 Sarah E. Little-Letsinger
Sarah E. Little-Letsinger Sara E. Hamilton
Sara E. Hamilton
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Endocrinol. , 10 May 2023
Sec. Bone Research
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1168552
This article is part of the Research Topic Insights in Bone Research: 2022 View all 6 articles
The skeletal and immune systems are intricately intertwined within the bone marrow microenvironment, a field of study termed osteoimmunology. Osteoimmune interactions are key players in bone homeostasis and remodeling. Despite the critical role of the immune system in bone health, virtually all animal research in osteoimmunology, and more broadly bone biology, relies on organisms with naïve immune systems. Drawing on insights from osteoimmunology, evolutionary anthropology, and immunology, this perspective proposes the use of a novel translational model: the dirty mouse. Dirty mice, characterized by diverse exposures to commensal and pathogenic microbes, have mature immune systems comparable to adult humans, while the naïve immune system of specific-pathogen free mice is akin to a neonate. Investigation into the dirty mouse model will likely yield important insights in our understanding of bone diseases and disorders. A high benefit of this model is expected for diseases known to have a connection between overactivation of the immune system and negative bone outcomes, including aging and osteoporosis, rheumatoid arthritis, HIV/AIDS, obesity and diabetes, bone marrow metastases, and bone cancers.
Osteoimmunology is a field of study concerning the interface between the skeletal and immune systems (1). Arron & Choi first coined this term in 2000 in a Letter to Nature discussing then-recently published work by Takayanagi et al. (1, 2) on T-cell mediated regulation of osteoclasts. At the time, the molecular mechanisms by which immune cells could regulate bone homeostasis had not yet been elucidated. In 1999, Wong et al. (3) discovered tumor necrosis factor-related activation-induced cytokine (TRANCE; also known as RANKL, OPGL) expressed on the surface of activated T cells stimulates osteoclast differentiation and activity. Wong et al.’s work led Arron & Choi to remark on an important observation: “T cells are working constantly to fight off the universe of foreign particles in which we live … What prevents these T cells from causing extensive bone loss?” (1, 3). Cue Takayanagi et al. – their 2000 Letter to Nature dissects a mechanism to prevent uncontrolled bone loss during inflammatory T-cell responses via IFNγ degradation of tumor necrosis factor receptor-associated factor 6 (TRAF6) in osteoclasts to prevent osteoclast differentiation and activity (2). Arron & Choi describe Takayanagi et al.’s work as “the answer”, but these studies, and the entirety of rodent research from the 1960s on, have relied on specific pathogen-free (SPF) mice with little to no exposure to this “universe of foreign particles.
A year prior to Arron & Choi and Takayanagi et al., Kung et al. (4) published a novel paradigm demonstrating T-cell-derived RANK is a crucial mediator of osteoclastogenesis in a rodent model of rheumatoid arthritis. Nearly a quarter of a century later, it is well established that T-cells are causal in inflammatory bone loss, including in HIV/AIDS, inflammatory bowel diseases, and osteoporosis in addition to rheumatoid arthritis (4–11). Indeed, the term ‘immunoporosis’ was recently coined in 2018 by Rupesh Srivastava and colleagues to highlight the importance of the immune system in the pathophysiology of osteoporosis (7). Immune cell activity is tightly linked to bone homeostasis and remodeling (12). This is perhaps best exemplified by osteoclasts, which are tissue-resident macrophages that resorb bone derived from the monocyte/macrophage lineage of hematopoietic stem cells. The monocyte/macrophage lineage is an instrumental component of the innate immune system, the first responders to invading pathogens and major producers of inflammatory cytokines.
Inflammatory cytokines are the communication molecules of the immune system – in other words, immune cell production, activation, and activity is coordinated through release and uptake of pro- and anti-inflammatory cytokines (13, 14). This means that production of inflammatory cytokines is a direct result of immune activation or detection of invading pathogens (13, 14). In relation to bone, there is an abundance of evidence describing inflammatory cytokines as key modulators, both positive and negative, of bone mass (15–29); interleukin (IL) 1, IL-6, IL-17, and TNFα are among the most commonly studied cytokines associated with inflammatory bone loss (7, 30, 31). Given the intertwining of the skeletal and immune systems, understanding and effectively modeling the immune system is critical for the study of osteoimmunology, and more broadly, bone biology.
Modern humans have a more gracile, or slender, skeleton than earlier human ancestors and other apes. This gracility and the associated reductions in relative strength greatly increase the lifelong risk of osteoporosis (32). Skeletal gracilization has been documented in the cortical shaft of long bones (e.g., Increased periosteal and endocortical expansion explain increased cortical thickness, cortical bone volume fraction, estimated polar moment of inertia; (33)) and in trabecular bone at metaphyses surrounding joints (34–37). Temporally, skeletal gracilization coincides with the Neolithic Revolution (33, 38), the transition from hunter-gatherer societies to agricultural communities (e.g. farming, domestication of livestock) beginning in approximately 10,000 BCE (see Lewis et al. (39) for an insightful and detailed explanation of pathogens and the Neolithic revolution). The Neolithic Revolution signifies an increase in infectious disease and pandemics globally. This rise in pathogen burden is attributed to sustained increases in population density, greater contact between humans and animals, and poor sanitary conditions.
Much of the literature surrounding skeletal gracilization posits increased sedentism, defined as living in a fixed residential location for a long period of time, in modern humans as the primary cause (see Madimenos (40) for a stellar review on the topic). According to the literature, increased sedentism implies an increase in sedentary behavior and thus reductions in biomechanical loading. While the transition from a nomadic lifestyle to one characterized by permanent dwellings certainly involved an increase in sedentism, an important distinction should be made that living in one place for a long period of time fails to inform physical activity levels, or biomechanical loading. We know today that agricultural communities, like the Shuar in Ecuador and Tsimane in Bolivia, have high rates of physical activity throughout their lives, as well as modern farmers (41–44). Exercise modes differentially impact skeletal geometry and microarchitecture (45, 46); here, differential loading patterns resulting from the transition from endurance-based distance running and walking to a more strength training-based agricultural labor is likely more explanatory than reduced physical activity levels. Indeed, the relative deficits in femoral strength but not humeral strength documented by Ruff and colleagues (38) may represent a change in habitual behavior resulting in greater mechanical loading of the upper limbs (i.e. through farming, carrying), rather than decreased loading of the lower limbs. Notably, femoral and humeral strength have stabilized in the past 2000 years (38), suggesting environmental and/or behavioral factors associated with the Neolithic revolution are relevant.
