 Yifei Song
Yifei Song Xinying Wang1†
Xinying Wang1† Wenxin Ma
Wenxin Ma Jiapan Sun
Jiapan Sun Xiaoyun Zhu
Xiaoyun Zhu Yang Tang
Yang Tang- 1Beijing University of Chinese Medicine, Beijing, China
- 2Centre for Evidence-Based Chinese Medicine, Beijing University of Chinese Medicine, Beijing, China
- 3Tongling Municipal hospital, Anhui, China
- 4Department of Geriatrics, Peking University Shenzhen Hospital, Shenzhen Peking University-The Hong Kong University of Science and Technology Medical Center, Shenzhen, Guangdong, China
- 5Guang’anmen Hospital, China Academy of Chinese Medical Sciences, Beijing, China
Graves’ disease (GD) is characterized by diffuse enlargement and overactivity of the thyroid gland, which may be accompanied by other physical symptoms. Among them, depression can dramatically damage patients’ quality of life, yet its prevalence in GD has not received adequate attention. Some studies have established a strong correlation between GD and increased risk of depression, though the data from current study remains limited. The summary of mechanistic insights regarding GD and depression has underpinned possible pathways by which GD contributes to depression. In this review, we first summarized the clinical evidence that supported the increased prevalence of depression by GD. We then concentrated on the mechanistic findings related to the acceleration of depression in the context of GD, as mounting evidence has indicated that GD promotes the development of depression through various mechanisms, including triggering autoimmune responses, inducing hormonal disorders, and influencing the thyroid-gut-microbiome-brain axis. Finally, we briefly presented potential therapeutic approaches to decreasing the risk of depression among patients with GD.
1 Introduction
Graves’ disease (GD) is an autoimmune condition of the thyroid caused by the excessive production of stimulatory antibodies (thyroid-stimulating antibodies, TSAb) against the thyroid-stimulating- hormone receptor (TSH-R). It is the most common cause of hyperthyroidism (1). Epidemiological studies indicate that the incidence of GD is 20–40 cases per 100,000 population per year (2). Research has associated hyperthyroidism with severe work disability (3), and the development of various diseases across different systems in the onset of GD has been well documented (4–6). These complications dramatically worsen the clinical outcomes of GD patients and are associated with increased mortality (7) and economic burden. With recent advances in neuroendocrinology, psychiatric disorders in GD patients are of growing interest to researchers.
Previous research has related overt hyperthyroidism with psychiatric disorders such as irritability, anxiety, mania, sleeplessness, and depression (8, 9). In recent years, increasing evidence has indicated the high prevalence of depression and related symptoms in patients with GD (10–12). As GD hyperthyroidism is more commonly manifested as emotional agitation and irritability (4, 13) and depression is usually linked with hypothyroidism instead of hyperthyroidism, the mechanism behind GD and depression seems far less clear compared with other moods disorders observed in GD. Studies have predicted that the development of depression in GD may be related to the exhaustion of noradrenergic transmission (14). Yet, this assumption has only considered the traditional theory of the cause of hyperthyroidism, meaning that more complicated mechanisms involving autoimmunity, hormone metabolism, gut dysbiosis, etc., may be ignored.
As a result of the inability to understand the mechanism of depression and related symptoms in GD, depression in GD may well be overlooked, which can lead to severe clinical consequences. Characterized by a persistent feeling of sadness and/or an inability to experience pleasure, depression is widely linked to serious consequences. The World Health Organization has ranked it the second most significant cause of disability worldwide (15–17). Thus, the development of depression will not only seriously impair the quality of life of GD patients but also aggravate the original condition of GD and affect its prognosis. In spite of this, depression, along with other mood disorders, has not been listed in GD guidelines as an intervention target (18). These realities call for more attention in understanding GD and depression, which we hope will provide a more systematic vision of the crosstalk between thyroid functions and the neuropsychiatric system, as well as benefiting from the improvement of treatment methods of GD and lowering its recurrence rate.
This article aims to provide mechanistic insights into depression in GD. Through summarizing current literature in this field, the driving role of GD in the development of depression has been revealed. In this article, we will first review epidemiological evidence supporting the increased risk of depression among GD patients. We will then highlight the pathogenesis links between GD and depression from three different aspects. Finally, we will briefly present potential preventative and treatment methods for depression in the context of GD. The main objective of this article is to outline the mechanism of depression in the context of GD, so as to provide references for future studies on the prevention and treatment of GD emotional complications.
2 Graves’ disease and increased risk of depression
GD is a prevalent autoimmune thyroid syndrome that has been linked to a wide range of illnesses, including cardiovascular diseases (19), hepatic dysfunction (20), neurological diseases (6), and so on. Among them, mental disorders, mainly depression and anxiety, are found to be closely related to GD (21).
Previous and updated epidemiological studies have uncovered the relationship between GD and depression. To review the bodies of evidence, we systematically screened on PubMed/MEDLINE, Cochrane Library, and Web of Science from the database inception to March 22, 2023. We combined generic terms for GD, hyperthyroidism, depression, and population-based study designs. After the exclusion of duplicates, 11 studies were screened, which included 5 cohort studies, 3 cross-sectional studies, and 3 case-control studies (Table 1).

Table 1 Clinical studies of Graves' disease related to depression.
Three studies, including 2 cohort studies and 1 case-control study, have pointed out the direct correlation between GD and increased risk or severities of depression. Chen et al. conducted a retrospective cohort study involving 20975 patients (4195 GD patients and 16780 non-GD patients) and discovered that GD and GD treatment are associated with an increased risk of depression in Asian patients (22). Another nationwide Swedish cohort study has also pointed out a significantly higher risk of depression in GD (23). Moreover, as GD is the most common cause of hyperthyroidism (4), studies on hyperthyroidism have also provided evidence for the correlation between GD and depression. A cross-sectional study of 2142 individuals has found an association between untreated hyperthyroidism and a higher risk of depression (24). Another Korean national cross-sectional data study concluded that subclinical hyperthyroidism was independently associated with depressive symptoms. Additionally, studies by Pápai A et al. (25) and Hamed SA et al. (12) have supported the correlation between hyperthyroidism and depression.
Although our screening results have already supported a strong link between GD and depression, the number of current clinical studies available remains very limited. Further prospective longitudinal studies will be needed to determine whether GD affects the pathophysiology of depression.
3 Graves' disease as a mechanistic driver of depression
Mounting evidence has indicated that GD promotes the development of depression through a variety of mechanisms, including triggering autoimmune responses, inducing hormonal disorders, and influencing the thyroid-gut-microbiome-brain axis. (Shown in Figure 1).
