 Jordan Teoli1,2,3
Jordan Teoli1,2,3 Delphine Mallet1,4
Delphine Mallet1,4 Lucie Renault5Claire-Lise Gay4,6Elsa Labrune3,5,7Patricia Bretones4,8Sandrine Giscard D’Estaing3,5,7Béatrice Cuzin9Frédérique Dijoud3,7,10
Lucie Renault5Claire-Lise Gay4,6Elsa Labrune3,5,7Patricia Bretones4,8Sandrine Giscard D’Estaing3,5,7Béatrice Cuzin9Frédérique Dijoud3,7,10 Florence Roucher-Boulez1,4,7,11
Florence Roucher-Boulez1,4,7,11 Ingrid Plotton1,3,4,5,7*
Ingrid Plotton1,3,4,5,7*- 1Service de Biochimie et Biologie Moléculaire, Unité Médicale de Biologie Endocrinienne, Centre de Biologie et Pathologie Est, Hospices Civils de Lyon, Bron, France
- 2Département des sciences biomédicales B, Institut des sciences pharmaceutiques et biologiques, Université Claude Bernard Lyon 1, Lyon, France
- 3Institut Cellule Souche et Cerveau (SBRI), Unité de Institut national de la recherche médicale (INSERM) 1208, Centre de Recherche INSERM, Bron, France
- 4Centre de Référence Maladies Rares du Développement Génital: du Fœtus à l’Adulte, Filière Maladies Rares Endocriniennes, Bron, France
- 5Service de médecine de la reproduction, Hôpital Femme-Mère-Enfant, Hospices Civils de Lyon, Bron, France
- 6Service d’endocrinologie pédiatrique, Institut Saint-Pierre, Palavas-Les-Flots, France
- 7Faculté de médecine, Université Claude Bernard Lyon 1, Lyon, France
- 8Service d’endocrinologie pédiatrique, Hôpital Femme-Mère-Enfant, Hospices Civils de Lyon, Bron, France
- 9Chirurgie Urologique, Centre Lyonnais d’Urologie Bellecour, Lyon, France
- 10Service d’Anatomie Pathologique, Centre de Biologie et de Pathologie Est, Hospices Civils de Lyon, Bron, France
- 11Institut Génétique, Reproduction & Développement (iGReD), Centre national de la recherche scientifique (CNRS), INSERM, Université Clermont Auvergne, Clermont–Ferrand, France
Background: Steroidogenic factor 1 (SF-1), encoded by the nuclear receptor subfamily 5 group A member 1 (NR5A1) gene, is a transcriptional factor crucial for adrenal and gonadal organogenesis. Pathogenic variants of NR5A1 are responsible for a wide spectrum of phenotypes with autosomal dominant inheritance including disorders of sex development and oligospermia–azoospermia in 46,XY adults. Preservation of fertility remains challenging in these patients.
Objective: The aim was to offer fertility preservation at the end of puberty in an NR5A1 mutated patient.
Case report: The patient was born of non-consanguineous parents, with a disorder of sex development, a small genital bud, perineal hypospadias, and gonads in the left labioscrotal fold and the right inguinal region. Neither uterus nor vagina was detected. The karyotype was 46,XY. Anti-Müllerian hormone (AMH) and testosterone levels were low, indicating testicular dysgenesis. The child was raised as a boy. At 9 years old, he presented with precocious puberty treated by triptorelin. At puberty, follicle-stimulating hormone (FSH), luteinising hormone (LH), and testosterone levels increased, whereas AMH, inhibin B, and testicular volume were low, suggesting an impaired Sertoli cell function and a partially preserved Leydig cell function. A genetic study performed at almost 15 years old identified the new frameshift variant NM_004959.5: c.207del p.(Phe70Serfs*5) at a heterozygous state. He was thus addressed for fertility preservation. No sperm cells could be retrieved from three semen collections between the ages of 16 years 4 months and 16 years 10 months. A conventional bilateral testicular biopsy and testicular sperm extraction were performed at 17 years 10 months of age, but no sperm cells were found. Histological analysis revealed an aspect of mosaicism with seminiferous tubules that were either atrophic, with Sertoli cells only, or presenting an arrest of spermatogenesis at the spermatocyte stage.
Conclusion: We report a case with a new NR5A1 variant. The fertility preservation protocol proposed at the end of puberty did not allow any sperm retrieval for future parenthood.
1 Introduction
Steroidogenic factor 1 (SF-1) is a transcription factor crucial for adrenal and testis organogenesis as well as steroidogenesis regulation with a dose-dependent effect (1–3).
SF-1 protein is encoded by the nuclear receptor subfamily 5 group A member 1 (NR5A1) gene located in chromosome 9 and composed of seven exons. SF-1 protein is characterised by a DNA-binding domain (DBD) in the amino-terminal region and by a ligand-binding domain (LBD) in the carboxy-terminal region separated by a hinge region, which can host post-translational changes (2, 4, 5).
To date, more than 180 putative pathogenic variants have been reported in NR5A1 coding regions and splice sites, spanning the whole gene and including missense variants (58% of variants), frameshift variants (18.6%), non-sense variants (12.3%), and splice variants (3.3%) in a heterozygous and isolated state in almost all cases. Variants were de novo in almost half of the cases and autosomal dominant inheritance in the others (4).
Pathogenic variants in NR5A1 are responsible for almost 20% of 46,XY differences or disorders of sex development (DSDs) (4). 46,XY DSD related to mutated NR5A1 is characterised by a wide spectrum of phenotypes, from male to female external genitalia and including partial or complete dysgenesis, genital ambiguity, micropenis, hypospadias, cryptorchidism, and asplenia with no clear genotype–phenotype correlation. Adrenal insufficiency is rarely associated with the picture (4, 6).
Severe oligospermia or azoospermia can be found in 46,XY patients carrying NR5A1 pathogenic variants; sometimes, these are the only symptoms (7, 8). Fertility care in 46,XY patients presenting azoospermia and carrying NR5A1 pathogenic variants has rarely been studied. Among these patients, only four cases who underwent testicular sperm extraction (TESE) have been reported, with inconsistent results (Table 1), underlining that preservation of fertility in such cases is challenging.
