 Haocheng Zhang
Haocheng Zhang Yan Gu4
Yan Gu4
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 07 July 2023
Sec. Diabetes: Molecular Mechanisms
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1166756
This article is part of the Research TopicA Year in Review: Discussions in Diabetes: Molecular MechanismsView all 8 articles
Type 2 diabetes (T2D) is a metabolic disease with an increasing rate of incidence worldwide. Despite the considerable progress in the prevention and intervention, T2D and its complications cannot be reversed easily after diagnosis, thereby necessitating an in-depth investigation of the pathophysiology. In recent years, the role of epigenetics has been increasingly demonstrated in the disease, of which N6-methyladenosine (m6A) is one of the most common post-transcriptional modifications. Interestingly, patients with T2D show a low m6A abundance. Thus, a comprehensive analysis and understanding of this phenomenon would improve our understanding of the pathophysiology, as well as the search for new biomarkers and therapeutic approaches for T2D. In this review, we systematically introduced the metabolic roles of m6A modification in organs, the metabolic signaling pathways involved, and the effects of clinical drugs on T2D.
Type 2 diabetes (T2D) is a chronic metabolic condition characterized by high blood glucose levels, insulin resistance (IR), and insulin secretion deficiency. Over the past two decades, the number of people living with diabetes has more than tripled, from 151 million in 2000 to 537 million in 2021 (1). Thus, diabetes is becoming one of the fastest-growing metabolic disorders of the 21st century. T2D is the most common type worldwide, constituting about 90% of all diabetes cases (2). In addition to the rising number of people suffering from the disease, there has been a dramatic increase in the costs to healthcare systems and individual financial burdens due to T2D (3). Nonetheless, since the detection of the sequence of the human genome for the first time in 2001 (4), we gained new insights into the development of diseases with the development of omics. Based on these technologies, the pathogenic mechanisms underlying the rising prevalence of T2D can be explored further for refined phenotyping, prevention and treatment.
Epigenetics has been defined as reversible and heritable changes in gene functions without altering the DNA/RNA sequence that comprises DNA/RNA methylation, histone modifications, and noncoding RNA-mediated processes (5). Among various post-transcriptional RNA modifications, N6-methyladenosine (m6A) is the most abundant modification on RNA molecules and the most prevalent methylated nucleoside presented in eukaryotes (6). The m6A modification plays a biological role regulated by “writers” methylases, “erasers” demethylases, and “readers” TYH domain families (7). Several studies have shown that m6A can bind to mRNA and affect the expression of target genes, thus regulating various physiological molecular mechanisms, such as cell cycle, energy metabolism, and inflammation (8). Importantly, m6A modification can respond to changes in homeostasis of the internal environment, which might regulate the sustained long-term expression of T2D-related pathogenic genes induced by prior hyperglycemia exposure (9). This mechanism could explain the early pathogenesis of T2D. In the present review, we describe the perspectives and the role of m6A modification in T2D and discuss the novel insight into the pathogenesis of T2D that could be translated into novel biomarkers and therapeutic modalities.
Post-transcriptional RNAs can be modified by more than 170 diverse modifications analogous to various biomacromolecules (10). Several mRNA modifications, including m6A, 5-methylcytidine (m5C), and N1-methyladenosine (m1A), have been reported based on a transcriptome-wide mapping approach (11). Among this, m6A is presently the most common modification in mammalian mRNA and long non-coding RNA, which account for more than 50% of eukaryotic methylation modification, and approximately 0.1%-0.4% of all adenosines have m6A modification (12). The m6A modification has the highest distribution at the 3’ end of the transcripts, near the end of coding regions and at the last exon of the non-coding regions. Specifically, its main manifestations are G-(m6A)-C (70%) and A-(m6A)-C (30%) (13), whose distribution on RNA is 37%: 28%: 20%: 12%: 3% for coding sequence: stop codon: 3’-untranslated region: transcription stop site: 5’-untranslated region (14). The m6A modification plays a vital role in modulating RNA processing, degradation, and stability, catalyzed by a multi-component enzyme complex (also called “writers”), reversed by demethylases in the nucleus (termed as “erasers”), and regulated by YTH domain family (also known as “readers”) (7). Accumulating evidence has shown that the regulation of target genes by m6A modification and its effect in T2D depends on two factors: I. The abnormal level of m6A modification in T2D mainly depends on the expression and activity of “writers” and “erasers.” II. Targets are critical genes affecting glucose and lipid metabolism, insulin secretion, and IR.
To date, most of the studies have focused on “writers.” The most decisive part of m6A modification “writers” is a methyltransferase complex, composed of METTL3, METTL14, WTAP, and VIRMA (KIAA1429). METTL3 and METTL14 form a heterodimer, which is the main methylase catalyzing the transfer of S-adenosyl methionine (SAM) to bind to specific RNA sites, a well-conserved motif DRACH (D = G/A/U, R = G/A, H = A/U/C) (15). WTAP is a critical regulatory subunit of the complex, recruiting METTL3/14 heterodimer into nuclear speckles and promoting its RNA binding ability. VIRMA is the complex’s main scaffold that promotes RNA anchoring and is involved in mRNA polyadenylation (16). In addition, other “writers” consist of RBM15/15B, ZC3H13, ZCCHC4, METTL16, METTL4, and METTL5. RBM15/15B and ZC3H13 interact with WTAP, promoting the complex catalytic function (17). ZCCHC4 is mainly associated with the methylation of human 28S rRNA (18). METTL16 differs from METTL3/14 due to its interaction with eIF3 and rRNAs to facilitate methylation (19). METTL4 is mainly related to m6A modification in internal U2 snRNA (20), and METTL5 is associated with m6A at adenosine 1832 of 18S rRNA (21).
Compared to “writers,” the components of demethylase “erasers,” FTO and ALKBH5, are much simpler. Both belong to the ALKB family of non-heme Fe(II)dioxygenases, and the former is not only involved in removing m6A modification but also contributes to human obesity (22), whereas the latter is a crucial demethylase located in nuclear-nascent RNAs, with maximal expression in the testis, heart, and kidney (23).
In addition to “writers” and “erasers” that can directly methylate/demethylate RNAs, “readers” recognize and combine the m6A modification, which includes the YTHDF family, TYHDC family, IGF2BP family, hnRNPs, FMRP, eIF3, and PRRC2A. Among these, the star molecules, TYHDF and THYDC families are widely studied. The TYHDF family entails TYHDF1–3, of which TYHDF2 effectuates target transcript degradation through the deadenylation pathway or the endoriboncleolytic pathway, while TYHDF1/3 has opposite functions, binding to eIF3 to facilitate the translation of target transcripts (24). TYHDC1, a TYHDC family protein, could anchor to the m6A modification of mRNA and mediates mRNA splicing, but this effect is currently found only in Drosophila (25). In addition, it accelerates nuclear mRNA export by binding to the SR protein family (26). While YTHDC2 is involved in mRNA degradation and translation initiation (27). The ribosomal proteins, include the IGF2BP family and FMRP, also function in m6A modification, strengthening mRNA stability (28) (Figure 1).
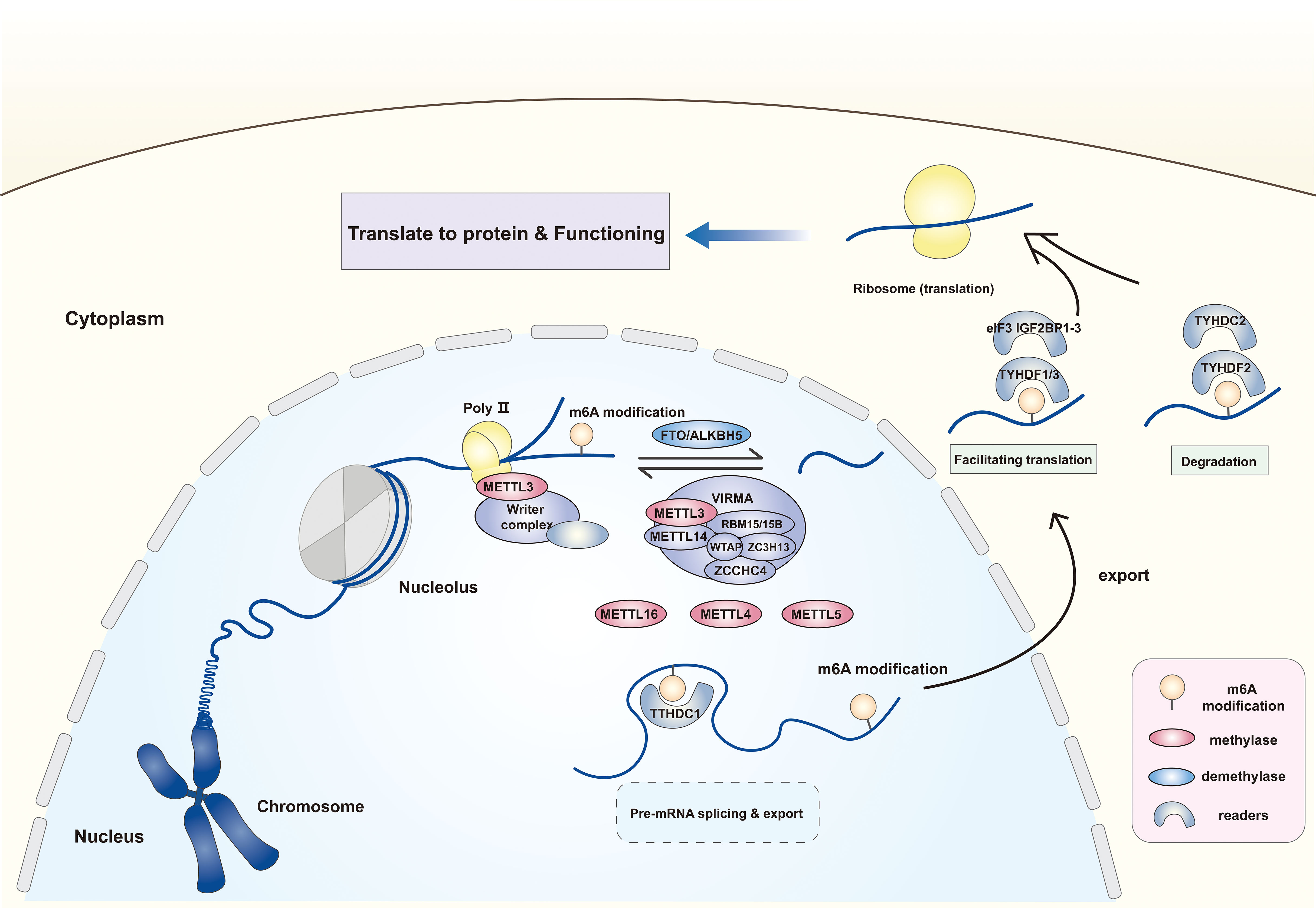
Figure 1 The expression location and form of function of m6A methylases, demethylases and readers. The m6A modification of RNA is dependent on the catalytic effect of methylases and demethylases. The former mainly includes methyltransferase complex with METTL3 as the core, METTL16, METTL4 and METTL5. The latter comprises the FTO and AKLKBH5. The m6A site on RNA can be recognized and combined by different “readers” to influence the RNA following effects by facilitating translation, splicing and affecting stability. METTL3/14/16/5/4, Methyltransferase-like 3/14/16/5/4; WTAP, Wilms tumor 1- associated protein; VIRMA, Vir-like m6A methyltransferase associated; RBM15/15B, RNA binding motif protein 15/15B; ZC3H13, ZCCHC4, zinc finger CCHC domain-containing protein 4; FTO, Fat mass and obesity-associated protein; ALKBH5, AlkB homologue 5; eIF3, Eukaryotic translation initiation factor 3; YTHDC1/2, YTH domain containing 1/2; YTHDF1-3, YTH N6-methyladenosine RNA binding protein 1-3; IGF2BP1-3, Insulin-like growth factor 2 mRNA binding protein 1-3.
