
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 25 July 2023
Sec. Pediatric Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1190670
This article is part of the Research TopicTransition to Adulthood in Turner SyndromeView all 10 articles
 Jenny Lam1*
Jenny Lam1* Sophie Stoppa-Vaucher2,3
Sophie Stoppa-Vaucher2,3 Maria Cristina Antoniou3
Maria Cristina Antoniou3 Thérèse Bouthors3Inge Ruiz3
Thérèse Bouthors3Inge Ruiz3 Nicole Sekarski4
Nicole Sekarski4 Tobias Rutz5
Tobias Rutz5 Sophie Fries6Pierre Alain Binz7Florence Niel Bütschi8
Sophie Fries6Pierre Alain Binz7Florence Niel Bütschi8 Nicolas Vulliemoz9
Nicolas Vulliemoz9 Aneta Gawlik10
Aneta Gawlik10 Nelly Pitteloud1,11
Nelly Pitteloud1,11 Michael Hauschild1,3
Michael Hauschild1,3 Kanetee Busiah1,3*
Kanetee Busiah1,3*Introduction: Turner syndrome association with multi-organ system comorbidities highlights the need for effective implementation of follow-up guidelines. We aimed to assess the adequacy of care with international guidelines published in 2007 and 2017 and to describe the phenotype of patients.
Methods: In this multicenter retrospective descriptive cohort study, we collected growth and pubertal parameters, associated comorbidities, treatment, and karyotype in patients diagnosed at age <18 years between 1993 and 2022. We assessed age-appropriate recommendation follow-up (children, adolescents and adults) according to the 2007 guidelines if the last visit was before 2017 (18 recommendations) and the 2017 guidelines if the last visit was after 2017 (19 recommendations).
Results: We included 68 patients followed at Lausanne University Hospital (n=64) and at Neuchatel Regional Hospital (RHNe) (n=4). 2.9% of patients underwent all recommended investigations.
Overall, 68.9 ± 22.5% and 78.5 ± 20.6% of the recommendations were followed, before and after 2017 respectively. High implementation rates were found for height, weight and BMI (100%), cardiac (80 to 100%) and renal (90 to 100%) imaging. Low implementation rates were found for Ear, Nose and Throat (ENT) (56.5%), skin (38.5%), dental (23.1%), ophthalmological (10%) and cholestasis (0 to 29%) assessments, depending on age and time of visit. In adults (n=33), the mean proportion of followed recommendations was lower before than after 2017: 63.5 ± 25.8% vs. 78.7 ± 23.4%, p=0.039.
Conclusion: Growth parameters, cardiac and renal imaging are well followed. However, efforts should be made for dental, ENT, ophthalmological, skin and cholestasis assessments. Adequacy of follow-up improved with the quality of transition to adult care.
Turner syndrome (TS), caused by the complete or partial absence of one of the two X chromosomes, is the most common sex chromosome disorder in females, affecting approximately 1 in 2,000 live-born females. Comorbidities of TS may involve the endocrine system (the main clinical features are short stature, hypogonadism due to ovarian dysgenesis, and thyroid disease), the cardiovascular system (congenital heart disease, aortopathy, vasculopathy, arterial hypertension) and neuropsychocognitive development (e.g. learning difficulties). Other possible manifestations include hearing loss, orthopedic disorders (hip dysplasia, scoliosis, osteoporosis), renal and urinary tract disorders, (metabolic syndrome (hypertension, insulin-resistance, diabetes, overweight) or autoimmune disorders (hypothyroidism, celiac disease). This explains the need for screening and lifelong multidisciplinary follow-up of these patients (1). As growth and pubertal disorders are the main complaints of patients with Turner syndrome, the pediatric endocrinologist is usually the first to start the workup. The transition of care to adult specialists will most often occur at the end of puberty.
International clinical practice guidelines for TS were first published in 2007 (2) and updated in 2017, with recommendations for care across the lifespan and covering all health issues and comorbidities (3). Based on these recommendations, the pediatric and adult endocrine units of the Lausanne University Hospital developed an in-house clinical care guideline in 2011. This document was upgraded in 2019 into a mobile, electronic health (m-health) tool called the TS health transition passport (Supplementary Data Sheet 1). The aim is to support patients’ understanding of their condition and improve effective transition to adult-oriented care.
The aims of our study were, first, to assess the adequacy of care according to published international recommendations in a tertiary center with pediatric and adult endocrine units (Lausanne University Hospital) and in a general hospital pediatric service (Neuchâtel Regional Hospital) and, second, to describe the clinical, biological and radiological phenotype of patients.
We conducted a retrospective study on patients affected with Turner syndrome.
We included patients with karyotype-confirmed TS, diagnosed before the age of 18 years and followed between 1993 and 2022 at the pediatric and adult endocrinology units of Lausanne University Hospital (CHUV) and the pediatric endocrinology unit of Neuchatel Regional Hospital (RHNe). No adult patients were followed at the RHNe. We excluded patients with less than 5% mosaic cells or with a written refusal.
We collected clinical, radiological and biological data obtained during follow-up from medical records including karyotype, growth parameters, growth laboratory tests (IGF-1 and IGFBP-3 concentrations), growth treatment information and pubertal parameters.
Height was expressed as standard deviation (SD) using healthy female growth charts. Serum IGF-1 and IGFBP-3 were routinely assayed using IGF-1 and IGFBP-3 immunoassay kits: Nichols Institute Diagnostics from 1995 to 2005; Immulite by Siemens thereafter. Reference values were from Le Bouc Y for the Nichols Institute Diagnostics kit (4) and from Elminger et al. (5) for the Immulite kit.
We recorded comorbidities as follows: number of surgical treatments, presence of heart disease, hearing impairment, liver disease, dysthyroidism, renal disease, bone disease and celiac disease, transthoracic echocardiography (TTE) and cardiac magnetic resonance (CMR), abdominal-pelvic US, total body bone mineral density and laboratory tests such as creatinine, urea, anti-transglutaminase antibody, Thyroid Stimulating Hormone (TSH), free Thyroxine (free T4), anti-TPO antibody, glycated hemoglobin (HbA1C), Alanine Aminotransferase (ALAT), Aspartate Aminotransferase (ASAT), Gamma-Glutamyl Transpeptidase (γGT) and Alkaline phosphatase (ALP) concentrations.