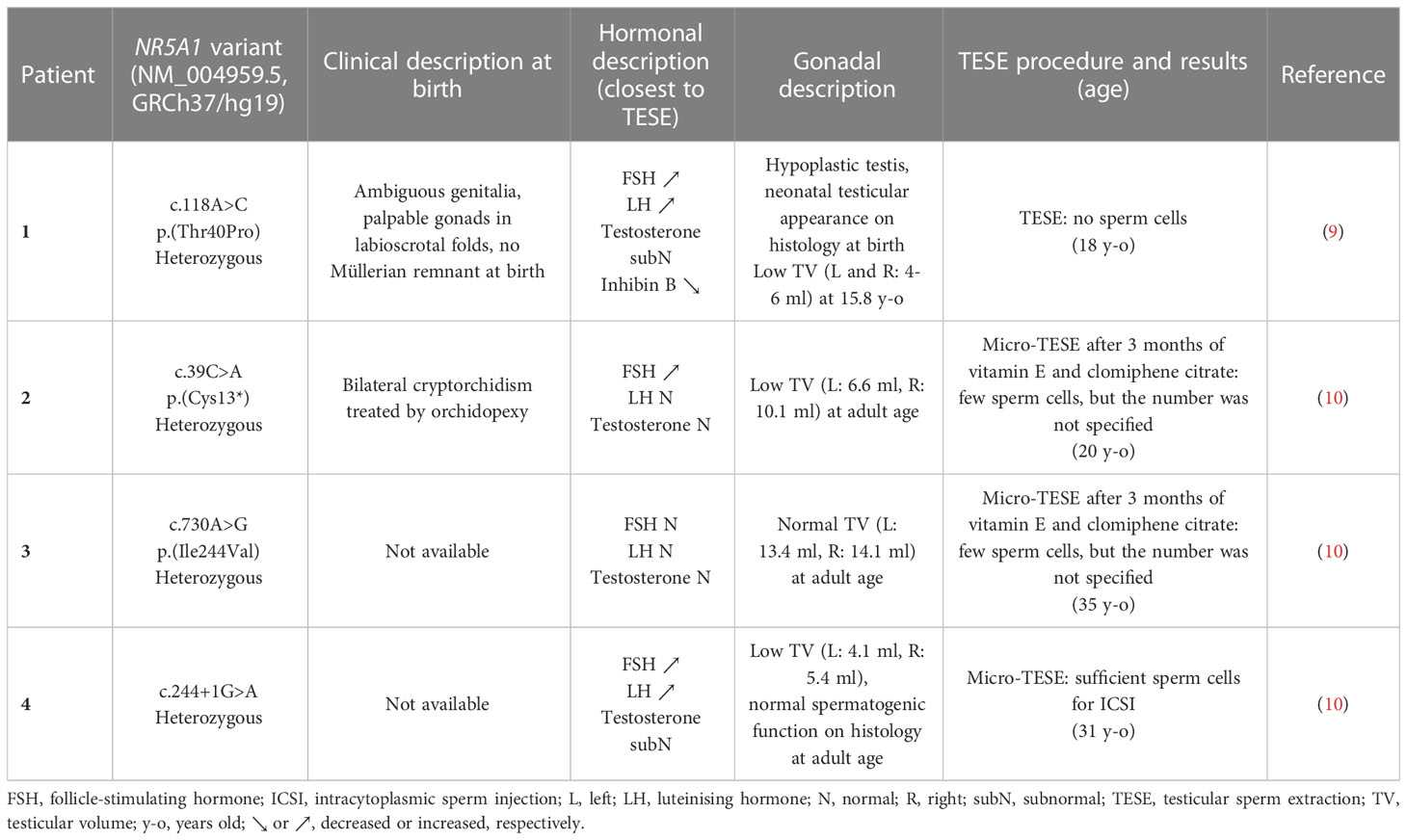
Table 1 Fertility preservation in mutated NR5A1 46,XY men presenting azoospermia.
Here, we report a longitudinal follow-up from birth to adulthood of a patient carrying a novel frameshift variant of NR5A1. We focused our follow-up on the physical and hormonal evaluations particularly during the puberty period, and also on testicular histology and semen collections.
2 Case report
The patient was born to non-consanguineous parents at 37 weeks of amenorrhea with a tetralogy of Fallot and a DSD. Longitudinal morphological and laboratory data are summarised in Table 2.
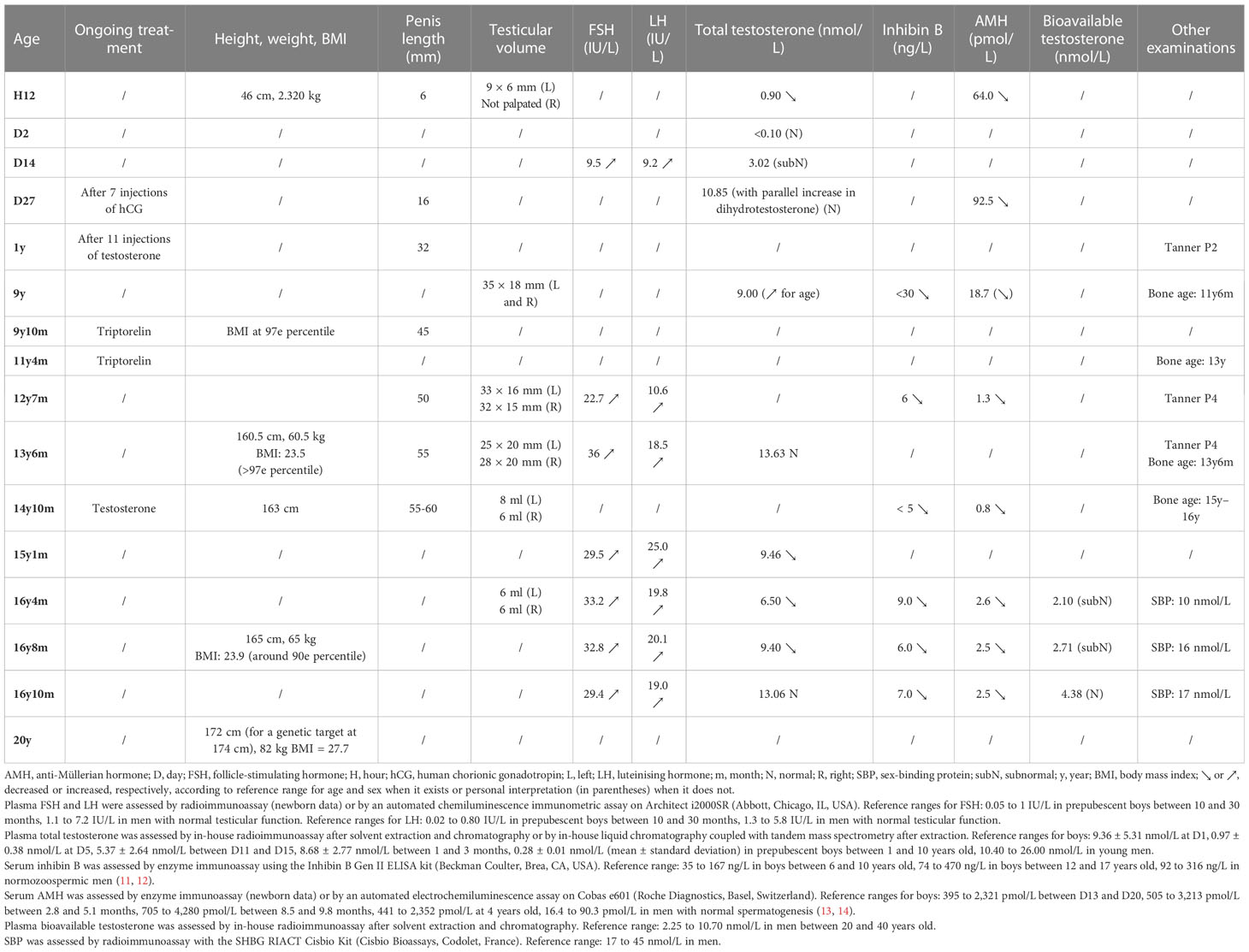
Table 2 Birth and follow-up morphological and laboratory data in a patient with a pathogenic NR5A1 (SF-1) frameshift variant.