To date, the detection methods for RNA modification are mainly based on mass spectrometry (MS) and high-throughput sequencing techniques. The former tends to be detected using liquid chromatography-MS (LC-MS) or tandem mass spectrometry (LC-MS/MS), which can detect the overall m6A abundance of mRNA and has high sensitivity and specificity as its detection is based on the physicochemical properties of m6A modification such as molecular mass and chromatographic retention time. However, it has higher requirements for sample handling, and if the sample has components of other RNAs, the source of m6A modification cannot be determined (29). In addition, LC-MS or LC-MS/MS does not provide sequence and localization information (28). High-throughput sequencing technology is another common method for m6A modification detection, which can determine the presence of mRNA m6A modification and its specific location in the transcriptome. But this approach relies on anti-m6A antibodies, which makes it impossible to obtain high-resolution information about the loci (30).
Typically, the development of T2D is the combination of impaired pancreas islet cells and metabolic disorders. Various causes of islet cell damage decrease the β-cell mass, leading to insulin deficiency. On the other hand, a massive accumulation of energy into adipose tissue, especially white adipose tissue, upregulates the adipose inflammatory factors, following which fat metabolites are transported into the liver and skeletal muscle leading to IR, further aggravating the metabolic disorder. These inflammatory factors also lead to islet cell stress, further damaging the islet cell biology (31). Therefore, we first described the role of m6A modification in the four major target organs of T2D (Figure 2).
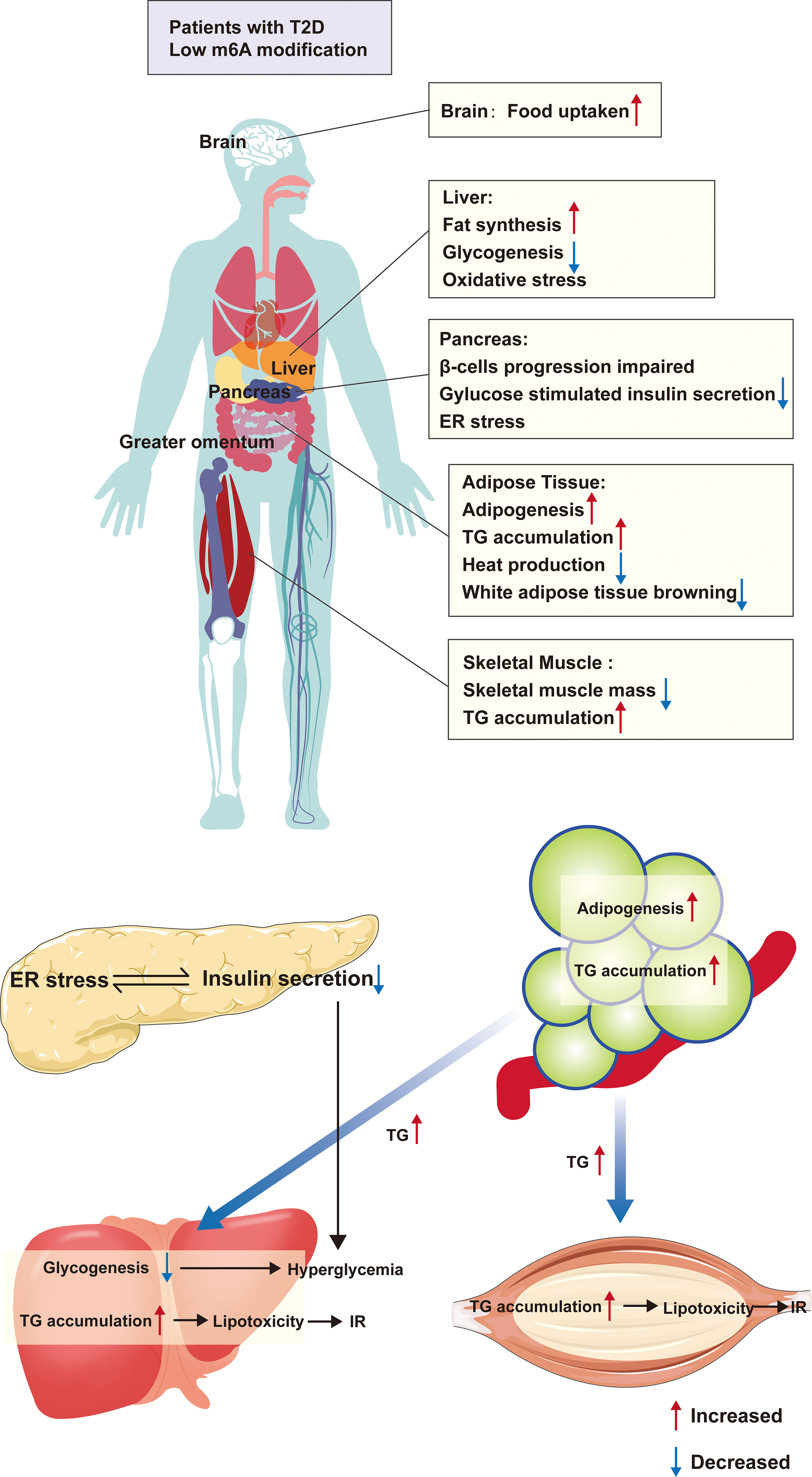
Figure 2 The m6A modification and organ metabolism in T2D. Patients with T2D are in a condition of low m6A modification, which causes functional abnormalities in multiple organs throughout the body. Firstly, in adipose tissue, there is excessive lipid droplet accumulation, increased white adipose tissue and decreased browning, which causes the release of large amounts of TG from adipose tissue into the circulation, and these TG absorbed by the liver and skeletal muscle, resulting in inflammation and IR due to lipotoxicity. Furthermore, the condition also directly affects the function of islet β-cells, leading to abnormal differentiation and ER stress, all of which cause a decrease in insulin secretion In addition, low m6A modification alters the expression of hepatic glycolipid genes, leading to hepatic steatosis as well as reduced glycogen synthesis. All of these further aggravates the hyperglycemic symptoms in T2D patients. ER, endoplasmic reticulum; TG, triglycerides; IR, insulin resistance.
As a response to endogenous or exogenous stimuli, such as glucose, lactose, and glucagon, the pancreatic β-cells release insulin, the only hypoglycemic hormone in the body. The loss of islet β-cell function is the decisive pathological mechanism leading to T2D (32). Accumulating evidence revealed that m6A modification is involved in β-cell dysfunction.
An in vivo study on the function of METTL14 in islet β-cells demonstrated that β-cell specific-knockout of Mettl14 mice leads to β-cells death and abnormal β-cell differentiation, contributing to low β-cell mass and insulin secretion (33). Another study (34) showed that EndoC-βH1 cell with Mettl3/14 knockdown induced G0/G1 cell cycle arrest and decreased insulin secretion. They also analyzed m6A hypomethylation-mediated transcription in vivo and in vitro, which mainly involved the cell cycle regulatory genes and insulin/insulin-like growth factor 1-serine/threonine kinase-pancreatic and duodenal homeobox 1 (insulin/IGF1-AKT-PDX1) pathway. Deleting Mettl3/14 decreased the insulin-induced AKT phosphorylation, resulting in low PDX1 expression, a vital transcription factor to maintain β-cells biology (35). Men et al. (36) further supplemented the role of METTL14 in β-cell biology. The results indicated that genes responsible for endoplasmic reticulum (ER) stress were upregulated in Mettl14 knockout islet β-cells, especially ER to nucleus signaling 1 (Ern1/Ire1α) and X-box protein binding 1 (Xbp-1). The former could respond to ER stress and unconventionally activate the latter mRNA, resulting in the overexpression of degraded proteins in the ER and slicing insulin mRNA (37). Wang et al. (38) reported that if endocrine progenitors lost Mettl3/14, they mature toward β-cells defectively, leading to early hyperglycemia. Moreover, the mechanism showed that loss of Mettl3/14 downregulates MAF bZIP transcription factor A (Mafa) mRNA stability. Interestingly, the abundance of MAFA protein decreased, and glucose-stimulated insulin secretion (GSIS) impaired the preceded loss of β-cell mass. Both studies observed m6A modification does not cause IR and rarely alters insulin sensitivity, indicating that dynamic m6A modification following internal environmental changes is involved in the early unknown pathophysiological development of T2D in the pancreas. This hypothesis could also be confirmed by the findings of Li et al. (39). Under inflammation and oxidative stress conditions, METTL3 level decreased in islet cells, and islet β-cell deficiency of Mettl3 further induces hyperglycemia. On the other hand, methylglyoxal (a precursor of the advanced glycation end product) treatment of β-cells downregulated the METTL3 level, affecting MAFA mRNA and protein levels and decreasing GSIS (40).In addition, a study illustrating the lack of WTAP in the T2D pancreas deserves our attention (41). Deficiency of Wtap in pancreas decreases the functional stability of MEETL3 protein, leading to pancreas hypomethylation and dysfunction. This implies that exploring other non-catalytic enzymes in the methylesterase complex to influence m6A levels might be another effective therapeutic option.
