 Jarrell Imamura1*
Jarrell Imamura1* Shinjini Ganguly1*
Shinjini Ganguly1* Andrew Muskara1
Andrew Muskara1 Ross S. Liao2
Ross S. Liao2 Jane K. Nguyen2,3
Jane K. Nguyen2,3 Christopher Weight2
Christopher Weight2 Christopher E. Wee1
Christopher E. Wee1 Shilpa Gupta1
Shilpa Gupta1 Omar Y. Mian1
Omar Y. Mian1- 1Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH, United States
- 2Glickman Urologic Institute, Cleveland Clinic, Cleveland, OH, United States
- 3Department of Pathology, Robert J. Tomsich Pathology and Laboratory Medicine Institute, Cleveland Clinic, Cleveland, OH, United States
Androgen deprivation therapy is a cornerstone of treatment for advanced prostate cancer, and the development of castrate-resistant prostate cancer (CRPC) is the primary cause of prostate cancer-related mortality. While CRPC typically develops through a gain in androgen receptor (AR) signaling, a subset of CRPC will lose reliance on the AR. This process involves genetic, epigenetic, and hormonal changes that promote cellular plasticity, leading to AR-indifferent disease, with neuroendocrine prostate cancer (NEPC) being the quintessential example. NEPC is enriched following treatment with second-generation anti-androgens and exhibits resistance to endocrine therapy. Loss of RB1, TP53, and PTEN expression and MYCN and AURKA amplification appear to be key drivers for NEPC differentiation. Epigenetic modifications also play an important role in the transition to a neuroendocrine phenotype. DNA methylation of specific gene promoters can regulate lineage commitment and differentiation. Histone methylation can suppress AR expression and promote neuroendocrine-specific gene expression. Emerging data suggest that EZH2 is a key regulator of this epigenetic rewiring. Several mechanisms drive AR-dependent castration resistance, notably AR splice variant expression, expression of the adrenal-permissive 3βHSD1 allele, and glucocorticoid receptor expression. Aberrant epigenetic regulation also promotes radioresistance by altering the expression of DNA repair- and cell cycle-related genes. Novel therapies are currently being developed to target these diverse genetic, epigenetic, and hormonal mechanisms promoting lineage plasticity-driven NEPC.
1 Introduction
In American men, prostate cancer (PCa) is among the most common malignancies and is projected to become the second leading cause of cancer related death in 2023, accounting for an estimated 29% of total new cases and 11% of total cancer related deaths (1). A rare but particularly lethal subtype is neuroendocrine PCa (NEPC), with a median survival of 7 months following diagnosis (2). The prevailing hypothesis behind the development of NEPC is the transdifferentiation of an adenocarcinoma to a neuroendocrine (NE) lineage (3). Generally, this NE phenotype is positive for markers of other high-grade NE carcinomas, such as chromogranin A(CHGA), neuron-specific enolase (NSE), synaptophysin (SYP), and CD56, and negative for luminal prostate differentiation markers (4). The transition to a NE phenotype is strongly associated with lineage plasticity. Lineage plasticity refers to the ability of cells to transition from one committed development program to another, allowing for multiple, varying phenotypes to arise from a singular genotype in response to environmental stimuli (5). This property is often maintained in cancer cells and is exploited to promote opportunistic adaptation and progression with plastic phenotypes being enriched in advanced stages of cancer. Therefore, lineage plasticity allows cancer cells to derive therapy resistance by reprogramming to a phenotype that is indifferent towards cellular pathways being targeted by therapy (3). Indeed, the lineage transition to a NE subtype is one mechanism by which PCa evades androgen receptor (AR)-targeting treatment by shifting its cellular phenotype from an AR-dependent adenocarcinoma to an AR-indifferent NE or small-cell carcinoma.
Unique to PCa is its reliance on the AR to regulate tumor progression via transcriptional regulation of AR targeted genes (6) and the activation of AR-targeted signaling pathways (7). Targeting the AR driven signaling cascade has proven effective in treating PCa (8) and has led to the emergence of androgen deprivation therapy (ADT) as a mainstay of treatment for locally advanced and metastatic PCa (9, 10). However, disease control using ADT alone is limited, and resistance is common, typically occurring after an average of 2-3 years of treatment (11). Recurrent tumors, known as castration-resistant prostate cancer (CRPC), are typically more aggressive, proliferating despite castrate levels of androgen. The emergence of a CRPC subtype is thought to be driven via androgen-dependent and androgen-independent mechanisms. The majority of CRPCs retain a degree of dependency on the AR and androgen signaling but escape ADT by a variety of mechanisms, including increased AR expression, expression of mutant AR variants, AR splice variant (AR-Vs) expression, and intratumoral steroid hormone synthesis (8). However, increased usage of AR-targeting treatments has been associated with an increased recognition of androgen-independent CRPC variants, with NEPC historically being the prototypic example of AR (-) CRPC (12, 13). Additionally, the AR’s role as a differentiation factor suggests its loss may play a significant role in lineage plasticity between CRPC subtypes (14), although numerous other factors have been implicated in driving PCa lineage plasticity. Notably, the development of CRPC may also promote resistance to radiotherapy (RT) as molecular pathways governing the transition to CRPC are often implicated in radioresistance (15–18). In this review, we discuss the genetic and epigenetic drivers of PCa lineage plasticity and link emergent phenotypes to treatment resistance. Specifically, we focus on the emergence of NEPC following ADT and the implications NE and other AR-indifferent phenotypes have on ADT and RT resistance.
2 Molecular mechanisms driving treatment emergent lineage plasticity
2.1 AR
For decades, androgens have been implicated in driving PCa development and progression through their interactions with the AR, a ligand-dependent nuclear transcription factor (6). With the androgen axis playing such a significant role in tumor progression, treatment plans frequently involve suppressing the androgen signaling axis via surgical or medical castration (19, 20). Several categories of modern ADT drugs can be used to accomplish medical castration, including luteinizing hormone-releasing hormone agonists/antagonists, AR antagonists, and androgen synthesis inhibitors (19, 20). First-generation AR antagonists were introduced in the late 1980s and include flutamide, nilutamide, and bicalutamide (21–23). These drugs are non-steroidal anti-androgens that act as selective competitive antagonists (22). Initial treatment with first-generation AR agonists is effective at reducing tumor burden and decreasing serum prostate-specific antigen levels; however, within a few years, many patients receiving ADT manifest early signs of biochemical progression, signaling the emergence of CRPC (11). Despite its classification, the majority of CRPC is still AR-dependent. As such, treatment with second-generation anti-androgens, such as enzalutamide, apalutamide, and darolutamide, have demonstrated clinical benefit, though typically for a limited duration. Treatment of CRPC with enzalutamide increased time to progression by 8.3 months in the AFFIRM trial, 11.2 months in the PREVAIL trial, and 19.4 months in the TERRAIN trial (21). Further progression following second-generation anti-androgen treatment can be mediated by the activation of the AR through secondary alterations to the AR gene, AR bypass or crosstalk mechanisms, or other means. However, increasing evidence suggests that a subset of CRPC tumors evade potent hormone therapy through a loss rather than a gain in AR function (24). These androgen-indifferent tumors become less dependent on AR signaling, a process associated with lineage plasticity, loss of luminal markers, and acquisition of NE features (24). Moreover, this AR-independent state is defined by a reduced reliance on the AR signaling pathway to promote tumor progression and the activation of other factors that promote cell survival such as MYCN, AURKA, ONECUT2, and BRN2 (3, 25, 26). Recently, it has been clarified that lineage plasticity is not a binary switch but occurs on a continuum with identifiable, preexisting clonal subsets. These subsets include AR activity-low tumors that exhibit decreased AR signaling despite continued AR expression, amphicrine tumors that express an active AR and NE program, double negative tumors that lack both AR and NE gene expression, and finally, NEPC tumors that lack AR expression and express a NE phenotype (12). Among these NE-negative subsets, NEPC is the dominant form, accounting for as much as 10-20% of CRPC (27, 28). Furthermore, NEPC incidence is expected to increase with widespread use of modern androgen receptor pathway inhibitors (ARPIs) (28).
2.2 RB1, TP53, and PTEN
Recent genomic analysis has revealed several alterations enriched in NEPC. Mutations in the tumor suppressor genes RB1, TP53, and PTEN are individually among the most common genetic alterations in PCa. However, the concomitant loss of more than one of these critical factors is enriched in the NE subtype (4, 29, 30). For instance, concurrent RB1 and TP53 loss was present in 53.3% of NE-CRPC biopsies, compared with 13.7% of castration-resistant adenocarcinoma (4). Emerging data places special emphasis on the loss of RB in promoting phenotypic plasticity towards NEPC (31). Interestingly, aggressive NE tumors from other organs also exhibit alterations in these tumor suppressor genes (32). Investigation of a combined knockdown of RB1 and TP53 in PTEN-null LNCaP cells resulted in reduced AR expression, enzalutamide resistance, and activation of a NEPC program. Additionally, SOX2 knockdown stopped the lineage plasticity program induced by RB1/TP53 loss, signaling the importance of lineage plasticity related transcription factors in driving the NEPC transition (33). In vivo comparison between PTEN loss (SKO), PTEN and RB1 loss (DKO), and PTEN, RB1, and TP53 loss (TKO) has demonstrated lower AR expression and activation of NEPC programming in the DKO and TKO groups. Notably, the DKO and TKO tumors exhibited increased expression of SOX2, a key downstream effector for NEPC lineage plasticity (34). Moreover, loss of RB1, TP53, and PTEN reportedly induce NE transdifferentiation by upregulating NE program-related transcription factors, such as SOX2, SOX11, and EZH2 (33–35). Evidently, RB1, TP53, and PTEN loss are important factors in the transition to NEPC; however, evidence suggests that these genetic alterations alone are insufficient to promote lineage plasticity. For instance, there are numerous instances of NEPC tumors not exhibiting these mutations, and non-NE CRPC tumors have been observed exhibiting these genetic alterations (4, 36). Moreover, RB1/TP53 knockouts of PTEN-deficient LNCaP cells resulted in decreased AR transcriptional activity but did not promote an increase of NEPC gene expression. This suggests necessity without sufficiency and that other reprogramming factors absent from these analyses may be required to induce a NE phenotype in PCa (36). These and other alterations that can be considered hallmarks of phenotypic lineage plasticity in prostate cancer are detailed schematically in Figure 1.
