 Hao Yu1,2†
Hao Yu1,2† Yuepeng Wang1,2,3†
Yuepeng Wang1,2,3† Yijun Li1Jin Du1Qinghua Guo1
Yijun Li1Jin Du1Qinghua Guo1 Weijun Gu1
Weijun Gu1 Zhaohui Lyu1
Zhaohui Lyu1 Jingtao Dou1
Jingtao Dou1 Yiming Mu1*
Yiming Mu1* Li Zang1*
Li Zang1*- 1Department of Endocrinology, The First Medical Center of Chinese PLA General Hospital, Beijing, China
- 2Department of Endocrinology, General Hospital of Northern Theater Command, Shenyang, China
- 3School of Medicine, Nankai University, Tianjin, China
Background: Castleman Disease (CD) is a group of diseases with characteristic lymph node histopathology, characterized by marked enlargement of deep or superficial lymph nodes. Adrenal CD is rarely reported, and an accurate preoperative diagnosis of adrenal CD is difficult.
Method: We report four cases of CD in the adrenal gland confirmed by pathology and review the characteristics of this rare disease, highlighting the necessity of diagnostic evaluation and follow-up of the patients.
Results: All of the patients sought medical advice because of adrenal incidentalomas. No significant abnormalities were presented in the biochemistry or endocrine systems. The imaging suggested a moderate-to-large mass with uneven moderate contrast enhancement of the adrenal region, similar to a pheochromocytoma. All cases were misdiagnosed as pheochromocytomas before operation and finally confirmed by histopathology. Three cases were pathologically diagnosed as hyaline vascular CD, and one case was diagnosed as plasma cell CD. All the patients are alive without recurrence after a median follow-up of 8 years.
Conclusion: The adrenal CD should be considered after excluding pheochromocytoma and malignancy in the adrenal region. The long-term prognosis of patients with complete resection of the mass is excellent.
Introduction
Castleman Disease (CD), also named Giant Lymphnode Hyperplasia, is a rare lymphoproliferative disease that can divide into hyaline vascular (HV), plasma cell (PC), and mixed variants on the basis of histological classification. The etiology of CD is still unclear and is associated with the monoclonal proliferation process (1) and increased interleukin-6 (IL-6) (2). A recent study revealed no clear associations between UCD and the activity of HHV-8 or HIV infection (3). The CD was divided into unicentric CD (UCD) and multicentric CD (MCD) according to single or multiple lymph node involvement (4, 5). It can involve all lymph nodes, typically in the mediastinum, cervical, retroperitoneal, and axillary areas, and around 4% of UCD occurs in the adrenal region (6–8). The clinical symptoms of CD are related to the number and location of the affected lymph nodes. The first manifestation is usually the discovery of a slow-growing tumor or systemic symptoms such as fever and weight loss, and the duration of systemic symptoms or lymph node enlargement varies from several weeks to several months. Adrenal UCD is rare and is often misdiagnosed as pheochromocytoma or adrenocortical carcinoma before operation due to its large size and lack of specificity in symptoms and laboratory examinations (9). CD is considered benign in its localized form but aggressive in the multicentric type (10). Because of the variability of clinical presentation, the selection of the appropriate therapeutic approach remains unclear. In the article, we present four cases of adrenal UCD and discuss the clinical manifestations, laboratory results, imaging and pathological features, management, and follow-up of this rare disorder.
Materials and methods
We performed a retrospective analysis of the clinical, biochemical, imaging, and pathological characteristics of four patients with a single focus on adrenal CD who were managed with surgical resection. Patients were diagnosed at the Chinese PLA General Hospital from September 2008 to February 2022. Each patient underwent surgery, and CD was then confirmed pathologically after surgery. Patients were followed up to supplement outcome information until December 1st, 2022. Our study was approved by the Ethics Committee of the First Medical Center of Chinese PLA General Hospital (approval number: S2021-554-01).
Results
Clinical characteristics
Four patients, two females and two males, between 26 and 58 years of age (mean age: 46 years), were diagnosed with adrenal incidentaloma. Two patients were transferred to the Department of Urology for surgery after detailed evaluation in the Department of Endocrinology (cases 1, 2), while two patients attended urology directly for surgery (cases 3, 4). After further consultation, one patient had prodromal symptoms of fever and palpitations (case 3). No one complained of headaches, sweating, abdominal pain, weight loss, or other typical symptoms of secondary hypertension. No patients presented with Cushing appearance. Physical examination revealed a soft abdomen without palpable mass or peripheral lymphadenopathy. One patient was treated with monotherapy for grade 2 hypertension (case 1); Another patient was found to have grade 1 hypertension and was not treated (case 2), and none of the patients had refractory hypertension. All patients denied any family history of cancer. One of the four cases had a family history of hypertension (case 2) (Table 1).
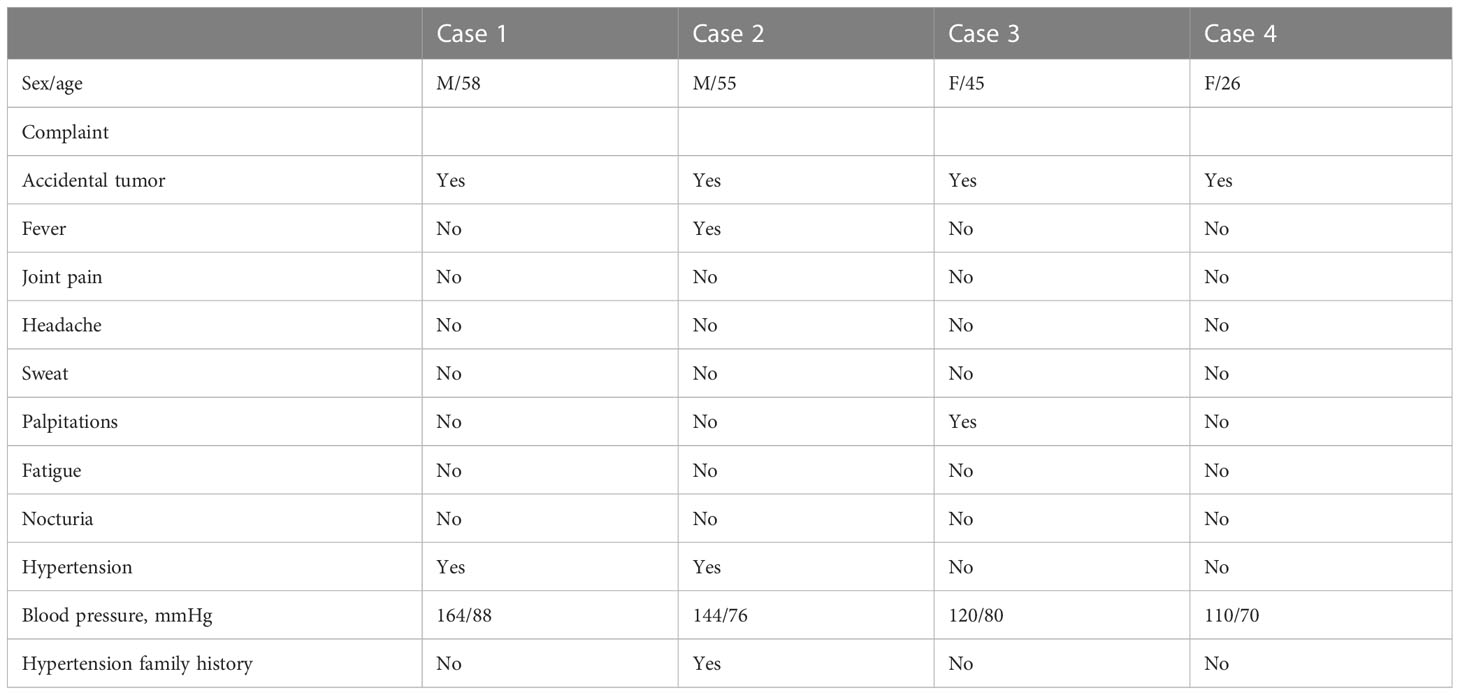
Table 1 Clinical features of four patients with adrenal CD.