“Erasers” are also involved in β-cells. Especially, FTO is an obesity gene but also partakes in the mRNA m6A modification of other core proteins. Several cohort studies (42–45) demonstrated that many variants of Fto and Igf2bp2 are related to an upregulated risk of T2D occurrence, and both are involved in islet β-cell biology. However, the studies on the role of FTO are controversial and have completely opposite results. Interestingly, the Fto knockdown in vitro inhibited insulin secretion, which was interpreted as FTO-enhancing β-cell exocytosis to increase the first phase insulin secretion rather than only enhancing insulin biosynthesis (46). Another recent study (47) indicated that silenced Fto stimulates glucose-induced insulin secretion by upregulating the gene expression maintaining the identity of β-cells, such as paired box 4 (Pax4), glucokinase (Gck) and solute carrier family 2 member 2 (Slc2a2/Glut2). In addition, the study reported that m6A modification is reduced in human T2D islets through the upregulated expression of Fto and Albhk5 mRNA and revised subcellular localization into the major m6A modification sites. In comparison with the essential functions of “writers” and “readers” in pancreatic growth/maturation/function, “erasers” in the pancreas seems to be more like an auto-regulator, which only alters the m6A modification of insulin secretion-related genes.
Similarly, “readers,” the major component of m6A RNA modification, also play a critical role in β-cell biology. Regue et al. (48) reported that mice with Igf2bp2 deletion showed decreased leanness, increased energy consumption, and reduced insulin secretion. Mechanically, the IGF2BP2 binding site is the Pdx1 mRNA m6A modification site, which improves the stability of Pdx1 mRNA, facilitating its translation. A cross-sectional study from China also verified that IGF2BP2 and IGF2BP3 were elevated in pancreatic samples from T2D patients (49). Additionally, TYHDC1 is also critical for pancreas function. TYHDC1 is significantly downregulated in T2D individuals, and lack of Tyhdc1 in β-cells leads to chronic inflammation, insulin secretion damaged and hyperglycemia (50). In a previous review (51), Dai concluded that the association between IGF2BP2 single nucleotide polymorphisms (SNPs) and T2D primarily concentrated on β-cell function deletion rather than reduced insulin sensitivity.
Accumulating evidence showed that the m6A modification components in β-cells might contribute to the pathophysiology of T2D before its onset, while the mRNA hypomethylation characteristics of genes in the insulin/IGF1-PDX1 pathway in T2D islets might be novel targets for the early diagnosis of T2D.
In addition to β-cell biology dysfunction and progressive insulin secretion deficiency, IR is a critical pathophysiological mechanism in T2D. Some studies suggested that the development of IR is closely associated with adipose tissue. Hyperglycemia contributes to the bioconversion of glucose to triglycerides (TGs) that accumulate in the white adipose tissue. Excessive energy accumulation in adipose tissue triggers the release of free fatty acids (FFAs) transported to the liver and skeletal muscle, leading to IR (52). On the other hand, inflammatory factors of adipose tissue reduce insulin sensitivity in the liver and skeletal muscle and aggravate IR (53). Accumulating evidence demonstrated that an essential role of m6A modification in stem cell transformation into preadipocytes and preadipocytes differentiation into mature adipocytes (54).
Yao et al. (55) demonstrated that METTL3 and TYHDF2 are essential for bone marrow stem cell adipose differentiation. Furthermore, Mettl3 silencing enhances the expression of janus kinase 1/signal transducer and activator of transcription 5/CCAAT enhancer binding protein beta (JAK1/STAT5/C/EBP β) pathway and mRNA levels of peroxisome proliferator-activated receptor gamma (Pparγ), CCAAT enhancer binding protein alpha (C/ebpα), and fatty acid binding protein 4 (Fabp4) in adipogenesis. Mechanistically, siMettle3 decreases the methylation abundance on Jak1 mRNA, reducing the degradation of TYHDF2 due to m6A sites reduced in Jak1 mRNA and upregulating JAK1 protein expression and STAT5 phosphorylation, which then bind to C/EBPβ promoter to promote adipogenesis. Moreover, an enrichment analysis showed that developmental genes were enriched in Mettl3 and Mettl14 targets (56). Mitotic clonal expansion (MCE) is a mandatory process in adipogenesis; a previous study reported that WTAP recruits METTL3/14 heterodimer to regulate adipocyte differentiation (57). In another study (58), 3T3-L1 cells with siMettl3 showed that cyclin D1 (Ccnd1), a conserved cell cycle gene, reduced the m6A modification level to avoid TYHDF2 degradation. Wang et al. (59) reported that knockout of Mettl3 aggravated high-fat diet (HFD)-induced IR and obesity. An in vitro experiment showed that large lipid droplets accumulate in Mettl3-knockout cells via suppression of the mRNA and protein expression of uncoupling protein 1 (UCP1), PPARG coactivator 1 alpha (PCG-1α/PPARGC1A), PPARγ, and PR/SET domain 16 (PRDM16). A recent study on IR in obstructive sleep apnea syndrome showed that downregulation of Mettl3 increased the TG levels, which released FFAs, upregulated glycerol, inhibited glucose utilization in the other organs, and induced IR (60). Since the IR induced by obstructive sleep apnea syndrome is similar to the IR pathogenesis in T2D, this study was included in our review.
Demethylase, especially FTO, plays a crucial role in adipogenesis and adipose metabolism. In a previous study, Merkestein et al. (61) reported that Fto deficiency on adipogenesis obstructed the MCE process by attenuating the short isoform of RUNX1 partner transcriptional co-repressor 1 (RUNX1T1), suppressing the levels of cell proliferation and cell cycle genes, such as Ccnd1, Ccnd3, Fabp4, and C/edpα during MCE process. RUNX1T1 has two functionally distinct spliceosomes comprising short and long isoforms; expression of the former enhances, whereas expression of the latter impairs adipogenesis (62). In addition, in obese individuals, TG flow causes macrophage to aggregate in adipose tissue, leading to chronic inflammation and facilitating IR. And FTO upregulates oxidized low density lipoprotein-induced AMP-activated protein kinase (AMPK) activation in macrophages to enhance TG flowing (63). Adipose tissue inflammation is an essential factor contributing to IR, in which macrophages play an important role. It will be interesting to explore how m6A modification involves in the biology of macrophages in lipid metabolism. A previous study on mice with adipose-specific knockout of autophagy-related 5 (Atg5) and Atg7 showed that the effect of anti-IR and anti-obesity could effectively regulate fat mass via autophagy (64). Furthermore, the study found that FTO overexpression significantly enhanced the levels of Atg5/7 mRNA and promoted adipogenesis and TG accumulation by modulating the ATG5/7-C/EBPβ pathway. In another study (65), Fto deletion elevated the browning of white adipose tissue, promoting the conversion of TG accumulation in white adipose tissue into energy consumption in brown adipose tissue. Subsequently, the results suggested that FTO increases the m6A modification sites of hypoxia-inducible factor 1 subunit alpha (Hif1-α) mRNA, which is later discerned by YTHDC2 that promotes protein translation. In a recent review by Azzam et al. (66), the overexpression of FTO was shown to upregulate CCND1, CCND3, PPARγ, and C/EBPα in adipocytes to enhance adipogenesis, as well as regulate the skeletal muscle lipid droplet accumulation by attenuating the AMPK pathway. Fto is also identified as the strongest factor associated with obesity by the first genome-wide association studies (GWAS); it plays a remarkable role in adipogenesis. The SNPs in the first intron of the gene were significantly related to the risk of obesity and T2D (67–69). There are two debates about how Fto SNPs increase the risk of these diseases: I. Fto SNPs increase the expression of FTO, and it acts as an auto-regulator function to promote lipid metabolism genes in an m6A dependent manner (as we mentioned above), thus causing body adipogenesis (66, 70). II. Fto SNPs regulate promoter of other genes to function, such as iroquois homeobox 3 and iroquois homeobox 5 (71). Ghrelin is an appetite stimulating hormone and its acylation is essential for appetite stimulation. Fto rs9939609 increases the expression of FTO, and increased the mRNA of ghrelin in an m6A-dependent manner, and increased the expression of acyl-ghrelin (72).
The studies on “readers” are widely scattered. YTHDF1 overexpression is related to enhanced adipogenesis (73), while YTHDF2 degrades the corresponding genes’ mRNA that recognize the m6A modification sites affected by methylases and demethylases (55, 58); only YTHDF3 has been reported to be associated with obesity (74). TYHDC2 increases the stability of Hif1-α mRNA to promote its expression (65). GWAS found that IGF2BP2 is functionally enriched in energy expenditure and FFA oxidation (75) and is associated with obesity-susceptible T2D (76).
Although studies on the role of m6A modification in T2D adipose tissue have been rarely reported, we speculated its role based on the low m6A modification level in T2D patients (77). T2D is closely associated with white adipose tissue, and adipose tissue hypomethylation level further leads to adipocyte differentiation and proliferation, which promotes FFA access to the liver and skeletal muscle, leading to IR and pre-T2D. Thus, whether hyperglycemia in the body leads to hypomethylation and obesity or whether obesity alters the methylation degree needs further investigation, which would improve our understanding of the pathogenesis of T2D.
The liver is one of the main metabolic organs that controls the metabolic homeostasis of the human body. Therefore, the metabolic disorders in the liver, especially the glucose and lipid metabolism disorders, are related to the pathological mechanism of T2D (78). Current studies have demonstrated that hepatic glucose and lipid metabolism disorders involved m6A modification.