High physical activity levels, relatively frequent periods of extreme caloric restriction and/or starvation, and increased pathogen burden are likely all key factors contributing to skeletal gracilization in modern humans – and particularly to the acceleration in skeletal gracilization coinciding with the Neolithic Revolution. While outside the scope of this paper, diet composition and caloric intake are highly relevant to understanding these changes and can be generally conceptualized during the Neolithic Revolution as including a loss of diversity and quality, as well as increased vulnerability to food shortages and periods of starvation (39, 47). Nutrition (quantity, quality) is a crucial component of both immune function and bone homeostasis, representing an area of study that would benefit from deep investigation along the same context described here.
Broad support for the concept that pathogen burden contributed to skeletal gracilization can be found in life history theory. Life history theory posits that an unfavorable environment, like one with hostile factors (e.g. pathogens), leads to defense-oriented metabolic programming in which energy is prioritized for protective responses at the expense of growth and reproduction. Caloric intake and energy availability, particularly when coupled with physical activity (48, 49), are primary regulators of bone mass accrual and remodeling – even when omitting pathogen exposure as a variable. From an evolutionary standpoint, immune defense would likely be prioritized at the expense of skeletal growth and bone mass; this is particularly likely when considering bone structurally remodels in such a way to compensate for size-induced detriments in bone strength (50). Among the Shuar (forager-horticulturalists of Ecuador), children re-route energy expenditure from growth and development to immune defense during active infections (51). The Tsimane (forager-horticulturalists of Bolivia) demonstrate significant prevalence of low bone mineral density despite habitually high physical activity levels and an absence of risk factors common in industrialized societies (52, 53). The Tsimane demonstrate a greater incidence of osteoporosis risk (23% Shuar vs. 49% Tsimane vs. 34% US) and fracture (women: 18% Tsimane vs. 9% US; men: 36% Tsimane vs. 11% US) than the Shuar and US citizens (54, 55). Notably, both the Shuar and Tsimane maintain moderate to vigorous physical activity levels across the lifespan and the Tsimane have a higher pathogen burden than the Shuar or Americans (52–56).
Our modern understanding of osteoimmune interactions and bone biology is built from studies conducted nearly exclusively with specific-pathogen free mice. To understand why this matters, let’s examine the work of Sjögren and colleagues (57) comparing conventionally raised and germ-free mice. Sjögren et al. demonstrated commensal (i.e., friendly or non-pathogenic) microbiota act as key mediators of bone health through modulation of the immune system (8, 57, 58). Germ-free SPF mice exhibited significantly greater bone mass at 7 weeks of age than conventionally raised SPF mice (8, 57, 58). Bone mass was causally reduced following reconstitution of microbiota in germ-free mice through expansion of the T-cell compartment and increased levels of inflammatory cytokines (8). Jones et al. went on to demonstrate “lack of immune cell activation”, and thereby decreased immune cell crosstalk via inflammatory cytokines, as the cause of elevated bone mass in germ-free mice (58). The work of Sjögren, Jones, and their colleagues led to a burst in research centering on the role of the gut microbiome in bone health. Still, the physiologic impact of commensal and pathogenic organisms remains untested. Presumably, as pathogenic microbes initiate a vigorous host immune response, whereas commensal microbes do not, the impact of pathogenic exposure would cause a more robust reduction in bone mass, or a greater attenuation in bone mass accrual.
Dirty mice (e.g., wild or pet store mice) are characterized by diverse commensal and pathogenic microbial exposures. Such diverse exposures to the “universe of foreign particles” induces recurrent immune activation (59–61). Recurrent immune activation stimulates persistent inflammatory signaling resulting in sustained and elevated production of inflammatory cytokines that suppress bone formation and enhance resorption (62, 63). Dirty mice can be produced in a laboratory by several different methods, including co-housing, fomite bedding exposure from dirty mice, and natural microbiota transfer or rewilding – the strengths and weakness of different methods have been reviewed by Hamilton et al. (59). Regardless of generation method, immune activation can be confirmed by testing for a wide range of pathogens [see (59) for common pathogens in dirty mice generated by three different methods] and analysis of serum (e.g. flow cytometry, blood cell panel) to confirm expected immune changes in response to infection (64). Rosshart and colleagues have also demonstrated significant differences in the microbiome (gut, skin, and vagina), gut mycobiome, and gut virome of SPF and dirty mice generated using a wilding model, where SPF C57Bl/6 embryos are transplanted into wild mice transferring pathogen-free microbiota (65, 66). Here, we describe the key features distinguishing specific-pathogen free mice (SPF) and dirty mice, focusing on dirty mouse models transferring both commensal and pathogenic microbes, and discuss the relevance to the field of osteoimmunology.
SPF mice maintain a high population of circulating naïve (~70%, CD62LhiCD44lo) CD8+ T cells and a low population of antigen-stimulated (~5%, CD62LloCD44hi) CD8+ T cells (60). This immune profile is consistent with that found in human neonates but stands in contrast to adult humans. Pet store mice demonstrate an immune profile consistent with adult humans; specifically, a low population of circulating naïve (~19%, CD62LhiCD44lo) and a high population of antigen-stimulated (~47%, CD62LloCD44hi) CD8+ T cells (60). Transfer of microbial exposures to SPF C57Bl/6 (B6) mice, via co-housing with pet store mice or their fomite bedding (64), induces sustained alterations to the basal immune profile like that seen in pet store mice and adult humans. While neither the total number of circulating immune cells nor the total number of CD4+ and CD8+ T cells differs between SPF and co-housed B6 mice, there are discrete differences in subtype, as in CD62L and CD44 expressing cells (60, 67). Co-housed B6 mice, in comparison to SPF B6 mice, exhibit elevations in circulating effector/effector memory CD4+ and CD8+ T cells, long-lived effector memory CD8+ T cells, monocytes, and neutrophils and reductions in B cells and natural killer cells (67).
In addition to circulating CD8+ T cells, Beura and colleagues demonstrate extensive changes to many innate and adaptive immune cell lineages in an array of tissues (60). To add further evidence, over 18,000 genes are differentially expressed between SPF and co-housed B6 mice; these data support gene expression profiles closely matching between immune naïve (i.e., SPF mice and neonates) and immune experienced (i.e., co-housed and pet store mice, adult humans) organisms (60). The transformation of the immune profile of the SPF B6 mouse following co-housing toward the profiles of pet store mice and adult humans, despite the differing genetic backgrounds, provides compelling causal evidence that immune system changes are due to environmental exposures.
As we know, inflammatory cytokines are the communication molecules of the immune system; therefore, given the immune cell changes in co-housed B6 we could reasonably expect altered levels of inflammatory cytokines. Indeed, co-housed B6 mice demonstrate robust elevations in over 20 inflammatory cytokines (Table 1), even two months after microbial exposure (67). These inflammatory cytokines largely promote osteoclast differentiation and activity while suppressing osteoblast differentiation and activity, either directly or indirectly; though elevated circulating levels of IL-5, IL-10, IL-13, and CCL5 were also detected in co-housed and pet-store mice, with these cytokines inhibiting osteoclastogenesis (102) or promoting osteogenesis (103, 104). Still, such robust and comprehensive elevation in systemic inflammation is significant, particularly given that it is a basal environmental feature in dirty mice as it is in humans (105). Our scientific understanding of how complex cytokine profiles affect bone homeostasis in health and disease would benefit from utilization of the dirty mouse model (Figure 1).