At birth, the patient had a small genital bud (6 mm long) with perineal hypospadias. A gonad of 9 × 6 mm was palpated in the left labioscrotal fold, and the right gonad was in the high inguinal region. The genitography did not show any uterus or vagina. Karyotype and fluorescence in situ hybridisation showed a normal 46,XY formula. Anti-Müllerian hormone (AMH) levels were low on the first day of life (64 pmol/L) and at the minipuberty (92.5 pmol/L). Testosterone level was also low at the 12th hour of life (0.90 nmol/L) and was stimulated to 10.85 nmol/L after a human chorionic gonadotropin (hCG) test (seven injections of 1,500 IU every 2 days). On the 14th day of life, follicle-stimulating hormone (FSH) and luteinising hormone (LH) levels were high (9.5 and 9.2 IU/L, respectively).
The patient received four injections of heptylate testosterone (two doses of 20 mg and then two doses of 25 mg, 15 days apart) and was declared male at 3 months of age. Several surgical treatments were performed, first for his hypospadias at 1 year of age, then for the undescended testis at 2 years 6 months, and finally for the correction of the penis curvature at 9 years. At 9 years, precocious puberty was suspected due to an increase in testicular volume (TV), and a gonadotropin-releasing hormone (GnRH) test confirmed a central origin. Magnetic resonance imaging of the hypothalamo-pituitary region was normal. GnRH analog treatment (triptorelin: one injection every 4 weeks and then every 3 weeks due to insufficient effectiveness) was introduced from 9 years 10 months to 11 years 4 months of age. From the age of 13 years 6 months to 14 years 10 months, testosterone enanthate (50 to 125 mg, one injection every 3 weeks) was undertaken since LH was high and to improve penis size prognosis but was interrupted because of its poor effectiveness. At 14 years old, a varicocele on the left side was observed and highlighted by testicular echography.
At 16 years 4 months of age, he was addressed for fertility preservation. TV was diminished (6 ml on both sides), and AMH and inhibin B levels were low. Three semen collections performed according to the 2010 World Health Organization criteria (15) between the ages of 16 years 4 months and 16 years 10 months retrieved no sperm cells. The varicocele on the left side was treated by embolisation at the age of 17 years 2 months. The patient was eligible for a testicular biopsy with testicular sperm extraction (conventional TESE) since no sperm cells were found in at least two sperm samples 3 months apart. TESE was practiced according to the procedure described previously (16) when the patient was 17 years 10 months old. Only one sperm cell was found on a right testis fraction, but this was insufficient for cryopreservation.
The histological analysis of biopsy fragments revealed a severely impaired spermatogenesis with an aspect of histological mosaicism using Johnsen score (17): the seminiferous tubules, of overall reduced diameter and lined by a thick basal membrane, were either atrophic, with Sertoli cells only, or with spermatogenesis arrest at the spermatocyte stage. The interstitial tissue was fibro-edematous with hyperplastic Leydig cells. No signs of malignancy or dysplasia were noticed (Figure 1).
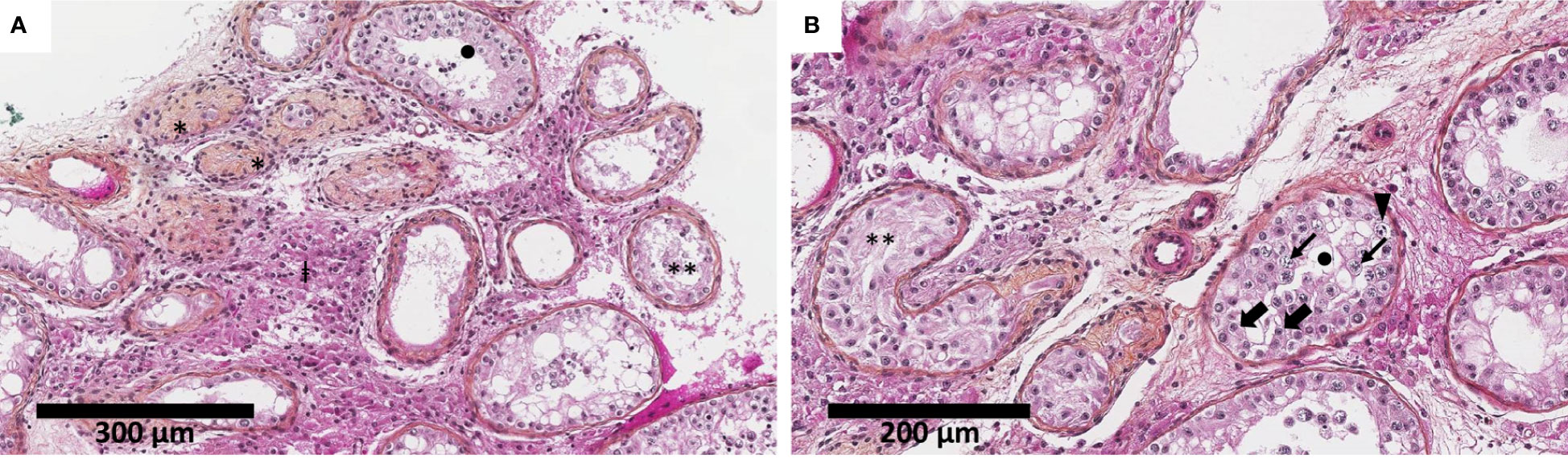
Figure 1 Testicular biopsy shows severely impaired spermatogenesis with an aspect of histological mosaicism. The seminiferous tubules were of overall reduced diameter (decreased by approximately 30%–50% compared to physiological adult pubertal seminiferous tubules of 150–250 µm in diameter) and lined by a thickened basal membrane. The seminiferous tubules were either atrophic (*), with Sertoli cells only (**), or presenting spermatogenesis arrest at the spermatocyte stage (•). The interstitial tissue was fibro-edematous with numerous Leydig cells (‡) (hematoxylin–eosin–saffron). Thick black arrow indicates Sertoli cells, thin black arrow indicates spermatocyte, and solid black triangle indicates spermatogonia. Biopsy fragments were fixed in alcohol, formalin, and acetic acid (AFA) and paraffin-embedded. Sections of 3 μm were stained by hematoxylin–phloxin–saffron. Slide evaluation was performed on a Leica DM2500 microscope. Two different scales: 300 µm (A) and 200 µm (B).