In a previous study, Xie et al. (79) reported that m6A modification and METTL3 were upregulated in the hepatic tissues of T2D patients and positively related to homeostatic model assessment (HOMA)-IR. In addition, hepatocyte-specific knockout of Mettle3 in high-fat diet (HFD)-fed mice improved insulin sensitivity and decreased fatty acid synthesis, which was mechanistically related to METTL3 targeting fatty acid synthase (Fasn) mRNA and increasing its stability. The role of METTL3 was supplemented in the study by Li et al. (80). They found that adeno-associated virus (AAV)-mediated hepatocyte-specific overexpression of Mettle3, which upregulated the HFD-induced liver metabolic disorders and IR. Furthermore, the mechanism showed that Mettl3 deletion advanced the mRNA half-life of the primary regulators of liver metabolism, such as Lpin1 and Lpin2. The inhibition of m6A modification via Mettl3 knockdown decreases Pparα mRNA m6A abundance and expression of sterol regulatory element-binding proteins-1c (Srebp-1c), Fasn, and acetyl-CoA carboxylase (Acc) mRNA and increases Pparα mRNA half-life and expression, thereby reducing lipid accumulation and alleviating IR in HepG2 cells in vitro (81). Yang et al. (82) innovated a new T2D mice model by specifically knocking out Tmeme30a in pancreatic islet β-cells and found that METTL3 and METTL14 were upregulated in vivo. Furthermore, the overexpression of METTL14 or METTL3 led to increased protein and mRNA levels of ATP citrate lyase (ACLY) and stearoyl-CoA desaturase 1 (SCD1) in vitro (83). In the inorganic arsenic (iAs)-induced T2DM, iAs treatment promoted translocation of METL14 and IGF2BP2 to the nucleus and activated m6A-mediated (METTL14-mediated) NOD-like receptor protein 3 (NLRP3) inflammasome. Moreover, METTL14 and IGF2BP2 enhanced the stability of Nlrp3 mRNA. Ultimately, iAs treatment induces early hepatic IR via m6A modification (84). Qin et al. (85) reported that METTL3 expression is elevated in obese mice hepatic macrophages, which would reduce DNA damage inducible transcript 4 (Ddit4) mRNA level in an m6A-dependent manner. Downregulated Ddit4 enhances the activation of mTORC1 and NF-κB signaling pathways, affecting hepatic oxidative stress, adipogenesis and glucose metabolism. Additionally, it has been shown that the high-glucose environment in the uterus of gestational diabetes changes the fetal epigenetic modification, making the generations more susceptible to T2D. Mechanistically, upgraded RBM15 inhibits the activation of insulin signaling pathway by regulating Claudin 4 in an m6A manner (86). Although all of the above studies showed elevated levels of hepatic methylase, the current view is generally that this is attributed to the possibility that some unclear feedback mechanism in the liver regulates the levels of methylase and demethylase. When high blood glucose-stimulated increases in Fto leads to low m6A modification in the liver, and as a feedback, methylase levels rise to maintain normal m6A abundance (77). However, an alternative explanation suggested that a high level of oxidative stress leads to increased m6A modification (81).
Yang et al. (77) demonstrated that the level of Fto mRNA in T2D patients was higher than that in healthy individuals and was positively correlated with fasting blood glucose. Furthermore, Fto knockout in high glucose-treated HepG2 cells showed some mRNA of glycolipid metabolism genes downregulated, such as forkhead box O1 (Foxo1), glucose-6-phosphatase catalytic subunit (G6pc), diacylglycerol O-acyltransferase 2 (Dgat2), and Fasn. The overexpression of Fto in vitro also upregulated the mRNA levels of these genes. As an inhibitor of FTO, Entacapone reduces fasting blood glucose and affects gluconeogenesis and fat thermogenesis in diet-induced obese mice by acting on the FTO-FOXO1 regulatory axis (87). In this study, the treatment of obese mice with FTO inhibition improved hepatic glucose tolerance and reduced white adipose tissue production. Considering that m6A methylase is essential for physiological function of the pancreas and that drug agonists are also more difficult to find than antagonists, therefore, FTO may be a better drug target for T2D. A recent study (88) showed that FTO affects sterol regulatory element binding transcription factor 1 (encode SREBP-1C) and carbohydrate responsive element binding protein, two adipogenesis transcription factors, enhancing lipid accumulation in a m6A-dependent manner. Since the role of FTO in the liver is not limited to glucose and lipid metabolism, Lim et al. (89) focused on the correlation between FTO and inflammation. The results demonstrated that Fto knockout relieved palmitic acid (PA)-induced oxidative stress, mitochondrial dysfunction, ER stress, and apoptosis in vitro. The high expression of FTO in T2D liver might be due to the high glucose environment stimulating insulin over-secretion. The binding of insulin to the insulin receptor in the liver causes insulin receptor (INSR) β to translocate to the nucleus and bind to the promoters of target genes, including Fto (90). This study suggests that hyperglycemia is a direct factor of hypomethylation in T2D patients, but the biology of this effect in IR still needs further investigation. Interestingly, in the liver both methylesterase and demethylases are most important in regulating lipid metabolism rather than carbohydrate metabolism. Nonetheless, the remaining demethylases, such as the ALKBH family, are poorly reported with T2D.
Zhou et al. (91) reported that TYHDC2 was significantly downregulated in the liver of obese mice, which resulted in the accumulation of hepatic TGs. Subsequently, Tyhdc2 overexpression in vivo ameliorated hepatic steatosis and IR. For further verification, ob/ob mice were injected with YTHDC2, which significantly lowered the blood glucose in fed/fasted states and improved the performance in the insulin tolerance test. The underlying mechanism is that YTHDC2 binds to the mRNA of lipogenic genes, including Srebp-1c, Fasn, Scd1, and Acc1 to decrease their mRNA stability and inhibit gene expression. Some studies have reported that YTHDF2 and eIF3G are upregulated in T2D model mice (83), high-glucose-induced HepG2 cells had upregulated Ythdf1 mRNA (92), and high level of ROS in vivo can increase the level of Ythdf2 mRNA (81), but there is no explanation for these results.
Skeletal muscle is a critical organ for maintaining the homeostasis of glucose metabolism throughout the whole body, and 80% of postprandial blood glucose regulation depends on skeletal muscle. Therefore, IR in skeletal muscle is a significant disease mechanism in T2D (93).
A past review (94) demonstrated the presence of accelerated loss of skeletal muscle in patients with T2D and a significant difference in total skeletal muscle mass between patients with and without T2D. A study of m6A modification and skeletal muscle growth in vivo (95) reported that METTL3 upregulation and increased m6A abundance implied physiological hypertrophy of skeletal muscle; the molecule is also required to maintain normal skeletal muscle function and muscle mass. Mechanistically, METTL3 regulates the abundance of methylation on mRNA of an activin receptor, activin type 2A receptor (Acvr2a), which is then recognized by YTHDF2 and degraded. As one of the negative regulators of skeletal muscle (96), ACVR2A phosphorylates SMAD3 protein, exerting anti-skeletal muscle hypertrophy effects. Similarly, another review concluded that METTL3/14 positively regulates skeletal muscle proliferation and differentiation (97). In the case of T2D, the mass of skeletal muscle and the IR of skeletal muscle are the major defects. Huang et al. (98) reported that PA-induced C2C12 myotubes resulted in IR-mediated reduction in Lpin1, which in turn increased the ceramide level in myotubes, directly promoting the expression of serine/threonine protein phosphatase 2A (PP2A) to dephosphorylate AKT that inhibits insulin signaling pathway. Although this study did not involve m6A modification, combined with the fact that Lpin1 increases half-life in the presence of hepatic hypomethylation (80) and the presence of METTL3 upregulation in PA-induced C2C12 cells (99), we suggested that the reduction of Lpin1 in this study is associated with m6A modification. At present, the role of methylases in skeletal muscle function is still unclear, especially the effect of methylases overexpression or silencing on skeletal muscle function deserve to be explored. Considering that skeletal muscle is the most essential glucose-consuming organ throughout the body, we believe that further exploration is necessary.
The normal expression of FTO is critical for skeletal muscle proliferation and differentiation. FTO ablation affects the mRNA expression of Pcg-1α and suppresses the mechanistic target of the rapamycin kinase (mTOR)-PCG-1α pathway, thereby decreasing the mitochondrial energy production and upregulated myogenin expression (100). In such a mechanism, FTO overexpression may enhance mitochondrial energy production leading to skeletal muscle IR, which needs to be explored experimentally. Bravard et al. (101) reported that patients with T2D showed a higher Fto level than nondiabetic subjects in skeletal muscle, which was positively correlated with glycosylated HbA1c and blood glucose. The overexpression of FTO in the skeletal muscle myotubes enhances lipogenesis and oxidative stress, leading to skeletal muscle IR. Similarly, the level of m6A modification is negatively associated with skeletal muscle lipid accumulation (102).
Furthermore, a few studies demonstrated that excessive energy causes FTO overexpression, reducing m6A modification. Notably, whether myocytes might secrete inflammatory molecules (103), irrespective of m6A modification, and exacerbate skeletal muscle IR needs further investigation.
The development of T2D is a complex process that cannot be separated from the role of intertwined pathway mechanisms. Herein, we focused on the role of m6A modification in different pathways to explore its profound role in T2D.
Phosphoinositide 3-kinase (PI3K)/AKT pathway regulates energy metabolism homeostasis, cell survival, proliferation, and progression, ensuring organisms’ normal growth. The upstream molecules of the PI3K/AKT pathway are mainly receptor tyrosine kinases, such as the epidermal growth factor receptor (EGFR) family, insulin, and IGF-1 receptor (104); hence, we focused on the pathway of insulin activation in the PI3K/AKT pathway. Initially, insulin binds to the α-subunit of the INSR on the cytosolic membrane, causing phosphorylation of the INSR intracellular β-subunit tyrosine residues. Subsequently, INSR attracts insulin receptor substrates (IRS) and phosphorylates the latter, after which it grips the downstream PI3K p85α subunit SH2 structural domain to bind to the IRS phosphorylated residues, recruiting and activating PI3K p110α subunit. Finally, activated PI3K phosphorylates phosphatidylinositol 4,5 biphosphate (PIP2) to form phosphatidylinositol 3,4,5 triphosphate (PIP3), which recruits and activates AKT in the presence of pyruvate dehydrogenase kinase 1 (PDK1) and mTOR2 (105).