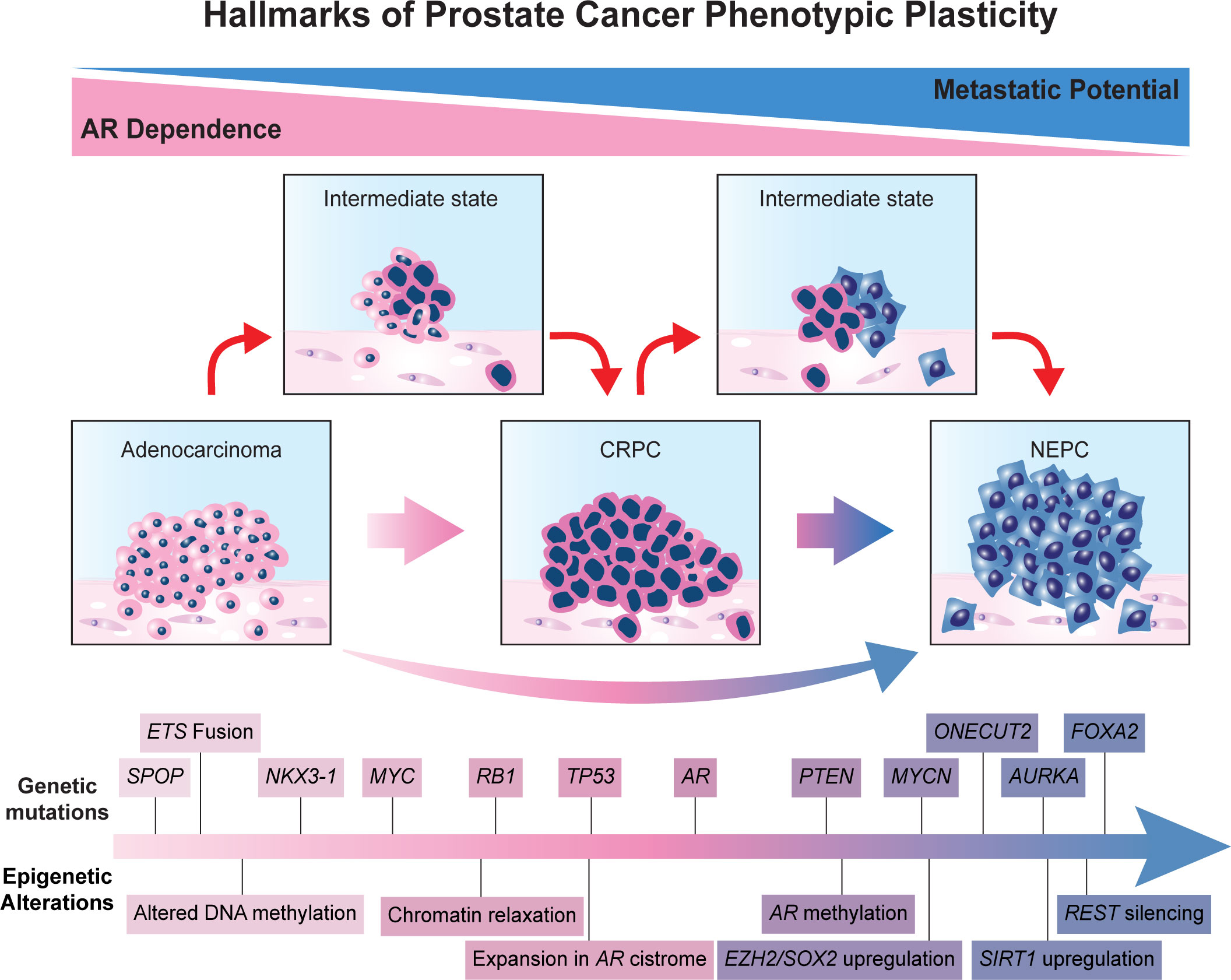
Figure 1 Hallmarks of prostate cancer phenotypic plasticity.
2.3 MYCN and AURKA
The phenotypic transition that leads to NEPC is also frequently associated with amplification of MYCN and AURKA, with concurrent amplification of these two genes occurring in 40% of NEPC compared to 5% of prostate adenocarcinoma (37). Preclinical models have found MYCN to be a key driver of NEPC phenotypes (37–39). MYCN overexpression in models with PTEN loss or AKT overexpression resulted in tumors with low AR expression and signaling, upregulation of NE markers, and resistance to AR targeting therapies (38, 39). Moreover, these tumors, along with MYCN overexpressing cell lines, demonstrated enriched expression of embryonic stem cell and active epithelial-mesenchymal transition (EMT) program gene sets (38, 39). Additionally, MYCN can be stabilized by dimerization with its allosteric protein partner, AURKA (40). Although pharmacological inhibition of MYCN is difficult, AURKA inhibitors (E.g., alisertib, CD532) can be used to indirectly target MYCN (38, 41, 42). MYCN protein levels decreased significantly in response to the anti-tumor activities of AURKA inhibitor treatment (39). Additionally, a mechanistic link between mutated TP53 and elevated AURKA levels has been described (43). Collectively these data point to MYCN as a central mediator of cellular plasticity during NE differentiation.
2.4 FOXA1 and FOXA2
Changes in the expression of the Forkhead Box A (FOXA) family of proteins, especially FOXA1 and FOXA2, have been implicated in NEPC development. The FOXA family is a group of pioneer transcription factors that can access condensed chromatin, facilitating the local binding of other regulatory factors (44). FOXA1 expression has been shown to be transiently increased in localized PCa but is lowered following progression to CRPC (45, 46) and further decreased in NEPC (47). Loss of FOXA1 promotes NE differentiation by upregulating CXCL8 and CXCL8-activated MAPK/ERK phosphorylation. Moreover, FOXA1 is reported to play a reprogramming role in lineage conversion (48), with FOXA1 mutations altering its pioneering function and perturbing normal luminal epithelial cell differentiation programs (49). FOXA1 interacts with the TET1 protein to mediate local DNA demethylation and histone 3 lysine 4 methylation (50). On the other hand, FOXA2 overexpression has been reported in genetically engineered mouse models of NEPC, a result consistent with clinical NEPC specimens (51, 52). However, FOXA2 expression is not detected in prostate adenocarcinoma samples (51). FOXA2 induces prostate adenocarcinoma-to-NE lineage transition through cooperation with HIF1 and REST (53, 54), and FOXA2 knockdown reverses this process. Interestingly, FOXA2 has been shown to positively regulate KIT expression in NEPC (55), a receptor tyrosine kinase that is positively correlated with NEPC score and SYP, ENO2, and EZH2 expression (55, 56). Inhibiting the KIT pathway resulted in NEPC growth suppression in both human and mouse NEPC, suggesting the potential utility of targeting KIT in the treatment of NEPC (55). Nevertheless, a better understanding of the roles FOXA1/FOXA2 hold in regulating lineage plasticity and their crosstalk with epigenetic modulators is still key to discover new therapeutic strategies.
3 Epigenetic dysregulation affecting lineage plasticity in PCa
Epigenetic modifications (e.g., DNA methylation, histone modification, and chromatin remodeling) are critical regulators of transcriptional states, are known drivers of carcinogenesis, and are tightly associated with NEPC lineage plasticity. Despite similarities in their genomic profiles, NEPC and prostate adenocarcinoma have markedly distinct epigenetic landscapes (4, 37, 57). Notably, AR signaling loss is strongly associated with epigenetic reprogramming and is essential for maintaining luminal cell differentiation in the prostate (58). Moreover, significant evidence demonstrates the importance of epigenetics in the transition to a NEPC phenotype. For instance, epigenetic dysregulation in NEPC has been reported in pathways involving cell-cell adhesion, development, epithelial-mesenchymal transition (EMT), and stem cell regulation and maintenance (4).
3.1 DNA methylation
DNA methylation is the most widely studied epigenetic modification and has implications in carcinogenesis, therapy resistance, and lineage plasticity. DNA methylation refers to the covalent attachment of a methyl group to DNA cytosine residues, forming 5-methylcytosine (5mC). De novo DNA methylation is catalyzed by the DNA methyltransferases (DNMT) DNMT3a and DNMT3b, while maintenance DNA methylation is catalyzed by DNMT1 (59). Inversely, DNA demethylation is mediated by ten eleven translocation (TET) enzymes. DNA methylation primarily occurs at CpG-rich regions called ‘CpG islands.’ Notably, CpG islands are preferentially located in the 5’ UTR of genes and make up approximately 60% of human gene promoters. Methylation of gene promoters predominantly results in gene silencing by either preventing or promoting the recruitment of regulatory elements (60). Global DNA hypomethylation is frequently observed in cancer and can lead to genomic instability and proto-oncogene activation (60). DNA hypomethylation is postulated to occur early in the development of many types of cancer, driving carcinogenesis. However, PCa breaks away from this common view, instead occurring late in disease progression and likely is involved in the formation and propagation of metastases (61). Site-specific CpG island hypermethylation leading to aberrant gene silencing is frequently observed in PCa (60, 62). DNA methylation is known to disrupt pathways involved in DNA damage repair, hormonal response, tumor-cell invasion and metastasis, and cell cycle control (63, 64). Additionally, DNA methylation can regulate lineage plasticity and therapy resistance through promoter hypermethylation of CRIP1, FLNC, RASGRF2, RUNX2, and HS3ST2. These genes are important for lineage commitment and differentiation and have been associated with tumor recurrence (65–68). As such, DNA methylation plays a significant role in driving NE transdifferentiation from an adenocarcinoma. For instance, in NEPC samples, ASXL3 and SPDEF genes were found to be hypermethylated, while the NEPC marker INSM1 and the plasticity gene CDH2 were hypomethylated (69).
DNA methylation has also been implicated in regulating AR signaling. Specifically, DNA hypermethylation of the AR promoter, driven by PTEN, RB1, and TP53 loss, can partially explain the inactivation of canonical AR signaling in NEPC (70, 71). For instance, in AR-null cell line models and patient tumor samples, DNA methylation has been implicated in regulating AR expression (73–75). Treatment with AR-negative cell lines, DuPro, TSU-PR1, and DU145, with the DNMT inhibitor 5-azacitidine resulted in AR re-expression and concomitant demethylation of the AR promoter (73, 75). DNMTs can also regulate AR signaling in a methylation independent manner. Prostate epithelial cells lacking RB1 demonstrate increased DNMT1 expression. RB1 negatively regulates the transcription factor E2F1, which can activate DNMT1 expression by interacting with the DNMT1 promoter. Overexpression of E2F1 was sufficient to repress AR expression and AR promoter-driven reporter genes (71). Similarly, DNMT1 knockdown renewed AR expression in AR-negative human prostate epithelial cells. DNMT1 directed ChIP analysis showed association of DNMT1 with the AR promoter; however, a lack of de novo methylation at the AR minimal promoter suggests that DNMT1 represses AR expression via a methylation-independent mechanism (72). These data present DNMT inhibitors as a promising class of drugs for the treatment of AR-null PCa.
3.2 Post translational histone modification
Significant evidence has revealed that post-translational histone modifications mediate numerous biological processes in cancer via alterations in chromatin conformation. These modifications include methylation, acetylation, phosphorylation, and ubiquitination; however, histone-related gene expression regulation is predominately regulated by histone methylation and acetylation. Histone acetylation is driven by histone acetyltransferases (HATs) and is deacetylated by histone deacetylases (HDACs). Similarly, histone methylation is catalyzed by histone methyltransferases (KMTs) and reversed by histone demethylases (KDMs). Histone acetylation is commonly associated with transcriptional activation, resulting from alterations in histone net charge. This leads to an open chromatin conformation that is more accessible to transcription factor binding and the regulation of gene transcription. Unlike histone acetylation, methylation does not exclusively lead to translational activation but is instead associated with either gene activation or repression.
3.2.1 Histone methylation and demethylation
AR expression loss has been linked to the repression of histone methylation. Enrichment of repressive histone modifications H3K27me3 and H3K9me2 has been observed in AR-null NEPC PDXs (76). Enhancer of Zeste Homologue 2 (EZH2), the catalytic subunit of the Polycomb Repressive Complex 2 (PRC2), is a master regulator of epigenetic rewiring in NEPC and is the KMT responsible for H3K27me3 marks. Upregulation of EZH2 is a well-established feature of NEPC (4, 37, 39, 77). Genes repressed by EZH2 are also downregulated in tumors from NEPC patients, with EZH2 inhibition reactivating AR signaling, decreasing NE target gene expression, and re-sensitizing tumors to enzalutamide (34). Mechanistically, EZH2 cooperates with lineage-guiding transcription factors to epigenetically regulate gene expression and lineage specificity. Notably, EZH2 has been shown to cooperate with MYCN to suppress AR signaling (39). EZH2 has also been shown to synergize with other epigenetic modifiers, regulating chromatin conformation to promote lineage plasticity. For instance, EZH2 has been linked to DNMT activity, potentially marking genes for de novo DNA methylation via a scaffolding mechanism mediated by the long ncRNA HOTAIR (78–80). Similarly, HOTAIR can scaffold EZH2 and lysine-specific demethylase 1 (LSD1), a H3K4 and H3K9 demethylase. EZH2:LSD1 coordinates to repress developmental genes, promoting a cellular state of elevated plasticity (81).
Several studies report that EZH2 mediates NE differentiation. For example, EZH2 can be activated by transcription factor 4 (TCF4), a key transcription factor involved in Wnt/-catenin signaling. Elevated Wnt signaling is a common feature in NEPC, and TCF4 inhibition has been shown to preclude the transdifferentiation of adenocarcinoma to NEPC, following ADT (82). Furthermore, EZH2 activity has been tied to cAMP-response element binding protein (CREB) activation. Inhibition of EZH2 can prevent CREB-induced H3K27 methylation and NE differentiation (83). Finally, MEK-ERK signaling has been shown to regulate EZH2 transcriptional upregulation and recruitment to the promoter of E-cadherin. Therefore, MEK-ERK signaling can facilitate epithelial-mesenchymal plasticity via EZH2 regulation (84, 85). These studies demonstrate EZH2’s role as a master regulator of NEPC lineage plasticity, and its inhibition is a promising strategy for novel NEPC treatment.
