 Elaine M. Duncan
Elaine M. Duncan Luca Vita
Luca Vita Bethany Dibnah
Bethany Dibnah Brian D. Hudson
Brian D. Hudson- Centre for Translational Pharmacology, University of Glasgow, Glasgow, United Kingdom
Metabolic disorders including obesity, diabetes and non-alcoholic steatohepatitis are a group of conditions characterised by chronic low-grade inflammation of metabolic tissues. There is now a growing appreciation that various metabolites released from adipose tissue serve as key signalling mediators, influencing this interaction with inflammation. G protein-coupled receptors (GPCRs) are the largest family of signal transduction proteins and most historically successful drug targets. The signalling pathways for several key adipose metabolites are mediated through GPCRs expressed both on the adipocytes themselves and on infiltrating macrophages. These include three main groups of GPCRs: the FFA4 receptor, which is activated by long chain free fatty acids; the HCA2 and HCA3 receptors, activated by hydroxy carboxylic acids; and the succinate receptor. Understanding the roles these metabolites and their receptors play in metabolic-immune interactions is critical to establishing how these GPCRs may be exploited for the treatment of metabolic disorders.
Introduction
Metabolic disorders, including metabolic syndrome, diabetes and non-alcoholic steatohepatitis (NASH) are a major global health and economic burden, with diabetes alone affecting an estimated 537 million adults globally (1). These conditions are characterised by chronic inflammation infiltrating metabolic tissues, resulting in a disruption to metabolic homeostasis and downstream complications. As obesity is a pertinent risk factor for many metabolic disorders, understanding the pathophysiology of these conditions is of utmost importance given its increasing global prevalence.
The relationship between chronic low-grade inflammation and obesity is well documented. Obesity has been associated with increased macrophage infiltration to adipose tissue in both mouse (2) and human (3). Release of pro-inflammatory cytokines by infiltrating macrophages is enhanced in obesity, contributing to impaired insulin sensitivity (4). The links between inflammation, obesity and insulin resistance have been reviewed extensively elsewhere (5, 6); however, many of the exact mechanisms involved in this interaction are yet to be elucidated.
It is now clear that several metabolic intermediates that are released from adipose tissue play important roles communicating with invading immune cells, in particular macrophages. Several of these metabolites are ligands for G protein-coupled receptors (GPCRs), and, importantly, some of these GPCRs are expressed in both adipocytes and the inflammatory cells that invade metabolic tissues in metabolic disorders (7). GPCRs are the most historically successful drug targets and understanding how these receptors control interactions between metabolism and inflammation may provide new avenues to treat metabolic disorders. Here we identify three metabolic intermediates: long chain fatty acids, hydroxy carboxylic acids, and succinate, that are released either directly or indirectly from adipocytes and signal through GPCRs expressed both on adipocytes themselves and on invading macrophages. In this review we discuss the roles the receptors for these metabolites play controlling interaction between adipocytes and macrophages, as well as how this may contribute to the development, progression, and, ultimately, treatment of metabolic disorders.
FFA4 free fatty acid receptor
FFA4, formerly GPR120, is a GPCR activated by long chain fatty acids and is reported to have an important role in interactions between metabolism and inflammation. FFA4 is primarily described as a Gαq coupled GPCR, but reports also link it to signalling through Gαi and Gαs, and it strongly engages with β-arrestin mediated pathways (8–10). There has been significant interest in FFA4, in part because FFA4-/- mice on a high-fat diet have increased body weight, accumulation of macrophages in their adipose, elevated fasting glucose levels and impaired insulin signalling (11). Furthermore, a deleterious variant of FFA4 (p.R254H/p.R270H) is associated with increased risk of obesity (11) and elevated fasting glucose (12). These findings have generated interested in FFA4 as a therapeutic target for the treatment of inflammatory metabolic disorders.
It is well known that certain long chain fatty acids, in particular the omega (n)-3 fatty acids, possess anti-inflammatory properties, with benefits reported in cardiovascular disease, diabetes, cancer, mental illness and dementia (13). Importantly, the n-3 fatty acids are known to be agonists of FFA4 (14–16). Early evidence also indicated many anti-inflammatory properties of dietary n-3 fatty acids were mediated by FFA4-β-arrestin signalling (17). However, several later studies have failed to reproduce this finding, suggesting instead that dietary n-3 fatty acids are protective regardless of FFA4 expression (18, 19). Therefore, although it is clear that n-3 fatty acids are agonists of this receptor and that dietary n-3 fats provide health benefits, the extent to which FFA4 contributes to the beneficial properties of these fatty acids in the diet remains unclear.
FFA4 is highly expressed in adipose tissue, and its expression increases following in vitro adipogenic differentiation of isolated primary human or murine 3T3-L1 adipocytes (20). It has been shown that FFA4 enhances adipogenesis, as its inhibition reduces lipid accumulation and expression of key adipogenic markers (20–22). Interestingly, it has been proposed that in preadipocyte cilia, FFA4 coupling to Gαs to increase ciliar cAMP may be an important trigger for adipogenesis (10). Adipocytes release long chain fatty acids through lipolysis, and it is established that fatty acid levels increase in culture medium during adipogenic differentiation. As these long chain fatty acids released through lipolysis are known agonists of FFA4 (16), this perhaps suggests FFA4 functions as part of a positive autocrine feedback loop reinforcing adipogenic differentiation.
The concept that FFA4 responds to fatty acids released by lipolysis is supported by the observation that conditioned medium from adipocytes treated with a β-adrenoceptor agonist to stimulate lipolysis is sufficient to activate FFA4 receptor signalling in vitro (23). In adipocytes, FFA4 couples strongly to the Gαi pathway to reduce cAMP levels (23), a signalling pathway that is well known to inhibit lipolysis (24). Consistent with this, FFA4 has been found to inhibit lipolysis both in vitro and in vivo (23, 25). These findings indicate FFA4 is part of a negative feedback loop contributing to the long-recognised ability of the fatty acids released from adipocytes to regulate lipolysis (26, 27). FFA4, likely through a Gαq pathway, also enhances GLUT4-mediated glucose uptake in both primary murine adipocytes and 3T3-L1 adipocytes (15, 17). To date, no studies have directly explored whether FFA4 regulation of glucose uptake is controlled in an autocrine fashion by long chain fatty acids released from adipocytes; this is clearly an area that needs future attention.
Chronic low-level inflammation of adipose tissue plays an important role in metabolic syndrome, particularly influencing the development of insulin resistance (4, 28). In immune cells, FFA4 is highly expressed in macrophages (17, 29, 30) and has been recently shown to be expressed in bovine neutrophils where it may play a role in the production of superoxides (31). In RAW 264.7 macrophages, activation of FFA4 through a β-arrestin pathway attenuated pro-inflammatory response to LPS, an effect which was abolished by FFA4-knockdown (17). Furthermore, it has been reported that FFA4 activation reduces macrophage infiltration of adipose tissue (32). Supporting this observation, macrophages displayed chemotaxis toward adipocyte conditioned medium, which was suppressed by FFA4 agonists (17, 32). No subsequent studies have investigated FFA4-regulated infiltration of adipose tissue macrophages, although a role for the receptor in migration is supported by observations that FFA4 activation attenuated the motility of alveolar macrophages (33) and migration of monocytes to atherosclerotic lesions (34). Given that adipocytes release fatty acids through lipolysis, these findings suggest that FFA4 may have an important paracrine signalling role between adipocytes and infiltrating macrophages. Although to date no studies have directly explored this, it will be important to establish if macrophage chemotaxis toward adipocyte conditioned medium is suppressed when the adipocytes are first exposed to a β-adrenoceptor agonists to stimulate lipolysis.