At 14 years 10 months of age, after his parents provide signed informed written consent for genetic testing, a molecular analysis of NR5A1 gene (Sanger sequencing on DNA extracted from whole blood) revealed the unreported heterozygous frameshift variant NM_004959.5: c.207del p.(Phe70Serfs*5) (GRCh37/hg19) (Figure 2). His parents and his sibling were not sequenced for NR5A1. According to the American College of Medical Genetics and Genomics (ACMG) criteria (18), this variant is classified as pathogenic. This variant had never been reported in Gnomad_v2, ClinVar, and dbSNP databases or in any literature to date. It is in the third exon of NR5A1 gene encoding the DBD of the SF-1 protein. Since it induces a frameshift with the manifestation of a premature stop codon, it should lead to either an inactive truncated protein (truncated DBD, absence of the hinge region and the LBD) or the absence of protein by the non-sense-mediated mRNA decay (NMD) mechanism (19). The coding regions of the androgen receptor (AR) gene were also studied; no pathogenic variant was found.
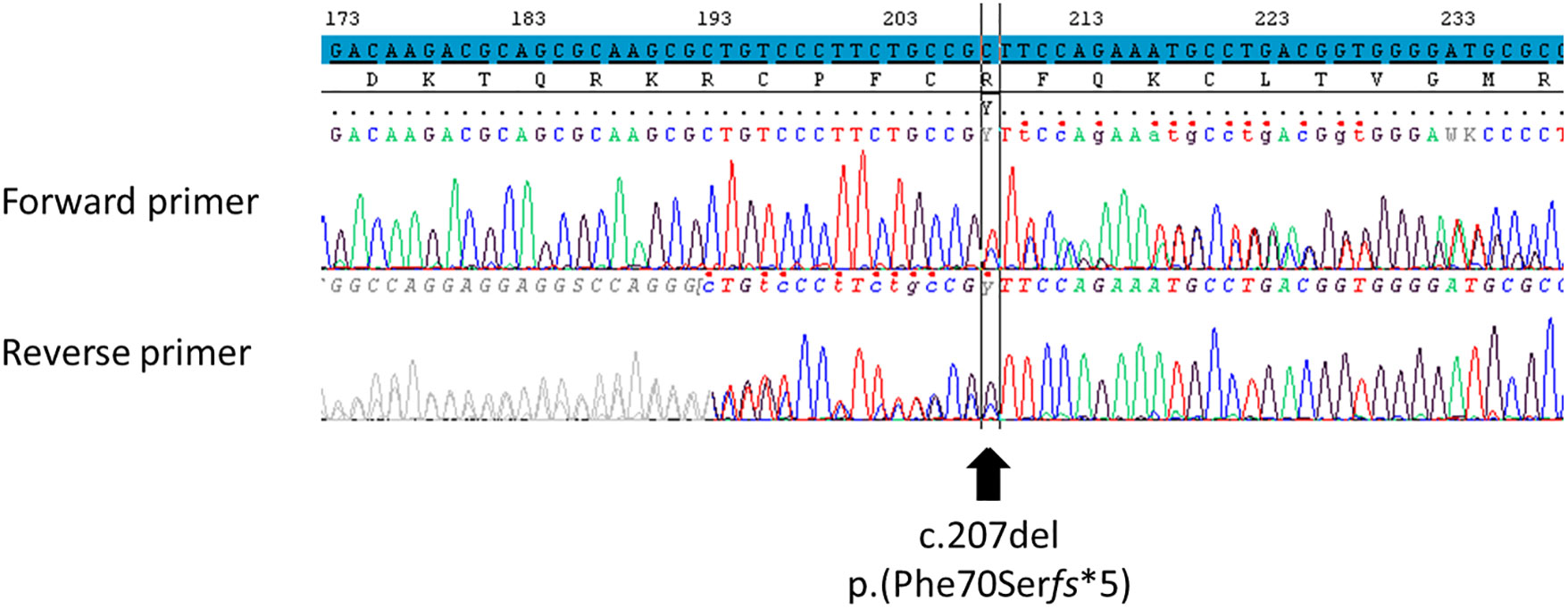
Figure 2 Identification of the NR5A1 variant using Sanger sequencing. Variation was identified using reference NM_004959.5 for NR5A1 transcript on GRCh37/hg19 human genome assembly, NP_004950.2 for SF-1 protein. The screenshot comes from SeqScape 3 software. The nucleotide reference sequence is highlighted in blue. This variant was classified as pathogenic according to the ACMG criteria (PVS1, PM1, and PM2). The polymorphism NM_004959.5: c.437G>C p.(Gly146Ala) was not found. In ACMG criteria: PM, pathogenic moderate; PVS, pathogenic very strong. “PVS1: null variant (non-sense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multi-exon deletion) in a gene where loss of function is a known mechanism of disease.” “PM1: located in a mutational hot spot and/or critical and well-established functional domain (e.g. active site of an enzyme) without benign variation.” “PM2: absent from controls (or at extremely low frequency if recessive) in Exome Sequencing Project, 1000 Genomes or ExAC.”.
Otherwise, the patient had some periods of overweight during childhood and early adulthood because of a lack of physical activity and overeating. The patient never presented with an adrenal crisis and the exploration of the hypothalamic–pituitary–adrenal axis was normal (cortisol and adrenocorticotropic hormone (ACTH)).
3 Discussion
We describe a 46,XY male patient carrying a new NR5A1 pathogenic variant with medical monitoring from birth to adulthood, at the beginning of which a testicular biopsy with TESE was performed.