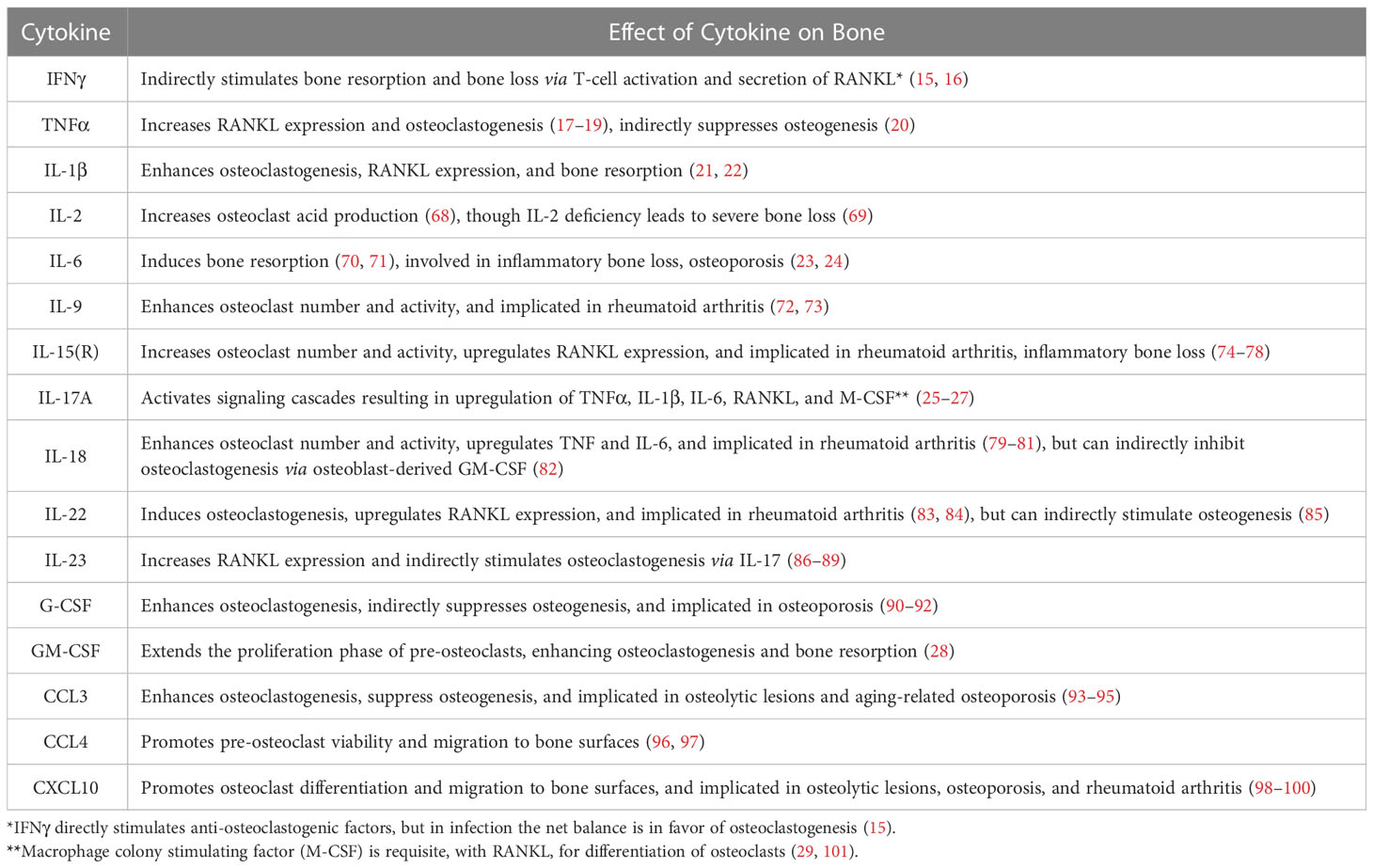
Table 1 Inflammatory cytokines directly and indirectly stimulate bone resorption and suppress bone formation.
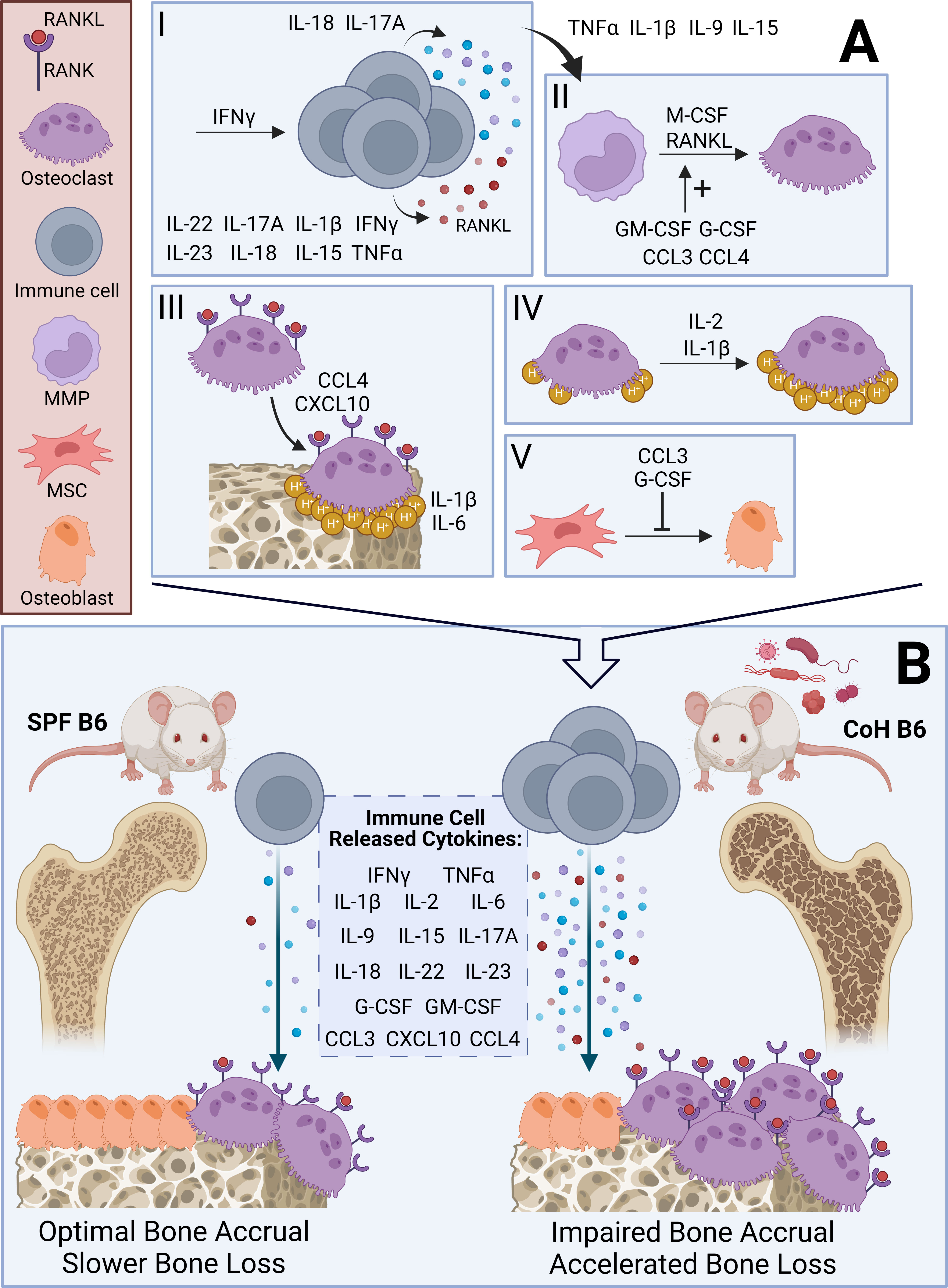
Figure 1 Hypothesized mechanisms and skeletal outcomes of osteoimmune interactions in SPF and Co-housed B6 mice. (A) Visual summary of effects of inflammatory cytokines outlined in Table 1 on the differentiation and activity of osteoclasts and osteoblasts. I – IFNγ promotes immune cell activation. TNFα, IFNγ, IL-1β, IL-15, IL-17A, IL-18, IL-22, and IL-23 augment immune cell-dependent RANKL secretion. IL-17A and IL-18 upregulate immune cell-dependent secretion of inflammatory cytokines [see Immune Cell Released Cytokines in (B)]. II –TNFα, IL-1β, IL-9, and IL-15 enhance osteoclastogenesis induced by immune cell-dependent cytokines, including RANKL. GM-CSF, G-CSF, CCL3, and CCL4 increase differentiation of macrophage-monocyte progenitors (MMP) into osteoclasts, which requires M-CSF and RANKL. III – CCL4 and CXCL10 promote migration of osteoclasts to bone surfaces. At the bone surface, IL-1β and IL-6 promote bone resorption. IV – IL-1β and IL-2 increase acid (H+) production by osteoclasts. V – G-CSF and CCL3 inhibit mesenchymal stem cell (MSC) differentiation into osteoblasts. (B) Visual summary of effect of diverse microbial exposures in co-housed (CoH) B6 mice compared to SPF B6 mice. CoH B6 mice have an expanded T cell compartment and greater levels of immune cell-dependent inflammatory cytokines (see Immune Cell Released Cytokines). We expect this to impair bone mass accrual during adolescence and accelerate bone loss during aging leading to an osteoporotic phenotype via mechanisms outlined in (A). We contrast CoH B6 mice with SPF B6 mice, which demonstrate a reduced T cell compartment and minimal production of inflammatory cytokines.
It is difficult to believe that the basal inflammatory environment in an organism with a mature immune system would not impair bone mass accrual prior to skeletal maturity and enhance bone resorption in aging. Even if no skeletal detriments occur from co-housing, this research will be valuable, potentially revealing novel osteoprotective therapeutic targets. In this event, one possible mechanism is in line with Takayanagi et al. (2): following T cell-mediated activation of the RANKL-TRAF6-NFκB-cSRC-JNK pathway that enhances osteoclast differentiation, activation, and survival, IFNy degrades TRAF6 within osteoclasts, preventing uncontrolled bone loss during inflammatory T-cell responses. Still, the work of Takayanagi et al. was performed in an organism with a naïve immune system, which