A critical question in understanding FFA4 autocrine or paracrine signalling will be to identify the specific types of fatty acids that are involved. There is evidence that different FFA4 signalling pathways may be activated by specific long chain fatty acids, with a particular difference observed between saturated and unsaturated fatty acids; both classes can activate Gαi and Gαq signalling pathways in vitro, however unsaturated fatty acids can also activate the Gαs pathway (35). Recent lipidomics data suggests that unstimulated mouse and human adipocytes typically release saturated fatty acids (stearic & palmitic acids), with an increase in unsaturated fatty acid release (linoleic & oleic acids) following β-adrenoceptor stimulated lipolysis (23). This may therefore indicate that the fatty acids released through basal adipocyte lipolysis will produce different FFA4 signalling profiles than the fatty acids released due to adrenergic stimulated lipolysis. Similarly, it will also be important to establish whether specific fatty acids from alternative sources, for example released from macrophages by phospholipase A2 (PLA2) hydrolysis of phospholipids, have a role modulating adipocytes/macrophages communications through FFA4. The main products of PLA2 hydrolysis are poly unsaturated n-3 and n-6 fatty acids, which are all known to activate FFA4 (16). Secreted PLA2 is expressed in adipose tissue macrophages, and the activity of PLA2 protects mice from diet induced obesity and adipose inflammation (36, 37). However, the potential for unsaturated fatty acids released by macrophage PLA2 contributing to autocrine or paracrine FFA4 signalling in adipose has not yet been investigated.
There has also been interest recently in the role of FFA4 in the progression and treatment of NASH. FFA4 has been suggested to have a protective role in NASH, as supplementation with omega-3 fatty acids reduces liver inflammation in an FFA4-dependent manner (38, 39). Furthermore, in an inflammatory mouse model of NASH, FFA4 appears to be upregulated in white adipose tissue and reduces the expression of pro-inflammatory IL-6 and TNF-α in this tissue (40). This may be particularly important therapeutically, given that FFA4 is also anti-lipolytic and thus may provide benefit to both the lipotoxity and inflammatory aspects of NASH.
Overall, there is substantial evidence that FFA4 is involved in the interaction between metabolism and inflammation and likely is a central player directly mediating communication between adipocytes and macrophages (Figure 1). However, our current lack of mechanistic understanding for how FFA4 signalling operates under physiological and pathophysiological conditions needs to be resolved before we can truly establish if this receptor is a viable target for metabolic disease.
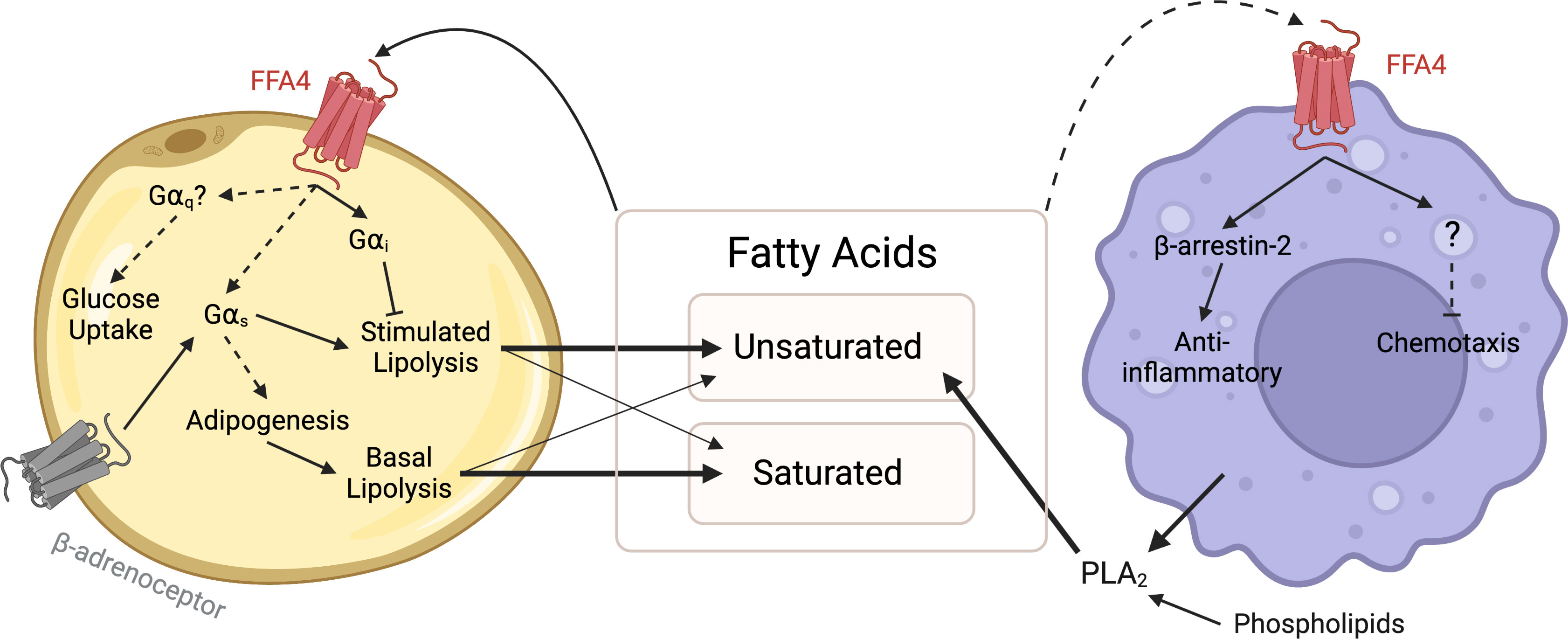
Figure 1 FFA4 signalling between adipocytes and macrophages. Saturated and unsaturated fatty acids are released from adipocytes by lipolysis, supplemented from the diet and may be produced through PLA2-mediated hydrolysis of phospholipids. These fatty acids act as ligands of FFA4 in an autocrine manner to inhibit lipolysis via Gαi signalling, stimulate glucose uptake via a Gαq pathway and stimulate or enhance adipogenesis via Gαs signalling. Fatty acids may also act in a paracrine manner to activate anti-inflammatory pathways in macrophages. Dashed lines represent interactions lacking direct mechanistic evidence, and thus further investigation is required. Created with BioRender.com.
Hydroxyl carboxylic acid receptors
The hydroxy-carboxylic acid (HCA) family of receptors form a cluster of class A GPCRs, which share significant sequence identity. The family includes three known receptors, HCA1, HCA2 and HCA3, formerly known as GPR81, GPR109A and GPR109B respectively (41). The receptors are activated by various different hydroxy-carboxylic acid metabolites whose plasma concentrations vary depending on the metabolic state of the organism, suggesting the receptors play a role in metabolic homeostasis. The HCA receptors are all expressed in adipose tissue and respond either directly or indirectly to metabolites released from adipocytes (42). In adipocytes all three receptors couple to Gαi to mediate anti-lipolytic effects via inhibition of cAMP (43), while HCA2 in particular has also been reported to signal through β-arrestins in some contexts (44) as well as through Gβγ subunits specifically in macrophages (45). Only HCA2 and HCA3 receptors are expressed on certain immune cells, including macrophages and neutrophils, suggesting these two receptors may have a role modulating inflammation in response to metabolic signals released from adipocytes.
The HCA2 receptor was first described as a receptor for the nutrient, nicotinic acid, in 2003, with evidence showing nanomolar affinity for the receptor (46, 47). Nicotinic acid has long been known to produce an antilipolytic effect in adipose, resulting in decreased plasma FFA concentrations (48). This effect of nicotinic acid is abolished in HCA2 knockout mice (46). However, as endogenous concentrations of nicotinic acid in the body are too low to activate the receptor, it is unlikely that this is the endogenous ligand of HCA2 (49). Instead, a second small carboxylic acid with activity at HCA2 has been identified, the ketone body β-hydroxybutyrate (BHB) (49). Like nicotinic acid, previous research had demonstrated BHB to have anti-lipolytic effects (50–52), which were found to be dependent on HCA2 (49). Critically, despite the fact that BHB only has high micromolar affinity for HCA2 (49), plasma concentrations of BHB increase to low millimolar levels during fasting (53–56), suggesting that BHB is a legitimate endogenous ligand of HCA2. The source of elevated BHB during fasting, involves first the lipolytic release of FFAs from adipose, followed by oxidation of these fatty acids in the liver to produce BHB (57–59). Given the anti-lipolytic effects of HCA2, this suggests a primary role for the receptor in a negative feedback loop to regulate lipolytic rate and preserve energy during fasting (60). Interestingly, a recent study has found that adipocytes also have the ability to produce and secrete BHB (61), suggesting that HCA2 signalling could also be activated directly through local autocrine or paracrine signalling in adipose, as well as the more traditional indirect activation via BHB production in liver.