We defined the last visit as the last endocrinology consultation and if there was none, the last specialist consultation for patients who were no longer followed up in the pediatric unit. The last visit was the date for assessing adequacy of follow-up. We defined good adequacy of recommendation if ≥65% of patients followed it.
To assess the adequacy of follow-up, patients were divided into 2 groups: those whose last visit was before 2017 and those whose last visit was after 2017. Their follow-up was compared with the appropriate guidelines at the time, i.e. the 2007 guidelines and the 2017 guidelines, according to their age and pubertal status. The results were as follows: children, adolescents (girls with a Tanner stage S2 stage or higher -spontaneous or with estrogen therapy- and <18 years old), and adults.
The diagnosis of TS was confirmed in all patients by karyotype using routine G-banding, including counting of at least 30 metaphases. We divided patients into 4 groups according to their karyotype: complete monosomy X (45,X); 45,X mosaicism (45,X/46,XX; 45,X/47,XXX; 45,X/46,XX/47,XXX; 45,X/46,XY); X structural rearrangement [45,X/46,X,del(Xp), 46,X,del(Xp); 45,X/46,X,del(Xq), 46,X,del(Xq); 45,X/46,X,i(Xq), 46,X,i(Xq); 45,X/46,X,r(X)], and Y structural rearrangement [45,X/46,X,idic (Y)]. We then described clinical and biochemical profiles according to karyotype.
Qualitative data were expressed as absolute number (percentage) and quantitative data as median (interquartile range - IQR) or as mean ± Standard Deviation (SD). We compared groups with Kruskall-Wallis, Chi2 or Fisher’s exact tests, using R statistical software. Values of p smaller than 0.05 were considered statistically significant.
This study was approved by the Local Ethics Committee (N°2021-00229).
We included 68 patients (Figure 1). Mean age at diagnosis was 6.3 ± 5.3 (range: 0 to 16.7) years. At last visit, there were 13 children, 22 adolescents and 33 adults. Mean age at last visit was 17.6 ± 7.5 (range: 1.2 to 40.5) years. We identified 17 patients (25%) who had their last visit before 2017 and 51 (75%) patients who had their last visit after 2017 (Table 1). All children had their last visit after 2017. In our clinic, we implemented 3 additional recommendations: IGFBP-3, thyroid US and bone age assessment, that are usually performed in children affected with growth retardation or dysthyroidism (Supplementary Table 1).
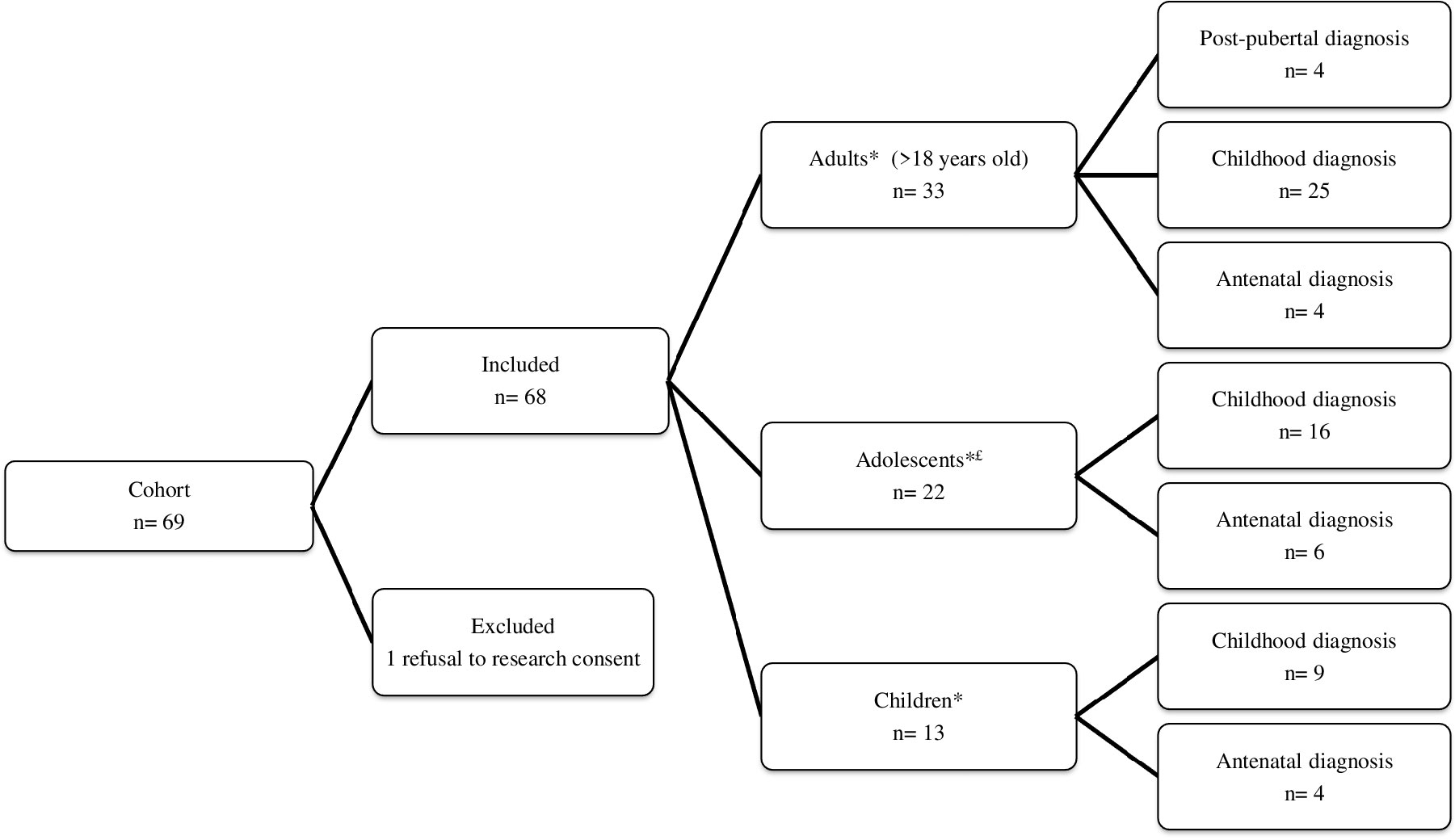
Figure 1 Patient flow chart. *Age at last visit: £Girls with a Tanner S2 stage or above (spontaneously or with estrogen therapy) and aged below 18 years old.