Laboratory investigations and endocrine parameters
Serum potassium, sodium, creatinine, and liver function were found to be almost normal in all four patients. One patient had increased urinary potassium excretion in 24 hours, but the serum potassium level was normal (case 2). The human immunodeficiency virus antibody was also negative in all patients. C-reactive protein was normal in cases 1 and 2. The diurnal cortisol rhythms of four patients were basically normal. 24-hour urine vanillin mandelic acid was normal in cases 3 and case 4. Blood dopamine, methoxyepinephrine, methoxynorepinephrine, epinephrine, norepinephrine, and methoxylamine were all in the normal range in case 1 and case 2. The Postural stimulation test showed that case 2 had elevated renin in supine and upright positions, but normal aldosterone levels. 24h urinary aldosterone was normal in all cases (Table 2).
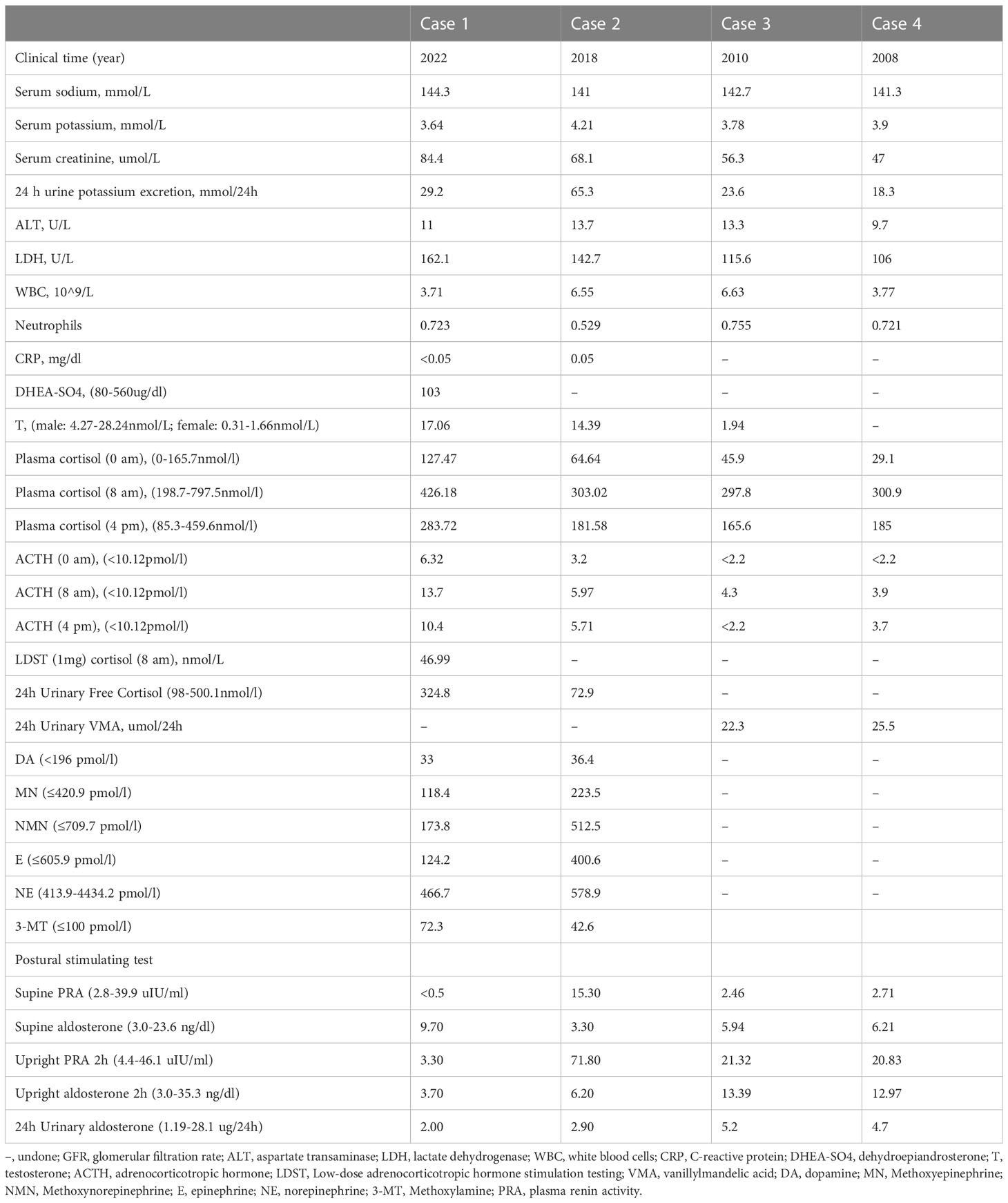
Table 2 Laboratory investigations of four patients with adrenal CD.
Imaging
Tumor size in the four patients determined by either CT or MRI ranged from 87 × 79 × 85 to 46 × 33 × 40 mm. In case 1, abdominal CT showed a solid-cystic mass with heterogeneous wall thickness in the right adrenal gland, and multiple mildly enlarged lymph nodes in the retroperitoneum. The enhanced scan showed marked enhancement of the wall and solid nodules. Positron emission tomography/computed tomography (PET-CT) suggested a cystic mass in the right adrenal gland with elevated metabolism in the solid portion, surrounded by scattered hypermetabolic lymph nodes. The maximum standardized uptake value (SUVmax) was 3.7. Imaging of the mass gave suspicion of malignancy or possibly pheochromocytoma. In case 2, enhanced CT showed a blood-rich mass of 46 × 33 × 40 mm in size with calcification and patchy unreinforced areas in the left adrenal gland with a CT attenuation value of 37.8 HU, which could indicate a pheochromocytoma.
Cases 1-4 shared similar MRI images: tumor size larger than 4 cm3, relatively clear boundary, slightly long T1 signal, and heterogeneous long T2 signal (Figures 1, 2). The lesion of case 1 manifested with a hypo-intensity signal on T1WI inside the tumor, and a hyper-intensity on T2WI characterized a “bulb sign” with the pseudocapsule. DWI images showed a hyper-intensity signal at the margin and internal septum of the lesion. Continuous mild to moderate enhancement of the margin and internal septum of the lesion was observed on the arterial and venous phases of T1WI, with no enhancement in the cystic center (Figure 1). Case 2-4 had similar features on MRI, represented with hypo-intensity signals on T1WI and hyper-intensity signals on T2WI. Enhanced images obtained in the arterial and venous phases showed moderate and uneven enhancement in mass (Figure 2). All the cases were considered to have imaging features of pheochromocytoma before surgery.