HCA2 is highly expressed in both human and murine white and brown adipose tissue, with expression increasing through adipogenesis of common adipocyte cell models (62, 63). To a lesser extent the receptor is also expressed in macrophages, with evidence showing that HCA2 expression is upregulated in the presence of proinflammatory stimuli, like lipopolysaccharide (LPS) and TNF-α (64, 65). A similar increase in HCA2 expression in response to LPS is also observed in cultured adipocyte models in vitro (66). In contrast, adipocyte HCA2 expression decreases in diet induced obese mice (66), and expression is decreased in subcutaneous adipose tissue of obese human subjects (67). These contrasting in vitro and in vivo findings may suggest that simple treatment of in vitro adipocytes with pro-inflammatory mediators does not accurately reproduce the chronic multicellular inflammatory responses observed in obese adipose (68). This highlights a need to develop more accurate in vitro approaches to allow for thorough investigation and to better understand how inflammation affects HCA2 expression and function.
The HCA3 receptor is closely related to HCA2, sharing 96% sequence identity. It is the result of a gene duplication that is present only in human and hominids (69). HCA3 has a very similar expression pattern to HCA2, being highly expressed in adipocytes, as well as several immune cells (47, 70). Despite the high level of similarity, the receptors do not share the same endogenous ligand. HCA3 is not activated by BHB, but instead by 3-hydroxyoctanoate (71). Like BHB, 3-hydroxyoctanoate is produced in the liver and muscles by β-oxidation of fatty acids produced by lipolysis and so HCA3 also appears to act as a negative feedback modulator of lipolysis during fasting (72). Like HCA2, addition of LPS has been found to significantly increase the expression of HCA3 in adipocytes and macrophages cultured in vitro (63).
Due to the increase in HCA2 and HCA3 expression with LPS treatment, recent studies have investigated whether these receptors play a role in modulating proinflammatory cytokine production. Activation of HCA2 and HCA3 with nicotinic acid and 1-isopropyl-1H-benzotriazole-5-carboxylic acid (IPBT) respectively, reduced the production of proinflammatory cytokines in SGBS adipocytes and THP-1 macrophages exposed to LPS (63). Likewise, HCA2 activation in primary murine macrophages had the same effect (73). Inflammatory cytokines have a key role in the development of metabolic syndrome, disrupting insulin and lipid signalling pathways (74). In addition, activation of HCA2 also has been found to suppress signalling responses in macrophages to key chemoattractant chemokines, CCL2, fMLF and RANTES (45, 75). Critically, these chemokines have important roles in the macrophage infiltration of adipose in metabolic disease (76). Together, this suggest HCA2 and HCA3 may be important regulators of inflammation in adipose tissue and potential targets for the treatment of metabolic disorders.
Interestingly HCA2 signalling differs in macrophages and adipocytes. In macrophages, although the receptor still couples to Gαi, this coupling results in increased intracellular calcium (likely via activation of Gβγ subunits) and is associated with the release of prostanoids (45). The HCA2 mediated release of prostanoids also appears to involve β-arrestin-1, as macrophages from β-arrestin-1-/- mice show reduced activation of cytosolic phospholipase A2, the first step in prostanoid release, when treated with an HCA2 agonist (44). Interestingly, prostanoid release from macrophages also seems to serve as an autocrine regulator for HCA2 signalling, resulting in an unexpected HCA2 mediated increase in cAMP levels through a Gαi-Gβγ pathway (45). To date the importance of this pathway has been explored only in relation to antiatherosclerotic properties of HCA2, and so it will be critical to establish what impact this pathway has on the regulation of adipose inflammation by HCA2.
The HCA receptors play an important role in controlling metabolic homeostasis through an anti-lipolytic negative feedback loop, while also playing an immunomodulatory role (Figure 2). Previously, production of endogenous HCA ligands was thought to occur exclusively in the liver and muscle, however recent research suggests that adipocytes are also capable of directly secreting certain HCA receptor ligands. It will be important for future work to address what, if any, contribution adipose secreted HCAs have to overall HCA receptor signalling, and how these receptors contribute to adipose-immune cell communication.
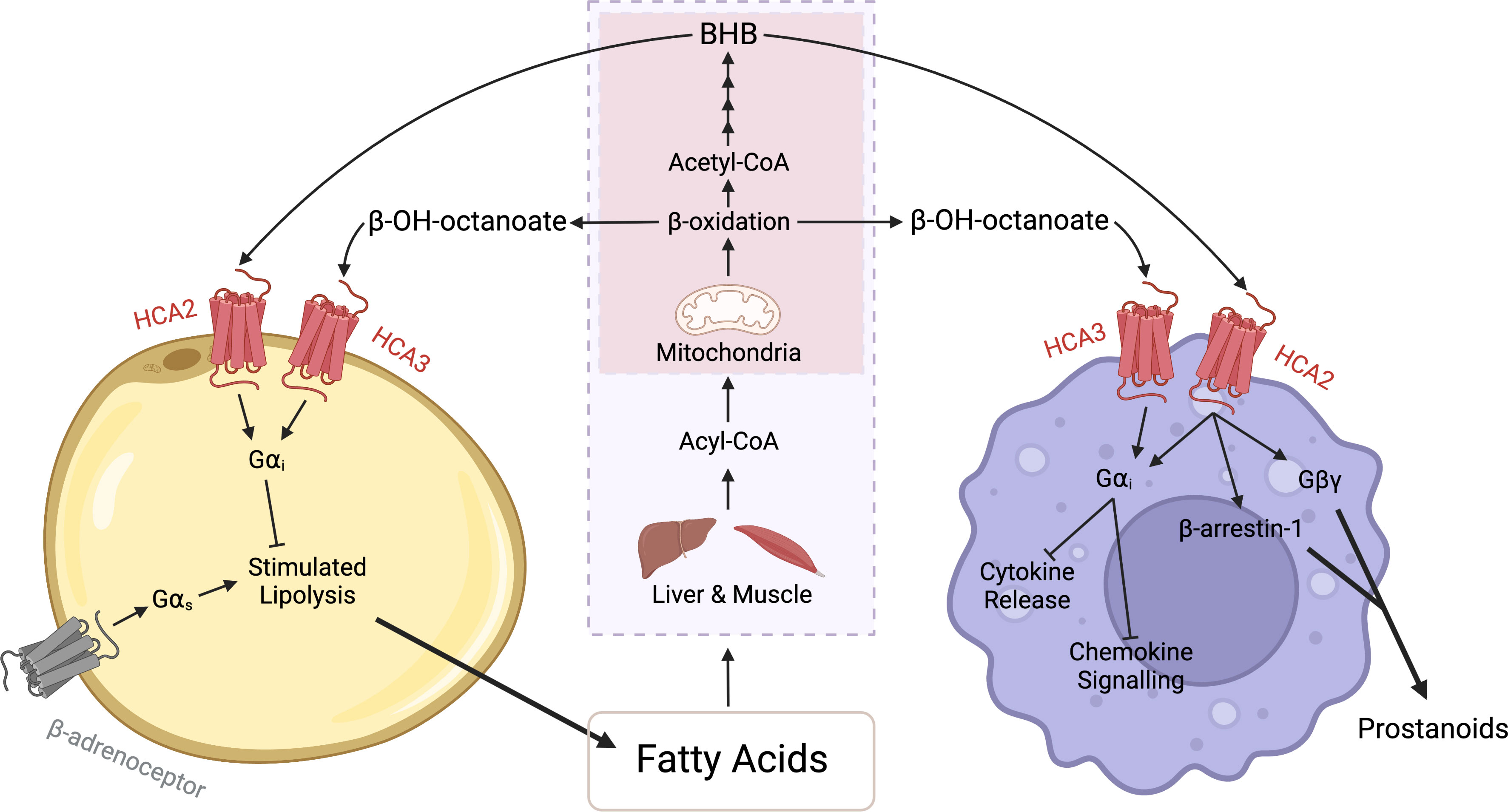
Figure 2 HCA receptor signalling between adipocytes and macrophages. During periods of fasting or exercise, there is an increase in lipolysis and subsequently an increased release of fatty acids from adipocytes. These fatty acids are converted in the liver or muscle (purple box) to Acyl-CoA and then transported into mitochondria (red box). Beta-oxidation in the mitochondria produces acetyl-CoA, which undergoes ketogenesis, a four-step process that can produce beta-hydroxybutyrate (BHB). BHB binds to and activates the HCA2 receptor and through Gαi signalling, evokes antilipolytic effects in adipocytes and anti-inflammatory effects in macrophages. The anti-inflammatory effects include a reduction in cytokine production and suppression of chemokine signalling. Additionally, in macrophages the HCA2 receptor signals via the Gβγ subunit and β-arrestin-1 to mediate the release of prostanoids. Similarly, beta-hydroxyoctanoate is produced via beta-oxidation of fatty acids. However, beta-hydroxyoctanoate is released as a result of incomplete beta-oxidation. Beta-hydroxyoctanoate binds to and activates the HCA3 receptor on adipocytes and macrophages and through its Gαi evokes anti-inflammatory and antilipolytic effects. Created with BioRender.com.
