 Sama Samadli
Sama Samadli Qiaoli Zhou
Qiaoli Zhou Bixia Zheng
Bixia Zheng Wei Gu
Wei Gu Aihua Zhang
Aihua Zhang- 1Nanjing Key Laboratory of Pediatrics, Children’s Hospital of Nanjing Medical University, Nanjing, China
- 2Department of Pediatric Diseases II, Azerbaijan Medical University, Baku, Azerbaijan
- 3Department of Endocrinology, Children’s Hospital of Nanjing Medical University, Nanjing, China
Monogenic diabetes gave us simplified models of complex molecular processes occurring within β-cells, which allowed to explore the roles of numerous proteins from single protein perspective. Constellation of characteristic phenotypic features and wide application of genetic sequencing techniques to clinical practice, made the major form of monogenic diabetes – the Maturity Onset Diabetes of the Young to be distinguishable from type 1, type 2 as well as neonatal diabetes mellitus and understanding underlying molecular events for each type of MODY contributed to the advancements of antidiabetic therapy and stem cell research tremendously. The functional analysis of MODY-causing proteins in diabetes development, not only provided better care for patients suffering from diabetes, but also enriched our comprehension regarding the universal cellular processes including transcriptional and translational regulation, behavior of ion channels and transporters, cargo trafficking, exocytosis. In this review, we will overview structure and function of MODY-causing proteins, alterations in a particular protein arising from the deleterious mutations to the corresponding gene and their consequences, and translation of this knowledge into new treatment strategies.
Introduction
The incidence of Maturity Onset Diabetes of the Young (MODY) is accounted for 1-2% of total diabetes cases and is expected to rise due to increasing awareness and better identification from more prevalent forms of diabetes by endocrinologists (1). Clinically, MODY patients present with following features: a strong family history of diabetes with an apparent autosomal-dominant inheritance, onset of diabetes before 25 years of age in at least one generation of the same family, sustained endogenous insulin production without insulin resistance and absence of β cell-specific autoantibodies (2). However, it often shares common features with the frequently seen forms of diabetes, which in turn creates challenges for making diagnosis. Luckily, wide application of genetic sequencing methods in clinical settings helps to avoid pitfalls in most cases (3). Despite of the fact that genetic testing methods are gold standard in the discovery of MODY-causing genes, the etiology of MODYX cases are yet to be identified (4). Besides, by means of more sensitive approaches, the later genotype-phenotype correlation studies revealed that some genes from the previous established list of 14 MODY genes should not be regarded as causative for MODY (5, 6).
Understanding molecular basis of MODY gave comprehensive knowledge concerning the physiological processes in functional β-cells during glucose-stimulated insulin secretion (GSIS) and insulin synthesis. Glucose sensing of β-cells is the first step in maintaining normoglycemia, characterized by transfer of glucose – which is entered into the cell through cell membrane related glucose-transporter-2 protein (GLUT-2) – to glucose-6-phosphate by glucokinase (GCK). Glucose-6-phosphate enters the Krebs cycle inside the mitochondria and results in enhanced production of ATP. Increased level of ATP induces closure of KATP channels and depolarize the cell membrane to approximately -30 mV. This in turn leads to opening of L-type voltage-gated calcium channels, increased entry of Ca2+ ions into the cell and subsequent release of insulin (7). Glucose homeostasis of an organism is also maintained by proper and adequate insulin synthesis. Insulin is translated in the cytosol as 110 amino acids-length preproinsulin, and then undergoes posttranslational modification to form proinsulin in the endoplasmic reticulum. Following transitioning of proinsulin to the Golgi apparatus culminates in formation of secretory granules composing of Zn2+ ion coupled insulin, proinsulin, C-peptide, amylin and regulatory proteins which are responsible for completion of “ready to use” insulin maturation (8, 9). Key steps of insulin biosynthesis and secretion are regulated by transcriptional network constituting of hierarchical, auto- and inter-regulatory complex of transcription factors (TFs), some of which involved in the pathogenesis of MODY (10) (Figure 1).
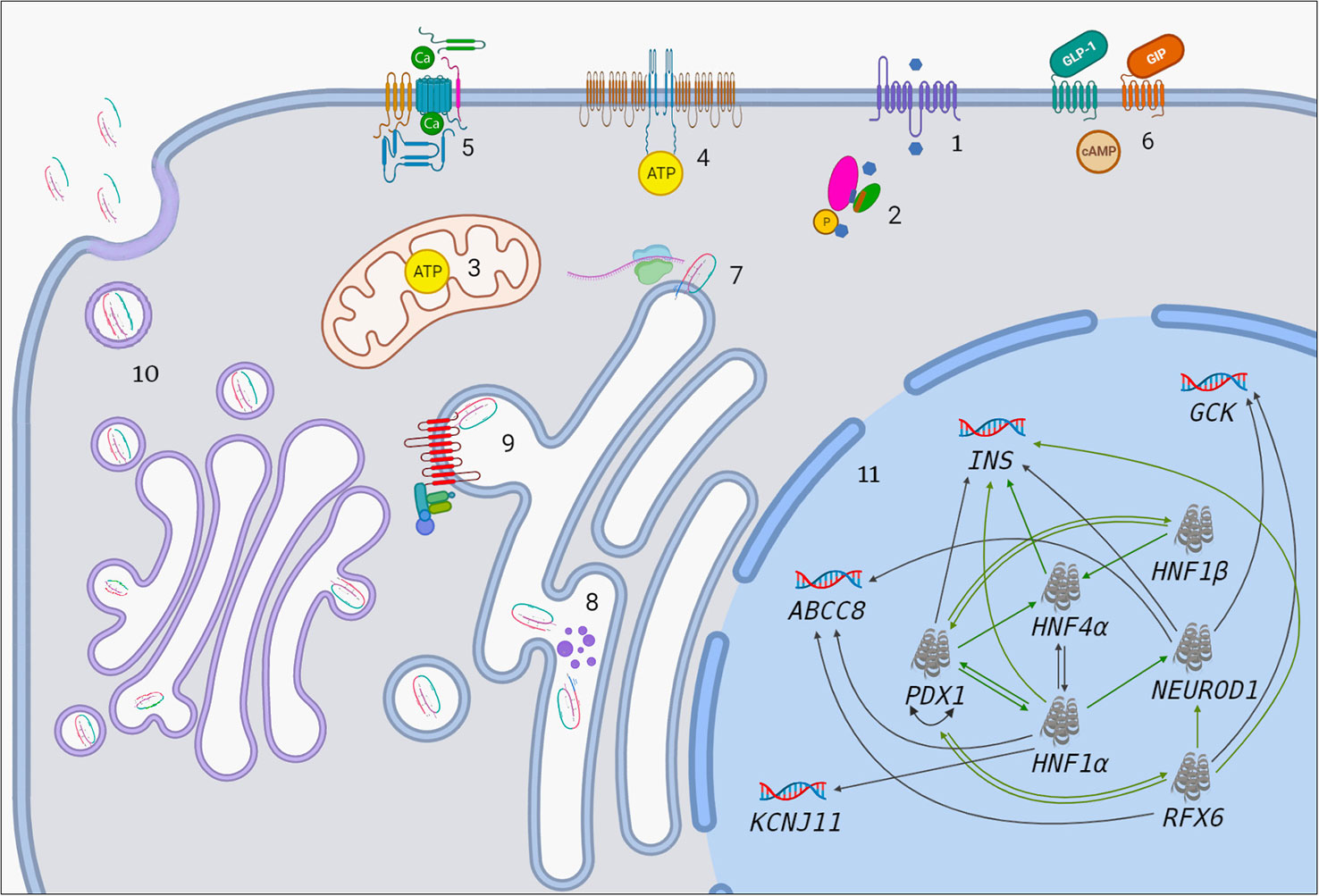
Figure 1 The association between MODY-causing proteins. 1. Influx of glucose to β-cell through GLUT-2. Blue hexagon represents glucose. 2. Transition of glucose to glucose-6-phosphate by GCK enzyme. Pink oval, green oval, purple cylinder and orange cylinder represent large domain, small domain, glucose-binding site and ATP-binding site of GCK, respectively. 3. Increased ATP production in mitochondria after the entry of G6P to Krebs cycle. 4. Closure of ATP-sensitive potassium channels. 5. Opening of voltage-gated calcium channels. 6. GIP and GLP-1 bind to their receptors and initiate number of events which result in enhanced insulin synthesis and secretion (There is apparent link between GIP/GLP-1 and MODY-causing molecules such as PDX1, RFX6. Molecular basis of this is unknown but it has therapeutic implications). 7. Preproinsulin biosynthesis in cytosol. 8. Preproinsulin processing in ER by endopeptidases (purple dots) and subsequent cleavage of signal peptide. 9. ER-membrane located wolframin interacts with proinsulin in one end and COP-II proteins in other end and helps formation of COP-II-coated proinsulin vesicles. 10. Proinsulin processing in Golgi apparatus, final vesicle formation, release of C-peptide from insulin in Golgi apparatus and inside the vesicles and exocytosis. 11. Transcriptional regulation in the nucleus. Black arrows indicate regulation of a given gene by the relevant TF in mature β-cell. Green arrows indicate that a given protein has binding site for the relevant TF and/or had been regulated by it at some point of pancreatic development.
Accumulated knowledge regarding the molecular events in the basis of MODY allows better management strategies and resultant improved quality of life in MODY patients (11). Nevertheless, increased understanding of core molecular processes offers benefits beyond the high-quality care for MODY patients. Considering the fact that TFs such as PDX1, NEUROD1, HNF family proteins, play a crucial role in the development and differentiation of embryogenic pancreas and in the maintenance of functional mature islet cells (Table 1), the use of these key regulatory proteins in the establishment of insulin-producing β-cells from stem cells paved the way for radical treatment of insulin-dependent forms of diabetes mellitus (12, 13). In this review, we will overview structure and function of MODY-causing proteins, alterations in a particular protein arising from the deleterious mutations to the corresponding gene and their consequences, and translation of this knowledge into new treatment strategies.