The novel variant NM_004959.5: c.207del p.(Phe70Serfs*5) found here was classified as pathogenic according to the ACMG criteria. Certain frameshift variants inducing a premature stop codon in the same position as those observed herein have been reported to lead to a protein with the same missing parts (truncated DBD, absence of the hinge region, and the LBD) if one is produced. The variant NM_004959.5: c.18del p.(Asp6Glufs*69) was found in a 46,XY patient born with ambiguous external genitalia and raised as female. At 27 years old, she had clitorimegaly, a blind-ended vagina with no uterus, severely hypoplastic inguinal testis, primary hypogonadism with high gonadotropin levels and low baseline testosterone level non-responsive to hCG stimulation, and no adrenal dysfunction (20). Another variant, NM_004959.5: c.70del p.(His24Thrfs*51), was found in a 46,XY patient with ambiguous external genitalia at birth. The patient presented with an absence of Müllerian ducts and no palpated gonads and was initially raised as female until 18 years old. The patient had high gonadotropin levels, normal testosterone concentration, and normal adrenal function at 18 years of age, and the patient’s testes were considered dysgenetics at 19 years old (21). The last variant, NM_004959.5: c.151del p.(Glu51Argfs*24), was found in a 46,XY adolescent patient raised as female and who presented with clitorimegaly, primary amenorrhea, and inguinal testes (22). The p.(Asp6Glufs*69) and p.(His24Thrfs*51) variants were explored by functional studies that showed a reduction in the transactivation capacity of SF-1 on the promoters of some genes coding for steroidogenic enzymes. Nevertheless, Western blotting performed on transfected cells could not detect any protein, due to either NMD or the non-ability of the technique to detect small peptides (20, 21). The p.(Phe70Serfs*5) variant identified herein should have similar consequences on the transactivation capacity of SF-1 than the p.(Asp6Glufs*69) and p.(His24Thrfs*51) variants.
The clinical and hormonal data recorded in the present patient could be integrated into the wide spectrum of phenotypes of mutated NR5A1 patients (reviewed in (4, 23)).
At the patient’s birth, AMH levels were low for a 46,XY newborn but too high for a 46,XX newborn, and total testosterone level was subnormal but increased properly after hCG stimulation in early childhood. These two parameters indicated the presence of dysgenetic testicular tissue with an impaired function more marked on Sertoli cells than on Leydig cells. However, the absence of the uterus indicated a sufficient secretion of AMH during the in utero life. As suggested elsewhere, foetal Sertoli cell function was thus sufficient to induce the regression of the Müllerian ducts but decreased after birth (23–25). The incompletely virilised external genitalia reported herein suggests that testosterone and dihydrotestosterone were probably insufficiently secreted during the in utero window of masculinisation to induce the complete development of external genitalia (26). Testosterone levels (basal or stimulated) vary greatly among 46,XY NR5A1 mutated patients (23).
At puberty, the patient showed an insufficient increase in TV, low AMH and inhibin B levels, and elevated FSH levels that indicated a severe primary Sertoli cell injury. The normal or subnormal testosterone levels with elevated LH levels suggested a compensated primary hypofunction of Leydig cells and explained the virilisation signs observed in the patient at puberty. This description is in line with that of Mönig et al., who studied 10 NR5A1 mutated patients during adolescence and puberty (24). Other authors also showed an impaired Sertoli cell function with a normal or subnormal Leydig cell function conserved at least until puberty (23, 25, 27).
Interestingly, the precocious increase in TV and testosterone levels herein suggested precocious puberty confirmed by a GnRH test. The occurrence of precocious puberty was surprising because SF-1 is expressed in pituitary cells in humans and is implicated in the formation of the ventromedial hypothalamic nucleus in mice (28) and because some mutated NR5A1 patients encountered difficulties when entering puberty spontaneously (29). However, Mönig et al. reported an early pubertal development with an early increase in testosterone levels in three out of 10 cases (24).
Amazingly, the spontaneous increase in testosterone levels and virilisation at puberty contrasted with the subnormal testosterone levels and incompletely virilised external genitalia at birth reported herein and elsewhere (23, 24). As demonstrated in mice, there is evidence in humans that testosterone synthesis during foetal life imply a coordinated action of foetal Sertoli cells and foetal Leydig cells (30). Therefore, a Sertoli cell dysfunction during foetal life may impair testosterone production by the foetal testis and lead to a lack of virilisation of the external genitalia at birth.
Furthermore, progressive degradation of testicular function with age was suggested in the literature based on several physical and hormonal observations. First, AMH was secreted during in utero life, but its levels were low at birth and in the neonatal period as discussed above (23–25), indicating gonadal dysgenesis. Herein, AMH levels were already low at birth. Then, a decrease in TV can occur during or after puberty (24); it was not significant herein perhaps because TV was initially too low. Finally, a progressive increase in the FSH and LH levels and a progressive decrease in testosterone and inhibin B levels with age were reported (9, 24, 25, 31). This pattern was observed herein mainly for FSH and LH levels but not inhibin B levels since the first value (at 12 years 7 months) was already too low. At 16 years 10 months, testosterone level was normal, but further degradation may not be excluded.
As expected given the low TV and low AMH and inhibin B levels, no sperm cells were retrieved in semen samples. Azoospermia was previously reported in NR5A1 mutated patients (7), but varicocele could aggravate the spermiological phenotype in this case (32). Spermatogenesis could be improved 3 to 6 months after varicocele treatment (32).