The m6A modification could affect the liver and skeletal muscle PI3K/AKT pathway to influence insulin sensitivity and gluconeogenesis. The upstream and downstream signals of PI3K/AKT pathway were directly affected through alteration of m6A modification. In skeletal muscle, low-abundance m6A modification is positively correlated with hypo-phosphorylation of IRS1, and the phosphorylation of IRS1 is enhanced in skeletal muscle overexpressing Mettl3 (99). Another hepatocytes experiment in vitro showed that Rbm15 silencing improved insulin sensitivity by increasing AKT phosphorylation, whereas Rbm15 overexpression had the opposite effect (86) (We need to emphasize here again that T2D patients are at low m6A modification overall, but high m6A abundance in the liver). As compared to PI3K/AKT upstream, downstream effector genes are regulated more obviously with m6A modifications, for example, Foxo1, G6pc, Fasn mRNA upregulation in the liver of T2D patients (77), leading to increased glycemia and hepatic steatosis (106). Furthermore, FTO inhibition-treatment increased m6A abundance of Foxo1 mRNA in the liver, reducing the expression of FOXO1 and gene G6pc, which reduces blood glucose by regulating gluconeogenesis (87). Moreover, aberrant energy supply of T2D also led to impaired mTOR and decreased the expression of downstream PCG-1α, thereby affecting the skeletal muscle function (100). Interestingly, the nuclear hormone receptor FOXO1 combines with PGC-1α and regulates gluconeogenesis (107). Notably, Sharabi et al. (108) increased the acetylation of PGC-1α to inhibit hepatic glucose production and alleviate T2D hyperglycemia. This phenomenon suggested that epigenetic modifications explain the mechanisms of the pathway; also, the dynamic and reversible epigenetic modifications could be utilized to modify the key genes of the pathway for disease treatment.
AMPK is a critical cellular energy sensor and a crucial regulatory factor for metabolic homeostasis, which could be activated by sensing the changes in the ratio of AMP/ATP and (or) ADP/ATP in the internal environment. AMPK is a multi-isomeric complex comprising α, β, and γ subunits, each of which has 2 or 3 isoforms, and each combined isomer has a unique cellular localization and biological function (109). Moreover, AMPK activation is also affected by PI3K/AKT and intracellular Ca2+.
On the one hand, some features of T2D, such as metabolic syndrome and chronic low-grade inflammation, directly impair the activation of AMPK pathway (110). On the other hand, m6A modification also influences AMPK activation, which lead to lipid accumulation in the liver and skeletal muscle. A hepatocyte study in vitro showed that LKB1, the upstream of AMPK, is regulated by the methylesterase WTAP and mediates AMPK phosphorylation in an m6A-dependent manner, and when Wtap was knocked down, hepatocytes’ AMPK phosphorylation levels were significantly increased (111). In this situation of diminished AMPK activation, downstream adipogenic genes also increase their mRNA stability in an m6A-dependent manner. The high expression of METTL3 and low expression of TYHDF2 in the liver of T2D increased the mRNA stability of Fasn, Acc1, and Srebp-1c, leading to hepatic steatosis and further reducing organ insulin sensitivity (79, 81, 91). Besides, alterations in AMPK also could in turn affect the m6A modification. In skeletal muscle, the knockdown of Ampkα2 decreases overall m6A abundance, leading to skeletal muscle lipid accumulation (102). Therefore, we suggest that the homeostasis of m6A modification is similar to that of AMPK and is an important modality for the regulation of energy homeostasis in the organism.
JAK/STAT is an indispensable signaling node for many cellular processes, a highly conserved signaling pathway in evolution. JAKs (JAK1, JAK2, JAK3, and TYK2) tend to bind to cytokine receptors and hormone receptors, mediated by cytokines and hormones to phosphorylate its tyrosine residues, and then recruit the downstream STATs (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6) to alter its conformation (112). Activated STATs enter the cell nucleus to bind to DNA and regulate gene transcription for specific biological processes, such as cell progression, cell differentiation, and lipid metabolism.
Inflammatory cytokines released by adipose tissue in T2D are well-established mediators to exacerbate T2D symptoms (113). These cytokines activate the JAK/STAT pathway in adipose tissue, while hypomethylation of T2D over-activates the JAK/STAT pathway by altering the stability of Jak mRNA, and promotes the expression of downstream lipogenic genes, such as C/RBPα/β, PPARγ, and FABP4 (55). Moreover, m6A modification could also regulate the expression of Suppressor of cytokine signaling 3, a JAK/STAT downstream gene, which may be associated with adipose tissue inflammation (114). However, further studies on the interactional node of m6A modification and JAK/STAT are still needed.
PPARs are members of nuclear transcription factors binding to retinoic X receptors, which could activate upon binding to ligands to regulate lipid metabolism, insulin sensitivity, and energy homeostasis (115). The PPARs superfamily consists of three isoforms: PPARα, PPARγ, and PPARβ/δ. PPARα is mainly located in high energy-consuming organs, such as the liver and skeletal muscle, and regulates energy homeostasis, PPARγ is primarily localized in the adipose tissue and modulates adipocyte formation and insulin sensitivity, and PPARβ/δ is detected in skeletal muscle and adipose tissue, wherein it controls fatty acid metabolism (116).
The PPAR family will increase adipogenesis according to the localization of its members to the liver and adipose tissue, which in turn contributes to the development of T2D. PPARα resembles an energy sensor that perceives circulating fatty acid metabolism, activated by endogenous fatty acid metabolic derivatives in the body (117). Its activation reduces plasma apolipoproteins, enhances β-oxidation of TGs, and increases lipoprotein lipase (LPL), lowering plasma TG and low-density lipoproteins and increasing high-density lipoproteins (118). A previous study showed that hepatic hypomethylation enhances Pparα expression, reduces hepatic steatosis, and increases insulin sensitivity (81). Moreover, m6A modification is more complicated in the regulation of PPARγ. In white adipose tissue, hypomethylation contributes to increased Pparγ expression, causing lipid accumulation and adipocytogenesis (55, 66); whereas in brown adipose tissue, it decreases Pparγ expression, impairing its maturation, which leads to loss of ability to resist obesity and aggravates systemic IR (59). Increasingly evidence suggests that high level of FTO act on the PPAR pathway to enhance adipogenesis, thereby promoting T2D.
Metformin has been used for >60 years to treat T2D, with clear anti-hyperglycemia and anti-obesity effects. Thus, it is the first-line drug for T2D recommended in the latest American Diabetes Association and European Association for the Study of Diabetes consensus (119, 120). A recent study showed (121) that metformin affects adipogenesis by altering adipose tissue m6A modification to decrease fat mass and alleviate IR in vivo. In the in vitro experiment, metformin-treated 3T3-L1 cells showed an increased G0/G1 phase ratio and a decreased ratio of S and G2/M phases. Mechanistically, metformin diminishes FTO expression, increasing m6A modification on Ccnd1 and cyclin-dependent kinase 2 (Cdk2) mRNAs. These manifestations enhance the degradation by TYHDF2, leading to adipocyte differentiation arrest in MCE.
Nowadays, it is proposed that metformin exerts its hypoglycemic effect by activating AMPK; also, m6A modification is involved in the AMPK signaling pathway (122). Thus, exploring how metformin regulates m6A modification might explain the underlying hypoglycemic mechanism.
Glucagon-like peptide-1 (GLP-1) is a gut-secreted hormone that enhances insulin in blood glucose regulation. The combination of GLP-1 and GLP-1 receptors enhances glucose-dependent insulin secretion and inhibits glucagon release; based on this function, several GLP-1 receptor agonists have been developed for T2D treatment (123). In a random controlled trial over three years (124), treatment with GLP-1 receptor agonist, exenatide, revealed that patients with T2D had β-cell biology function. Zhou et al. (125) demonstrated that exenatide reduces the apoptotic effect of oxidative stress on β-cells via augmented Mettl3 level in primary pancreatic islet cells and NIT-1 cells under H2O2 treatment. Regrettably, only anti-apoptotic and pro-apoptotic biomarkers were detected, and the regulatory mechanism was not explored.
Thiazolidinedione is an insulin sensitizer and binds to PPARγ to improve insulin sensitivity in the liver, skeletal muscle, pancreas, and other organs to maintain normal blood glucose levels, in contrast to lowering blood glucose directly (126). The common clinical agents include rosiglitazone and pioglitazone. Previous studies (101) have shown that rosiglitazone reduces skeletal muscle Fto expression in T2D patients, improving insulin sensitivity in T2D patients. Another study (127) showed that rosiglitazone diminished Fto mRNA in subcutaneous adipose tissue and improved glucose utilization in female patients with T2D. However, TZD is now less commonly used clinically, as many safety concerns exist, such as cardiovascular, bone fracture, and bladder cancer risks (128).
To date, only a few studies have addressed pre- and post-treatment methylation changes in T2D. Thus, the drug could modulate the m6A modification in different body organs to affect T2D metabolism, which might be a novel target for T2D treatment.
The incidence of the metabolic disease T2D is increasing throughout the world and represents a significant burden for world health. Regrettably, there is still no overall cure for T2D and the molecular pathogenesis of the disease requires full elucidation.
Therefore, we attempted the further understanding of T2D from an epigenetic perspective. We investigated the role of m6A methylation, the most common epigenetic modification in humans, in T2D. Reduced m6A modification was observed in T2D patients, specifically affecting genes involved in glucose and lipid metabolism and pancreatic β-cell biology, thus contributing further insight into the pathophysiology of T2D. Thus, monitoring the m6A modification of critical metabolic genes may represent a new diagnostic target for the assessment of pre-T2D. Furthermore, manipulation of m6A levels may offer a new direction for T2D treatment. Ultimately, novel drugs for the treatment of T2D could be developed to adjust m6A modification, which would further deepen our understanding of the metabolic processes involved in the disease. Taken together, the findings indicated that m6A modification plays a significant role in the pathogenesis of T2D. Further investigation into the key factors and signaling pathways involved in m6A modification, as well as the investigation of potential drugs targeting these modifications, is required to provide new perspectives for T2D treatment.
Conceptualization, XH and HZ; validation, XH and HZ; investigation, YG, QG, JH and QX; writing—original draft preparation, HZ; writing—review and editing, XH. All authors contributed to the article and approved the submitted version.
This research was funded by Nation Natural Science Foundation of China, grant number 81273568.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. IDF DIABETES ATLAS. Brussels: International Diabetes Federation (2021). Available at: https://diabetesatlas.org/atlas/tenth-edition/.
