
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 07 July 2023
Sec. Cancer Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1191040
This article is part of the Research TopicNew Insights into Multiple Endocrine Neoplasia Type 1View all 6 articles
 Laura Pierotti1
Laura Pierotti1 Elena Pardi1
Elena Pardi1 Elisa Dinoi1
Elisa Dinoi1 Paolo Piaggi2Simona Borsari1
Paolo Piaggi2Simona Borsari1 Simone Della Valentina1Chiara Sardella3
Simone Della Valentina1Chiara Sardella3 Angela Michelucci4Maria Adelaide Caligo4
Angela Michelucci4Maria Adelaide Caligo4 Fausto Bogazzi1
Fausto Bogazzi1 Claudio Marcocci1,3
Claudio Marcocci1,3 Filomena Cetani3*
Filomena Cetani3*Background: Multiple Endocrine Neoplasia type 1 is a rare genetic syndrome mainly caused by mutations of MEN1 gene and characterized by a combination of several endocrine and non-endocrine manifestations. The objective of this study was to describe cutaneous lesions and other non-endocrine manifestations of MEN1 in a cohort of patients with familial (F) and sporadic (S) MEN1, compare the prevalence of these manifestations between the two cohorts, and investigate the correlation with MEN1 mutation status.
Methods: We collected phenotypic and genotypic data of 185 patients with F-MEN1 and S-MEN1 followed from 1997 to 2022. The associations between F-MEN1 and S-MEN1 or MEN1 mutation-positive and mutation-negative patients and non-endocrine manifestations were determined using chi-square or Fisher’s exact tests or multivariate exact logistic regression analyses.
Results: The prevalence of angiofibromas was significantly higher in F-MEN1 than in S-MEN1 in both the whole (p < 0.001) and index case (p = 0.003) cohorts. The prevalence of lipomas was also significantly higher in F-MEN1 than in S-MEN1 (p = 0.009) and in MEN1 mutation-positive than in MEN1 mutation-negative (p = 0.01) index cases. In the whole cohort, the prevalence of lipomas was significantly higher in MEN1 mutation-positive compared to MEN1 mutation-negative patients (OR = 2.7, p = 0.02) and in F-MEN1 than in S-MEN1 (p = 0.03), only after adjustment for age. No significant differences were observed for the other non-endocrine manifestations between the two cohorts. Hibernoma and collagenoma were each present in one patient (0.5%) and meningioma and neuroblastoma in 2.7% and 0.5%, respectively. Gastric leiomyoma was present in 1.1% of the patients and uterine leiomyoma in 14% of women. Thyroid cancer, breast cancer, lung cancer, basal cell carcinoma, melanoma, and colorectal cancer were present in 4.9%, 2.7%, 1.6%, 1.6%, 2.2%, and 0.5% of the whole series, respectively.
Conclusions: We found a significantly higher prevalence of angiofibromas and lipomas in F-MEN1 compared with S-MEN1 and in MEN1 mutation-positive compared to MEN1 mutation-negative patients. In patients with one major endocrine manifestation of MEN1, the presence of cutaneous lesions might suggest the diagnosis of MEN1 and a possible indication for genetic screening.
Multiple endocrine neoplasia type 1 (MEN1) is a rare hereditary syndrome with an estimated prevalence of approximately one to three in 100,000 inhabitants (1). In most patients (90%), MEN1 occurs in a familial form with an autosomal dominant inheritance. Mutations of MEN1 gene are identified in up to 90% of index cases with familial disease and in up to 30% of sporadic cases (2). MEN1 gene, consisting of 10 exons, is located on the long arm of chromosome 11 (11q13). The gene product, menin, plays a key role in the regulation of cell proliferation and differentiation by interacting directly or indirectly with more than 50 different proteins involved in cell adhesion, cell cycle progression, cell division, DNA repair, and other several signaling pathways (3–6). More than 1,500 mutations have been identified in familial and sporadic cases, without a correlation between genotype and phenotype (7, 8). Heterozygous germline inactivating mutation in MEN1 gene represents the first hit usually followed by a second hit represented by the loss of a large chromosomal region (11q13) of the normal copy of the gene (LOH) in MEN1-associated endocrine tissues or another somatic mutation. Such events lead to the complete loss of function of the encoded protein menin, according to Knudson’s model of tumor suppressor genes (9).
A combination of more than 20 endocrine and non-endocrine manifestations has been reported in the affected subjects. Clinical variability has also been observed in subjects within the same family, suggesting that epigenetic regulation may contribute to the clinical phenotype of MEN1 (10). MEN1 syndrome most frequently involves the parathyroids, pancreatic islets, and pituitary (11). The most common manifestation is primary hyperparathyroidism (PHPT), which occurs in 99% of patients by the age of 50, usually caused by benign, uniglandular or multiglandular, synchronous or asynchronous, parathyroid involvement and extremely rarely by parathyroid carcinoma (5, 12). PHPT is the first manifestation of MEN1 in approximately 80%–85% of patients. Tumors of the gastroenteropancreatic (GEP) tract, present in 30%–70% of patients, typically occur after 40 years of age as non-functioning or functioning lesions secreting gastrin, insulin, glucagon, or vasoactive intestinal peptide (5). Pituitary involvement is present in 30%–40% of cases, mostly due to prolactinomas (5).
MEN1 mutations seem to be a discriminating factor associated with the classic phenotype of MEN1 syndrome, whereas most of the MEN1 mutation-negative patients have a different phenotype and clinical course of the disease, representing the so-called phenocopies (13). These patients, mainly affected by MEN1 without a familial history, have a later onset of the first manifestation, a lower likelihood of developing a third MEN1-related lesion, and a life expectancy comparable with that of the general population (2, 8, 13–15). In these cases, mutations of other genes might be responsible for a MEN1-like phenotype. In particular, mutations of CDKN1B gene encoding the cyclin-dependent kinase inhibitor p27 are responsible for the MEN4 syndrome (16, 17), whereas mutations of other cyclin-dependent kinase inhibitor (CDKI) genes—CASR, AIP, and CDC73 genes—can also found in rare cases of MEN1-like phenotypes (2, 18, 19).
Several non-endocrine manifestations such as lipomas, angiofibromas, collagenomas, hibernomas, leiomyomas, and central nervous system tumors (meningiomas and ependymomas) have been reported in MEN1 patients (20–24). The association between cutaneous lesions and MEN1 syndrome was first reported in 1997 (20), and the prevalence of these manifestations differs in different series with a prevalence of 22%–88% of multiple facial angiofibromas, 0%–72% of collagenomas, and 5%–34% of lipomas (20–22, 25, 26). The finding of these lesions in association with endocrine tumors suggests the diagnosis of MEN1 syndrome. In particular, the occurrence of multiple angiofibromas as isolate cutaneous manifestation has the highest specificity, whereas a combination of multiple angiofibromas and any collagenomas has the highest sensitivity and specificity for MEN1 (22). Therefore, a thorough skin examination should be performed in patients with PHPT, GEP, and pituitary tumors, and the finding of cutaneous lesions raises suspicion of MEN1.
Increased risk and an early-onset of breast cancer have been reported in MEN1-mutated women than in the general population (27–29), and therefore, breast cancer surveillance should be started 10 years earlier in the former than in the latter women (27).
The objective of this study was to describe cutaneous lesions and other non-endocrine manifestations of MEN1 in a well-characterized cohort of patients with familial and sporadic MEN1 syndrome, compare their prevalence in the two cohorts, and seek a correlation between these manifestations and the MEN1 mutational status.
Clinical data of 106 index cases with MEN1 syndrome followed up at the Endocrine Unit of Pisa from January 1997 to May 2022 were retrospectively collected. Seventy-nine relatives carrying MEN1 mutations were also evaluated. Data obtained up to 2015 were already reported (2). The diagnosis of MEN1 syndrome was made according to the criteria established by the latest International guidelines, namely, i) familial MEN1 (F-MEN1): the presence of at least two MEN1 major lesions in the index case, with a first-degree relative with at least one major lesion; 2) sporadic MEN1 (S-MEN1) in the absence of a family history of MEN1-related manifestations; 3) atypical MEN1 by the association of a single major lesion with one or more uncommon MEN1-related manifestations (5).
All patients underwent a total skin examination by the attending endocrinologist on the first visit to our center and repeated each follow-up visit. The clinical criterion for the diagnosis of angiofibroma was a dome-shaped, skin-colored to red papule located on the central face, usually around the nose and on the malar eminences (30). Angiofibromas were considered multiple when more than three lesions were detected. Collagenomas are benign connective tissue nevi and usually present as asymptomatic, firm, round to oval hypopigmented, or skin-colored papules preferentially located on the trunk and upper part of the arms. Lipomas were defined as non-painful, round, mobile masses, with a characteristic soft, doughty feel localized at a subcutaneous or visceral site (31). The diagnosis of cutaneous lipomas was made by clinical examination and/or ultrasonography (US), while visceral lipomas were mostly diagnosed with contrast-enhanced computed tomography (CECT), magnetic resonance imaging (MRI), or US. Lipomas that were surgically removed underwent histological examination. We also included angiolipoma, which is a variant of lipoma with co-existing vascular proliferation (32). Hibernomas are rare benign tumors originating from the brown adipose tissue, usually located in the thigh, shoulder, and back. In most cases, they are asymptomatic, although occasionally a “pressure” type of pain may be present. Hibernomas are typically mobile and pliable. Imaging plays a key role in their diagnosis (33). We detected hibernoma during clinical exams and then confirmed it by CECT. The presence of suspicious melanoma was confirmed histologically after surgical excision.
Uterine leiomyomas, if not referred to medical history, were identified by complete abdominal ultrasound and/or CECT or MRI, including uterus evaluation, routinely performed in women of any age according to the MEN1 guidelines (5). Meningiomas were incidentally identified by pituitary MRI performed during the regular follow-up and screening of pituitary adenoma or by CECT or whole-brain MRI (available in 35 out of 185 patients of the whole cohort) performed for other purposes. Breast cancer was diagnosed with US or mammography during breast cancer screening according to general population guidelines or as an incidental finding on the chest CECT scan performed for the screening of the MEN1-related tumors.
Gastric leiomyomas appeared as a well-defined solid mass with smooth contours and low homogeneous contrast enhancement on CECT (34). The diagnosis was confirmed by endoscopic ultrasound.
The study was part of the regular patient follow-up with retrospective analysis of data on the basis of the written informed consent routinely obtained from the overall patient population in the institution.
