inflammation and remodeling (21, 22). BMP4 and BMP7 act their function by binding onto the receptors including bone morphogenetic protein receptor 2 (BMPR2). BMPR2-deficient adipocytes were susceptible to pyroptosis and apoptosis (23). Qian et al. observed that the fatty acid–binding protein 4 (FABP4)–Cre-BMPR2fl/fl mice, with the adipocyte-specific knockout of BMPR2, begin to weaken and die after 3 weeks of weaning, with an adult survival rate of only 25% (23). However, it is not clear whether adipose tissue dysfunction in FABP4-Cre-BMPR2fl/fl mice impacts the lungs. Our study aims to investigate the effect of adipocyte dysfunction on lung inflammation and lung injury and hopes to develop a better understanding of the molecular targets for COPD therapy.
We reviewed the clinical studies of “COPD” and “obesity.” Then, we summarized their clinical characteristics and relationships with obesity from the cohort of our center. We downloaded raw data for gene expression profile from GEO database, which included subcutaneous adipose tissues (GSE11906) (24) and small airways (GSE151839) (25). The differentially expressed genes (DEGs) were obtained and were further subjected to gene set enrichment analysis (GSEA), Kyoto Encyclopedia of Genes and Genomes (KEGG), and protein–protein interaction (PPI) network analysis. At the same time, weighted gene co-expression network analysis (WGCNA) was conducted to explore the specific key modules. Finally, we validated lung injury and emphysema in the lungs of FABP4-Cre-BMPR2fl/fl mice by hematoxylin–eosin (H&E) stain and immunostaining, revealing partial results consistent with public database analysis. Re-analysis of the RNA sequencing and prediction of the related genes between FABP4 and BMPR2 were assessed to illustrate the role of adipocytokines.
To investigate the clinical relationship between COPD and obesity, we searched PubMed (http://www.ncbi.nlm.nih.gov/pubmed) for relevant studies (March 2013 to March 2023), using keywords “COPD” and “obesity.” The following critical elements were extracted and recorded, including study population, obesity-related definition, with or without interference, outcome, and statistical method.
To compare the difference among patients with COPD with different BMI, we re-analyzed the clinical data within a prospective cohort. Detailed description of this cohort has been published in our previous studies (26, 27). In brief, we recruited patients hospitalized for AECOPD (acute exacerbation of COPD) in the Department of Pulmonary Medicine of Shanghai Zhongshan Hospital between January 2015 and July 2017, collected their clinical information, and followed up their status about recurrent AE (re-AE) and survival within 1 year after discharge. Inclusion criterion was that patients with a clearly recorded COPD history had deteriorated respiratory symptoms, which was categorized as AECOPD by two separate pulmonologists. Exclusion criteria were exacerbations induced by other respiratory diseases, such as asthma, bronchiectasis, congestive heart failure, pleuritis, pneumothorax, pulmonary embolism, and restrictive lung disease. Clinical outcomes were days of in-hospital stay, hospitalized death, and re-AE within 1, 3, 6, and 12 months. Re-AE was defined as the new worsening of respiratory symptoms lasting for over 2 days, which required additional medical intervention. The prospective cohort was approved by Institutional Review Board (IRB) of Shanghai Zhongshan Hospital (Certificate No. B2015-119R).
We identified the shared DEGs of COPD and obesity. Gene expression datasets of subcutaneous adipose tissues (GSE11906) (24) and small airways (GSE151839) (25) were retrieved from publicly available GEO database. Obesity-associated differential gene analysis was performed in white adipose tissues from between overweight volunteers (n = 10) and normal weight volunteers (n = 10). COPD-associated DEGs were analyzed between non-smokers (n = 24) and patients with COPD (n = 36) by performing the R package “limma.” Genes with an adjusted P-value <0.05 and log2 fold change >1.2 were considered as being differentially expressed. Volcano plot was performed using the R package ggplot2 version 3.2.1 to visualize the DEGs. R package “venneuler” was used to draw a Venn diagram. The shared DEGs were retained for further analysis.
GSEA of the DEGs was conducted to investigate the biological characteristics of COPD and obesity. False discovery rate (FDR) <0.25, Nominal (NOM) p-value <0.05, and Normalized enrichment score (NES) >1 were considered significant enrichment. Furthermore, we used R package “clusterProfiler” to conduct KEGG analyses of the DEGs. P < 0.05 was considered statistically significant. We used the R software “ggplot2” to visualize above results.
To identify hub regulatory genes and to examine the interactions between DEGs, PPI network was generated with an open-source software Cytoscape (version 3.9.1), which is based on the results of Search Tool for the Retrieval of Interacting Genes/Proteins (STRING; https://string-db.org/) (28). These interactions among proteins with a combined score >0.4 were considered statistically significant. Furthermore, hub genes were selected with Molecular Complex Detection (MCODE), which is a free plugin in Cytoscape. In addition, the parameters were set as degree cutoff = 2, haircut on, node score cutoff = 0.2, k-core = 2, and maximum depth = 100.
We preformed WGCNA to find the key modules. We used the “WGCNA” package in R to build a co-expression network targeting DEGs to construct specific modules and analyze the module–phenotype relationship as previously described (29). Subsequently, the adjacency matrix was transformed into a topological overlap matrix (TOM), and average linkage hierarchical clustering was used to construct a clustering dendrogram of the TOM matrix. Then, gene hierarchical clustering dendrogram was made to identify co-expression modules, and the module eigengene (ME) was calculated. The minimal gene module size was set to 30 to obtain appropriate modules, and the threshold to merge similar modules was set to 0.25. Finally, we predicted correlation between ME and phenotype to reveal the clinical traits.
FABP4 is an adipocyte-specific promoter. FABP4-Cre-BMPR2fl/fl (CKO) mice are characterized by adipocyte-specific knockout of BMPR2. As Qian et al. reported (23), mice bearing both FABP4-cre and BMPR2 flox/flox genotypes were first crossed to breed FABP4-Cre-BMPR2fl/− (CKO heterozygote) mice. Then, CKO heterozygote mice were further crossed to breed FABP4-Cre-BMPR2fl/fl (CKO homozygote) mice, which were identified by PCR and DNA electrophoresis. Primers of FABP4-Cre are GGTCGATGCAACGAG TGATGAGGT and CAGCATTGCTGTCACTTGGTCGTG. Primers of BMPR2-flox are TTATTGTAAGTACACTGTTGCT and GGCAGACTCTGACTTTGACGC. Qian et al. previously isolated cells from inguinal white adipose tissue of wild-type (WT) and CKO mice and performed Western blots to successfully confirm decreased levels of BMPR2 in CKO mice’s adipose tissue (23, 30).
Qian et al. kindly granted us three paraffin-embedded lung tissues, including one WT and two CKO mice, all of which were sacrificed and collected on days 19–22. We made serial sections of these tissues and performed H&E staining and digital photographic scan on every eight slides. CaseViewer software (3D HISTECH Ltd., Hungary) was used to measure the indices of H&E stain. Lung injury score was divided into five grades: normal as grade 1, focal interstitial congestion as grade 2, diffuse congestion as grade 3, focal consolidation as grade 4, and diffuse consolidation as grade 5 (at ×5 magnification).
Detailed protocols of immunohistochemical stain and immunofluorescence stain were reported in our previous study (31). We used the Super Plus™ High Sensitive and Rapid Immunohistochemical Kit (E-IR-R221, Elabscience®, China) and the Immunofluorescence Application Solutions Kit (12727, Cell Signaling Technology (CST), USA). The list of antibodies is shown in Supplementary Table 1.
The one-step terminal deoxynucleotidyl transferase (TdT)–mediated Deoxyuridine Triphosphate (dUTP)-biotin nick end labeling (TUNEL) In Situ Apoptosis Kit (E-CK-A321, Elabscience®, China) was used to detect apoptotic cells in lung tissues. After dewaxing and hydrating, the paraffin sections were incubated with Proteinase K for permeation. Positive control was extra incubated with Deoxyribonuclease I (DNase I) buffer. Working solution was prepared by mixing TdT equilibration buffer, labeling solution, and TdT enzyme and then added onto the paraffin sections for 1 h. After sufficient washing and nuclear staining, we sealed the paraffin sections and recorded photographs using fluorescence microscope.
The single protein name (“Bone Morphogenetic Protein Receptor Type 2” or “Fatty Acid Binding Protein 4”) and organism (“Homo sapiens”) were searched at STRING website. The main parameters were set as minimum required interaction score [“low confidence (0.150)”], meaning of network edges (“evidence”), maximum number of interactors to show (“no more than 50 interactors” in the first shell), and active interaction sources (“experiments”). Finally, we obtained the available experimentally determined BMPR2-binding proteins and FABP4-binding proteins.
Categorical variables were showed as number (percentage) and compared by the Chi-square test. Continuous variables were expressed as the mean and standard deviation, which were analyzed by Kruskal–Wallis test for three different groups or Mann–Whitney test for two groups. We used GraphPad Prism 6 (GraphPad Software, CA, USA) and IBM SPSS statistics 23 (SPSS Inc., Chicago, IL, USA) to perform the above statistical analysis and defined the two-tailed P < 0.05 as statistical significance.