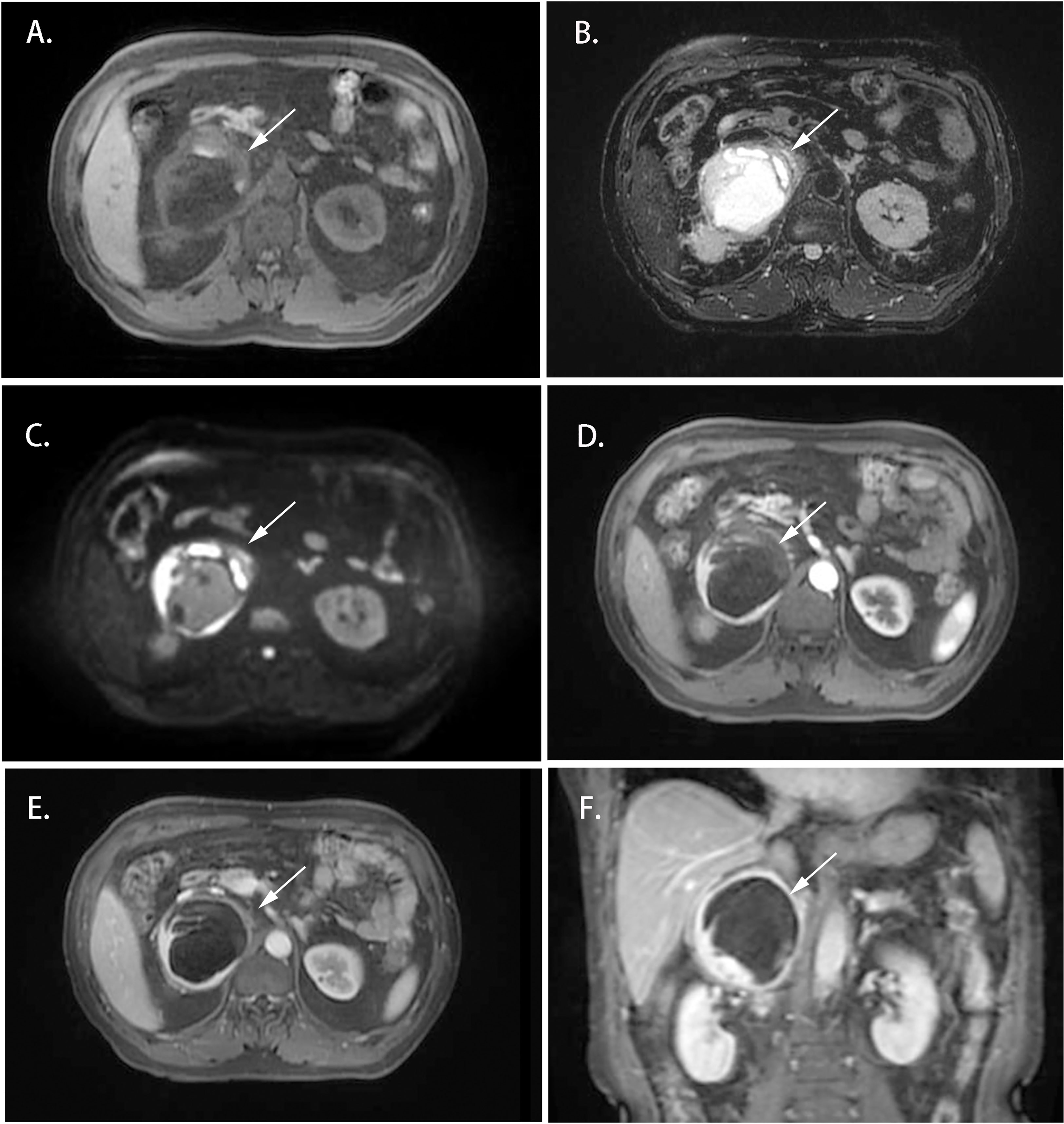
Figure 1 Magnetic resonance imaging of the Adrenal CD of Case 1 (white arrow). (A) Axial T1WI; (B) Axial T2WI; (C) DWI; (D) Enhanced T1WI (cortex phase); (E) Enhanced T1WI (delayed phase); (F) Coronal Enhanced T1WI. MRI scan and dynamic enhancement showed an 87 × 79 × 85 mm mass with partial hyper-intensity on T2WI and mild to moderate heterogeneous enhancement at the edge and septum of the lesion in the right adrenal region.
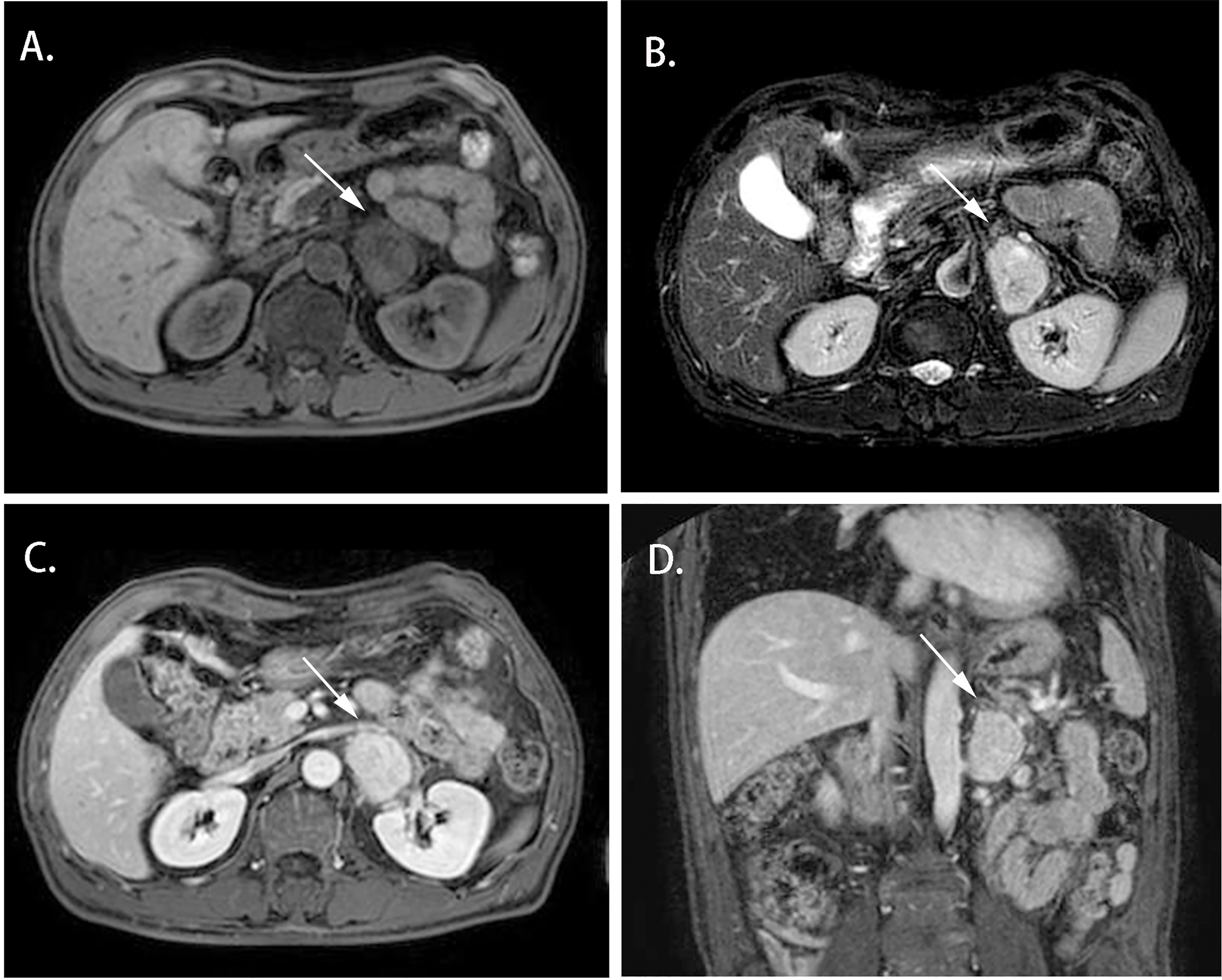
Figure 2 Magnetic resonance imaging of the Adrenal CD of Case 2 (white arrow). (A) Axial T1WI; (B) Axial T2WI; (C) Enhanced T1W1; (D) Coronal Enhanced T1W1. MRI scan and dynamic enhancement showed a 46× 33× 40 mm mass with hypo-intensity signal on T1WI and hyper-intensity on T2WI and with mild to moderate heterogeneous enhancement in the left adrenal region.