DNA was extracted from index patients’ peripheral leucocytes with Maxwell16 Instrument according to the manufacturer’s instructions (Promega Corp., Madison, WI, USA). The entire coding region and intron/exon boundaries of MEN1 (GenBank entry NM_130799.2), CDKN1B (NM_004064.5), and AIP (NM_003977.4) genes were first investigated by sequencing germline DNA from all index patients. PCR-amplified DNA was sequenced in forward and reverse directions by direct cycle-sequencing using BigDye Sequencing Reaction kit v.1.1 (Applied Biosystems, Foster City, CA, USA) and run-on ABI 3130XL automated sequencer (Applied Biosystems). In kindreds carrying MEN1 mutation, the mutational analysis of the region of interest was extended to first-degree relatives of the index case.
Multiplex ligation-dependent probe amplification (MLPA) analysis has been performed on the DNA of the index cases that resulted negative by sequencing analysis to detect possible large monoallelic deletions or amplifications in MEN1, AIP, and CDKN1B genes. We used the SALSA MLPA probemix kit P244-C1 (MRCHolland, Amsterdam, The Netherlands). The assay was performed according to the manufacturer’s instructions, as previously reported (35). Every experiment included almost three reference DNA blood samples derived from healthy subjects that are not expected to have any copy number changes in the region of interest and a negative control sample with no DNA, as well as appropriate positive controls.
Quantitative variables with normal data distribution were expressed as mean and standard deviation (SD). The associations between F-MEN1 and S-MEN1 or MEN1 mutation-positive and mutation-negative patients and dichotomous variables (e.g., presence or absence of MEN1-related non-endocrine tumors) were determined using chi-square or Fisher’s exact tests, as appropriate. Multivariate exact logistic regression analyses were performed to evaluate the previously mentioned associations after adjustment for age at the last visit. A value of p < 0.05 was considered statistically significant.
The whole series included 185 MEN1 patients: 106 index cases and 79 relatives. This cohort included 119 (64%) women and 66 (36%) men (female-to-male ratio of 1.8:1), with a mean age at the first manifestation of 41 years (SD ± 16, range 6–88 years). Fifty (47%) index cases were classified as F-MEN1 and 55 (53%) as S-MEN1. The remaining patient was not classified as familial or sporadic because she was adopted.
Familial MEN1 (n = 129): This group included 50 index cases and 79 relatives, with a female-to-male ratio of 1.4:1 (Table 1). The mean age at first diagnosis was 37 years (SD ± 17, range 6–88 years).
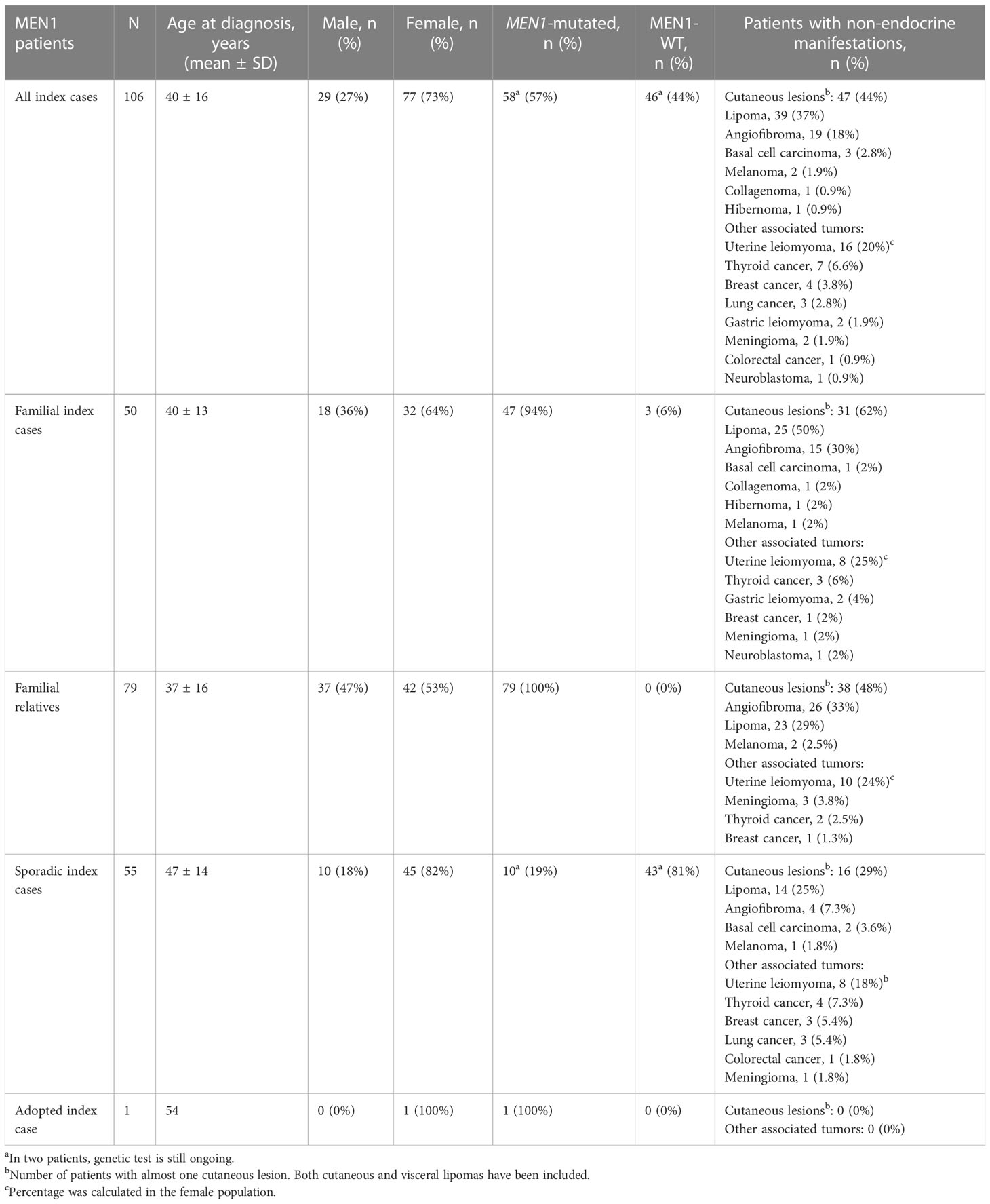
Table 1 Demographic, MEN1 mutation status, and non-endocrine manifestations of MEN1 patients.
Nine patients (mean age 23 years, range 8–62 years) had no clinical manifestations but carried MEN1 gene mutation.
Sporadic MEN1 (n = 55): In this cohort, we observed a ± higher female-to-male ratio (4:1), with a mean age at diagnosis of 47 years (SD 14, range 17–70 years) (Table 1).
PHPT was present in 175 (95%) patients, GEP tumors in 116 (63%), pituitary adenomas in 85 (45%), and adrenal lesions in 64 (35%). Nine family members were MEN1 gene carriers with no abnormalities in biochemical analyses or instrumental evidence of main endocrine-associated tumors. Two of them only presented non-endocrine manifestations (one angiofibroma and one melanoma).
The classical triad of MEN1-related tumors (PHPT, GEP, and pituitary) was present in 49 (28%), PHPT and GEP tumors in 67 (38%), and PHPT and pituitary tumors in 35 (20%), whereas PHPT alone or associated with minor tumors (adrenal lesions, lung and thymic carcinoids, gastrointestinal stromal tumors, and pheochromocytomas) was observed in 24 (14%) affected patients of the whole series. Thirty-one (29%) index cases presented the classical triad of MEN1-related tumors, 37 (35%), PHPT and GEP tumors, and 31 (29%) PHPT and pituitary adenomas. Seven index cases (7%) had PHPT alone or were associated with minor tumors.
Familial MEN1 (n = 129): In the whole series, PHPT was present in 119 (92%) patients, GEP tumors in 90 (70%), pituitary adenoma in 45 (35%), and adrenal lesions in 43 (33%). Fifty-three (44%) patients developed PHPT and GEP tumors; 37 (31%) PHPT, pituitary, and GEP tumors; and 7 (6%) PHPT and pituitary adenomas. Twenty-two (18%) patients had PHPT with or without minor tumors, and one relative (1%) had only pituitary adenoma. Nine patients had no clinical manifestations but carried MEN1 gene mutation.
The phenotype of the index cases consisted of the classical triad in 19 (38%), PHPT and GEP tumors in 23 (46%), and PHPT and pituitary adenomas in 3 (6%) patients. Five (10%) patients had PHPT with or without minor tumors.
Sporadic MEN1 (n = 55): All patients had PHPT, alone or in association with other main MEN1-related tumors, 71% had pituitary adenoma, 45% had GEP tumors, and 38% had adrenal lesions. The classical triad was present in 12 (21%), PHPT and pituitary tumors in 28 (51%) patients, PHPT and GEP tumors in 14 (25%), and PHPT with or without minor tumors in 2 (4%).
Whole series (n = 185): Eighty-five (46%) patients had at least one cutaneous lesion, lipoma (cutaneous or visceral), and/or hibernoma. Lipoma, observed in 62 patients, was the most common non-endocrine manifestation (33.5%), which was cutaneous in 46 cases, visceral in 8, and both cutaneous and visceral in 8. Angiofibromas were present in 45 (24%) patients. Twenty-three patients had both lipomas and angiofibromas. Melanomas were present in four (2.2%) patients, two index cases, and two relatives. Basal cell carcinoma was present in three (1.6%). Collagenomas and hibernomas were each present only in one patient (0.5%). Lipomas represented the main non-endocrine manifestation in the index cases (37%), followed by angiofibromas, detected in 18% (Table 1).
Familial MEN1 cohort (n = 129): Sixty-nine (53%) patients had at least one cutaneous lesion, lipoma, and/or hibernoma. Angiofibromas were observed in 40 (31%) patients, equally distributed between index cases (30%) and relatives (33%). In 28 patients, angiofibromas were multiple and preferentially (64%) localized to the face (upper lip and nose) (Table 2). A representative example of angiofibroma is shown in Figure 1. Lipomas were observed in 48 (37%) cases, mostly detected in index cases rather than relatives (50% vs. 29%). Thirty-six patients had only cutaneous lipomas, five had both visceral and cutaneous, and seven had only visceral. Cutaneous lipomas had multiple localizations and were preferentially located at the thorax and the upper and lower limbs. A representative example of visceral lipoma is shown in Figure 2. Seventeen lipomas were surgically removed, and in one case, the histology was consistent with a malignant liposarcoma. Twenty-one (16%) patients had both angiofibromas and lipomas (visceral and cutaneous). The frequency and distribution of angiofibromas and lipomas are summarized in Table 2. Melanomas were present in three (2.3%) patients. Collagenoma or hibernoma was present in one (0.8%) index case.