2. Ke C, Narayan KMV, Chan JCN, Jha P, Shah BR. Pathophysiology, phenotypes and management of type 2 diabetes mellitus in Indian and Chinese populations. Nat Rev Endocrinol (2022) 18(7):413–32. doi: 10.1038/s41574-022-00669-4
3. Tinajero MG, Malik VS. An update on the epidemiology of type 2 diabetes: a global perspective. Endocrinol Metab Clin North Am (2021) 50(3):337–55. doi: 10.1016/j.ecl.2021.05.013
4. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. The sequence of the human genome. Science (2001) 291(5507):1304–51. doi: 10.1126/science.1058040
5. Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab (2019) 29(5):1028–44. doi: 10.1016/j.cmet.2019.03.009
6. Wang A, Tao W, Tong J, Gao J, Wang J, Hou G, et al. m6A modifications regulate intestinal immunity and rotavirus infection. eLife (2022) 11:e73628. doi: 10.7554/eLife.73628
7. Deng LJ, Deng WQ, Fan SR, Chen MF, Qi M, Lyu WY, et al. m6A modification: recent advances, anticancer targeted drug discovery and beyond. Mol Cancer (2022) 21(1):52. doi: 10.1186/s12943-022-01510-2
8. An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer (2022) 21(1):14. doi: 10.1186/s12943-022-01500-4
9. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol (2019) 15(6):327–45. doi: 10.1038/s41581-019-0135-6
10. Wiener D, Schwartz S. The epitranscriptome beyond m6A. Nat Rev Genet (2020) 22(2):119–31. doi: 10.1038/s41576-020-00295-8
11. Song P, Tayier S, Cai Z, Jia G. RNA Methylation in mammalian development and cancer. Cell Biol Toxicol (2021) 37(6):811–31. doi: 10.1007/s10565-021-09627-8
12. Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell (2012) 149(7):1635–46. doi: 10.1016/j.cell.2012.05.003
13. Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell (2017) 169(7):1187–200. doi: 10.1016/j.cell.2017.05.045
14. Li M, Zha X, Wang S. The role of N6-methyladenosine mRNA in the tumor microenvironment. Biochim Biophys Acta (BBA) - Rev Cancer (2021) 1875(2):188522. doi: 10.1016/j.bbcan.2021.188522
15. Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal transduction targeted Ther (2021) 6(1):74–4. doi: 10.1038/s41392-020-00450-x
16. Liu L, Li H, Hu D, Wang Y, Shao W, Zhong J, et al. Insights into N6-methyladenosine and programmed cell death in cancer. Mol Cancer (2022) 21(1):32. doi: 10.1186/s12943-022-01508-w
17. Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in cancer progression. Mol Cancer (2020) 19(1):88–8. doi: 10.1186/s12943-020-01204-7
18. Ma H, Wang X, Cai J, Dai Q, Natchiar SK, Lv R, et al. N(6-)Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat Chem Biol (2019) 15(1):88–94. doi: 10.1038/s41589-018-0184-3
19. Su R, Dong L, Li Y, Gao M, He PC, Liu W, et al. METTL16 exerts an m 6 a-independent function to facilitate translation and tumorigenesis. Nat Cell Biol (2022) 24(2):205. doi: 10.1038/s41556-021-00835-2
20. Goh YT, Koh C, Sim DY, Roca X, Goh W. METTL4 catalyzes m6Am methylation in U2 snRNA to regulate pre-mRNA splicing. Nucleic Acids Res (2020) 48(16):9250–61. doi: 10.1093/nar/gkaa684
21. Rong B, Zhang Q, Wan J, Xing S, Dai R, Li Y, et al. Ribosome 18S m(6)A methyltransferase METTL5 promotes translation initiation and breast cancer cell growth. Cell Rep (2020) 33(12):108544. doi: 10.1016/j.celrep.2020.108544
22. Zhao X, Yang Y, Sun BF, Shi Y, Yang X, Xiao W, et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res (2014) 24(12):1403–19. doi: 10.1038/cr.2014.151
23. Qu J, Yan H, Hou Y, Cao W, Liu Y, Zhang E, et al. RNA Demethylase ALKBH5 in cancer: from mechanisms to therapeutic potential. J Hematol Oncol (2022) 15(1):8–8. doi: 10.1186/s13045-022-01224-4
24. Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell (2019) 74(4):640–50. doi: 10.1016/j.molcel.2019.04.025
25. Kan L, Grozhik AV, Vedanayagam J, Patil DP, Pang N, Lim K, et al. The m6A pathway facilitates sex determination in drosophila. Nat Commun (2017) 8(1):15737. doi: 10.1038/ncomms15737
26. Roundtree IA, Luo GZ, Zhang Z, Wang X, Zhou T, Cui Y, et al. YTHDC1 mediates nuclear export of N(6)-methyladenosine methylated mRNAs. eLife (2017) 6:e31311. doi: 10.7554/eLife.31311
27. Widagdo J, Anggono V, Wong JJ. The multifaceted effects of YTHDC1-mediated nuclear m(6)A recognition. Trends Genet (2022) 38(4):325–32. doi: 10.1016/j.tig.2021.11.005
28. Zaccara S, Ries RJ, Jaffrey SR. Reading, writing and erasing mRNA methylation. Nat Rev Mol Cell Biol (2019) 20(10):608–24. doi: 10.1038/s41580-019-0168-5
29. Moshitch-Moshkovitz S, Dominissini D, Rechavi G. The epitranscriptome toolbox. Cell (2022) 185(5):764–76. doi: 10.1016/j.cell.2022.02.007
30. Wiener D, Schwartz S. The epitranscriptome beyond m(6)A. Nat Rev Genet (2021) 22(2):119–31. doi: 10.1038/s41576-020-00295-8
31. Mastrototaro L, Roden M. Insulin resistance and insulin sensitizing agents. Metabolism (2021) 125:154892. doi: 10.1016/j.metabol.2021.154892
32. Eizirik DL, Pasquali L, Cnop M. Pancreatic beta-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol (2020) 16(7):349–62. doi: 10.1038/s41574-020-0355-7
33. Liu J, Luo G, Sun J, Men L, Ye H, He C, et al. METTL14 is essential for beta-cell survival and insulin secretion. Biochim Biophys Acta Mol Basis Dis (2019) 1865(9):2138–48. doi: 10.1016/j.bbadis.2019.04.011
34. De Jesus DF, Zhang Z, Kahraman S, Brown NK, Chen M, Hu J, et al. M 6 a mRNA methylation regulates human β-cell biology in physiological states and in type 2 diabetes. Nat Metab (2019) 1(8):765. doi: 10.1038/s42255-019-0089-9
35. De Jesus DF, Kulkarni RN. “Omics” and “epi-omics” underlying the beta-cell adaptation to insulin resistance. Mol Metab (2019) 27S:S42–8. doi: 10.1016/j.molmet.2019.06.003
36. Men L, Sun J, Luo G, Ren D. Acute deletion of METTL14 in beta-cells of adult mice results in glucose intolerance. Endocrinology (2019) 160(10):2388–94. doi: 10.1210/en.2019-00350
37. Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ. Therapeutic opportunities for pancreatic beta-cell ER stress in diabetes mellitus. Nat Rev Endocrinol (2021) 17(8):455–67. doi: 10.1038/s41574-021-00510-4
38. Wang Y, Sun J, Lin Z, Zhang W, Wang S, Wang W, et al. m(6)A mRNA methylation controls functional maturation in neonatal murine beta-cells. Diabetes (2020) 69(8):1708–22. doi: 10.2337/db19-0906
39. Li X, Jiang Y, Sun X, Wu Y, Chen Z. METTL3 is required for maintaining beta-cell function. Metabolism (2021) 116:154702. doi: 10.1016/j.metabol.2021.154702
40. Cheng Y, Yao XM, Zhou SM, Sun Y, Meng XJ, Wang Y, et al. The m(6)A methyltransferase METTL3 ameliorates methylglyoxal-induced impairment of insulin secretion in pancreatic beta cells by regulating MafA expression. Front Endocrinol (Lausanne) (2022) 13:910868. doi: 10.3389/fendo.2022.910868
41. Li X, Yang Y, Li Z, Wang Y, Qiao J, Chen Z. Deficiency of WTAP in islet beta cells results in beta cell failure and diabetes in mice. Diabetologia (2023) 66(6):1084–96. doi: 10.1007/s00125-023-05900-z
42. Horikawa Y, Miyake K, Yasuda K, Enya M, Hirota Y, Yamagata K, et al. Replication of genome-wide association studies of type 2 diabetes susceptibility in Japan. J Clin Endocrinol Metab (2008) 93(8):3136–41. doi: 10.1210/jc.2008-0452
43. Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, et al. And the development of type 2 diabetes. N Engl J Med (2008) 359(21):2220–32. doi: 10.1056/NEJMoa0801869
44. Caro-Gomez MA, Naranjo-Gonzalez CA, Gallego-Lopera N, Parra-Marin MV, Valencia DM, Arcos EG, et al. Association of native American ancestry and common variants in ACE, ADIPOR2, MTNR1B, GCK, TCF7L2 and FTO genes with glycemic traits in Colombian population. Gene (2018) 677:198–210. doi: 10.1016/j.gene.2018.07.066
45. Robiou-du-Pont S, Bonnefond A, Yengo L, Vaillant E, Lobbens S, Durand E, et al. Contribution of 24 obesity-associated genetic variants to insulin resistance, pancreatic beta-cell function and type 2 diabetes risk in the French population. Int J Obes (Lond) (2013) 37(7):980–5. doi: 10.1038/ijo.2012.175
46. Taneera J, Prasad RB, Dhaiban S, Mohammed AK, Haataja L, Arvan P, et al. Silencing of the FTO gene inhibits insulin secretion: an in vitro study using GRINCH cells. Mol Cell Endocrinol (2018) 472:10–7. doi: 10.1016/j.mce.2018.06.003
47. Bornaque F, Delannoy CP, Courty E, Rabhi N, Carney C, Rolland L, et al. Glucose regulates m(6)A methylation of RNA in pancreatic islets. Cells (2022) 11(2):291. doi: 10.3390/cells11020291
48. Regue L, Zhao L, Ji F, Wang H, Avruch J, Dai N. RNA m6A reader IMP2/IGF2BP2 promotes pancreatic beta-cell proliferation and insulin secretion by enhancing PDX1 expression. Mol Metab (2021) 48:101209. doi: 10.1016/j.molmet.2021.101209