Surgery and pathology
All four cases underwent adrenal mass resection. They received α-receptor blockers according to preoperative preparations because pheochromocytoma/paraganglioma could not be ruled out. 2 cases were treated with the Da Vinci robot (cases 1, 2), while the operation was converted to laparotomy because of the large tumor volume and severe adhesion in case 1, and the other 2 cases had laparoscopic surgery (cases 3, 4). All operations were successful without complications. All cases were intra-adrenal. The tumors were all well-circumscribed, firm, and grayish-white to yellowish-brown in color (Table 3). The hematoxylin and eosin (H&E) stain of case 1 presented with adrenal lymphoid hyperplasia, and atretic germinal centers were traversed by sclerotic penetrating vessels and hyalinization, like lollipop follicles. Mantle zones were thickened with lymphocytes arranged in layers. Mantle zones may fuse and contain more than 1 germinal center, and follicular dendritic cells show dysplastic features. Interfollicular areas and medulla contain sheets of small, mature plasma cells, and it showed histological features of a plasma cell type (Figures 3A–C). Immunohistochemical staining revealed that CD138 (plasma cell), CD38 (plasma cell), CD20 (B cell) (Figures 3D–F), CD3 (T cell), Bcl-2, and Ki-67 (germinal centers +90%) of case 1 were positive. No metastases from three retroperitoneal lymph nodes were detected by freezing during the operation. In cases 2-4, histopathology revealed lymphoid follicles with atrophic germinal centers, multiple germinal centers within the same mantle zone, an “onion skin” appearance at the periphery of the follicular, and increased vascularity with hyalinization. Immunohistochemical staining revealed that CD31 and CD34 were positive in cases 2–4. Pathology indicated that case 1 was consistent with plasma cell type, and the other 3 cases were diagnosed as hyaline vascular type (cases 2-4).
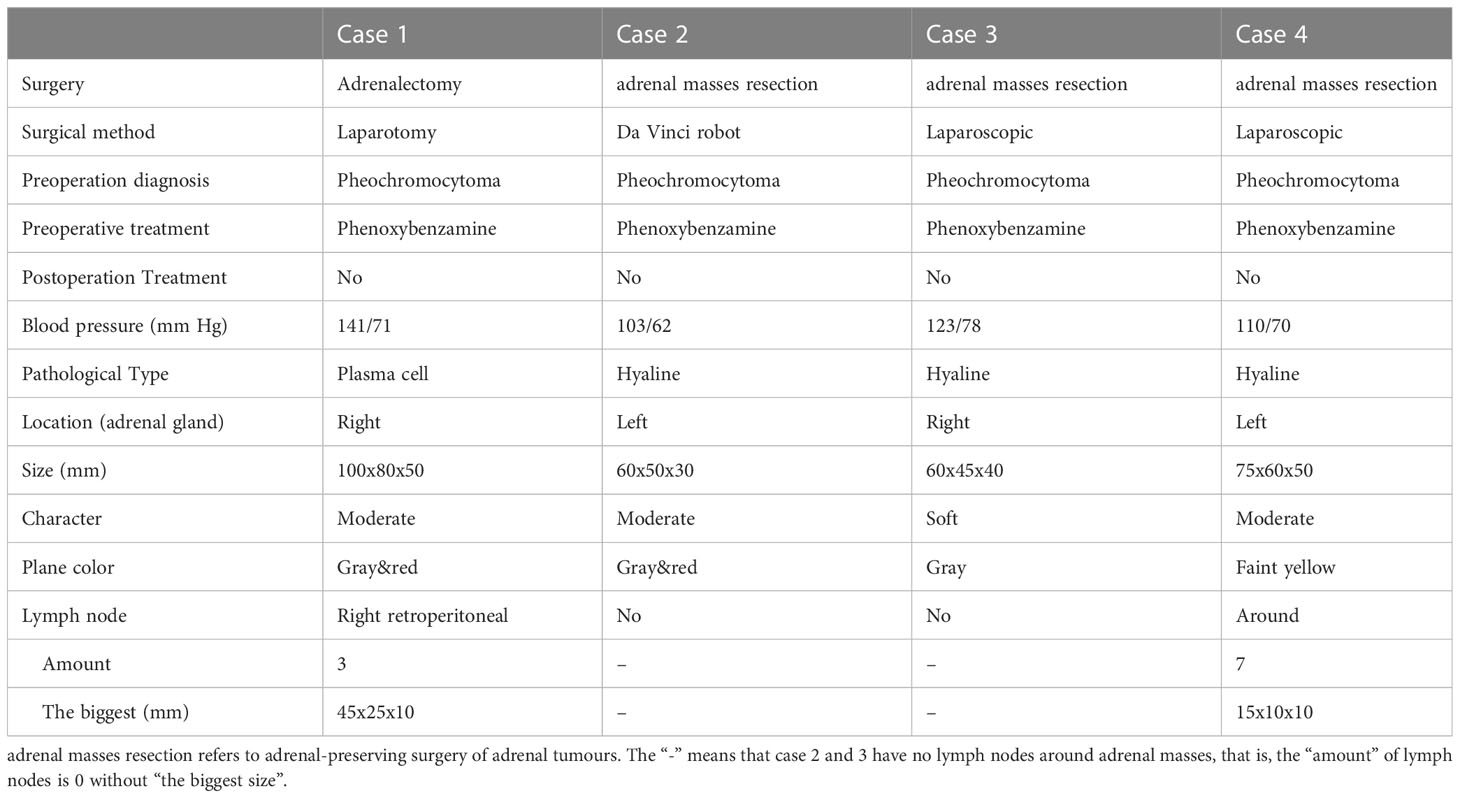
Table 3 Surgery and pathology of four patients with adrenal CD.