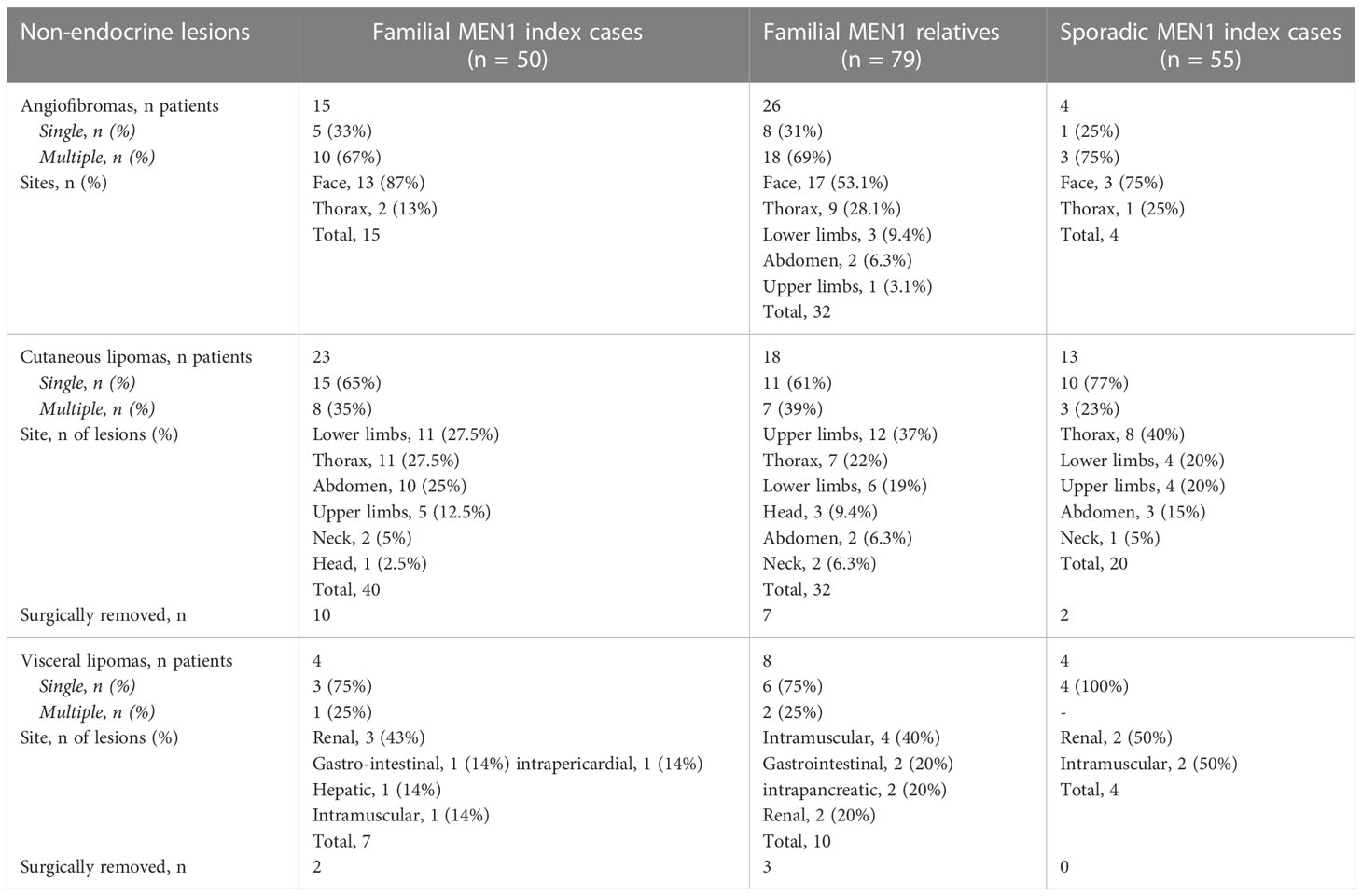
Table 2 Frequency and distribution of angiofibromas and lipomas in familial and sporadic MEN1 patients.
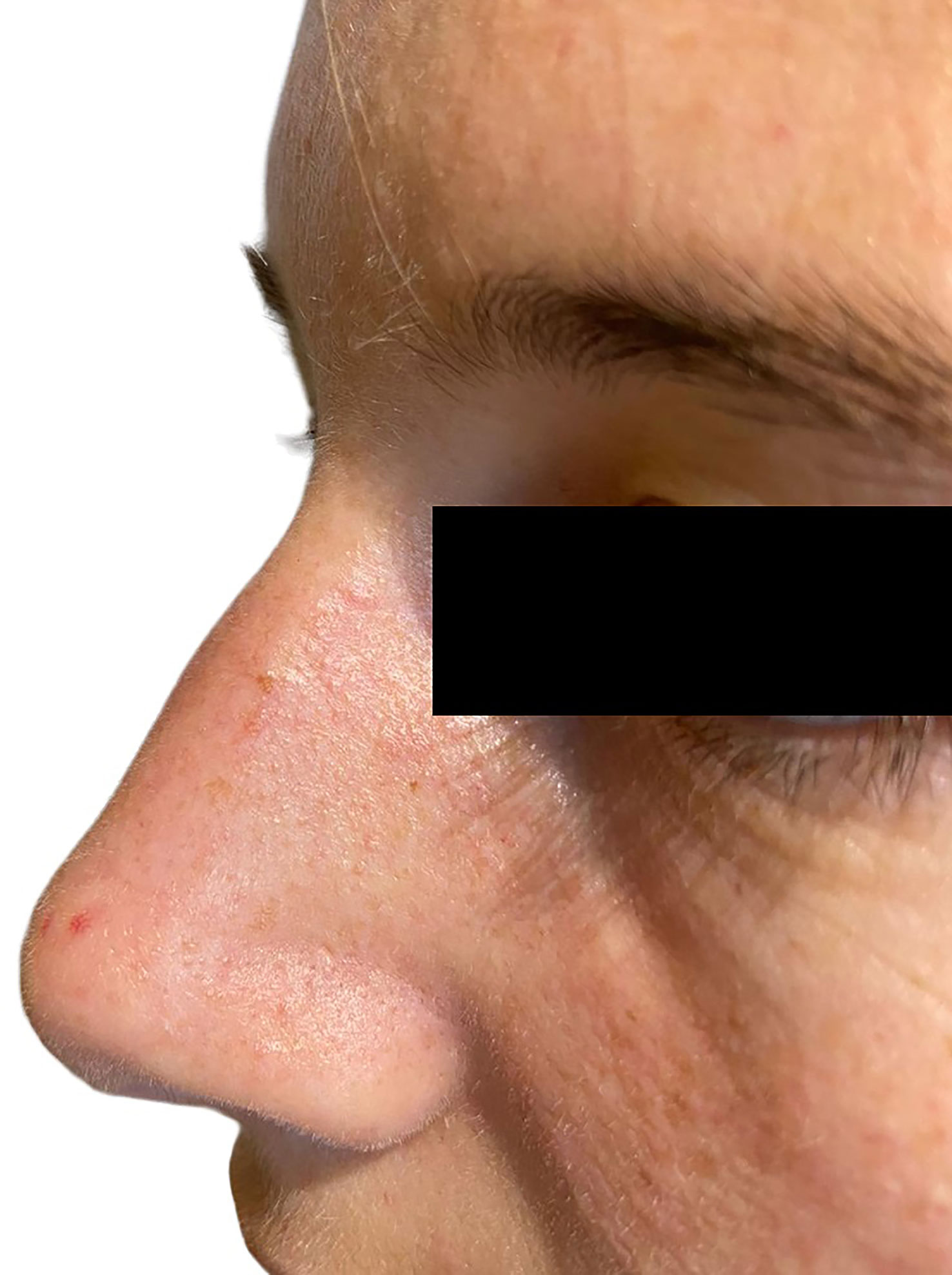
Figure 1 A facial angiofibroma in a 22-year-old woman with familial MEN1 is shown. It appears as a dome-shaped, skin-colored to red papule located on the nose.
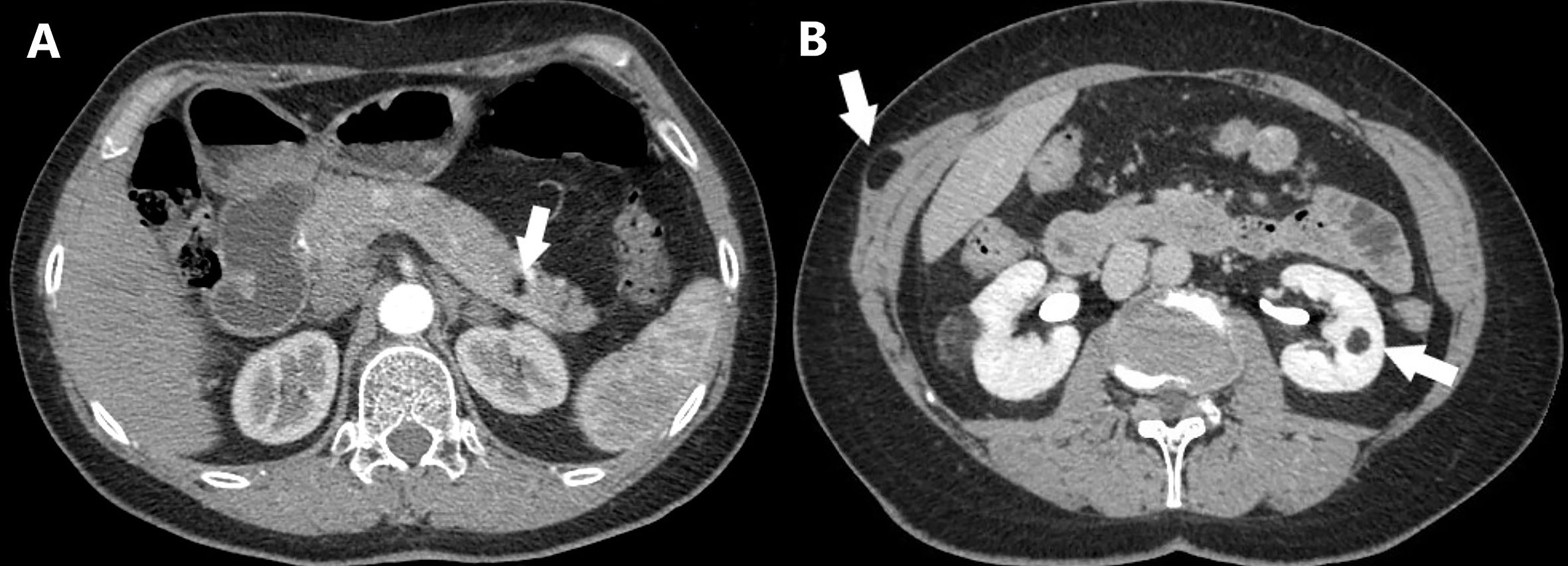
Figure 2 Axial CT scan of a MEN1 patient with multiple lipomatosis. (A) Intrapancreatic lipoma is shown (arrow). (B) Intramuscular lipoma and left renal angiomyolipoma are shown (arrows).