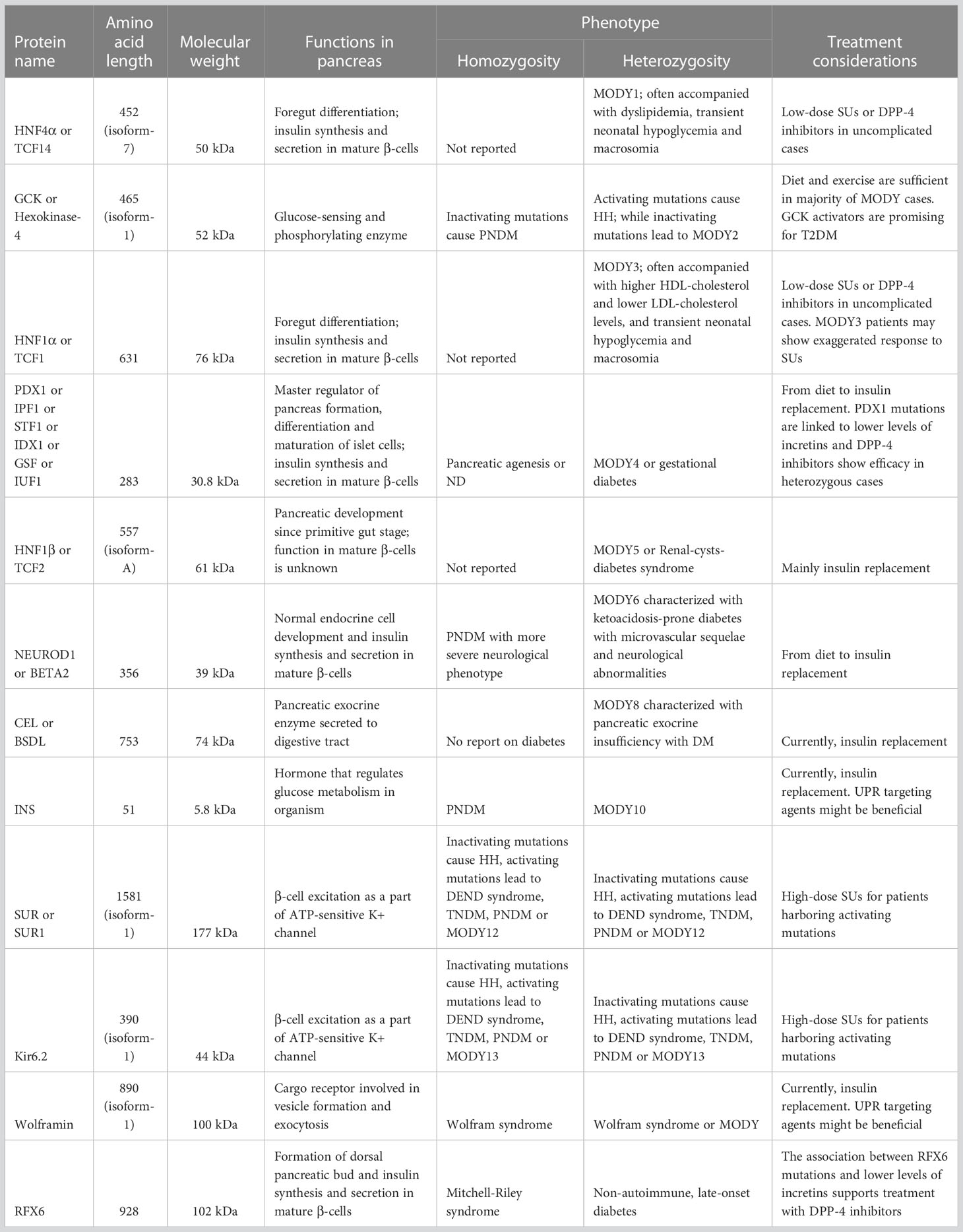
Table 1 The characteristics of MODY-causing proteins.
Impaired glucose sensing
GCK mutations (MODY2)
GCK is a glycolytic enzyme initiates glucose utilization and acts as a glucose-sensor through controlling glucose phosphorylation in β-cells (14). Heterozygous inactivating mutations in GCK gene are responsible for MODY2, which constitutes the highest proportion of all MODY cases (15). Heterozygous activating GCK mutations lead to HH, while inactivating mutations in both alleles cause permanent neonatal diabetes occurring in the first 6 months of life, explaining the vital role of GCK enzyme in glucose homeostasis (Table 1) (16).
Pancreatic glucokinase is composed of large and small domains separated by glucose binding active site, also known as deep cleft (Figure 2) (17). Lower affinity for glucose in its passive super-open form and lack of inhibition by the end product are their main properties, allowing to exhibit its rate-limiting catalytic activity. According to the mnemonic model hypothesis (18), under the influence of 5 mM or higher glucose concentration, super-open form transforms into open form, which directly binds to ATP and changes into closed form. Ongoing influx of glucose, sustains the transition between open and closed forms and this step is termed as fast cycle. On the contrary, lower level of glucose makes the molecule to regain its super-open form in a slow cycle fashion (19, 20). To date, more than 600 missense and nonsense mutations were discovered in the GCK gene of families suffering from MODY2 which alter glucose and ATP-binding abilities of the enzyme through directly impairing its kinetic parameters, or interfering with either structural stability or posttranslational regulation of the protein (16, 21). Certain missense mutations impede kinetic activity of GCK enzyme via stabilizing its inactive form or causing electrostatic repulsion of particles within active (closed) configuration (22). On the other hand, some mutations are associated with thermal instability affecting the three-dimensional structure of GCK (23). Functional analysis found that mutations remote to the active site of GCK enzyme prevented GCK-containing granules translocate to the cytoplasm by unidentified posttranslational regulator in insulin-producing pancreatic cells (24). Lastly, a rare mutation in the GCK β-cell promoter revealed Sp1 as one of the human transcription factors, although it is considered that inclusion of promoter analysis to routine sequencing methods would yield higher detection of such mutations (25).
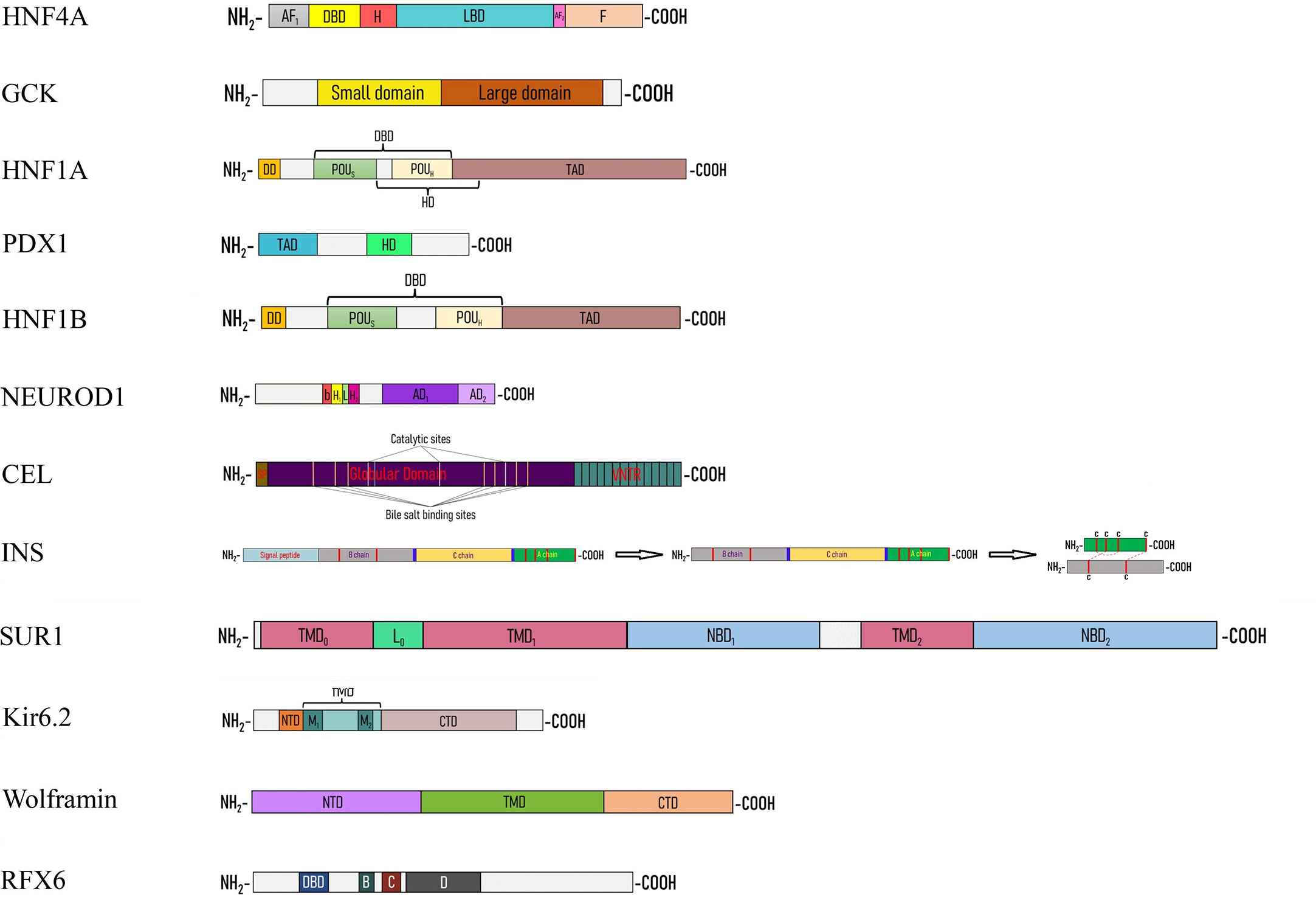
Figure 2 Linear cartoon structure of MODY-causing proteins. AF1(2) – activation function 1(2), bHLH – basic helix-loop-helix, CTD – C-terminal domain, DBD – DNA-binding domain, DD – dimerization domain, H – hinge, HD – homeodomain, L0 – loop, LBD – ligand binding domain, NBD – nucleotide binding domain, NTD – N-terminal domain, TAD – transactivation domain, TMD – transmembrane domain.
Taking advantage of central role of GCK in glucose homeostasis, its activators – which are able to bind to the allosteric site, change kinetic parameters of GCK, and render the molecule more sensitive to glucose – have gained a lot of attention for better management of Type 2 Diabetes Mellitus (T2DM). While majority of these agents failed to show sustained glycemic control (26), brand-new drug – dorzagliatin passed phase 3 trial successfully and is hoped to fulfill the need for more effective hypoglycemic agent with less undesirable events (27). Some MODY2 mutations may decrease efficient targeting of GCK by these agents (28). Nevertheless, since MODY2 mutations cause GCK to be hyposensitive rather than insensitive to glucose, most MODY2 patients have mild fasting hyperglycemia and lower chance of complications and they achieve good control over glucose levels for several years with only lifestyle modifications (16).
Channelopathies
KATP channelopathies
KATP channel are composed of four pore-forming Potassium inward rectifier 6.2 (Kir6.2) subunits and four regulatory sulfonylurea receptor 1 (SUR1) subunits, which are encoded by two neighboring genes: ABCC8 and KCNJ11, respectively (29). SUR-1 shows both stimulatory and inhibitory effect on Kir6.2 through sensitizing or desensitizing Kir6.2 to ATP, which are best represented by the impact of activating and inactivating mutations on this channel. Gain-of-function mutations of these genes cause DEND syndrome, permanent or transient NDM, MODY or T2DM susceptibility depending on the degree of metabolic impairment within β-cells and other cell types (30). Loss-of-function mutations, on the other hand, produce HH attributed to either loss of channels on the cell surface or decreased channel activity (Table 1) (31). Compelling amount of knowledge regarding the molecular basis of KATP channelopathies extrapolated from the mutational analysis of NDM or HH. MODY emerges as a mild form of NDM due to lesser increase in KATP channel current (30) or as a consequence of exhausted β-cells in HH (32).
ABCC8 mutations (MODY12)
SUR1 subunit comprised of a functionally active five-helix transmembrane domain 0 (TMD0), six-helix TMD1 and TMD2, a cytosolic loop (L0) between TMD0 and TMD1 and cytosolic nucleotide-binding domains NBD1 and NBD2 (Figure 2). NBDs act as cytosolic sensors and channel openers and are modified by Mg-nucleotides to create 2 nucleotide-binding sites. Nucleotide-binding sites 1 and 2 are formed by Walker A and B motifs of one NBD with the signature sequence located between Walker A and B of the head-to-tail aligned opposite NBD. Dimerization of nucleotide-binding sites is translated into conformational change of the TMD1 and TMD2, then subsequent transformation of Lasso motif of the L0 loop and TMD0, and eventually, clockwise rotation of Kir6.2 cytoplasmic domains to open the pore (33). Substitutions in the linker region is associated with impaired transduction, rather than altered Mg-nucleotide-binding and hydrolysis. This indicates NBDs interact with TMDs via linker region (34). Mutations that elicit the impairment of Mg-nucleotide-binding and/or catalytic hydrolyzation at the nucleotide-binding sites abolish stimulatory effect of SUR1 on Kir6.2 and result in closure of channels (35, 36). On the other hand, activating mutations of NBD1 and NBD2 hyperpolarize pancreatic β-cells and lead to hyperglycemia owing to reduced KATP channel inhibition caused by slow deactivation of Mg-ADP from its bound state (37–39), although nucleotide-binding sites show asymmetry with their distinct preference to binding and hydrolysis of nucleotides (40). Interestingly, the effects of the interaction of Mg-nucleotides with SUR1 on Kir6.2 are best represented in the reports, which same residues cause opposing phenotypes – either diabetes or hyperinsulinism depending on the amino acid substitution (41). NDM mutations at TMDs are characterized by disrupting the transduction and abrogating the stimulatory effect of NBDs on Kir6.2 (42, 43). Moreover, unusual phenotype of alternating hypoglycemia and hyperglycemia are reported in a patient with homozygous ABCC8 mutation, even though heterozygous inheritance of the same mutation caused severe trafficking defect (44). Above reports indicate that slight differences in the molecular mechanism of ABCC8 mutations manifest with completely distinct clinical picture.