we know demonstrates profound differences in gene expression and circulating immune cells and cytokines compared to an organism with a mature immune system. Much remains to be understood about the contributions of differing immune cell populations and immune cell-dependent inflammatory cytokine secretion in the context of bone biology and the bone marrow microenvironment. Experiments comparing bone homeostasis and remodeling between SPF and dirty mice are needed. Research areas linked to overactivation of the immune system, including aging and osteoporosis, rheumatoid arthritis, HIV/AIDS, obesity and diabetes, bone marrow metastases and bone cancers, will likely benefit from insights derived from the dirty mouse model.
Adoption of the SPF model was intended to reduce the complexity of in vivo experimental systems, but the complexity arising from diverse microbial exposures isn’t noise. In the words of Maizels and Nussey, it “represents the genetic and environmental framework in which the immune system evolved and functions” (106). While the decades of experimental research using SPF mice have undoubtedly been valuable, it is time to embrace the complexity. Drawing on insights from osteoimmunology, evolutionary anthropology, and immunology in the above sections, we argue that the dirty mouse model will improve our understanding and treatment of bone loss caused by inflammation and immune activation. With rates of osteoporosis exceeding 1 in 3 women and 1 in 5 men globally, as well as the steep economic and health costs of an osteoporotic fracture, new therapeutic targets and interventions are needed (107). To identify these new targets, novel paradigms and models will need to be considered.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
SL-L conceptualized and drafted the manuscript, table, and figure. SH critically evaluated and edited the manuscript, table, and figure. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arron JR, Choi Y. Bone versus immune system. nat 2000 4086812 (2000). Available at: https://www.nature.com/articles/35046196.
2. Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, et al. T-Cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-γ. nat 2000 4086812 (2000). Available at: https://www.nature.com/articles/35046102.
3. Wong BR, Josien R, Choi Y. TRANCE is a TNF family member that regulates dendritic cell and osteoclast function. J Leukoc Biol (1999) 65(6):715–24. doi: 10.1002/jlb.65.6.715. cited 2023 Jan 3.
4. Kung YY, Felge U, Sarosi I, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. nat 1999 4026759 (1999). Available at: https://www.nature.com/articles/46303.