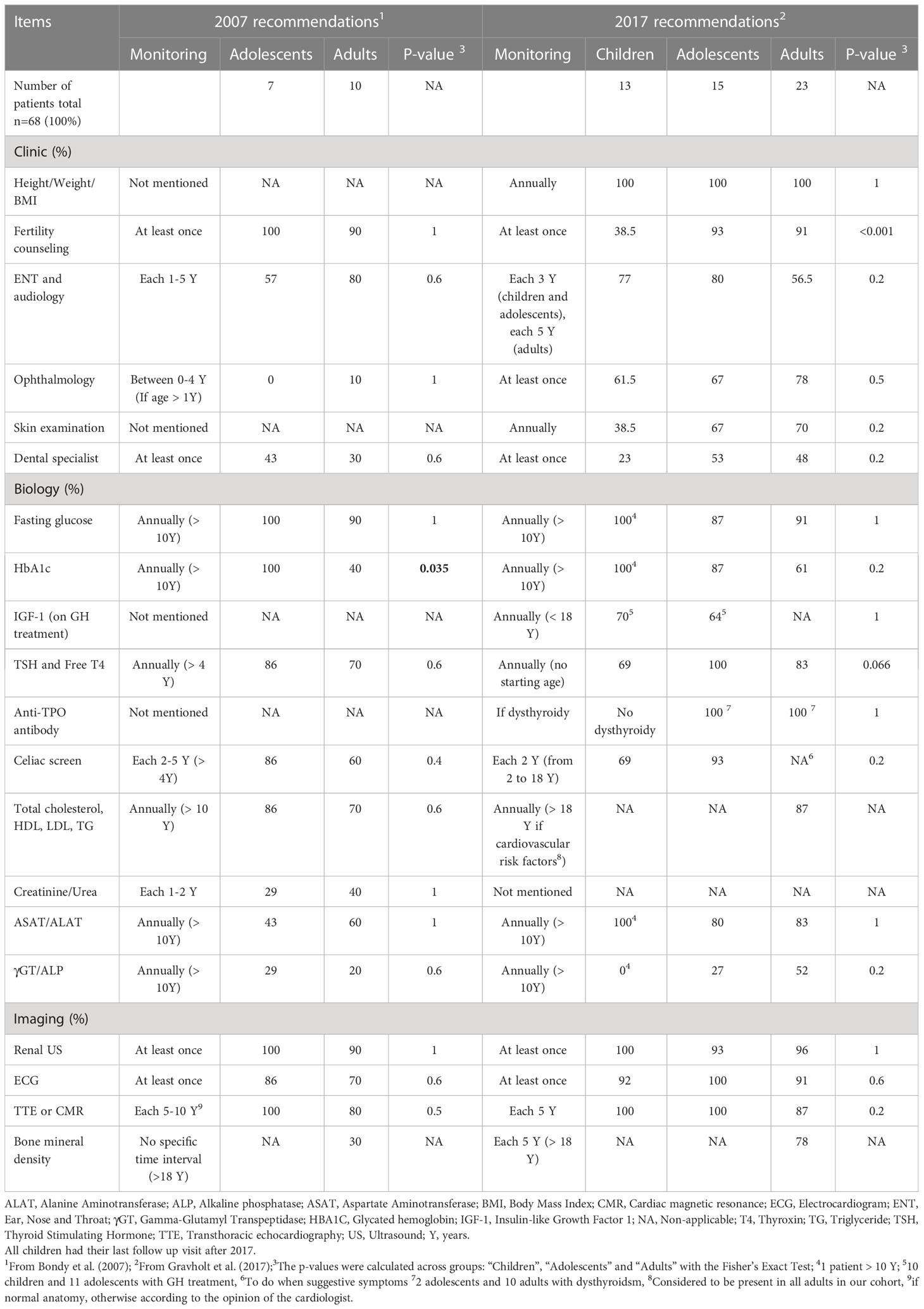
Table 1 Adequacy of recommendations of care according to the 2007 and the 2017 international guidelines.
Overall, 2.9% (n=2/68, one child and one adult last seen after 2017) of the patients underwent all recommended investigations. The overall mean proportion of recommendations followed was 76.1 ± 21.4%: 68.9 ± 22.5% of the 2007 recommendations for patients last seen before 2017 and 78.5 ± 20.6% of the 2017 recommendations for patients last seen after 2017 (Table 1 and Supplementary Table 1).
The recommendations with the highest implementation rate were height, weight and BMI (100%), and cardiac (range: 80 to 100%) and renal (range: 90 to 100%) imaging. The recommendations with the lowest implementation rate were bone mineral density (in adults last seen before 2017: 30%), skin examination (in children: 38.5%), ENT (in adolescents last seen before 2017: 57% and in adults last seen after 2017: 56.5%), ophthalmological (in adolescents and adults last seen before 2017: respectively: 0% and 10%, and children: 61.5%) and dental consultations for the whole cohort (Table 1). Liver function biomarkers were often not assayed, especially ASAT and ALAT for adolescents last seen before 2017 (43%), and γGT and ALP for the whole cohort (Table 1).
We found a difference for HbA1C between adolescents and adults last seen before 2017 (100% vs. 40% p=0.035) and for fertility counseling among all patients last seen after 2017 (38.5% of children, 93.3% of adolescents and 91.3% of adults, p<0.001).