In Table 1, 15 studies on COPD and obesity are listed. Four studies reported that overweight or obesity was beneficial to the survival for patients with COPD, and three studies showed that patients with overweight or obesity had a decreased risk of acute exacerbation. However, three other studies reported that obesity could impair exercise capacity of patients with COPD. Some large-scale cross-sectional studies demonstrated that people with overweight or obesity had increased incidence of COPD, which might be associated with aggravated response to cigarette smoke or pollutants. Thus, the effect of overweight or obesity on COPD is inconsistent among current studies.
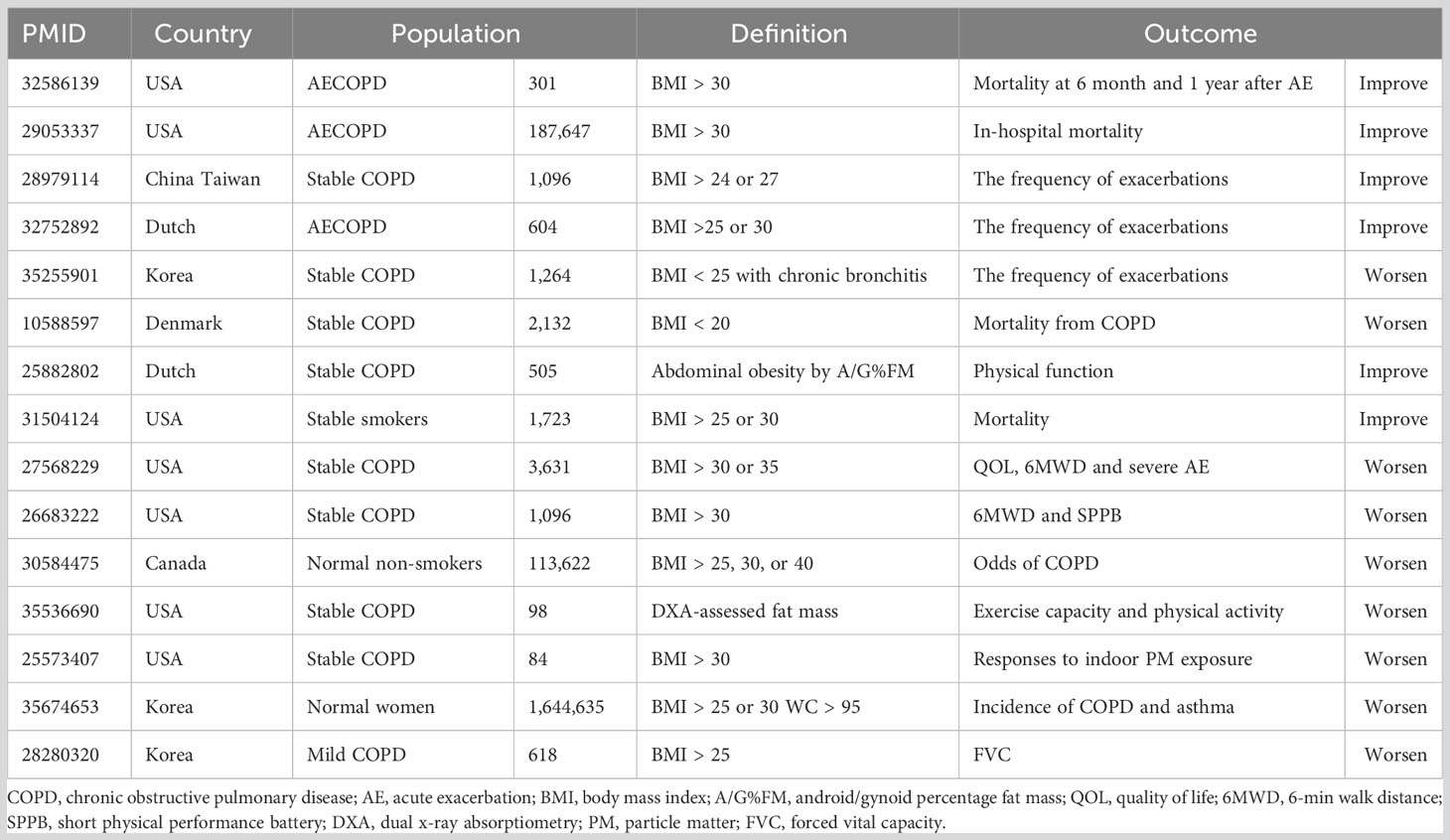
Table 1 A literature review of clinical studies on COPD and obesity.
To enrich the clinical data of Chinese people on obesity and COPD, 120 patients hospitalized for AECOPD were enrolled in our study and divided into three groups of underweight, normal weight, and overweight based on BMI. As shown in Table 2, some lung function indices (FEV1% and FEV1/FVC) of overweight group were statistically superior to those of normal weight and overweight group, suggesting that proper weight gain might help slow the rate of decline in the lung function. In addition, diffusing capacity of the lungs for carbon monoxide and COPD assessment test also showed a positive trend in the overweight group. Despite the lack of statistical significance, overweight group had lower percentage of re-AE in 3 months and 1 year, indicting the better prognosis.
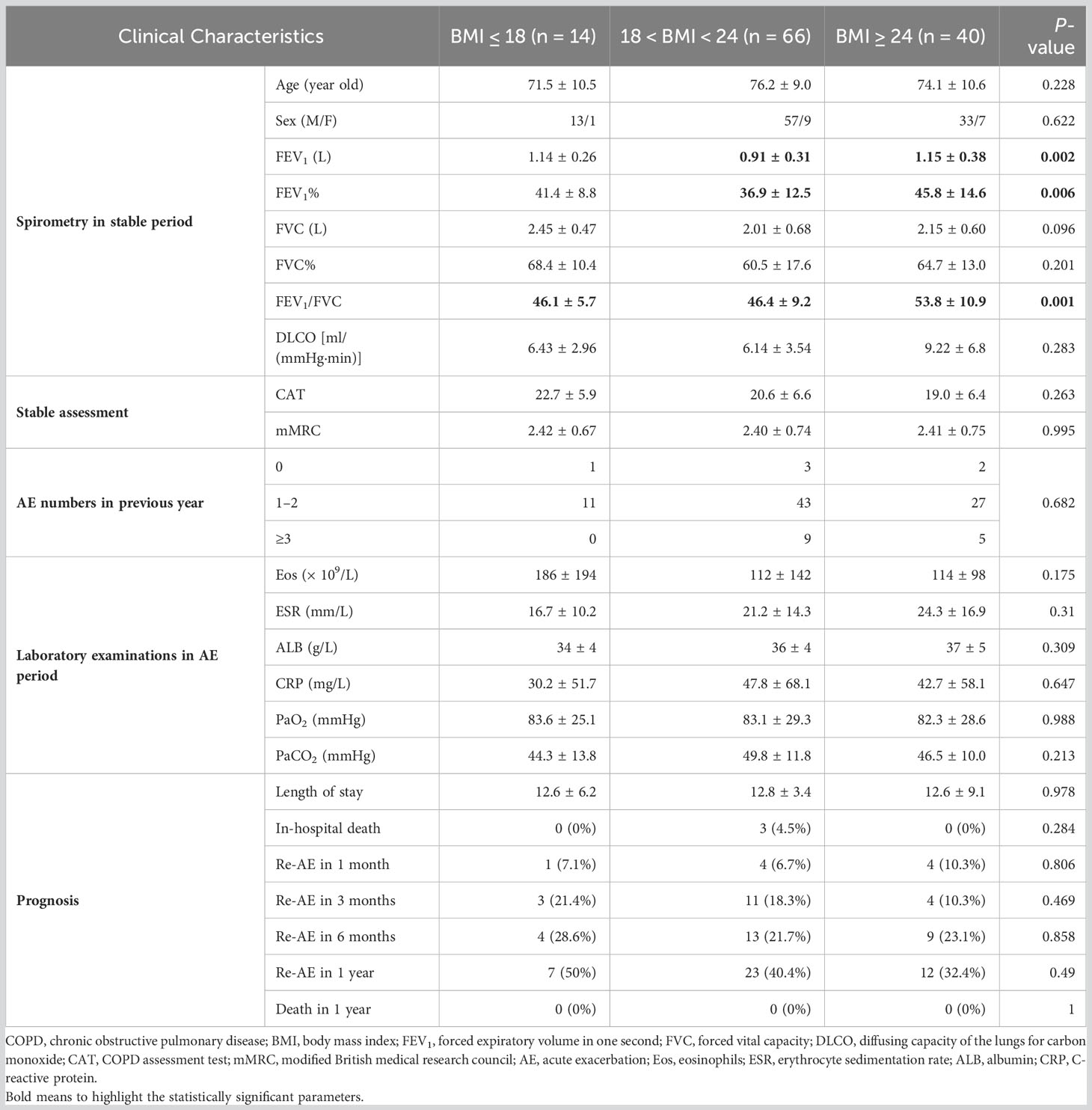
Table 2 Clinical characteristics of patients with COPD with different BMI.