5. Manavalan JS, Arpadi S, Tharmarajah S, Shah J, Zhang CA, Foca M, et al. Abnormal bone acquisition with early-life HIV infection: role of immune activation and senescent osteogenic precursors. J Bone Miner Res (2016) 31(11):1988–96. doi: 10.1002/jbmr.2883
6. Toben D, Schroeder I, El Khassawna T, Mehta M, Hoffmann JE, Frisch JT, et al. Fracture healing is accelerated in the absence of the adaptive immune system. J Bone Miner Res (2011) 26(1):113–24. doi: 10.1002/jbmr.185
7. Srivastava RK, Dar HY, Mishra PK. Immunoporosis: immunology of osteoporosis-role of T cells. Front Immunol (2018) 9(APR):1–12. doi: 10.3389/fimmu.2018.00657
8. Ohlsson C, Sjögren K. Effects of the gut microbiota on bone mass. Trends Endocrinol Metab (2015) 26(2):69–74. doi: 10.1016/j.tem.2014.11.004
9. Cline-Smith A, Axelbaum A, Shashkova E, Chakraborty M, Sanford J, Panesar P, et al. Ovariectomy activates chronic low-grade inflammation mediated by memory T cells, which promotes osteoporosis in mice. J Bone Miner Res (2020) 35(6):1174–87. doi: 10.1002/jbmr.3966
10. Szafors P, Che H, Barnetche T, Morel J, Gaujoux-Viala C, Combe B, et al. Risk of fracture and low bone mineral density in adults with inflammatory bowel diseases. a systematic literature review with meta-analysis. Osteoporos Int (2018) 29(11):2389–97. doi: 10.1007/s00198-018-4586-6
11. Bernstein CN, Blanchard JF, Leslie W, Wajda A, Yu BN. The incidence of fracture among patients with inflammatory bowel disease. a population-based cohort study. Ann Intern Med (2000) 133(10):795–99. doi: 10.7326/0003-4819-133-10-200011210-00012
12. Walsh MC, Takegahara N, Kim H, Choi Y. Updating osteoimmunology: regulation of bone cells by innate and adaptive immunity. Nat Rev Rheumatol (2017) 14(3):146–56. doi: 10.1038/nrrheum.2017.213
13. Kany S, Vollrath JT, Relja B. Cytokines in inflammatory disease. Int J Mol Sci (2019) 20(23). doi: 10.3390/ijms20236008
14. Zhang JM, An J. Cytokines, inflammation and pain. Int Anesthesiol Clin (2007) 45(2):27. doi: 10.1097/AIA.0b013e318034194e
15. Gao Y, Grassi F, Ryan MR, Terauchi M, Page K, Yang X, et al. IFN-γ stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J Clin Invest (2007) 117(1):122–32. doi: 10.1172/JCI30074
16. Tang M, Tian L, Luo G, Yu X. Interferon-Gamma-Mediated osteoimmunology. Front Immunol (2018) 9(June). doi: 10.3389/fimmu.2018.01508
17. Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, et al. Tumor necrosis factor α stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med (2000) 191(2):275–85. doi: 10.1084/jem.191.2.275
18. Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest (2000) 106(12):1481–8. doi: 10.1172/JCI11176
19. Nanes MS. Tumor necrosis factor-α: molecular and cellular mechanisms in skeletal pathology. Gene (2003) 321(1–2):1–15. doi: 10.1016/S0378-1119(03)00841-2
20. Kaneki H, Guo R, Chen D, Yao Z, Schwarz EM, Zhang YE, et al. Tumor necrosis factor promotes Runx2 degradation through up-regulation of Smurf1 and Smurf2 in osteoblasts. J Biol Chem (2006) 281(7):4326–33. doi: 10.1074/jbc.M509430200
21. Shiratori T, Kyumoto-Nakamura Y, Kukita A, Uehara N, Zhang J, Koda K, et al. IL-1β induces pathologically activated osteoclasts bearing extremely high levels of resorbing activity: a possible pathological subpopulation of osteoclasts, accompanied by suppressed expression of kindlin-3 and talin-1. J Immunol (2018) 200(1):218–28. doi: 10.4049/jimmunol.1602035
22. Leite FRM, de Aquino SG, Guimarães MR, Cirelli JA, Junior CR. RANKL expression is differentially modulated by TLR2 and TLR4 signaling in fibroblasts and osteoblasts. Immunol Innov (2014) 2(1):1. doi: 10.7243/2053-213X-2-1
23. Scheidt-Nave C, Bismar H, Leidig-Bruckner G, Woitge H, Seibel MJ, Ziegler R, et al. Serum interleukin 6 is a major predictor of bone loss in women specific to the first decade past menopause. J Clin Endocrinol Metab (2001) 86(5):2032–42. doi: 10.1210/jc.86.5.2032
24. Le Goff B, Blanchard F, Berthelot JM, Heymann D, Maugars Y. Role for interleukin-6 in structural joint damage and systemic bone loss in rheumatoid arthritis. Jt Bone Spine (2010) 77(3):201–5. doi: 10.1016/j.jbspin.2010.03.002
25. Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJJ, Kolls JK, et al. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-κB Ligand/Osteoprotegerin balance. J Immunol (2003) 170(5):2655–62. doi: 10.4049/jimmunol.170.5.2655
26. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T Cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med (1996) 183(6):2593–603. doi: 10.1084/jem.183.6.2593
27. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol [Internet] (1998) 160(7):3513–21. doi: 10.4049/jimmunol.160.7.3513
28. Ruef N, Dolder S, Aeberli D, Seitz M, Balani D, Hofstetter W. Granulocyte-macrophage colony-stimulating factor-dependent CD11c-positive cells differentiate into active osteoclasts. Bone (2017) 97:267–77. doi: 10.1016/j.bone.2017.01.036
29. Mun SH, Park PSU, Park-Min KH. The m-CSF receptor in osteoclasts and beyond. Exp Mol Med (2020) 52(8):1239–54. doi: 10.1038/s12276-020-0484-z
30. Coury F, Peyruchaud O, Machuca-Gayet I. Osteoimmunology of bone loss in inflammatory rheumatic diseases. Front Immunol (2019) 10(APR):1–9. doi: 10.3389/fimmu.2019.00679
31. Redlich K, Smolen JS. Inflammatory bone loss: pathogenesis and therapeutic intervention. Nat Rev Drug Discovery (2012) 11(3):234–50. doi: 10.1038/nrd3669
32. Weaver CM, Gordon CM, Janz KF, Kalkwarf HJ, Lappe JM, Lewis R, et al. The national osteoporosis foundation’s position statement on peak bone mass development and lifestyle factors: a systematic review and implementation recommendations. Osteoporos Int (2016) 27(4):1281–386. doi: 10.1007/s00198-015-3440-3