In addition to histone methylation, histone demethylation also plays a significant role in regulating NEPC. LSD1 is known to demethylate H3K4 and lysine residues on several non-histone proteins, including, TP53, E2F1, DNMT1, and HIF-1. Notably, LSD1 is an important regulator of AR transcriptional activity, suppressing the transcription of AR target genes (86). Indirect demethylation of H3K9 on AR target genes and cell cycle genes can also be facilitated by LSD1 (87). Interactions between LSD1 and another master epigenetic regulator SRRM4 can produce a neuronal-specific isoform of LSD1, called LSD1 + 8a, that is exclusively expressed in NEPC as opposed to adenocarcinoma. SRRM4 is overexpressed in NEPC and is a powerful driver of transdifferentiation in NEPC (88). This splice variant of LSD1 may act as a biomarker for NEPC and contribute to lineage plasticity in NEPC (28). These epigenetic factors may provide novel targets for NEPC treatment.
3.2.2 Histone acetylation, deacetylation, and chromatin remodeling
Recent studies have shown that the use of modern ARPIs can result in widespread changes in chromatin structure and contribute to lineage plasticity. Histone acetylation and deacetylation are two factors that affect chromatin accessibility (89–92). Notably, the HDAC SIRT1 is upregulated in NEPC, and its overexpression has been shown to promote lineage plasticity (93). The precise mechanism by which SIRT1 promotes lineage plasticity is unknown; however, in breast cancer (94), liver cancer stem cells (95), and bone marrow-derived mesenchymal stem cells (96), SIRT has been shown to promote upregulation of SOX2. SIRT1 mediated SOX2 upregulation may be an avenue whereby SIRT1 promotes lineage plasticity in NEPC, though evidence remains preliminary and evolving.
Other chromatin remodelers in PCa can affect lineage plasticity. CDH1 loss, for example, has been shown to promote neuronal differentiation and enzalutamide resistance (92). CDH1 loss resulted in global changes in chromatin accessibility, notably, increasing accessibility for four transcription factors associated with non-luminal lineage program activation. Deletion of each transcription factor by CRISPR-Cas9 knockout re-sensitized cells to enzalutamide (97). The restrictive element-1 silencing transcription factor (REST) cooperates with AR and corepressors such as EZH2 and LSD1 to suppress neuronal differentiation (98, 99). REST silencing attenuated AR signaling and increased NE lineage markers (100). Moreover, loss of REST activity via a splicing-in event by SRRM3/4 can promote expression of BAF53B rather than BAF53A, subunits of the chromatin remodelers, neuron specific BAF (nBAF) complex and canonical BAF complex respectively (12). Exchanging the subunits BAF53A and BAF45A for BAF53B and BAF45B in the nBAF complex has been shown to promote a chromatin switch to a differentiated neuronal phenotype in post-mitotic neurons (101). Moreover, BAF53B and BAF45B are both highly expressed in NEPC and absent in adenocarcinoma (90). Lastly, it has been shown that BET bromodomain proteins are important in NEPC. Specifically, the BET bromodomain protein BRD4, which recognizes H3K27 acetylation, can cooperate with E2F1 to induce lineage plasticity. BET bromodomain inhibition suppressed the induction of lineage plasticity driving genes and suppressed the growth of treatment emergent NEPC cells high in E2F1 (89). All together, these data demonstrate the importance of epigenetic factors in driving lineage plasticity toward a NEPC phenotype.
4 Early therapeutic resistance in localized PCa
4.1 Early hormone therapy resistance
As previously described, early hormone therapy failure often results in AR-dependent CRPC. Several mechanisms promote the progression to AR-dependent CRPC, including increased AR amplification and mutation, AR-Vs expression, and intra-tumoral steroid hormone synthesis (8).
Increased AR expression allows PCa to circumvent low androgen levels by hypersensitizing tumor cells to androgens, promoting disease progression (102). Several factors can promote increased AR expression, including gene amplification, increased translation, and decreased degradation (8). Moreover, AR expression can be enhanced by increased protein stability via the E3 ligase MID1 (103). AR amplification has been identified in a significant proportion of CRPC cell lines, ranging from 30-80% (104, 105). Several point mutations in the AR gene have been identified that can result in increased AR activity, despite low androgen levels. For instance, mutations in the AR ligand binding domain can decrease ligand specificity, allowing alternate steroid hormones, such as -DHT, progesterone, or DHEA, to activate AR signaling (106). Notably, other mutations that confer resistance to second generation AR agonists, such as F876L, have been identified (107).
Several alternative AR-Vs that confer ADT resistance have been identified. These splice variants often emerge in late stages of PCa, are constitutively active due to the loss of the C-terminus ligand binding domain, and often correlate with a poor prognosis (108, 109). Several common AR-Vs are ARv1, ARv7, and ARv567, with ARv7 being the most well studied (110, 111). ARv7 is constitutively active because it lacks the canonical AR ligand binding site (111). Moreover, ARv7 can heterodimerize with AR to translocate to the nucleus where it acts as a transcription factor (112). ARv7 has been shown to regulate both AR-regulated genes as well as a unique set of AR-independent genes (111). Given the correlation between AR splice variant expression and poor prognosis, the targeting of AR variants may offer novel CRPC treatment with the ARv7 inhibitor niclosamide being recently tested (113).
Another key mechanism contributing to AR-dependent CRPC is the intra tumoral synthesis of androgen, including testosterone and dihydrotestosterone (DHT), which originate from adrenal precursor steroids such as dehydroepiandrosterone (DHEA) (114, 115). Several enzymes and genes involved in intra tumoral androgen synthesis have been identified as potential targets for diagnosis and treatment. One such target is 3β-hydroxysteroid dehydrogenase isoenzyme-1 (3βHSD1, encoded by HSD3B1), which catalyzes the rate-limiting step in the metabolic conversion from DHEA to testosterone and DHT in the adrenal gland. A specific germline missense-encoding variant of HSD3B1(1245A>C) leads to a divergence of enzyme level and downstream androgen synthesis. HSD3B1(1245A) is known as an adrenal-restrictive allele as it codes for an enzyme that is degraded more rapidly, while HSD3B1(1245C), an adrenal-permissive allele, codes for a stable enzyme resistant to proteasomal degradation that promotes robust conversion from DHEA to DHT (116–118). Numerous studies have shown that prostate cancer patients carrying the HSD3B1(1245C) variant are more likely to become resistant to ADT and progress to CRPC and have worse survival outcomes. However, to date there is no known disparity of overall survival time related to HSD3B1.
The upregulation of glucocorticoid receptors (GR) is one AR-independent mechanism known to mediate ADT resistance in PCa. GRs are a part of the nuclear receptor superfamily of transcription factors (119). Like AR, GR undergoes a conformational change in the cytoplasm and is translocated to the nucleus after binding with androgens or glucocorticoids. Once inside the nucleus, GR can bind to androgen responsive elements and regulate the expression of target genes (120). Notably, AR and GR bind to nearly identical DNA motifs (121). One implication of this finding is that amplified GR signaling may be redirected to circumvent AR loss of function under castrate conditions (18). Notably, GR regulates a significant number of genes that are AR pathway-specific (122), and GR activity has been found to replace AR activity in CRPC, following enzalutamide treatment (123, 124).
4.2 Radioresistance in PCa
Radiotherapy is a cornerstone of curative PCa treatment, and its combination with ADT has been shown to significantly increase patients’ overall survival (125). Nevertheless, approximately 30-50% of high-risk, clinically localized PCa cases recur within five years of curative intent radiotherapy. While the majority of these recurrences are out of field metastases, PSMA PET scans are shedding increasing light on non-trivial local recurrence rates and offer an opportunity for deeper study of mechanisms driving resistance to radiotherapy/hormone therapy combinations (126). The intra-tumoral genetic and epigenetic heterogeneity observed in PCa has been theorized as a source of resistant subpopulations (127). Moreover, the selective pressure applied by RT may trigger evolutionary adaptations resulting in acquired radioresistance promoted by epigenetic and transcriptional reprogramming (128). Both acquired and intrinsic radioresistance involves changes in biological mechanisms controlling DNA repair, hypoxia response, cell proliferation and survival, EMT, apoptosis inhibition, and autophagy (129). As described below, extensive experimental evidence also supports the role of an aberrant epigenome in driving a radioresistant phenotype.
4.2.1 Alternative AR signaling driving PCa radioresistance
One inference that can be drawn from the significant clinical benefit observed with ADT and RT combinations is that their anti-neoplastic effects are at least additive and more likely synergistic. It has been demonstrated that AR signaling promotes radioresistance by upregulating the transcription of DNA double strand break (DSB) repair genes. Treatment with second-generation anti-androgens was shown to downregulate DNA repair genes in a CRPC model. Moreover, primary PCa was observed to displays a wide spectrum of AR transcriptional output that correlates with the expression of a set of DNA repair genes. The addition of androgens to ionizing radiation (IR) treated PCa cells enhanced DNA damage repair and treatment with anti-androgens increased DNA damage and decreased clonogenic survival (130). Similarly, increased AR expression and activity was observed following RT in several human PCa models, both in vitro and in vivo. AR expression was also correlated with survival in vitro and time to tumor progression in vivo. Finally, AR pathway upregulation was found in nearly 20% of patients following RT (131). Taken together, these data demonstrate the strong synergistic effect of ADT and RT. This underscores the important role of AR signaling in therapeutic resistance to genotoxic treatments.
Given the association between AR signaling and radioresistance in localized hormone sensitive disease, it follows that castrate-resistance is similarly tied to radiation response. A possible pathway leading to radioresistance in CRPC is the emergence of AR-Vs. One study has shown that AR-Vs can promote non-homologous end joining (NHEJ) repair when canonical AR signaling is blocked by ARPI (132). Similarly, others have used CRISPR-engineered PCa cell lines to show that AR-Vs can desensitize cells to IR when canonical AR signaling is disturbed (133). ARv7 is the most clinically relevant of the AR-Vs and was shown to significantly promote the DNA damage response (DDR) of PCa cells following severe DNA damage. ARv7 expression was sufficient to upregulate both homologous recombination (HR) and NHEJ in PCa by forming a positive regulatory loop with poly ADP-ribose polymerase 1 (PARP-1) (17). Interestingly, epigenetics seem to play a role in ARv7 expression in PCa via the inclusion of a cryptic exon. The KDM Jumonji domain containing (JMJD) 1A, an eraser of H3K9 methylation, can complex with the splicing factor hnRPNF. This complex binds to methylated H3K9 regions of the cryptic exon located in the intronic sequence between exons 3 and 4, favoring the inclusion of the cryptic exon and ARv7 production. Furthermore, JMJD1A knockdown has been shown to reduce ARv7 levels (134). With AR splice variants promoting radioresistance, inhibiting their expression may re-sensitize tumors to IR.
4.2.2 Epigenetic aberrations promote PCa radioresistance
Several epigenetic events affecting the DDR and cell cycle that can promote tumor radioresistance. Key proteins, such as ataxia-telangiectasia mutated (ATM), DNA dependent protein kinases, -H2AX, breast cancer gene 1/2 (BRCA 1/2), PARP-1, and RAD51, are important in DDR induction (135). Defective DDR pathways are frequently described as drivers of PCa tumorigenesis, with 15-30% of cases displaying DDR instability (136). Several genes, involved in DNA damage are frequently hypermethylated in PCa, including GPX3, MGMT, and ASC (137). Moreover, ATM may be hypermethylated, as its functional loss far exceeds its rate of mutation (138). The methylation of DDR genes frequently results in tumorigenesis due to increased chromosomal instability, but impaired DDR can leave tumors more susceptible to RT. For example, patients with ATM and BRCA1/2 mutations exhibited superior tumor response following RT (139).