To explore the inherent pattern of gene expression and potential association between COPD and obesity, we analyzed DEGs in GEO public datasets. After filtering the duplicate and non-sense genes, a total of 342 DEGs in COPD and 223 DEGs in obesity were identified, including 18 overlapping DEGs (Figure 1A). Volcano maps of the DEGs are plotted in Supplementary Figure 1A and Supplementary Figure 1B. The inflammatory factors [C-X-C Motif Chemokine Ligand 8 (CXCL8), CXCL2, and matrix metalloproteinase 9 (MMP9)] were significantly increased and the lipid metabolism regulators (Apolipoprotein B (APOB) and Cholesteryl ester transfer protein (CETP)) were significantly decreased in patients with obesity. As for patients with COPD, the Activin A Receptor Type 2B (ACVR2B) and Transforming Growth Factor Beta 1 (TGFβ1), which were the key modulators of BMPR2 pathway, were significantly downregulated, as well as the obesity-related gene Apelin Receptor Early Endogenous Ligand (APELA). However, the BMP4 was remarkably upregulated. The identified 18 overlapping DEGs in obesity and COPD were referred to Supplementary Tables 2, 3. Of these genes, we found that the expression level of protein kinase C beta (PRKCB) was high in obesity but low in COPD. Expression of phosphoinositide-3-kinase regulatory subunit 2 (PIK3R2) and Reduced nicotinamide adenine dinucleotide phosphate (NADPH) quinone oxidoreductase 1 (NOQ1) genes was increased in obesity and COPD, respectively, compared with the controls (Figure 1B).
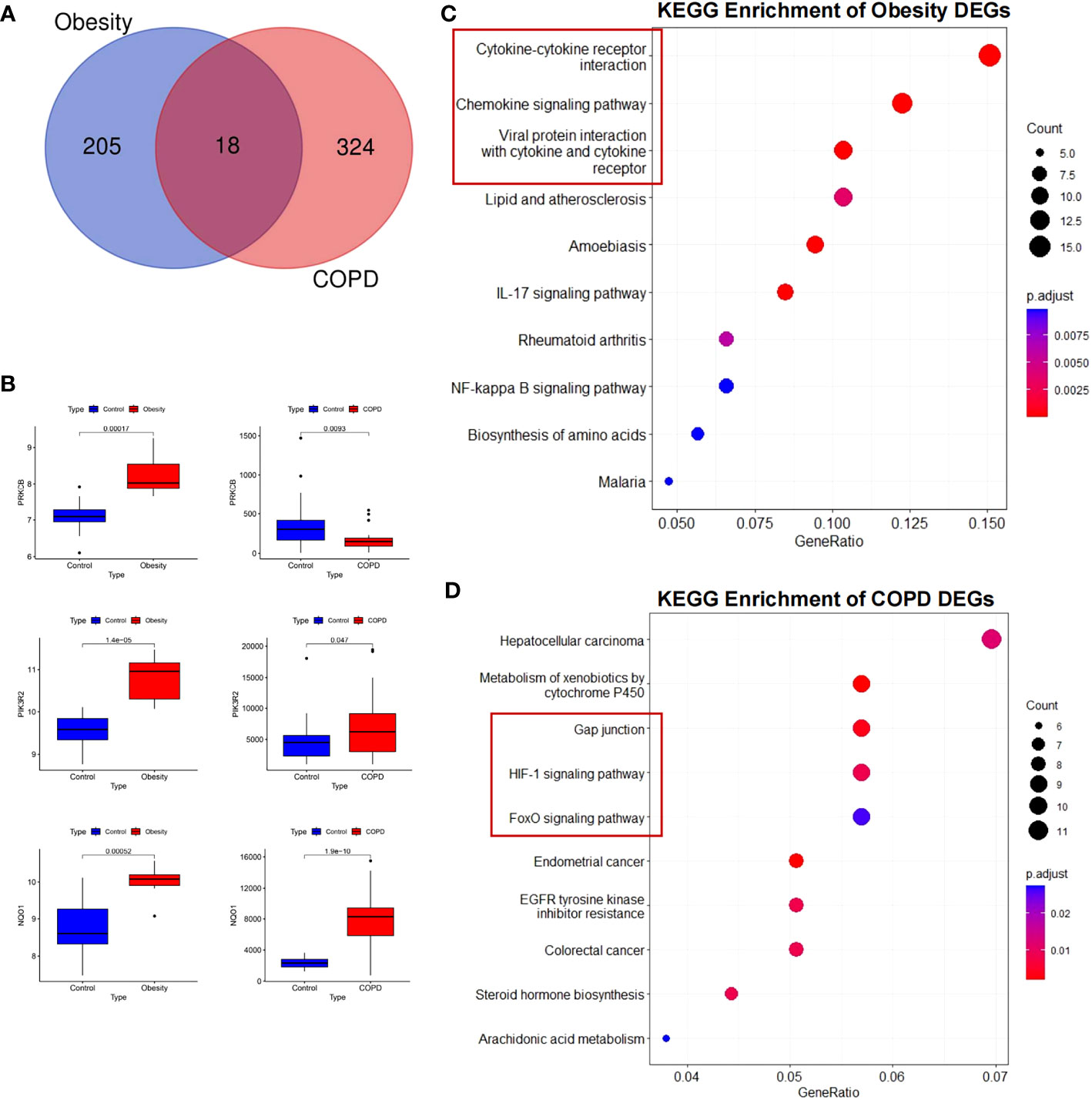
Figure 1 Differentially expressed genes and KEGG enrichment of obesity and COPD. (A) Differential genes in the three datasets. (B) Validation of differential expression of three crucial genes in GEO public datasets. Increased expression of PRKCB, PIK3R2, and NQO1 in obesity; increased expression of PIK3R2 and NQO1, decreased expression of PRKCB in COPD. (C) KEGG enrichment of obesity DEGs. (D) KEGG enrichment of COPD DEGs. KEGG, Kyoto Encyclopedia of Genes and Genomes; COPD, chronic obstructive pulmonary disease; PRKCB, protein kinase C beta; PIK3R2, phosphoinositide-3-kinase regulatory subunit 2; NOQ1, NADPH quinone oxidoreductase 1; DEGs, differentially expressed genes.
KEGG annotation analysis of DEGs in obesity revealed that the pathway mainly included cytokine-cytokine receptor interaction (P = 4.17E-05), viral protein interaction with cytokine and cytokine receptor (P = 9.32E-06), chemokine signaling pathway (P = 4.17E-05), and lipid and atherosclerosis (P = 0.0029) (Figure 1C). As for COPD, KEGG pathway analyses of DEGs were significantly enriched for gap junction (P = 0.003734), metabolism of xenobiotics by cytochrome P450 (P = 0.002103), Hypoxia-inducible factor 1-alpha (HIF-1) signaling pathway (P = 0.008497), and Forkhead box O (FoxO) signaling pathway (P =0.02568) (Figure 1D). Similarly, through GSEA, we confirmed that the cytokine and cytokine receptor genes were simultaneously upregulated in COPD and obesity but exhibited an opposite trend in the two pathological statuses (Supplementary Figures 1C, D).
To determine the interactions among the DEGs obtained above, we also generated PPI network by STRING analysis. It discovered that adipocytokines and proteases such as CXCL8, MMP9, and C-C Motif Chemokine Receptor 5 (CCR5) were hub genes in white adipose tissue of patients with obesity. Moreover, cell-junction proteins and growth factors such as Catenin beta-1 (CTNNB1), Epidermal growth factor (EGF), Epidermal growth factor receptor (EGFR), and Brain-derived neurotrophic factor (BDNF) were the hub genes in airway epithelial cells in COPD. Our results indicated that inflammation and injury repairment were the main and shared inherent patterns in obesity and COPD.
To describe the co-expression relationship of genes, WGCNA analysis was performed to identify the highly correlated gene modules closely related to obesity and COPD, respectively. The co-expression modules were showed by cluster dendrogram. Obesity and control groups revealed 19 gene modules, referred to leaf and branches in Figure 2A. The MEbrown was most significant correlated to obesity (correlation value, 0.84; P = 4E-06). The DEGs and hub gene were mainly distributed in MEorange (correlation value, −0.64; P = 0.002) and MEdarkturquoise (correlation value, 0.77; P = 8E-05) modules, and BMPR2 was distributed in MEmidnightblue module (correlation value, 0.44; P = 0.05) (Figure 2C). Fifteen gene modules could be enriched in the COPD and control groups (Figure 2B) among which MEtan showed the most significant correlation (correlation value, −0.51; P = 9E-05). The DEGs and Hub gene were mainly distributed in MEgreen (correlation value, −0.87; P = 2E-17), and BMPR2 was located in ME turquoise (correlation value, 0.095; P = 0.5) (Figure 2D).