33. Ruff CB, Trinkaus E, Walker A, Larsen CS. Postcranial robusticity in homo. I: temporal trends and mechanical interpretation. Am J Phys Anthropol (1993) 91(1):21–53. doi: 10.1002/ajpa.1330910103
34. Chirchir H, Kivell TL, Ruff CB, Hublin JJ, Carlson KJ, Zipfel B, et al. Recent origin of low trabecular bone density in modern humans. Proc Natl Acad Sci U.S.A. (2015) 112(2):366–71. doi: 10.1073/pnas.1411696112
35. Hernandez CJ, Loomis DA, Cotter MM, Schifle AL, Anderson LC, Elsmore L, et al. Biomechanical allometry in hominoid thoracic vertebrae. J Hum Evol (2009) 56(5):462–70. doi: 10.1016/j.jhevol.2008.12.007
36. Cotter MM, Loomis DA, Simpson SW, Latimer B, Hernandez CJ, Larsen CS. Human evolution and osteoporosis-related spinal fractures. PloS One (2011) 6(10):e26658. doi: 10.1371/journal.pone.0026658
37. Ryan TM, Shaw CN. Gracility of the modern homo sapiens skeleton is the result of decreased biomechanical loading. Proc Natl Acad Sci U.S.A. (2015) 112(2):372–7. doi: 10.1073/pnas.1418646112
38. Ruff CB, Holt B, Niskanen M, Sladek V, Berner M, Garofalo E, et al. Gradual decline in mobility with the adoption of food production in Europe. Proc Natl Acad Sci U S A (2015) 112(23):7147–52. doi: 10.1073/pnas.1502932112
39. Cecil M, Lewis J, Akinyi MY, DeWitte SN, Stone AC. Ancient pathogens provide a window into health and well-being. Proc Natl Acad Sci (2023) 120(4):e2209476119. doi: 10.1073/pnas.2209476119
40. Madimenos FC. An evolutionary and life-history perspective on osteoporosis. Annu Rev Anthropol (2015) 44(1):189–206. doi: 10.1146/annurev-anthro-102214-013954
41. Pontzer H, Wood BM, Raichlen DA. Hunter-gatherers as models in public health. Obes Rev (2018) 19:24–35. doi: 10.1111/obr.12785
42. Loughman TM, Flaherty GT, Houlihan A, Dunne D. A cross-sectional analysis of physical activity patterns, aerobic capacity and perceptions about exercise among Male farmers in the mid-West region of Ireland. J Agromedicine (2022) 27(1):87–97. doi: 10.1080/1059924X.2021.1879699
43. Bassett DR, Schneider PL, Huntington GE. Physical activity in an old order Amish community. Med Sci Sports Exerc (2004) 36(1):79–85. doi: 10.1249/01.MSS.0000106184.71258.32
44. Dufour DL, Piperata BA. Energy expenditure among farmers in developing countries: what do we know? Am J Hum Biol (2008) 20(3):249–58. doi: 10.1002/ajhb.20764
45. Mohammad Rahimi GR, Smart NA, Liang MTC, Bijeh N, Albanaqi AL, Fathi M, et al. The impact of different modes of exercise training on bone mineral density in older postmenopausal women: a systematic review and meta-analysis research. Calcif Tissue Int (2020) 106(6):577–90. doi: 10.1007/s00223-020-00671-w
46. Martyn-St James M, Carroll S. A meta-analysis of impact exercise on postmenopausal bone loss: the case for mixed loading exercise programmes. Br J Sports Med (2009) 43(12):898–908. doi: 10.1136/bjsm.2008.052704
47. Larsen CS. Animal source foods and human health during evolution. J Nutr (2003) 133(11):3893S–7S. doi: 10.1093/jn/133.11.3893S
48. Little-Letsinger SE, Pagnotti GM, McGrath C, Styner M. Exercise and diet: uncovering prospective mediators of skeletal fragility in bone and marrow adipose tissue. Curr Osteoporos Rep (2020) 18(6):774–89. doi: 10.1007/s11914-020-00634-y
49. Papageorgiou M, Dolan E, Elliott-Sale KJ, Sale C. Reduced energy availability: implications for bone health in physically active populations. Eur J Nutr (2018) 57(3):847–59. doi: 10.1007/s00394-017-1498-8
50. Hart NH, Nimphius S, Rantalainen T, Ireland A, Siafarikas A, Newton RU. Mechanical basis of bone strength: influence of bone material, bone structure and muscle action. J Musculoskelet Neuronal Interact (2017) 17(3):114–39.
51. Urlacher SS, Ellison PT, Sugiyama LS, Pontzer H, Eick G, Liebert MA, et al. Tradeoffs between immune function and childhood growth among Amazonian forager-horticulturalists. Proc Natl Acad Sci (2018) 115(17):E3914–21. doi: 10.1073/pnas.1717522115
52. Stieglitz J, Madimenos F, Kaplan H, Gurven M. Calcaneal quantitative ultrasound indicates reduced bone status among physically active adult forager-horticulturalists. J Bone Miner Res (2016) 31(3):663–71. doi: 10.1002/jbmr.2730
53. Stieglitz J, Beheim BA, Trumble BC, Madimenos FC, Kaplan H, Gurven M. Low mineral density of a weight-bearing bone among adult women in a high fertility population. Am J Phys Anthropol (2015) 156(4):637–48. doi: 10.1002/ajpa.22681
54. Madimenos FC, Liebert MA, Cepon-Robins TJ, Urlacher SS, Josh Snodgrass J, Sugiyama LS, et al. Disparities in bone density across contemporary Amazonian forager-horticulturalists: cross-population comparison of the tsimane and shuar. Am J Phys Anthropol (2020) 171(1):50–64. doi: 10.1002/ajpa.23949
55. Stieglitz J, Trumble BC, Finch CE, Dong L, Budoff MJ, Kaplan H, et al. Computed tomography shows high fracture prevalence among physically active forager-horticulturalists with high fertility. Elife (2019) 8. doi: 10.7554/eLife.48607
56. Blackwell AD, Gurven MD, Sugiyama LS, Madimenos FC, Liebert MA, Martin MA, et al. Evidence for a peak shift in a humoral response to helminths: age profiles of IgE in the shuar of Ecuador, the tsimane of Bolivia, and the U.S. NHANES. PloS Negl Trop Dis (2011) 5(6):e1218. doi: 10.1371/journal.pntd.0001218
57. Sjögren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, et al. The gut microbiota regulates bone mass in mice. J Bone Miner Res (2012) 27(6):1357–67. doi: 10.1002/jbmr.1588
58. Jones RM, Mulle JG, Paci R. Osteomicrobiology: the influence of gut microbiota on bone in health and disease. Bone (2018) 115:59–67. doi: 10.1016/j.bone.2017.04.009