Sporadic MEN1 cohort (n = 55): Sixteen (29%) patients had at least one cutaneous lesion and/or lipoma. Lipomas were present in 25% of the patients. None of them was surgically excised. Angiofibromas, mostly multiple, were present in 7% of the patients. Half of the patients bearing angiofibromas also had cutaneous lipomas. The frequency and distribution of angiofibromas and lipomas are summarized in Table 2. Melanoma was present in one (1.8%) patient (Table 1).
Whole series (n = 185): Uterine leiomyomas were present in 26 (22%) out of 119 women and gastric leiomyomas in two (1.1%) patients of the whole series.
Familial MEN1 (n = 129): Uterine leiomyomas were observed in 18 (24%) out of 119 women, and such percentage was similar in both index cases and relatives (Table 1). Gastric leiomyomas were present in two (1.5%) index cases.
Sporadic MEN1 (n = 55): Uterine leiomyomas were present in nine (20%) out of 45 women.
Whole series (n = 185): Meningiomas were detected in five (2.7%) patients. Neuroblastoma was present in only one patient (0.5%).
Familial MEN1 (n = 129): Meningiomas were present in four (3.1%) patients. Neuroblastoma was present in only one index case (0.8%).
Sporadic MEN1 (n = 55): Meningioma was present in one (1.8%) patient (Table 1).
In the whole series, breast cancer was present in five (2.7%) patients: one familial index case, one relative, and three sporadic cases. The median age of diagnosis was 49 years, and only one case was triple-negative invasive ductal carcinoma with lymph node metastasis with recurrence occurring 15 years later. All the remaining were unilateral and unifocal ductal carcinoma in situ. All patients had PHPT; however, none of them had prolactinoma or insulinoma. Seventy-five percent of them expressed the estrogen receptor. In one case, these data were not available.
In the whole series, thyroid cancers (one Hurthle cell, seven papillary, and one medullary) were found in nine (4.9%), lung cancers in three (1.6%), and colorectal cancer in one (0.5%). Lung and colorectal cancers were exclusively found in sporadic cases (Table 1).
Some of the MEN1 mutations identified were previously described (2). Fifty-eight index cases (56%) carried germline MEN1 variants. DNA of two patients of the S-MEN1 cohort was not available because the patients refused the genetic test. Fifty-four variants were identified by direct sequencing and mainly localized in exons 2 (26%), 10 (17%), and 9 (14%). No mutations were detected in exon 5, and only one was detected in exons 6 and 8 (Figure 3). Four germline MEN1 large deletions were identified in two F-MEN1 and two S-MEN1 cases (Figure 4) (2). Eight variants recurred in two or more index cases (14%). Eighty-one percent of all detected variants were identified in familial cases, being identified in 94% of F-MEN1 and 19% of S-MEN1 (p < 0.00001). Sixty percent of the variants were frameshift, non-sense, or splice site junction mutations, leading to a truncated menin protein (Figure 4). All but three mutations had been already described and had a proven or predicted pathogenicity. The variant p.A25V is considered a variant of unknown significance by ClinVar, although in silico tools (Fathmm, MutationTaster, PolyPhen-2, and Align-GVGD) all predicted a likely pathogenic role. A putative pathogenic role was also predicted for the two novel missense mutations (p.L37R and p.H317D), whose codons were already described to be affected by different substitutions (36, 37). The classifications for these three variants were further analyzed using the standards and guidelines published by the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) that propose a clue for the interpretation of missense variants dividing them into five categories based on evidence obtained through population data, computational, functional, and segregation data and expert opinion, and workgroup (38). Varsome, according to the ACMG/AMP guidelines, reported the variant p.A25V with moderate evidence of pathogenicity (PM1 and PM2), p.L37R as likely pathogenic (PP3, PM1, and PM2), and p.H317D as pathogenic (PM5, PP3, PM1, and PM2).
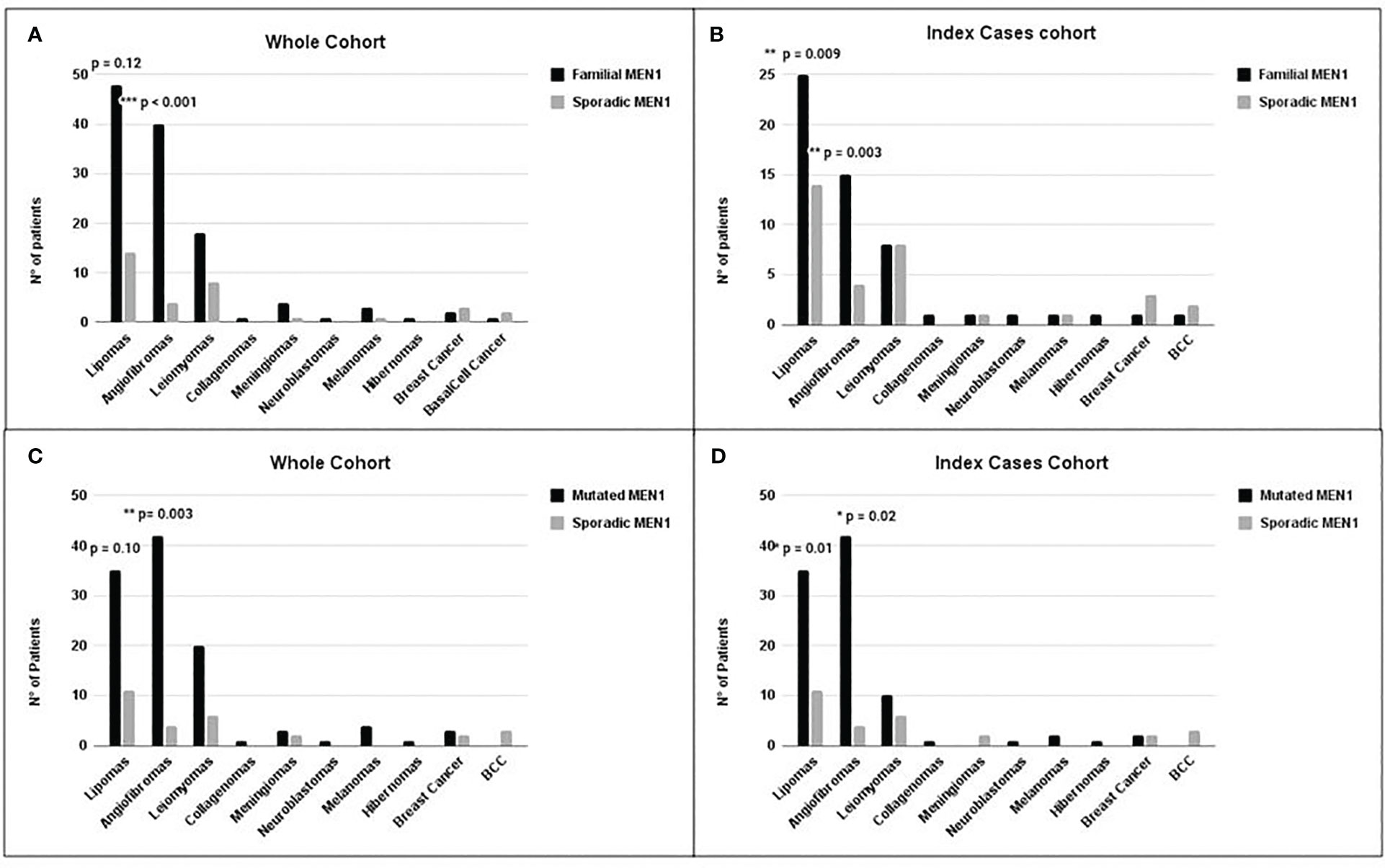
Figure 3 Distribution of the main non-endocrine manifestations in MEN1 cohorts. Comparison between non-endocrine manifestations in the F-MEN1 vs. S-MEN1 in the whole cohort (A) and index cases (B) and in MEN1 mutation-positive vs. MEN1 mutation-negative in the whole cohort (C) and index cases (D). Statistical significance was determined by Fisher’s or chi-square test. *p < 0.05, **p < 0.01 and ***p < 0.001.
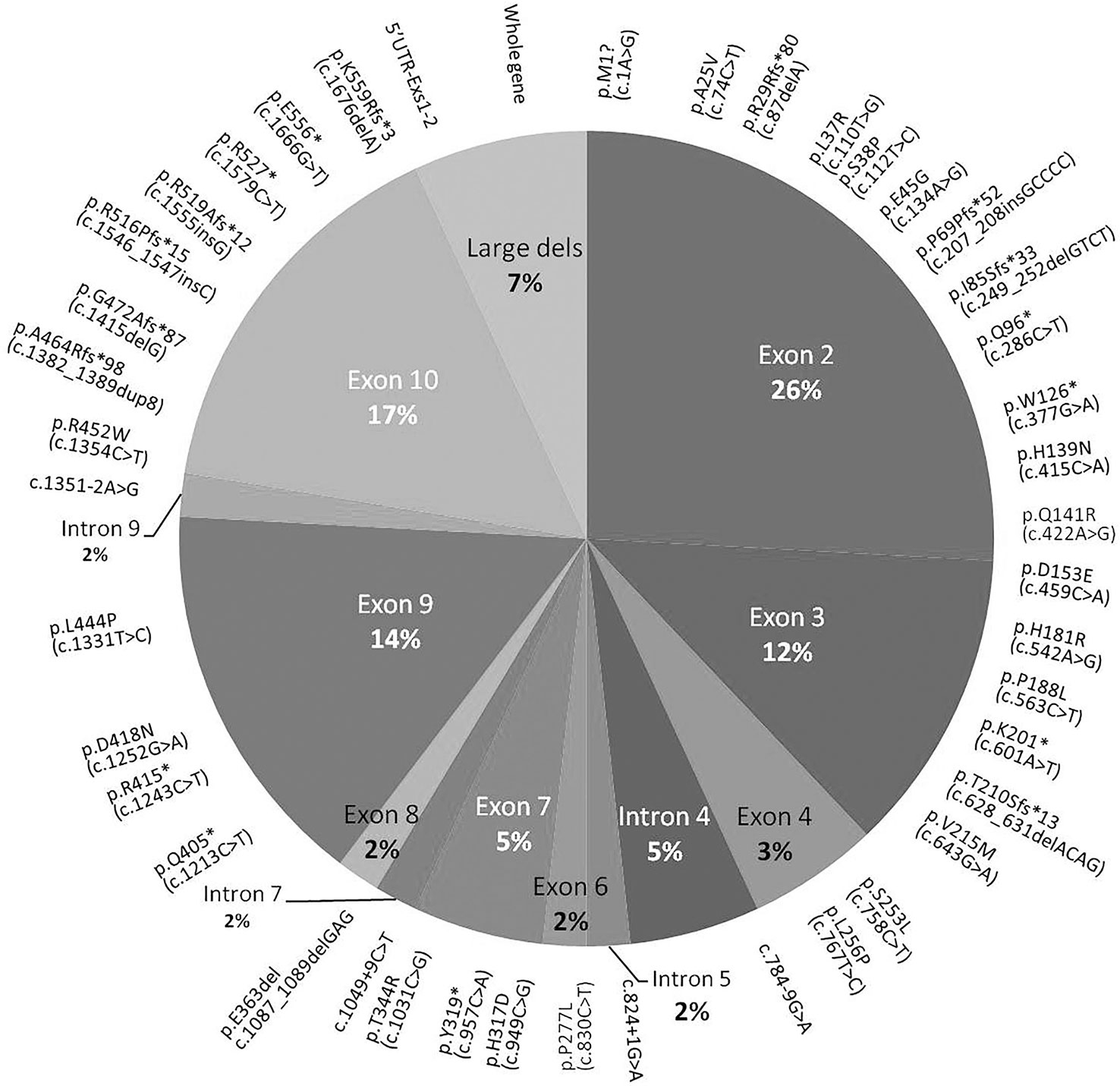
Figure 4 Pie chart showing the distribution of MEN1 mutations through the exonic and intronic portions of MEN1 gene. Mutations detected in more than one patient or one or more sporadic patients are reported in parentheses. F, familial; S, sporadic.