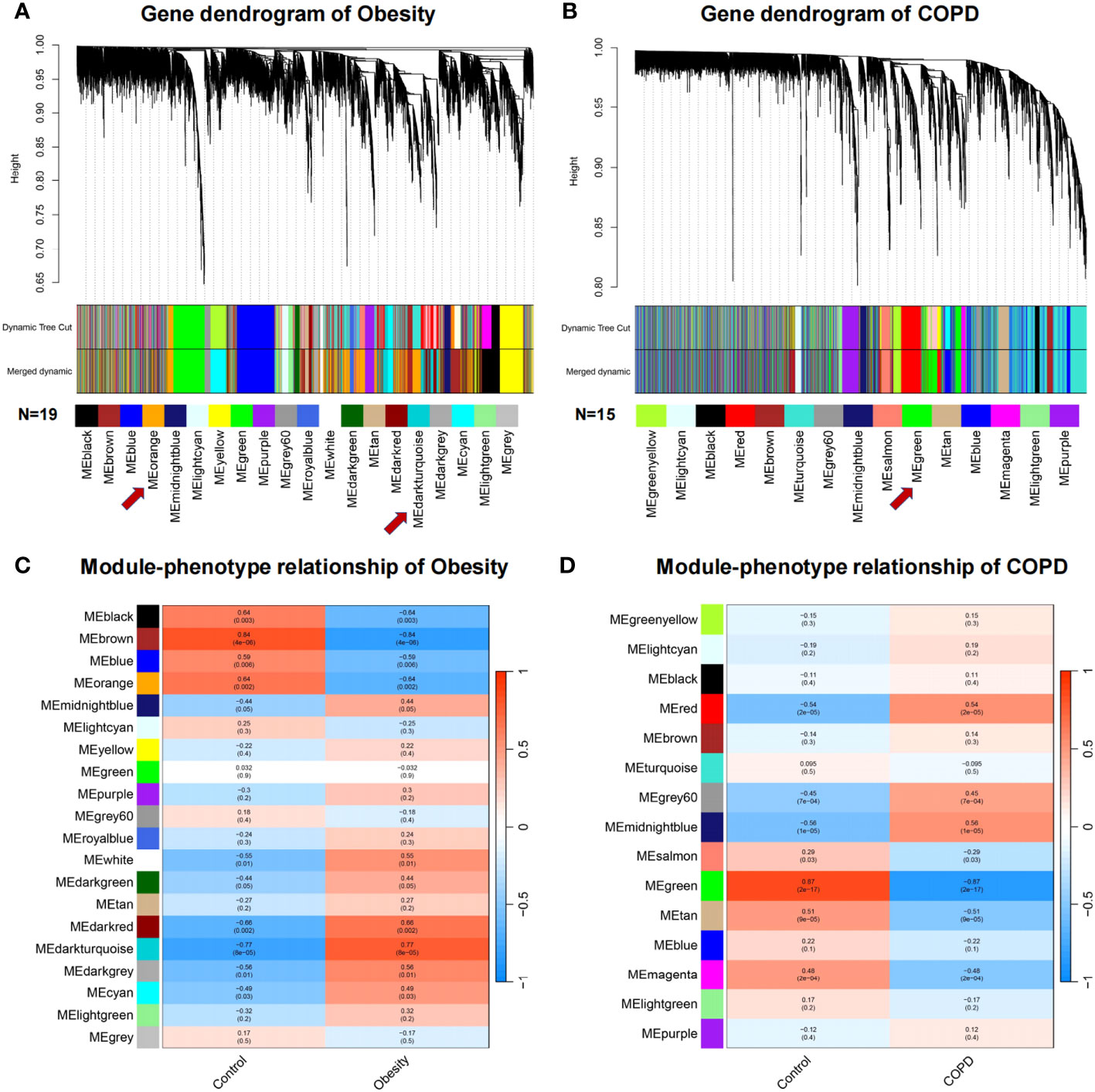
Figure 2 Weighted gene co-expression network analysis of obesity and COPD. (A, B) Network heatmap plot in the co-expression modules of obesity and COPD. Each leaf and branch on the tree represent a gene and a co-expression module. Each color below represents a co-expression gene module. (C, D) Heatmap between the correlation between modules and obesity, modules and COPD, respectively. Each cell contained the correlation coefficient and corresponding P-value. COPD, chronic obstructive pulmonary disease.
BMPR2 is pivotal in maintaining pulmonary vascular homeostasis and the development of PAH, and BMPs are its ligands. As one of the 18 overlapping DEGs (shown in Supplementary Tables 2, 3) in obesity and COPD, PRKCB (encoded PKCβprotein) had the PPI with BMPR2. Secreted Phosphoprotein 1 (SPP1) (encoded osteopontin) also had interactions with BMPs. In addition, from the results of PPI network of obesity and COPD (Supplementary Figures 2A, B), the hub genes in COPD-PPI had close relationship with BMPR2. Therefore, BMPR2 might play a vital role in COPD-related lung injury and emphysema.
We demonstrated that FABP4-Cre-BMPR2fl/fl (CKO) mice presented with spontaneous lung inflammation and significantly increased lung injury score, in comparison with WT mice (Figures 3A, B). Peripheral emphysema was also observed in CKO mice, with elevated mean linear intercept in the lung periphery (Figures 3A, C). In addition, Figures 3D–F showed that walls of small vessels became moderately thickened, along with the hypertrophy of pulmonary artery smooth muscle cells (PASMCs). As for inflammatory cells, we observed that CD68-marked macrophages and MPO-marked neutrophils were evidently upregulated in the alveolus of CKO mice (Figures 3G, H). Interestingly, lung inflammation, emphysema, and vascular remodeling are also pathological traits of COPD. We hypothesized that exacerbated inflammation and small artery remodeling synergistically resulted in right heart dysfunction, yet mice under 3 weeks old lacked mature compensatory mechanism and were prone to death.
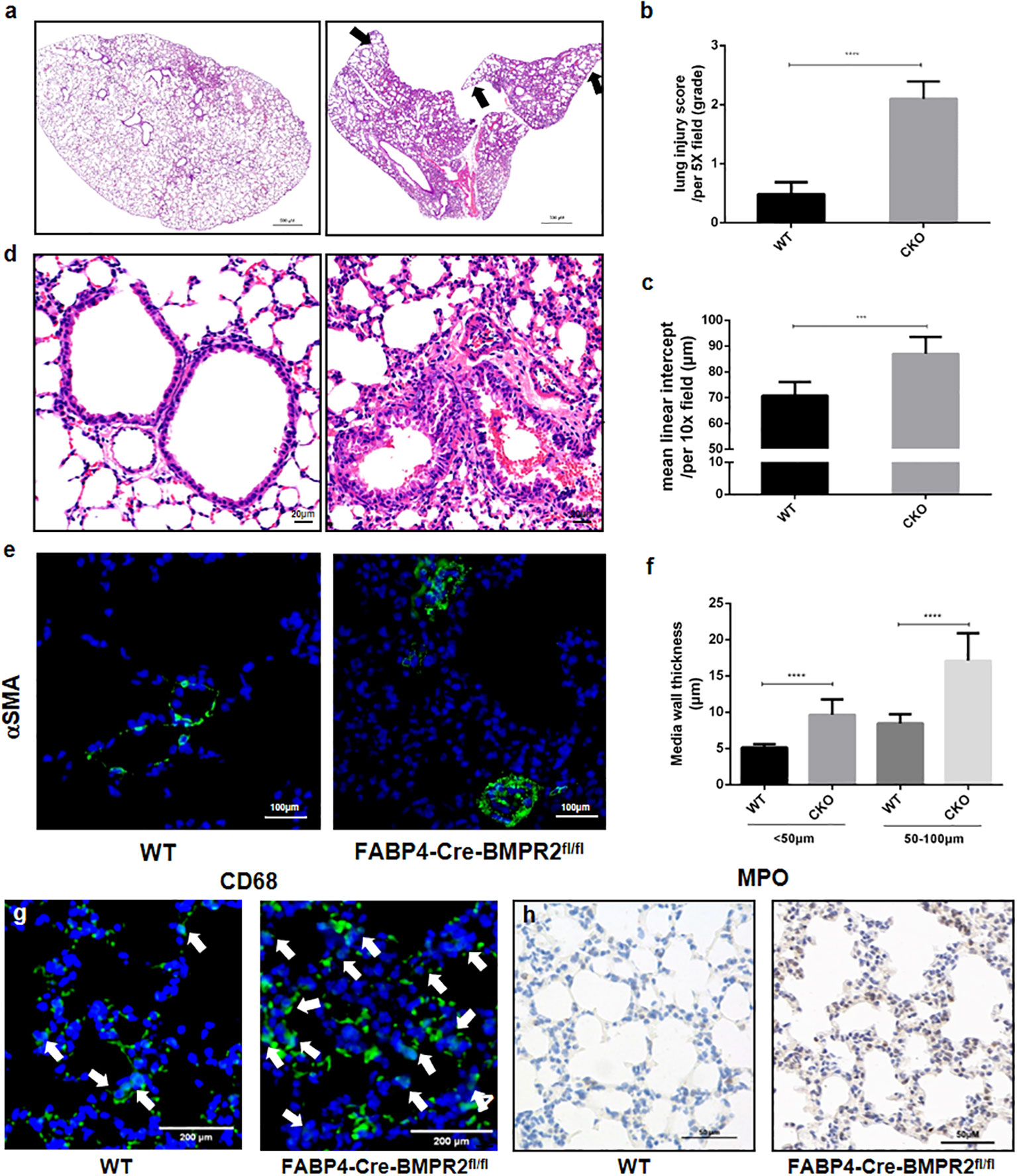
Figure 3 FABP4-Cre-BMPR2fl/fl mice were susceptible to spontaneous lung inflammation, emphysema and vascular remodeling. (A) FABP4-Cre-BMPR2fl/fl (CKO) mice had excessive inflammation in the whole lung and peripheral emphysema (black arrows, at 1x magnification). (B, C) Lung injury score and mean linear intercept (MLI) were calculated by the mean of 3-4 fields per slice and 5-6 slices per mouse. (D) CKO mice showed thicken vascular walls (at 20x magnification). (E) Immunofluorescent stain of α-SMA in the small lung arteries. (F) Media wall thickness of small vessels with different diameters was calculated by the mean of 2-3 fields per slice and 5-7 slices per mouse. (G, H) Infiltration of macrophages (CD68) and neutrophils (MPO) in the alveolus. ***P<0.001; ****P<0.0001. FABP4, fatty acid–binding protein 4; BMPR2, bone morphogenetic protein type II receptor; WT, wild type; CKO, conditional knockout; MPO, myeloperoxidase.