In children (n=13), the overall mean proportion of recommendations followed was 75.5 ± 19.1% (Table 1). We found good adequacy (i.e., ≥65% of recommendations followed) for 11/16 (69%) of the recommendations in children. In adolescents (n=22), we found no difference between overall followed recommendations for patients last seen before 2017 compared to patients last seen after 2017 (76.6 ± 15.1% vs. 80.9 ± 18.3%, p=0.306). We found good adequacy for 9/15 (60%) of the recommendations for adolescents last seen before 2017 and 14/17 (82%) of the recommendations for adolescents last seen after 2017. In contrast, in adults (n=33), the mean proportion of overall followed recommendations was lower before than after 2017: 63.5 ± 25.8% vs. 78.7 ± 23.4%, respectively, p=0.039. We found good adequacy for 8/16 (50%) of the recommendations for adults last seen before 2017 and for 13/17 (76%) of the recommendations for adults last seen after 2017.
All children and adolescents had cardiac imaging, whereas 20% and 13% of adults last seen before and after 2017 respectively did not have any cardiac imaging; the difference was not significant.
We then compared patients’ clinical, biological and radiological findings according to karyotypes, that were as follows: monosomy 45,X (n=24, 35.3%); 45,X mosaicism (n=18, 26.5%); X chromosome structural rearrangement (n=24, 35.3%); and Y chromosome structural rearrangement (n=2, 2.9%).
Height and height velocity at diagnosis were significantly different across the groups (p=0.031 and p=0.019, respectively) (Table 2). Nevertheless, final height was similar across the groups (p=0.106).
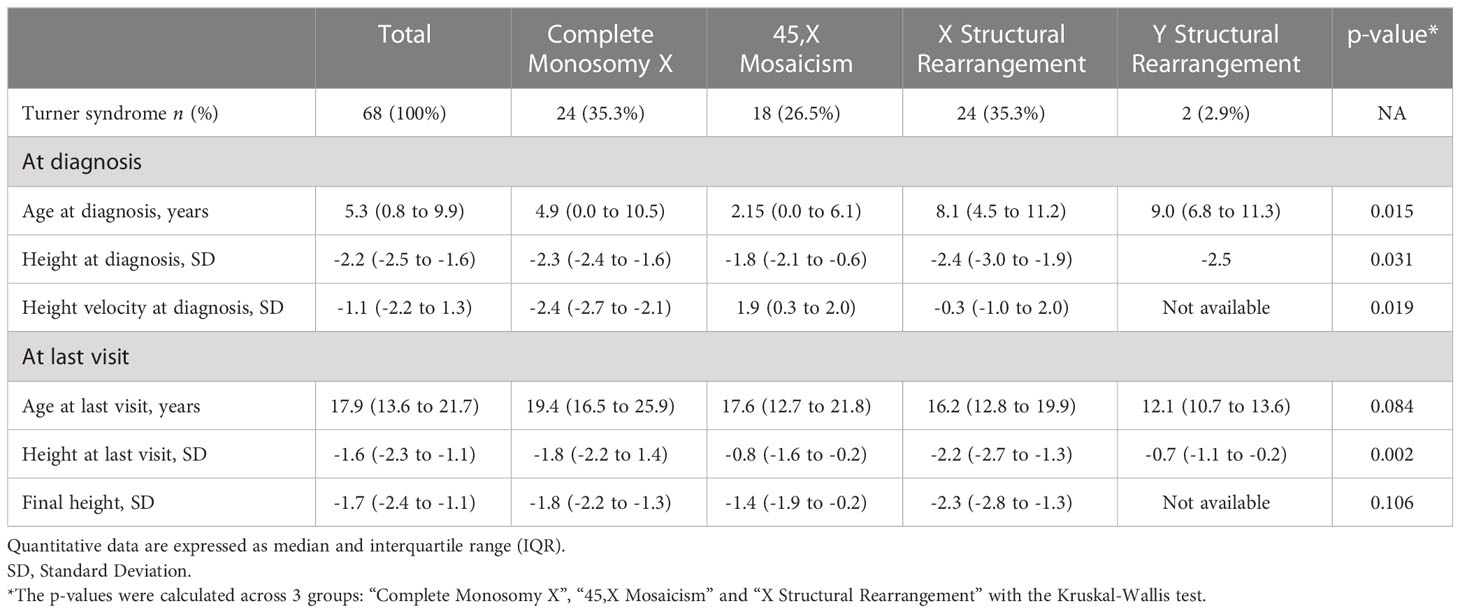
Table 2 Anthropometric characteristics according to karyotype.
Among the 86.8% of patients who had GH treatment, height and height velocity at the start of GH were significantly different across the groups (p=0.017 and 0.006 respectively) (Table 3). One year after the start of GH treatment, height velocity was no longer different across the groups (p=0.971) while the height remained significantly different (p=0.021). Bone age delay of more than 1 year was found in 23/46 (50%) of patients at the start of GH treatment and in 12/46 (26.1%) of patients at the end of GH. In these 12 patients, GH therapy was discontinued despite the bone delay, because of patients’ willingness.