Succinate receptor
GPR91 was identified in 2001 as an orphan GPCR sequence located on chromosome 3 with homology to purinergic receptors (77). Subsequent work demonstrated that GPR91 is activated by the citric acid cycle intermediate, succinate (78), and the receptor was subsequently renamed the succinate receptor, but is also commonly referred to by its gene name, SUCNR1 (79). Initial expression studies found high levels of SUCNR1 in kidney, liver and spleen (78), while the receptor has also been found in adipose (80), and in various immune cells including dendritic cells and macrophages (81). Since its discovery, SUCNR1 has received significant attention for its role in the pathophysiology of, and potential treatment for, a variety of conditions including hypertension, cardiovascular disease, obesity and insulin resistance, NASH, macular degeneration and inflammatory bowel diseases (82).
Although succinate is normally found primarily in the mitochondrial matrix, under conditions of hypoxic and metabolic stress, succinate dehydrogenase, which converts succinate to fumarate as part of the citric acid cycle, reverses its function, resulting in a build-up of succinate in the mitochondria (83). This excess succinate is transported out of the mitochondria through a mitochondrial dicarboxylic acid carrier (84), before ultimately being released from the cell through solute carrier transporters (85). Extracellular succinate is then able to bind to and activate SUCNR1. While this process occurs in many cell types, it may be particularly important in adipocytes, which become hypoxic with chronic low-level inflammation in obesity, metabolic syndrome and diabetes (86).
SUCNR1 is primarily described as a Gαi coupled GPCR inhibiting production of cAMP, while a few studies have also observed Gαq mediated SUCNR1 signalling (78, 87, 88). With this in mind, SUCNR1 has been shown to inhibit adipocyte lipolysis both in vitro and ex vivo in a Gαi dependent manner (80). Subsequent studies have also demonstrated anti-lipolytic effects of SUCNR1 in vivo (89), and specifically linked these effects to adipocyte expression of a dicarboxylic acid carrier responsible for transporting succinate out of mitochondria (84). Together, these findings support an autocrine signalling pathway mediated by succinate released from adipocytes to control lipolysis.
Mice lacking SUCNR1 have a variety of disfunctions in their metabolic phenotype when fed a high fat diet, including: hyperglycemia, reduced weight gain, and impaired glucose clearance (89). The fact that these metabolic phenotypes in SUCNR1-/- mice are only observed with a HFD, suggests a need for metabolic stress and/or hypoxia in the adipose for SUCNR1 pathways to be active. Indeed, there is clear evidence that plasma succinate levels are elevated in human patients with obesity, metabolic syndrome and diabetes (90–92). Given that each of these conditions is associated with chronic inflammation of adipose, and that SUCNR1 is also expressed in immune cells, this implicates SUCNR1 as a potential mediator of adipocyte-immune cell communication. Providing support for this possibility, abnormal metabolic phenotypes have been observed in mice with a myeloid specific SUCNR1 knockout (93), suggesting a key role of SUCNR1 in macrophages metabolic dysfunction.
While there is now clear evidence that succinate-SUCNR1 signalling plays an important role mediating communication between adipose tissue and macrophages, it remains controversial whether SUCNR1 is pro- or anti-inflammatory. Studies in SUCNR1-/- macrophages have indicated that this receptor mediates chemotaxis towards succinate released from hypoxic adipocytes (90), suggesting a pro-inflammatory function. This is consistent with earlier work reporting a pro-inflammatory role of SUCNR1 in macrophages in various other inflammatory disorders, including arthritis (94), arthrosclerosis (95), and inflammatory gut conditions (96, 97). In contrast, others have reported pro-inflammatory cytokine secretion to be enhanced in SUCNR1-/- macrophages (98), and adipose tissue inflammation to be exacerbated in myeloid cell specific SUCNR1 knockout mice fed a high fat diet (93), suggesting an anti-inflammatory role of the receptor. Consistent with this, a recent transcriptomic study found that succinate, acting through an SUCNR1-Gαq pathway, hyperpolarized human macrophages toward the M2 anti-inflammatory phenotype (87). Adding a further layer of complexity, studies in adipose taken from lean vs obese patients found that while succinate was anti-inflammatory in the lean adipose, it enhanced IL-1β and TNF expression in obese adipose (93), suggesting cell or context dependent SUCNR1 control of inflammation. Developing a clear understanding of the role of SUCNR1 in inflammation will be key to establishing whether agonism or antagonism of the receptor will have therapeutic benefit.
In addition to the release of succinate from hypoxic adipocytes, macrophages themselves also directly release succinate in response to inflammatory signals (99). While there is evidence that succinate released from macrophages does act in an autocrine fashion through SUCNR1 to affect inflammatory responses (94), to date it is not clear whether macrophage released succinate mediates communication to adipocytes through this receptor. In addition, circulating succinate levels are utilised and cleared by uncoupling protein 1 (UCP1) expressed in brown and beige adipose (100, 101), and genetic disruption of UCP1 leads to SUCNR1 mediated liver inflammation (100). This raises interesting questions about the potential use of SUCNR1 in treating liver diseases like NASH, given that, like many other receptors that inhibit lipolysis in adipose including both FFA4 and HCA2/3, SUCNR1 actions in adipose have protective effects on liver lipotoxicity (84). This indicates that like its control of inflammation more broadly, the potential of SUCNR1 in treating metabolic and inflammatory mediated liver disease is complex.
SUCNR1 signalling has an important role in regulating metabolism and inflammation in the context of metabolic disease (Figure 3). Succinate, acting through SUCNR1, is an important mediator of stress signalling to affect immune cell function and inflammation. However, given the conflicting findings on this receptor as either pro- or anti-inflammatory, there is a real need to establish more physiologically relevant approaches to better understand the role of SUCNR1 and determine if and how we can target this receptor for the treatment of disease.