To investigate further mechanisms, we compared the difference of proliferation and apoptosis of lung’ cells between the WT and CKO groups, by performing immunological stain of proliferating cell nuclear antigen (PCNA) and TUNEL stain for the lung slices. PCNA was mainly expressed in the airways and did not differ in two groups (Figure 4A). Slightly increased number of apoptotic cells was observed in the CKO group (Figure 4B). Interestingly, BMPR2 and its targeted protein Id2 were declined in small arteries of CKO mice but not in small airways (Figures 4C, D). It demonstrated that the deficiency of BMPR2 in adipose tissue could influence its expression in other organs, especially in pulmonary vasculature. In addition, pro-contraction endothelin was upregulated in the small airways and arterioles of CKO mice (Figure 4E), but endothelin-converting enzyme 1 was similar in two groups (data not shown). Consistent with inflammatory cells, MMP2 was also increased in the alveolus of CKO mice (Figure 4F).
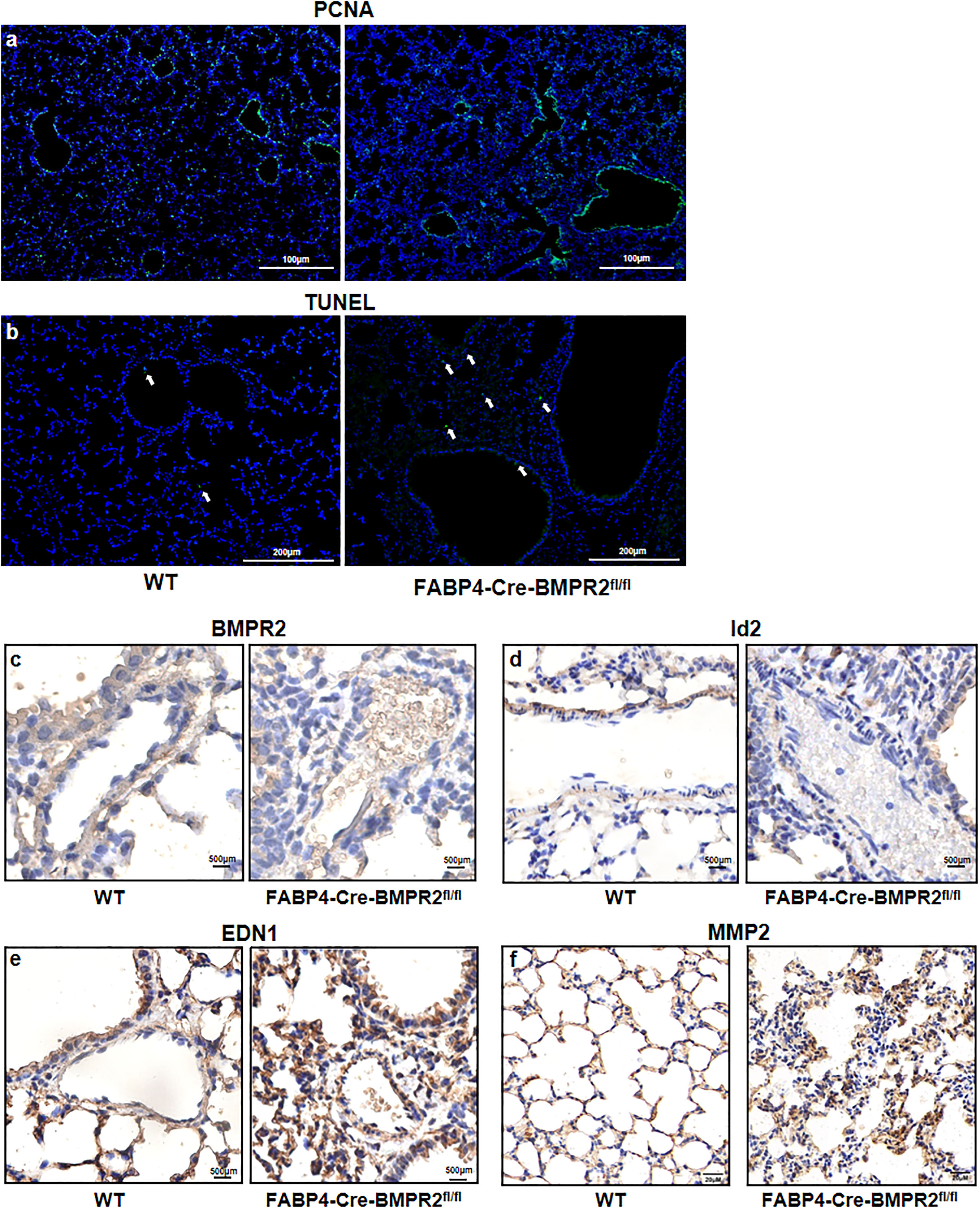
Figure 4 Preliminary exploration of the mechanisms of pathological changes in the lungs of FABP4-Cre-BMPR2fl/fl mice. (A) Immunofluorescent stain of PCNA was not evident in the small lung arteries and not different in the whole lung between two groups. (B) TUNEL stain showed slightly increased numbers of apoptotic cells (white arrows) in the lungs of CKO mice. (C, D) Immunohistochemistry of BMPR2 and Id2 in the lung arteries. (E) Immunohistochemistry of EDN1 in the small airways and arterioles. (F) Immunohistochemistry of MMP2 in the alveolus. PCNA, proliferating cell nuclear antigen; TUNEL, terminal deoxynucleoitidyl transferase–mediated nick end labeling; FABP4, fatty acid–binding protein 4; BMPR2, bone morphogenetic protein type II receptor; Id2, inhibitor of DNA binding 2; EDN1, endothelin; MMP2, matrix metalloproteinase 2.
To preliminarily explore the roles of BMPR2-defiecient adipocytes, we re-analyzed the RNA sequence data of adipocytes in WT and CKO mice, which was uploaded by Qian et al. (Sequence Read Archive (SRA) accession: PRJNA611934) (23). Adipocytes of each group were pooled from four individual mice’s inguinal adipose tissue. We compared the differential expression of multiple adipocytokines and listed those with fragments per kilobase of exon model per million mapped fragments >1 and fold change >1.5 (Figure 5A). We observed the dramatically decreased apelin and slightly increased adipsin in the CKO group (Figure 5A). Moreover, we searched the top 50 BMPR2-associated proteins and FABP4-associated proteins and identified as the sole conjunct gene (Figure 5B). Thus, we postulated that apelin, adipsin, and growth differentiation factor 5 (GDF5) might participate in the pathological process that BMPR2-deficient adipocytes influenced lung structure.