KCNJ11 mutations (MODY13)
Kir6.2 subunit is formed by N- and C-terminal cytoplasmic domains, TMD containing M1 and M2, between which pore loop and selectivity filter locate, and tether helix linking TMD to CTD (Figure 2). It possesses ATP-binding domain which plays crucial role in coupling channel inhibition to metabolic activation (45). Disrupted ATP-binding and hydrolysis, altered intersubunit interactions and morphology of the channel, transduction abnormalities, deranged allosteric regulation of ATP sensitivity and disconnection with SUR1 interfere with proper functioning of Kir6.2 and lead to imbalance between KATP inhibition and high glucose levels (46–51). ATP interacts with Kir6.2 as proposed: G334 residue of the solvent-exposed helical segment with α-phosphate of ATP, K185 residue of the C-terminal β-sheet with both α- and β-phosphates and R50 residue of the N-terminal peptide with γ-phosphate. ATP brings NTD and CTD of Kir6.2 subunits with L0 of SUR1 together and locks the channel in closed state (45). The mutations in these residues are well documented in patients suffering from ND and DEND syndrome (49). On the other hand, mutations which are located far from ATP-binding site could also prevent normal ATP-binding and hydrolysis. Some substitutions to opposite charged amino acid increase steric repulsion between two adjacent ATP-binding pocket monomers (50), while others derange ATP-binding site, which make them less flexible for ATP to be able to fit (47). Impaired transduction from ATP-binding pocket to gate is another mechanism for Kir6.2 related diabetes, mostly associated with the mutations of slide helix (48). Structural integrity is key for effective channel behavior, which could be disrupted by the mutations through compression of the ion conducting part of the channel or halted intersubunit interactions (50). Mutations that cause disconnection between Kir6.2 and SUR1 decrease ATP sensitivity of the channel, even though intrinsic gating properties are unchanged (46). In addition to nucleotides, other allosteric regulators such as PIP2 also play major role in gating. Hypersensitivity to PIP2 due to cysteine substitutions increases open probability of KATP via enhancing S-palmitoylation (51). Furthermore, understanding underlying molecular mechanisms of channelopathies gives clue to why identical mutations show phenotypic heterogeneity. In MODY13 patients, late occurrence of the disease is not just accounted for lesser half maximal inhibitory concentration of ATP (52) and also additional trafficking defect of the channel which compensate for the increased open probability (53).
Molecular basis of MODY12 and MODY13 warranted to switch treatment option from insulin to sulfonylurea, which has 90% success rate (54, 55). Cryo-EM structure analysis revealed that glibenclamide binds to its binding site, which is formed by helices 6-8,11 of TMD1 and helices 16,17 of TMD2 of SUR1, and stabilize the interactions between N-terminus of Kir6.2 and L0 and TMD0 of SUR1 to prevent opening of the channel (45). Treatment failure is partly explained by the type of the mutation (56). It is obvious that mutations causing morphological changes in pore-forming subunit (50) or abnormal SUR1-Kir6.2 transduction (57) render the channel insensitive to SUs. There is no report on the SU binding site defect related treatment failure.
Impaired insulin trafficking
INS mutations (MODY10)
Mature insulin (Table 1) is a small molecule composed of A and B chains linked with 3 disulfide bonds. After synthesizing in the cytosol, preproinsulin is subjected to SRP facilitated translocation to ER, where it is cleaved to form proinsulin. Proinsulin in turn is liberated from C peptide via the recruitment of ER chaperons (Figures 1, 2) (58). Dominant and recessive mutations to the INS gene give rise to wide range of phenotypes varying from late-onset mild hyperglycemia to severe neonatal diabetes. Type of mutation is the main determinant of clinical phenotype (59) and as the best characterized monogenic mutations, analysis of insulin substitutes provided with broad knowledge regarding the insulin biosynthesis, action, and tight control of all these processes.
Cysteine substitutions interrupting formation of the disulfide bonds (B7-A7, B19-A20 and A6-A11) are classic examples of protein misfolding which lead to: 1) ER retention of both mutant and wild-type protein due to inappropriate interactions between unpaired cysteine of mutant protein and wild-type molecule (60); 2) impairment of β-cell proliferation by subsequent ER stress (61). Besides, any substitution to cysteine outside of the disulfide bonds produce similar effect (62). However, non-cysteine substitutions are also able to perturb normal folding of proinsulin (63). Preproinsulin recognition by signal peptidases and translocation of it to ER are initiating events in insulin maturation, which are disrupted by mutations to the signal peptide of preproinsulin (64). In order to fold and deliver properly to Golgi apparatus, proinsulin must be cleaved by endopeptidases at the junctions between C-peptide and each of the chain (58). Notably, mutations to these junctions underlie mild and late-onset diabetes accompanied by hyperproinsulinemia (65). Furthermore, some mutations may also interfere with insulin binding to its receptor. Resultant hyperinsulinemia is explained by disrupted degradation of insulin which fails to uptake by hepatocytes (65). Apart from the coding regions, the mutations affecting regulatory sequences are also described. Mutations to the promoter region of INS gene preventing the TFs to bind to its cis-elements; substitutions at the start codon abolishing translation; and alterations at the untranslated sites of mRNA sensitizing the molecule to RNA decay – all fall into this category (66).
As expected, MODY10 predominantly is managed by insulin injections (67), albeit understanding of underlying molecular mechanisms of INS mutations offered new perspectives for better therapeutic options. Regarding this, rescuing trapped wild-type proinsulin from ER through either degradation of mutant molecule or acceleration of oxidative folding is suggested by some laboratories (68, 69).
WFS1 mutations (Newly proposed MODY type)
Wolframin (Table 1) is a cargo-protein receptor, located in the ER membrane and contains two hydrophilic N- and C-terminal domains and the hydrophobic transmembrane domain between them (Figure 2) (70). Patients bearing homozygous and compound heterozygous mutations of WFS1 gene present with different combinations of young-onset diabetes mellitus, diabetes insipidus, optic atrophy and hearing loss, which are classical features of Wolfram syndrome (71). Wide application of sequencing techniques in diverse populations brought to light significant number of heterozygous WFS1 gene mutations contributing to MODY phenotype (72–75), although homozygosity is not always exception in MODY incidence (76). The knock-out mice generated with CRISPR-Cas9 technique highlighted the role of wolframin in proinsulin trafficking and vesicle formation. The study showed that NTD mutations disrupt interactions with Coat Protein Complex II (COPII) vesicle subunits, while CTD mutations interrupt wolframin-proinsulin bonds, both of which activated unfolded protein response pathway (70).
In the recent years, induced pluripotent stem cell models derived from MODY patients, have gained a lot of attention as an exciting tool to deepen the understanding of cellular and molecular processes of mature and developing β-cells in healthy and pathological states of pancreas, as well as to introduce new treatment strategies (77). β-cell iPSC models derived from patients with Wolfram syndrome helped to demonstrate that curative treatments such as CRISPR-Cas9 correction of a mutant gene or chemical chaperons targeting UPR pathways are not far-fetched for patients suffering from variety of ER stress causing diseases (78, 79).
Impaired transcriptional regulation
TFs are the group of proteins bind to cis-acting elements of the promoter of particular genes and regulate their transcriptional activity. MODY-related TFs (PDX1, NEUROD1, HNF1α, HNF4α, HNF1β, RFX6) are indispensable for the development of pancreas, β-cell identity and function of mature β-cells (Table 1) (80) and they all have direct binding sites at the insulin promoter (Figure 3) (81–84). Recently, detailed in silico analysis of available published data revealed that two TF genes, namely KLF11 and PAX4 – which previously enlisted as MODY-causing proteins – can not be included in the MODY list due to lack of co-segregation with diabetes and higher rate of same mutant genes in healthy population in addition to two other non-TF genes –BLK and APPL1 (5, 6). Thus, accurate co-segregation analysis of genetic test results is crucial to avoid misinterpretations.

Figure 3 Schematic illustration of the binding sites of insulin promoter for MODY-related transcription factors.  HNF1α
HNF1α  HNF4α
HNF4α  NEUROD1/E47
NEUROD1/E47  PDX1
PDX1  RFX6.
RFX6.
HNF4α mutations (MODY1)
HNF4α (Table 1) has a N-terminal AF1 domain that shows ligand-independent transactivation ability, a highly-conserved DNA-binding domain containing zinc-finger motif, a lipophilic ligand-binding domain, and a regulatory F domain (Figure 2). The domains are interrelated in such a way that the remote residues may have allosteric modulation over other sites owing to domain-domain interactions (85). Hyperglycemia in MODY1 is frequently accompanied by dyslipidemia and preceding transient neonatal diabetes with macrosomia (86). The latest report on MODY3 patient-derived iPSC lines solved the long debate of biphasic nature of disease by determining direct binding sites of ABCC8 and KCNJ11 genes for HNF4α (87). Detailed analysis of biphasic MODY1-causing LBD mutations revealed that dimerization of HNF4α allows it to conform to more structurally stable form and to expose more hydrophobic sites at the surface so that LBD may able to be occupied by its lipid ligands to the fullest extent (88). In DNA-binding motif, negatively charged amino acid substitutions for serine residues result in both lesser DNA-binding and transcriptional activity. Additionally, these findings also discovered the inhibitory effect of PKA-phosphorylation on DNA-binding and transactivation of HNF4α (89). While most single amino acid substitutions lead to structural and/or binding defect, some of them induce proteolytic degradation which turn the protein into a smaller truncated form (90). In addition to coding region, MODY1 is also originates from the mutations in the pancreas-specific P2 promoter (91). Functional studies found the binding sites of the P2 promoter for the TFs including HNF1α, HNF1β, PDX1 (91) along with direct binding of HNF4α to the INS, and HNF1α promoters (82, 92, 93), all of which indicate that β-cell function is controlled by complex and multifaceted hierarchical and interregulatory network.
MODY1-derived iPSC lines provided with model that represents the early stages of formation of human hepatopancreatic tissues. According to these models, insufficient amount of HNF4α downregulates foregut-specifying genes while upregulating hindgut markers and impacts normal foregut endodermal cell fate acquisition (94).
HNF1α mutations (MODY3)
HNF1α (Table 1) is a homeodomain-containing transcription factor of HNF family and it consists of a dimerization domain, a DNA-binding domain with POU-like and homeodomain-like motifs, and a transactivation domain in an order from amino-terminus to carboxy-terminus (Figure 2) (95). HNF1α binds to promoters of different genes including PDX1, INS, GLUT-2 and controls glucose sensing, mitochondrial metabolism, insulin secretion and exocytosis in β-cells (96–99). MODY3 mutations are dominant loss-of-function mutations (100) that cause the clinical phenotype identical to MODY1, if higher levels of HDL-cholesterol are excepted (86). Mutations affecting dimerization domain are characterized with more severe disruption of DNA-binding abilities which manifest with younger age at onset due to thermodynamic destabilization and structural abnormality of the protein (100, 101). In silico analysis in combination with in vitro methods revealed the characteristics and functions of POU-like and a homeodomain-like motifs of HNF1α in detail. According to the results of the study (102), the substitutions located in the residues of DNA-binding domain give rise to direct (through interrupted hydrogen bonds) and indirect (through perturbed local environment) disruption of bindings between DNA and HNF1α, impaired interdomain interactions, hindered nuclear translocation, and structural instability, which in turn cause accumulation of misfolding protein. Mutations to transactivation domain destabilize interactions with co-activators or other regulatory proteins and reduce transactivation with or without diminishing DNA-binding activities (100).