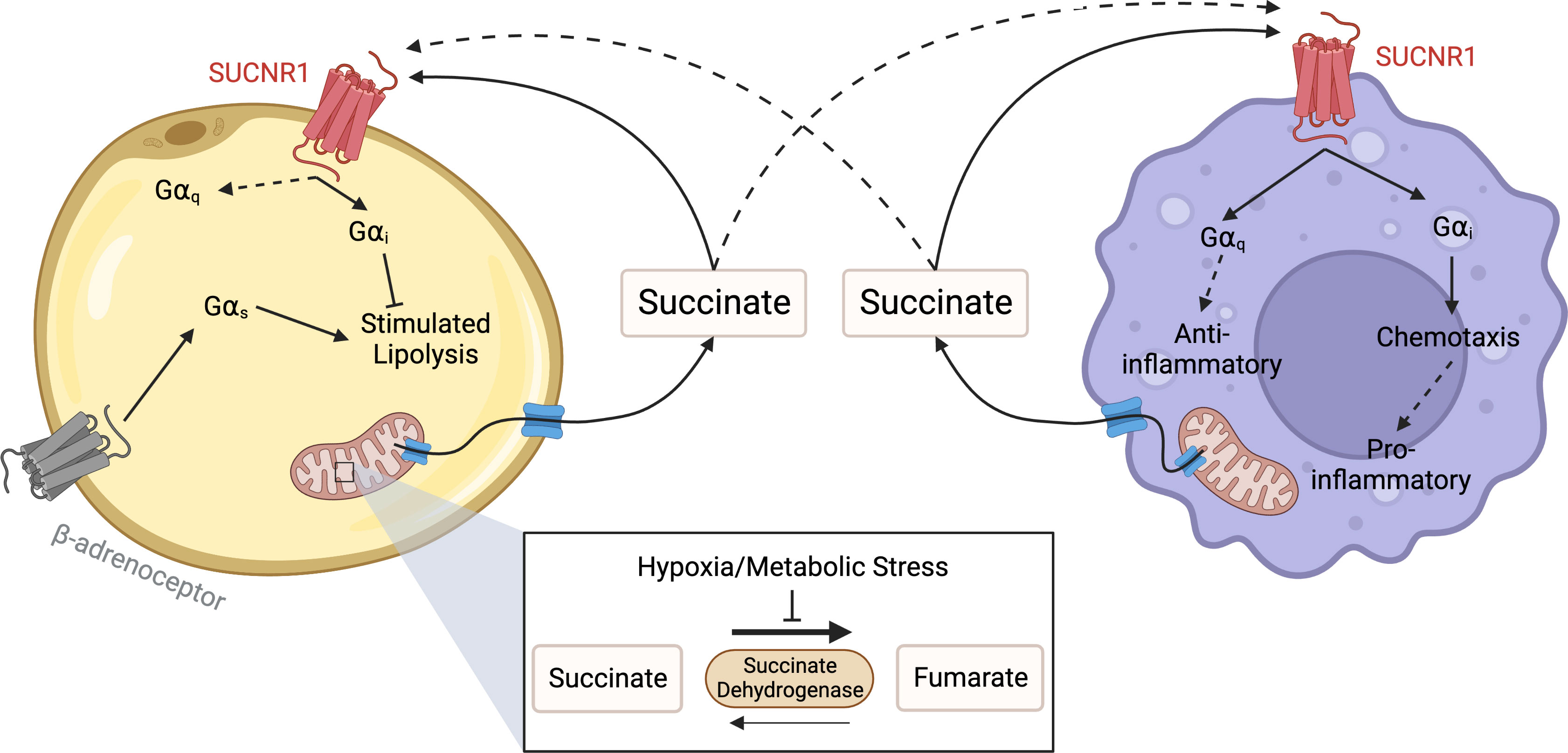
Figure 3 Succinate Receptor signalling between adipocytes and macrophages. Under conditions of hypoxic or metabolic stress succinate dehydrogenase reverses function, leading to a build-up of succinate in the mitochondria. When concentrations become elevated, succinate leaves the mitochondria through a dicarboxylate carrier, before exiting the cell through other solute carriers. Once outside the cell, succinate binds to and activates SUCNR1 on the surface of adipocytes and macrophages to inhibit lipolysis and control inflammation respectively. In macrophages, SUCNR1 has been reported to have both pro-inflammatory and anti-inflammatory effects. These include mediating chemotaxis towards hypoxic adipose, likely through Gαi activation, as well as hyperpolarisation to the M2 macrophage phenotype through a Gαq mediated pathway. Created with BioRender.com.
Conclusion
Various metabolites released by adipocytes act through either direct or indirect signalling pathways to activate metabolite-sensing GPCRs both in adipocytes themselves and in the immune cells that infiltrate adipose in metabolic disorders. These receptors play a variety of functional roles, but commonly both regulate inflammation in immune cells and lipolytic pathways in adipose. The complex, multicellular nature of these signalling pathways and networks has made mechanistically dissecting this signalling quite difficult in vitro, and it will be critical to develop and establish more robust experimental models and approaches to achieve this.
Not surprisingly, given the functions of the FFA4, HCA and SUCNR1 receptors in controlling metabolism and inflammation, each of these has received interest as a potential target in the treatment of metabolic disorders including dyslipidaemia, diabetes and NASH. HCA2 is the most well developed, with its naturally occurring ligand, niacin, having been widely used clinically to control dyslipidaemia through a mechanism that is at least partly mediated by HCA2 (102). However, these clinical studies have also demonstrated that niacin, also via HCA2, produces an unwanted flushing effect in skin (103). Efforts to eliminate this side effect led to the development of a synthetic partial HCA2 agonist, MK-0354 that maintains lipid lowering effects, but without producing flushing in animal models (104). These findings led to MK-0354 entering phase I and II clinical trials, where although it did not produce flushing, it also did not improve lipid levels following chronic treatment (105). Despite this failure, medicinal chemistry efforts have continued around HCA2, but without any yet reaching the clinic (106). Similarly, FFA4 has received significant attention from both academic and industrial drug discovery programmes, primarily focused on developing agonists for type 2 diabetes and/or NASH, however to date no molecules have entered the clinic (107). While also receiving some attention as a therapeutic target, the conflicting data around the benefits of agonism vs antagonism of SUCNR1, as well as the relatively wide spread expression pattern for SUCNR1 in multiple tissues and cell types (82) has perhaps led to a somewhat slower pace of development for this receptor.
Ultimately, understanding the signalling networks of these metabolite-sensing GPCRs will help us better understand how interactions between metabolism and inflammation drive metabolic disease. Developing this understanding is likely to open new opportunities for the treatment of a variety of metabolic disorders, including obesity, diabetes and NASH.
Author contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This work was supported by Medical Research Scotland (BH, LV), and by the EPSRC and SFI Centre for Doctoral Training in Engineered Tissues for Discovery, Industry and Medicine, Grant Number EP/S02347X/1 (ED).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. International Diabetes Federation. IDF diabetes atlas 2021, in: IDF diabetes atlas (2021). Available at: https://diabetesatlas.org/atlas/tenth-edition/ (Accessed 23, 2023).
2. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi: 10.1172/JCI19246
3. Harman-Boehm I, Blüher M, Redel H, Sion-Vardy N, Ovadia S, Avinoach E, et al. Macrophage infiltration into omental versus subcutaneous fat across different populations: effect of regional adiposity and the comorbidities of obesity. J Clin Endocrinol Metab (2007) 92:2240–7. doi: 10.1210/JC.2006-1811
4. Gasmi A, Noor S, Menzel A, Doşa A, Pivina L, Bjørklund G. Obesity and insulin resistance: associations with chronic inflammation, genetic and epigenetic factors. Curr Med Chem (2020) 28:800–26. doi: 10.2174/0929867327666200824112056
5. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi: 10.1146/ANNUREV-PHYSIOL-021909-135846
6. Artemniak-Wojtowicz D, Pyrżak B, Kucharska AM. Obesity and chronic inflammation crosslinking. Cent Eur J Immunol (2020) 45:461. doi: 10.5114/CEJI.2020.103418
7. Husted AS, Trauelsen M, Rudenko O, Hjorth SA, Schwartz TW. GPCR-mediated signaling of metabolites. Cell Metab (2017) 25:777–96. doi: 10.1016/J.CMET.2017.03.008
8. Milligan G, Alvarez-Curto E, Hudson BD, Prihandoko R, Tobin AB. FFA4/GPR120: pharmacology and therapeutic opportunities. Trends Pharmacol Sci (2017) 38:809–21. doi: 10.1016/j.tips.2017.06.006
9. Ghislain J, Poitout V. Targeting lipid GPCRs to treat type 2 diabetes mellitus - progress and challenges. Nat Rev Endocrinol (2021) 17:162–75. doi: 10.1038/S41574-020-00459-W
10. Hilgendorf KI, Johnson CT, Mezger A, Rice SL, Norris AM, Demeter J, et al. Omega-3 fatty acids activate ciliary FFAR4 to control adipogenesis. Cell (2019) 179:1289–1305.e21. doi: 10.1016/J.CELL.2019.11.005/ATTACHMENT/5F9119C4-FBB0-4AA0-9E9C-ADD67FC28599/MMC6.XLSX
11. Ichimura A, Hirasawa A, Poulain-Godefroy O, Bonnefond A, Hara T, Yengo L, et al. Dysfunction of lipid sensor GPR120 leads to obesity in both mouse and human. Nature (2012) 483:350–4. doi: 10.1038/nature10798
12. Bonnefond A, Lamri A, Leloire A, Vaillant E, Roussel R, Lévy-Marchal C, et al. Contribution of the low-frequency, loss-of-function p.R270H mutation in FFAR4 (GPR120) to increased fasting plasma glucose levels. J Med Genet (2015) 52:595–8. doi: 10.1136/JMEDGENET-2015-103065
13. Shahidi F, Ambigaipalan P. Omega-3 polyunsaturated fatty acids and their health benefits(2018) 9:345–381. doi: 10.1146/ANNUREV-FOOD-111317-095850.
14. Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, et al. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med (2005) 11:90–4. doi: 10.1038/nm1168
15. Hudson BD, Shimpukade B, Mackenzie AE, Butcher AJ, Pediani JD, Christiansen E, et al. The pharmacology of TUG-891, a potent and selective agonist of the free fatty acid receptor 4 (FFA4/GPR120), demonstrates both potential opportunity and possible challenges to therapeutic agonism. Mol Pharmacol (2013) 84:710–25. doi: 10.1124/MOL.113.087783
16. Christiansen E, Watterson KR, Stocker CJ, Sokol E, Jenkins L, Simon K, et al. Activity of dietary fatty acids on FFA1 and FFA4 and characterisation of pinolenic acid as a dual FFA1/FFA4 agonist with potential effect against metabolic diseases. Br J Nutr (2015) 113:1677–88. doi: 10.1017/S000711451500118X
17. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan WQ, et al. GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell (2010) 142:687–98. doi: 10.1016/j.cell.2010.07.041
18. Bjursell M, Xu X, Admyre T, Böttcher G, Lundin S, Nilsson R, et al. The beneficial effects of n-3 polyunsaturated fatty acids on diet induced obesity and impaired glucose control do not require Gpr120. PloS One (2014) 9:e114942. doi: 10.1371/JOURNAL.PONE.0114942
19. Pærregaard SI, Agerholm M, Serup AK, Ma T, Kiens B, Madsen L, et al. FFAR4 (GPR120) signaling is not required for anti-inflammatory and insulin-sensitizing effects of omega-3 fatty acids. Mediators Inflamm (2016) 2016:1536047. doi: 10.1155/2016/1536047
20. Gotoh C, Hong Y-H, Iga T, Hishikawa D, Suzuki Y, Song S-H, et al. The regulation of adipogenesis through GPR120. Biochem Biophys Res Commun (2007) 354:591–7. doi: 10.1016/J.BBRC.2007.01.028
21. Song T, Zhou Y, Peng J, Tao YX, Yang Y, Xu T, et al. GPR120 promotes adipogenesis through intracellular calcium and extracellular signal-regulated kinase 1/2 signal pathway. Mol Cell Endocrinol (2016) 434:1–13. doi: 10.1016/J.MCE.2016.06.009
22. Watterson KR, Hansen SVF, Hudson BD, Alvarez-Curto E, Raihan SZ, Azevedo CMG, et al. Probe-dependent negative allosteric modulators of the long-chain free fatty acid receptor FFA4. Mol Pharmacol (2017) 91:630–41. doi: 10.1124/mol.116.107821
23. Husted AS, Ekberg JH, Tripp E, Nissen TAD, Meijnikman S, O’Brien SL, et al. Autocrine negative feedback regulation of lipolysis through sensing of NEFAs by FFAR4/GPR120 in WAT. Mol Metab (2020) 42:101103. doi: 10.1016/J.MOLMET.2020.101103
24. Langin D. Adipose tissue lipolysis as a metabolic pathway to define pharmacological strategies against obesity and the metabolic syndrome. Pharmacol Res (2006) 53:482–91. doi: 10.1016/J.PHRS.2006.03.009
25. Satapati S, Qian Y, Wu MS, Petrov A, Dai G, Wang SP, et al. GPR120 suppresses adipose tissue lipolysis and synergizes with GPR40 in antidiabetic efficacy. J Lipid Res (2017) 58:1561–78. doi: 10.1194/jlr.M075044
26. Rodbell M. MODULATION OF LIPOLYSIS IN ADIPOSE TISSUE BY FATTY ACID CONCENTRATION IN FAT CELL. Ann N Y Acad Sci (1965) 131:302–14. doi: 10.1111/J.1749-6632.1965.TB34798.X
27. Burns TW, Langley PE, Terry BE, Robinson GA. The role of free fatty acids in the regulation of lipolysis by human adipose tissue cells. Metabolism (1978) 27:1755–62. doi: 10.1016/0026-0495(78)90261-5
28. Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: review of the underlying molecular mechanisms. J Cell Physiol (2019) 234:8152–61. doi: 10.1002/JCP.27603
29. Li X, Yu Y, Funk CD. Cyclooxygenase-2 induction in macrophages is modulated by docosahexaenoic acid via interactions with free fatty acid receptor 4 (FFA4). FASEB J (2013) 27:4987–97. doi: 10.1096/FJ.13-235333
30. Liu Y, Chen LY, Sokolowska M, Eberlein M, Alsaaty S, Martinez-Anton A, et al. The fish oil ingredient, docosahexaenoic acid, activates cytosolic phospholipase A2 via GPR120 receptor to produce prostaglandin E2 and plays an anti-inflammatory role in macrophages. Immunology (2014) 143:81–95. doi: 10.1111/IMM.12296
31. Olmo I, Larrazabal C, Alarcon P, Raipane F, Burgos RA, Hidalgo MA. Docosahexaenoic acid and TUG-891 activate free fatty acid-4 receptor in bovine neutrophils. Vet Immunol Immunopathol (2019) 209:53–60. doi: 10.1016/J.VETIMM.2019.02.008
32. Oh DY, Walenta E, Akiyama TE, Lagakos WS, Lackey D, Pessentheiner AR, et al. A Gpr120-selective agonist improves insulin resistance and chronic inflammation in obese mice. Nat Med (2014) 20:942–7. doi: 10.1038/nm.3614
33. Su XL, Liu YG, Shi M, Zhao YY, Liang XY, Zhang LJ, et al. The GPR120 agonist TUG-891 inhibits the motility and phagocytosis of mouse alveolar macrophages. BioMed Res Int (2020) 2020:1706168. doi: 10.1155/2020/1706168
34. Shewale SV, Brown AL, Bi X, Boudyguina E, Sawyer JK, Alexander-Miller MA, et al. In vivo activation of leukocyte GPR120/FFAR4 by PUFAs has minimal impact on atherosclerosis in LDL receptor knockout mice. J Lipid Res (2017) 58:236–46. doi: 10.1194/JLR.M072769
35. Mao C, Xiao P, Tao X-N, Qin J, He Q-T, Zhang C, et al. Unsaturated bond recognition leads to biased signal in a fatty acid receptor. Science (2023) 380(6640). doi: 10.1126/SCIENCE.ADD6220
36. Sato H, Taketomi Y, Ushida A, Isogai Y, Kojima T, Hirabayashi T, et al. The adipocyte-inducible secreted phospholipases PLA2G5 and PLA2G2E play distinct roles in obesity. Cell Metab (2014) 20:119–32. doi: 10.1016/j.cmet.2014.05.002
37. Jeong H, Lee C, Cheng C, Chou HC, Yang HJ, Bae H. Targeting of adipose tissue macrophages by bee venom phospholipase A2 attenuates high-fat diet-induced obesity. Int J Obes (2021) 45:1656–67. doi: 10.1038/s41366-021-00823-4
38. Raptis DA, Limani P, Jang JH, Ungethüm U, Tschuor C, Graf R, et al. GPR120 on kupffer cells mediates hepatoprotective effects of ω3-fatty acids. J Hepatol (2014) 60:625–32. doi: 10.1016/J.JHEP.2013.11.006
39. Nakamoto K, Shimada K, Harada S, Morimoto Y, Hirasawa A, Tokuyama S. DHA supplementation prevent the progression of NASH via GPR120 signaling. Eur J Pharmacol (2018) 820:31–8. doi: 10.1016/J.EJPHAR.2017.11.046
40. Nakamoto K, Tokuyama S. Docosahexaenoic acid attenuates the progression of nonalcoholic steatohepatitis by suppressing the adipocyte inflammation via the G protein-coupled receptor 120/Free fatty acid receptor 4 pathway. Pharmacology (2022) 107:330–8. doi: 10.1159/000522117
41. Offermanns S, Colletti SL, Lovenberg TW, Semple G, Wise A, Ijzerman AP. International union of basic and clinical pharmacology. LXXXII: nomenclature and classification of hydroxy-carboxylic acid receptors (GPR81, GPR109A, and GPR109B). Pharmacol Rev (2011) 63:269–90. doi: 10.1124/PR.110.003301
42. Ahmed K, Tunaru S, Offermanns S. GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol Sci (2009) 30:557–62. doi: 10.1016/J.TIPS.2009.09.001
43. Blad CC, Ahmed K, IJzerman AP, Offermanns S. Biological and pharmacological roles of HCA receptors. Adv Pharmacol (2011) 62:219–50. doi: 10.1016/B978-0-12-385952-5.00005-1