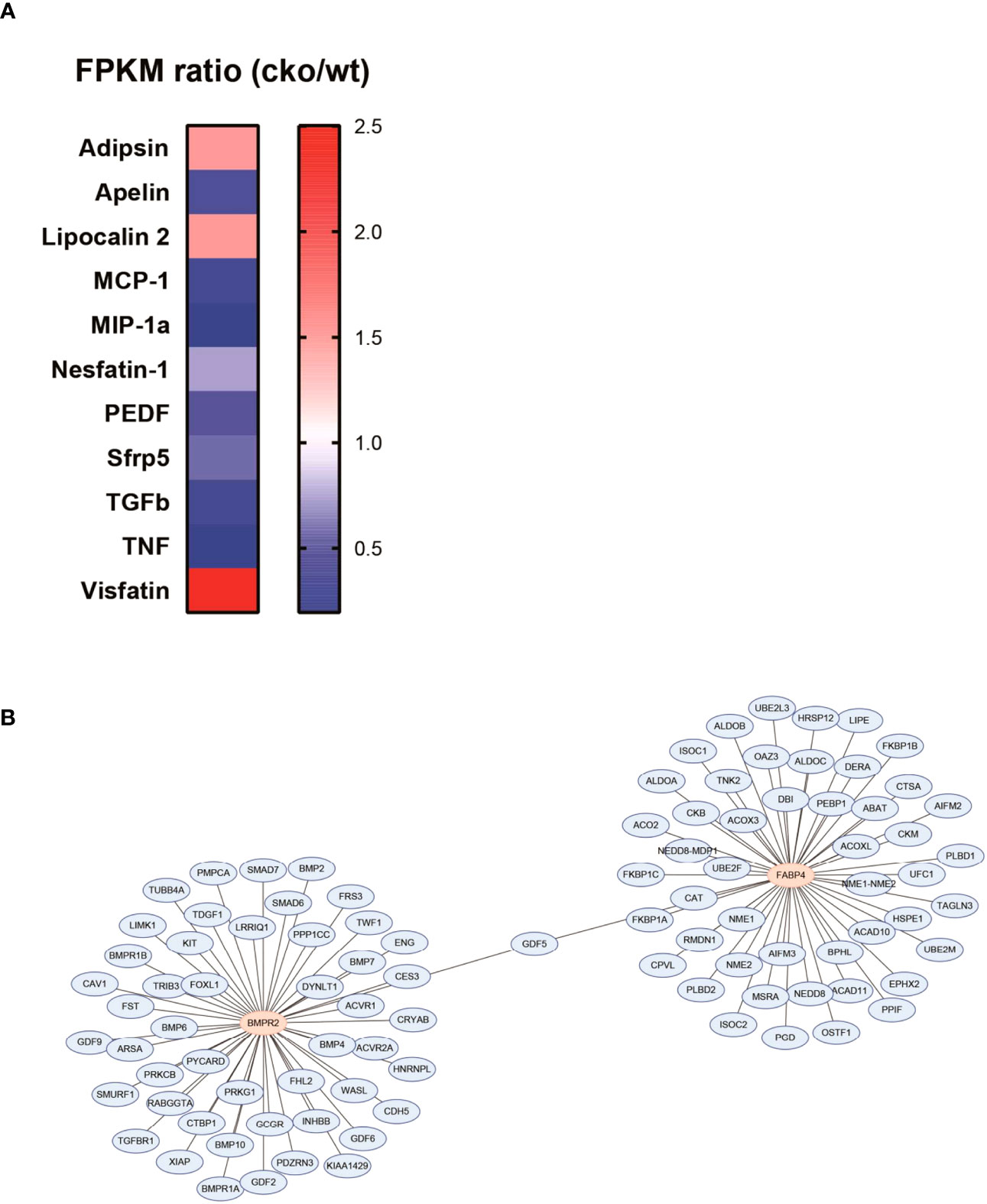
Figure 5 Adipocytokines involved the pathogenesis of lung injury in BMPR2-deficient adipocytes. (A) Differently expressed adipocytokines in the adipocytes were calculated by the FPKM ratio of CKO and WT group. (B) PPI network of BMPR2-related proteins and FABP4-related proteins. FPKM, fragments per kilobase of exon model per million mapped fragments; WT, wild type; CKO, conditional knockout; PPI, protein–protein interaction; FABP4, fatty acid–binding protein 4; BMPR2, bone morphogenetic protein type II receptor.
In our study, we discovered that patients with obesity with COPD had improved prognosis and lung function. In addition, several potential inflammatory hub genes (CXCL8, MMP9, and CCR5) and adipocytokine hub genes (PRKCB, PIK3R2, NOQ1, BMP4, ACVR2B, and TGFβ1) were identified. Further KEGG analysis, GSEA, PPI network, and WGCNA showed that inflammation and injury repairment were the main and shared inherent patterns in obesity and COPD. We also found the adipocyte-specific BMPR2 knockout mice to spontaneously develop lung injury and emphysema, in which adipsin and GDF5 might be involved in the pathogenesis. Our study indicated that adipocytes dysfunction might promote lung injury, which could provide therapeutic targets and biomarkers for patients with COPD.
Our cohort analysis of patients with COPD showed that the group with BMI ≥24 had decreased percentage of re-AE in 3 months and 1 year, indicating a favorable prognosis. Similar findings were also shown in the previous studies (32–34). BMI is the most frequently used anthropometric tool to measure the fatness. Although it is convenient to access, BMI is a very crude measurement of obesity (35). It could not distinguish between fat mass and free-fat mass (36). BMI is hard to identify the fat distribution like the abdominal fat or gluteo-femoral fat (37). Further studies are needed to explore the relationship between different body components and prognosis of COPD.
Table 1 demonstrates the inconsistent effects of obesity on COPD. However, multiple confounding factors between COPD and obesity influence the prognosis of patients. For example, patients with overweight and obesity were more likely to be misdiagnosed with COPD and be prescribed with inhaled medications due to dyspnea (38). In addition, patients with obesity often had complications that led to dyspnea and hypoxemia, such as functional restriction, congenital heart failure, and obstructive sleep apnea (39).
In recent years, bioinformatic tools have been widely used for identifying DEGs and novel markers in COPD (40–42). In our bioinformatic analysis, we found several DEGs including inflammatory factors (CXCL8, CXCL2, and MMP9) and lipid metabolism regulators (APOB and CETP), which were also reported in some previous studies of COPD (43–45). Similarly, some DEGs of the COPD dataset, like the regulator of BMPR2 pathway and APELA, are also the obesity-related genes. NOQ1, one of antioxidant response elements (46), was significantly increased in both obesity and COPD, which is consistent with the previous results (41). We also observed an upregulation of PIK3R2 in COPD and obesity. Inhibition of PIK3R2 could increase the levels of tight junction protein and protect the function of epithelial cells (47). The expression levels of PRKCB differed in obese and COPD datasets. PRKCB plays multiple roles in regulating cell survival and apoptosis (48), indicating that it might determine different cellular fate in adipocytes and bronchial epithelial cells. GSEA and KEGG analysis of obesity showed an enhancement of cytokine and chemokine pathways, whereas the results of COPD showed an upregulation of repair-related genes such as cell junctions, HIF1α, and FOXO3. On the basis of the above results, we speculate white adipose tissue to be a potential endocrine organ that influences lung inflammation and injury repair, by secreting systemic cytokines and chemokines.
Mutation and deficiency of BMPR2 leads to an obvious perivascular inflammation and muscularization in pulmonary hypertension (49). To date, there are limited studies on BMPR2 and COPD. BMPR2 is significantly decreased in lung tissue of patients with COPD (50). Moreover, BMPR2 mutation could increase the susceptibility of COPD (51). Hence, deficiency of BMPR2 may aggravate the progression of COPD. Our study utilized adipocyte-specific BMPR2 knockout mice and found that CKO mice presented with a COPD phenotype, with emphysema, lung injury, and arteriole remodeling. On the basis of the previous study by Qian et al., it is possibly due to elevated level of serum TNFα, leading to increased infiltration of inflammatory cells into lung and alveolar destruction (23). Our results do not show the disbalance of proliferation and apoptosis to be a major cause of lung structural damage.
Adipocytokines are bioactive mediators secreted by adipocytes, and we screened out three potential adipocytokines in our study. Apelin was decreased in the white adipose tissues of the CKO group. Consistently, APELA was downregulated in small airways of patients with COPD. Similar results have been reported to decrease apelin in patients with pulmonary artery hypertension secondary to COPD (52). Apelin plays protective role in obesity, by regulating the inflammation and oxidative stress (53). Moreover, adipsin was increased in white adipose tissues of the CKO group. It was regarded as a novel serum biomarker of COPD-associated pulmonary hypertension (54). Some adipocytokines like adipsin were similarly elevated in patients with chronic bronchitis (55). GDF5 was predicted by PPI network as a core protein between BMPR2- and FABP4-interating network. GDF5 was also known as BMP14, belonging to family of BMPs. Its function is still unknown in COPD, but administration of GDF5 could promote cartilage repair by inhibition of inflammation in osteoarthritis models (56). Moreover, FABP4-GDF5 transgenic mice showed increased sensitivity to insulin on a high-diet food, probably by promoting thermogenesis of white adipose tissue (57).