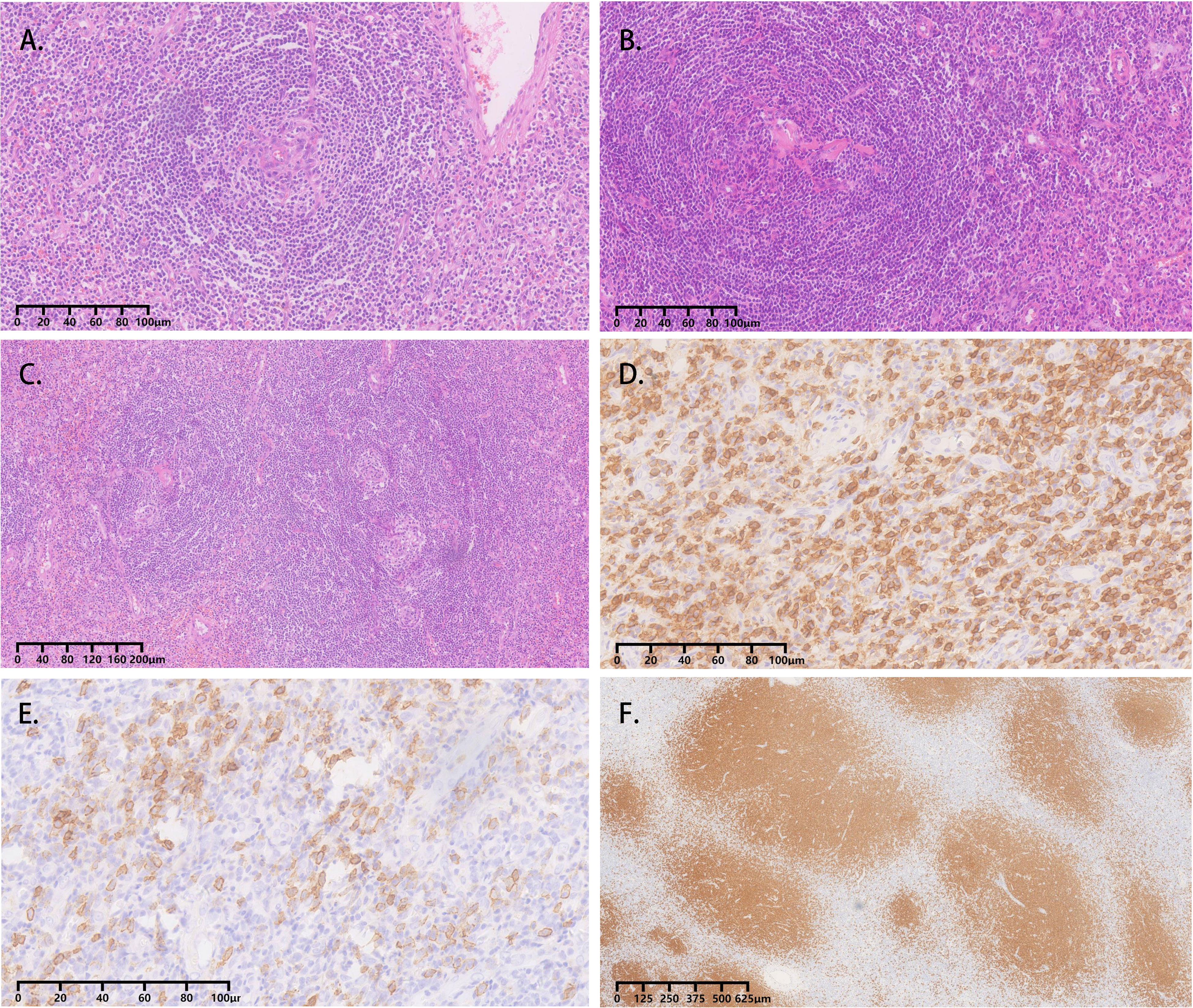
Figure 3 Adrenal CD, plasma cell variant histopathology in case 1. (A-C) Atretic germinal centers were traversed by sclerotic penetrating vessels and hyalinization, like lollipop follicles. Mantle zones were thickened with lymphocytes arranged in layers. Mantle zones may fuse and contain more than one germinal center, and follicular dendritic cells showed dysplastic features. Interfollicular areas and medulla contain sheets of small, mature plasma cells; (D) Clusters of plasma cells in the interfollicular areas were positive for CD138; (E) Immunohistochemical stain for CD38 identifies plasma cells; (F) The CD20 staining showed the expanded mantle zones.
Follow-up
All four cases are alive without relapse after a median follow-up of 8 years, and no chemotherapy or steroid replacement therapy was administered after the operation.
Discussion
Over the past few decades, there has been a significant increase in the use of diagnostic imaging techniques, resulting in a subsequent rise in the incidental discovery of adrenal masses. Nonfunctional adrenocortical adenomas are the most common adrenal incidentalomas, followed by hormone-producing adenomas, and adrenocortical carcinomas (11–13). Adrenal CD is relatively rare, and the features of adrenal CD are less well described. Our case series report illustrated the baseline characteristics and outcomes of adrenal UCD from the Chinese PLA General Hospital, a type of adrenal incidentaloma lacking specific clinical and biochemical features. These patients underwent adrenal mass resection, and indicated a favorable prognosis.
The CD was first described by Benjamin Castleman in 1954 in a group of patients with mediastinal lymph node enlargement (14). Epidemiological studies show that the prevalence of CD is about 21-25 cases per million people, with a median age of onset of 34–40years (6). UCD can involve many sites, commonly in the neck, abdomen, and mediastinum (8, 15). Retroperitoneum is also a common site of UCD, and the number of the adrenal UCD lesions was found to be about 12% in retroperitoneal UCD (7). Patients with adrenal UCD ranged in age from 16 to 70 years with an equal sex ratio and localization (Table 4). Patients with UCD usually have no obvious clinical symptoms or abdominal discomfort, and their conventional biochemical and adrenal function examinations do not show obvious abnormalities (16–25). In other reports, the patients were accompanied by hypertension, weight loss, paraneoplastic pemphigus, and pheochromocytoma (18, 26–30). In a rare case report, hyperandrogenism was found in a female patient with adrenal UCD, and the author attributed this phenomenon to enhanced 5α-reductase activity associated with IL-6 (28). However, adrenal UCD usually has a relatively large volume, which is different from a nonfunctional adrenocortical adenoma (33). In the present study, cases 1–4 had adrenal incidentalomas found on imaging with no local or systemic symptoms.
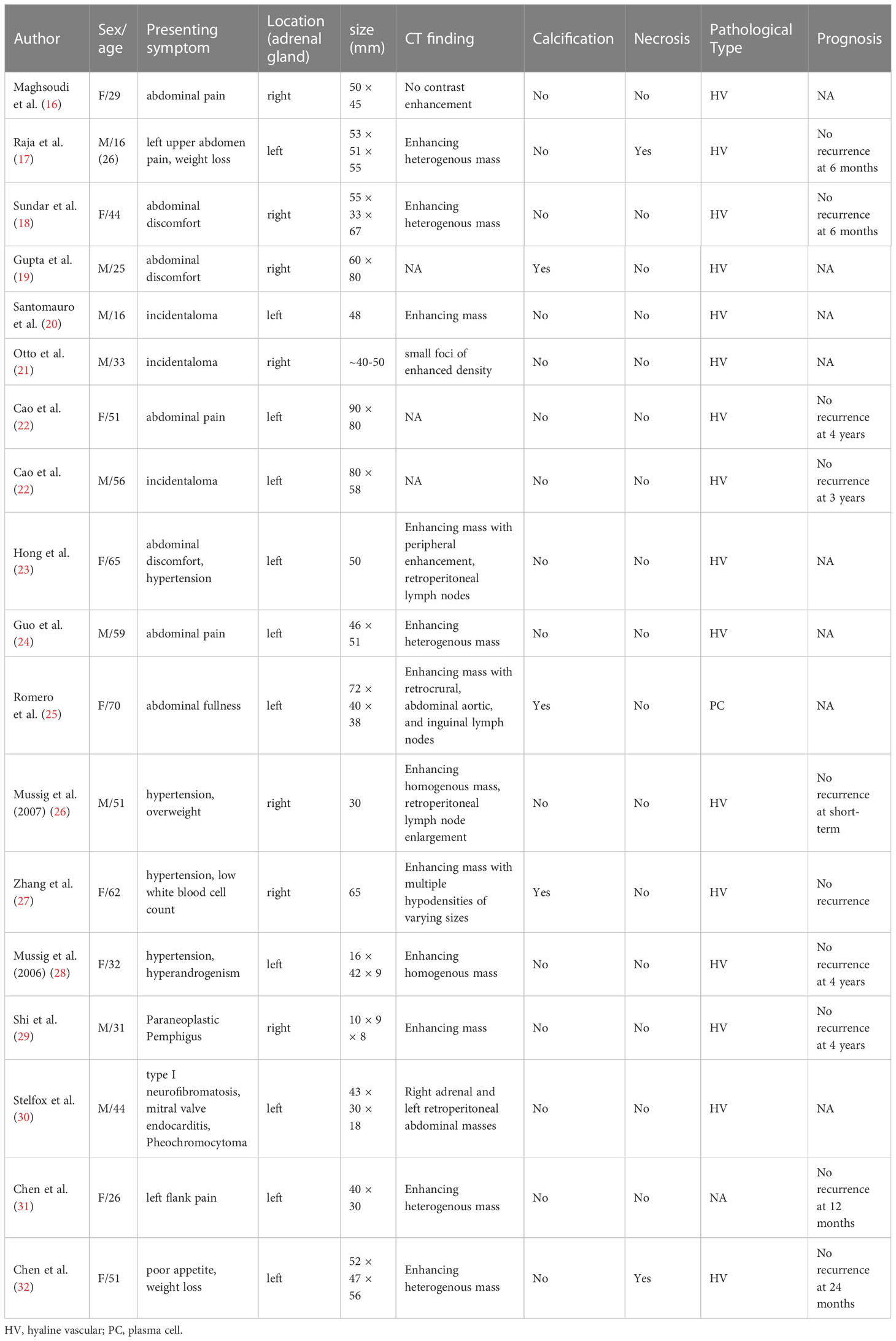
Table 4 Reported cases of adrenal Castleman’s disease.