Interdependency of HNF1α and HNF4α is in consistent with the similarities in the clinical phenotype and treatment option of MODY1 and MODY3 (4, 82, 91). Currently, the first-line treatment of both types is SU and/or incretin-based agents (103, 104). The exaggerated sensitivity of MODY3 patients to SUs (87) and negative role of HNF1α mutations on glucagon secretion and incretin levels favored the treatment with DPP-4 inhibitors (105). Comparative study is required to define the best therapeutic agent(s) in accordance with the mutational characteristics of MODY1 and MODY3, as well as HNF1α and HNF4α variants of T2DM.
PDX1 mutations (MODY4)
PDX1 (Table 1) is a homeodomain-containing transcription factor, possesses a N-terminal transactivation domain, a C-terminal domain and a DNA-binding homeodomain (HD), which are hot-spot regions for mutations (Figure 2) (106). Depending upon the mode of inheritance, location and penetrance of the mutation, PDX1 gene alterations result in partial and total pancreatic agenesis (107), ND without exocrine insufficiency (108), gestational diabetes (93), and MODY with variable age at onset and severity (106). Since PDX1 is a master regulator of embryonic pancreas development, islet formation and β-cell differentiation (109), it is not surprising that its homozygous and compound heterozygous missense and frameshift mutations underlie the pancreatic agenesis (107). In the recent study, MODY4 patient-derived iPSC line was differentiated into pancreatic progenitors and genome-wide analysis of transcriptional targets of PDX1 was carried out in different stages of pancreatic cells. The analysis indicates that PDX1-binding sites for target genes evolve during embryonic development and change significantly. For instance, while PDX1 activates HNF1α, HNF1β and RFX6 during early developmental stage, KCNJ11 regulation becomes apparent only after islet formation (110). Recently, interesting case of ductal pancreatic agenesis is reported in a family carrying heterozygous frameshift mutation (111). We assume that because PDX1 is activated by different factors in ventral and dorsal pancreatic bud (Hex1 in ventral bud, Hb9 in dorsal bud) (10), mutations may affect interaction between PDX1 and its upstream regulators differently in an early developmental stage. Further work is needed to examine this assumption.
At least 33 MODY4 related mutations were reported so far (4, 106, 112) and their phenotypes are explained by dysregulation of glucose stimulated INS promoter activity at the transcriptional level by PDX-1 (113). Structural stability is vital for PDX1 in order to be capable of binding to gene promoter and the mutations to both NTD and HD are known to cause structural instability (93, 114). Reduced transcriptional activity is mostly derived from NTD mutations interfering with protein-protein interactions (93, 115). Although the function of CTD is still unclear, functional studies indicate that CTD mutations lead to diminished transactivation (115). However, according to the in vitro study with unnaturally generated CTD mutations, it also plays pivotal role in subnuclear localization of PDX1 through phosphorylation (116).
Management of MODY4 ranges from diet to insulin replacement depending on the degree of insulin synthesis defect. Recent studies propose DPP-4 inhibitors as a best treatment option since it is proved that PDX1 mutations are also responsible for lower levels of incretins (106).
HNF1β mutations (MODY5)
Similar to HNF1α, HNF1β (Table 1) has a N-terminal dimerization domain, a bipartite DNA-binding POU domain and a C-terminal transactivation domain (Figure 2). Its highly conserved dimerization domain and DBD permit HNF1β to be able to bind to DNA as a homodimer and heterodimer with HNF1α, while flexible transactivation domain and interdomain linkers allow it to cooperate with wide range of variable proteins in diverse tissues (117). MODY5 or renal cyst-diabetes syndrome occurs due to loss-of-function mutations and is characterized by phenotypic heterogeneity regarding the organ involvement and higher penetrance of renal anomalies than diabetes even among same family members (118). Intron 2 is the hot spot for splice-site mutations, which result in complete loss of exon 2 through yet-unidentified mechanisms and consequent truncated inactive protein (119). Most single amino acid substitutions are located in the DBD and disrupt the bonds with DNA directly or indirectly via perturbing local environment such as creating superfluous interactions with neighboring residues. Besides, some mutations to the same region affect protein structure and stability and leads to misfolded protein response (117).
Although the pancreatic development by HNF1β at the primitive gut stage is well demonstrated (120), the results of the studies indicating whether HNF1β is involved in GSIS and even is present or absent in mature β-cells are ambiguous (121, 122). Despite of the 70% homology in sequence identities of HNF1α and HNF1β (116), the management of MODY5 is different from MODY3. Unlike MODY3, MODY5 shows poor response to SUs and insulin replacement is the only option in majority of cases (123).
NEUROD1 mutations (MODY6)
With the aid of its basic helix-loop-helix and transactivation domains, NEUROD1 (Table 1) displays DNA-binding, dimerization and transactivation abilities (Figure 2) (124). NEUROD1 is highly expressed in neuroendocrine tissues and its central function in their development is evident from the mutations causing ketoacidosis-prone diabetes with microvascular sequelae and neurological abnormalities such as cerebellar hypoplasia, hearing and visual impairments, low IQ (125). Up to now, at least 26 missense and nonsense mutations found in NEUROD1 gene, which four of them were homozygous mutations (125–129). Homozygous inheritance shows more severe phenotype, earlier onset and higher penetrance than heterozygous state (130). The analysis of the first case of MODY6 revealed that NEUROD1 heterodimerize with another bHLH (basic Helix-Loop-Helix) transcription factor – E47 and binds to E-box element of the insulin promoter to induce its synthesis through the interaction between p-300 and C-terminal glutamine-rich domain (131). Two distinct transactivation domains, namely AD1 and AD2, have different transactivation capabilities (132) and whether regulators other than p300 interacts with NEUROD1 is unclear, since studies related to the structure and function of this protein are scarce. Moreover, the role of disease-causing substitutions outside of the bHLH and C-terminal domains (132) needs to be investigated.
Treatment of MODY6 is not specific and varies from diet to insulin replacement depending upon the severity of the disease (125).
RFX6 mutations (Newly proposed MODY type)
RFX6 (Table 1) is a winged-helix transcription factor and encompasses DBD, dimerization domain D and extended dimerization domains B and C according to the homology modeling of RFX protein family (Figure 2) (133). Homozygous and compound heterozygous truncating mutations lead to Mitchell-Riley syndrome, the syndrome characterized with neonatal/early-onset diabetes, hypoplastic/annular pancreas, gallbladder hypoplasia/agenesis, intestinal atresia with or without intestinal malrotation (134). On the other hand, patients harboring heterozygous mutations suffer from late-onset diabetes, which show reduced penetrance (135). As an upstream regulator of PDX-1 and NEUROD1, RFX6 is vital for islet formation (81). Consistent with the pancreatic anomalies observed in patients bearing RFX6 mutations, iPSC line derived from the family suffering from Mitchell-Riley syndrome exhibited the absence of body and tail of pancreas. Besides, the study also proposed the possible regulatory loop between PDX1 and RFX6 in the pancreatic progenitor stage (136). RFX6 is able to homodimerize and heterodimerize with RFX3 to bind to X-box motif of target promoter and its role in GSIS is linked to modulatory effect on SUR1, GCK and L-type Ca2+ channels (84, 137). While substitutions in the conserved region of DBD completely abrogate DNA-binding, alterations in other regions partially interfere with DNA-binding abilities (81). Since RFX6 is a newly discovered protein, there is a hope that the molecular characterization of RFX6-MODY will provide with valuable knowledge regarding its structure and activity.
The association between RFX6 mutations and GIP levels has been demonstrated (134). Patients with RFX6-MODY are responsive to treatment with DPP-4 inhibitors (138).
MODY beyond the β-cell
CEL mutations (MODY8)
Carboxyl ester lipase (Table 1) is a pancreatic enzyme produced in acinar cells, but not in β-cells (139). It has signal peptide, bile-salt binding sites and catalytic sites containing globular domain, and multiple O-glycosylation sites containing polymorphic variable number tandem repeats (Figure 2) (140). Gain-of-function mutations to CEL gene give rise to pancreatic exocrine insufficiency during adolescence or young adulthood and a couple of decades later, to insulin-dependent diabetes (141). Recent few preliminary studies shed light into the impact of external CEL protein to β-cells (142–145). O-glycosylation sites at the VNTR render the CEL enzyme soluble and frameshift mutations at the VNTR turn the molecule into positively charged insoluble aggregates which is prone to bind with negatively charged structures including cell and organelle membranes (142). In acinar cells, mutant cell enzyme causes ER retention and UPR activation. In spite of acinar cells induce yet-to-be-dentified degradative machinery, the acinar cells can not overcome the excess amount of misfolded protein (144). Modified misfolded CEL protein gain access to extracellular space through induction of exocytosis and then forms insoluble aggregates (143). Cross-talk between acinar and islet cells allows the insoluble aggregates to be re-uptaken by neighboring β-cells via endocytosis (145). Eventually, the capability of β-cells to degrade endocytic substrates through lysosomal pathways depletes (145). We assume that “sticky” CEL aggregates interact with the membranes inside the cell and this in turn triggers dysfunction of different organelles displayed as altered mitochondrial activity and ER stress (Figure 4) (143). In fact, direct interaction of another toxic misfolded protein product – Islet Amyloid Polypeptide Oligomers with the membranes in β-cells had been demonstrated by electron microscopy (146). On the other hand, other possible toxic pathways such as expression of “disallowed” genes or β-cell-senescence can not be ruled out (145). However, structural characteristics of CEL aggregates, mechanism of exocytosis and endocytosis, behavior of internalized macromolecules within β-cells and involved pathways are largely unknown and further in vitro studies using variety of approaches are highly demanded, which may also have therapeutic implications.