The number of clinical cases in our study being relatively low is a major limitation. Further studies on body composition and nutrition of COPD are needed. Moreover, the inclusion criterion was hospitalized patients with AECOPD. Because Shanghai Zhongshan Hospital is a grade-A tertiary hospital, participants might have several comorbidities, such as hypertension, diabetes, and coronary heart disease. Larger, multi-center studies will be conducted in the future. Because of premature death of FABP4-Cre-BMPR2fl/fl mice after weaning, it was difficult to measure the pulmonary function parameters. We plan to breed FABP4-CreERT2-BMPR2fl/fl mice and administered tamoxifen via intraperitoneal injection or gavage in adulthood to induce the knockout of BMPR2 in adipocytes. We are also planning to confirm whether TNFα or apelin is the main culprit of lung injury in our future research. Co-culture of mouse pulmonary endothelial cells and BMPR2-knockdown adipocytes will be performed and supplemented with anti–TNFα-neutralizing antibody or apelin receptor agonist. Mouse adipocytes were differentiated from 3T3-L1 cell line. More experiments are needed to be designed and conducted to further validate the mechanism of protective role of GDF5 in obesity and COPD.
In summary, inflammation and abnormal repair might be potential mechanisms of the pathological association between obesity and COPD. Moreover, this study innovatively supplemented a pathogenic link between adipocyte dysfunction and lung injury, indicating adipocytes potentially to be a new class of key cells in the development of COPD. This provides new insight into the pathological mechanism and a promising therapy of COPD.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The studies involving humans were approved by Institutional Ethics Committee of Shanghai Zhongshan Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by Institutional Ethics Committee of Shanghai Zhongshan Hospital. The study was conducted in accordance with the local legislation and institutional requirements. The clinical study of patients with AECOPD has been approved by Institutional Ethics Committee of Shanghai Zhongshan Hospital (No. B2015-119R). Informed written consent for clinical data collection and anonymous publication has been acquired from each patient on admission.
Conception and design: S-JZ and W-PH; experiment: S-JZ, B-FH, and W-PH; analysis and interpretation: X-ZQ and JZho; clinical study design: W-PH and JZha; drafting the manuscript: S-JZ, SS, and W-PH. All authors read and approved the final manuscript.
This study was funded by the National Natural ScienceFoundation of China (grant number 82270039 and 82300047).
We sincerely acknowledge Dr. Shu-wen QIAN for generously providing the paraffin-embedded lung tissues of WT and FABP4-Cre-BMPR2fl/fl mice. We appreciate Dr. Jie-yu GUO for converting raw RNA-sequencing data into processible FPKM data. We thank Dr. Bhavana Rajbanshi for her kind suggestions on our manuscript.
Author SS was employed by the company Om Asha Hospital Pvt. Ltd.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1204744/full#supplementary-material
1. Fang X, Wang X, Bai C. COPD in China: the burden and importance of proper management. Chest (2011) 139(4):920–9. doi: 10.1378/chest.10-1393
2. Adeloye D, Chua S, Lee C, Basquill C, Papana A, Theodoratou E, et al. Global and regional estimates of COPD prevalence: Systematic review and meta-analysis. J Global Health (2015) 5(2):020415. doi: 10.7189/jogh.05.020415
3. Rosenberg SR, Kalhan R, Mannino DM. Epidemiology of chronic obstructive pulmonary disease: prevalence, morbidity, mortality, and risk factors. Semin Respir Crit Care Med (2015) 36(4):457–69. doi: 10.1055/s-0035-1555607
4. Mannino DM, Buist AS. Global burden of COPD: risk factors, prevalence, and future trends. Lancet (London England) (2007) 370(9589):765–73. doi: 10.1016/S0140-6736(07)61380-4
5. Di Cesare M, Bentham J, Stevens GA, Zhou B, Danaei G, Lu Y, et al Trends in adult body-mass index in 200 countries from 1975 to 2014: a pooled analysis of 1698 population-based measurement studies with 19·2 million participants. Lancet (London England) (2016) 387(10026):1377–96. doi: 10.1016/S0140-6736(16)30054-X
6. Huxley R, Mendis S, Zheleznyakov E, Reddy S, Chan J. Body mass index, waist circumference and waist:hip ratio as predictors of cardiovascular risk–a review of the literature. Eur J Clin Nutr (2010) 64(1):16–22. doi: 10.1038/ejcn.2009.68
7. Kulkarni K, Karssiens T, Kumar V, Pandit H. Obesity and osteoarthritis. Maturitas (2016) 89:22–8. doi: 10.1016/j.maturitas.2016.04.006
8. Gray N, Picone G, Sloan F, Yashkin A. Relation between BMI and diabetes mellitus and its complications among US older adults. South Med J (2015) 108(1):29–36. doi: 10.14423/SMJ.0000000000000214
9. Frybova B, Drabek J, Lochmannova J, Douda L, Hlava S, Zemkova D, et al. Cholelithiasis and choledocholithiasis in children; risk factors for development. PloS One (2018) 13(5):e0196475. doi: 10.1371/journal.pone.0196475
10. Zammit C, Liddicoat H, Moonsie I, Makker H. Obesity and respiratory diseases. Int J Gen Med (2010) 3:335–43. doi: 10.2147/IJGM.S11926
11. Li J, Zhu L, Wei Y, Lv J, Guo Y, Bian Z, et al. Association between adiposity measures and COPD risk in Chinese adults. Eur Respir J (2020) 55(4). doi: 10.1183/13993003.01899-2019
12. Spelta F, Fratta Pasini AM, Cazzoletti L, Ferrari M. Body weight and mortality in COPD: focus on the obesity paradox. Eating weight Disord EWD (2018) 23(1):15–22. doi: 10.1007/s40519-017-0456-z
13. Guenette JA, Jensen D, O'Donnell DE. Respiratory function and the obesity paradox. Curr Opin Clin Nutr Metab Care (2010) 13(6):618–24. doi: 10.1097/MCO.0b013e32833e3453
14. Kurosaki H, Ishii T, Motohashi N, Motegi T, Yamada K, Kudoh S, et al. Extent of emphysema on HRCT affects loss of fat-free mass and fat mass in COPD. Internal Med (Tokyo Japan) (2009) 48(1):41–8. doi: 10.2169/internalmedicine.48.1102
15. Divo MJ. The adipose tissue and lung health: like many things in life, the extremes are not good. Eur Respir J (2020) 55(4). doi: 10.1183/13993003.00107-2020
16. Bellou V, Belbasis L, Konstantinidis AK, Tzoulaki I, Evangelou E. Prognostic models for outcome prediction in patients with chronic obstructive pulmonary disease: systematic review and critical appraisal. BMJ (Clinical Res ed) (2019) 367:l5358. doi: 10.1136/bmj.l5358
17. Cao C, Wang R, Wang J, Bunjhoo H, Xu Y, Xiong W. Body mass index and mortality in chronic obstructive pulmonary disease: a meta-analysis. PloS One (2012) 7(8):e43892. doi: 10.1371/journal.pone.0043892
18. Dubois-Deruy E, Rémy G, Alard J, Kervoaze G, Chwastyniak M, Baron M, et al. Modelling the impact of chronic cigarette smoke exposure in obese mice: metabolic, pulmonary, intestinal, and cardiac issues. Nutrients (2020) 12(3). doi: 10.3390/nu12030827
19. Maia LA, Cruz FF, de Oliveira MV, Samary CS, Fernandes MVS, Trivelin SAA, et al. Effects of obesity on pulmonary inflammation and remodeling in experimental moderate acute lung injury. Front Immunol (2019) 10:1215. doi: 10.3389/fimmu.2019.01215
20. Wu SW, Peng CK, Wu SY, Wang Y, Yang SS, Tang SE, et al. Obesity attenuates ventilator-induced lung injury by modulating the STAT3-SOCS3 pathway. Front Immunol (2021) 12:720844. doi: 10.3389/fimmu.2021.720844
21. Medoff BD. Fat, fire and muscle–the role of adiponectin in pulmonary vascular inflammation and remodeling. Pulmonary Pharmacol Ther (2013) 26(4):420–6. doi: 10.1016/j.pupt.2012.06.006
22. Fasshauer M, Blüher M. Adipokines in health and disease. Trends Pharmacol Sci (2015) 36(7):461–70. doi: 10.1016/j.tips.2015.04.014
23. Qian S, Pan J, Su Y, Tang Y, Wang Y, Zou Y, et al. BMPR2 promotes fatty acid oxidation and protects white adipocytes from cell death in mice. Commun Biol (2020) 3(1):200. doi: 10.1038/s42003-020-0928-y