59. Hamilton SE, Badovinac VP, Beura LK, Pierson M, Jameson SC, Masopust D, et al. New insights into the immune system using dirty mice. J Immunol (2020) 205(1):3–11. doi: 10.4049/jimmunol.2000171
60. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature (2016) 532(7600):512–6. doi: 10.1038/nature17655
61. Sjaastad FV, Pierson M, Huggins MA, Danahy DB, Badovinac VP, Griffith TS, et al. Physiological microbial exposure has the capacity to substantially influence inflammatory responses in vivo. J Immunol (2018) 200(1 Supplement):108–9. doi: 10.4049/jimmunol.200.Supp.108.9
62. Schett G, Kiechl S, Weger S, Pederiva A, Mayr A, Petrangeli M, et al. High-sensitivity c-reactive protein and risk of nontraumatic fractures in the bruneck study. Arch Intern Med (2006) 166(22):2495–501. doi: 10.1001/archinte.166.22.2495
63. Takayanagi H. Osteoimmunology: shared mechanisms and crosstalk between the immune and bone systems. Nat Rev Immunol (2007) 7:292–304. doi: 10.1038/nri2062
64. Pierson M, Merley A, Hamilton SE. Generating mice with diverse microbial experience. Curr Protoc (2021) 1(2):e53. doi: 10.1002/cpz1.53
65. Rosshart SP, Herz J, Vassallo BG, Hunter A, Wall MK, Badger JH, et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Sci (80- ) (2019) 365(6452). doi: 10.1126/science.aaw4361
66. Rosshart SP, Vassallo BG, Angeletti D, Hutchinson DS, Morgan AP, Takeda K, et al. Wild mouse gut microbiota promotes host fitness and improves disease resistance. Cell (2017) 171(5):1015–28.e13. doi: 10.1016/j.cell.2017.09.016
67. Huggins MA, Sjaastad FV, Pierson M, Kucaba TA, Swanson W, Staley C, et al. Microbial exposure enhances immunity to pathogens recognized by TLR2 but increases susceptibility to cytokine storm through TLR4 sensitization. Cell Rep (2019) 28(7):1729–43.e5. doi: 10.1016/j.celrep.2019.07.028
68. Ries WL, Seeds MC, Key LL. Interleukin-2 stimulates osteoclastic activity: increased acid production and radioactive calcium release. J Periodontal Res (1989) 24(4):242–6. doi: 10.1111/j.1600-0765.1989.tb01788.x
69. Ashcroft AJ, Cruickshank SM, Croucher PI, Perry MJ, Rollinson S, Lippitt JM, et al. Colonic dendritic cells, intestinal inflammation, and T cell-mediated bone destruction are modulated by recombinant osteoprotegerin. Immunity (2003) 19(6):849–61. doi: 10.1016/S1074-7613(03)00326-1
70. Ishimi Y, Miyaura C, Jin CH, Akatsu T, Abe E, Nakamura Y, et al. IL-6 is produced by osteoblasts and induces bone resorption. J Immunol (1990) 145:3297–303. doi: 10.4049/jimmunol.145.10.3297
71. Wong PKK, Quinn JMW, Sims NA, Van Nieuwenhuijze A, Campbell IK, Wicks IP. Interleukin-6 modulates production of T lymphocyte-derived cytokines in antigen-induced arthritis and drives inflammation-induced osteoclastogenesis. Arthritis Rheumatol (2006) 54(1):158–68. doi: 10.1002/art.21537
72. Chakraborty S, Schneider J, Mitra DK, Kubatzky KF. Mechanistic insight of interleukin-9 induced osteoclastogenesis. Immunology (2023), 1–14. doi: 10.1111/imm.13630
73. Kar S, Gupta R, Malhotra R, Sharma V, Farooque K, Kumar V, et al. Interleukin-9 facilitates osteoclastogenesis in rheumatoid arthritis. Int J Mol Sci (2021) 22(19):10397. doi: 10.3390/ijms221910397
74. Takeda H, Kikuchi T, Soboku K, Okabe I, Mizutani H, Mitani A, et al. Effect of IL-15 and natural killer cells on osteoclasts and osteoblasts in a mouse coculture. Inflammation (2014) 37(3):657–69. doi: 10.1007/s10753-013-9782-0
75. Djaafar S, Pierroz DD, Chicheportiche R, Zheng XX, Ferrari SL, Ferrari-Lacraz S. Inhibition of T cell–dependent and RANKL-dependent osteoclastogenic processes associated with high levels of bone mass in interleukin-15 receptor–deficient mice. Arthritis Rheum (2010) 62(11):3300–10. doi: 10.1002/art.27645
76. Okabe I, Kikuchi T, Mogi M, Takeda H, Aino M, Kamiya Y, et al. IL-15 and RANKL play a synergistically important role in osteoclastogenesis. J Cell Biochem (2017) 118(4):739–47. doi: 10.1002/jcb.25726
77. Feng S, Madsen SH, Viller NN, Neutzsky-Wulff AV, Geisler C, Karlsson L, et al. Interleukin-15-activated natural killer cells kill autologous osteoclasts via LFA-1, DNAM-1 and TRAIL, and inhibit osteoclast-mediated bone erosion in vitro. Immunology (2015) 145(3):367–79. doi: 10.1111/imm.12449
78. Ferrari-Lacraz S, Zanelli E, Neuberg M, Donskoy E, Kim YS, Zheng XX, et al. Targeting IL-15 receptor-bearing cells with an antagonist mutant IL-15/Fc protein prevents disease development and progression in murine collagen-induced arthritis. J Immunol (2004) 173(9):5818–26. doi: 10.4049/jimmunol.173.9.5818
79. Dai SM, Nishioka K, Yudoh K. Interleukin (IL) 18 stimulates osteoclast formation through synovial T cells in rheumatoid arthritis: comparison with IL1β and tumour necrosis factor α. Ann Rheum Dis (2004) 63(11):1379–86. doi: 10.1136/ard.2003.018481
80. Gracie JA, Forsey RJ, Chan WL, Gilmour A, Leung BP, Greer MR, et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J Clin Invest (1999) 104(10):1393–401. doi: 10.1172/JCI7317
81. Olee T, Hashimoto S, Quach J, Lotz M. IL-18 is produced by articular chondrocytes and induces proinflammatory and catabolic responses. J Immunol (1999) 162(2):1096–100. doi: 10.4049/jimmunol.162.2.1096
82. Udagawa N, Horwood NJ, Elliott J, Mackay A, Owens J, Okamura H, et al. Interleukin-18 (Interferon-γ–inducing factor) is produced by osteoblasts and acts Via Granulocyte/Macrophage colony-stimulating factor and not Via interferon-γ to inhibit osteoclast formation. J Exp Med (1997) 185(6):1005–12. doi: 10.1084/jem.185.6.1005
83. Min HK, Won JY, Kim BM, Lee KA, Lee SJ, Lee SH, et al. Interleukin (IL)-25 suppresses IL-22-induced osteoclastogenesis in rheumatoid arthritis via STAT3 and p38 MAPK/IκBα pathway. Arthritis Res Ther (2020) 22(1):222. doi: 10.1186/s13075-020-02315-8