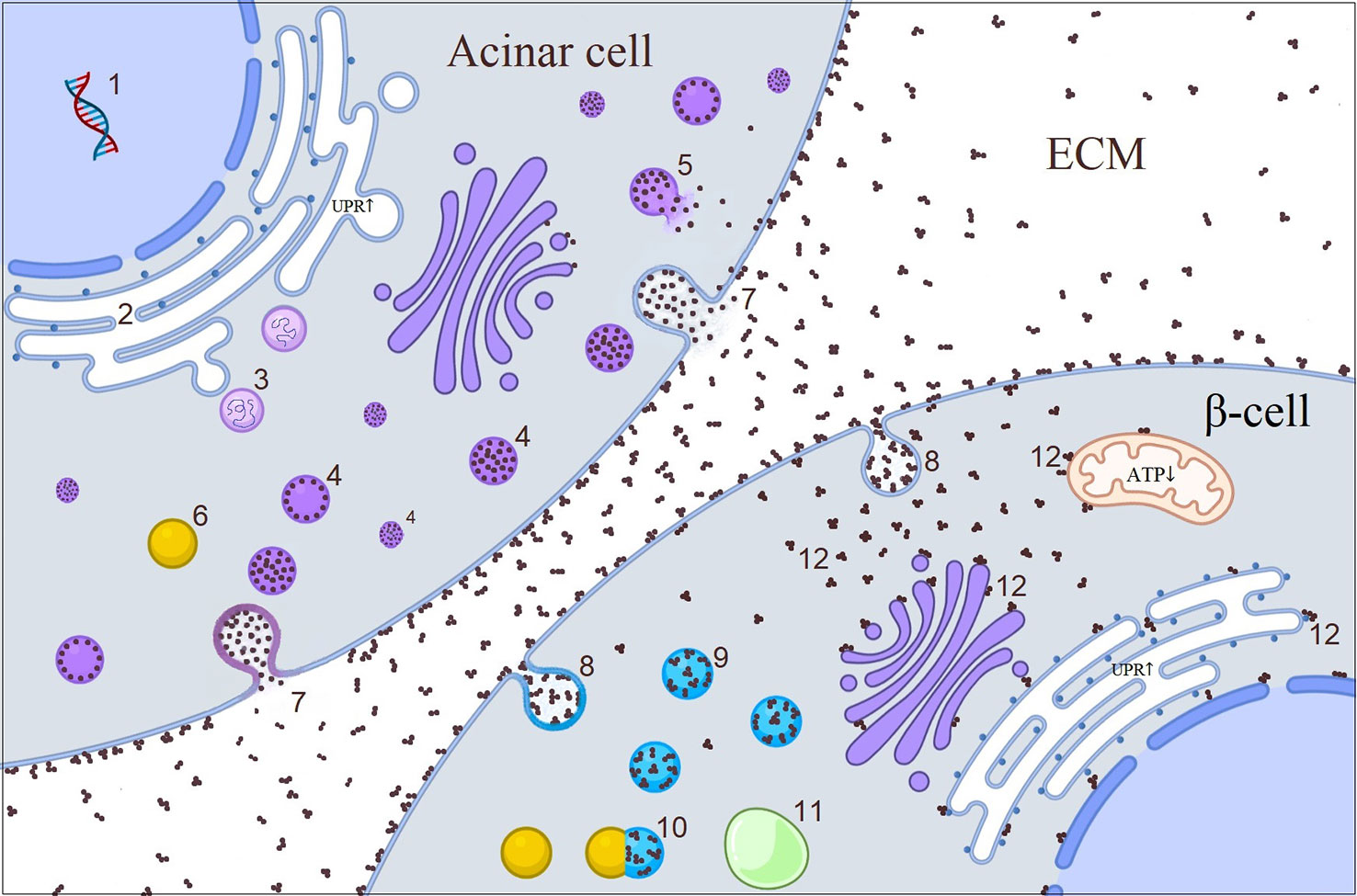
Figure 4 Illustrative picture of the cross-talk between acinar cell and β-cell in CEL-mutated pancreas. 1. CEL synthesis in acinar cell; 2. Misfolded CEL protein causing increased UPR in ER during post-translational modification; 3. UPR initiated retro-translocation of misfolded CEL protein; 4. CEL protein containing vesicles; 5. Disrupted vesicle; 6. Lysosome; 7. Exocytosis of CEL protein through either fusion of vesicles with plasma membrane or direct accumulation of the protein in the plasma membrane which later becomes core of CEL aggregates in the ECM; 8. Endocytosis of CEL aggregates; 9. Endosomes; 10. Fusion of lysosome to the endosome; 11. Autophagosome; 12. Interaction of free aggregates with the membranes (?). CEL, carboxyl ester lipase; ECM, extracellular matrix; ER, endoplasmic reticulum; UPR, unfolded protein response.
Summary
Monogenic diabetes gave us simplified models of complex molecular processes occurring within β-cells, which allowed to explore the roles of numerous proteins from single protein perspective. Clarification of the structural and functional abnormality of a particular protein, which arose from a mutation to its gene, helps to define the crystal structure, function and interaction with other molecules of this protein. The characteristics of the mutations also help to identify phenotypical heterogeneity and have implications for personalized medicine. Most importantly, all of these enlightenments pave the way for the new treatment strategies. In fact, understanding of the kinetic parameters of GCK enzyme brought in new antidiabetic agents which have potential to fulfill the need for an optimum drug to control glucose levels efficiently with minimum side effects in the management of T2DM (27). KATP channel is the target for SUs and switching therapy to SUs free the patients from the burden of insulin therapy (54, 55). SUs are also effective in the management of MODY1 and MODY3, probably due to regulation of KATP channel subunits by HNF1α (87, 104). On the other hand, insulin requirement in almost all cases of MODY5 might be explained by the central role of HNF1β in the early pancreatic development rather than its involvement in glucose homeostasis (123). Clinically observed association between GLP-1/GIP and some MODY-related genes (at least PDX1 and RFX6), of which molecular basis is unknown, made the treatment with GLP-1 analogs or DPP-4 inhibitors to be favorable (106, 134). INS, WFS1 and CEL mutations that lead to ER retention and UPR (61, 70, 145), might be rescued by therapies targeting these toxic pathways. Moreover, extrapolation of data related to the aforementioned transcription factors into the stem cell research has potential to provide the radical treatment for T1DM patients.
In the last century, the approach to MODY was based on the investigation of the previously known proteins (147) and this approach broadened our comprehension regarding their role in developing and mature β-cells. The application of sequencing methods to experimental and clinical medicine (148) introduced new molecules involved in β-cell development, identity, and function. Extensive exploration of these proteins not only resulted in better medical care to diabetes patients, but also enriched our understanding about the universal cellular processes including transcriptional and translational regulation, behavior of ion channels and transporters, cargo trafficking, exocytosis. Noteworthy, β-cells are important model to test similar events in other cells. In the recent years, state-of-the-art technique – the establishment of MODY patient-derived stem cell lines – has been added to ongoing diabetes research and is expected to yield breakthrough treatment options in the near future, of which benefits might exceed diabetes management.
Author contributions
AZ provided the first draft of the manuscript. WG and BZ carried out literature search and designed the table and the figures. SS and QZ wrote the manuscript. All authors revised and approved the final version of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
bHLH, Basic Helix-Loop-Helix; CTD, C-terminal domain; HD, Homeodomain; HH, Hyperinsulinaemic Hypoglycemia; L0, Loop; LBD, Ligand-binding domain; NBD, Nucleotide-binding domain; NDM, Neonatal Diabetes Mellitus; NTD, N-terminal domain; SU(s), Sulfonylurea(s); TMD, Transmembrane domain.
References
1. Nkonge KM, Nkonge DK, Nkonge TN. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin Diabetes Endocrinol (2020) 6(1):20. doi: 10.1186/s40842-020-00112-5
2. Thanabalasingham G, Owen KR. Diagnosis and management of maturity onset diabetes of the young (MODY). BMJ (Online) (2011) 343(7828):1–9. doi: 10.1136/bmj.d6044
3. Pang L, Colclough KC, Shepherd MH, McLean J, Pearson ER, Ellard S, et al. Improvements in awareness and testing have led to a threefold increase over 10 years in the identification of monogenic diabetes in the U.K. Diabetes Care (2022) 45(3):642–9. doi: 10.2337/dc21-2056
4. Aydogan HY, Gul N, Demirci DK, Mutlu U, Gulfidan G, Arga KY, et al. Precision diagnosis of maturity-onset diabetes of the young with next-generation sequencing: findings from the MODY-IST study in adult patients. Omics A J Integr Biol (2022) 26(4):218–35. doi: 10.1089/omi.2022.0006
5. Ivanoshchuk DE, Shakhtshneider EV, Rymar OD, Ovsyannikova AK, Mikhailova SV, Orlov PS, et al. Analysis of appl1 gene polymorphisms in patients with a phenotype of maturity onset diabetes of the young. J Personalized Med (2020) 10(3):1–9. doi: 10.3390/jpm10030100
6. Laver TW, Wakeling MN, Knox O, Colclough K, Wright CF, Ellard S, et al. Evaluation of evidence for pathogenicity demonstrates that BLK, KLF11, and PAX4 should not be included in diagnostic testing for MODY. Diabetes (2022) 71(5):1128–36. doi: 10.2337/db21-0844
7. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. New Engl J Med (2001) 345(13):971–80. doi: 10.1056/nejmra002168
8. Hutton JC. Insulin secretory granule biogenesis and the proinsulin-processing endopeptidases. Diabetologia (1994) 37(2 Supplement):S48–56. doi: 10.1007/BF00400826
9. Boland BB, Rhodes CJ, Grimsby JS. The dynamic plasticity of insulin production in b -cells. Mol Metab (2017) 6(9):958–73. doi: 10.1016/j.molmet.2017.04.010
10. Servitja JM, Ferrer J. Transcriptional networks controlling pancreatic development and beta cell function. Diabetologia (2004) 47(4):597–613. doi: 10.1007/s00125-004-1368-9
11. Naylor RN, John PM, Winn AN, Carmody D, Greeley SAW, Philipson LH, et al. Cost-effectiveness of MODY genetic testing: translating genomic advances into practical health applications. Diabetes Care (2014) 37(1):202–9. doi: 10.2337/dc13-0410
12. Hogrebe NJ, Maxwell KG, Augsornworawat P, Millman JR. Generation of insulin-producing pancreatic β cells from multiple human stem cell lines. Nat Protoc (2021) 16(9):4109–43. doi: 10.1038/s41596-021-00560-y
13. Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat Cell Biol (2019) 21(February):263–74. doi: 10.1038/s41556-018-0271-4
14. Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes. Diabetes (1990) 39(6):647–52. doi: 10.2337/diabetes.39.6.647
15. Passanisi S, Salzano G, Bombaci B, Lombardo F. Clinical and genetic features of maturity-onset diabetes of the young in pediatric patients: a 12-year monocentric experience. Diabetol Metab Syndrome (2021) 13(1):1–8. doi: 10.1186/s13098-021-00716-6
16. Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, et al. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat (2009) 30(11):1512–26. doi: 10.1002/humu.21110
17. Miller SP, Anand GR, Karschnia EJ, Bell GI, LaPorte DC, Lange AJ. Characterization of glucokinase mutations associated with maturity- onset diabetes of the young type 2 (MODY-2): different glucokinase defects lead to a common phenotype. Diabetes (1999) 48(8):1645–51. doi: 10.2337/diabetes.48.8.1645
18. Ricard J, Meunier JC, Buc J. Regulatory behavior of monomeric enzymes: 1. the mnemonical enzyme concept. Eur J Biochemistry 49(1) (1974) pp:195–208. doi: 10.1111/j.1432-1033.1974.tb03825.x
19. Kamata K, Mitsuya M, Nishimura T, Eiki J, Nagata Y. Structural basis for allosteric regulation of the monomeric allosteric enzyme human glucokinase. Structure (2004) 12(3):429–38. doi: 10.1016/j.str.2004.02.005
20. Liu S, Ammirati MJ, Song X, Knafels JD, Zhang J, Greasley SE, et al. Insights into mechanism of glucokinase activation: observation of multiple distinct protein conformations. J Biol Chem (2012) 287(17):13598–610. doi: 10.1074/jbc.M111.274126
21. Galán M, Vincent O, Roncero I, Azriel S, Boix-Pallares P, Delgado-Alvarez E, et al. Effects of novel maturity-onset diabetes of the young (MODY)-associated mutations on glucokinase activity and protein stability. Biochem J (2006) 393(1):389–96. doi: 10.1042/BJ20051137
22. Langer S, Waterstradt R, Hillebrand G, Santer R, Baltrusch S. The novel GCK variant p.Val455Leu associated with hyperinsulinism is susceptible to allosteric activation and is conducive to weight gain and the development of diabetes. Diabetologia (2021) 64(12):2687–700. doi: 10.1007/s00125-021-05553-w
23. Liu L, Liu Y, Ge X, Liu X, Chen C, Wang Y, et al. Insights into pathogenesis of fi ve novel GCK mutations identi fi ed in Chinese MODY patients. Metabolism (2018) 89:8–17. doi: 10.1016/j.metabol.2018.09.004
24. Ding SY, Tribble ND, Kraft CA, Markwardt M, Gloyn AL, Rizzo MA. Naturally occurring glucokinase mutations are associated with defects in posttranslational s-nitrosylation. Mol Endocrinol (2010) 24(1):171–7. doi: 10.1210/me.2009-0138
25. Gašperíková D, Tribble ND, Staník J, Hucková M, Misovicová N, van de Bunt M, et al. Identification of a novel β-cell glucokinase (GCK) promoter mutation (-71G>C) that modulates GCK gene expression through loss of allele-specific Sp1 binding causing mild fasting hyperglycemia in humans. Diabetes (2009) 58(8):1929–35. doi: 10.2337/db09-0070