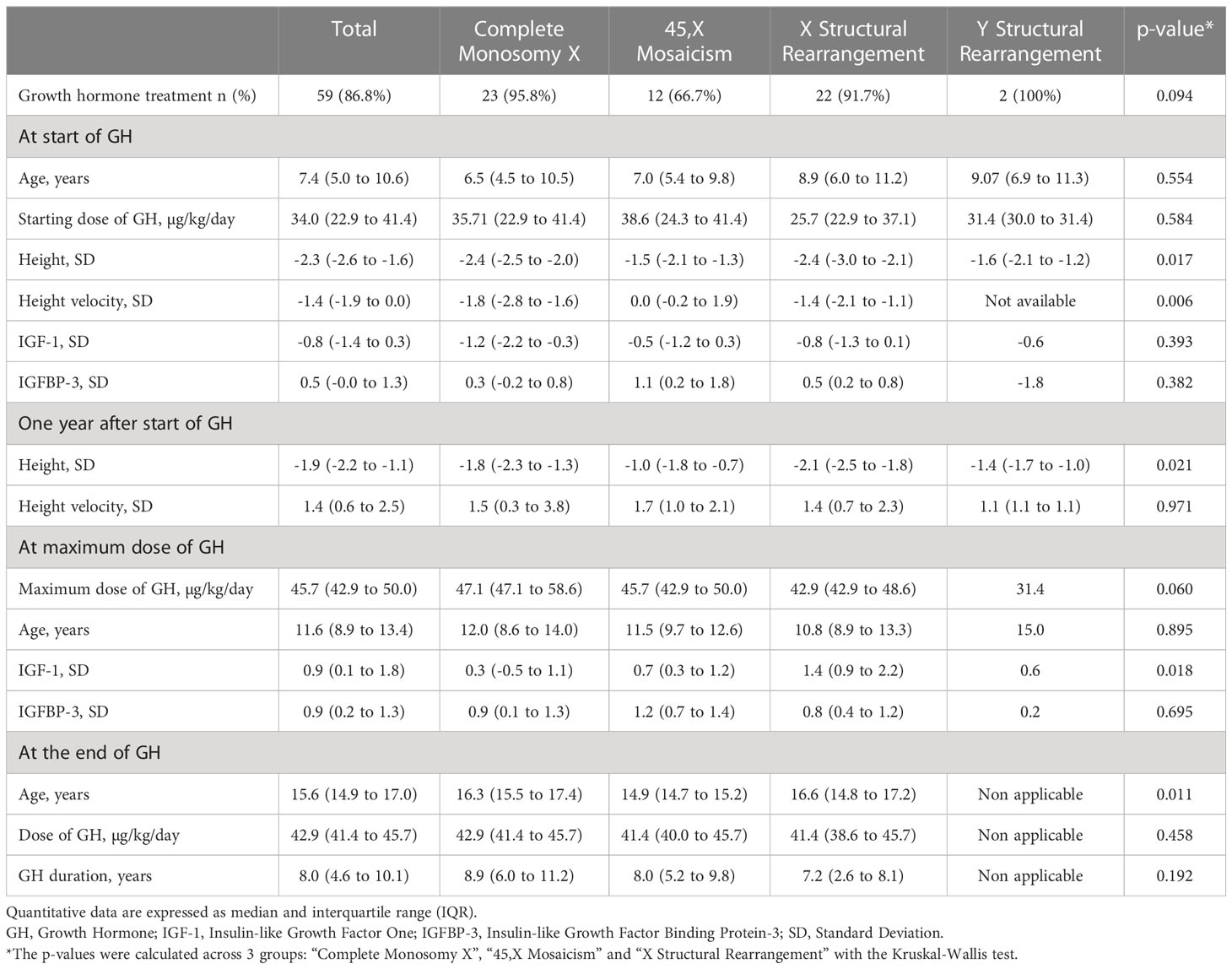
Table 3 Main clinical and laboratory characteristics on growth hormone treatment according to karyotype.
Puberty was spontaneous in 3 (13.6%) 45,X patients (Table 4). Puberty was induced in 28 (50.9%) patients, mainly in the 45,X group (p<0.001), and after 12 years of age for 20/28 (71.4%) patients. 40 patients received estrogen therapy, 29 by oral administration route and 11 by transdermal administration route.
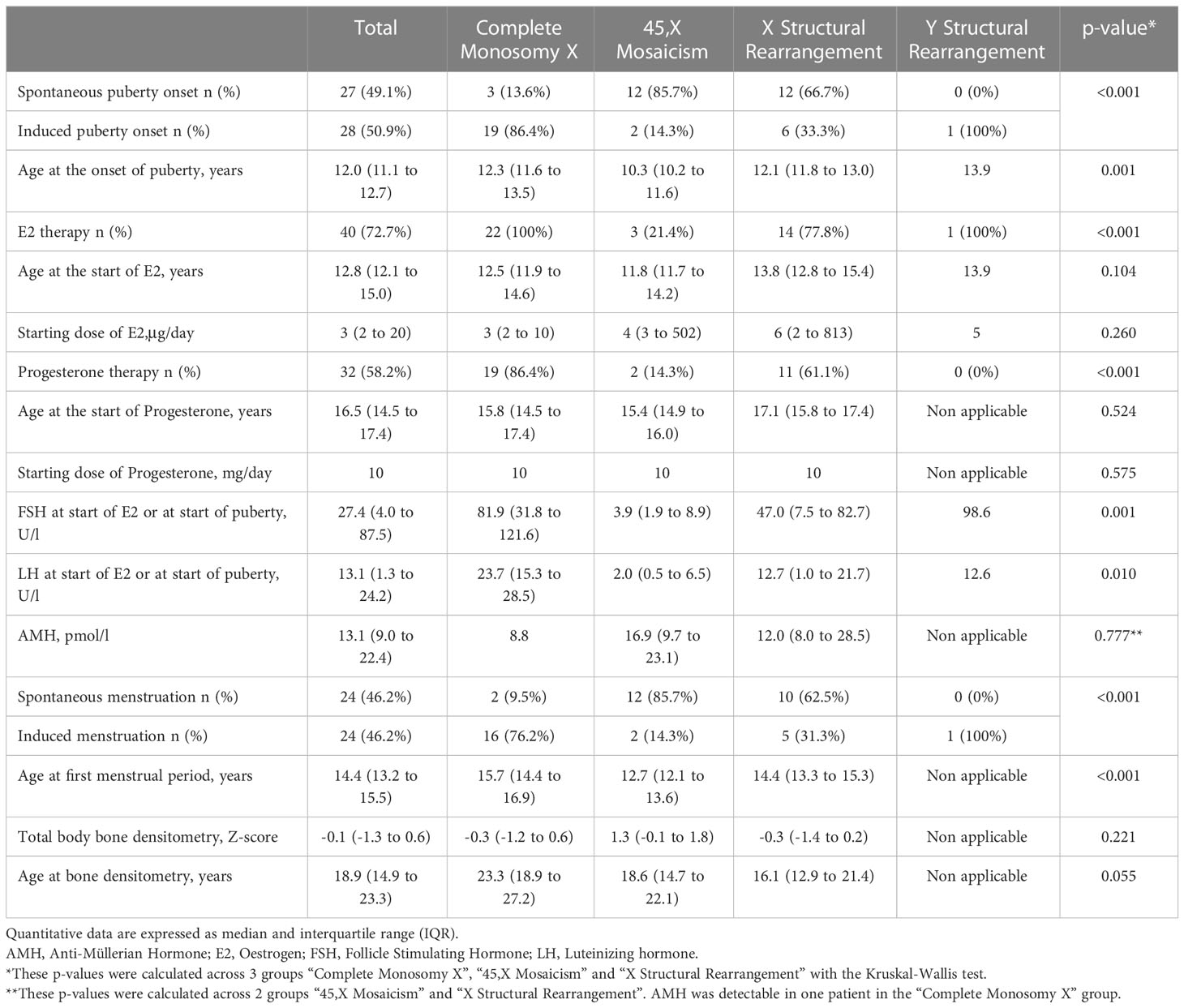
Table 4 Clinical and biological puberty characteristics according to karyotype.