Interestingly, some sperm cells were collected in the semen of some 46,XY mutated NR5A1 patients (7, 8), and certain patients even fathered children naturally (25, 33–35). Among the latter, one patient had two children even though he carried an NR5A1 pathogenic variant in a mosaic state in DNA extracted from blood leukocytes (25). One had two children at 30 and 33 years old but refused further investigations (35). One fathered five children before the age of 32 years and presented with increased FSH levels and undetectable AMH and inhibin B levels at 57 years old. However, no hormonal data were available when he was 32, and no sperm data were available for him or his boys (33). From the perspective of progressive hormonal function alteration, some authors suggested a progressive degradation of spermatogenesis with age that allows paternity in young men before spermatogenesis collapses (7, 23, 25, 31). However, this hypothesis remains to be confirmed by longitudinal sperm counts in NR5A1 mutated 46,XY patients in whom spermatogenesis is preserved. Consequently, men carrying NR5A1 pathogenic variants should be addressed for fertility preservation as early as possible after puberty; if mature sperm cells are retrieved, cryopreservation can thus be performed to ensure a timely medically assisted reproduction. A TESE was proposed to the patient herein when he was 17 years 10 months. Although it was performed sufficiently long enough after varicocele treatment to allow for the potential restoration of spermatogenesis, only one sperm cell was retrieved, thus preventing cryopreservation and intracytoplasmic sperm injection (ICSI) to be performed. In the literature (Table 1), one team failed to retrieve sperm cells in an 18-year-old man using TESE (9), while another retrieved sperm cells in three men (20, 31, and 35 years old) using micro-TESE after 3 months of vitamin E and clomiphene citrate for two of them (10). This discrepancy in TESE outcomes could be explained by different situations. First, based on the hypothesis of progressive spermatogenesis degradation, the age when TESE was performed may have impacted the outcomes. However, TESE retrieved sperm cells in the three older patients but failed in the youngest. Second, the TESE procedure performed: micro-TESE did not show better results for retrieving sperm cells than conventional TESE in men with non-obstructive azoospermia in a recent meta-analysis (36). Third, the wide spectrum of the disease without a clear phenotype–genotype relation likely impacts TESE outcomes. The fact that the same NR5A1 pathogenic variant can cause different phenotypes in patients belonging to the same family (9, 33, 35, 37, 38) may suggest a possible polygenic inheritance or the intervention of additional epigenetic or environmental factors in the phenotype severity. In the case of polygenic inheritance, whole genome sequencing would be of great interest to find another mutated gene and to understand the spectrum of NR5A1-related diseases. Finally, features correlated with spermatogenesis function (39–41) like hormonal markers (FSH, LH, testosterone, and inhibin B), TV, and history of cryptorchidism are likely to affect TESE outcomes here: one patient in whom TESE retrieved sperm cells had a normal hormonal profile and normal TV. Unfortunately, we did not have access to the clinical description at birth or the follow-up of inhibin B levels since adolescence in all patients who underwent TESE to be able to suggest a relationship between the severity of the DSD and the TESE outcomes (9, 10).
The hormonal anomalies, sperm sampling, and TESE outcomes herein were consistent with the results of the pathological analysis of the biopsied testicular fragments. The pathological aspect observed herein was also consistent with the wide spectrum of testicular biopsy descriptions found elsewhere in 46,XY NR5A1 mutated adults (8, 23, 42). The possible degradation of testicular function in terms of hormonal and sperm parameters with aging discussed above might parallel a progressive degradation of testis structure observable on the testicular biopsy, as suggested by Camats et al. (21). Nevertheless, if testicular biopsy finds germ cells and functional seminiferous tubules, future techniques of fertility preservation, such as the emerging in vitro spermatogenesis technology (43, 44), would be of great interest in NR5A1 mutated patients with azoospermia.
Although overweight in NR5A1 mutated patients has already been described (20, 45, 46), this feature was not found in all patients (46). Interestingly, the homozygous deletion of NR5A1 in an SF-1 knock-out mouse model induced obesity (47). In line with this finding, some authors suggested the intervention of SF-1 in the development of the ventromedial hypothalamic nucleus, a central player in appetite regulation in humans (20, 46).
Finally, the presence of tetralogy of Fallot is surprising and was not reported elsewhere in association with a mutated SF-1. However, we could not exclude an additional genetic anomaly in other genes associated with DSD and/or tetralogy of Fallot, GATA4 and ZFPM2/FOG2 genes, for example (48, 49).
4 Conclusion
We report a case with a new NR5A1 pathogenic variant addressed for fertility care. The physical, hormonal, and histological description of the testis could be integrated into the wide spectrum of 46,XY DSD related to mutated NR5A1. The patient presented with azoospermia since the first semen analysis when he was 16 years 4 months. A conventional TESE was performed at 17 years 10 months, but this procedure did not retrieve sufficient sperm cells for cryopreservation to perform an ICSI for future parenthood. These data extend the knowledge regarding fertility in NR5A1 mutated patients. Further investigations in NR5A1 mutated patients would help define a fertility care protocol in order to increase their chances of fertility.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by Ethics committee of Lyon University Hospital. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin. Written informed consent was obtained from the participant/patient(s) for the publication of this case report.
Author contributions
JT wrote the manuscript. IP, DM, FD, and FR-B supervised the laboratory procedures. JT, DM, LR, FD, FR-B, and IP interpreted the data. Patient care was performed by CG, EL, PB, SGD’E, BC, and IP. All authors contributed to the article and approved the submitted version.
Acknowledgments
We thank the technicians of our unit, who participated in the generation of the results, and Véréna Landel (DRS, Hospices Civils de Lyon) for reading the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACMG, American College of Medical Genetics and genomics; AMH, anti-Müllerian hormone; DBD, DNA-binding domain; DSD, disorder of sex development; FSH, follicle-stimulating hormone; GnRH, gonadotropin-releasing hormone; hCG, human chorionic gonadotropin; ICSI, intracytoplasmic sperm injection; LH, luteinising hormone; LBD, ligand-binding domain; NMD, non-sense-mediated mRNA decay; NR5A1, nuclear receptor subfamily 5 group A member 1; SF-1, steroidogenic factor 1; TESE, testicular sperm extraction.