26. Toulis KA, Nirantharakumar KPourzitaki C, Barnett AH, Tahrani AA. Glucokinase activators for type 2 diabetes: challenges and future developments. Drugs (2020) 80(5):467–75. doi: 10.1007/s40265-020-01278-z
27. Zhu D, Li X, Ma J, Zeng J, Gan S, Dong X, et al. Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med (2022) 28(5):965–73. doi: 10.1038/s41591-022-01802-6
28. Sagen JV, Odili S, Bjørkhaug L, Zelent D, Buettger C, Kwagh J, et al. From clinicogenetic studies of maturity-onset diabetes of the young to unraveling complex mechanisms of glucokinase regulation. Diabetes (2006) 55(6):1713–22. doi: 10.2337/db05-1513
29. Clement IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, et al. Association and stoichiometry of K(ATP) channel subunits. Neuron (1997) 18(5):827–38. doi: 10.1016/S0896-6273(00)80321-9
30. Ashcroft FM. ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest (2005) 115(8):2047–58. doi: 10.1172/JCI25495
31. De Franco E, Saint-Martin C, Brusgaard K, Johnson AEK, Aguilar-Bryan L, Bowman P, et al. Update of variants identified in the pancreatic β-cell KATP channel genes KCNJ11 and ABCC8 in individuals with congenital hyperinsulinism and diabetes. Hum Mutat (2020) 41(5):884–905. doi: 10.1002/humu.23995
32. Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S, Perry A, Abu-Libdeh A, Glaser B, et al. Novel De novo mutation in sulfonylurea receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes (2008) 57(7):1935. doi: 10.2337/DB08-0159
33. Li N, Wu J, Ding D, Cheng J, Gao N, Chen L. Structure of a pancreatic ATP-sensitive potassium article structure of a pancreatic ATP-sensitive potassium channel. Cell (2017) 168(1–2):101–110.e10. doi: 10.1016/j.cell.2016.12.028
34. Matsuo M, Dabrowski M, Ueda K, Ashcroft FM. Mutations in the linker domain of NBD2 of SUR inhibit transduction but not nucleotide binding. EMBO J (2002) 21(16):4250–8. doi: 10.1093/emboj/cdf419
35. Tanizawa Y, Matsuda K, Matsuo M, Ohta Y, Ochi N, Adachi M, et al. Genetic analysis of Japanese patients with persistent hyperinsulinemic hypoglycemia of infancy: nucleotide-binding fold-2 mutation impairs cooperative binding of adenine nucleotides to sulfonylurea receptor 1. Diabetes (2000) 49(1):114–20. doi: 10.2337/diabetes.49.1.114
36. Nessa A, Aziz QH, Thomas AM, Harmer SC, Tinker A, Hussain K. Molecular mechanisms of congenital hyperinsulinism due to autosomal dominant mutations in ABCC8. Hum Mol Genet (2015) 24(18):5142–53. doi: 10.1093/hmg/ddv233
37. Ueda K, Inagaki N, Seino S. MgADP antagonism to Mg2+-independent ATP binding of the sulfonylurea receptor SUR1. J Biol Chem (1997) 272(37):22983–6. doi: 10.1074/jbc.272.37.22983
38. De Wet H, Rees MG, Shimomura K, Aittoniemi J, Patch A, Flanagan SE, et al. Increased ATPase activity produced by mutations at arginine-1380 in nucleotide-binding domain 2 of ABCC8 causes neonatal diabetes. Proc Natl Acad Sci USA (2007) 104(48):18988–92. doi: 10.1073/pnas.0707428104
39. de Wet H, Proks P, Lafond M, Aittoniemi J, Sansom MSP, Flanagan SE, et al. A mutation (R826W) in nucleotide-binding domain 1 of ABCC8 reduces ATPase activity and causes transient neonatal diabetes. EMBO Rep (2008) 9(7):648–54. doi: 10.1038/embor.2008.71
40. Campbell JD, Sansom MSP, Ashcroft FM. Potassium channel regulation. structural insights into the function of the nucleotide-binding domains of the human sulphonylurea receptor. EMBO Rep (2003) 4(11):1038–42. doi: 10.1038/sj.embor.7400003
41. Männikkö R, Flanagan SE, Sim X, Segal D, Hussain KEllard S. Mutations of the same conserved glutamate residue in NBD2 of the sulfonylurea receptor 1 subunit of the KATP channel can result in either hyperinsulinism or neonatal diabetes. Diabetes (2011) 60(6):1813–22. doi: 10.2337/db10-1583
42. Tarasov AI, Nicolson TJ, Riveline J, Taneja TK, Baldwin SA, Baldwin JM, et al. A rare mutation in ABCC8/SUR1 leading to altered ATP-sensitive K + channel activity and β-cell glucose sensing is associated with type 2 diabetes in adults. Diabetes (2008) 57(6):1595–604. doi: 10.2337/db07-1547
43. Riveline JP, Rousseau E, Reznik Y, Fetita S, Philippe J, Dechaume A, et al. Clinical and metabolic features of adult-onset diabetes caused by ABCC8 mutations. Diabetes Care (2012) 35(2):248–51. doi: 10.2337/dc11-1469
44. Harel S, Cohen ASA, Hussain K, Flanagan SE, Schlade-Bartusiak K, Patel M, et al. Alternating hypoglycemia and hyperglycemia in a toddler with a homozygous p.R1419H ABCC8 mutation: an unusual clinical picture. J Pediatr Endocrinol Metab (2015) 28(3–4):345–51. doi: 10.1515/jpem-2014-0265
45. Martin GM, Kandasamy B, DiMaio F, Yoshioka C, Shyng S. Anti-diabetic drug binding site in a mammalian KATP channel revealed by cryo-EM. eLife (2017) 6:1–27. doi: 10.7554/eLife.31054.001
46. Tammaro P, Proks P, Ashcroft FM. Functional effects of naturally occurring KCNJ11 mutations causing neonatal diabetes on cloned cardiac KATP channels. J Physiol (2006) 571(1):3–14. doi: 10.1113/jphysiol.2005.099168
47. Koster JC, Kurata HT, Enkvetchakul D, Nichols CG. DEND mutation in Kir6.2 (KCNJ11) reveals a flexible n-terminal region critical for ATP-sensing of the KATP channel. Biophys J (2008) 95(10):4689–97. doi: 10.1529/biophysj.108.138685
48. Winkler M, Lutz R, Russ U, Quast U, Bryan J. Analysis of two KCNJ11 neonatal diabetes mutations, V59G and V59A, and the analogous KCNJ8 I60G substitution: differences between the channel subtypes formed with SUR1. J Biol Chem (2009) 284(11):6752–62. doi: 10.1074/jbc.M805435200
49. Shimomura K, de Nanclares GP, Foutinou C, Caimari M, Castaño L, Ashcroft FM. The first clinical case of a mutation at residue K185 of Kir6.2 (KCNJ11): a major ATP-binding residue. Diabetic Med (2010) 27(2):225–9. doi: 10.1111/j.1464-5491.2009.02901.x
50. Liu L, Nagashima K, Yasuda T, Liu Y, Hu H, He G, et al. Mutations in KCNJ11 are associated with the development of autosomal dominant, early-onset type 2 diabetes. Diabetologia (2013) 56(12):2609–18. doi: 10.1007/s00125-013-3031-9
51. Yang HQ, Martinez-Ortiz W, Hwang J, Fan X, Cardozo TJ, Coetzee WA. Palmitoylation of the KATP channel Kir6.2 subunit promotes channel opening by regulating PIP2 sensitivity. Proc Natl Acad Sci USA (2020) 117(19):10593–602. doi: 10.1073/pnas.1918088117
52. Vedovato N, Cliff E, Proks P, Poovazhagi V, Flanagan SE, Ellard S, et al. Neonatal diabetes caused by a homozygous KCNJ11 mutation demonstrates that tiny changes in ATP sensitivity markedly affect diabetes risk. Diabetologia (2016) 59(7):1430–6. doi: 10.1007/s00125-016-3964-x
53. Lin YW, Li A, Grasso V, Battaglia D, Crinò A, Colombo C, et al. Functional characterization of a novel KCNJ11 in frame mutation-deletion associated with infancy-onset diabetes and a mild form of intermediate DEND: a battle between KATP gain of channel activity and loss of channel expression. PLoS One (2013) 8(5):e63758. doi: 10.1371/journal.pone.0063758
54. Pearson ER, Flechtner I, Njølstad PR, Malecki MT, Flanagan SE, Larkin B, et al. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. New Engl J Med (2006) 355(5):467–77. doi: 10.1056/nejmoa061759
55. Rafiq M, Flanagan SE, Patch A, Shields BM, Ellard S, Hattersley AT. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care (2008) 31(2):204–9. doi: 10.2337/dc07-1785
56. Garcin L, Mericq V, Fauret-Amsellem A, Cave H, Polak M, Beltrand J. Neonatal diabetes due to potassium channel mutation: response to sulfonylurea according to the genotype. Pediatr Diabetes (2020) 21(6):932–41. doi: 10.1111/pedi.13041
57. Martin GM, Sung MW, Shyng SL. Pharmacological chaperones of ATP-sensitive potassium channels: mechanistic insight from cryoEM structures. Mol Cell Endocrinol (2020) 502:110667. doi: 10.1016/j.mce.2019.110667
58. Dodson G, Steiner D. The role of assembly in insulin’s biosynthesis. Curr Opin Struct Biol (1998) 8(2):189–94. doi: 10.1016/S0959-440X(98)80037-7
59. Liu M, Sun J, Cui J, Chen W, Guo H, Barbetti F, et al. INS -gene mutations : from genetics and beta cell biology to clinical disease. Mol Aspects Med (2015) 42:3–18. doi: 10.1016/j.mam.2014.12.001
60. Støy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, et al. Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci USA (2007) 104(38):15040–4. doi: 10.1073/pnas.0707291104
61. Balboa D, Saarimäki-Vire J, Borshagovski D, Survila M, Lindholm P, Galli E, et al. Insulin mutations impair beta-cell development in a patient-derived iPSC model of neonatal diabetes. eLife (2018) 7:e38519. doi: 10.7554/eLife.38519
62. Edghill EL, Flanagan SE, Patch A, Boustred C, Parrish A, Shields B, et al. Insulin mutation screening in 1,044 patients with diabetes mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes (2008) 57(4):1034–42. doi: 10.2337/db07-1405
63. Colombo C, Porzio O, Liu M, Massa O, Vasta M, Salardi S, et al. Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J Clin Invest (2008) 118(6):2148–56. doi: 10.1172/JCI33777
64. Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, et al. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes (2010) 59(3):653–61. doi: 10.2337/db09-1091
65. Steiner DF, Tager HS, Chan SJ, Nanjo K, Sanke T, Rubenstein AH, et al. Lessons learned from molecular biology of insulin-gene mutations. Diabetes Care (1990) 13(6):600–9. doi: 10.2337/diacare.13.6.600
66. Garin I, Edghill EL, Akerman I, Rubio-Cabezas O, Rica I, Locke JM, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA (2010) 107(7):3105–10. doi: 10.1073/pnas.0910533107
67. Aarthy R, Aston-Mourney K, Mikocka-Walus A, Radha V, Amutha A, Anjana RM, et al. Clinical features, complications and treatment of rarer forms of maturity-onset diabetes of the young (MODY) - a review. J Diabetes its Complications (2021) 35(1):107640. doi: 10.1016/j.jdiacomp.2020.107640