Large adrenal gland mass usually requires a systematic evaluation of adrenal endocrine workup to assist in diagnosing the mass from the perspective of endocrinology diagnosis and treatment (11). Our data showed that two patients had elevated midnight cortisol and one patient had mildly elevated ACTH, but no obvious abnormalities in aldosterone or catecholamine metabolism was found. These biochemical abnormalities are not representative, and we did not see significant adrenal insufficiency, or excess production of adrenal hormones either, even in the case of a larger adrenal CD. This phenomenon is plausible, given that the cases were all unilateral non-functional adrenal lymphocytic hyperplasia. It is worth noting that hypertension may be present in some patients with adrenal UCD (23, 26–28). This may be attributed to the differential diagnosis of adrenal mass, since adrenal mass is a major cause of secondary hypertension.
Imaging findings of adrenal UCD are related to discriminating features and also with the pathological types. Hyaline vascular adrenal CD usually appears as a soft tissue mass with uniform density and well-defined borders on CT and with uneven density in a few lesions (16–18, 20, 21, 23, 24, 26–32). The plasma cell type adrenal UCD was rare, and had only been reported in 1 previous case, according to our review of the literature (25). Despite the controversy, the plasma cell type UCD is less common, larger in size, and more likely to have necrosis, peripheral enhancement, and the presence of enlarged retroperitoneal lymph nodes than the hyaline vascular type, contributing to malignant morphological features (25, 34–36). In our report, the imaging results in plasma cell type adrenal UCD in case 1 showed a large mass with apparent necrosis and peripheral enhancement. Cases 2-4 were hyaline vascular type adrenal UCD; their mass bodies were relatively smaller and the mass density was relatively uniform, compared with case 1.
As a less common disease, adrenal UCD is easily misdiagnosed. Physicians often need to differentiate adrenal CD from pheochromocytoma, adrenal lymphoma, and cortical carcinoma. Pheochromocytoma patients usually present with sweating, headache, tachycardia, and hypertension (37). However, the presence of silent pheochromocytoma or pheochromocytoma with less typical signs should also be considered, which may also cause adverse cardiovascular events, as well as severe intraoperative blood pressure fluctuations (38). Pheochromocytoma with a rich blood supply presents with a hyper-intensity signal on T2WI, strong enhancement after contrast agent administration, and liquefaction necrosis frequently occurring. Adrenal CD, especially plasma cell variant adrenal UCD, manifested with an isohypointense T1W1 signal and a characteristically hyperintense T2W1 signal and is sometimes accompanied by necrosis or cystification in larger adrenal CD, making it more difficult to distinguish from pheochromocytoma and adrenal cortical carcinoma. Hyperandrogenism and cortisol autonomous hypersecretion with tumor metastasis often occur in adrenocortical carcinoma, which can help distinguish it from adrenal CD. Fever with lymphadenopathy makes it difficult to differentiate adrenal CD from adrenal lymphoma, while the latter often presents with an adrenal mass with uniform density involving bilateral adrenal glands.
PET-CT has a certain diagnostic value in differentiating pheochromocytoma from UCD. A study by Jiang et al. reported the major points of 18F fluorodeoxyglucose (FDG) PET-CT to differentiate paragangliomas from UCD. Paragangliomas usually have higher SUV values, and the SUVmax > 7.75 may be used as a cut-off point to differentiate paragangliomas from UCD. In this study, the SUVmax of adrenal UCD in case 1 was 3.7, which is consistent with Jiang’s findings (9). Plasma free metanephrines or urine fractionated metanephrines also have certain differential diagnosis values, but there are still some pheochromocytomas or paragangliomas that cannot be distinguished by biochemical detection (39). All cases in this report were misdiagnosed as pheochromocytomas with preoperative imaging, and phenoxybenzamine was used for preoperative preparation in all 4 cases. There are a few report of patients with both pheochromocytoma and UCD (30, 40). Therefore, it is prudent to apply alpha-blockers preoperatively to avoid severe intraoperative complications with the omission of pheochromocytoma (9).
A definitive diagnosis of adrenal CD or lymphoma is important and challenging because the preferred therapies for lymphoma are usually not surgical, and CD may be a pre-disease form of lymphoma (35, 41). Adrenal lymphomas are usually large and well-defined masses, and some have bilateral involvement of the adrenal glands. Adrenal lymphomas have homogeneous enhancement and are less likely to have calcification on CT imaging. On a CT scan, the hyaline vascular type of adrenal UCD appears more similar to adrenal lymphoma than the plasma cell type of adrenal UCD. However, it is possible to differentiate between hyaline vascular type adrenal UCD and lymphoma by observing the presence of calcification and heterogeneous enhancement in the former (19, 25, 27, 35). In our case series, one case (case 2) was found to have a blood-rich occupancy with calcification, but the high SUVmax values in PET-CT raised the suspicion of lymphoma. Patients with adrenal lymphoma often present with systemic symptoms, including back pain, fever, night sweats, and weight loss. Bilateral adrenal involvement may also lead to adrenal insufficiency in affected individuals. Higher LDH and β2-microglobulin levels in the biochemistry tests imply the diagnosis of adrenal lymphoma; in contrast, the adrenal CD is usually unilateral with no definitive biochemical abnormalities (42, 43). An adrenal puncture biopsy can be considered in cases where a definitive diagnosis cannot be made based on imaging and clinical features (25, 44). Further accumulation of clinical and imaging features of plasma cell type adrenal UCD is required.