Conversely, the methylation of genes involved in cell cycle regulation are more often associated with increased radioresistance. For instance, the cyclin-dependent kinase inhibitors (CDKs), CDKN1A (p21Cip1), CDKN1B (p27Kip1), and CDKN2A (p16Ink4A), reprimo (RPRM), stratifin (SFN), and CDK1 are all frequently hypermethylated and play various roles in tumor radioresistance (140). The CDKNs and their associated pathways have a variety of complex interactions regulating tumor suppression, DDR activation, cell cycle arrest, and apoptosis control (141). This regulation is accomplished by inhibiting cyclin-dependent kinases, with p21 and p27 inhibiting CDK2 and p16 inhibiting CDK4/6. Moreover, activation of p21 signaling has been found to promote radioresistance (142). Similarly, demethylation of p21 and p53 promoters in nasopharyngeal carcinoma restored and activated p21 and p53 activity, leading to cell cycle arrest and apoptosis, resulting in increased radioresistance (143). Cell phase change, due to decreased expression or loss of p27, has been shown to confer radioresistance in esophageal carcinoma and luminal breast cancer (144, 145). Depletion of p16 in cervical cancer led to increased radioresistance and higher self-renewal capability (146). Moreover, wild-type p16 expression was consistent with radiosensitivity in malignant melanoma cells when compared to homozygous p16 deficient cell lines (147). The tumor suppressor RPRM has previously been implicated in triggering p53-mediated G2 cell cycle arrest following DNA damage (148, 149) and negatively regulates ATM levels (150). Moreover, RPRM loss has been shown to confer radioresistance both in vitro and in vivo, marking RPRM as a potential target for cancer therapy and radiation protection (149, 150). SFN is known to mediate G2 arrest by preventing the nuclear translocation of the CDC2-cyclin B1 complex in response to DNA damage (151). In contrast to other hypermethylated cell-cycle related genes, the hypermethylation of SFN is not associated with greater radioresistance. Instead, cells that lose SFN frequently undergo mitotic catastrophe following DNA damage (151, 152). Interestingly, one study found the expression of SFN to increase with tumor progression; however, the driving mechanism behind the increased expression has not been investigated (151). Still, this group of cell cycle-regulating genes offers many novel targets for radiation protection, and the transient nature of these epigenetic changes may allow for the re-sensitization of radioresistant tumors. Nevertheless, more data are needed before these findings can be effectively translated into the clinic.
Post-translational histone modification is another class of epigenetic modifications that can affect PCa radioresistance. For instance, EZH2 is frequently upregulated and suppresses transcription via H3K27 methylation (153). In addition to driving lineage plasticity, EHZ2 has several cellular functions pertaining to the DDR, including regulating DDR elements and chromatin conformation following IR-induced damage. IR promotes H3K27 trimethylation by EZH2 around DNA damage sites. The resulting chromatin compaction is necessary for efficient DNA damage repair (154). Additionally, EZH2 can cooperate with BRCA1 to maintain a cancer stem cell signature, promoting radioresistance. Treatment with a global histone methylation inhibitor downregulates BRCA1 and EZH2 expression, inhibiting tumorigenicity and radioresistance (155). Similarly, in glioblastoma multiforme cells, EZH2 inhibition has been shown to significantly reduce H3K27 methylation and increased residual H2AX foci following IR. This significantly increased cell cycle arrest at the G2 checkpoint and apoptotic cell death (156). Finally, EZH2 inhibition has been shown to robustly downregulate DDR genes by preventing EZH2-mediated FOXA1 direct methylation (157). Another histone modifier that is frequently overexpressed in PCa is the histone methyltransferase KMT2D. KMT2D serves as an oncogene, promoting tumor growth and metastasis; however, it also has functions regarding radioresistance. Silencing of KMT2D has been shown to promote ROS-mediated DNA damage, leading to apoptosis and senescence. Moreover, KMT2D loss reduced enhancer activity markers H3K4me1 and H3K27ac, which blocked the binding of FOXO3, an important mediator of the cellular oxidative stress response, resulting in suppressed antioxidant gene expression (158). Additionally, KMT2D overexpression can also epigenetically activate PI3K/Akt and upregulate EMT and oncogenic pathways, promoting tumor radioresistance (159).
The PI3K/Akt/mTOR pathway can affect radioresistance in other ways. The Ras family of GTPases plays a key role in various basic cellular functions such as controlling cell proliferation, differentiation, and apoptosis, with Ras stimulations having a wide range of downstream signaling pathways. Phosphoinositide 3-kinases (PI3Ks) are one of the best characterized Ras effector groups, and their activation leads to the induction of the PI3K/Akt/mTOR pathway that affects cell growth, cell cycle, and cell survival (160). Uncontrolled activation of this signaling pathway has been associated with the development of cancer and tumor radioresistance. Radioresistant cell lines have demonstrated an increased activation in the PI3K/Akt/mTOR pathway (161). Moreover, dual inhibition of PI3K and mTOR has resulted in a significant reduction in radioresistance, increasing apoptosis, arrest of the G2/M phase, increased double strand breaks, and reduced cell cycle checkpoint inactivation and autophagy both in-vitro and in-vivo (162, 163). DNA methylation can promote PI3K/Akt/mTOR pathway-driven radioresistance via the hypermethylation of negative regulators of this pathway (164, 165). Moreover, H3 and H4 acetylation have been shown to activate Akt/mTOR, resulting in increased PCa progression and therapy resistance (166).
5 Therapeutic implications
With lineage plasticity driven CRPC posing a challenge for clinical management, novel therapeutic options targeting NEPC are needed. Several clinical trials focusing on agents that target NEPC have been reported, with one of the earliest Phase 2 trials testing the AURKA inhibitor alisertib. Investigators tested alisertib in NEPC patients as AURKA appears to be important in stabilizing MYCN (40). It should be noted that this trial did not require MYCN or AURKA upregulation for enrollment. Although this study did not meet its primary end point of 6-months progression free survival, the subpopulation of patients that exhibited increased AURKA expression (16%) did appear to have longer overall survival (167). Another NEPC targeting agents is rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate. This agent was tested in a Phase 1/2 trial that included 18 patients with NEPC, and a 10% objective response rate was observed (168). A comprehensive list of therapies targeting NEPC from 2017 to 2023 can be found in Table 1.
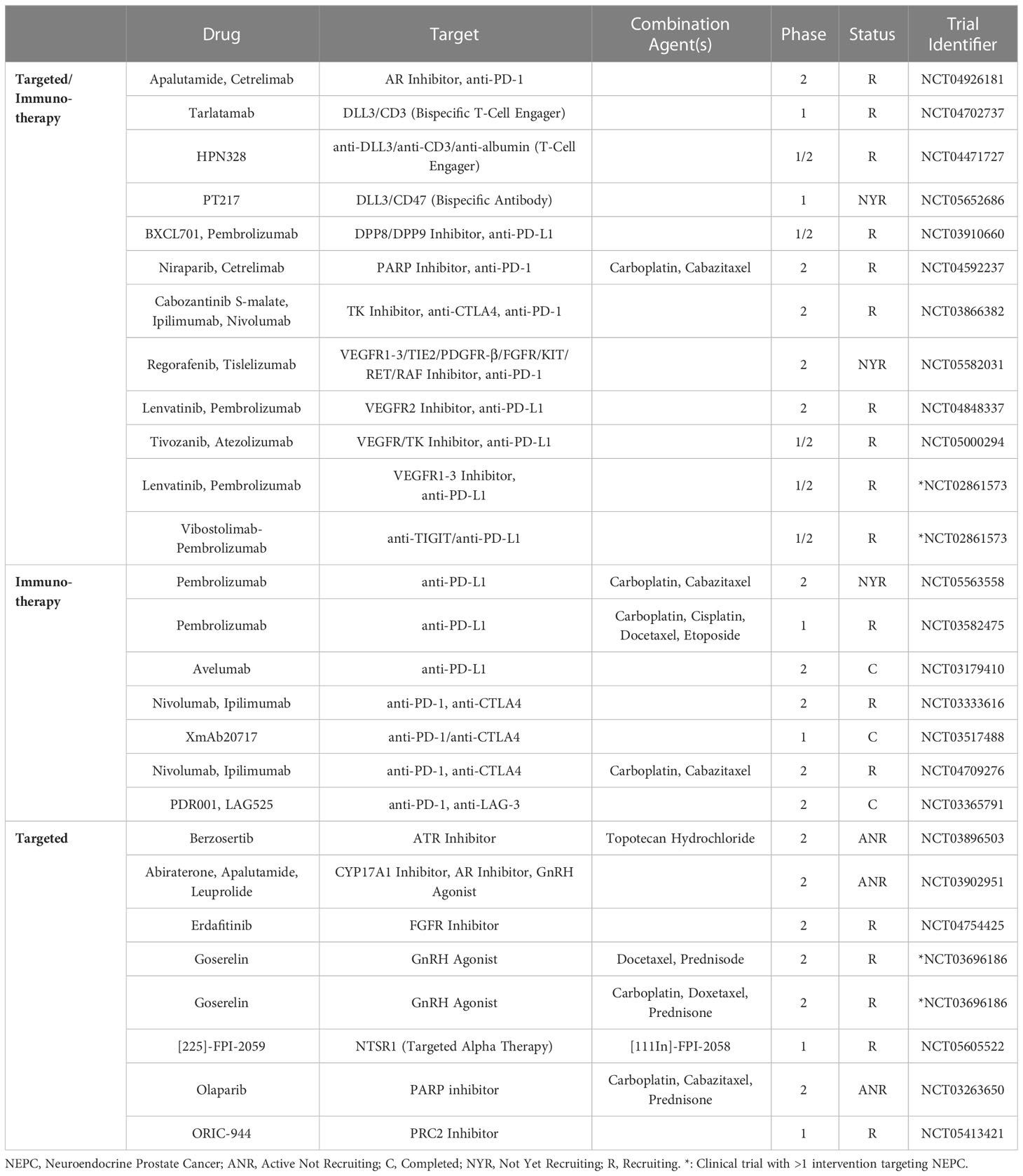
Table 1 Clinical trials of non-epigenetic therapies targeting neuroendocrine prostate cancer (2017-2023).
In addition to NEPC targeted therapies, drugs that target the NEPC epigenome and delay or reverse NE differentiation are promising. EZH2 is the most well studied epigenetic factor that is dysregulated in NEPC (39, 77). Preclinical NEPC models treated with EZH2 inhibitors have demonstrated attenuation of MYCN driven NEPC phenotypes and re-sensitization to ARPI treatment (39, 169). For instance, the EZH2 inhibitor, PF-06821497, is currently being tested in a Phase 1 study in metastatic CRPC patients (NCT03460977). Similarly, ORIC-944 is being tested in a Phase 1/1b trial in patients with metastatic CRPC (NCT05413421).
BET bromodomain proteins also offer a potential route for epigenetic targeting of NEPC. Preclinically, treatment with BRD4 inhibitors, either in monotherapy or in combination with ARPIs, have exhibited anti-tumor properties in PCa (170–172). Early investigation into the BET inhibitor ZEN-3694 has found that patients with NEPC tumors that exhibit high E2F1/BRD4 activity may be more susceptible to BET inhibition (89, 173). ZEN-3694 is now being tested in combination with enzalutamide and pembrolizumab in a Phase II trial, comprised of treatment emergent NEPC patients (NCT04471974). Similarly, the combination of ZEN-3694 and enzalutamide vs. enzalutamide alone is being tested in patients whose disease responded poorly to abiraterone, with the hope that this will enrich the cohort for patients with tumors that have undergone lineage plasticity (NCT04986423). A comprehensive overview of the current clinical trials for epigenetic drugs targeting NEPC can be found in Table 2.