49. Wu X, Wang W, Fan S, You L, Li F, Zhang X, et al. U-Shaped association between serumIGF2BP3 and T2DM: a cross-sectional study in Chinese population. J Diabetes (2023) 15(4):349–61. doi: 10.1111/1753-0407.13378
50. Li X, Yang Y, Chen Z. Downregulation of the m(6)A reader protein YTHDC1 leads to islet beta-cell failure and diabetes. Metabolism (2023) 138:155339. doi: 10.1016/j.metabol.2022.155339
51. Dai N. The diverse functions of IMP2/IGF2BP2 in metabolism. Trends Endocrinol Metab (2020) 31(9):670–9. doi: 10.1016/j.tem.2020.05.007
52. Smith U, Kahn BB. Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids. J Intern Med (2016) 280(5):465–75. doi: 10.1111/joim.12540
53. Pellegrinelli V, Rodriguez-Cuenca S, Rouault C, Figueroa-Juarez E, Schilbert H, Virtue S, et al. Dysregulation of macrophage PEPD in obesity determines adipose tissue fibro-inflammation and insulin resistance. Nat Metab (2022) 4(4):476–94. doi: 10.1038/s42255-022-00561-5
54. Wu R, Wang X. Epigenetic regulation of adipose tissue expansion and adipogenesis by N(6) -methyladenosine. Obes Rev (2021) 22(2):e13124. doi: 10.1111/obr.13124
55. Yao Y, Bi Z, Wu R, Zhao Y, Liu Y, Liu Q, et al. METTL3 inhibits BMSC adipogenic differentiation by targeting the JAK1/STAT5/C/EBPbeta pathway via an m(6)A-YTHDF2-dependent manner. FASEB J (2019) 33(6):7529–44. doi: 10.1096/fj.201802644R
56. Wang Y, Li Y, Toth JI, Petroski MD, Zhang Z, Zhao JC. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat Cell Biol (2014) 16(2):191–8. doi: 10.1038/ncb2902
57. Kobayashi M, Ohsugi M, Sasako T, Awazawa M, Umehara T, Iwane A, et al. The RNA methyltransferase complex of WTAP, METTL3, and METTL14 regulates mitotic clonal expansion in adipogenesis. Mol Cell Biol (2018) 38(16):e00116–18. doi: 10.1128/MCB.00116-18
58. Liu Q, Zhao Y, Wu R, Jiang Q, Cai M, Bi Z, et al. ZFP217 regulates adipogenesis by controlling mitotic clonal expansion in a METTL3-m6A dependent manner. RNA Biol (2019) 16(12):1785–93. doi: 10.1080/15476286.2019.1658508
59. Wang Y, Gao M, Zhu F, Li X, Yang Y, Yan Q, et al. METTL3 is essential for postnatal development of brown adipose tissue and energy expenditure in mice. Nat Commun (2020) 11(1):1648. doi: 10.1038/s41467-020-15488-2
60. Huang X, Huang X, Guo H, Li J, Zhou C, Huang Y, et al. Intermittent hypoxia-induced METTL3 downregulation facilitates MGLL-mediated lipolysis of adipocytes in OSAS. Cell Death Discovery (2022) 8(1):352. doi: 10.1038/s41420-022-01149-4
61. Merkestein M, Laber S, McMurray F, Andrew D, Sachse G, Sanderson J, et al. FTO influences adipogenesis by regulating mitotic clonal expansion. Nat Commun (2015) 6:6792. doi: 10.1038/ncomms7792
62. Yang Z, Yu GL, Zhu X, Peng TH, Lv YC. Critical roles of FTO-mediated mRNA m6A demethylation in regulating adipogenesis and lipid metabolism: implications in lipid metabolic disorders. Genes Dis (2022) 9(1):51–61. doi: 10.1016/j.gendis.2021.01.005
63. Zhu X, Tang H, Yang M, Yin K. N6-methyladenosine in macrophage function: a novel target for metabolic diseases. Trends Endocrinol Metab (2023) 34(2):66–84. doi: 10.1016/j.tem.2022.12.006
64. Wang X, Wu R, Liu Y, Zhao Y, Bi Z, Yao Y, et al. m(6)A mRNA methylation controls autophagy and adipogenesis by targeting Atg5 and Atg7. Autophagy (2020) 16(7):1221–35. doi: 10.1080/15548627.2019.1659617
65. Wu R, Chen Y, Liu Y, Zhuang L, Chen W, Zeng B, et al. m6A methylation promotes white-to-beige fat transition by facilitating Hif1a translation. EMBO Rep (2021) 22(11):e52348. doi: 10.15252/embr.202052348
66. Azzam SK, Alsafar H, Sajini AA. FTO m6A demethylase in obesity and cancer: implications and underlying molecular mechanisms. Int J Mol Sci (2022) 23(7):3800. doi: 10.3390/ijms23073800
67. Loos RJ, Yeo GS. The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol (2014) 10(1):51–61. doi: 10.1038/nrendo.2013.227
68. Di Renzo L, Cioccoloni G, Falco S, Abenavoli L, Moia A, Sinibaldi SP, et al. Influence of FTO rs9939609 and Mediterranean diet on body composition and weight loss: a randomized clinical trial. J Transl Med (2018) 16(1):308. doi: 10.1186/s12967-018-1680-7
69. Ling C. Epigenetic regulation of insulin action and secretion – role in the pathogenesis of type 2 diabetes. J Intern Med (2020) 288(2):158–67. doi: 10.1111/joim.13049
70. Berulava T, Horsthemke B. The obesity-associated SNPs in intron 1 of the FTO gene affect primary transcript levels. Eur J Hum Genet (2010) 18(9):1054–6. doi: 10.1038/ejhg.2010.71
71. Sobreira DR, Joslin AC, Zhang Q, Williamson I, Hansen GT, Farris KM, et al. Extensive pleiotropism and allelic heterogeneity mediate metabolic effects of IRX3 and IRX5. Science (2021) 372(6546):1085–91. doi: 10.1126/science.abf1008
72. Karra E, O Daly OG, Choudhury AI, Yousseif A, Millership S, Neary MT, et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Invest. (2013) 123(8):3539–51. doi: 10.1172/JCI44403
73. Jiang Q, Sun B, Liu Q, Cai M, Wu R, Wang F, et al. MTCH2 promotes adipogenesis in intramuscular preadipocytes via an m6A-YTHDF1-dependent mechanism. FASEB J (2019) 33(2):2971–81. doi: 10.1096/fj.201801393RRR
74. Ronningen T, Dahl MB, Valderhaug TG, Cayir A, Keller M, Tonjes A, et al. m6A regulators in human adipose tissue - depot-specificity and correlation with obesity. Front Endocrinol (Lausanne) (2021) 12:778875. doi: 10.3389/fendo.2021.778875
75. Huang LO, Rauch A, Mazzaferro E, Preuss M, Carobbio S, Bayrak CS, et al. Genome-wide discovery of genetic loci that uncouple excess adiposity from its comorbidities. Nat Metab (2021) 3(2):228–43. doi: 10.1038/s42255-021-00346-2
76. Alvarez-Dominguez JR, Winther S, Hansen JB, Lodish HF, Knoll M. An adipose lncRAP2-Igf2bp2 complex enhances adipogenesis and energy expenditure by stabilizing target mRNAs. iScience (2022) 25(1):103680. doi: 10.1016/j.isci.2021.103680
77. Yang Y, Shen F, Huang W, Qin S, Huang J, Sergi C, et al. Glucose is involved in the dynamic regulation of m6A in patients with type 2 diabetes. J Clin Endocrinol Metab (2019) 104(3):665–73. doi: 10.1210/jc.2018-00619
78. Chen YS, Ouyang XP, Yu XH, Novak P, Zhou L, He PP, et al. N6-adenosine methylation (m(6)A) RNA modification: an emerging role in cardiovascular diseases. J Cardiovasc Transl Res (2021) 14(5):857–72. doi: 10.1007/s12265-021-10108-w
79. Xie W, Ma LL, Xu YQ, Wang BH, Li SM. METTL3 inhibits hepatic insulin sensitivity via N6-methyladenosine modification of fasn mRNA and promoting fatty acid metabolism. Biochem Bioph. Res Co. (2019) 518(1):120–6. doi: 10.1016/j.bbrc.2019.08.018
80. Li Y, Zhang Q, Cui G, Zhao F, Tian X, Sun B, et al. m6A regulates liver metabolic disorders and hepatogenous diabetes. Genomics Proteomics Bioinf (2020) 18(4):371–83. doi: 10.1016/j.gpb.2020.06.003
81. Zhong X, Yu J, Frazier K, Weng X, Li Y, Cham CM, et al. Circadian clock regulation of hepatic lipid metabolism by modulation of m6A mRNA methylation. Cell Rep (2018) 25(7):1816–1828.e4. doi: 10.1016/j.celrep.2018.10.068
82. Yang Y, Sun K, Liu W, Li X, Tian W, Shuai P, et al. The phosphatidylserine flippase β-subunit Tmem30a is essential for normal insulin maturation and secretion. Mol Ther (2021) 29(9):2854–72. doi: 10.1016/j.ymthe.2021.04.026
83. Yang Y, Cai J, Yang X, Wang K, Sun K, Yang Z, et al. Dysregulated m6A modification promotes lipogenesis and development of non-alcoholic fatty liver disease and hepatocellular carcinoma. Mol Ther (2022) 30(6):2342–53. doi: 10.1016/j.ymthe.2022.02.021
84. Qiu T, Wu C, Yao X, Han Q, Wang N, Yuan W, et al. AS3MT facilitates NLRP3 inflammasome activation by m 6 a modification during arsenic-induced hepatic insulin resistance. Cell Biol Toxicol (2022). doi: 10.1007/s10565-022-09703-7
85. Qin Y, Li B, Arumugam S, Lu Q, Mankash SM, Li J, et al. m6A mRNA methylation-directed myeloid cell activation controls progression of NAFLD and obesity. Cell Rep (2021) 37(6):109968. doi: 10.1016/j.celrep.2021.109968
86. Fang J, Wu X, He J, Zhang H, Chen X, Zhang H, et al. RBM15 suppresses hepatic insulin sensitivity of offspring of gestational diabetes mellitus mice via m6A-mediated regulation of CLDN4. Mol Med (2023) 29(1):23. doi: 10.1186/s10020-023-00615-8
87. Peng S, Xiao W, Ju D, Sun B, Hou N, Liu Q, et al. Identification of entacapone as a chemical inhibitor of FTO mediating metabolic regulation through FOXO1. Sci Transl Med (2019) 11(488):eaau7116. doi: 10.1126/scitranslmed.aau7116
88. Tang Z, Sun C, Yan Y, Niu Z, Li Y, Xu X, et al. Aberrant elevation of FTO levels promotes liver steatosis by decreasing the m6A methylation and increasing the stability of SREBF1 and ChREBP mRNAs. J Mol Cell Biol (2022) 14(9):mjac061. doi: 10.1093/jmcb/mjac061