Heart disease, kidney disease, celiac disease and surgery were significantly different according to their karyotype (Table 5). Osteopenia, defined as a BMD Z-score <-1, was present in 7/24 (29%) patients.
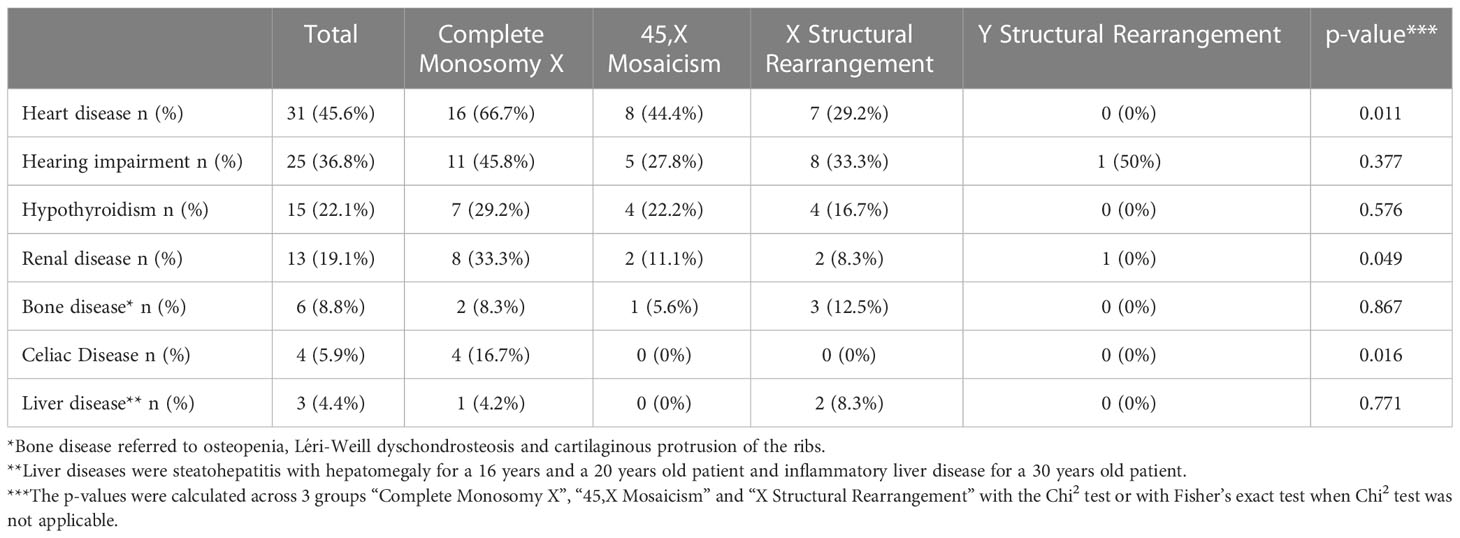
Table 5 Frequency of co-morbidities and associated features according to karyotype.
In this cohort of children, adolescents and adults patients with TS, a small minority of patients had a complete follow-up according to the international guidelines. However, the most important and potentially serious comorbidities were well followed-up, especially growth parameters, cardiac assessment and renal ultrasound. Adequacy of follow-up improved with quality of transition to adult care.
Four studies evaluated the adequacy of care according to international guidelines: three compared the adequacy of follow-up with the 2007 guidelines, and one with the 2017 guidelines (6–9). Two studies from France and Poland found that less than 5% of adult patients received all the medical investigations recommended in the 2007 guidelines. They showed that liver enzymes were often not assayed (6, 7). The prevalence of liver disease is higher in adults with TS, especially with elevated γGT rather than transaminases (10). Reported complications include non-alcoholic steatohepatitis, hepatic architectural changes such as cirrhosis, and biliary lesions such as sclerosing cholangitis (1, 11). An American study evaluated the medical care of girls with TS compared to the 2007 guidelines. In our study, adherence to recommendations was higher for follow-up of lipid levels, liver enzymes, blood glucose, thyroid function, ENT assessments, fertility counselling, celiac screening and bone mineral density and cardiac imaging, depending on the study (6–8). However, celiac screening, ENT and ophthalmological assessments were lower than in the study published by Hoag and colleagues (9). This suggests an improvement in the management of patients with TS. There is still room for improvement in care coordination. The implementation of new models of care coordination could help (12).