84. Kim KW, Kim HR, Park JY, Park JS, Oh HJ, Woo YJ, et al. Interleukin-22 promotes osteoclastogenesis in rheumatoid arthritis through induction of RANKL in human synovial fibroblasts. Arthritis Rheum (2012) 64(4):1015–23. doi: 10.1002/art.33446
85. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-γt+ CD3+CD4–CD8– entheseal resident T cells. Nat Med (2012) 18(7):1069–76. doi: 10.1038/nm.2817
86. Shin HS, Sarin R, Dixit N, Wu J, Gershwin E, Bowman EP, et al. Crosstalk among IL-23 and DNAX activating protein of 12 kDa–dependent pathways promotes osteoclastogenesis. J Immunol (2015) 194(1):316–24. doi: 10.4049/jimmunol.1401013
87. Iwakura Y, Ishigame H. The IL-23/IL-17 axis in inflammation. J Clin Invest (2006) 116(5):1218–22. doi: 10.1172/JCI28508
88. Yago T, Nanke Y, Kawamoto M, Furuya T, Kobashigawa T, Kamatani N, et al. IL-23 induces human osteoclastogenesis via IL-17 in vitro, and anti-IL-23 antibody attenuates collagen-induced arthritis in rats. Arthritis Res Ther (2007) 9(5):1–12. doi: 10.1186/ar2297
89. Adamopoulos IE, Tessmer M, Chao CC, Adda S, Gorman D, Petro M, et al. IL-23 is critical for induction of arthritis, osteoclast formation, and maintenance of bone mass. J Immunol (2011) 187(2):951–9. doi: 10.4049/jimmunol.1003986
90. Lee MY, Fukunaga R, Lee TJ, Lottsfeldt JL, Nagata S. Bone modulation in sustained hematopoietic stimulation in mice. Blood (1991) 77(10):2135–41. doi: 10.1182/blood.V77.10.2135.2135
91. Takamatsu Y, Simmons PJ, Moore RJ, Morris HA, To LB, Leívesque JP. Osteoclast-mediated bone resorption is stimulated during short-term administration of granulocyte colony-stimulating factor but is not responsible for hematopoietic progenitor cell mobilization. Blood (1998) 92(9):3465–73. doi: 10.1182/blood.V92.9.3465
92. Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood (2005) 106(9):3020–7. doi: 10.1182/blood-2004-01-0272
93. Vallet S, Pozzi S, Patel K, Vaghela N, Fulciniti MT, Veiby P, et al. A novel role for CCL3 (MIP-1α) in myeloma-induced bone disease via osteocalcin downregulation and inhibition of osteoblast function. Leuk 2011 257 (2011) 25(7):1174–81. doi: 10.1038/leu.2011.43
94. Yu D, Zhang S, Ma C, Huang S, Xu L, Liang J, et al. CCL3 in the bone marrow microenvironment causes bone loss and bone marrow adiposity in aged mice. JCI Insight (2023) 8(1). doi: 10.1172/jci
95. Fu R, Liu H, Zhao S, Wang Y, Li L, Gao S, et al. Osteoblast inhibition by chemokine cytokine ligand3 in myeloma-induced bone disease. Cancer Cell Int (2014) 14(1):1–8. doi: 10.1186/s12935-014-0132-6
96. Xuan W, Feng X, Qian C, Peng L, Shi Y, Xu L, et al. Osteoclast differentiation gene expression profiling reveals chemokine CCL4 mediates RANKL-induced osteoclast migration and invasion via PI3K pathway. Cell Biochem Funct (2017) 35(3):171–7. doi: 10.1002/cbf.3260
97. Lee D, Shin KJ, Kim DW, Yoon KA, Choi YJ, Lee BNR, et al. CCL4 enhances preosteoclast migration and its receptor CCR5 downregulation by RANKL promotes osteoclastogenesis. Cell Death Dis (2018) 9(5):1–12. doi: 10.1038/s41419-018-0562-5
98. Sun GJ, Guo T, Chen Y, Xu B, Guo JH, Zhao JN. Significant pathways detection in osteoporosis based on the bibliometric network. Eur Rev Med Pharmacol Sci (2013) 17(1):1–7.
99. Lee JH, Kim HNHH, Kim KO, et al. CXCL10 promotes osteolytic bone metastasis by enhancing cancer outgrowth and osteoclastogenesis (2012). Available at: https://aacrjournals.org/cancerres/article/72/13/3175/575894/CXCL10-Promotes-Osteolytic-Bone-Metastasis-by.
100. Han BK, Ha H, Kim HN, Lee JH, Hun SK, Lee S, et al. Reciprocal cross-talk between RANKL and interferon-γ–inducible protein 10 is responsible for bone-erosive experimental arthritis. Arthritis Rheum (2008) 58(5):1332–42. doi: 10.1002/art.23372
101. Ross FP. M-CSF, c-fms, and signaling in osteoclasts and their precursors. Ann N Y Acad Sci (2006) 1068(1):110–6. doi: 10.1196/annals.1346.014
102. Zhou P, Zheng T, Zhao B. Cytokine-mediated immunomodulation of osteoclastogenesis. Bone (2022) 164. doi: 10.1016/j.bone.2022.116540
103. Wintges K, Beil FT, Albers J, Jeschke A, Schweizer M, Claass B, et al. Impaired bone formation and increased osteoclastogenesis in mice lacking chemokine (C-c motif) ligand 5 (Ccl5). J Bone Miner Res (2013) 28(10):2070–80. doi: 10.1002/jbmr.1937
104. Liu YC, Kao YT, Huang WK, Lin KY, Wu SC, Hsu SC, et al. CCL5/RANTES is important for inducing osteogenesis of human mesenchymal stem cells and is regulated by dexamethasone. Biosci Trends (2014) 8(3):138–43. doi: 10.5582/bst.2014.01047
105. Kleiner G, Marcuzzi A, Zanin V, Monasta L, Zauli G. Cytokine levels in the serum of healthy subjects. Mediators Inflammation (2013) 2013. doi: 10.1155/2013/434010
106. Maizels RM, Nussey DH. Into the wild: digging at immunology’s evolutionary roots. Nat Immunol (2013) 14(9):879–83. doi: 10.1038/ni.2643
Keywords: dirty mice, inflammatory bone loss, immune cells, skeletal gracilization, osteoporosis, bone homeostasis
Citation: Little-Letsinger SE and Hamilton SE (2023) Leveraging mice with diverse microbial exposures for advances in osteoimmunology. Front. Endocrinol. 14:1168552. doi: 10.3389/fendo.2023.1168552
Received: 17 February 2023; Accepted: 21 April 2023;
Published: 10 May 2023.
Edited by:
Jonathan H. Tobias, University of Bristol, United KingdomReviewed by:
Vikram Khedgikar, Brigham and Women’s Hospital and Harvard Medical School, United StatesCopyright © 2023 Little-Letsinger and Hamilton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sarah E. Little-Letsinger, c2FyYWguZS5saXR0bGVAZHVrZS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.