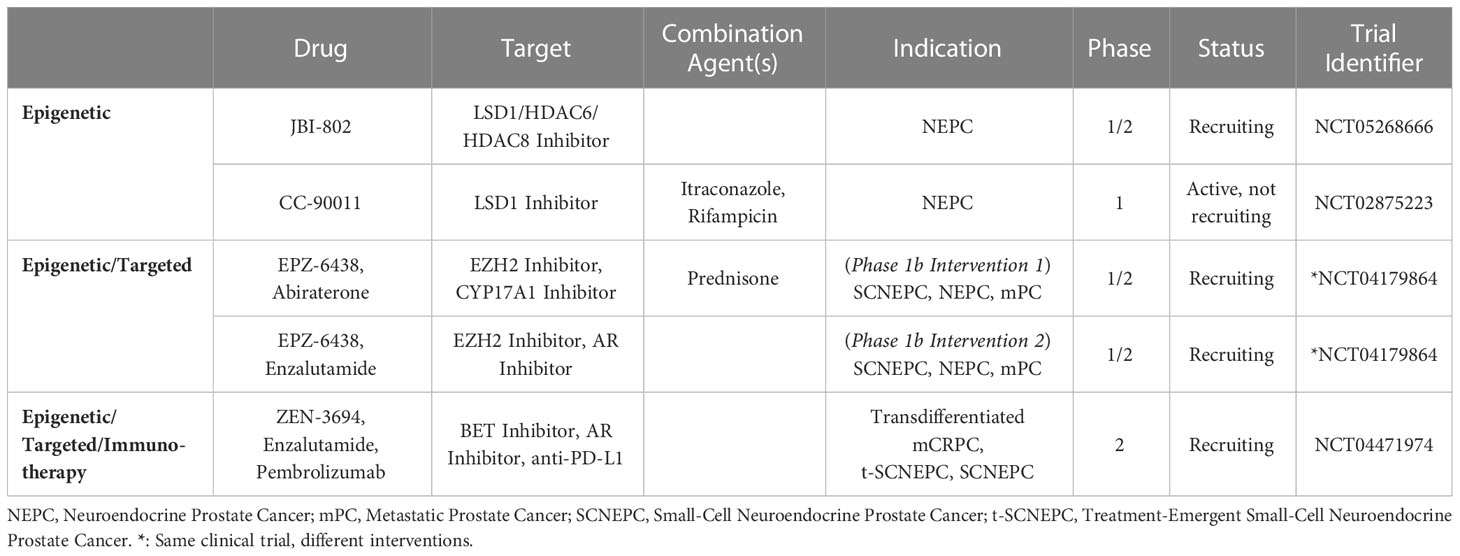
Table 2 Clinical trials of epigenetic therapies targeting neuroendocrine prostate cancer.
Other potential avenues for epigenetic targeting are LSD1 and the DNMTs. LSD1 is a KDM that is overexpressed in androgen-independent PCa and modulates FOXA1-dependent, AR-associated lineage plasticity and stem cell-associated gene expression (174, 175). Several clinical studies testing LSD1 inhibition have been proposed but were later terminated for various reasons (NCT02712905, NCT02217709, and NCT01253642); however, recruitment for Phase 1/2 testing of JBI-802, an LSD1/HDAC6 inhibitor, is currently ongoing (NCT05268666). Preclinical testing of DNMT inhibitors have shown promising results, with DNMT inhibition re-sensitizing resistant NE-like cell lines to ARPIs (176, 177). Notably, the DNMT inhibitors decitabine and azacytidine already have FDA approval for the treatment of myelodysplastic syndromes and could be repurposed for the treatment of NEPC. However, Phase 2 testing of DNMT inhibitors in CRPC did not show strong anti-tumor activity (178, 179).
Drugs that inhibit lineage plasticity by targeting the epigenome may also be used as radiosensitizers in the future. For instance, one pre-clinical study showed that EZH2 inhibition dramatically enhanced CRPC cells to genotoxic stress, demonstrating the potential utility in EZH2 inhibitors in sensitizing cancers that overexpress EZH2-activated DDR genes to genotoxic agents (157). Moreover, BRD4 is essential for the repair of DNA DSBs by mediating NHEJ, and BRD4 is negatively associated with outcome following RT (180). LSD1 also offers a potential option for radiosensitizing PCa, as pre-clinical data has shown that LSD1 knockdown can significantly enhance radiosensitivity (181). Finally, DNMT inhibitors and HDAC inhibitors have also shown radiosensitizing effects (182, 183). However, evidence supporting the efficacy of epigenetic targeting in radiosensitizing PCa has not progressed beyond pre-clinical investigation to date.
Despite the potential of new epigenetic and NE-targeting therapies in NEPC treatment, their development and clinical deployment has proven challenging. One major hurdle for many novel epigenetic therapies is their lack of specificity, often affecting a wide range of transcriptional networks leading to undesired off-target results and side-effects (10). Nevertheless, epigenetic targeting in NEPC offers a promising therapeutic avenue with potential to bring new options to the clinic.
6 Conclusion
Prostate cancer encompasses a range of natural histories, spanning from indolent disease to treatment-refractory CRPC/NEPC. Therapy-induced NEPC is marked by lineage plasticity, loss of luminal markers, and acquisition of NE features, such as SYP and CHGA. While genetic mutations, such as loss of PTEN, TP53, and RB1, and MYCN amplification have been implicated in this lineage transition, they are insufficient to induce the transdifferentiation of adenocarcinoma to NEPC. Instead, increasing evidence points to an aberrant epigenome as a major driver of lineage plasticity. Hyper-methylation of the AR promoter may partially explain the loss of canonical AR signaling observed in NEPC. Similarly, abnormal function of master epigenetic regulators, such as EZH2, promote NE lineage programs. Epigenetic dysregulation can also affect PCa radioresistance by altering the expression of genes involved in DDR and cell cycle. NEPC’s reliance on epigenetic machinery has provided a new avenue for treating a disease that, so far, has been nearly incurable. Epigenome-targeting drugs have shown promise for treating NEPC in preclinical and early-clinical studies, and similar mechanisms are beginning to be exploited to re-sensitize tumors to RT. Despite their potential, numerous challenges remain before these epigenetic therapies can be successfully implemented in modern treatment regimens for NEPC. In summary, understanding the mechanisms that promote lineage plasticity, from tumorigenesis to the emergence of treatment resistance, is imperative in contemporary prostate cancer research and represents a critical step in the discovery of new targets and more effective therapies to prevent and treat lethal PCa.
Author contributions
JI and SGa did the literature review and wrote the initial draft, made the figure, and worked on the revisions. AM constructed the tables. RL wrote the abstract. JN, CEW, CW, and SGu provided valuable suggestions for the manuscript. OM reviewed the final version and approved the final version for submission. All authors contributed to the article and approved the submitted version.
Funding
This work has been supported by grants from the American Cancer Society (134805-RSG-20-070-01-TBG to OM), U.S. National Institutes of Health/National Cancer Institute (L30 CA220908 to OM), VeloSano Foundation (OM), National Comprehensive Cancer Network Foundation (OM).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin (2023) 73(1):17–48. doi: 10.3322/caac.21763
2. Yamada Y, Beltran H. Clinical and biological features of neuroendocrine prostate cancer. Curr Oncol Rep (2021) 23(2). doi: 10.1007/s11912-020-01003-9
3. Davies AH, Beltran H, Zoubeidi A. Cellular plasticity and the neuroendocrine phenotype in prostate cancer. Nat Rev Urol (2018) 15(5):271–86. doi: 10.1038/nrurol.2018.22
4. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22(3):298–305. doi: 10.1038/nm.4045
5. Nijhout HF. Development and evolution of adaptive polyphenisms. Evol Dev (2003) 5(1):9–18. doi: 10.1046/j.1525-142X.2003.03003.x
6. Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev (2004) 25(2):276–308. doi: 10.1210/er.2002-0032
7. Leung JK, Sadar MD. Non-genomic actions of the androgen receptor in prostate cancer. Front Endocrinol (Lausanne) (2017) 8. doi: 10.3389/fendo.2017.00002
8. Vellky JE, Ricke WA. Development and prevalence of castration-resistant prostate cancer subtypes. Neoplasia (2020) 22(11):566–75. doi: 10.1016/j.neo.2020.09.002
9. Huggins C, Hodges CV. Studies on prostatic cancer i. the effect of castration, of estrogen and of androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res (1941) 1(4):293–7. doi: 10.3322/canjclin.22.4.232
10. Ge R, Wang Z, Montironi R, Jiang Z, Cheng M, Santoni M, et al. Epigenetic modulations and lineage plasticity in advanced prostate cancer. Annals Oncol (2020) 31(4):470–9.
11. Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res (2006) 12(6):1665–71. doi: 10.1158/1078-0432.CCR-06-0067
12. Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B, et al. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest (2019) 129(10):4492–505. doi: 10.1172/JCI128212
13. Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R, et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell (2017) 32(4):474–89. doi: 10.1016/j.ccell.2017.09.003
14. Cunha GR, Vezina CM, Isaacson D, Ricke WA, Timms BG, Cao M, et al. Development of the human prostate. Differentiation (2018) 103:24–45. doi: 10.1016/j.diff.2018.08.005
15. Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl (2014) 16(3):378–86. doi: 10.4103/1008-682X.122876
16. Chang L, Graham PH, Ni J, Hao J, Bucci J, Cozzi PJ, et al. Targeting PI3K/Akt/mTOR signaling pathway in the treatment of prostate cancer radioresistance. Crit Rev Oncol Hematol (2015) 96(3):507–17. doi: 10.1016/j.critrevonc.2015.07.005
17. Luo H, Liu Y, Li Y, Zhang C, Yu B, Shao C. Androgen receptor splicing variant 7 (ARv7) promotes DNA damage response in prostate cancer cells. FASEB J (2022) 36(9). doi: 10.1096/fj.202200190R
18. Chen X, Chen F, Ren Y, Weng G, Keng PC, Chen Y, et al. Glucocorticoid receptor upregulation increases radioresistance and triggers androgen independence of prostate cancer. Prostate (2019) 79(12):1386–98. doi: 10.1002/pros.23861
19. Loblaw DA, Virgo KS, Nam R, Somerfield MR, Ben-Josef E, Mendelson DS, et al. Initial hormonal management of androgen-sensitive metastatic, recurrent, or progressive prostate cancer: 2006 update of an American society of clinical oncology practice guideline. J Clin Oncol (2007) 25(12):1596–605. doi: 10.1200/JCO.2006.10.1949
20. Mohler JL, Antonarakis ES, Armstrong AJ, D’Amico AV, Davis BJ, Dorff T, et al. Prostate cancer, version 2.2019. J Natl Compr Cancer Netw (2019) 17(5):479–505. doi: 10.6004/jnccn.2019.0023
21. Penson DF, Armstrong AJ, Concepcion R, Agarwal N, Olsson C, Karsh L, et al. Enzalutamide versus bicalutamide in castration-resistant prostate cancer: the STRIVE trial. J Clin Oncol (2016) 34(18):2098–106. doi: 10.1200/JCO.2015.64.9285
22. Simard J, Singh M, Labrie F. Comparison of in vitro effects of the pure antiandrogens OH-flutamide, casodex, and nilutamide on androgen-sensitive parameters. Urology (1997) 49:580–9. doi: 10.1016/S0090-4295(97)00029-0
23. Kolvenbag G, Furr B, Blackledge G. Receptor affinity and potency of non-steroidal antiandrogens: translation of preclinical findings into clinical activity. Prostate Cancer Prostatic Dis (1998) 1:307–14. doi: 10.1038/sj.pcan.4500262
24. Vlachostergios PJ, Puca L, Beltran H. Emerging variants of castration-resistant prostate cancer. Curr Oncol Rep (2017) 19(5). doi: 10.1007/s11912-017-0593-6
25. Guo H, Ci X, Ahmed M, Hua JT, Soares F, Lin D, et al. ONECUT2 is a driver of neuroendocrine prostate cancer. Nat Commun (2019) 10(1). doi: 10.1038/s41467-018-08133-6
26. Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The master neural transcription factor BRN2 is an androgen receptor–suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discovery (2017) 7(1):54–71. doi: 10.1158/2159-8290.CD-15-1263
27. Parimi V, Goyal R, Poropatich K, Yang XJ. Neuroendocrine differentiation of prostate cancer: a review. Am J Clin Exp Urol (2014) 2(4):273–85.