68. Ushioda R, Hoseki J, Araki K, Jansen G, Thomas DY, Nagata K. ERdj5 is required as a disulfide reductase for degradation of misfolded proteins in the ER. Science (2008) 321(5888):569–72. doi: 10.1126/science.1159293
69. Wright J, Birk J, Haataja L, Liu M, Ramming T, Weiss MA, et al. Endoplasmic reticulum oxidoreductin-1α(Ero1α) improves folding and secretion of mutant proinsulin and limits mutant proinsulin-induced endoplasmic reticulum stress. J Biol Chem (2013) 288(43):31010–8. doi: 10.1074/jbc.M113.510065
70. Wang L, Liu H, Zhang X, Song E, Wang Y, Xu T, et al. WFS1 functions in ER export of vesicular cargo proteins in pancreatic β-cells. Nat Commun (2021) 12(1):6996. doi: 10.1038/s41467-021-27344-y
71. Inoue H, et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome). Nat Genet (1998) 20(2):143–8. doi: 10.1038/2441
72. Bansal V, Boehm BO, Darvasi A. Identification of a missense variant in the WFS1 gene that causes a mild form of wolfram syndrome and is associated with risk for type 2 diabetes in ashkenazi Jewish individuals. Diabetologia (2018) 61(10):2180–8. doi: 10.1007/s00125-018-4690-3
73. Mohan V, Radha V, Nguyen TT, Stawiski EW, Pahuja KB, Goldstein LD, et al. Comprehensive genomic analysis identifies pathogenic variants in maturity-onset diabetes of the young (MODY) patients in south India. BMC Med Genet (2018) 19(1):1–10. doi: 10.1186/s12881-018-0528-6
74. Billings LK, Shi Z, Resurreccion WK, Wang C, Wei J, Pollin TI, et al. Statistical evidence for high-penetrance MODY-causing genes in a large population-based cohort. Endocrinology Diabetes Metab (2022) 5(6):e372. doi: 10.1002/edm2.372
75. Saint-Martin C, Bouvet D, Bastide M, Bellanné-Chantelot C. Gene panel sequencing of patients with monogenic diabetes brings to light genes typically associated with syndromic presentations. Diabetes (2022) 71(3):578–84. doi: 10.2337/db21-0520
76. Bansal V, Gassenhuber J, Phillips T, Oliveira G, Harbaugh R, Villarasa N, et al. Spectrum of mutations in monogenic diabetes genes identified from high-throughput DNA sequencing of 6888 individuals. BMC Med (2017) 15(1):1–14. doi: 10.1186/s12916-017-0977-3
77. Teo AKK, Windmueller R, Johansson BB, Dirice E, Njolstad PR, Tjora E, et al. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J Biol Chem (2013) 288(8):5353–6. doi: 10.1074/jbc.C112.428979
78. Maxwell KG, Augsornworawat P, Velazco-Cruz L, Kim MH, Asada R, Hogrebe NJ, et al. Gene-edited human stem cell–derived β cells from a patient with monogenic diabetes reverse preexisting diabetes in mice. Sci Trans Med (2020) 12(540):eaax9106. doi: 10.1126/scitranslmed.aax9106
79. Kitamura RA, Maxwell KG, Ye W, Kries K, Brown CM, Augsornworawat P, et al. Multidimensional analysis and therapeutic development using patient iPSC–derived disease models of wolfram syndrome. JCI Insight (2022) 7(18):e156549. doi: 10.1172/jci.insight.156549
80. Balakrishnan S, Dhavamani S, Prahalathan C. β-cell specific transcription factors in the context of diabetes mellitus and β-cell regeneration. Mech Dev (2020) 163:103634. doi: 10.1016/j.mod.2020.103634
81. Okita K, Yang Q, Yamagata K, Hangenfeldt KA, Miyagawa J, Kajimoto Y, et al. Human insulin gene is a target gene of hepatocyte nuclear factor-1α (HNF-1α) and HNF-1β. Biochem Biophys Res Commun (1999) 263(2):566–9. doi: 10.1006/bbrc.1999.1412
82. Ohneda K, Mirmira RG, Wang J, Johnson JD, German MS. The homeodomain of PDX-1 mediates multiple protein-protein interactions in the formation of a transcriptional activation complex on the insulin promoter. Mol Cell Biol (2000) 20(3):900. doi: 10.1128/MCB.20.3.900-911.2000
83. Bartoov-Shifman R, Hertz R, Wang H, Wollheim CB, Bar-Tana J, Walker MD. Activation of the insulin gene promoter through a direct effect of hepatocyte nuclear factor 4α. J Biol Chem (2002) 277(29):25914–9. doi: 10.1074/jbc.M201582200
84. Chandra V, Albagli-Curiel O, Hastoy B, Piccand J, Randriamampita C, Vaillant E, et al. RFX6 regulates insulin secretion by modulating Ca2+ homeostasis in human β cells. Cell Rep (2014) 9(6):2206–18. doi: 10.1016/j.celrep.2014.11.010
85. Chandra V, Huang P, Potluri N, Wu D, Kim Y, Rastinejad F. Multidomain integration in the structure of the HNF-4α nuclear receptor complex. Nature (2013) 495(7441):394–8. doi: 10.1038/nature11966
86. Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic β-cell diabetes. Nat Clin Pract Endocrinol Metab (2008) 4(4):200–13. doi: 10.1038/ncpendmet0778
87. Hermann FM, Kjærgaard MFTian C, Tiemann U, Jackson A, Olsen LR. An insulin hypersecretion phenotype precedes pancreatic β cell failure in MODY3 patient-specific cells. Cell Stem Cell (2023) 30(1):38–51.e8. doi: 10.1016/j.stem.2022.12.001
88. Singh P, Tung S, Han EH, Lee I, Chi Y. Dimerization defective MODY mutations of hepatocyte nuclear factor 4α. Mutat Res - Fundam Mol Mech Mutagenesis (2019) 814(September 2018):1–6. doi: 10.1016/j.mrfmmm.2019.01.002
89. Oxombre B, Kouach M, Moerman E, Formstecher P, Laine B. The G115S mutation associated with maturity-onset diabetes of the young impairs hepatocyte nuclear factor 4α activities and introduces a PKA phosphorylation site in its DNA-binding domain. Biochem J (2004) 383(3):573–80. doi: 10.1042/BJ20040473
90. Navas MA, Munoz-Elias EJ, Kim J, Shih D, Stoffel M. Functional characterization of the MODY1 gene mutations HNF4(R127W), HNF4(V255M), and HNF4(E276Q). Diabetes (1999) 48(7):1459–65. doi: 10.2337/diabetes.48.7.1459
91. Thomas H, Jaschkowitz K, Bulman M, Frayling TM, Mitchell SM, Roosen S, et al. A distant upstream promoter of the HNF-4α gene connects the transcription factors involved in maturity-onset diabetes of the young. Hum Mol Genet (2001) 10(19):2089–97. doi: 10.1093/hmg/10.19.2089
92. Gragnoli C, Lindner T, Cockburn BN, Kaisaki PJ, Gragnoli F, Marozzi G, et al. Maturity-onset diabetes of the young due to a mutation in the hepatocyte nuclear factor-4α binding site in the promoter of the hepatocyte nuclear factor-1α gene. Diabetes (1997) 46(10):1648–51. doi: 10.2337/DIACARE.46.10.1648
93. Gragnoli C, Stanojevic V, Gorini A, Preussenthal GMV, Thomas MK, Habener JF, et al. IPF-1/MODY4 gene missense mutation in an Italian family with type 2 and gestational diabetes. Metabolism: Clin Exp (2005) 54(8):983–8. doi: 10.1016/j.metabol.2005.01.037
94. Jin NH, Jasmen JB, Lim CS, Lau HH, Krishnan VG, Kadiwala J, et al. HNF4A haploinsufficiency in MODY1 abrogates liver and pancreas differentiation from patient- derived induced pluripotent stem cells HNF4A haploinsufficiency in MODY1 abrogates liver and pancreas differentiation from patient-derived induced pluripotent stem cells. ISCIENCE (2019) 16:192–205. doi: 10.1016/j.isci.2019.05.032
95. Bjørkhaug L, Bratland A, Njølstad PR, Molven A. Functional dissection of the HNF-1alpha transcription factor: a study on nuclear localization and transcriptional activation. DNA Cell Bio (2005) 24(11):661–9. doi: 10.1089/DNA.2005.24.661
96. Wang H. Dominant-negative suppression of HNF-1alpha function results in defective insulin gene transcription and impaired metabolism-secretion coupling in a pancreatic beta -cell line. EMBO J (1998) 17(22):6701–13. doi: 10.1093/emboj/17.22.6701
97. Gerrish K, Cissell MA, Stein R. The role of hepatic nuclear factor 1α and PDX-1 in transcriptional regulation of the pdx-1 gene. J Biol Chem (2001) 276(51):47775–84. doi: 10.1074/jbc.M109244200
98. Fukui K, Yang Q, Cao Y, Takahashi N, Hatakeyama H, Wang H, et al. The HNF-1 target collectrin controls insulin exocytosis by SNARE complex formation. Cell Metab (2005) 2(6):373–84. doi: 10.1016/j.cmet.2005.11.003
99. Low BSJ, Lim CS, Ding SSL, Tan YS, Ng NHJ, Krishnan VG, et al. Decreased GLUT2 and glucose uptake contribute to insulin secretion defects in MODY3/HNF1A hiPSC-derived mutant β cells. Nat Commun (2021) 12(1):1–20. doi: 10.1038/s41467-021-22843-4
100. Çubuk H, Yalçın Çapan Ö. A review of functional characterization of single amino acid change mutations in HNF transcription factors in MODY pathogenesis. Protein J (2021) 40(3):348–60. doi: 10.1007/s10930-021-09991-8
101. Bellanné-Chantelot C, Carette C, Riveline J, Valéro R, Gautier J, Larger E, et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes (2008) 57(2):503–8. doi: 10.2337/db07-0859
102. Chi YI, Frantz JD, Oh B, Hansen L, Dhe-Paganon S, Shoelson SE. Diabetes mutations delineate an atypical POU domain in HNF-1α. Mol Cell (2002) 10(5):1129–37. doi: 10.1016/S1097-2765(02)00704-9
103. Østoft SH, Bagger JI, Hansen T, Pedersen OFaber J, Holst JJ, et al. Glucose-lowering effects and low risk of hypoglycemia in patients with maturity-onset diabetes of the young when treated with a GLP-1 receptor agonist: a double-blind, randomized, crossover trial. Diabetes Care (2014) 37(7):1797–805. doi: 10.2337/dc13-3007
104. Christensen AS, Hædersdal S, Støy J, Storgaard HKampmann U, Forman JL, et al. Efficacy and safety of glimepiride with or without linagliptin treatment in patients with hnf1a diabetes (Maturity-onset diabetes of the young type 3): a randomized, double-blinded, placebo-controlled, crossover trial (glimlina). Diabetes Care (2020) 43(9):2025–33. doi: 10.2337/dc20-0408
105. Haliyur R, Tong X, Sanyoura M, Shrestha S, Lindner J, Saunders DC, et al. Human islets expressing HNF1A variant have defective β cell transcriptional regulatory networks. J Clin Invest (2019) 129(1):246–51. doi: 10.1172/JCI121994
106. Yoshiji S, Horikawa Y, Kubota S, Enya M, Iwasaki Y, Keidai Y, et al. First Japanese family with PDX1-MODY (MODY4): a novel PDX1 frameshift mutation, clinical characteristics, and implications. J Endocrine Soc (2022) 6(1):bvab159. doi: 10.1210/jendso/bvab159
107. Fajans SS, Bell GI, Paz VP, Below JE, Cox NJ, Martin C, et al. Obesity and hyperinsulinemia in a family with pancreatic agenesis and MODY caused by the IPF1 mutation Pro63fsX60. Trans research : J Lab Clin Med (2010) 156(1):7–14. doi: 10.1016/j.trsl.2010.03.003