One germline missense variant in exon 1 of the AIP gene (p.R9Q) was already reported in one S-MEN1 proband (2). Due to the conflicting interpretation of pathogenicity based on structural, functional, and in silico studies, this variant is reported as having uncertain clinical significance (PP5, BP1, and BP4, according to the ACMG/AMP guidelines).
The female-to-male ratio differs between the two cohorts (p = 0.001) with a higher female-to-male ratio in the sporadic than in the familial cohort (4.5:1 vs. 1.4:1). This different ratio was confirmed also in the index cases cohort (4.5:1 vs. 1.6:1).
We compared the prevalence of non-endocrine manifestations in F-MEN1 and S-MEN1 in the whole and index case cohorts. The prevalence of angiofibromas was significantly higher in the whole F-MEN1 cohort compared to S-MEN1 (OR = 5.9, p < 0.001) even after adjustment for age (adj. p = 0.002, data not shown). Such difference was statistically significant even when considering only index cases (OR = 5.5, p = 0.003) (Figures 3A, B). The prevalence of lipomas did not differ between the two whole cohorts (p = 0.12) (Figure 3A), but the difference between F-MEN1 and S-MEN1 became significant after adjustment for age (OR = 2.4, adj. p = 0.03, data not shown) or if we only considered the index cases cohort (OR = 2.9, p = 0.009) (Figure 3B). The prevalence of other minor MEN1-related non-endocrine manifestations did not differ between the two cohorts (Figure 3).
In this analysis, we compared the prevalence of non-endocrine manifestations in MEN1 mutation-positive and MEN1 mutation-negative index cases and the whole series independently of whether they were classified as F-MEN1 or S-MEN1.
In the whole cohort of familial cases, we observed a higher prevalence of angiofibromas in MEN1 mutation-positive compared to MEN1 mutation-negative (OR = 4.5, p = 0.003) (Figure 3C), also after adjustment for age. The prevalence of lipomas did not differ between the two whole cohorts (p = 0.10): however, after adjustment for age, such prevalence in MEN1 mutation-positive was significantly higher compared to MEN1 mutation-negative (OR = 2.7, p = 0.02, data not shown). No difference was found in the prevalence of other manifestations between the two cohorts.
In the index cases cohort, we observed a significantly higher prevalence of angiofibromas (OR = 3.7, p = 0.02) and lipomas (OR = 3.0, p = 0.01) in the MEN1 mutation-positive compared to MEN1 mutation-negative index cases (Figure 3D). No difference was found in the prevalence of other minor MEN1-related non-endocrine manifestations between the two cohorts.
We further conducted two additional analyses: we compared the occurrence of non-endocrine lesions in MEN1 mutation-positive index cases of F-MEN1 (n = 47) vs. S-MEN1 (n = 10) and in MEN1 mutation-positive (n = 10) vs. mutation-negative (n = 43) index cases of S-MEN1. No significant differences were found in the prevalence of any non-endocrine lesions in both comparisons.
The association between a variety of cutaneous lesions (i.e., angiofibromas, collagenomas, and lipomas) and MEN1 syndrome was first reported in 1997 by Darling et al. (20) and has been confirmed in subsequent studies (39, 40). Biopsies of facial angiofibromas, lipomas, and collagenomas from patients harboring germline MEN1 variants exhibited an allelic deletion of chromosome 11 including the MEN1 locus (24). This alteration was specific to these lesions and absent in other skin lesions found in the same patients but not typically associated with the syndrome (24). Rusconi et al. showed that in a patient carrying germline heterozygous MEN1 mutation, the somatic inactivation of the wild-type allele arose in different MEN1-related tumors (pituitary adenoma and lipoma) of the same patient by distinct mechanisms, i.e., loss of heterozygosity and balanced translocation (41).
Cutaneous manifestations are often underestimated or considered ancillary findings not related to the classical clinical spectrum of the MEN1 syndrome, as they are typically benign and do not usually require specific treatments. Only six previous studies evaluated the frequency of cutaneous lesions in a series of patients with MEN1 (20–22, 25, 42, 43). In a prospective study including 110 consecutive patients with gastrinomas, either sporadic or in the context of MEN1 syndrome, the combined presence of multiple angiofibromas and any collagenomas was considered the best cutaneous diagnostic criterion for MEN1 with a specificity of 95% and a sensitivity of 75% (22).
In our study, 46% of patients of the whole series, including relatives carrying MEN1 mutation, had at least one cutaneous lesion, lipoma, and/or hibernoma. These lesions were significantly more frequent in familial vs. sporadic cohorts. An overall prevalence of cutaneous lesions in MEN1 patients reported in the literature ranges from 22% to 55% (21, 42, 43). Our results are consistent with those reported by Vidal and colleagues (13) in nine MEN1 mutation-positive patients. They compared the prevalence of cutaneous lesions (angiofibromas, collagenomas, melanosis guttaca, lipomas, melanomas, and “café-au-lait macules”) with 20 non-carrier relatives and found a significantly higher prevalence of any cutaneous lesions in the former compared to the latter group (55.5% vs. 25%; p = 0.029) (21).
Angiofibromas, mostly multiple (69%) and localized to the face (upper lip and nose), were present in about one-quarter (24%) of the whole series and 18% of the index cases. The prevalence of angiofibromas in MEN1 among different studies ranges from 0% to 88% (12–14, 17, 32, 33). This wide difference might be due to the expertise of the skin examiner. Of interest, the three studies in which experienced dermatologists performed a thorough skin evaluation reported a prevalence of angiofibromas of 88%, 65%, and 22% (20–22).
The prevalence of angiofibromas in our MEN1 mutation-positive patients was significantly higher compared to that of MEN1 mutation-negative. This finding supports the hypothesis that mutations in MEN1 gene could lead to the abnormal proliferation of some cutaneous cells and the development of skin lesions reported in patients with the syndrome (24). The significantly higher prevalence of angiofibromas in our familial compared to sporadic MEN1 cohort, observed in both the whole and index cases, was expected due to the association between MEN1 mutations and familial MEN1 cases (94% F-MEN1 mutation-positive vs. 19% S-MEN1 mutation-positive, p < 0.00001).
The presence of lipomas in patients with MEN1 has been reported since the first description of the syndrome (11, 44). An in vitro study that matched normal and menin-deficient adipocytes from wild-type and menin-null mouse embryonic stem cells showed that menin deficiency led to fat-cell hypertrophy, supporting the causal relation between MEN1 gene alterations and the onset of lipomas (45). In our study, the prevalence of lipomas (33.5%) is in agreement with that reported in the literature (20–22, 43, 46–50). We found a significantly higher prevalence of lipomas in MEN1 mutation-positive compared to MEN1 mutation-negative (p = 0.01) and in familial compared to sporadic (p = 0.009) cohorts if only index cases were considered. Although a clear correlation between the prevalence of lipomas and patient age or disease duration was not demonstrated through literature (22), our data suggest that the young age (46 ± 19 vs. 57 ± 14) and/or a short follow-up (11 vs. 21 years) of relatives compared to index cases might contribute to the underestimation of the prevalence of these lesions.
The lack of a significant difference in the prevalence of any non-endocrine lesions between MEN1 mutation-positive index cases of the F-MEN1 (n = 47) and the S-MEN1 (n = 10) cohort seems to confirm previous findings, suggesting a putative role of MEN1 mutations in the classic phenotype of MEN1 syndrome, including the occurrence of related non-endocrine manifestations.
The observed difference in the prevalence of two main non-endocrine manifestations between MEN1 mutation-positive and mutation-negative patients, as well as familial and sporadic index cases, raises the central issue about the existence of MEN1 phenocopies. Among the group of S-MEN1 patients who tested negative for MEN1 mutations (80%), 82% had two main MEN1-related lesions (70% of them had PHPT and pituitary adenoma, and 30% had PHPT and GEP), whereas the remaining 18% had the classical triad. These patients may have the so-called MEN1 phenocopies, which are characterized by a later onset of the first manifestation, a lower likelihood of developing a third MEN1-related lesion, and a life expectancy comparable with that of the general population (2, 8, 13–15). Mutations of other genes (CDKN1B and other CDKI genes, CASR, AIP, and CDC73) might be responsible for a MEN1-like phenotype (2, 16–19). For this reason, all patients also underwent Sanger sequencing for CDKN1B and AIP genes. Only one S-MEN1 patient carried an already reported germline missense mutation (p.R9Q) in AIP gene (2, 51, 52). This patient presented a triad consisting of multiglandular PHPT, non-functional GEP, and a pituitary adenoma co-secreting PRL and GH, a type of pituitary tumor common in AIP mutation-positive patients with familial isolated pituitary adenoma (53). Due to the conflicting interpretation of pathogenicity based on structural, functional, and in silico studies, this variant is reported of uncertain clinical significance.