Surgical resection is the primary treatment option for UCD (8), and the prognosis is usually prospective if the lesion can be completely removed irrespective of the pathology, clinical presentation, and highly expressed Ki67 index (17, 18, 22, 26–29, 31, 32, 45). Anti-IL-6 antibody therapy or radiotherapy can be considered for those unresectable UCD masses because of their size or location (46). Talat et al. summarized 278 cases of UCD and found that the 3-year and 5-year postoperative progression-free survival rates were 89.7% and 81.2%, respectively, and the 10-year overall survival rate was 95.3% (8). Similar results also been reported in research by Zhang et al. (47). However, an increased risk of paraneoplastic pemphigus, bronchiolitis obliterans, AA amyloidosis, and lymphoma has been reported in CD patients (35, 48, 49), suggesting that adrenal CD patients with persisting constitutional symptoms still need further follow-up despite complete excision.
Conclusions
Adrenal Castleman disease is a proliferative disease of the lymphoid tissue occurred in the adrenal region and is difficult to distinguish from pheochromocytoma and lymphoma on imaging. MRI, 18F PET-CT, and adrenal function assessment are helpful in the diagnosis of adrenal UCD. Surgery is the mainstay of treatment, and the prognosis is usually promising.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Ethics statement
The studies involving human participants were reviewed and approved by the Ethics Committee of the First Medical Center of Chinese PLA General Hospital. The patients provided their written informed consent to participate in this study.
Author contributions
Conceptualization and writing: HY, YW, and LZ. Review and editing: YM, and LZ. Instrumental in revising the manuscript: YL, JD, QG, WG, ZL, and JTD. All authors contributed to the article and approved the submitted version.
Funding
This research did not receive any specific grant from any funding agency in the public, commercial or not-for-profit sector.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chang K-C, Wang Y-C, Hung L-Y, Huang W-T, Tsou J-H, M Jones D, et al. Monoclonality and cytogenetic abnormalities in hyaline vascular castleman disease. Mod Pathol an Off J United States Can Acad Pathol Inc (2014) 27:823–31. doi: 10.1038/modpathol.2013.202
2. Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, et al. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative castleman’s disease. Am J Pathol (1997) 151:1517–22.
3. Nabel CS, Sameroff S, Shilling D, Alapat D, Ruth JR, Kawano M, et al. Virome capture sequencing does not identify active viral infection in unicentric and idiopathic multicentric castleman disease. PloS One (2019) 14:e0218660. doi: 10.1371/journal.pone.0218660
4. Castleman B, Iverson L, Menendez VP.Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer (1956) 9:822–30. doi: 10.1002/1097-0142(195607/08)9:4<822::aid-cncr2820090430>3.0.co;2-4
5. Gaba AR, Stein RS, Sweet DL, Variakojis D. Multicentric giant lymph node hyperplasia. Am J Clin Pathol (1978) 69:86–90. doi: 10.1093/ajcp/69.1.86
6. Wong RSM. Unicentric castleman disease. Hematol Oncol Clin North Am (2018) 32:65–73. doi: 10.1016/j.hoc.2017.09.006
7. Wang W, Dong D, Wen J, Li H. A 10-year observational single-center study of retroperitoneal unicentric castleman disease. Med (Baltimore) (2021) 100:e25088. doi: 10.1097/MD.0000000000025088
8. Talat N, Belgaumkar AP, Schulte K-M. Surgery in castleman’s disease: a systematic review of 404 published cases. Ann Surg (2012) 255:677–84. doi: 10.1097/SLA.0b013e318249dcdc
9. Jiang Y, Hou G, Zhu Z, Huo L, Cheng W, Li F. The value of multiparameter (18)F-FDG PET/CT imaging in differentiating retroperitoneal paragangliomas from unicentric castleman disease. Sci Rep (2020) 10:12887. doi: 10.1038/s41598-020-69854-7
10. Dispenzieri A, Fajgenbaum DC. Overview of castleman disease. Blood (2020) 135:1353–64. doi: 10.1182/blood.2019000931
11. Fassnacht M, Arlt W, Bancos I, Dralle H, Newell-Price J, Sahdev A, et al. Management of adrenal incidentalomas: European society of endocrinology clinical practice guideline in collaboration with the European network for the study of adrenal tumors. Eur J Endocrinol (2016) 175:G1–G34. doi: 10.1530/EJE-16-0467
12. Jing Y, Hu J, Luo R, Mao Y, Luo Z, Zhang M, et al. Prevalence and characteristics of adrenal tumors in an unselected screening Population: a cross-sectional study. Ann Intern Med (2022) 175:1383–91. doi: 10.7326/M22-1619
13. Patrova J, Jarocka I, Wahrenberg H, Falhammar H. CLINICAL OUTCOMES IN ADRENAL INCIDENTALOMA: EXPERIENCE FROM ONE CENTER. Endocr Pract Off J Am Coll Endocrinol Am Assoc Clin Endocrinol (2015) 21:870–7. doi: 10.4158/EP15618.OR
14. Castleman B., Towne V. CASE records of the Massachusetts general hospital weekly clinicopathological exercises: case 40011. N Engl J Med (1954) 250:26–30. doi: 10.1056/NEJM195401072500107
15. Wang X-Q, Zhong N-N, Sun Q, Yan S-C, Xu G-C, Wang Y-G, et al. Comprehensive analysis of 65 patients with castleman disease in a single center in China. Sci Rep (2022) 12:8694. doi: 10.1038/s41598-022-12797-y
16. Maghsoudi R, Shakiba B, Panahi M, Moradi A. Castleman disease mimicking an adrenal tumor: a case report. Urol Case Rep (2022) 40:101876. doi: 10.1016/j.eucr.2021.101876
17. Raja A, Malik K, Mahalingam S. Adrenal castleman’s disease: case report and review of literature. J Indian Assoc Pediatr Surg (2022) 27:109–11. doi: 10.4103/jiaps.JIAPS_282_20
18. Sundar P, Bijalwan P, Pooleri GK. Castleman’s disease: a suprarenal surprise! Indian J Surg Oncol (2018) 9:254–5. doi: 10.1007/s13193-018-0737-7
19. Gupta NP, Ansari MS, Chopra P, Dinda AK. Castleman’s disease masquerading as an adrenal tumor. J Urol (2002) 168:2524. doi: 10.1016/S0022-5347(05)64182-1
20. Santomauro M, Choe C, Heimbigner J, Roberts J, Auge B. Castleman’s disease in the left suprarenal region, mimicking an adrenal neoplasm. Urology (2011) 78:319. doi: 10.1016/j.urology.2010.12.031
21. Otto M, Wieprzowski L, Dzwonkowski J, Ziarkiewicz-Wróblewska B. Castleman’s disease - an unusual indication for laparoscopic adrenalectomy. Wideochirurgia i inne Tech maloinwazyjne = Videosurgery other miniinvasive Tech (2012) 7:50–4. doi: 10.5114/wiitm.2011.25620
22. Cao D-H, Liu L-R, Xu R-H, Jiang D, Lv X, Yuan H-C, et al. Retroperitoneal laparoscopic management method of castleman’s disease in the adrenal gland: two cases report and review of the literature. Int J Clin Exp Med (2014) 7:5909–12.