28. Storck WK, May AM, Westbrook TC, Duan Z, Morrissey C, Yates JA, et al. The role of epigenetic change in therapy-induced neuroendocrine prostate cancer lineage plasticity. Front Endocrinol (2022) 13. doi: 10.3389/fendo.2022.926585
29. Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, et al. Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J Clin Oncol (2018) 36:2492–503. doi: 10.1200/JCO.2017
30. Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature (2012) 487(7406):239–43. doi: 10.1038/nature11125
31. Venkadakrishnan VB, Yamada Y, Weng K, Idahor O, Beltran H. Significance of RB loss in unlocking phenotypic plasticity in advanced cancers. Mol Cancer Res (2023) 21(6):497–510. doi: 10.1158/1541-7786.MCR-23-0045
32. Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer (2017) 17(12):725–37. doi: 10.1038/nrc.2017.87
33. Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53-and RB1-deficient prostate cancer. Sci (1979) (2017) 355(6320):84–8. doi: 10.1126/science.aah4307
34. Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Sci (1979) (2017) 355(6320):78–83. doi: 10.1126/science.aah4199
35. Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discovery (2017) 7(7):736–49. doi: 10.1158/2159-8290.CD-16-1174
36. Nyquist MD, Corella A, Coleman I, de Sarkar N, Kaipainen A, Ha G, et al. Combined TP53 and RB1 loss promotes prostate cancer resistance to a spectrum of therapeutics and confers vulnerability to replication stress. Cell Rep (2020) 31(8). doi: 10.1016/j.celrep.2020.107669
37. Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discovery (2011) 1(6):487–95. doi: 10.1158/2159-8290.CD-11-0130
38. Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell (2016) 29(4):536–47. doi: 10.1016/j.ccell.2016.03.001
39. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell (2016) 30(4):563–77. doi: 10.1016/j.ccell.2016.09.005
40. Otto T, Horn S, Brockmann M, Eilers U, Schüttrumpf L, Popov N, et al. Stabilization of n-myc is a critical function of aurora a in human neuroblastoma. Cancer Cell (2009) 15(1):67–78. doi: 10.1016/j.ccr.2008.12.005
41. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, et al. Drugging MYCN through an allosteric transition in aurora kinase a. Cancer Cell (2014) 26(3):414–27. doi: 10.1016/j.ccr.2014.07.015
42. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of n-myc in childhood neuroblastoma. Cancer Cell (2013) 24(1):75–89. doi: 10.1016/j.ccr.2013.05.005
43. Li Z, Sun Y, Chen X, Squires J, Nowroozizadeh B, Liang C, et al. P53 mutation directs AURKA overexpression via miR-25 and FBXW7 in prostatic small cell neuroendocrine carcinoma. Mol Cancer Res (2015) 13(3):584–91. doi: 10.1158/1541-7786.MCR-14-0277-T
44. Golson ML, Kaestner KH. Fox transcription factors: from development to disease. Development (2016) 143(24):4558–70. doi: 10.1242/dev.112672
45. Jin HJ, Zhao JC, Wu L, Kim J, Yu J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat Commun (2014) 5. doi: 10.1038/ncomms4972
46. Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature (2011) 474(7351):390–7. doi: 10.1038/nature10006
47. Kim J, Jin H, Zhao JC, Yang YA, Li Y, Yang X, et al. FOXA1 inhibits prostate cancer neuroendocrine differentiation. Oncogene (2017) 36(28):4072–80. doi: 10.1038/onc.2017.50
48. Sahu B, Laakso M, Ovaska K, Mirtti T, Lundin J, Rannikko A, et al. Dual role of FoxA1 in androgen receptor binding to chromatin, androgen signalling and prostate cancer. EMBO J (2011) 30(19):3962–76. doi: 10.1038/emboj.2011.328
49. Adams EJ, Karthaus WR, Hoover E, Liu D, Gruet A, Zhang Z, et al. FOXA1 mutations alter pioneering activity, differentiation and prostate cancer phenotypes. Nature (2019) 571(7765):408–12. doi: 10.1038/s41586-019-1318-9
50. Yang YA, Zhao JC, Fong KW, Kim J, Li S, Song C, et al. FOXA1 potentiates lineage-specific enhancer activation through modulating TET1 expression and function. Nucleic Acids Res (2016) 44(17):8153–64. doi: 10.1093/nar/gkw498
51. Park JW, Lee JK, Witte ON, Huang J. FOXA2 is a sensitive and specific marker for small cell neuroendocrine carcinoma of the prostate. Modern Pathol (2017) 30(9):1262–72. doi: 10.1038/modpathol.2017.44
52. Chiaverotti T, Couto SS, Donjacour A, Mao JH, Nagase H, Cardiff RD, et al. Dissociation of epithelial and neuroendocrine carcinoma lineages in the transgenic adenocarcinoma of mouse prostate model of prostate cancer. Am J Pathol (2008) 172(1):236–46. doi: 10.2353/ajpath.2008.070602
53. Lin TP, Chang YT, Lee SY, Campbell M, Wang TC, Shen SH, et al. REST reduction is essential for hypoxia-induced neuroendocrine differentiation of prostate cancer cells by activating autophagy signaling. Oncotarget (2016) 7(18):26137–51. doi: 10.18632/oncotarget.8433
54. Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell (2010) 18(1):23–38. doi: 10.1016/j.ccr.2010.05.024
55. Han M, Li F, Zhang Y, Dai P, He J, Li Y, et al. FOXA2 drives lineage plasticity and KIT pathway activation in neuroendocrine prostate cancer. Cancer Cell (2022) 40(11):1306–1323.e8. doi: 10.1016/j.ccell.2022.10.011
56. Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci (2019) 116(23):11428–36. doi: 10.1073/pnas.1902651116
57. Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res (2014) 74(4):1272–83. doi: 10.1158/0008-5472.CAN-13-2921-T
58. Formaggio N, Rubin MA, Theurillat JP. Loss and revival of androgen receptor signaling in advanced prostate cancer. Oncogene (2021) 40(7):1205–16. doi: 10.1038/s41388-020-01598-0
59. Breiling A, Lyko F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenet Chromatin (2015) 8(1). doi: 10.1186/s13072-015-0016-6
60. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis (2009) 31:27–36. doi: 10.1093/carcin/bgp220
61. Yegnasubramanian S, Haffner MC, Zhang Y, Gurel B, Cornish TC, Wu Z, et al. DNA Hypomethylation arises later in prostate cancer progression than CpG island hypermethylation and contributes to metastatic tumor heterogeneity. Cancer Res (2008) 68(21):8954–67. doi: 10.1158/0008-5472.CAN-07-6088
62. Zelic R, Fiano V, Grasso C, Zugna D, Pettersson A, Gillio-Tos A, et al. Global DNA hypomethylation in prostate cancer development and progression: a systematic review. Prostate Cancer Prostatic Dis (2015) 18(1).
63. Sutton LP, Jeffreys SA, Phillips JL, Taberlay PC, Holloway AF, Ambrose M, et al. DNA Methylation changes following DNA damage in prostate cancer cells. Epigenetics (2019) 14(10):989–1002. doi: 10.1080/15592294.2019.1629231
64. Li LC, Okino ST, Dahiya R. DNA Methylation in prostate cancer. Biochim Biophys Acta - Rev Cancer (2004) 1704:87–102. doi: 10.1016/j.bbcan.2004.06.001
65. Ruiz S, Santos E, Bustelo XR. RasGRF2, a guanosine nucleotide exchange factor for ras GTPases, participates in T-cell signaling responses. Mol Cell Biol (2007) 27(23):8127–42. doi: 10.1128/MCB.00912-07
66. Ebihara T, Song C, Ryu SH, Plougastel-Douglas B, Yang L, Levanon D, et al. Runx3 specifies lineage commitment of innate lymphoid cells. Nat Immunol (2015) 16(11):1124–33. doi: 10.1038/ni.3272
67. Brunskill EW, Sequeira-Lopez MLS, Pentz ES, Lin E, Yu J, Aronow BJ, et al. Genes that confer the identity of the renin cell. J Am Soc Nephrol (2011) 22(12):2213–25. doi: 10.1681/ASN.2011040401
68. Mahapatra S, Klee EW, Young CYF, Sun Z, Jimenez RE, Klee GG, et al. Global methylation profiling for risk prediction of prostate cancer. Clin Cancer Res (2012) 18(10):2882–95. doi: 10.1158/1078-0432.CCR-11-2090
69. Beltran H, Romanel A, Conteduca V, Casiraghi N, Sigouros M, Franceschini GM, et al. Circulating tumor DNA profile recognizes transformation to castration-resistant neuroendocrine prostate cancer. J Clin Invest (2020) 130(4):1653–68. doi: 10.1172/JCI131041
70. Greenberg NM, Demayo F, Finegold MJ, Medina D, Tilleyv WD, Aspinalls JO, et al. Prostate cancer in a transgenic mouse. Med Sci (1995) 92:3439–43. doi: 10.1073/pnas.92.8.3439
71. Mccabe MT, Davis JN, Day ML. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res (2005) 65(9):3624–32. doi: 10.1158/0008-5472.CAN-04-2158
72. Valdez CD, Davis JN, Odeh HM, Layfield TL, Cousineau CS, Berton TR, et al. Repression of androgen receptor transcription through the E2F1/DNMT1 axis. PloS One (2011) 6(9). doi: 10.1371/journal.pone.0025187
73. Jarrard DF, Kinoshita H, Shi Y, Sandefur C, Hoff D, Meisner LF, et al. Methylation of the androgen receptor promoter CpG island is associated with loss of androgen receptor expression in prostate cancer cells. Cancer Res (1998) 58(23):5310–4.
74. Kinoshita H, Shi Y, Sandefur C, Meisner LF, Chang C, Choon A, et al. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res (2000) 60(13):3623–30.