89. Lim A, Zhou J, Sinha RA, Singh BK, Ghosh S, Lim K, et al. Hepatic FTO expression is increased in NASH and its silencing attenuates palmitic acid-induced lipotoxicity. Biochem Bioph Res Co (2016) 479(3):476–81. doi: 10.1016/j.bbrc.2016.09.086
90. Hancock ML, Meyer RC, Mistry M, Khetani RS, Wagschal A, Shin T, et al. Insulin receptor associates with promoters genome-wide and regulates gene expression. Cell (2019) 177(3):722–736.e22. doi: 10.1016/j.cell.2019.02.030
91. Zhou B, Liu C, Xu L, Yuan Y, Zhao J, Zhao W, et al. N(6) -methyladenosine reader protein YT521-b homology domain-containing 2 suppresses liver steatosis by regulation of mRNA stability of lipogenic genes. Hepatology (2021) 73(1):91–103. doi: 10.1002/hep.31220
92. Jiang H, Yao Q, An Y, Fan L, Wang J, Li H. Baicalin suppresses the progression of type 2 diabetes-induced liver tumor through regulating METTL3/m(6)A/HKDC1 axis and downstream p-JAK2/STAT1/clevaged Capase3 pathway. Phytomedicine (2022) 94:153823. doi: 10.1016/j.phymed.2021.153823
93. Sylow L, Tokarz VL, Richter EA, Klip A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab (2021) 33(4):758–80. doi: 10.1016/j.cmet.2021.03.020
94. Al-Ozairi E, Alsaeed D, Alroudhan D, Voase N, Hasan A, Gill J, et al. Skeletal muscle and metabolic health: how do we increase muscle mass and function in people with type 2 diabetes? J Clin Endocrinol Metab (2021) 106(2):309–17. doi: 10.1210/clinem/dgaa835
95. Petrosino JM, Hinger SA, Golubeva VA, Barajas JM, Dorn LE, Iyer CC, et al. The m(6)A methyltransferase METTL3 regulates muscle maintenance and growth in mice. Nat Commun (2022) 13(1):168. doi: 10.1038/s41467-021-27848-7
96. Rodgers BD, Ward CW. Myostatin/Activin receptor ligands in muscle and the development status of attenuating drugs. Endocr Rev (2022) 43(2):329–65. doi: 10.1210/endrev/bnab030
97. Li J, Pei Y, Zhou R, Tang Z, Yang Y. Regulation of RNA N(6)-methyladenosine modification and its emerging roles in skeletal muscle development. Int J Biol Sci (2021) 17(7):1682–92. doi: 10.7150/ijbs.56251
98. Huang S, Huang S, Wang X, Zhang Q, Liu J, Leng Y. Downregulation of lipin-1 induces insulin resistance by increasing intracellular ceramide accumulation in C2C12 myotubes. Int J Biol Sci (2017) 13(1):1–12. doi: 10.7150/ijbs.17149
99. Jiao Y, Williams A, Wei N. Quercetin ameliorated insulin resistance via regulating METTL3-mediated N6-methyladenosine modification of PRKD2 mRNA in skeletal muscle and C2C12 myocyte cell line. Nutr Metab Cardiovasc Dis (2022) 32(11):2655–68. doi: 10.1016/j.numecd.2022.06.019
100. Wang X, Huang N, Yang M, Wei D, Tai H, Han X, et al. FTO is required for myogenesis by positively regulating mTOR-PGC-1alpha pathway-mediated mitochondria biogenesis. Cell Death Dis (2017) 8(3):e2702. doi: 10.1038/cddis.2017.122
101. Bravard A, Lefai E, Meugnier E, Pesenti S, Disse E, Vouillarmet J, et al. FTO is increased in muscle during type 2 diabetes, and its overexpression in myotubes alters insulin signaling, enhances lipogenesis and ROS production, and induces mitochondrial dysfunction. Diabetes (2011) 60(1):258–68. doi: 10.2337/db10-0281
102. Wu W, Feng J, Jiang D, Zhou X, Jiang Q, Cai M, et al. AMPK regulates lipid accumulation in skeletal muscle cells through FTO-dependent demethylation of N(6)-methyladenosine. Sci Rep (2017) 7:41606. doi: 10.1038/srep41606
103. Wu H, Ballantyne CM. Skeletal muscle inflammation and insulin resistance in obesity. J Clin Invest. (2017) 127(1):43–54. doi: 10.1172/JCI88880
104. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Semin Cancer Biol (2019) 59:125–32. doi: 10.1016/j.semcancer.2019.07.009
105. Schultze SM, Hemmings BA, Niessen M, Tschopp O. PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Rev Mol Med (2012) 14:e1. doi: 10.1017/S1462399411002109
106. Li Y, Ma Z, Jiang S, Hu W, Li T, Di S, et al. A global perspective on FOXO1 in lipid metabolism and lipid-related diseases. Prog Lipid Res (2017) 66:42–9. doi: 10.1016/j.plipres.2017.04.002
107. Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature (2003) 423(6939):550–5. doi: 10.1038/nature01667
108. Sharabi K, Lin H, Tavares C, Dominy JE, Camporez JP, Perry RJ, et al. Selective chemical inhibition of PGC-1alpha gluconeogenic activity ameliorates type 2 diabetes. Cell (2017) 169(1):148–160.e15. doi: 10.1016/j.cell.2017.03.001
109. Trefts E, Shaw RJ. AMPK: restoring metabolic homeostasis over space and time. Mol Cell (2021) 81(18):3677–90. doi: 10.1016/j.molcel.2021.08.015
110. Jeon SM. Regulation and function of AMPK in physiology and diseases. Exp Mol Med (2016) 48(7):e245. doi: 10.1038/emm.2016.81
111. Li G, Deng L, Huang N, Cui Z, Wu Q, Ma J, et al. m6A mRNA methylation regulates LKB1 to promote autophagy of hepatoblastoma cells through upregulated phosphorylation of AMPK. Genes (2021) 12(11):1747. doi: 10.3390/genes12111747
112. Hu X, Li J, Fu M, Zhao X, Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther (2021) 6(1):402. doi: 10.1038/s41392-021-00791-1
113. Perry RJ, Camporez JG, Kursawe R, Titchenell PM, Zhang D, Perry CJ, et al. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell (2015) 160(4):745–58. doi: 10.1016/j.cell.2015.01.012
114. Zhang B, Yuan L, Chen G, Chen X, Yang X, Fan T, et al. Deciphering obesity-related gene clusters unearths SOCS3 immune infiltrates and 5mC/m6A modifiers in ossification of ligamentum flavum pathogenesis. Front Endocrinol (Lausanne) (2022) 13:861567. doi: 10.3389/fendo.2022.861567
115. Shi Y, Zou Y, Shen Z, Xiong Y, Zhang W, Liu C, et al. Trace elements, PPARs, and metabolic syndrome. Int J Mol Sci (2020) 21(7):2612. doi: 10.3390/ijms21072612
116. Wagner N, Wagner KD. The role of PPARs in disease. Cells (2020) 9(11):2367. doi: 10.3390/cells9112367
117. Nakamura MT, Yudell BE, Loor JJ. Regulation of energy metabolism by long-chain fatty acids. Prog Lipid Res (2014) 53:124–44. doi: 10.1016/j.plipres.2013.12.001
118. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol (2015) 62(3):720–33. doi: 10.1016/j.jhep.2014.10.039
119. Davies MJ, Aroda VR, Collins BS, Gabbay RA, Green J, Maruthur NM, et al. Management of hyperglycemia in type 2 diabetes, 2022. a consensus report by the American diabetes association (ADA) and the European association for the study of diabetes (EASD). Diabetes Care (2022) 45(11):2753–86. doi: 10.2337/dci22-0034
120. Foretz M, Guigas B, Viollet B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat Rev Endocrinol (2019) 15(10):569–89. doi: 10.1038/s41574-019-0242-2
121. Liao X, Liu J, Chen Y, Liu Y, Chen W, Zeng B, et al. Metformin combats obesity by targeting FTO in an m6A-YTHDF2-dependent manner. J Drug Target (2022) 30(9):983–91. doi: 10.1080/1061186X.2022.2071906
122. Zhang CS, Li M, Ma T, Zong Y, Cui J, Feng JW, et al. Metformin activates AMPK through the lysosomal pathway. Cell Metab (2016) 24(4):521–2. doi: 10.1016/j.cmet.2016.09.003
123. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab (2018) 27(4):740–56. doi: 10.1016/j.cmet.2018.03.001
124. van Raalte DH, Bunck MC, Smits MM, Hoekstra T, Corner A, Diamant M, et al. Exenatide improves beta-cell function up to 3 years of treatment in patients with type 2 diabetes: a randomised controlled trial. Eur J Endocrinol (2016) 175(4):345–52. doi: 10.1530/EJE-16-0286
125. Zhou S, Sun Y, Xing Y, Wang Z, Wan S, Yao X, et al. Exenatide ameliorates hydrogen peroxide-induced pancreatic β-cell apoptosis through regulation of METTL3-mediated m6A methylation. Eur J Pharmacol (2022) 924:174960–0. doi: 10.1016/j.ejphar.2022.174960
126. Nanjan MJ, Mohammed M, Prashantha Kumar BR, Chandrasekar MJN. Thiazolidinediones as antidiabetic agents: a critical review. Bioorg. Chem (2018) 77:548–67. doi: 10.1016/j.bioorg.2018.02.009
127. Bravard A, Veilleux A, Disse E, Laville M, Vidal H, Tchernof A, et al. The expression of FTO in human adipose tissue is influenced by fat depot, adiposity, and insulin sensitivity. Obes (Silver Spring Md.) (2013) 21(6):1165–73. doi: 10.1002/oby.20110
Keywords: insulin resistance, metabolism, m6A modification, signaling pathway, type 2 diabetes
Citation: Zhang H, Gu Y, Gang Q, Huang J, Xiao Q and Ha X (2023) N6-methyladenosine RNA modification: an emerging molecule in type 2 diabetes metabolism. Front. Endocrinol. 14:1166756. doi: 10.3389/fendo.2023.1166756
Received: 15 February 2023; Accepted: 16 June 2023;
Published: 07 July 2023.
Edited by:
Marcia Hiriart, Universidad Nacional Autonoma De Mexico, MexicoReviewed by:
Jianru Yi, Sichuan University, ChinaCopyright © 2023 Zhang, Gu, Gang, Huang, Xiao and Ha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoqin Ha, aGF4aWFvcWluMjAxM0AxNjMuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.