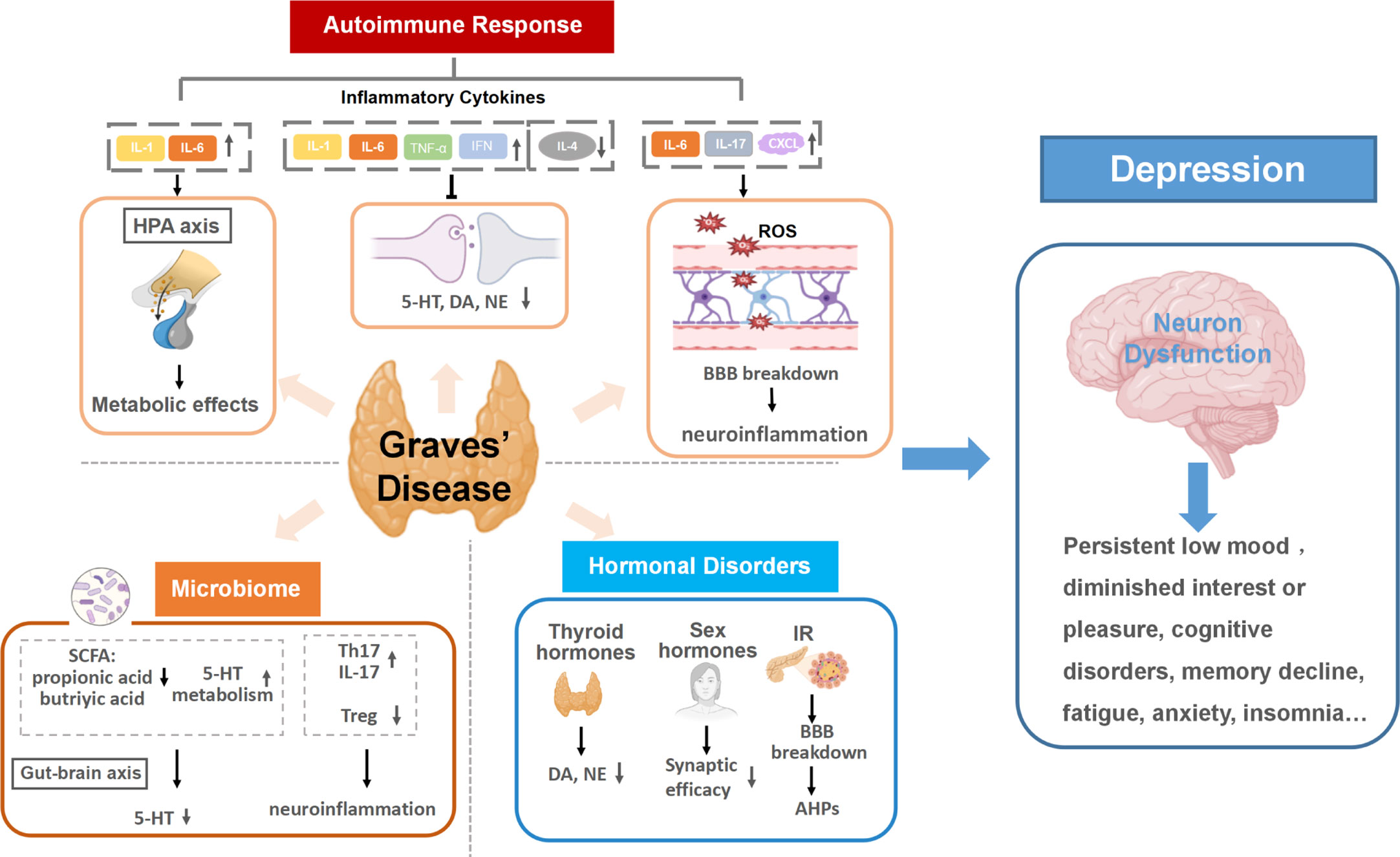
Figure 1 Graves’ disease as a mechanistic driver of depression. Specific pathways of autoimmune responses, hormonal disorders and microbiota dysbiosis in GD trigger neuron dysfunction in the brain and promote the development of depression. (1) Autoimmune responses in GD cause altered inflammatory cytokines levels. Continuous activation of the HPA axis induced by persistent inflammatory cytokines stimulation can damage neurons and activate continuing central inflammation, along with affecting the expression and action of hormone receptors. Inflammatory cytokines can also disrupt monoamine neurotransmitter metabolism, resulting in lower accessibility to these neurotransmitters. The increase of BBB permeability in the inflammatory state of GD allows inflammatory cytokines to enter the brain and may lead to neuroinflammation. (2) Multiple hormonal disorders can develop as a result of GD hyperthyroidism, including thyroid hormones, insulin and sex hormones. The increase of thyroid hormones can reduce the secretions of dopamine (DA) and norepinephrine (NE). Insulin resistance (IR) in GD can increase both blood and intracellular glucose level, inducing neuronal tissue damage and afterhyperpolarization (AHP). Additionally, sex hormone disorders in GD are likely to lower Synaptic efficacy, which is related to the pathogenesis of depression. (3) Gut dysbiosis in GD can result in lower accessibility to 5-HT and activate host immune responses, increasing the risk of depression. HPA axis, the hypothalamic-pituitary-adrenal axis; 5-HT, serotonin; IR, insulin resistance; DA, dopamine; NE, norepinephrine; BBB, the blood-brain barrier; SCFA, short-chain fatty acids; AHP, afterhyperpolarization; 5-HT, 5-hydroxytryptamine.
3.1 Autoimmune and inflammatory responses in GD increase the risk of depression
GD is a common autoimmune disease in which excessive production of stimulatory autoantibodies disrupts critical physiological processes inside and outside the thyroid gland in GD, resulting in cellular and humoral immune disorders (26). Earlier studies have demonstrated the relationship between depression and immune disorders, with a focus on the role of inflammation and proinflammatory cytokines (27). A more recent study by Fam et al. observed significantly higher TSH receptor antibodies (TRAbs) concentration in depressed patients than in healthy controls, which suggested that autoimmunity responses in GD may exert neuropsychiatric effects, which increase susceptibility to depression (28).
Cytokines play an important role in autoimmune responses and are often used as indicators to predict the occurrence and progression of diseases (29). Previous studies have shown that cytokines can enter and influence the central nervous system through nerves, body fluids, the blood-brain barrier(BBB), cytokine receptors, and other pathways (30). As indispensable components of immunity, lymphocytes, and their specific cytokines are shown to play a pathogenic role during the course of GD by previous studies (31). These cytokines can lead to various subsequent neuropsychiatric effects, including disrupting neurotransmitter metabolism, inducing BBB dysfunction, and dysfunction of the hypothalamic-pituitary-adrenal (HPA) axis, playing a vital role in the development of depression.
3.1.1 Disrupting neurotransmitter metabolism
Neurotransmitters are vital chemicals that transmit information between neurons or neurons and effectors. Studies have shown that depression may involve abnormalities of numerous transmitter systems and molecular mechanisms across specific neural brain structures. For example, the mesolimbic dopaminergic pathway composed of dopaminergic (DA) neurons in the ventral tegmental area (VTA) and their projections to the nucleus accumbent (NAc) is crucial for the recognition of emotionally salient stimuli such as reward and aversion. Decreased DA neuron activity in VTA is associated with depression-related behaviors (32). As cytokines can regulate the metabolism and availability of neurotransmitters, they may play a driving role in the above pathological processes.
Changes in levels of various cytokines, which have potential effects on neurotransmitter metabolism, have been widely observed in the thyroid and orbital tissues of GD patients (33). These changes can be mainly summarized as increased pro-inflammatory cytokines and decreased anti-inflammatory cytokines. Ujhely et al. compared the levels of proinflammatory cytokines in tears between GD patients and healthy controls and discovered that the release of IL-1β, IL-6, IL-13, IL-17A, IL-18, TNF-α, and RANTES significantly increased in patients with GD and Graves ophthalmopathy (GO) (34). Elevated concentration of circulating IL-6/sIL-6R was also discovered in patients with GD (35). Additionally, Ruiz-Riol et al. (36) found overexpression of interferon (IFN) signal after bioinformatics analysis of the thyroid gland in GD. By contrast, a study by Kallmann et al. discovered that the release of IL-4, a typical anti-inflammatory cytokine, decreased in GD (37).