75. Nakayama T, Watanabe M, Suzuki H, Toyota M, Sekita N, Hirokawa Y, et al. Epigenetic regulation of androgen receptor gene expression in human prostate cancers. Lab Invest (2000) 80(12):1789–96. doi: 10.1038/labinvest.3780190
76. Kleb B, Estécio MRH, Zhang J, Tzelepi V, Chung W, Jelinek J, et al. Differentially methylated genes and androgen receptor re-expression in small cell prostate carcinomas. Epigenetics (2016) 11(3):184–93. doi: 10.1080/15592294.2016.1146851
77. Clermont PL, Lin D, Crea F, Wu R, Xue H, Wang Y, et al. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin Epigenet (2015) 7(1). doi: 10.1186/s13148-015-0074-4
78. Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The polycomb group protein EZH2 directly controls DNA methylation. Nature (2006) 439(7078):871–4. doi: 10.1038/nature04431
79. Schlesinger Y, Straussman R, Keshet I, Farkash S, Hecht M, Zimmerman J, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet (2007) 39(2):232–6. doi: 10.1038/ng1950
80. Xiang ST, Zou PL, Tang Q, Zheng F, Wu JJ, Chen ZQ, et al. HOTAIR-mediated reciprocal regulation of EZH2 and DNMT1 contribute to polyphyllin I-inhibited growth of castration-resistant prostate cancer cells in vitro and in vivo. BBA - Gen Subj (2018) 1862:589–99. doi: 10.1016/j.bbagen.2017.12.001
81. Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, et al. Long noncoding RNA as modular scaffold of histone modification complexes. Sci (1979) (2010) 329(5992):689–93. doi: 10.1126/science.1192002
82. Shan J, Al-Muftah MA, Al-Kowari MK, Abuaqel SWJ, Al-Rumaihi K, Al-Bozom I, et al. Targeting Wnt/EZH2/microRNA-708 signaling pathway inhibits neuroendocrine differentiation in prostate cancer. Cell Death Discovery (2019) 5(1). doi: 10.1038/s41420-019-0218-y
83. Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N, et al. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat Commun (2018) 9(1). doi: 10.1038/s41467-018-06177-2
84. Nolan KD, Franco OE, Hance MW, Hayward SW, Isaacs JS. Tumor-secreted Hsp90 subverts polycomb function to drive prostate tumor growth and invasion. J Biol Chem (2015) 290(13):8271–82. doi: 10.1074/jbc.M115.637496
85. Cai H, Memarzadeh S, Stoyanova T, Beharry Z, Kraft AS, Witte ON. Collaboration of kras and androgen receptor signaling stimulates EZH2 expression and tumor-propagating cells in prostate cancer. Cancer Res (2012) 72(18):4672–81. doi: 10.1158/0008-5472.CAN-12-0228
86. Perillo B, Tramontano A, Pezone A, Migliaccio A. LSD1: more than demethylation of histone lysine residues. Exp Mol Med (2020) 52(12):1936–47. doi: 10.1038/s12276-020-00542-2
87. He Y, Zhao Y, Wang L, Bohrer LR, Pan Y, Wang L, et al. LSD1 promotes s-phase entry and tumorigenesis via chromatin co-occupation with E2F1 and selective H3K9 demethylation. Oncogene (2018) 37(4):534–43. doi: 10.1038/onc.2017.353
88. Coleman DJ, Sampson DA, Sehrawat A, Kumaraswamy A, Sun D, Wang Y, et al. Alternative splicing of LSD1 + 8a in neuroendocrine prostate cancer is mediated by SRRM4. Neoplasia (United States) (2020) 22(6):253–62. doi: 10.1016/j.neo.2020.04.002
89. Kim DH, Sun D, Storck WK, Leng KW, Jenkins C, Coleman DJ, et al. BET bromodomain inhibition blocks an AR-repressed, E2F1-activated treatment-emergent neuroendocrine prostate cancer lineage plasticity program. Clin Cancer Res (2021) 27(17):4923–36. doi: 10.1158/1078-0432.CCR-20-4968
90. Cyrta J, Augspach A, De Filippo MR, Prandi D, Thienger P, Benelli M, et al. Role of specialized composition of SWI/SNF complexes in prostate cancer lineage plasticity. Nat Commun (2020) 11(1). doi: 10.1038/s41467-020-19328-1
91. Pomerantz MM, Qiu X, Zhu Y, Takeda DY, Pan W, Baca SC, et al. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat Genet (2020) 52(8):790–9. doi: 10.1038/s41588-020-0664-8
92. Zhang Z, Zhou C, Li X, Barnes SD, Deng S, Hoover E, et al. Loss of CHD1 promotes heterogeneous mechanisms of resistance to AR-targeted therapy via chromatin dysregulation. Cancer Cell (2020) 37(4):584–598.e11. doi: 10.1016/j.ccell.2020.03.001
93. Ruan L, Wang L, Wang X, He M, Yao X. SIRT1 contributes to neuroendocrine differentiation of prostate cancer. Oncotarget (2018) 9(2):2002–16. doi: 10.18632/oncotarget.23111
94. Jia Y, Zhao J, Yang J, Shao J, Cai Z. miR-301 regulates the SIRT1/SOX2 pathway via CPEB1 in the breast cancer progression. Mol Ther Oncolytics (2021) 22:13–26. doi: 10.1016/j.omto.2021.03.007
95. Liu L, Liu C, Zhang Q, Shen J, Zhang H, Shan J, et al. SIRT1-mediated transcriptional regulation of SOX2 is important for self-renewal of liver cancer stem cells. Hepatology (2016) 64(3):814–27. doi: 10.1002/hep.28690
96. Yoon DS, Choi Y, Jang Y, Lee M, Choi WJ, Kim SH, et al. SIRT1 directly regulates SOX2 to maintain self-renewal and multipotency in bone marrow-derived mesenchymal stem cells. Stem Cells (2014) 32(12):3219–31. doi: 10.1002/stem.1811
97. Augello MA, Liu D, Deonarine LD, Robinson BD, Huang D, Stelloo S, et al. CHD1 loss alters AR binding at lineage-specific enhancers and modulates distinct transcriptional programs to drive prostate tumorigenesis. Cancer Cell (2019) 35(4):603–617.e8. doi: 10.1016/j.ccell.2019.03.001
98. Schoenherr CJ, Anderson DJ, Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Sci (1979) (1995) 267:1360–3. doi: 10.1126/science.7871435
99. Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell (2005) 121:645–57. doi: 10.1016/j.cell.2005.03.013
100. Svensson C, Ceder J, Iglesias-Gato D, Chuan YC, Pang ST, Bjartell A, et al. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res (2014) 42(2):999–1015. doi: 10.1093/nar/gkt921
101. Olave I, Wang W, Xue Y, Kuo A, Crabtree GR. Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev (2002) 16(19):2509–17. doi: 10.1101/gad.992102
102. Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, et al. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet (1995) 9(4):401–6. doi: 10.1038/ng0495-401
103. Köhler A, Demir Ü, Kickstein E, Krauss S, Aigner J, Aranda-Orgillés B, et al. A hormone-dependent feedback-loop controls androgen receptor levels by limiting MID1, a novel translation enhancer and promoter of oncogenic signaling. Mol Cancer (2014) 13(1). doi: 10.1186/1476-4598-13-146
104. Liu W, Xie CC, Zhu Y, Li T, Sun J, Cheng Y, et al. Homozygous deletions and recurrent amplications implicate new genes involved in prostate cancer. Neoplasia (2008) 10(8):897–907. doi: 10.1593/neo.08428
105. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell (2010) 18(1):11–22. doi: 10.1016/j.ccr.2010.05.026
106. Steketee K, Timmerman L, Ziel-Van Der Made ACJ, Doesburg P, Brinkmann AO, Trapman J. Broadened ligand responsiveness of androgen receptor mutants obtained by random amino acid substitution of H874 and mutation hot spot T877 in prostate cancer. Int J Cancer (2002) 100(3):309–17. doi: 10.1002/ijc.10495
107. Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife (2013) 2013(2). doi: 10.7554/eLife.00499
108. Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res (2008) 68(13):5469–77. doi: 10.1158/0008-5472.CAN-08-0594
109. Hörnberg E, Ylitalo EB, Crnalic S, Antti H, Stattin P, Widmark A, et al. Expression of androgen receptor splice variants in prostate cancer bone metastases is associated with castration-resistance and short survival. PloS One (2011) 6(4). doi: 10.1371/journal.pone.0019059
110. Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res (2009) 69(6):2305–13. doi: 10.1158/0008-5472.CAN-08-3795
111. Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res (2009) 69(1):16–22. doi: 10.1158/0008-5472.CAN-08-2764
112. Xu D, Zhan Y, Qi Y, Cao B, Bai S, Xu W, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res (2015) 75(17):3663–71. doi: 10.1158/0008-5472.CAN-15-0381
113. Sobhani N, Generali D, D’Angelo A, Aieta M, Roviello G. Current status of androgen receptor-splice variant 7 inhibitor niclosamide in castrate-resistant prostate-cancer. Invest New Drugs (2018) 36(6):1133–7. doi: 10.1007/s10637-018-0653-2
114. Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur Urol (2015) 67(3):470–9. doi: 10.1016/j.eururo.2014.09.049
115. Sharifi N. Minireview: androgen metabolism in castration- resistant prostate cancer. Mol Endocrinol (2013) 27(5):708–14. doi: 10.1210/me.2013-1007
116. Evaul K, Li R, Papari-Zareei M, Auchus RJ, Sharifi N. 3β-hydroxysteroid dehydrogenase is a possible pharmacological target in the treatment of castration-resistant prostate cancer. Endocrinology (2010) 151(8):3514–20. doi: 10.1210/en.2010-0138
117. Naelitz BD, Sharifi N. Through the looking-glass: reevaluating DHEA metabolism through HSD3B1 genetics. Trends Endocrinol Metab (2020) 31(9):680–90. doi: 10.1016/j.tem.2020.05.006
118. Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, et al. XA gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell (2013) 154(5):1074–84. doi: 10.1016/j.cell.2013.07.029
119. Zhou J, Cidlowski JA. The human glucocorticoid receptor: one gene, multiple proteins and diverse responses. Steroids (2005) 70:407–17. doi: 10.1016/j.steroids.2005.02.006
120. Beato M. Gene regulation by steroid hormones. Cell (1989) 56(3):335–44. doi: 10.1016/0092-8674(89)90237-7
121. Shaffer PL, Jivan A, Dollins DE, Claessens F, Gewirth DT, Richardson JS. Structural basis of androgen receptor binding to selective androgen response elements. PNAS (2004) 6(14):4758–63. doi: 10.1073/pnas.0401123101
122. Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res (2013) 73(5):1570–80. doi: 10.1158/0008-5472.CAN-12-2350
123. Isikbay M, Otto K, Kregel S, Kach J, Cai Y, vander Griend DJ, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer (2014) 5(2):72–89. doi: 10.1007/s12672-014-0173-2
124. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell (2013) 155(6):1309–22. doi: 10.1016/j.cell.2013.11.012
125. Bolla M, Collette L, Blank L, Warde P, Dubois JB, Mirimanoff RO, et al. Long-term results with immediate androgen suppression and external irradiation in patients with locally advanced prostate cancer (an EORTC study): a phase III randomised trial. Lancet (2002) 360(9327):103–8. doi: 10.1016/S0140-6736(02)09408-4
126. Smith CP, Xiang M, Armstrong WR, Nickols NG, Steinberg ML, Reiter RE, et al. Patterns of failure in men with radiorecurrent prostate cancer: a post-hoc analysis of three prospective Ga-68-PSMA PET/CT imaging trials. Int J Radiat Oncol Biol Phys (2023). doi: 10.1016/j.ijrobp.2023.02.039
127. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
128. Wang J-s, Wang H-j, Qian H-l. Biological effects of radiation on cancer cells. Mil Med Res (2018) 5(1).
129. Belli M, Tabocchini MA. Ionizing radiation-induced epigenetic modifications and their relevance to radiation protection. Int J Mol Sci (2020) 21(17):1–34. doi: 10.3390/ijms21175993
130. Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discovery (2013) 3(11):1245–53. doi: 10.1158/2159-8290.CD-13-0172
131. Spratt DE, Evans MJ, Davis BJ, Doran MG, Lee MX, Shah N, et al. Androgen receptor upregulation mediates radioresistance after ionizing radiation. Cancer Res (2015) 75(22):4688–96. doi: 10.1158/0008-5472.CAN-15-0892
132. Yin Y, Li R, Xu K, Ding S, Li J, Baek GH, et al. Androgen receptor variants mediate DNA repair after prostate cancer irradiation. Cancer Res (2017) 77(18):4745–54. doi: 10.1158/0008-5472.CAN-17-0164
133. Kounatidou E, Nakjang S, McCracken SRC, Dehm SM, Robson CN, Jones D, et al. A novel CRISPR-engineered prostate cancer cell line defines the AR-V transcriptome and identifies PARP inhibitor sensitivities. Nucleic Acids Res (2019) 47(11):5634–47. doi: 10.1093/nar/gkz286
134. Fan L, Zhang F, Xu S, Cui X, Hussain A, Fazli L, et al. Histone demethylase JMJD1A promotes alternative splicing of AR variant 7 (AR-V7) in prostate cancer cells. Proc Natl Acad Sci U S A (2018) 115(20):E4584–93. doi: 10.1073/pnas.1802415115
135. De Felice F, Tombolini V, Marampon F, Musella A, Marchetti C. Defective DNA repair mechanisms in prostate cancer: impact of olaparib. Drug Des Devel Ther (2017) 11:547–52. doi: 10.2147/DDDT.S110264
136. Warner EW, Yip SM, Chi KN, Wyatt AW. DNA Repair defects in prostate cancer: impact for screening, prognostication and treatment. BJU Int (2019) 123(5):769–76. doi: 10.1111/bju.14576
137. Kamińska K, Białkowska A, Kowalewski J, Huang S, Lewandowska MA. Differential gene methylation patterns in cancerous and non-cancerous cells. Oncol Rep (2019) 42(1):43–54. doi: 10.3892/or.2019.7159
138. Jette NR, Kumar M, Radhamani S, Arthur G, Goutam S, Yip S, et al. ATM-Deficient cancers provide new opportunities for precision oncology. Cancers (Basel) (2020) 12(3). doi: 10.3390/cancers12030687
139. Kim KH, Kim HS, Kim SS, Shim HS, Yang AJ, Bock Lee JJ, et al. Increased radiosensitivity of solid tumors harboring ATM and BRCA1/2 mutations. Cancer Res Treat (2022) 54(1):54–64. doi: 10.4143/crt.2020.1247
140. Wu Y, Sarkissyan M, Vadgama JV. Epigenetics in breast and prostate cancer. Methods Mol Biol (2015) 1238:425–66. doi: 10.1007/978-1-4939-1804-1_23
141. Amani J, Gorjizadeh N, Younesi S, Najafi M, Ashrafi AM, Irian S, et al. Cyclin-dependent kinase inhibitors (CDKIs) and the DNA damage response: the link between signaling pathways and cancer. DNA Repair (Amst) (2021) 102. doi: 10.1016/j.dnarep.2021.103103
142. Xie H, Li C, Dang Q, Chang LS, Li L. Infiltrating mast cells increase prostate cancer chemotherapy and radiotherapy resistances via modulation of p38/p53/p21 and ATM signals. Oncotarget (2015) 7:1342–52. doi: 10.18632/oncotarget.6372
143. Wu C, Guo E, Ming J, Sun W, Nie X, Sun L, et al. Radiation-induced DNMT3B promotes radioresistance in nasopharyngeal carcinoma through methylation of p53 and p21. Mol Ther Oncolytics (2020) 17:306–19. doi: 10.1016/j.omto.2020.04.007
144. Tong Q, Zhang W, Jin S, Li S, Chen Z. The relationship between p27kip1 expression and the change of radiosensitivity of esophageal carcinoma cells. Scand J Gastroenterol (2011) 46(2):173–6. doi: 10.3109/00365521.2010.522721
145. Berton S, Cusan M, Segatto I, Citron F, D’Andrea S, Benevol S, et al. Loss of p27kip1 increases genomic instability and induces radio-resistance in luminal breast cancer cells. Sci Rep (2017) 7(1). doi: 10.1038/s41598-017-00734-3
146. Fu HC, Chuang IC, Yang YC, Chuang PC, Lin H, Ou YC, et al. Low P16 INK4A expression associated with high expression of cancer stem cell markers predicts poor prognosis in cervical cancer after radiotherapy. Int J Mol Sci (2018) 19:2541. doi: 10.3390/ijms19092541
147. Matsumuraa Y, Yamagishi N, Miyakoshi J, Imamura S, Takebe H. Increase in radiation sensitivity of human malignant melanoma cells by expression of wild-type pl6 gene. ELSEVIER Cancer Lett (1997) 115:91–6. doi: 10.1016/s0304-3835(97)04714-9
148. Wichmann IA, Zavala K, Hoffmann FG, Vandewege MW, Corvalán AH, Amigo JD, et al. Evolutionary history of the reprimo tumor suppressor gene family in vertebrates with a description of a new reprimo gene lineage. Gene (2016) 592(1):245–54. doi: 10.1016/j.gene.2016.07.036
149. Li Z, Zhou Z, Tian S, Zhang K, An G, Zhang Y, et al. RPRM deletion preserves hematopoietic regeneration by promoting EGFR-dependent DNA repair and hematopoietic stem cell proliferation post ionizing radiation. Cell Biol Int (2022) 46(12):2158–72. doi: 10.1002/cbin.11900
150. Zhang Y, Ou G, Ye Z, Zhou Z, Cao Q, Li M, et al. RPRM negatively regulates ATM levels involving its phosphorylation mediated by CDK4/CDK6. bioRxiv (2021). doi: 10.1101/2021.11.10.468148
151. Cheng L, Pan CX, Zhang JT, Zhang S, Kinch MS, Li L, et al. Loss of 14-3-3 in prostate cancer and its precursors. Clin Cancer Res (2004) 10(9):3064–8.