23. Hong SB, Lee NK, Kim S, Han GJ, Ha HK, Ku JY, et al. Adrenal castleman’s disease mimicking other adrenal neoplasms: a case report. J Korean Soc Radiol (2017) 76:73–7. doi: 10.3348/jksr.2017.76.1.73
24. Guo L, Sang Z, Du J, Zhang Q, Meng H, Shi B, et al. A case report of adrenal castleman’s disease. SN Compr Clin Med (2019) 1:479–82. doi: 10.1007/s42399-019-00069-2
25. Mindiola-Romero AE, Seigne JD, Ornstein DL. Plasma cell variant of castleman lymphadenopathy presenting as an adrenal mass. Urol Case Rep (2021) 36:101583. doi: 10.1016/j.eucr.2021.101583
26. Müssig K, Horger M, Wehrmann M. Adrenal castleman’s disease. Ann Hematol (2007) 86:63–5. doi: 10.1007/s00277-006-0200-7
27. Zhang J, Song N, Liu B, Hua L, Wang Z, Yin C, et al. A case report of unusual retroperitoneal castleman’s disease in an old woman. Urol Int (2012) 89:369–72. doi: 10.1159/000341691
28. Müssig K, Gallwitz B, Machicao F, Horger M, Häring H-U, Kaiserling E. Paraadrenal castleman disease presenting with adrenal hyperandrogenism. J Endocrinol Invest (2006) 29:172–6. doi: 10.1007/BF03344093
29. Shi B, Li H, Zhao L, Sun Q, Fan H, Li H, et al. Paraneoplastic pemphigus caused by castleman’s disease masquerading as an adrenal neoplasm. J Clin Endocrinol Metab (2009) 94:1841–2. doi: 10.1210/jc.2009-0100
30. Stelfox HT, Stewart AK, Bailey D, Harrison D. Castleman’s disease in a 44-year-old male with neurofibromatosis and pheochromocytoma. Leuk Lymphoma (1997) 27:551–6. doi: 10.3109/10428199709058324
31. Chen J, Yang C, Liang C-Z. Detection of a unicentric type of castleman-like mass at the site of adrenal grand: a case report and review of literature. World J Clin cases (2018) 6:683–7. doi: 10.12998/wjcc.v6.i13.683
32. Chen J-H, Yu C-Y, Pai C-Y, Chan D-C, Chen C-J, Yu J-C, et al. Castleman’s disease in the left upper retroperitoneal space mimicking an adrenal neoplasm: report of a case and literature review. Jpn J Clin Oncol (2005) 35:353–6. doi: 10.1093/jjco/hyi092
33. Elhassan YS, Alahdab F, Prete A, Delivanis DA, Khanna A, Prokop L, et al. Natural history of adrenal incidentalomas with and without mild autonomous cortisol excess: a systematic review and meta-analysis. Ann Intern Med (2019) 171:107–16. doi: 10.7326/M18-3630
34. Bonekamp D, Horton KM, Hruban RH, Fishman EK. Castleman disease: the great mimic. Radiogr Rev Publ Radiol Soc North Am Inc (2011) 31:1793–807. doi: 10.1148/rg.316115502
35. Hill AJ, Tirumani SH, Rosenthal MH, Shinagare AB, Carrasco RD, Munshi NC, et al. Multimodality imaging and clinical features in castleman disease: single institute experience in 30 patients. Br J Radiol (2015) 88:20140670. doi: 10.1259/bjr.20140670
36. Zhao S, Wan Y, Huang Z, Song B, Yu J. Imaging and clinical features of castleman disease. Cancer Imaging Off Publ Int Cancer Imaging Soc (2019) 19:53. doi: 10.1186/s40644-019-0238-0
37. Falhammar H, Kjellman M, Calissendorff J. Initial clinical presentation and spectrum of pheochromocytoma: a study of 94 cases from a single center. Endocr Connect (2018) 7:186–92. doi: 10.1530/EC-17-0321
38. Mannelli M, Lenders JWM, Pacak K, Parenti G, Eisenhofer G. Subclinical phaeochromocytoma. Best Pract Res Clin Endocrinol Metab (2012) 26:507–15. doi: 10.1016/j.beem.2011.10.008
39. Eisenhofer G, Peitzsch M. Laboratory evaluation of pheochromocytoma and paraganglioma. Clin Chem (2014) 60:1486–99. doi: 10.1373/clinchem.2014.224832
40. Maia R, Couto J, Diogo J, Torre E, Guerra D. Unicentric castleman disease and pheochromocytoma. Eur J Case Rep Intern Med (2022) 9:3068. doi: 10.12890/2022_003068
41. Larroche C, Cacoub P, Soulier J, Oksenhendler E, Clauvel J-P, Piette J-C, et al. Castleman’s disease and lymphoma: report of eight cases in HIV-negative patients and literature review. Am J Hematol (2002) 69:119–26. doi: 10.1002/ajh.10022
42. Wang Y, Ren Y, Ma L, Li J, Zhu Y, Zhao L, et al. Clinical features of 50 patients with primary adrenal lymphoma. Front Endocrinol (Lausanne) (2020) 11:595. doi: 10.3389/fendo.2020.00595
43. Zeng J, Yan F, Chen Y, Zang L, Chen K, Lyu Z, et al. Primary adrenal lymphoma: two case series from China. Front Endocrinol (Lausanne) (2021) 12:778984. doi: 10.3389/fendo.2021.778984
44. Zhou L, Peng W, Wang C, Liu X, Shen Y, Zhou K. Primary adrenal lymphoma: radiological; pathological, clinical correlation. Eur J Radiol (2012) 81:401–5. doi: 10.1016/j.ejrad.2010.11.026
45. Wang S, Chen S, Xu J, Cai S. Clinicopathological characteristics of unicentric retroperitoneal castleman’s disease: a study of 14 cases. World J Surg Oncol (2016) 14:3. doi: 10.1186/s12957-015-0756-6
46. Chan K-L, Lade S, Prince HM, Harrison SJ. Update and new approaches in the treatment of castleman disease. J Blood Med (2016) 7:145–58. doi: 10.2147/JBM.S60514
47. Zhang X, Rao H, Xu X, Li Z, Liao B, Wu H, et al. Clinical characteristics and outcomes of castleman disease: a multicenter study of 185 Chinese patients. Cancer Sci (2018) 109:199–206. doi: 10.1111/cas.13439
48. Barry KK, Plumptre I, Bazewicz CG, Green BP, Treat JR, Hughes M, et al. Paraneoplastic pemphigus associated with castleman disease: a multicenter case series. Pediatr Dermatol (2023) 40:90–5. doi: 10.1111/pde.15138
49. Fayand A, Boutboul D, Galicier L, Kahn J-E, Buob D, Boffa J-J, et al. Epidemiology of castleman disease associated with AA amyloidosis: description of 2 new cases and literature review. Amyloid Int J Exp Clin Investig Off J Int Soc Amyloidosis (2019) 26:197–202. doi: 10.1080/13506129.2019.1641078
Keywords: castleman disease, adrenal gland, lymphoma, adrenal incidentaloma, pheochromocytoma
Citation: Yu H, Wang Y, Li Y, Du J, Guo Q, Gu W, Lyu Z, Dou J, Mu Y and Zang L (2023) Analysis of characteristics of four patients with adrenal unicentric Castleman disease. Front. Endocrinol. 14:1181929. doi: 10.3389/fendo.2023.1181929
Received: 08 March 2023; Accepted: 03 May 2023;
Published: 17 May 2023.
Edited by:
Henrik Falhammar, Karolinska Institutet (KI), SwedenReviewed by:
C. Christofer Juhlin, Karolinska Institutet (KI), SwedenChristina Bothou, University Hospital Zurich, Switzerland
Copyright © 2023 Yu, Wang, Li, Du, Guo, Gu, Lyu, Dou, Mu and Zang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zang, emFuZ2xpMzAxQDE2My5jb20=; Yiming Mu, bXV5aW1pbmdAMzAxaG9zcGl0YWwuY29tLmNu
†These authors have contributed equally to this work and share first authorship