Subsequently, the alteration of these cytokines is involved in the disruption of monoamine neurotransmitters, including serotonin (5-HT), dopamine (DA), and norepinephrine (NE) (38, 39). In the metabolism processes of 5-HT, Indoleamine 2-dioxygenase (IDO) is a critical enzyme that catabolizes L-tryptophan (TRP) into L-kynurenine (KYN) by cytokine-induced activation. KYN is then converted into quinolinic acid (QUIN) and kynurenic acid (KA), which are neuroactive and may contribute to the behavioral changes experienced by some patients during exposure to inflammatory stimuli (40). Pro-inflammatory cytokines represented by IL-1, IL-6, TNF- α, and IFN can activate IDO, while anti-inflammatory cytokines such as IL-4 can down-regulate the enzyme. With increased pro-inflammatory cytokines and decreased anti-inflammatory cytokines in GD, IDO can be activated, resulting in greater tryptophan consumption and decreased 5-HT accessibility (41, 42). In addition to interfering with 5-HT metabolism, proinflammatory cytokines have also been reported to disrupt the synthesis, release, and reuptake of DA. By suppressing striatal DA release, INF-α may contribute to multiple depressive symptoms (43–45). TNF-α can induce delayed and progressive loss of DA neurons in the substantia nigra (SN) (46). Moreover, central inflammatory signals can activate mitogen-activated protein kinase (MAPK), which in turn increases the number and activity of presynaptic reuptake pumps, resulting in reduced synaptic availability of monoamine neurotransmitters, including norepinephrine (NE) (47, 48).
As neurotransmitters play a critical role in the function of the central nervous system, the disrupted metabolism of monoamine neurotransmitters can increase the risk of depressive behaviors. On the one hand, disruption of these neurotransmitter metabolisms can result in the loss of innervation of monoamine neurons in the brain and induces depressive behaviors such as mood disorders (45, 49, 50). On the other hand, inflammation, as previously indicated, triggers an increase in QUIN, which binds to the N-methyl-D-aspartate receptor (NMDAR), inducing the release of glutamate (48, 51). As an excitatory neurotransmitter, high level of glutamate can alter the strength of certain groups of glutamatergic synapses in a variety of brain regions, such as the prefrontal cortex (PFC), hippocampus, and nucleus accumbens (NAc), causing dysfunction of cortico-mesolimbic reward circuitry that underlies many manifestations of depression (52).
3.1.2 Triggering neuroinflammation following BBB disruption
Neuroinflammation was mediated by the activation of microglial cells (53), the primary resident immune cells in the central nervous system, which in turn activates nuclear factor-kappaB (NF-κB) and produces inflammatory cytokines (54, 55). Its vital role in depression is increasingly recognized (56). Converging lines of evidence indicated that activation of immuno-inflammatory pathways is highly associated with developing various neuropsychiatric diseases (57). Studies also found that persistent low-grade inflammatory activation is related to the severity of depressive symptoms and the probability of treatment response (57).
The blood-brain barrier (BBB) is formed by endothelial cells sealed by tight junction proteins, pericytes, and astrocytes. The specific multicellular structure of BBB controls its major functions, which include maintaining brain homeostasis, regulating influx and efflux transport, and providing protection from injury (58). Various factors in GD can increase BBB’s permeability, which allows multiple inflammatory cytokines to enter the brain and contributes to the development of neuroinflammation. For instance, IL-17 and IL-6, which are typical cytokines observed in GD patients and animal models (35, 59), can disrupt the normal structure and function of the BBB. The effect of IL-17 on BBB disruption has been thoroughly documented (35, 59). Huppert et al. found that IL-17 increased reactive oxygen species (ROS) production and activated oxidative stress, activating endothelial contractile machinery. Activation of the contractile apparatus is responsible for the loss and disorganization of dense tight junction (TJ) proteins, which consecutively lead to BBB breakdown (60). Apart from IL-17, researchers also found that circulating IL-6 could act on the main cell adhesion component of endothelial cells, tight junction protein claudin5 (Cldn5), which alters BBB permeability (61). In addition to these two cytokines, lymphocytes in the thyroid secrete diverse pathogenic cytokines into the blood, resulting in circulating cytokine disorders and increased BBB’s permeability.
As a result of BBB disruption, inflammatory factors are more prone to entering the central nervous system and subsequently activate neuroinflammation. One typical pathway is through activating microglia. Microglia are inherent immune effector cells in the central nervous system, which play an essential role in regulating neuronal development, neuron apoptosis, and other physiological processes (62). A previous experiment showed that the up-regulation of IL-17 induced the activation of microglia in the hippocampus, amygdala, and prefrontal cortex ofmodel mice, resulting in depression-like behaviors in early adulthood than those in single or dual stress groups (63). Activated and polarized microglia typically exhibit the pro-inflammatory M1 phenotype and produce pro-inflammatory cytokines, contributing to neuronal dysfunction and reinforcing disease-related behaviors, all of which are related to psychiatric disorders (64, 65).
Another pathway is through recruiting immune cells by chemokines. Antonelli et al. observed elevated chemokine levels in patients with GD (66). C-X-C motif chemokine ligand (CXCL), a typical chemokine, can recruit immune cells to infiltrate the cerebrovascular system and brain parenchyma before initiating a variety of pathways leading to neuronal death (67). In addition, Th17 cells recruited by IL-17 can induce neuronal death by directly interacting with neurons or inducing elevated Ca2+ levels (68). Neuronal death thus causes the reduction of brain volume and hinders neuro-progression, which in turn contributes to depression. The atrophy of brain regions such as the hippocampus and prefrontal cortex was observed in patients with depression (69), which may be the result of decreased neuron number as a result of neuronal death. Research has also shown that neuronal death, neurodegeneration, decreased neurogenesis, and reduced neuroplasticity can jointly contribute to impaired neuro-progression (70). Impaired neuro-progression, which results in somatic symptoms including anhedonia, anxious behavior, fatigue, discomfort, and psychiatric symptoms such as mild cognitive impairment (MCI), is associated with the progression of depression (71, 72).
In summary, abnormal levels of GD-related inflammatory factors can disrupt the BBB before mediating the formation of neuroinflammation through multiple pathways, such as activation of central microglia and recruitment of immune cells, which can cause nerve damage and increase susceptibility to depression.
3.1.3 Dysregulation of the HPA axis
The HPA axis is one of the critical physiological systems that regulate systemic inflammatory response and immune response (73). It secretes three main classes of hormones, which are regulated hierarchically. Sensory signals control the release of hypothalamic corticotropin-releasing hormone (CRH) and vasopressin (AVP) from the paraventricular nucleus and into the pituitary portal circulation, where they reach the adenohypophysis and stimulate the release of adrenocorticotropic hormone (ACTH), which in turn promotes adrenocortical production and cortisol release (74). From an evolutionary perspective, the activation of the ACTH/HPA axis and sympathetic spinal system, along with the diminished function of the prefrontal cortex, results in a diminished ability to seek and experience pleasure, reduced food intake, decreased sexual activity, and sleep apnea. These are transient adaptive changes the body makes in response to stress (75).