44. Walters RW, Shukla AK, Kovacs JJ, Violin JD, DeWire SM, Lam CM, et al. β-Arrestin1 mediates nicotinic acid–induced flushing, but not its antilipolytic effect, in mice. J Clin Invest (2009) 119:1312–21. doi: 10.1172/JCI36806
45. Gaidarov I, Chen X, Anthony T, Maciejewski-Lenoir D, Liaw C, Unett DJ. Differential tissue and ligand-dependent signaling of GPR109A receptor: implications for anti-atherosclerotic therapeutic potential. Cell Signal (2013) 25:2003–16. doi: 10.1016/J.CELLSIG.2013.06.008
46. Tunaru S, Kero J, Schaub A, Wufka C, Blaukat A, Pfeffer K, et al. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat Med (2003) 9:352–5. doi: 10.1038/nm824
47. Wise A, Foord SM, Fraser NJ, Barnes AA, Elshourbagy N, Eilert M, et al. Molecular identification of high and low affinity receptors for nicotinic acid *. J Biol Chem (2003) 278:9869–74. doi: 10.1074/JBC.M210695200
48. Obert R, Nopp HK. Drug treatment of lipid disorders. N Engl J Med (1999) 341:498–511. doi: 10.1056/NEJM199908123410707
49. Taggart AKP, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, et al. (d)-β-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem (2005) 280:26649–52. doi: 10.1074/JBC.C500213200
50. Björntorp P. The effect of beta-hydroxybutyric acid on glycerol outflow from adipose tissue. in vitro. Metab (1966) 15:191–3. doi: 10.1016/0026-0495(66)90016-3
51. Björntorp P. Effect of ketone bodies on lipolysis in adipose tissue. in vitro J Lipid Res (1966) 7:621–6. doi: 10.1016/S0022-2275(20)39242-7
52. Van Hove JLK, Grünewald S, Jaeken J, Demaerel P, Declercq PE, Bourdoux P, et al. D,L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD). Lancet (2003) 361:1433–5. doi: 10.1016/S0140-6736(03)13105-4
53. Senior B, Loridan L. Direct regulatory effect of ketones on lipolysis and on glucose concentrations in man. Nature (1968) 219:83–4. doi: 10.1038/219083A0
54. Laffel L. Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab Res Rev (1999) 15:412–26. doi: 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8
55. Owen OE, Reichard GA. Ketone body metabolism in normal, obese and diabetic subjects. Isr J Med Sci (1975) 11:560–70.
56. Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids (2004) 70:243–51. doi: 10.1016/J.PLEFA.2003.11.001
57. Cuenoud B, Hartweg M, Godin J-P, Croteau E, Maltais M, Castellano C-A, et al. Metabolism of exogenous d-Beta-Hydroxybutyrate, an energy substrate avidly consumed by the heart and kidney. Front Nutr (2020) 7:13. doi: 10.3389/fnut.2020.00013
58. Newman JC, Verdin E. β-hydroxybutyrate: a signaling metabolite. Annu Rev Nutr (2017) 37:51–76. doi: 10.1146/annurev-nutr-071816-064916
59. Houten SM, Wanders RJA. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis (2010) 33:469–77. doi: 10.1007/s10545-010-9061-2
60. Offermanns S, Schwaninger M. Nutritional or pharmacological activation of HCA2 ameliorates neuroinflammation. Trends Mol Med (2015) 21:245–55. doi: 10.1016/J.MOLMED.2015.02.002
61. Nishitani S, Fukuhara A, Tomita I, Kume S, Shin J, Okuno Y, et al. Ketone body 3-hydroxybutyrate enhances adipocyte function. Sci Rep (2022) 12:1–13. doi: 10.1038/s41598-022-14268-w
62. Jeninga EH, Bugge A, Nielsen R, Kersten S, Hamers N, Dani C, et al. Peroxisome proliferator-activated receptor γ regulates expression of the anti-lipolytic G-protein-coupled receptor 81 (GPR81/Gpr81). J Biol Chem (2009) 284:26385–93. doi: 10.1074/JBC.M109.040741
63. Mandrika I, Tilgase A, Petrovska R, Klovins J. Hydroxycarboxylic acid receptor ligands modulate proinflammatory cytokine expression in human macrophages and adipocytes without affecting adipose differentiation. Biol Pharm Bull (2018) 41:1574–80. doi: 10.1248/BPB.B18-00301
64. Benyó Z, Gille A, Kero J, Csiky M, Suchánková MC, Nüsing RM, et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid-induced flushing. J Clin Invest (2005) 115:3634–40. doi: 10.1172/JCI23626
65. Zandi-Nejad K, Takakura A, Jurewicz M, Chandraker AK, Offermanns S, Mount D, et al. The role of HCA2 (GPR109A) in regulating macrophage function. FASEB J (2013) 27:4366. doi: 10.1096/FJ.12-223933
66. Wanders D, Graff EC, Judd RL. Effects of high fat diet on GPR109A and GPR81 gene expression. Biochem Biophys Res Commun (2012) 425:278–83. doi: 10.1016/J.BBRC.2012.07.082
67. Al Mahri S, Okla M, Rashid M, Malik SS, Iqbal J, Al IM, et al. Profiling of G-protein coupled receptors in adipose tissue and differentiating adipocytes offers a translational resource for Obesity/Metabolic research. Cells (2023) 12:377. doi: 10.3390/CELLS12030377/S1
68. Kawai T, Autieri MV, Scalia R. Inflammation: from cellular mechanisms to immune cell education: adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol (2021) 320:C375. doi: 10.1152/AJPCELL.00379.2020
69. Peters A, Krumbholz P, Jäger E, Heintz-Buschart A, Çakir MV, Rothemund S, et al. Metabolites of lactic acid bacteria present in fermented foods are highly potent agonists of human hydroxycarboxylic acid receptor 3. PloS Genet (2019) 15. doi: 10.1371/JOURNAL.PGEN.1008145
70. Knowles HJ, Te PR, Workman P, Harris AL. Niacin induces PPARγ expression and transcriptional activation in macrophages via HM74 and HM74a-mediated induction of prostaglandin synthesis pathways. Biochem Pharmacol (2006) 71:646–56. doi: 10.1016/J.BCP.2005.11.019
71. Davenport AP, Alexander SPH, Sharman JL, Pawson AJ, Benson HE, Monaghan AE, et al. International union of basic and clinical pharmacology. LXXXVIII. G protein-coupled receptor list: recommendations for new pairings with cognate ligands. Pharmacol Rev (2013) 65:967. doi: 10.1124/PR.112.007179
72. Ahmed K, Tunaru S, Langhans CD, Hanson J, Michalski CW, Kölker S, et al. Deorphanization of GPR109B as a receptor for the β-oxidation intermediate 3-OH-octanoic acid and its role in the regulation of lipolysis. J Biol Chem (2009) 284:21928. doi: 10.1074/JBC.M109.019455
73. Zhou E, Li Y, Yao M, Wei Z, Fu Y, Yang Z. Niacin attenuates the production of pro-inflammatory cytokines in LPS-induced mouse alveolar macrophages by HCA2 dependent mechanisms. Int Immunopharmacol (2014) 23:121–6. doi: 10.1016/J.INTIMP.2014.07.006
74. Shi J, Fan J, Su Q, Yang Z. Cytokines and abnormal glucose and lipid metabolism. Front Endocrinol (Lausanne) (2019) 10:703. doi: 10.3389/FENDO.2019.00703
75. Shi Y, Lai X, Ye L, Chen K, Cao Z, Gong W, et al. Activated niacin receptor HCA2 inhibits chemoattractant-mediated macrophage migration via Gβγ/PKC/ERK1/2 pathway and heterologous receptor desensitization. Sci Rep (2017) 7:1–14. doi: 10.1038/srep42279
76. Bai Y, Sun Q. Macrophage recruitment in obese adipose tissue. Obes Rev (2015) 16:127–36. doi: 10.1111/OBR.12242
77. Wittenberger T, Schaller HC, Hellebrand S. An expressed sequence tag (EST) data mining strategy succeeding in the discovery of new G-protein coupled receptors. J Mol Biol (2001) 307:799–813. doi: 10.1006/jmbi.2001.4520
78. He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, et al. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature (2004) 429:188–93. doi: 10.1038/nature02488