108. De Franco E, Shaw-Smith C, Flanagan SE, Edghill EL, Wolf J, Otte V, et al. Biallelic PDX1 (insulin promoter factor 1) mutations causing neonatal diabetes without exocrine pancreatic insufficiency. Diabetic Med (2013) 30(5):197–200. doi: 10.1111/dme.12122
109. McKinnon CM, Docherty K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia (2001) 44(10):1203–14. doi: 10.1007/s001250100628
110. Wang X, Sterr M, Burtscher I, Chen S, Hieronimus A, Machicao F, et al. Genome-wide analysis of PDX1 target genes in human pancreatic progenitors. Mol Metab (2018) 9:57–68. doi: 10.1016/j.molmet.2018.01.011
111. Caetano LA, Santana LS, Costa-Riquetto AD, Lerario AM, Nery M, Nogueira GF, et al. PDX1 -MODY and dorsal pancreatic agenesis: new phenotype of a rare disease. Clin Genet (2018) 93(2):382–6. doi: 10.1111/cge.13044
112. Park G-M, Lee SJ, Seo JY, Lim KI. A case of maturity-onset diabetes of the young type 4 in Korea. Ann Pediatr Endocrinol Metab (2022). doi: 10.6065/apem.2142188.094
113. Andrali SS, Sampley ML, Vanderford NL, Ozcan S. Glucose regulation of insulin gene expression in pancreatic β-cells. Biochem J (2008) 415(1):1–10. doi: 10.1042/BJ20081029
114. Deng M, Xiao X, Zhou L, Wang T. First case report of maturity-onset diabetes of the young type 4 pedigree in a Chinese family. Front Endocrinol (2019) 10:406(JULY). doi: 10.3389/fendo.2019.00406
115. Hani EH, Stoffers DA, Chèvre JC, Durand E, Stanojevic V, Dina C, et al. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J Clin Invest (1999) 104(9):R41–8. doi: 10.1172/JCI7469
116. An R, Xavier GS, Semplici F, Vakhshouri S, Hao H, Rutter J, et al. Pancreatic and duodenal homeobox 1 (PDX1) phosphorylation at serine-269 is HIPK2-dependent and affects PDX1 subnuclear localization. Biochem Biophys Res Commun (2010) 399(2):155–61. doi: 10.1016/j.bbrc.2010.07.035
117. Lu P, Geun BR, Chi YI. Structural basis of disease-causing mutations in hepatocyte nuclear factor 1β. Biochemistry (2007) 46(43):12071–80. doi: 10.1021/bi7010527
118. Chen YZ, Gao Q, Zhao X, Chen Y, Bennett CL, Xiong X, et al. Systematic review of TCF2 anomalies in renal cysts and diabetes syndrome/maturity onset diabetes of the young type 5. Chin Med J (2010) 123(22):3326–33. doi: 10.3760/cma.j.issn.0366-6999.2010.22.029
119. Harries LW, Ellard S, Jones RWA, Hattersley AT, Bingham C. Abnormal splicing of hepatocyte nuclear factor-1 beta in the renal cysts and diabetes syndrome. Diabetologia (2004) 47(5):937–42. doi: 10.1007/s00125-004-1383-x
120. De Vas MG, Kopp JL, Heliot C, Sander M, Cereghini S, Haumaitre C. Hnf1b controls pancreas morphogenesis and the generation of Ngn3+ endocrine progenitors. Dev (Cambridge) (2015) 142(5):871–82. doi: 10.1242/dev.110759
121. Maestro MA, Boj SF, Luco RF, Pierreux CE, Cabedo J, Servitja JM, et al. Hnf6 and Tcf2 (MODY5) are linked in a gene network operating in a precursor cell domain of the embryonic pancreas. Hum Mol Genet (2003) 12(24):3307–14. doi: 10.1093/hmg/ddg355
122. Wang L, Coffinier C, Thomas MK, Gresh L, Eddu G, Manor T, et al. Selective deletion of the Hnf1β (MODY5) gene in β-cells leads to altered gene expression and defective insulin release. Endocrinology (2004) 145(8):3941–9. doi: 10.1210/en.2004-0281
123. Pearson ER, Badman MK, Lockwood CR, Clark PM, Ellard S, Bingham C, et al. Contrasting diabetes phenotypes associated with hepatocyte nuclear factor-1α and -1β mutations. Diabetes Care (2004) 27(5):1102–7. doi: 10.2337/diacare.27.5.1102
124. Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet (1999) 23(3):323–8. doi: 10.1038/15500
125. Horikawa Y, Enya M. Genetic dissection and clinical features of MODY6 (NEUROD1-MODY). Curr Diabetes Rep (2019) 19(3):12. doi: 10.1007/s11892-019-1130-9
126. de Santana LS, Caetano LA, Costa-Riquetto AD, Franco PC, Dotto RP, Reis AF, et al. Targeted sequencing identifies novel variants in common and rare MODY genes. Mol Genet Genomic Med (2019) 7(12):e962. doi: 10.1002/mgg3.962
127. Ming-Qiang Z, Yang-Li D, Ke H, Wei W, Jun-Fen F, Chao-Chun Z, et al. Maturity onset diabetes of the young (MODY) in Chinese children: genes and clinical phenotypes. J Pediatr Endocrinol Metab (2019) 32(7):759–65. doi: 10.1515/JPEM-2018-0446/MACHINEREADABLECITATION/RIS
128. Brodosi L, Baracco B, Mantovani V, Pironi L. NEUROD1 mutation in an Italian patient with maturity onset diabetes of the young 6: a case report. BMC Endocrine Disord (2021) 21(1):202. doi: 10.1186/s12902-021-00864-w
129. Lezzi M, Aloi C, Salina A, Fragola M, Bassi M, Strati MF, et al. Diabetes mellitus diagnosed in childhood and adolescence with negative autoimmunity: results of genetic investigation. Front Endocrinol (2022) 13:894878. doi: 10.3389/fendo.2022.894878
130. Demirbilek H, Hatipoglu N, Gul U, Tatli ZU, Ellard S, Flanagan SE, et al. Permanent neonatal diabetes mellitus and neurological abnormalities due to a novel homozygous missense mutation in NEUROD1. Pediatr Diabetes (2018) 19(5):898–904. doi: 10.1111/pedi.12669
131. Qiu Y, Sharma A, Stein R. p300 mediates transcriptional stimulation by the basic helix-Loop-Helix activators of the insulin gene. Mol Cell Biol (1998) 18(5):2957–64. doi: 10.1128/mcb.18.5.2957
132. Chapla A, Mruthyunjaya MD, Asha HS, Varghese D, Varshney M, Vasan SK, et al. Maturity onset diabetes of the young in India - a distinctive mutation pattern identified through targeted next-generation sequencing. Clin Endocrinol (2015) 82(4):533–42. doi: 10.1111/cen.12541
133. Aftab S, Semenec L, Chu JS, Chen N. Identification and characterization of novel human tissue-specific RFX transcription factors. BMC Evolutionary Biol (2008) 8(1):226. doi: 10.1186/1471-2148-8-226
134. Mitchell J, Punthakee Z, Lo B, Bernard C, Chong K, Newman C, et al. Neonatal diabetes, with hypoplastic pancreas, intestinal atresia and gall bladder hypoplasia: search for the aetiology of a new autosomal recessive syndrome. Diabetologia (2004) 47(12):2160–7. doi: 10.1007/s00125-004-1576-3
135. Patel KA, Kettunen J, Laakso M, Stančáková A, Laver TW, Colclough K, et al. Heterozygous RFX6 protein truncating variants are associated with MODY with reduced penetrance. Nat Commun (2017) 8(1):888. doi: 10.1038/S41467-017-00895-9
136. Trott J, Alpagu Y, Tan EK, Shboul M, Dawood Y, Elsy M, et al. Mitchell-Riley Syndrome iPSCs exhibit reduced pancreatic endoderm differentiation due to a mutation in RFX6. Dev (Cambridge) (2020) 147(21):dev194878. doi: 10.1242/dev.194878
137. Piccand J, Strasser P, Hodson DJ, Meunier A, Ye T, Keime C, et al. Rfx6 maintains the functional identity of adult pancreatic β cells. Cell Rep (2014) 9(6):2219–32. doi: 10.1016/j.celrep.2014.11.033
138. Artuso R, Provenzano A, Mazzinghi B, Giunti L, Palazzo V, Andreucci E, et al. Therapeutic implications of novel mutations of the RFX6 gene associated with early-onset diabetes. Pharmacogenomics J (2015) 15(1):49–54. doi: 10.1038/tpj.2014.37
139. Hui DY, Howles PN. Carboxyl ester lipase: structure-function relationship and physiological role in lipoprotein metabolism and atherosclerosis. J Lipid Res (2002) 43(12):2017–30. doi: 10.1194/jlr.R200013-JLR200
140. Gravdal A, Xiao X, Cnop M, Jellas KE, Johansson S, Njølstad PR, et al. The position of single-base deletions in the VNTR sequence of the carboxyl ester lipase (CEL) gene determines proteotoxicity. J Biol Chem (2021) 296:100661. doi: 10.1016/j.jbc.2021.100661
141. Kondoh T, Nakajima Y, Yokoi K, Matsumoto Y, Inagaki H, Kato T, et al. Identification of a novel mutation in carboxyl ester lipase gene in a patient with MODY-like diabetes. Tohoku J Exp Med (2022) 256(1):37–41. doi: 10.1620/tjem.256.37
142. Johansson BB, Torsvik J, Bjørkhaug L, Vesterhus M, Ragvin A, Tjora E, et al. Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene-maturity onset diabetes of the young (CEL-MODY): a protein misfolding disease. J Biol Chem (2011) 286(40):34593–605. doi: 10.1074/jbc.M111.222679
143. Torsvik J, Johansson BB, Dalva M, Marie M, Fjeld K, Johansson S, et al. Endocytosis of secreted carboxyl ester lipase in a syndrome of diabetes and pancreatic exocrine dysfunction. J Biol Chem (2014) 289(42):29097–111. doi: 10.1074/jbc.M114.574244
144. Xiao X, Jones G, Sevilla WA, Stolz DB, Magee KE, Haughney M, et al. A carboxyl ester lipase (CEL) mutant causes chronic pancreatitis by forming intracellular aggregates that activate apoptosis. J Biol Chem (2016) 291(44):23224–36. doi: 10.1074/jbc.M116.734384
145. Kahraman S, Dirice E, Basile G, Diegisser D, Alam J, Johansson BB, et al. Abnormal exocrine–endocrine cell cross-talk promotes β-cell dysfunction and loss in MODY8. Nat Metab (2022) 4(1):76–89. doi: 10.1038/s42255-021-00516-2
146. Gurlo T, Ryazantsev S, Huang C, Yeh MW, Reber HA, Hines OJ, et al. Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am J Pathol (2010) 176(2):861–9. doi: 10.2353/ajpath.2010.090532
147. Stoffel M, Patel P, Lo YM, Hattersley AT, Lucassen AM, Page R, et al. Missense glucokinase mutation in maturity-onset diabetes of the young and mutation screening in late-onset diabetes. Nat Genet (1992) 2(2):153–6. doi: 10.1038/ng1092-153
Keywords: MODY, maturity onset diabetes of the young, glucokinase, channelopathy, unfolded protein response, transcription factors, carboxy ester lipase
Citation: Samadli S, Zhou Q, Zheng B, Gu W and Zhang A (2023) From glucose sensing to exocytosis: takes from maturity onset diabetes of the young. Front. Endocrinol. 14:1188301. doi: 10.3389/fendo.2023.1188301
Received: 17 March 2023; Accepted: 28 April 2023;
Published: 15 May 2023.
Edited by:
Manami Hara, The University of Chicago, United StatesReviewed by:
Meihang Li, McGill University Health Centre, CanadaTiago Santos, Centro Hospitalar Universitário do Porto (CHUP), Portugal
Copyright © 2023 Samadli, Zhou, Zheng, Gu and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aihua Zhang, emhhaWh1YUBuam11LmVkdS5jbg==; Bixia Zheng, Yml4aWEuemhlbmdAbmptdS5lZHUuY24=
†These authors share first authorship