In the pathological state, due to the persistent stimulation of the central cytokine system, the CRH/HPA axis and the noradrenergic system will be continuously activated. IL-1 and IL-6, two proinflammatory cytokines elevated in GD (76–78), are typical cytokines involved in stimulating CRH/HPA axis hyperactivity. After binding to the IL-1 type I receptor, IL-1β could mediate the activation of the HPA axis in the rodent model of depression (79), which provides evidence for IL-1’s role in activating the HPA axis. As IL-1 has known effects on hypothalamic biogenic amines, the main source of corticotropin-releasing factor (CRF), it can regulate on pituitary ACTH and promote the production and release of cortisol (80), which may be the underlying mechanism of the HPA axis activation by IL-1. Apart from IL-1, the role of IL-6 in activating the HPA axis has also been found (81, 82).
The HPA axis is involved in the regulation of many physiological functions, such as energy balance, sleep-wake transition, and hippocampal neuron survival (83). Therefore, the disruption of the HPA axis is the pathological basis of many diseases. HPA axis abnormalities are related to the pathophysiology of cognitive impairment (84). Study data show that more than half of depressed patients have other HPA axis disorders, such as hypercortisolemia or altered cortisol circadian rhythm (85). The activated HPA axis is involved in the pathogenesis of depression by damaging neurons and continuing central inflammation. Adrenocortical steroids enter the brain and have widespread effects, particularly impairing hippocampal function (86). Long-term exposure to the hormone environment can decrease the secretion of derived neurotrophic factor (BDNF) and interfere with neural development (87, 88). Adrenocortical hormones not only destroy the plasticity and structure of neurons, but also exert cytotoxic effects by regulating neurotransmitters.
In addition to mediating HPA axis activation resulting in abnormal hormone secretion, cytokines also affect the expression and action of hormone receptors. The number of adrenergic receptors expressed in IL-1Ra gene knockout (IL-1RA KO) mice showed complex stage changes (89). The levels of IL-6, IL-10, and TNF-α in patients with depression can also affect the sensitivity of glucocorticoid receptors.
It has also been found that there is an interaction between the two major stress response systems in depression—The hypothalamic-pituitary-adrenal axis and the immune system (90). Glucocorticoids secreted by the HPA axis are biphasic immunomodulatory substances that balance anti-inflammatory and pro-inflammatory (91). In the absence of inflammation, basal levels of glucocorticoid receptor signaling promote the expression of PRRs, cytokine receptors, and complement factors, sensitizing cells to noxious stimuli and promoting the induction of inflammatory responses after tissue injury. However, in the inflammatory state, glucocorticoid concentrations suppress the immune response mainly by inhibiting the transmission of PRR, Fc receptors, and cytokine signaling, thereby shortening the duration of the immune response (92, 93). In depressed patients, the expression and availability of glucocorticoid receptors are decreased, which prevents the HPA axis from exerting anti-inflammatory effects and promotes persistent inflammation (94, 95). The increased activity of pro-inflammatory transcription factors and NF-κB has also been associated with decreased glutamate receptor activity (96).
Therefore, as an autoimmune disease, GD can cause systemic immune activation and high levels of inflammatory factors in the circulation, leading to the activation of the HPA axis and hypersecretion of related hormones. Continuous activation of the HPA axis weakens its anti-inflammatory functions, causing a persistent state in the body and increasing the risk of depression.
3.2 Multiple hormonal disorders in GD may increase the risk of depression
GD is not only an autoimmune disease but also an endocrine disease characterized by excessive secretion of thyroid hormones. The extracellular portion of TSHR on the surface of thyroid cells is recognized by GD autoantibodies - TRAbs, which causes hyperthyroidism (97). Subsequent abnormalities of various endocrine hormones are developed as a result of hyperthyroidism. Studies have confirmed that thyroid function will affect HPA axis activity, insulin sensitivity of glucose, lipid metabolism, and ovarian secretion function, resulting in hypercortisolism, insulin resistance, and sexual dysfunction (78, 98–102). On the other hand, patients with depression have manifested abnormalities of numerous hormones, including thyroid hormones, insulin, and sex hormones. This suggests that endocrine disorders may be involved in the pathology of depression (103).
Although currently, there are few studies about the effect of GD on depression from the perspective of endocrine disorders, evidence has found that hormonal abnormalities are closely related to the clinical manifestations of depression. Therefore, we hypothesized that several hormonal abnormalities in GD could increase the risk of depression, and we analyzed the specific mechanism regarding hormonal abnormality of the HPT axis, insulin metabolism, and sex hormones.
3.2.1 Hormonal abnormalities in the HPT axis
The hypothalamus-pituitary-thyroid axis (HPT) is a hormone axis that regulates thyroid function by grading and feedback regulation of thyrotropin-releasing hormone (TRH), thyrotropin (TSH), thyroxine (T4), and triiodothyronine (T3). It interacts with several other hypothalamic-pituitary-target axis systems and plays a key role in maintaining homeostasis (104). Autoantibodies produced in GD can activate the TSHR pathologically and overcomes the physiological HPT negative feedback loop, leading to elevated thyroid hormones despite low levels of serum TSH (105). As the HPT axis has long been involved in the research of depression (106), it is reasonable to believe that the dysfunction of the HPT axis in GD, with its effect of disrupting neurotransmitter metabolism, inducing oxidative stress, contributing to other complications and so forth, has a potentially vital effect on the development of depression.
On the one hand, the increase in thyroid hormones can affect the metabolism of neurotransmitters in the brain. In the physiological state, thyroid hormones regulate the expression of genes needed for nerve cell differentiation, migration, and myelin formation. This plays a vital role in nerve development and signal transmission. Nevertheless, with the excessive production of thyroid hormones in GD, changes in the levels of several neurotransmitters, particularly NE and DA, will disrupt the brain’s regular signal route and thus increase the risk of depression. A significant decrease in NE and DA in the cerebral cortex of hyperthyroidism rats has been observed in experiments (107), which can further lead to depression (108, 109). On the other hand, long-term exposure to high levels of thyroid hormones is associated with oxidative stress, which is related to depression-related neurodegenerative changes or even neuro-death in the brain (110). Neuroimaging studies have found that patients with hyperthyroidism had reduced grey matter volume in portions of the occipital lobe (lateral occipital sulcus and fusiform gyrus) in the left hemisphere, the frontal and parietal lobes (insula, paracentral, precuneus, superior frontal cingulate, superior parietal and postcentral) in the right hemisphere. It has been clearly demonstrated that altered circulating thyroid hormone is associated with various mental signs and symptoms, such as emotional disturbances (impulsiveness, irritability), cognitive deficits (impairments in memory, concentration, attention, planning, and productivity), and affective symptoms (anxiety, depression, mania), which increase the incidence of neuropsychiatric disorder like depression (111). Apart from the increased thyroid hormones, clinical studies have found that decreased level of TSH is an important risk factor for depression in the elderly (112), although its underlying mechanism remains unknown.