152. Yeh CN, Pang ST, Jung SM, Chen TW, Jan YY, Chen MF. Cytoplasmic overexpression with membrane accentuation of stratifin in intrahepatic mass-forming cholangiocarcinoma after hepatectomy: correlation with clinicopathologic features and patient survival. J Surg Oncol (2010) 102(6):608–14. doi: 10.1002/jso.21604
153. Ngollo M, Lebert A, Daures M, Judes G, Rifai K, Dubois L, et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer (2017) 17(1). doi: 10.1186/s12885-017-3256-y
154. Peng Q, Weng K, Li S, Xu R, Wang Y, Wu Y. A perspective of epigenetic regulation in radiotherapy. Front Cell Dev Biol (2021) 9. doi: 10.3389/fcell.2021.624312
155. Gorodetska I, Lukiyanchuk V, Peitzsch C, Kozeretska I, Dubrovska A. BRCA1 and EZH2 cooperate in regulation of prostate cancer stem cell phenotype. Int J Cancer (2019) 145(11):2974–85. doi: 10.1002/ijc.32323
156. Sak A, Kübler D, Bannik K, Groneberg M, Strunz S, Kriehuber R, et al. Epigenetic silencing and activation of transcription: influence on the radiation sensitivity of glioma cell lines. Int J Radiat Biol (2017) 93(5):494–506. doi: 10.1080/09553002.2017.1270472
157. Liao Y, Chen CH, Xiao T, de la Peña Avalos B, v. DE, Cai C, et al. Inhibition of EZH2 transactivation function sensitizes solid tumors to genotoxic stress. Proc Natl Acad Sci (2022) 119(3). doi: 10.1073/pnas.2105898119
158. Lv S, Wen H, Shan X, Li J, Wu Y, Yu X, et al. Loss of KMT2D induces prostate cancer ROS-mediated DNA damage by suppressing the enhancer activity and DNA binding of antioxidant transcription factor FOXO3. Epigenetics (2019) 14(12):1194–208. doi: 10.1080/15592294.2019.1634985
159. Lv S, Ji L, Chen B, Liu S, Lei C, Liu X, et al. Histone methyltransferase KMT2D sustains prostate carcinogenesis and metastasis via epigenetically activating LIFR and KLF4. Oncogene (2018) 37(10):1354–68. doi: 10.1038/s41388-017-0026-x
160. Sarker D, Reid AHM, Yap TA, De Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res (2009) 15(15):4799–805. doi: 10.1158/1078-0432.CCR-08-0125
161. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. PI3K/Akt/mTOR pathway inhibitors enhance radiosensitivity in radioresistant prostate cancer cells through inducing apoptosis, reducing autophagy, suppressing NHEJ and HR repair pathways. Cell Death Dis (2014) 5(10). doi: 10.1038/cddis.2014.415
162. Chang L, Graham PH, Hao J, Ni J, Bucci J, Cozzi PJ, et al. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis (2013) 4(10). doi: 10.1038/cddis.2013.407
163. Potiron VA, Abderrhamani R, Giang E, Chiavassa S, di Tomaso E, Maira SM, et al. Radiosensitization of prostate cancer cells by the dual PI3K/mTOR inhibitor BEZ235 under normoxic and hypoxic conditions. Radiother Oncol (2013) 106(1):138–46. doi: 10.1016/j.radonc.2012.11.014
164. Ge YZ, Xu LW, Jia RP, Xu Z, Feng YM, Wu R, et al. The association between RASSF1A promoter methylation and prostate cancer: evidence from 19 published studies. Tumour Biol (2014) 35(4):3881–90. doi: 10.1007/s13277-013-1515-3
165. Krop I, Player A, Tablante A, Taylor-Parker M, Lahti-Domenici J, Fukuoka J, et al. Frequent HIN-1 promoter methylation and lack of expression in multiple human tumor types. Mol Cancer Res (2004) 2(9):489–94. doi: 10.1158/1541-7786.489.2.9
166. Makarević J, Tawanaie N, Juengel E, Reiter M, Mani J, Tsaur I, et al. Cross-communication between histone H3 and H4 acetylation and akt-mTOR signalling in prostate cancer cells. J Cell Mol Med (2014) 18(7):1460–6. doi: 10.1111/jcmm.12299
167. Beltran H, Oromendia C, Danila DC, Montgomery B, Hoimes C, Szmulewitz RZ, et al. A phase II trial of the aurora kinase a inhibitor alisertib for patients with castration-resistant and neuroendocrine prostate cancer: efficacy and biomarkers. Clin Cancer Res (2019) 25(1):43–51. doi: 10.1158/1078-0432.CCR-18-1912
168. Mansfield AS, Hong DS, Hann CL, Farago AF, Beltran H, Waqar SN, et al. A phase I/II study of rovalpituzumab tesirine in delta-like 3–expressing advanced solid tumors. Precis Oncol (2021) 5(1). doi: 10.1038/s41698-021-00214-y
169. Berger A, Brady NJ, Bareja R, Robinson B, Conteduca V, Augello MA, et al. N-myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J Clin Invest (2019) 129(9):3924–40. doi: 10.1172/JCI127961
170. Asangani IA, Dommeti VL, Wang X, Malik R, Cieslik M, Yang R, et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature (2014) 510(7504):278–82. doi: 10.1038/nature13229
171. Asangani IA, Wilder-Romans K, Dommeti VL, Krishnamurthy PM, Apel IJ, Escara-Wilke J, et al. BET bromodomain inhibitors enhance efficacy and disrupt resistance to AR antagonists in the treatment of prostate cancer. Mol Cancer Res (2016) 14(4):324–31. doi: 10.1158/1541-7786.MCR-15-0472
172. Welti J, Sharp A, Yuan W, Dolling D, Rodrigues DN, Figueiredo I, et al. Targeting bromodomain and extra-terminal (BET) family proteins in castration-resistant prostate cancer (CRPC). Clin Cancer Res (2018) 24(13):3149–62. doi: 10.1158/1078-0432.CCR-17-3571
173. Aggarwal RR, Schweizer MT, Nanus DM, Pantuck AJ, Heath EI, Campeau E, et al. A phase Ib/IIa study of the pan-BET inhibitor ZEN-3694 in combination with enzalutamide in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res (2020) 26(20):5338–47. doi: 10.1158/1078-0432.CCR-20-1707
174. Sehrawat A, Gao L, Wang Y, Bankhead A, McWeeney SK, King CJ, et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc Natl Acad Sci U S A (2018) 115(18):E4179–88. doi: 10.1073/pnas.1719168115
175. Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, et al. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep (2014) 9(5):1618–27. doi: 10.1016/j.celrep.2014.11.008
176. Reina-Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, et al. Increased serine and one-carbon pathway metabolism by PKCλ/ι deficiency promotes neuroendocrine prostate cancer. Cancer Cell (2019) 35(3):385–400.e9. doi: 10.1016/j.ccell.2019.01.018
177. Gravina GL, Festuccia C, Millimaggi D, Dolo V, Tombolini V, de Vito M, et al. Chronic azacitidine treatment results in differentiating effects, sensitizes against bicalutamide in androgen-independent prostate cancer cells. Prostate (2008) 68(7):793–801. doi: 10.1002/pros.20748
178. Sonpavde G, Aparicio AM, Zhan F, North B, Delaune R, Garbo LE, et al. Azacitidine favorably modulates PSA kinetics correlating with plasma DNA LINE-1 hypomethylation in men with chemonaïve castration-resistant prostate cancer. Urol Oncol (2011) 29(6):682–9. doi: 10.1016/j.urolonc.2009.09.015
179. Thibault A, Figg WD, Bergan RC, Lush RM, Myers CE, Tompkins A, et al. A phase II study of 5-aza-2’deoxycytidine (decitabine) in hormone independent metastatic (D2) prostate cancer. Tumori (1998) 84(1):87–9. doi: 10.1177/030089169808400120
180. Li X, Baek GH, Ramanand SG, Sharp A, Gao Y, Yuan W, et al. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep (2018) 22(3):796–808. doi: 10.1016/j.celrep.2017.12.078
181. Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim HJ, Majid MC, et al. The histone demethylase LSD1/KDM1A promotes the DNA damage response. J Cell Biol (2013) 203(3):457–70. doi: 10.1083/jcb.201302092
182. Jonsson M, Ragnum HB, Julin CH, Yeramian A, Clancy T, Frikstad KAM, et al. Hypoxia-independent gene expression signature associated with radiosensitisation of prostate cancer cell lines by histone deacetylase inhibition. Br J Cancer (2016) 115(8):929–39. doi: 10.1038/bjc.2016.278
Keywords: neuroendocrine prostate cancer, lineage plasticity, epigenetic dysregulation, hormone therapy resistance, radioresistance
Citation: Imamura J, Ganguly S, Muskara A, Liao RS, Nguyen JK, Weight C, Wee CE, Gupta S and Mian OY (2023) Lineage plasticity and treatment resistance in prostate cancer: the intersection of genetics, epigenetics, and evolution. Front. Endocrinol. 14:1191311. doi: 10.3389/fendo.2023.1191311
Received: 21 March 2023; Accepted: 12 June 2023;
Published: 30 June 2023.
Edited by:
Carlos Pérez-Plasencia, National Autonomous University of Mexico, MexicoReviewed by:
Mauricio Rodriguez-Dorantes, National Institute of Genomic Medicine (INMEGEN), MexicoSusan Kasper, University of Cincinnati, United States
Copyright © 2023 Imamura, Ganguly, Muskara, Liao, Nguyen, Weight, Wee, Gupta and Mian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jarrell Imamura, aW1hbXVyajJAY2NmLm9yZw==; Shinjini Ganguly, Z2FuZ3Vsc0BjY2Yub3Jn