Nevertheless, a possible reason for false-negative genetic tests might be due to technical problems: Sanger sequencing, theoretically a very sensitive method for heterozygous germline mutations, might have missed mutations, especially those present in the sequencing traces as well as genetic mosaicisms (54). Moreover, although the prevalence of alterations in 5′ and 3′ untranslated regions as well as in introns seems to be very low (55, 56), we cannot exclude such events. Of note, three F-MEN1 patients had no mutations of MEN1, CDKN1B, and AIP genes. One of them had the classical triad, namely, PHPT, pituitary adenoma secreting GH, pancreatic non-secreting tumor, adrenal adenoma and angiofibroma, lipoma, meningioma, and uterine leiomyomas. The remaining two index cases had PHPT and prolactinoma, and one of them also had an adrenal adenoma. We can hypothesize that the genetic standard analyses may have missed the rare anomalies in non-codifying DNA regions (highly likely for the first patient) or that they represent MEN1 phenocopies.
To address the question of whether the higher female-to-male ratio in the sporadic than in the familial cohort might account for the difference in the prevalence of skin lesions, we checked for a potential gender difference in the prevalence of angiofibroma and lipoma in the general population. No gender difference in the prevalence of angiofibroma was reported in the literature (57). To our knowledge, no data on gender differences were available for lipoma. We evaluated the prevalence of lipomas in men and women in our cohort of 185 patients. We found no statistical difference in the prevalence of lipoma between gender (women, 26%; men, 35%; p = 0.17). Thus, we may conclude that the different female-to-male ratios observed between familial and sporadic cohorts would not account for the statistical difference in the prevalence of such lesions.
Of interest, in one familial MEN1 mutation-positive index case, the histologic examination of an apparently resected benign lipoma was consistent with the diagnosis of liposarcoma. To our knowledge, only two cases of liposarcoma were reported in patients with MEN1 (58, 59).
We found one case of hibernoma under the left scapula in a relative of familial MEN1 mutation-positive. Only seven cases of hibernoma in MEN1 patients have been reported in the literature (60–65), and a relationship between this manifestation and the syndrome seems to be supported by genetic analysis (60, 64). The discovery of deletions of MEN1 gene in resected hibernomas of MEN1 and non-MEN1 patients supported the role of this gene in the development of these lesions (66). Later studies underlined that deletions involved a large region on 11q13 also including the gene encoding aryl hydrocarbon receptor-interacting protein (AIP), and the concomitant loss of MEN1 and AIP were supposed to be involved in the pathogenesis of hibernoma (67, 68).
We also reported only one case of collagenoma in a familial MEN1 index case. The prevalence of collagenoma in MEN1 reported in the literature ranges from 0% to 72% (20–22). Pack and colleagues found 11q13 loss of heterozygosity in collagenomas resected from MEN1 patients, which was not present in other skin lesions unrelated to the syndrome, supporting the association of this manifestation with MEN1 (24).
Three MEN1 mutation-positive and one MEN1 mutation-negative patients had melanomas. Melanomas were reported in other MEN1 cohorts, but a clear relationship between these lesions and MEN1 gene alteration has not been proved (39, 40). Although some studies reported a tumor suppressor role for MEN1 in sporadic melanomas, the relatively high prevalence of this malignancy in the general population and the lack of evidence of a somatic hit in resected melanoma of MEN1 patients suggest that the occurrence of melanomas in MEN1 syndrome may be incidental (69, 70).
Neoplasms of the central nervous system, i.e., meningiomas, ependymomas, and schwannomas, have been reported as clinical manifestations of MEN1 syndrome (5). Asgharian et al. suggested that the increased occurrence of meningiomas in MEN1, observed in previous studies, was not accidental. They found that meningiomas were 11 times more frequent in patients with MEN1 and Zollinger–Ellison (ZES) syndrome than with ZES alone and demonstrated that allelic loss at MEN1 but not at NF2 gene locus and frequent alterations in sporadic meningiomas play a role in the pathogenesis of MEN1-associated meningioma (71). Herein, meningioma was present in 5/185 (2.7%) cases: three familial MEN1 mutation-positive, one familial, and one sporadic MEN1 mutation-negative patients. The co-occurrence of meningioma and pituitary adenoma, especially GH-omas, has been reported (72, 73). A genetic predisposition, i.e., a germline MEN1 mutation, seems to explain the high rate of the simultaneous development of these two benign tumors of the central nervous system (74). In our study, only one patient with meningioma also had a GH-oma.
Leiomyomas are benign mesenchymal smooth muscle tumors that arise throughout the body but most commonly affect the uterus, representing the most common neoplasms of reproductive-aged women (75). Although an association between leiomyoma and MEN1 had been previously suggested by several case reports, Vortmeyer and colleagues first demonstrated the inactivation of MEN1 gene in the esophageal leiomyoma tissue of a MEN1 patient, suggesting that this neoplasm could share a common molecular cause with the main MEN1-associated tumors (76). Loss of heterozygosity at MEN1 locus was also found in esophageal and uterine leiomyomas in four of five F-MEN1 patients, whereas such loss seemed not to play a role in sporadic uterine leiomyomas (77). In our study, 22% of women developed uterine leiomyomas. A systematic review of the general population reported an incidence range between 5.4% and 77% in women of reproductive age (78). Due to this wide range, a comprehensive comparison between our data and those of the literature is difficult. Nevertheless, we calculated the cumulative incidence of leiomyoma in our MEN1 patients across age groups. An increase in the cumulative incidence of leiomyoma was observed according to the age groups (2.9% in women <30 years, 5.7% between 30 and 39 years, 13.9% between 40 and 49 years, and 27% >50 years). A similar trend of increase but with higher cumulative-incidence events was reported by Baird et al. in Caucasian women (79). Therefore, we may speculate that the association between MEN1 syndrome and uterine leiomyoma remains still to be established.
Only one study has carried out a systematic evaluation of such lesions and reported a prevalence of 12.6% in the MEN1 female population (43).
Five women in our whole series (4.2% of the female population) developed breast cancer. In recent years, there has been a growing interest in the relationship between breast cancer and MEN1 syndrome (27, 28, 40, 80). To date, breast cancer (approximately ninety cases) has been reported only in women with MEN1 (40). Among four unrelated cohorts from Holland, the United States, Tasmania, and France, which respectively included 190, 68, 71, and 536 women affected by MEN1 with a follow-up from 5 to 27 years, breast cancer has 2.3- to 2.8-fold penetrance than the control population, obtained from the respective national cancer registries (81). A reduced menin staining in breast cancer samples and loss of heterozygosity at the MEN1 locus in one-third of MEN1-mutated patients with breast cancer was reported, suggesting a possible role of the gene in breast carcinogenesis (81). Given the increased risk, a Dutch study suggested starting breast cancer screening from the age of 40 in MEN1 women (27). Since there is currently no dedicated screening program for women with MEN1, it is important to inform them about the potential increased risk and provide appropriate counseling.
Our study has several strengths: i) a large cohort of consecutive patients with MEN1 syndrome having full clinical, biochemical, instrumental, and genetic characterization followed up at a single Italian endocrine outpatient clinic; ii) inclusion of a large cohort of sporadic MEN1 cases; iii) comparison between the clinical characteristics of patients with sporadic and familial MEN1 syndrome; iii) inclusion of a relatively high number of MEN1 mutation-negative patients that allowed a statistical comparison of the prevalence of non-endocrine manifestations between MEN1 mutation-positive and MEN1 mutation-negative to strengthen the putative pathogenic role of MEN1 mutations in the development of some cutaneous lesions. However, our study does have some limitations: i) difficulty in assessing the true incidence of de novo mutations in the sporadic MEN1 cohort due to the unavailability of genetic data of index case’s parents; ii) the lack of systematic evaluation of the skin manifestations by a dermatologist that might have underestimated the presence of some cutaneous lesions, i.e., collagenomas; iii) the lack of the study of the whole brain with MRI or CECT might have underestimated the presence of meningiomas or ependymomas.
In conclusion, the results of our study contribute to increasing knowledge regarding non-endocrine manifestations of MEN1 syndrome. We found a significantly higher prevalence of angiofibromas and lipomas in F-MEN1 compared with S-MEN1. Both these manifestations were significantly more frequent in MEN1 mutation-positive compared to MEN1 mutation-negative patients.
In patients presenting with one major endocrine manifestation of MEN1, the presence of cutaneous lesions, i.e., angiofibromas and lipomas, might suggest the diagnosis of MEN1 and a possible indication of genetic screening. In these patients, it is advisable to perform an accurate physical examination, which includes a total skin examination to identify the presence of these manifestations.
Further studies are necessary to describe all non-endocrine manifestations of the syndrome and tailor a personalized approach during the follow-up and screening of some malignancies such as breast cancer.
The original contributions presented in the study are publicly available. Genomic data can be found in LOVD v. 3.0 (https://www.lovd.nl/), gnomAD v. 2.1 (https://gnomad.broadinstitute.org/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), dbVAR (https://www.ncbi.nlm.nih.gov/dbvar/). The raw data supporting the conclusions of this article will be available from the corresponding author on reasonable request.
The studies involving human participants were reviewed and approved by Comitato Etico Regionale per la sperimentazione clinica - Sezione autonoma Area Vasta Nord Ovest (CEAVNO). Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
LP, CM, and FC contributed to the study conception and design and wrote the first draft of the manuscript. AM, MC, EP, and SB performed the genetic analyses. LP, EP, ED, SB, SD, CS, and FB collected all the data. PP performed the statistical analyses. LP, EP, CM, and FC revised the manuscript. All authors read and approved the final manuscript. FC is a co-coordinator of the Mineral and Bone Club of the Italian Society of Endocrinology (part of research projects). All authors contributed to the article and approved the submitted version.
This work was supported by Clinical Protocol Veristat PaTH Forward TCP-201.