We focused on more recommendations from the 2007 and 2017 guidelines than these studies, especially dental, eye and skin examinations. Lack of compliance with these follow-up visits can lead to reduced quality of life. This highlights the importance of awareness among clinicians. Dermatological screening aims to detect lymphedema, dermatitis, eczema, psoriasis and multiple pigmented nevi (1, 13). Eye disorders include high rates of strabismus, visual impairment such as myopia, or sight-threatening abnormalities such as papilledema (14). The time interval recommended by the 2007 guidelines for seeing an ophthalmologist was very restrictive. This might explain the poor follow-up adequacy of ophthalmic consultations for patients with last visit before 2017. Dental disorders include a wide range of manifestations from micrognathia to abnormal dental development (1). The fact that dental consultations are not covered by the Swiss National Health Insurance may explain the low number of dental consultations in our cohort. The distribution of karyotypes in our cohort showed slightly higher proportion of X structural rearrangement than previously published (15). The comorbidities and their distribution between the different karyotypes in our cohort were globally consistent with the literature (1, 16–18). In our study, we found lower proportions of heart and liver diseases but similar proportions of thyroid and celiac diseases (15). Nevertheless, the number of comorbidities should be correlated with the adherence to recommended follow-up.
All recommendations, except HbA1c for patients last seen before 2017 and fertility counselling for patients last seen after 2017, were monitored equally between children, adolescents and adults. This may reflect the structured transition clinic between pediatric and adult endocrinological care that we have developed at the CHUV. This transition endocrine clinic has improved, as suggested by a better follow-up of adults last seen before 2017 compared to those last seen after 2017. As all RHNe patients are children or adolescents, they did not have transition. Our group has shown the usefulness of integrating transition passports as a usable, understandable health tool for patients and physicians, to reduce gaps in transition from pediatric to adult-oriented care (19). Our study suggests that the CHUV pediatric and adult TS transition passport could provide patients with a better understanding of their follow-up, of treatment, fertility care and comorbidities.
GH treatment followed the 2017 guidelines in terms of starting age, doses and IGF-1 monitoring. Estrogen treatment was started at low doses and increased over 2 to 3 years as recommended. However, the majority of patients started later than recommended. This discrepancy may be explained by the fact that only five patients started treatment after the publication of the 2017 guidelines (3). Late initiation of estrogen therapy can be detrimental to bone and uterine health (20). Data are consistent with no change in adult height when low-doses estrogen is started before the age of 12, as recommended.
The strengths of our study include the comparison of long-term medical follow-up of girls and women with TS between the two published international guidelines. Studies on the adequacy of medical follow-up in girls and women with TS are limited (6–9), and most have compared the adequacy of follow-up with the 2007 guideline. We divided our cohort into 3 different age groups, as in the study by Hoag and colleagues (9). We also included additional important recommendations such as dental and ophthalmological consultations and skin examinations, which are rarely investigated, and we could perform a karyotype in all patients. This study looked at patients with TS followed at a large university center (CHUV) with many experienced specialists, including cardiologists, radiologists, ear, nose and throat specialists, adult endocrinologists, and others. As a result, follow-up of these patients may not be the same throughout Switzerland. Moreover, in other studies, most adult patients with TS were followed up by general practitioners, who were sometimes unaware of the TS diagnosis. For this reason, we developed a patient oriented electronic health (m-health) TS health transition passport, to avoid loss of medical information (Supplementary Data Sheet 1).
A limitation of our study is its retrospective design, which is justified by the rarity of the disease. However, we minimized missing data through in-depth analysis of medical records.
Our study highlights the importance of improving awareness among patients themselves and primary care physicians of the broad spectrum and variability of TS presentation at different ages. We should aim to reduce health inequalities by making multidisciplinary clinics and comprehensive care available and accessible. It is also important to ensure adequate medical and social support for transition of young adults and care of adults with TS. We also should involve the patient, who gains autonomy and responsibility for her health care during adolescence and young adulthood. Our TS education program, launched in 2011 and improved in 2019, aims to address these challenges. We aim to serve as a regional resource for the community and for physicians in our community.
In conclusion, complete guideline adherence in TS patient care and follow-up should be improved, especially in bone mineral density, liver, ophthalmic, ENT, dermatological and dental assessment. Our results open a field for possible future research on patient education and healthcare organizations: how to understand the lack of awareness in TS, how to improve structural problems, how to implement a complete work-up or how to spread these guidelines among non-endocrinologists, especially pediatricians and general practitioners. A better follow-up has already been observed compared to the studies before 2017, which makes us optimistic for the future.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by Commission cantonale (VD) d’éthique de la recherche sur l’être humain (CER-VD). Written informed consent from the participants’ legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements.
JL collected the data, performed the data analysis and interpretation, performed the statistical analysis, wrote, and critically revised the manuscript. KB designed the study, was responsible for the data analysis and interpretation, and for the statistical analysis, wrote, and critically revised the manuscript. NV and MH interpreted the data and critically revised the manuscript. SS-V, MA, TB, IR, NS, TR, SF, NP, MH, and KB followed the patients, PB was responsible for hormonal assay, FB was responsible for genetic investigation. AG provided feedback on the study design. All authors contributed to the article and approved the submitted version.
Open access funding by University of Lausanne.
We are indebted to the patients and their families for their participation in the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1190670/full#supplementary-material
AMH, Anti-Müllerian Hormone; Anti-TPO, Anti-Thyroperoxydase; BMI, Body Mass Index; CMR, Cardiac Magnetic Resonance; ECG, Electrocardiogram; ENT, Ear, Nose and Throat; FSH, Follicle-Stimulating Hormone; GH, Growth Hormone; IGF-1, Insulin-like Growth Factor One; IGFBP-3, Insulin-like Growth Factor Binding Protein-3; LH, Luteinizing Hormone; MRI, Magnetic Resonance Imaging; SD, Standard deviation; TTE, Transthoracic Echocardiography; TS, Turner Syndrome; US, Ultrasound.