In addition to direct effects from the hormonal abnormality of the HPT axis, complications of HPT dysfunction can also contribute to depression. A mendelian randomization study has found that hyperthyroidism can cause hemodynamic changes, which results in cardiovascular diseases represented by atrial fibrillation (AF) (113, 114). As studies have uncovered AF as a risk factor for depression (114, 115), the development of AF in the context of HPT dysfunction may increase the risk of depression. Additionally, hyperthyroidism can also cause insulin resistance (IR) and diabetes. Microvascular dysfunction is widespread in people with diabetes, which can affect the brain. A growing body of evidence suggests that microvascular dysfunction is one of the key underlying mechanisms of an increased risk of developing certain brain or mental disorders in diabetes. The microvasculature is involved in the regulation of many cerebral processes. When it is impaired, patients are more susceptible to lacunar and hemorrhagic stroke, cognitive dysfunction, and depression (116). A population-based cohort study demonstrated that vascular damage caused by microvascular dysfunction in frontal and subcortical brain regions, which are involved in mood regulation, might lead to depression in older individuals (117). Besides, cerebral microvascular dysfunction can increase BBB permeability (116). Data from human and animal studies provide strong evidence of neurovascular unit dysfunction with BBB hyperpermeability in association with oxidative stress and neuroinflammation (118), which can increase the susceptibility to neurological disorders such as depression.
In summary, elevated thyroid hormones in GD hyperthyroidism can directly increase the risk of depression. It is also noteworthy that complications of HPT dysfunction in GD can also increase susceptibility to depression.
3.2.2 Insulin metabolism abnormalities
Insulin is a protein hormone secreted by islet β cells in the pancreas, stimulated by endogenous or exogenous substances such as glucose, lactose, glucagon, etc. It is the only hormone that can lower blood glucose levels. Although specific mechanisms remain unclear, thyroid hormone has the function of regulating blood glucose level in the body (119, 120). Previous studies have shown that excessive thyroid hormones can lead to IR and abnormal glucose metabolism (121).
IR refers to the inability of insulin to effectively promote glucose uptake in peripheral tissues or to inhibit hepatic glucose output, which leads to increased blood glucose. High intracellular glucose concentration develops when hyperthyroidism induces IR in GD patients, because brain cells like cerebral microvascular endothelial cells, pericytes, and astrocytes are unable to slow down their glucose transport rate (122, 123). It can then cause dysfunction of these brain cells, including increasing microvascular endothelial cells’ permeability, leucocyte adhesion, procoagulant activity, and reducing the availability of nitric oxide through various biochemical pathways initiated by mitochondrial overproduction of reactive oxygen species (124). Certain regions of the brain can become dysfunctional as a result of malfunctioning cells, which raises the risk of mental diseases. According to the vascular depression hypothesis, vascular damage in the frontal and subcortical brain regions, which are involved in mood regulation, might lead to depression in older individuals (117). On the other hand, chronic hyperglycemia due to IR leads to increased extracellular and intracellular formation of advanced glycation end products (AGEs), leading to detrimental effects, including increased oxidative stress and production of inflammatory cytokines in these brain cells (125). Oxidative stress and inflammatory cytokines can directly disrupt the BBB and damage neuronal tissue, which is likely to cause cognitive dysfunction and increase the risk of depression (116).
Furthermore, insulin has the function of enhancing memory and mediating signal transduction in plasticity and stress responses (126). Decreased insulin receptor sensitivity plays an important role in the mechanism of neuropsychiatric diseases, including depression. Insulin sensitivity in patients with hyperthyroidism has been reported to be reduced (127). The loss of insulin sensitivity of hippocampal CA1 pyramidal neurons can subsequently cause long-term Ca 2+-dependent afterhyperpolarizations (AHPs), which can produce significant cognitive and memory impairment in the short term and throughout life, contributing to depression (122, 123).
3.2.3 Sex hormones abnormalities
Many research has discovered sex differences in the prevalence of both GD and depression, with a reported male-to-female ratio of 1:4-6 in GD and a ratio of roughly 1:2 in major depressive disorder (122, 128, 129). Moreover, women are more susceptible to both of these diseases during pregnancy and the perinatal period (130). As sex hormone levels differ greatly between significantly genders and during different physiological events of women, the evidence has pointed to the essential role that sex hormones play in both diseases.
Both estrogen(E2) and androgen can increase the risk of GD through different mechanisms. B cell activating factor (BAFF) is a B cell survival factor that promotes autoreactive B lymphocytes and inhibits their deletion (131). BAFF expression is closely linked with autoimmunity, and upregulated BAFF activity was found to contribute to the occurrence of multiple AIDs, including GD. E2 might be involved in modulating BAFF levels, resulting in a higher incidence of AITD in women (132–134). Cheng et al. (135) compared the serum BAFF levels between genders in clinical samples and discovered higher BAFF levels in women in both GD and control groups. Additionally, in the experiment of SAT mice, exogenous E2 treatment increased serum BAFF levels in male SAT mice, and higher thyroid BAFF transcripts levels were found in female SAT mice regardless of E2 treatment. Thus, abnormal estrogen metabolism, which results in increased E2 levels, may contribute to a higher occurrence of GD. On the other hand, evidence shows that androgen can inhibit the production of antigen-presenting cells in the thyroid gland, as well as the uptake and transport of autoantibodies, thereby preventing the occurrence and progression of autoimmune thyroid diseases (136). Thus, androgen depletion would increase the risk of the thyroid being subjected to an autoimmune attack, leading to an increased susceptibility to GD.
Similar effects of sex hormones were found in patients with depression, which may be related to synaptic efficacy (137). As the biological basis of learning and memory, synaptic efficacy is regulated by sex hormones. Female estrogen changes periodically, affecting the structure and function of the brain, resulting in female-unique brain plastic changes and related vulnerability (130). As one of the most basic and essential functions of the brain, synaptic plasticity affects the ability to perceive, evaluate and store complex information and guides the body to make appropriate adaptive responses to subsequent stimuli. It plays a crucial role in short-term and long-term memory. Many factors, such as the interruption of the estradiol cycle and the increase of inflammatory cytokines, will lead to changes in synaptic function and morphology, which may be one of the pathogenesis of depression (138).
It is noteworthy that when sex hormone disorders cause GD, it may also contribute to depression, resulting in the comorbidity of both diseases. Furthermore, sex hormone disorders during the onset of GD are likely to affect the normal metabolism of sex hormones, which might lead to or aggravate the depressive state of GD patients. Further research into this field will help provide deeper insight into the interaction between the two diseases.
3.3 A possible thyroid-gut-microbiome-brain axis increases the risk of depression
The human microbiota has been found to be a key regulator of health and diseases, and more and more evidence has pointed out their relevance to many human diseases (139). As the primary interface, the gastrointestinal tract contains about 66% of the human microbiota (139), including bacteria, viruses, fungi, and so on (140, 141). The gut microbiota has been implicated in numerous human diseases (142). The bidirectional pathways between the gut and the brain, known as the gut-microbiome-brain axis, have long been recognized. Research has shown a strong correlation between the gut microbiota and the pathophysiology of depression and anxiety (143–146). In recent years, there has been increasing awareness of the role of microbiota in the pathogenesis of GD (139, 147–149). Current evidence has pointed to shared microbiota changes in GD and depression, indicating that gut microbiome dysbiosis in GD may act as a trigger for depression (26, 150–154). A prospective clinical study with 39 participants with GD and 17 without GD found that the levels of bacilli, Lactobacillales, Prevotella, Megamonas (154). Hou et al. (155) summarize the studies of 263 Graves Disease(GD)/239 Health Control(HC) samples until July 2021. In general, about 29 gut microbiota taxa are different in GDs compared with HCs. Though results of some microbiota taxa are not consistent in different studies, several taxa, including Prevotellaceae and Veillonellaceae at the family level and Bacteroides, Lactobacillus, Prevotella, and Veillonella at the genus level were identified in three or more studies. Among these studies, it is noteworthy that Prevotellaceae and Prevotella were identified as the core microbiome of the GD group and were closely associated with GD patients. On the other hand, the microbiota alterations in depression include the decline of both the microbiota diversity and richness (150, 156). Interestingly, research have revealed that the abundance of Prevotellaceae and Prevotella increased in depressed patients, indicating that gut microbiome dysbiosis in GD is likely to contribute to the microbiota alteration in depression. Given the above correlation, we predict that a possible thyroid-gut-microbiome-brain axis is involved in the pathogenesis of mental disorders, thereby increasing the risk of depression.