References
1. El-Khairi R, Achermann J. Steroidogenic factor-1 and human disease. Semin Reprod Med (2012) 30:374–81. doi: 10.1055/s-0032-1324720
2. Hoivik EA, Lewis AE, Aumo L, Bakke M. Molecular aspects of steroidogenic factor 1 (SF-1). Mol Cell Endocrinol (2010) 315:27–39. doi: 10.1016/j.mce.2009.07.003
3. Lala DS, Rice DA, Parker KL. Steroidogenic factor I, a key regulator of steroidogenic enzyme expression, is the mouse homolog of fushi tarazu-factor I. Mol Endocrinol (1992) 6:1249–58. doi: 10.1210/mend.6.8.1406703
4. Fabbri-Scallet H, Sousa LM, Maciel-Guerra AT, Guerra-Júnior G, Mello MP. Mutation update for the NR5A1 gene involved in DSD and infertility. Hum Mutat (2020) 41:58–68. doi: 10.1002/humu.23916
5. Wong M, Ramayya MS, Chrousos GP, Driggers PH, Parker KL. Cloning and sequence analysis of the human gene encoding steroidogenic factor 1. J Mol Endocrinol (1996) 17:139–47. doi: 10.1677/jme.0.0170139
6. Achermann JC, Ito M, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet (1999) 22:125–6. doi: 10.1038/9629
7. Bashamboo A, Ferraz-de-Souza B, Lourenço D, Lin L, Sebire NJ, Montjean D, et al. Human Male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am J Hum Genet (2010) 87:505–12. doi: 10.1016/j.ajhg.2010.09.009
8. Röpke A, Tewes A-C, Gromoll J, Kliesch S, Wieacker P, Tüttelmann F. Comprehensive sequence analysis of the NR5A1 gene encoding steroidogenic factor 1 in a large group of infertile males. Eur J Hum Genet (2013) 21:1012–5. doi: 10.1038/ejhg.2012.290
9. Werner R, Mönig I, Lünstedt R, Wünsch L, Thorns C, Reiz B, et al. New NR5A1 mutations and phenotypic variations of gonadal dysgenesis. PloS One (2017) 12:e0176720. doi: 10.1371/journal.pone.0176720
10. An M, Liu Y, Zhang M, Hu K, Jin Y, Xu S, et al. Targeted next-generation sequencing panel screening of 668 Chinese patients with non-obstructive azoospermia. J Assist Reprod Genet (2021) 38:1997–2005. doi: 10.1007/s10815-021-02154-9
11. Barbotin A-L, Ballot C, Sigala J, Ramdane N, Duhamel A, Marcelli F, et al. The serum inhibin b concentration and reference ranges in normozoospermia. Eur J Endocrinol (2015) 172:669–76. doi: 10.1530/EJE-14-0932
12. Crofton PM, Evans AEM, Groome NP, Taylor MRH, Holland CV, Kelnar CJH. Inhibin b in boys from birth to adulthood: relationship with age, pubertal stage, FSH and testosterone. Clin Endocrinol (Oxf) (2002) 56:215–21. doi: 10.1046/j.0300-0664.2001.01448.x
13. Benderradji H, Barbotin A-L, Leroy-Billiard M, Prasivoravong J, Marcelli F, Decanter C, et al. Defining reference ranges for serum anti-müllerian hormone on a Large cohort of normozoospermic adult men highlights new potential physiological functions of AMH on FSH secretion and sperm motility. J Clin Endocrinol Metab (2022) 107:1878–87. doi: 10.1210/clinem/dgac218
14. Plotton I, Raverot V, Roche E, Jean Marc L, Morel Y. Évaluation de la trousse AMH (anti-mullerienne hormone) de roche® dans les indications pédiatriques: détermination des valeurs de références chez le garçon. Annales d’Endocrinologie (2015) 76:367–8. doi: 10.1016/j.ando.2015.07.214
15. World Health Organization. WHO laboratory manual for the examination and processing of human semen FIFTH EDITION (2010). Available at: https://www.who.int/docs/default-source/reproductive-health/srhr-documents/infertility/examination-and-processing-of-human-semen-5ed-eng.pdf (Accessed December 16, 2021).
16. Plotton I, Giscard d’Estaing S, Cuzin B, Brosse A, Benchaib M, Lornage J, et al. Preliminary results of a prospective study of testicular sperm extraction in young versus adult patients with nonmosaic 47,XXY klinefelter syndrome. J Clin Endocrinol Metab (2015) 100:961–7. doi: 10.1210/jc.2014-3083
17. Johnsen SG. Testicular biopsy score count - a method for registration of spermatogenesis in human testes: normal values and results in 335 hypogonadal males. Hormones (1970) 1:2–25. doi: 10.1159/000178170
18. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
19. Frischmeyer PA. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet (1999) 8:1893–900. doi: 10.1093/hmg/8.10.1893
20. Hasegawa T, Fukami M, Sato N, Katsumata N, Sasaki G, Fukutani K, et al. Testicular dysgenesis without adrenal insufficiency in a 46,XY patient with a heterozygous inactive mutation of steroidogenic factor-1. J Clin Endocrinol Metab (2004) 89:5930–5. doi: 10.1210/jc.2004-0935
21. Camats N, Pandey AV, Fernández-Cancio M, Andaluz P, Janner M, Torán N, et al. Ten novel mutations in the NR5A1 gene cause disordered sex development in 46,XY and ovarian insufficiency in 46,XX individuals. J Clin Endocrinol Metab (2012) 97:E1294–306. doi: 10.1210/jc.2011-3169
22. Philibert P, Leprieur E, Zenaty D, Thibaud E, Polak M, Frances A-M, et al. Steroidogenic factor-1 (SF-1) gene mutation as a frequent cause of primary amenorrhea in 46,XY female adolescents with low testosterone concentration. Reprod Biol Endocrinol (2010) 8:28. doi: 10.1186/1477-7827-8-28
23. Pedace L, Laino L, Preziosi N, Valentini MS, Scommegna S, Rapone AM, et al. Longitudinal hormonal evaluation in a patient with disorder of sexual development, 46,XY karyotype and one NR5A1 mutation. Am J Med Genet (2014) 164:2938–46. doi: 10.1002/ajmg.a.36729
24. Mönig I, Schneidewind J, Johannsen TH, Juul A, Werner R, Lünstedt R, et al. Pubertal development in 46,XY patients with NR5A1 mutations. Endocrine (2022) 75:601–13. doi: 10.1007/s12020-021-02883-y
25. Philibert P, Polak M, Colmenares A, Lortat-Jacob S, Audran F, Poulat F, et al. Predominant sertoli cell deficiency in a 46,XY disorders of sex development patient with a new NR5A1/SF-1 mutation transmitted by his unaffected father. Fertility Sterility (2011) 95:1788.e5–1788.e9. doi: 10.1016/j.fertnstert.2010.11.035
26. Grinspon RP, Bergadá I, Rey RA. Male Hypogonadism and disorders of sex development. Front Endocrinol (2020) 11:211. doi: 10.3389/fendo.2020.00211
27. Faienza MF, Chiarito M, Baldinotti F, Canale D, Savino C, Paradies G, et al. NR5A1 gene variants: variable phenotypes, new variants, different outcomes. Sex Dev (2019) 13:258–63. doi: 10.1159/000507411