1. Gravholt CH, Viuff MH, Brun S, Stochholm K, Andersen NH. Turner syndrome: mechanisms and management. Nat Rev Endocrinol (2019) 15:601–14. doi: 10.1038/s41574-019-0224-4
2. Bondy CA. Care of girls and women with turner syndrome: A guideline of the turner syndrome study group. J Clin Endocrinol Metab (2007) 92:10–25. doi: 10.1210/jc.2006-1374
3. Gravholt CH, Andersen NH, Conway GS, Dekkers OM, Geffner ME, Klein KO, et al. Clinical practice guidelines for the care of girls and women with Turner syndrome: proceedings from the 2016 Cincinnati International Turner Syndrome Meeting. Eur J Endocrinol (2017) 177:G1–G70. doi: 10.1530/EJE-17-0430
4. Le Bouc Y. Insulin-like growth factors (IGF) and their binding proteins (IGFBP). Physiological and pathological regulation. Journ Annu Diabetol Hotel Dieu (1995), 29–38.
5. Elmlinger MW, Kühnel W, Weber MM, Ranke MB. Reference ranges for two automated chemiluminescent assays for serum insulin-like growth factor I (IGF-I) and IGF-binding protein 3 (IGFBP-3). Clin Chem Lab Med CCLM (2004) 42:654–664. doi: 10.1515/CCLM.2004.112
6. Devernay M, Ecosse E, Coste J, Carel J-C. Determinants of medical care for young women with turner syndrome. J Clin Endocrinol Metab (2009) 94:3408–13. doi: 10.1210/jc.2009-0495
7. Gawlik A, Kaczor B, Kaminska H, Zachurzok-Buczynska A, Gawlik T, Malecka-Tendera E. Quality of medical follow-up of young women with turner syndrome treated in one clinical center. Horm Res Paediatr (2012) 77:222–8. doi: 10.1159/000337780
8. Nabhan ZM, Eugster EA. Medical care of girls with turner syndrome: where are we lacking? Endocr Pract (2011) 17:747–52. doi: 10.4158/EP11059.OR
9. Hoag BD, Tsai SL, Williams DD, Cernich JT. International guideline adherence in girls with turner syndrome: multiple subspecialty clinics versus coordinated multidisciplinary clinic. Endocr Pract (2022) 28:1203–9. doi: 10.1016/j.eprac.2022.08.011
10. Bertrand A-M, Theuriet L, Colle M, Pienkowski C, Soskin S, Richard O, et al. Syndrome de Turner et fonction hépatique chez l’enfant et l’adolescente. In: Le syndrome de Turner. Paris: Springer Paris (2009). p. 123–34. doi: 10.1007/978-2-287-87855-8_12
11. Roulot D. Liver involvement in Turner syndrome. Liver Int (2013) 33:24–30. doi: 10.1111/liv.12007
12. Walton H, Simpson A, Ramsay AIG, Hudson E, Hunter A, Jones J, et al. Developing a taxonomy of care coordination for people living with rare conditions: a qualitative study. Orphanet J Rare Dis (2022) 17:171. doi: 10.1186/s13023-022-02321-w
13. Viuff MH, Stochholm K, Juul S, Gravholt CH. Disorders of the eye, ear, skin, and nervous system in women with Turner syndrome –a nationwide cohort study. Eur J Hum Genet (2022) 30:229–36. doi: 10.1038/s41431-021-00989-5
14. Huang J, Basith SST, Patel S, Goetsch Weisman A, Brickman W, Mets MB, et al. Ocular findings in pediatric turner syndrome. Ophthalmic Genet (2022) 43:1–4. doi: 10.1080/13816810.2022.2045512
15. Gravholt CH, Viuff M, Just J, Sandahl K, Brun S, van der Velden J, et al. The changing face of turner syndrome. Endocr Rev (2023) 44:33–69. doi: 10.1210/endrev/bnac016
16. Cleemann Wang A, Hagen CP, Nedaeifard L, Juul A, Jensen RB. Growth and adult height in girls with turner syndrome following IGF-1 titrated growth hormone treatment. J Clin Endocrinol Metab (2020) 105:2566–74. doi: 10.1210/clinem/dgaa274
17. Hamza RT, Mira MF, Hamed AI, Ezzat T, Sallam MT. Anti-Müllerian hormone levels in patients with turner syndrome: Relation to karyotype, spontaneous puberty, and replacement therapy. Am J Med Genet A (2018) 176:1929–34. doi: 10.1002/ajmg.a.40473
18. Hankus M, Soltysik K, Szeliga K, Antosz A, Drosdzol-Cop A, Wilk K, et al. Prediction of spontaneous puberty in turner syndrome based on mid-childhood gonadotropin concentrations, karyotype, and ovary visualization: A longitudinal study. Horm Res Paediatr (2018) 89:90–7. doi: 10.1159/000485321
19. Dwyer AA, Héritier V, Llahana S, Edelman L, Papadakis GE, Vaucher L, et al. Navigating disrupted puberty: development and evaluation of a mobile-health transition passport for klinefelter syndrome. Front Endocrinol (2022) 13:909830. doi: 10.3389/fendo.2022.909830
Keywords: Turner syndrome, international guidelines, follow-up, transition, recommendations, care coordination, comorbidities
Citation: Lam J, Stoppa-Vaucher S, Antoniou MC, Bouthors T, Ruiz I, Sekarski N, Rutz T, Fries S, Binz PA, Bütschi FN, Vulliemoz N, Gawlik A, Pitteloud N, Hauschild M and Busiah K (2023) Turner syndrome: skin, liver, eyes, dental and ENT evaluation should be improved. Front. Endocrinol. 14:1190670. doi: 10.3389/fendo.2023.1190670
Received: 21 March 2023; Accepted: 27 June 2023;
Published: 25 July 2023.
Edited by:
George Paltoglou, National and Kapodistrian University of Athens, GreeceReviewed by:
Dimitrios T. Papadimitriou, National and Kapodistrian University of Athens, GreeceCopyright © 2023 Lam, Stoppa-Vaucher, Antoniou, Bouthors, Ruiz, Sekarski, Rutz, Fries, Binz, Bütschi, Vulliemoz, Gawlik, Pitteloud, Hauschild and Busiah. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jenny Lam, amVubnkubGFtNDBAZ21haWwuY29t; Kanetee Busiah, S2FuZXRlZS5CdXNpYWhAY2h1di5jaA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.