3.3.1 Gut microbiome dysbiosis in GD induces unbalanced neurotransmitters in the brain
Neurotransmitters like serotonin [also known as 5-hydroxytryptamine (5-HT), dopamine (DA), and noradrenaline (NE)] have been implicated in the pathophysiology of depression for many years as essential components of the brain’s function. Under normal conditions, the gut microbiota secretes neurotransmitters (e.g., Gamma-Amino Butyric Acid - GABA, serotonin) and trophic factors (e.g., brain-derived neurotrophic factor - BDNF), which is related to neuronal communication, maintenance, and survival of the brain through neurotrophic support. Although the exact mechanism of how gut microbiota influences neurotransmitters in depression is still unclear, some hypotheses and research have pointed out that neurotransmitter imbalance might play a vital role in the process. In the monoaminergic neurotransmitter deficiency hypothesis, it is posited that depressive symptoms arise from insufficient levels of monoamine neurotransmitters such as 5-HT, norepinephrine (NE), and/or dopamine (DA) (157). Subsequent research has added that the hyperactivity of glutamatergic system (158) and acetylcholine system (159), the inhibition of the gamma-aminobutyric acid (GABA) system (160) also contribute to depression.
Among the neurotransmitters related to depression, GABA is the main inhibitory mediator in the CNS, which is crucial in controlling various psychological and physiological processes. These processes include antianxiety, diuresis, and neurotransmission (161). Multiple neurological and psychiatric illnesses, including epilepsy, depression, and Alzheimer’s disease, are linked to the etiology of abnormalities in GABA levels and GABA receptor functioning in the central nervous system (162). Researchers have discovered that the GABA system is inhibited in the pathogenesis of depression (51, 163, 164), resulting in less secretion of GABA in the brain. The gut microbiome is an important regulator of GABA synthesis, with Lactobacillus brevis being the most effective enteral flora generator of GABA (165).
Research has reported the reduction of Lactobacillus in the onset of depression, resulting in less production of GABA, which is likely to contribute to depression (166). On the other hand, three studies have reported the increase of Lactobacillus in GD patients and the positive correlation between TRAb level and Lactobacillus. While Lactobacillus is often not harmful in the human body and may exhibit an antidepressant effect at the start of GD, some research has revealed that Lactobacillus might cause macrophages to release pro-inflammation cytokines including IL-6 and TNF-α (167). Thus, according to the cytokine hypothesis, it is reasonable to believe that these pro-inflammatory cytokines can then increase the risk of depression through ways such as inhibiting the negative feedback of the HPA axis, increasing the permeability of the BBB, and so on. Whether the increase of Lactobacillus in GD plays a protective or pathogenic role in the pathogenesis of depression remains to be further studied.
Other neurotransmitters related to gut microbiota may also contribute to depression in the onset of GD. Serotonin, or 5-HT, is a tryptophan (Trp) metabolite that plays a significant role in GI (gastrointestinal) regulation and has a variety of physiological effects on both humans and animals (168). Serotonin exists and is stored in two distinct pools in the human body, namely the peripheral and the central pool. Unlike peripheral 5-HT, central 5-HT performs critical control functions in the brain. It is a key regulator of behaviors such as emotion, stress response, appetite, addiction, and pain (169). It is also an important participant in the modulation of neuronal differentiation and migration, axonal outgrowth, myelination, and synapse formation (170). Decreased availability of 5-HT in the brain is a key characteristic of depression (170). Since peripheral 5-HT cannot cross the BBB, central serotonin is synthesized from Trp, which is transported through the BBB from blood circulation (170). As a crucial role in supplying our body with the required amount of Trp, the gut microbiota can regulate the CNS by modulating the central 5-HT level.
Research has revealed several pathways in which gut microbiota alterations in GD may contribute to a decreased level of central serotonin, thereby increasing the susceptibility to depression. On the one hand, reduced short-chain fatty acids (SCFAs) in GD patients may contribute to the reduction of central serotonin concentration. According to a research on gut microbiota in GD patients by gas chromatography-mass spectrometry (GC-MS), propionic acid and butyric acid, two prominent SCFAs, were considerably lower in GD patients (153). SCFA-producing Bacteroides fragilis YCH46 strain (B.f.S) has also been discovered to be significantly reduced in GD patients (153). SCFAs, particularly butyrate, which can be transported into blood circulation, are reported to increase the brain serotonin concentration, providing neuroprotective benefits to stressed mice (171). Consequently, reduced level of SCFAs in GD patients is likely to result in decreased brain serotonin concentration, which increases the risk of depression. On the other hand, tryptophan and serotonin metabolism alterations in GD patients are also likely to lower the central 5-HT level. Several recent studies have pointed out that alterations of the gut microbial tryptophan metabolism influence peripheral Trp availability, affecting central tryptophan levels and, thereby, leading to changes in the central serotonin metabolism (171, 172). Some other research discovered that enhanced serotonin metabolism may also contribute to decreased central serotonin levels following gut dysbiosis (173). It should be noted that the exact mechanism of tryptophan and serotonin metabolism alterations in GD still needs to be clarified. Thus, further studies into this aspect will establish a more profound correlation between GD and depression induced by 5-HT reduction.
3.3.2 Gut microbiome dysbiosis in GD activates host immune response
The gut microbiota plays a vital role in the formation and function of the host’s immune system. As mentioned in the previous sections, autoimmune responses play a critical role in the development of depression in GD. However, it is not yet clear how autoimmune responses of GD are activated. Given the gut microbiota’s critical role in the immune system, gut dysbiosis can induce immune responses and contribute to the autoimmune responses of GD, which is correlated with an increased risk of depression.
The main immune response is the imbalance between Th17 and regulatory T cells (Tregs). Under healthy conditions, gut microbiota can maintain the balance between Th17/Tregs. But in GD patients, dysbiosis may increase Th17 cells and suppress Treg production, manifested by the decrease of CD4+Foxp3+ Treg cells and the increase of CD4+IL-17+ Th17 cells in circulation (153, 174). Additionally, Bacteroides fragilis YCH46 strain (B.f.S), which can increase Tregs, IL-10, reduce Th17 cells and IL-17A levels in peripheral blood mononuclear cells(PBMCs) from healthy individuals, significantly decreased in GD patients, resulting in decreased IL-10 and elevated IL-17A levels (153).