We wish to thank all the patients and the members of the families who graciously agreed to collaborate in the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Al-Salameh A, Cadiot G, Calender A, Goudet P, Chanson P. Clinical aspects of multiple endocrine neoplasia type 1. Nat Rev Endocrinol (2021) 17(4):207–24. doi: 10.1038/s41574-021-00468-3
2. Pardi E, Borsari S, Saponaro F, Bogazzi F, Urbani C, Mariotti S, et al. Mutational and large deletion study of genes implicated in hereditary forms of primary hyperparathyroidism and correlation with clinical features. PloS One (2017) 12(10):e0186485. doi: 10.1371/journal.pone.0186485
3. La P, Desmond A, Hou Z, Silva AC, Schnepp RW, Hua X. Tumor suppressor menin: the essential role of nuclear localization signal domains in coordinating gene expression. Oncogene (2006) 25(25):3537–46. doi: 10.1038/sj.onc.1209400
4. Hendy GN, Kaji H, Canaff L. Cellular functions of menin. In: Balogh K, Patocs A editors. SuperMEN1. Advances in experimental medicine and biology. New York, NY: Springer (2009). p. 37–50. doi: 10.1007/978-1-4419-1664-8_4
5. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab (2012) 97(9):2990–3011. doi: 10.1210/jc.2012-1230
6. Balogh K, Patócs A, Hunyady L, Rácz K. Menin dynamics and functional insight: take your partners. Mol Cell Endocrinol (2010) 326(1–2):80–4. doi: 10.1016/j.mce.2010.04.011
7. Vierimaa O, Ebeling TML, Kytölä S, Bloigu R, Eloranta E, Salmi J, et al. Multiple endocrine neoplasia type 1 in northern finland; clinical features and genotype–phenotype correlation. Eur J Endocrinol (2007) 157(3):285–94. doi: 10.1530/EJE-07-0195
8. Kövesdi A, Tóth M, Butz H, Szücs N, Sármán B, Pusztai P, et al. True MEN1 or phenocopy? evidence for geno-phenotypic correlations in MEN1 syndrome. Endocrine (2019) 65(2):451–9. doi: 10.1007/s12020-019-01932-x
9. Thakker RV, Bouloux P, Wooding C, Chotai K, Broad PM, Spurr NK, et al. Association of parathyroid tumors in multiple endocrine neoplasia type 1 with loss of alleles on chromosome 11. N Engl J Med (1989) 321(4):218–24. doi: 10.1056/NEJM198907273210403
10. Chung YJ, Hwang S, Jeong JJ, Song SY, Kim SH, Rhee Y. Genetic and epigenetic analysis in Korean patients with multiple endocrine neoplasia type 1. Endocrinol Metab (2014) 29(3):270. doi: 10.3803/EnM.2014.29.3.270
11. Wermer P. Genetic aspects of adenomatosis of endocrine glands. Am J Med (1954) 16(3):363–71. doi: 10.1016/0002-9343(54)90353-8
12. Song A, Yang Y, Liu S, Nie M, Jiang Y, Li M, et al. Prevalence of parathyroid carcinoma and atypical parathyroid neoplasms in 153 patients with multiple endocrine neoplasia type 1: case series and literature review. Front Endocrinol (Lausanne) (2020) 11:557050. doi: 10.3389/fendo.2020.557050
13. de Laat JM, van der Luijt RB, Pieterman CRC, Oostveen MP, Hermus AR, Dekkers OM, et al. MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med (2016) 14(1):1–9. doi: 10.1186/s12916-016-0708-1
14. Hai N, Aoki N, Shimatsu A, Mori T, Kosugi S. Clinical features of multiple endocrine neoplasia type 1 (MEN1) phenocopy without germline MEN1 gene mutations: analysis of 20 Japanese sporadic cases with MEN1. Clin Endocrinol (Oxf) (2000) 52(4):509–18. doi: 10.1046/j.1365-2265.2000.00966.x
15. Turner JJO, Christie PT, Pearce SHS, Turnpenny PD, Thakker RV. Diagnostic challenges due to phenocopies: lessons from multiple endocrine neoplasia type1 (MEN1). Hum Mutat (2010) 31(1):1089–101. doi: 10.1002/humu.21170
16. Alrezk R, Hannah-Shmouni F, Stratakis CA. MEN4 and CDKN1B mutations: the latest of the MEN syndromes. Endocr Relat Cancer (2017) 24(10):T195–208. doi: 10.1530/ERC-17-0243
17. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Höfler H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci USA (2006) 103(42):15558–63. doi: 10.1073/pnas.0603877103
18. Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab (2009) 94(5):1826–34. doi: 10.1210/jc.2008-2083
19. Backman S, Bajic D, Crona J, Hellman P, Skogseid B, Stålberg P. Whole genome sequencing of apparently mutation-negative MEN1 patients. Eur J Endocrinol (2019) 182(1):35–45. doi: 10.1530/EJE-19-0522
20. Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol (1997) 133(7):853–7. doi: 10.1001/archderm.1997.03890430067009
21. Vidal A, Iglesias MJ, Fernández B, Fonseca E, Cordido F. Cutaneous lesions associated to multiple endocrine neoplasia syndrome type 1. J Eur Acad Dermatol Venereol (2008) 22(7):835–8. doi: 10.1111/j.1468-3083.2008.02578.x
22. Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT. Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab (2004) 89(11):5328–36. doi: 10.1210/jc.2004-0218
23. Raj R, Elshimy G, Mishra R, Jha N, Joseph V, Bratman R, et al. Dermatologic manifestations of endocrine disorders. Cureus (2021) 27. doi: 10.7759/cureus.18327
24. Pack S, Turner ML, Zhuang Z, Vortmeyer AO, Böni R, Skarulis M, et al. Cutaneous tumors in patients with multiple endocrine neoplasia type 1 show allelic deletion of the MEN1 gene. J Invest Dermatol (1998) 110(4):438–40. doi: 10.1046/j.1523-1747.1998.00140.x
25. Sakurai A, Matsumoto K, Ikeo Y, Nishio S-I, Kakizawa T, Arakura F, et al. Frequency of facial angiofibromas in Japanese patients with multiple endocrine neoplasia type 1. Endocr J (2000) 47(5):569–73. doi: 10.1507/endocrj.47.569
26. Yoshimoto K, Saito S. Clinical characteristics in multiple endocrine neoplasia type 1 in Japan: a review of 106 patients. Folia Endocrinol Jpn (1991) 67(7):764–74. doi: 10.1507/endocrine1927.67.7_764
27. van Leeuwaarde RS, Dreijerink KM, Ausems MG, Beijers HJ, Dekkers OM, de Herder WW, et al. MEN1-dependent breast cancer: indication for early screening? results from the Dutch MEN1 study group. J Clin Endocrinol Metab (2017) 102(6):2083–90. doi: 10.1210/jc.2016-3690
28. Jeong Y, Oh H, Bong JG. Multiple endocrine neoplasia type 1 associated with breast cancer. A Case Rep Rev literature (2014) 1:230–4. doi: 10.3892/ol.2014.2144
29. Marx SJ. Recent topics around multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (2018) 103(4):1296–301. doi: 10.1210/jc.2017-02340
30. Roberts BJ. Lumps and bumps: benign and malignant tumors. In: Zaoutis LB, Chiang VW editors. Comprehensive pediatric hospital medicine. Elsevier Inc. (2007). p. 947–53. doi: 10.1016/B978-032303004-5.50153-8
31. Mancoll JS. Lipomas treated with liposuction. In: Liposuction. Berlin, Heidelberg: Springer Berlin Heidelberg (2006). p. 481–4.
33. Furlong MA, Fanburg-Smith JC, Miettinen M. The morphologic spectrum of hibernoma: a clinicopathologic study of 170 cases. Am J Surg Pathol (2001) 25:809–14. doi: 10.1097/00000478-200106000-00014
34. Yang HK, Kim YH, Lee YJ, Park JH, Kim JY, Lee KH, et al. Leiomyomas in the gastric cardia: CT findings and differentiation from gastrointestinal stromal tumors. Eur J Radiol (2015) 84(9):1694–700. doi: 10.1016/j.ejrad.2015.05.022
35. Pardi E, Marcocci C, Borsari S, Saponaro F, Torregrossa L, Tancredi M, et al. Aryl hydrocarbon receptor interacting protein (AIP) mutations occur rarely in sporadic parathyroid adenomas. J Clin Endocrinol Metab (2013) 98(7):2800–10. doi: 10.1210/jc.2012-4029
36. Görtz B, Roth J, Krähenmann A, De Krijger RR, Muletta-Feurer S, Rütimann K, et al. Mutations and allelic deletions of the MEN1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol (1999) 154(2):429–36. doi: 10.1016/S0002-9440(10)65289-3
37. Wautot V, Vercherat C, Lespinasse J, Chambe B, Lenoir GM, Zhang CX, et al. Germline mutation profile of MEN1 in multiple endocrine neoplasia type 1: search for correlation between phenotype and the functional domains of the MEN1 protein. Hum Mutat (2002) 20(1):35–47. doi: 10.1002/humu.10092
38. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J. Standards and guidelines for the interpretation of sequence variants. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
39. Băicoianu-Niţescu L-C, Gheorghe A-M, Carsote M, Dumitrascu MC, Sandru F. Approach of multiple endocrine neoplasia type 1 (MEN1) syndrome–related skin tumors. Diagnostics (2022) 12(11):2768. doi: 10.3390/diagnostics12112768
40. Waguespack SG. Beyond the “3 ps”: a critical appraisal of the non-endocrine manifestations of multiple endocrine neoplasia type 1. Front Endocrinol (Lausanne) (2022) 13. doi: 10.3389/fendo.2022.1029041
41. Rusconi D, Valtorta E, Rodeschini O, Giardino D, Lorenzo I, Predieri B, et al. Combined characterization of a pituitary adenoma and a subcutaneous lipoma in a MEN1 patient with a whole gene deletion. Cancer Genet (2011) 204(6):309–15. doi: 10.1016/j.cancergen.2011.03.006
42. Febrero B, Segura P, Ruiz-Manzanera JJ, Teruel E, Ros I, Ríos A, et al. Uncommon tumors in multiple endocrine neoplasia (MEN) type 1: do they have a relationship with the prognosis of these patients? J Endocrinol Invest (2021) 44(6):1327–30. doi: 10.1007/s40618-020-01414-2
43. Marini F, Giusti F, Brandi ML. Multiple endocrine neoplasia type 1: extensive analysis of a large database of Florentine patients. Orphanet J Rare Dis (2018) 13(1):1–18. doi: 10.1186/s13023-018-0938-8
44. Ballard HS, Fame B, Hartsock RJ. Familial multiple endocrine adenoma-peptic ulcer complex. Med (Baltimore) (1964) 43:481–516. doi: 10.1097/00005792-196407000-00003