79. Alexander SP, Christopoulos A, Davenport AP, Kelly E, Mathie A, Peters JA, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: G protein-coupled receptors. Br J Pharmacol (2021) 178 Suppl 1:S27–S156. doi: 10.1111/bph.15538
80. Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell (2008) 135:561–71. doi: 10.1016/j.cell.2008.08.040
81. Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, et al. Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol (2008) 9:1261–9. doi: 10.1038/ni.1657
82. Li X, Xie L, Qu X, Zhao B, Fu W, Wu B, et al. GPR91, a critical signaling mechanism in modulating pathophysiologic processes in chronic illnesses. FASEB J (2020) 34:13091–105. doi: 10.1096/FJ.202001037R
83. Ariza AC, Deen PM, Robben JH. The succinate receptor as a novel therapeutic target for oxidative and metabolic stress-related conditions. Front Endocrinol (Lausanne) (2012) 3:22. doi: 10.3389/fendo.2012.00022
84. An YA, Chen S, Deng Y, Wang ZV, Funcke JB, Shah M, et al. The mitochondrial dicarboxylate carrier prevents hepatic lipotoxicity by inhibiting white adipocyte lipolysis. J Hepatol (2021) 75:387–99. doi: 10.1016/j.jhep.2021.03.006
85. de Castro Fonseca M, Aguiar CJ, da Rocha Franco JA, Gingold RN, Leite MF. GPR91: expanding the frontiers of Krebs cycle intermediates. Cell Commun Signal (2016) 14:3. doi: 10.1186/s12964-016-0126-1
86. Engin A. Adipose tissue hypoxia in obesity and its impact on preadipocytes and macrophages: hypoxia hypothesis. Adv Exp Med Biol (2017) 960:305–26. doi: 10.1007/978-3-319-48382-5_13
87. Trauelsen M, Hiron TK, Lin D, Petersen JE, Breton B, Husted AS, et al. Extracellular succinate hyperpolarizes M2 macrophages through SUCNR1/GPR91-mediated gq signaling. Cell Rep (2021) 35:109246. doi: 10.1016/j.celrep.2021.109246
88. Robben JH, Fenton RA, Vargas SL, Schweer H, Peti-Peterdi J, Deen PM, et al. Localization of the succinate receptor in the distal nephron and its signaling in polarized MDCK cells. Kidney Int (2009) 76:1258–67. doi: 10.1038/ki.2009.360
89. McCreath KJ, Espada S, Galvez BG, Benito M, de Molina A, Sepulveda P, et al. Targeted disruption of the SUCNR1 metabolic receptor leads to dichotomous effects on obesity. Diabetes (2015) 64:1154–67. doi: 10.2337/db14-0346
90. van Diepen JA, Robben JH, Hooiveld GJ, Carmone C, Alsady M, Boutens L, et al. SUCNR1-mediated chemotaxis of macrophages aggravates obesity-induced inflammation and diabetes. Diabetologia (2017) 60:1304–13. doi: 10.1007/s00125-017-4261-z
91. Ceperuelo-Mallafre V, Llaurado G, Keiran N, Benaiges E, Astiarraga B, Martinez L, et al. Preoperative circulating succinate levels as a biomarker for diabetes remission after bariatric surgery. Diabetes Care (2019) 42:1956–65. doi: 10.2337/dc19-0114
92. Osuna-Prieto FJ, Martinez-Tellez B, Ortiz-Alvarez L, Di X, Jurado-Fasoli L, Xu H, et al. Elevated plasma succinate levels are linked to higher cardiovascular disease risk factors in young adults. Cardiovasc Diabetol (2021) 20:151. doi: 10.1186/s12933-021-01333-3
93. Keiran N, Ceperuelo-Mallafre V, Calvo E, Hernandez-Alvarez MI, Ejarque M, Nunez-Roa C, et al. SUCNR1 controls an anti-inflammatory program in macrophages to regulate the metabolic response to obesity. Nat Immunol (2019) 20:581–92. doi: 10.1038/s41590-019-0372-7
94. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med (2016) 213:1655–62. doi: 10.1084/jem.20160061
95. Xu J, Zheng Y, Zhao Y, Zhang Y, Li H, Zhang A, et al. Succinate/IL-1beta signaling axis promotes the inflammatory progression of endothelial and exacerbates atherosclerosis. Front Immunol (2022) 13:817572. doi: 10.3389/fimmu.2022.817572
96. Macias-Ceja DC, Ortiz-Masia D, Salvador P, Gisbert-Ferrandiz L, Hernandez C, Hausmann M, et al. Succinate receptor mediates intestinal inflammation and fibrosis. Mucosal Immunol (2019) 12:178–87. doi: 10.1038/s41385-018-0087-3
97. Bauset C, Lis-Lopez L, Coll S, Gisbert-Ferrandiz L, Macias-Ceja DC, Seco-Cervera M, et al. SUCNR1 mediates the priming step of the inflammasome in intestinal epithelial cells: relevance in ulcerative colitis. Biomedicines (2022) 10:532. doi: 10.3390/biomedicines10030532
98. Harber KJ, de Goede KE, Verberk SGS, Meinster E, de Vries HE, van Weeghel M, et al. Succinate is an inflammation-induced immunoregulatory metabolite in macrophages. Metabolites (2020) 10:372. doi: 10.3390/metabo10090372
99. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi: 10.1038/nature11986
100. Mills EL, Harmon C, Jedrychowski MP, Xiao H, Garrity R, Tran NV, et al. UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat Metab (2021) 3:604–17. doi: 10.1038/s42255-021-00389-5
101. Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, et al. Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature (2018) 560:102–6. doi: 10.1038/s41586-018-0353-2
102. Boden WE, Sidhu MS, Toth PP. The therapeutic role of niacin in dyslipidemia management. J Cardiovasc Pharmacol Ther (2014) 19:141–58. doi: 10.1177/1074248413514481
103. Benyó Z, Gille A, Kero J, Csiky M, Suchánková MC, Nüsing RM, et al. GPR109A (PUMA-G/HM74A) mediates nicotinic acid–induced flushing. J Clin Invest (2005) 115:3634–40. doi: 10.1172/JCI23626
104. Semple G, Skinner PJ, Gharbaoui T, Shin YJ, Jung JK, Cherrier MC, et al. 3-(1H-tetrazol-5-yl)-1,4,5,6-tetrahydro-cyclopentapyrazole (MK-0354): a partial agonist of the nicotinic acid receptor, G-protein coupled receptor 109a, with antilipolytic but no vasodilatory activity in mice. J Med Chem (2008) 51:5101–8. doi: 10.1021/JM800258P
105. Lai E, Waters MG, Tata JR, Radziszewski W, Perevozskaya I, Zheng W, et al. Effects of a niacin receptor partial agonist, MK-0354, on plasma free fatty acids, lipids, and cutaneous flushing in humans. J Clin Lipidol (2008) 2:375–83. doi: 10.1016/J.JACL.2008.08.445
106. Kothawade PB, Thomas AB, Chitlange SS. Novel niacin receptor agonists: a promising strategy for the treatment of dyslipidemia. Mini-Reviews Medicinal Chem (2021) 21:2481–96. doi: 10.2174/1389557521666210125144921
Keywords: free fatty acid, succinate, hydroxy carboxylic acids, G protein-coupled receptor, adipose, inflammation, metabolite signalling
Citation: Duncan EM, Vita L, Dibnah B and Hudson BD (2023) Metabolite-sensing GPCRs controlling interactions between adipose tissue and inflammation. Front. Endocrinol. 14:1197102. doi: 10.3389/fendo.2023.1197102
Received: 30 March 2023; Accepted: 13 June 2023;
Published: 06 July 2023.
Edited by:
Kanhaiya Singh, Purdue University Indianapolis, United StatesReviewed by:
Dayoung Oh, University of Texas Southwestern Medical Center, United StatesHuh Jin Young, Sogang University, Republic of Korea
Copyright © 2023 Duncan, Vita, Dibnah and Hudson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Brian D. Hudson, QnJpYW4uSHVkc29uQEdsYXNnb3cuYWMudWs=
†These authors have contributed equally to this work and share first authorship