Although the role of Th17 in depression has not been fully clarified, and research on the correlation between IL-17 and depression is still limited, accumulating evidence have pointed to the involvement of Th17 and IL-17 in depression. Chen et al. have observed increased circulating Th17 cells in depressed patients (175). The exact influence of circulating Th17 cells on depression remains unclear, but since Th17 cells can infiltrate the brain parenchyma without requiring VLA4 signals, it can be hypothesized that Th17 cells may reach the brain and synthesize IL-17 (68). On the other hand, two studies have found that circulating IL-17A is elevated in depressed patients (175, 176), and the administration of IL-17A is able to promote depressive-like behaviors (177). As evidence has shown that IL-17 can increase depression and depression-like behaviors through the activation of microglia and inducing neuronal death, we can speculate that increased Th17 cells and IL-17A, induced by gut dysbiosis during the onset of GD are involved in the immune response of depression, exacerbating depressive symptoms. More complex immune interactions between gut dysbiosis in GD and depression will require further investigations in this field.
4 Therapeutic approaches for decreasing depression risk among individuals with GD
Reports have covered that GD is often complicated with psychiatric symptoms and diseases, such as depression, anxiety, and schizophrenia (178, 179). These neuropsychiatric symptoms and illnesses not only seriously impair the quality of life of patients with GD, but also aggravate the original condition of GD and affect its prognosis (180, 181).
In view of this, more attention should be paid to the mental and psychological status of GD patients, and preventive measures should be taken to avoid the occurrence of depression during GD. Although current guidelines do not specifically address the management of depression in GD patients, several approaches can be adopted in light of the current understanding of both conditions. Close monitoring and routine evaluations of the patient’s mental state are necessary to prevent the detrimental effects of GD on mental health. Previous studies have applied the standard Mini-International Neuropsychiatric Interview or the Hamilton Depression Rating Scale and the Beck Depression Inventory for evaluating the mood state of GD patients, which can be potential methods for assessing the patients' mental state (182).
In terms of treatment options, it is notable that early diagnosis and treatment, as well as GD progression management using standard methods recommended by guidelines might lower the occurrence of mental disorders. Besides, previous research has pointed out differences in gut microbiome in patients with depression and indicated the potential treatment method with probiotics and symbiotics (183). Given the shared microbiota changes in GD and depression (26, 150–154), as well as the potential effects of microbiota dysbiosis in GD on depression, probiotics and symbiotics may exert beneficial effects in improving the intestinal flora environment of patients with GD, thus reducing the risk of depression. Additionally, another research reveals that methimazole (MMI) combined with probiotics improves the level of TRAb, which will play a positive role in lowering the recurrence rate of GD and is better suited to sustaining the positive emotion of GD patients (184). Since they are rarely implied in the current clinical treatment of GD, we recommend that probiotics and symbiotics should be utilized to improve depressive disorders in GD patients. Medicines used in the treatment of hyperthyroidism have also been shown to have potential beneficial effects in relieving depressive symptoms. Clinical practice suggests that antithyroid drugs combined with β-adrenergic receptor antagonists can be used as effective drugs for the treatment of mental disorders and psychiatric symptoms caused by hyperthyroidism, as the excessive activation of the adrenal system induced by hyperthyroidism can lead to somatic symptoms (14).
Additionally, psychosomatic synchronization therapy is considered to be suitable for GD patients with psychiatric symptoms (185), as research has shown that the combination of antithyroid, antipsychotic drugs and psychotherapy contribute to the prognosis of the disease (186). Also, psychological treatment can be supplemented when patients exhibit mental symptoms such as depression. However, due to the complexity of their pathophysiological mechanisms, treatments and interventions for depressive symptoms in GD patients is still under development, and further research is needed to assess the efficacy of these therapies (187).
5 Conclusions
GD, as the most common cause of hyperthyroidism, is often manifested as emotional agitation and irritability. In recent years, increasing evidence has pointed out the high occurrence of depression in GD, but it has not received adequate attention. Although currently only a few clinical studies have been conducted on the relationship between GD and depression, evidence has been able to point out the strong correlation between GD and depression. The pathophysiological mechanisms, including inducing autoimmune responses, hormonal disorders, and exerting an effect on the possible thyroid-gut-microbiome-brain axis, have underpinned the role of GD as a crucial driver in the development of depression and depressive symptoms. This can illuminate better management and treatment methods in terms of avoiding depression in GD patients and treating patients with depressive complications. We hope that through reviewing the potential mechanism of depression in GD, more attention can be drawn to the prevention and treatment of psychiatric complications of GD, so as to improve the treatment and prognosis of the disease.
Author contributions
YS and XW wrote the manuscript. WM, JS, XZ, YT, and YY contributed to the provided guidance of the whole manuscript. WM, XZ and YT reviewed and revised the manuscript. JS, YS, and SY prepared the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grants from the National Natural Science Foundation of China, Youth Project (#82004337).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Zimmermann MB, Boelaert K. Iodine deficiency and thyroid disorders. Lancet Diabetes Endocrinol (2015) 3(4):286–95. doi: 10.1016/s2213-8587(14)70225-6
2. Davies TF, Andersen S, Latif R, Nagayama Y, Barbesino G, Brito M, et al. Graves' disease. Nat Rev Dis Primers (2020) 6(1):52. doi: 10.1038/s41572-020-0184-y
3. Brandt F, Thvilum M, Hegedüs L, Brix TH. Hyperthyroidism is associated with work disability and loss of labour market income. a Danish register-based study in singletons and disease-discordant twin pairs. Eur J Endocrinol (2015) 173(5):595–602. doi: 10.1530/eje-15-0306
4. De Leo S, Lee SY, Braverman LE. Hyperthyroidism. Lancet (London England) (2016) 388(10047):906–18. doi: 10.1016/s0140-6736(16)00278-6
5. Zhang W, Wang X, Li P, Zhang W, Wang H, Tang B. Evaluating hyperthyroidism-induced liver injury based on in situ fluorescence imaging of glutathione and phosphate via nano-mofs sensor. Anal Chem (2020) 92(13):8952–8. doi: 10.1021/acs.analchem.0c00925
6. Folkestad L, Brandt F, Lillevang-Johansen M, Brix TH, Hegedüs L. Graves' disease and toxic nodular goiter, aggravated by duration of hyperthyroidism, are associated with alzheimer's and vascular dementia: a registry-based long-term follow-up of two large cohorts. Thyroid Off J Am Thyroid Assoc (2020) 30(5):672–80. doi: 10.1089/thy.2019.0672
7. Naser JA, Pislaru S, Stan MN, Lin G. Incidence, risk factors, natural history and outcomes of heart failure in patients with graves' disease. Heart (British Cardiac Society) (2022) 108(11):868–74. doi: 10.1136/heartjnl-2021-319752
8. Bunevicius R, Prange AJ Jr. Thyroid disease and mental disorders: cause and effect or only comorbidity? Curr Opin Psychiatry (2010) 23(4):363–8. doi: 10.1097/YCO.0b013e3283387b50
9. Fukao A, Takamatsu J, Arishima T, Tanaka M, Kawai T, Okamoto Y, et al. Graves' disease and mental disorders. J Clin Transl Endocrinol (2020) 19:100207. doi: 10.1016/j.jcte.2019.100207
10. Bové KB, Watt T, Vogel A, Hegedüs L, Bjoerner JB, Groenvold M, et al. Anxiety and depression are more prevalent in patients with graves' disease than in patients with nodular goitre. Eur Thyroid J (2014) 3(3):173–8. doi: 10.1159/000365211
11. Dai F, Yuan L, Fang J, Zhang Q, Wang K. Impaired decision making under risky conditions in the acute phase of graves' thyroitoxicosis. Neurosci Lett (2017) 661:1–4. doi: 10.1016/j.neulet.2017.08.058
12. Hamed SA, Attiah FA, Abdulhamid SK, Fawzy M. Behavioral assessment of children and adolescents with graves' disease: a prospective study. PloS One (2021) 16(4):e0248937. doi: 10.1371/journal.pone.0248937
13. Fischer S, Ehlert U. Hypothalamic-pituitary-thyroid (HPT) axis functioning in anxiety disorders. A systematic review. Depress Anxiety (2018) 35(1):98–110. doi: 10.1002/da.22692
14. Bunevicius R, Prange AJ Jr. Psychiatric manifestations of graves' hyperthyroidism: pathophysiology and treatment options. CNS Drugs (2006) 20(11):897–909. doi: 10.2165/00023210-200620110-00003
15. Henriksson MM, Aro HM, Marttunen MJ, Heikkinen ME, Isometsä ET, Kuoppasalmi KI, et al. Mental disorders and comorbidity in suicide. Am J Psychiatry (1993) 150(6):935–40. doi: 10.1176/ajp.150.6.935
16. National Center for Health S. Health, United States. Health, united states, 2016: with chartbook on long-term trends in health. Hyattsville (MD: National Center for Health Statistics (US (2017) p. 2017–1232.