45. Parekh VI, Modali SD, Desai SS, Agarwal SK. Consequence of menin deficiency in mouse adipocytes derived by In vitro differentiation. Int J Endocrinol (2015) 2015:1–10. doi: 10.1155/2015/149826
46. Trump D, Farren B, Wooding C, Pang JT, Besser GM, Buchanan KD, et al. Clinical studies of multiple endocrine neoplasia type 1 (MEN1). QJM (1996) 89(9):653–70. doi: 10.1093/qjmed/89.9.653
47. Gibril F, Schumann M, Pace A, Jensen RT. Multiple endocrine neoplasia type 1 and zollinger-Ellison syndrome. Med (Baltimore) (2004) 83(1):43–83. doi: 10.1097/01.md.0000112297.72510.32
48. Bassett JHD, Forbes SA, Pannett AAJ, Lloyd SE, Christie PT, Wooding C, et al. Characterization of mutations in patients with multiple endocrine neoplasia type 1. Am J Hum Genet (1998) 62(2):232–44. doi: 10.1086/301729
49. Kouvaraki MA. Genotype-phenotype analysis in multiple endocrine neoplasia type 1. Arch Surg (2002) 137(6):641. doi: 10.1001/archsurg.137.6.641
50. Carty SE, Helm AK, Amico JA, Clarke MR, Foley TP, Watson CG, et al. The variable penetrance and spectrum of manifestations of multiple endocrine neoplasia type 1. Surgery (1998) 124(6):1106–14. doi: 10.1067/msy.1998.93107
51. Oriola J, Lucas T, Halperin I, Mora M, Perales MJ, Alvarez-Escolá C, et al. Germline mutations of AIP gene in somatotropinomas resistant to somatostatin analogues. Eur J Endocrinol (2013) 168(1):9–13. doi: 10.1530/EJE-12-0457
52. Cazabat L, Bouligand J, Salenave S, Bernier M, Gaillard S, Parker F, et al. Germline AIP mutations in apparently sporadic pituitary adenomas: prevalence in a prospective single-center cohort of 443 patients. J Clin Endocrinol Metab (2012) 97(4):663–70. doi: 10.1210/jc.2011-2291
53. Korbonits M, Storr H, Kumar AV. Familial pituitary adenomas - who should be tested for AIP mutations? Clin Endocrinol (Oxf) (2012) 77(3):351–6. doi: 10.1111/j.1365-2265.2012.04445.x
54. Beijers HJBH, Stikkelbroeck NML, Mensenkamp AR, Pfundt R, van der Luijt RB, Timmers HJLM, et al. Germline and somatic mosaicism in a family with multiple endocrine neoplasia type 1 (MEN1) syndrome. Eur J Endocrinol (2019) 180(2):K15–9. doi: 10.1530/EJE-18-0778
55. Lagarde A, Mougel G, Coppin L, Haissaguerre M, Le CL, Mohamed A, et al. Systematic detection of mosaicism by using digital NGS reveals three new MEN1 mosaicisms. Endocr Connect (2022) 11(11). doi: 10.1530/EC-22-0093
56. Carvalho RA, Urtremari B, Jorge AAL, Santana LS, Quedas EPS, Sekiya T, et al. Germline mutation landscape of multiple endocrine neoplasia type 1 using full gene next-generation sequencing. Eur J Endocrinol (2018) 179(6):391–407. doi: 10.1530/EJE-18-0430
57. Nasimi M, Kamyab K, Moradi A, Dasdar S, Kianfar N. Clinical and histopathological evaluation of cutaneous angiofibromas. J Cutan Pathol (2021) 48(10):1262–5. doi: 10.1111/cup.14060
58. Duro T, Gonzales KL. Adrenal liposarcoma: a novel presentation of multiple endocrine neoplasia type 1. AACE Clin Case Rep (2023) 9(1):10–2. doi: 10.1016/j.aace.2022.11.003
59. Johnson GJ, Summerskill WHJ, Anderson VE, Keating FR. Clinical and genetic investigation of a Large kindred with multiple endocrine adenomatosis. N Engl J Med (1967) 277(26):1379–85. doi: 10.1056/NEJM196712282772601
60. Dong Q, Debelenko LV, Chandrasekharappa SC, Emmert-Buck MR, Zhuang Z, Guru SC, et al. Loss of heterozygosity at 11q13: analysis of pituitary tumors, lung carcinoids, lipomas, and other uncommon tumors in subjects with familial multiple endocrine neoplasia type 1. J Clin Endocrinol Metab (1997) 82(5):1416–20. doi: 10.1210/jcem.82.5.3944
61. Ognong Boulemo A, Roch J-A, Ricard F, Fontaine Hommell J, Cotton F. Hibernoma: don’t be caught out by a PET scan! Diagn Interv Imaging (2013) 94(6):649–51. doi: 10.1016/j.diii.2013.02.002
62. Karrouz W, Kamoun M, Odou M-F, Pigny P, Caiazzo R, Pattou F, et al. Hibernoma and type 1 multiple endocrine neoplasia (MEN1)? a metabolic link? Ann Endocrinol (Paris) (2013) 74(2):157. doi: 10.1016/j.ando.2013.03.013
63. Hedayati V, Thway K, Thomas JM, Moskovic E. MEN1 syndrome and hibernoma: an uncommonly recognised association? Case Rep Med (2014) 2014:1–4. doi: 10.1155/2014/804580
64. Marchand L, Decaussin-Petrucci M, Giraud S, Cotton F, Thivolet C, Simon C. Hibernoma and multiple endocrine neoplasia type 1 syndrome: a non-fortuitous association? a case report and literature review. Ann Endocrinol (Paris) (2017) 78(3):194–7. doi: 10.1016/j.ando.2017.03.001
65. Deguidi G, Mirandola S, Nottegar A, Tsvetkova V, Bianchi B, Pellini F. Axillary hibernoma in woman with lobular breast cancer and MEN1 syndrome: a case report. Int J Surg Case Rep (2020) 77:834–8. doi: 10.1016/j.ijscr.2020.11.073
66. Gisselsson D, Höglund M, Mertens F, Dal Cin P, Mandahl N. Hibernomas are characterized by homozygous deletions in the multiple endocrine neoplasia type I region. Am J Pathol (1999) 155(1):61–6. doi: 10.1016/S0002-9440(10)65099-7
67. Nord KH, Magnusson L, Isaksson M, Nilsson J, Lilljebjörn H, Domanski HA, et al. Concomitant deletions of tumor suppressor genes MEN1 and AIP are essential for the pathogenesis of the brown fat tumor hibernoma. Proc Natl Acad Sci (2010) 107(49):21122–7. doi: 10.1073/pnas.1013512107
68. Magnusson L, Hansen N, Saba KH, Nilsson J, Fioretos T, Rissle P, et al. Loss of the tumor suppressor gene AIP mediates the browning of human brown fat tumors. J Pathol (2017) 243(2):160–4. doi: 10.1002/path.4945
69. Fang M, Xia F, Mahalingam M, Virbasius C-M, Wajapeyee N, Green MR. MEN1 is a melanoma tumor suppressor that preserves genomic integrity by stimulating transcription of genes that promote homologous recombination-directed DNA repair. Mol Cell Biol (2013) 33(13):2635–47. doi: 10.1128/MCB.00167-13
70. Nord B, Platz A, Smoczynski K, Kytölä S, Robertson G, Calender A, et al. Malignant melanoma in patients with multiple endocrine neoplasia type 1 and involvement of the MEN1 gene in sporadic melanoma. Int J Cancer (2000) 87(4):463–7. doi: 10.1002/1097-0215(20000815)87:4<463::AID-IJC1>3.0.CO;2-8
71. Asgharian B, Chen YJ, Patronas NJ, Peghini PL, Reynolds JC, Vortmeyer A, et al. Meningiomas may be a component tumor of multiple endocrine neoplasia type 1. Clin Cancer Res (2004) 10(3):869–80. doi: 10.1158/1078-0432.CCR-0938-3
72. Honegger J, Buchfelder M, Schrell U, Adams EF, Fahlbusch R. The coexistence of pituitary adenomas and meningiomas: three case reports and a review of the literature. Br J Neurosurg (1989) 3(1):59–69. doi: 10.3109/02688698909001027
73. Herrero-Ruiz A, Villanueva-Alvarado HS, Corrales-Hernández JJ, Higueruela-Mínguez C, Feito-Pérez J, Recio-Cordova JM. Coexistence of GH-producing pituitary macroadenoma and meningioma in a patient with multiple endocrine neoplasia type 1 with hyperglycemia and ketosis as first clinical sign. Case Rep Endocrinol (2017) 2017:1–5. doi: 10.1155/2017/2390797
74. Zhu H, Miao Y, Shen Y, Guo J, Xie W, Zhao S, et al. Germline mutations in MEN1 are associated with the tumorigenesis of pituitary adenoma associated with meningioma. Oncol Lett (2020) 20(1):561–8. doi: 10.3892/ol.2020.11601
75. Stewart EA, Laughlin-Tommaso SK, Catherino WH, Lalitkumar S, Gupta D, Vollenhoven B. Uterine fibroids. Nat Rev Dis Prim (2016) 2(1):16043. doi: 10.1038/nrdp.2016.43
76. Vortmeyer A, Lubensky IA, Skarulis M, Li G, Moon YW, Park WS, et al. Multiple endocrine neoplasia type 1: atypical presentation, clinical course, and genetic analysis of multiple tumors. Mod Pathol (1999) 12(9):919–24.
77. McKeeby JL, Li X, Zhuang Z, Vortmeyer AO, Huang S, Pirner M, et al. Multiple leiomyomas of the esophagus, lung, and uterus in multiple endocrine neoplasia type 1. Am J Pathol (2001) 159(3):1121–7. doi: 10.1016/S0002-9440(10)61788-9
78. Stewart E, Cookson C, Gandolfo R, Schulze-Rath R. Epidemiology of uterine fibroids: a systematic review. BJOG Int J Obstet Gynaecol (2017) 124(10):1501–12. doi: 10.1111/1471-0528.14640
79. Day Baird D, Dunson DB, Hill MC, Cousins D, Schectman JM. High cumulative incidence of uterine leiomyoma in black and white women: ultrasound evidence. Am J Obstet Gynecol (2003) 188(1):100–7. doi: 10.1067/mob.2003.99
80. Dean PG, van Heerden JA, Farley DR, Thompson GB, Grant CS, Harmsen WS, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg (2000) 24(11):1437–41. doi: 10.1007/s002680010237
Keywords: angiofibroma, lipoma, primary hyperparathyroidism, pituitary, adrenal, pancreas, GEP, cutaneous lesions
Citation: Pierotti L, Pardi E, Dinoi E, Piaggi P, Borsari S, Della Valentina S, Sardella C, Michelucci A, Caligo MA, Bogazzi F, Marcocci C and Cetani F (2023) Cutaneous lesions and other non-endocrine manifestations of Multiple Endocrine Neoplasia type 1 syndrome. Front. Endocrinol. 14:1191040. doi: 10.3389/fendo.2023.1191040
Received: 21 March 2023; Accepted: 13 June 2023;
Published: 07 July 2023.
Edited by:
Natalia Simona Pellegata, Helmholtz Association of German Research Centres (HZ), GermanyReviewed by:
Thierry C. Brue, Aix-Marseille Université, FranceCopyright © 2023 Pierotti, Pardi, Dinoi, Piaggi, Borsari, Della Valentina, Sardella, Michelucci, Caligo, Bogazzi, Marcocci and Cetani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Filomena Cetani, Y2V0YW5pQGVuZG9jLm1lZC51bmlwaS5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.