28. Ikeda Y, Luo X, Abbud R, Nilson JH, Parker KL. The nuclear receptor steroidogenic factor 1 is essential for the formation of the ventromedial hypothalamic nucleus. Mol Endocrinol (1995) 9:478–86. doi: 10.1210/mend.9.4.7659091
29. Lin L, Philibert P, Ferraz-de-Souza B, Kelberman D, Homfray T, Albanese A, et al. Heterozygous missense mutations in steroidogenic factor 1 (SF1/Ad4BP, NR5A1) are associated with 46,XY disorders of sex development with normal adrenal function. J Clin Endocrinol Metab (2007) 92:991–9. doi: 10.1210/jc.2006-1672
30. O’Donnell L, Whiley PAF, Loveland KL. Activin a and sertoli cells: key to fetal testis steroidogenesis. Front Endocrinol (2022) 13:898876. doi: 10.3389/fendo.2022.898876
31. Tantawy S, Lin L, Akkurt I, Borck G, Klingmuller D, Hauffa BP, et al. Testosterone production during puberty in two 46,XY patients with disorders of sex development and novel NR5A1 (SF-1) mutations. Eur J Endocrinol (2012) 167:125–30. doi: 10.1530/EJE-11-0944
32. Chiba K, Fujisawa M. Clinical outcomes of varicocele repair in infertile men: a review. World J Mens Health (2016) 34:101. doi: 10.5534/wjmh.2016.34.2.101
33. Ciaccio M, Costanzo M, Guercio G, De Dona V, Marino R, Ramirez PC, et al. Preserved fertility in a patient with a 46,XY disorder of sex development due to a new heterozygous mutation in the NR5A1/SF-1 gene: evidence of 46,XY and 46,XX gonadal dysgenesis phenotype variability in multiple members of an affected kindred. Horm Res Paediatr (2012) 78:119–26. doi: 10.1159/000338346
34. Rocca MS, Ortolano R, Menabò S, Baronio F, Cassio A, Russo G, et al. Mutational and functional studies on NR5A1 gene in 46,XY disorders of sex development: identification of six novel loss of function mutations. Fertility Sterility (2018) 109:1105–13. doi: 10.1016/j.fertnstert.2018.02.123
35. Yagi H, Takagi M, Kon M, Igarashi M, Fukami M, Hasegawa Y. Fertility preservation in a family with a novel NR5A1 mutation. Endocr J (2015) 62:289–95. doi: 10.1507/endocrj.EJ14-0340
36. Corona G, Minhas S, Giwercman A, Bettocchi C, Dinkelman-Smit M, Dohle G, et al. Sperm recovery and ICSI outcomes in men with non-obstructive azoospermia: a systematic review and meta-analysis. Hum Reprod Update (2019) 25:733–57. doi: 10.1093/humupd/dmz028
37. Hattori A, Zukeran H, Igarashi M, Toguchi S, Toubaru Y, Inoue T, et al. A novel c-terminal truncating NR5A1 mutation in dizygotic twins. Hum Genome Var (2017) 4:17008. doi: 10.1038/hgv.2017.8
38. Warman DM, Costanzo M, Marino R, Berensztein E, Galeano J, Ramirez PC, et al. Three new SF-1 (NR5A1) gene mutations in two unrelated families with multiple affected members: within-family variability in 46,XY subjects and low ovarian reserve in fertile 46,XX subjects. Horm Res Paediatr (2011) 75:70–7. doi: 10.1159/000320029
39. Jensen TK, Andersson A-M, Hjollund NHI, Scheike T, Kolstad H, Giwercman A, et al. Inhibin b as a serum marker of spermatogenesis: correlation to differences in sperm concentration and follicle-stimulating hormone levels. a study of 349 Danish men. J Clin Endocrinol Metab (1997) 82:4059–63. doi: 10.1210/jcem.82.12.4456
40. Kumanov P, Nandipati K, Tomova A, Agarwal A. Inhibin b is a better marker of spermatogenesis than other hormones in the evaluation of male factor infertility. Fertility Sterility (2006) 86:332–8. doi: 10.1016/j.fertnstert.2006.01.022
42. Ferlin A, Rocca MS, Vinanzi C, Ghezzi M, Di Nisio A, Foresta C. Mutational screening of NR5A1 gene encoding steroidogenic factor 1 in cryptorchidism and male factor infertility and functional analysis of seven undescribed mutations. Fertility Sterility (2015) 104:163–169.e1. doi: 10.1016/j.fertnstert.2015.04.017
43. Kanbar M, de Michele F, Poels J, Van Loo S, Giudice MG, Gilet T, et al. Microfluidic and static organotypic culture systems to support ex vivo spermatogenesis from prepubertal porcine testicular tissue: a comparative study. Front Physiol (2022) 13:884122. doi: 10.3389/fphys.2022.884122
44. Perrard M-H, Sereni N, Schluth-Bolard C, Blondet A, d’Estaing SG, Plotton I, et al. Complete human and rat ex vivo spermatogenesis from fresh or frozen testicular tissue. Biol Reprod (2016) 95:89–9. doi: 10.1095/biolreprod.116.142802
45. Laan M, Kasak L, Timinskas K, Grigorova M, Venclovas Č, Renaux A, et al. NR5A1 c.991-1G > c splice-site variant causes familial 46,XY partial gonadal dysgenesis with incomplete penetrance. Clin Endocrinol (2021) 94:656–66. doi: 10.1111/cen.14381
46. Malikova J, Camats N, Fernández-Cancio M, Heath K, González I, Caimarí M, et al. Human NR5A1/SF-1 mutations show decreased activity on BDNF (Brain-derived neurotrophic factor), an important regulator of energy balance: testing impact of novel SF-1 mutations beyond steroidogenesis. PloS One (2014) 9:e104838. doi: 10.1371/journal.pone.0104838
47. Majdic G, Young M, Gomez-Sanchez E, Anderson P, Szczepaniak LS, Dobbins RL, et al. Knockout mice lacking steroidogenic factor 1 are a novel genetic model of hypothalamic obesity. Endocrinology (2002) 143:607–14. doi: 10.1210/endo.143.2.8652
48. Bashamboo A, Brauner R, Bignon-Topalovic J, Lortat-Jacob S, Karageorgou V, Lourenco D, et al. Mutations in the FOG2/ZFPM2 gene are associated with anomalies of human testis determination. Hum Mol Genet (2014) 23:3657–65. doi: 10.1093/hmg/ddu074
Keywords: testicular sperm extraction, gonadal dysgenesis, spermatogenesis, male infertility, congenital, disorder of sex development, hypospadias, azoospermia
Citation: Teoli J, Mallet D, Renault L, Gay C-L, Labrune E, Bretones P, Giscard D’Estaing S, Cuzin B, Dijoud F, Roucher-Boulez F and Plotton I (2023) Case Report: Longitudinal follow-up and testicular sperm extraction in a patient with a pathogenic NR5A1 (SF-1) frameshift variant: p.(Phe70Serfs*5). Front. Endocrinol. 14:1171822. doi: 10.3389/fendo.2023.1171822
Received: 22 February 2023; Accepted: 26 May 2023;
Published: 20 June 2023.
Edited by:
Yasmin Jayasinghe, The University of Melbourne, AustraliaReviewed by:
Alberto Ferlin, University of Padua, ItalyRyoma Yoneda, Saitama Medical University, Japan
Copyright © 2023 Teoli, Mallet, Renault, Gay, Labrune, Bretones, Giscard D’Estaing, Cuzin, Dijoud, Roucher-Boulez and Plotton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ingrid Plotton, aW5ncmlkLnBsb3R0b25AY2h1LWx5b24uZnI=