24. Raman T, O'Connor TP, Hackett NR, Wang W, Harvey BG, Attiyeh MA, et al. Quality control in microarray assessment of gene expression in human airway epithelium. BMC Genomics (2009) 10:493. doi: 10.1186/1471-2164-10-493
25. Walker JM, Garcet S, Aleman JO, Mason CE, Danko D, Butler D, et al. Obesity and ethnicity alter gene expression in skin. Sci Rep (2020) 10(1):14079. doi: 10.1038/s41598-020-70244-2
26. Hu WP, Lhamo T, Zhang FY, Hang JQ, Zuo YH, Hua JL, et al. Predictors of acute cardiovascular events following acute exacerbation period for patients with COPD: a nested case-control study. BMC Cardiovasc Disord (2020) 20(1):518. doi: 10.1186/s12872-020-01803-8
27. Hu WP, Lhamo T, Liu D, Hang JQ, Zhang FY, Zuo YH, et al. Development of a nomogram to predict the risk of 30-day re-exacerbation for patients hospitalized for acute exacerbation of chronic obstructive pulmonary disease. Copd (2019) 16(2):160–7. doi: 10.1080/15412555.2019.1606187
28. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf (2003) 4:2. doi: 10.1186/1471-2105-4-2
29. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinf (2008) 9:559. doi: 10.1186/1471-2105-9-559
30. Beppu H, Lei H, Bloch KD, Li E. Generation of a floxed allele of the mouse BMP type II receptor gene. Genesis (New York NY 2000) (2005) 41(3):133–7. doi: 10.1002/gene.20099
31. Jiang T, Hu W, Zhang S, Ren C, Lin S, Zhou Z, et al. Fibroblast growth factor 10 attenuates chronic obstructive pulmonary disease by protecting against glycocalyx impairment and endothelial apoptosis. Respir Res (2022) 23(1):269. doi: 10.1186/s12931-022-02193-5
32. DeLapp DA, Glick C, Furmanek S, Ramirez JA, Cavallazzi R. Patients with obesity have better long-term outcomes after hospitalization for COPD exacerbation. Copd (2020) 17(4):373–7. doi: 10.1080/15412555.2020.1781805
33. Goto T, Hirayama A, Faridi MK, Camargo CA Jr., Hasegawa K. Obesity and severity of acute exacerbation of chronic obstructive pulmonary disease. Ann Am Thorac Society (2018) 15(2):184–91. doi: 10.1513/AnnalsATS.201706-485OC
34. Wei YF, Tsai YH, Wang CC, Kuo PH. Impact of overweight and obesity on acute exacerbations of COPD - subgroup analysis of the Taiwan Obstructive Lung Disease cohort. Int J Chronic Obstructive Pulmonary Disease (2017) 12:2723–9. doi: 10.2147/COPD.S138571
35. Toplak H, Leitner DR, Harreiter J, Hoppichler F, Wascher TC, Schindler K, et al. ["Diabesity"-Obesity and type 2 diabetes (Update 2019)]. Wiener klinische Wochenschrift (2019) 131(Suppl 1):71–6. doi: 10.1007/s00508-018-1418-9
36. Merchant RA, Seetharaman S, Au L, Wong MWK, Wong BLL, Tan LF, et al. Relationship of fat mass index and fat free mass index with body mass index and association with function, cognition and sarcopenia in pre-frail older adults. Front Endocrinol (2021) 12:765415. doi: 10.3389/fendo.2021.765415
37. Bastien M, Poirier P, Lemieux I, Després JP. Overview of epidemiology and contribution of obesity to cardiovascular disease. Prog Cardiovasc Diseases (2014) 56(4):369–81. doi: 10.1016/j.pcad.2013.10.016
38. Collins BF, Ramenofsky D, Au DH, Ma J, Uman JE, Feemster LC. The association of weight with the detection of airflow obstruction and inhaled treatment among patients with a clinical diagnosis of COPD. Chest (2014) 146(6):1513–20. doi: 10.1378/chest.13-2759
39. Vecchié A, Dallegri F, Carbone F, Bonaventura A, Liberale L, Portincasa P, et al. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur J Internal Med (2018) 48:6–17. doi: 10.1016/j.ejim.2017.10.020
40. Regan EA, Hersh CP, Castaldi PJ, DeMeo DL, Silverman EK, Crapo JD, et al. Omics and the search for blood biomarkers in chronic obstructive pulmonary disease. Insights COPDGene Am J Respir Cell Mol Biol (2019) 61(2):143–9. doi: 10.1165/rcmb.2018-0245PS
41. Lin Z, Xu Y, Guan L, Qin L, Ding J, Zhang Q, et al. Seven ferroptosis-specific expressed genes are considered as potential biomarkers for the diagnosis and treatment of cigarette smoke-induced chronic obstructive pulmonary disease. Ann Trans Med (2022) 10(6):331. doi: 10.21037/atm-22-1009
42. Zhu M, Ye M, Wang J, Ye L, Jin M. Construction of potential miRNA-mRNA regulatory network in COPD plasma by bioinformatics analysis. Int J chronic obstructive pulmonary disease (2020) 15:2135–45. doi: 10.2147/COPD.S255262
43. Ha H, Debnath B, Neamati N. Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics (2017) 7(6):1543–88. doi: 10.7150/thno.15625
44. Ghosh S, Rihan M, Ahmed S, Pande AH, Sharma SS. Immunomodulatory potential of apolipoproteins and their mimetic peptides in asthma: Current perspective. Respir Med (2022) 204:107007. doi: 10.1016/j.rmed.2022.107007
45. Wells JM, Gaggar A, Blalock JE. MMP generated matrikines. Matrix Biol J Int Soc Matrix Biol (2015) 44-46:122–9. doi: 10.1016/j.matbio.2015.01.016
46. Cen M, Ouyang W, Zhang W, Yang L, Lin X, Dai M, et al. MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol (2021) 41:101936. doi: 10.1016/j.redox.2021.101936
47. Zhou Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Chang E, et al. Exosomes from endothelial progenitor cells improve outcomes of the lipopolysaccharide-induced acute lung injury. Crit Care (London England) (2019) 23(1):44. doi: 10.1186/s13054-019-2339-3
48. Bononi A, Agnoletto C, De Marchi E, Marchi S, Patergnani S, Bonora M, et al. Protein kinases and phosphatases in the control of cell fate. Enzyme Res (2011) 2011:329098. doi: 10.4061/2011/329098
49. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J (2022) 43(38):3618–731. doi: 10.1093/eurheartj/ehac237
50. Wang J, Zhang C, Zhang Z, Zheng Z, Sun D, Yang Q, et al. A functional variant rs6435156C > T in BMPR2 is associated with increased risk of chronic obstructive pulmonary disease (COPD) in Southern Chinese population. EBioMedicine (2016) 5:167–74. doi: 10.1016/j.ebiom.2016.02.004
51. Llinàs L, Peinado VI, Ramon Goñi J, Rabinovich R, Pizarro S, Rodriguez-Roisin R, et al. Similar gene expression profiles in smokers and patients with moderate COPD. Pulmonary Pharmacol Ther (2011) 24(1):32–41. doi: 10.1016/j.pupt.2010.10.010
52. Hu Y, Zong Y, Jin L, Zou J, Wang Z. Reduced Apela/APJ system expression in patients with pulmonary artery hypertension secondary to chronic obstructive pulmonary disease. Heart Lung J Crit Care (2023) 59:8–15. doi: 10.1016/j.hrtlng.2023.01.008
53. Chwalba A, Machura E, Ziora K, Ziora D. The role of adipokines in the pathogenesis and course of selected respiratory diseases. Endokrynologia Polska (2019) 70(6):504–10. doi: 10.5603/EP.a2019.0051
54. Zhang Y, Lin P, Hong C, Jiang Q, Xing Y, Tang X, et al. Serum cytokine profiles in patients with chronic obstructive pulmonary disease associated pulmonary hypertension identified using protein array. Cytokine (2018) 111:342–9. doi: 10.1016/j.cyto.2018.09.005
55. Khudiakova AD, Polonskaya YV, Shramko VS, Shcherbakova LV, Striukova EV, Kashtanova EV, et al. Blood adipokines/cytokines in young people with chronic bronchitis and abdominal obesity. Biomolecules (2022) 12(10). doi: 10.3390/biom12101502
56. Takahata Y, Hagino H, Kimura A, Urushizaki M, Yamamoto S, Wakamori K, et al. Regulatory mechanisms of prg4 and gdf5 expression in articular cartilage and functions in osteoarthritis. Int J Mol Sci (2022) 23(9). doi: 10.3390/ijms23094672
Keywords: adipocyte dysfunction, Chronic Obstructive Pulmonary Disease, lung inflammation, aberrant repair, adipocytokines
Citation: Zhang S-j, Qin X-z, Zhou J, He B-f, Shrestha S, Zhang J and Hu W-p (2023) Adipocyte dysfunction promotes lung inflammation and aberrant repair: a potential target of COPD. Front. Endocrinol. 14:1204744. doi: 10.3389/fendo.2023.1204744
Received: 12 April 2023; Accepted: 18 September 2023;
Published: 10 October 2023.
Edited by:
Zhu Cuiling, Tongji University, ChinaReviewed by:
Dong Hang, Nanjing Medical University, ChinaCopyright © 2023 Zhang, Qin, Zhou, He, Shrestha, Zhang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei-ping Hu, d3BodUBmb3htYWlsLmNvbQ==; Jing Zhang, emhhbmcuamluZ0B6cy1ob3NwaXRhbC5zaC5jbg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.