17. Greenberg PE, Fournier A-A, Sisitsky T, Pike CT, Kessler RC. The economic burden of adults with major depressive disorder in the united states (2005 and 2010). J Clin Psychiatry (2015) 76(2):155–62. doi: 10.4088/JCP.14m09298
18. Kahaly GJ, Bartalena L, Hegedüs L, Leenhardt L, Poppe K, Pearce SH. European Thyroid association guideline for the management of graves' hyperthyroidism. Eur Thyroid J (2018) 7(4):167–86. doi: 10.1159/000490384
19. Gawałko M, Balsam P, Lodziński P, Grabowski M, Krzowski B, Opolski G, et al. Cardiac arrhythmias in autoimmune diseases. Circ J (2020) 84(5):685–94. doi: 10.1253/circj.CJ-19-0705
20. Yorke E. Hyperthyroidism and liver dysfunction: a review of a common comorbidity. Clin Med Insights Endocrinol Diabetes (2022) 15:11795514221074672. doi: 10.1177/11795514221074672
21. Ritchie M, Yeap BB. Thyroid hormone: influences on mood and cognition in adults. Maturitas (2015) 81(2):266–75. doi: 10.1016/j.maturitas.2015.03.016
22. Chen HH, Yeh SY, Lin CL, Chang SN, Kao CH. Increased depression, diabetes and diabetic complications in graves' disease patients in Asia. QJM (2014) 107(9):727–33. doi: 10.1093/qjmed/hcu069
23. Leone M, Kuja-Halkola R, Leval A, Butwicka A, Skov J, Zhang RY, et al. Genetic and environmental contribution to the co-occurrence of endocrine-metabolic disorders and depression: a nationwide Swedish study of siblings. Am J Psychiatry (2022) 179(11):824–32. doi: 10.1176/appi.ajp.21090954
24. Ittermann T, Volzke H, Baumeister SE, Appel K, Grabe HJ. Diagnosed thyroid disorders are associated with depression and anxiety. Soc Psychiatry Psychiatr Epidemiol (2015) 50(9):1417–25. doi: 10.1007/s00127-015-1043-0
25. Pápai A, Coşa LE, Cozma MM, Mihai A. [The role of coping strategies, depression and anxiety in thyroid disease]. Orv Hetil (2021) 162(7):262–8. doi: 10.1556/650.2021.31951
26. Chang SC, Lin SF, Chen ST, Chang PY, Yeh YM, Lo FS, et al. Alterations of gut microbiota in patients with graves' disease. Front Cell Infect Microbiol (2021) 11:663131. doi: 10.3389/fcimb.2021.663131
27. Anders S, Tanaka M, Kinney DK. Depression as an evolutionary strategy for defense against infection. Brain Behav Immun (2013) 31:9–22. doi: 10.1016/j.bbi.2012.12.002
28. Fam J, Rush AJ, Burt T, Chan ES, Siddiqui FJ, Assam PN, et al. Thyroid autoimmune antibodies and major depressive disorder in women. Ann Acad Med Singap (2015) 44(8):284–9. doi: 10.47102/annals-acadmedsg.V44N8p284
29. Ehlers M, Schott M, Allelein S. Graves' disease in clinical perspective. Front Biosci (Landmark Ed) (2019) 24(1):35–47. doi: 10.2741/4708
30. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol (2006) 27(1):24–31. doi: 10.1016/j.it.2005.11.006
31. Li Q, Wang B, Mu K, Zhang J-A. The pathogenesis of thyroid autoimmune diseases: new T lymphocytes - cytokines circuits beyond the Th1-Th2 paradigm. J Cell Physiol (2019) 234(3):2204–16. doi: 10.1002/jcp.27180
32. Chaudhury D, Liu H, Han M-H. Neuronal correlates of depression. Cell Mol Life Sci (2015) 72(24):4825–48. doi: 10.1007/s00018-015-2044-6
33. Gianoukakis AG, Khadavi N, Smith TJ. Cytokines, graves' disease, and thyroid-associated ophthalmopathy. Thyroid (2008) 18(9):953–8. doi: 10.1089/thy.2007.0405
34. Ujhelyi B, Gogolak P, Erdei A, Nagy V, Balazs E, Rajnavolgyi E, et al. Graves' orbitopathy results in profound changes in tear composition: a study of plasminogen activator inhibitor-1 and seven cytokines. Thyroid (2012) 22(4):407–14. doi: 10.1089/thy.2011.0248
35. Salvi M, Pedrazzoni M, Girasole G, Giuliani N, Minelli R, Wall JR, et al. Serum concentrations of proinflammatory cytokines in graves' disease: effect of treatment, thyroid function, ophthalmopathy and cigarette smoking. Eur J Endocrinol (2000) 143(2):197–202. doi: 10.1530/eje.0.1430197
36. Ruiz-Riol M, Barnils Mdel P, Colobran Oriol R, Pla AS, Borràs Serres FE, Lucas-Martin A, et al. Analysis of the cumulative changes in graves' disease thyroid glands points to IFN signature, plasmacytoid DCs and alternatively activated macrophages as chronicity determining factors. J Autoimmun (2011) 36(3-4):189–200. doi: 10.1016/j.jaut.2011.01.002
37. Kallmann BA, Hüther M, Tubes M, Feldkamp J, Bertrams J, Gries FA, et al. Systemic bias of cytokine production toward cell-mediated immune regulation in IDDM and toward humoral immunity in graves' disease. Diabetes (1997) 46(2):237–43. doi: 10.2337/diab.46.2.237
38. Currier MB, Nemeroff CB. Depression as a risk factor for cancer: from pathophysiological advances to treatment implications. Annu Rev Med (2014) 65:203–21. doi: 10.1146/annurev-med-061212-171507
39. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry (2009) 65(9):732–41. doi: 10.1016/j.biopsych.2008.11.029
40. Raison CL, Dantzer R, Kelley KW, Lawson MA, Woolwine BJ, Vogt G, et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-alpha: relationship to CNS immune responses and depression. Mol Psychiatry (2010) 15(4):393–403. doi: 10.1038/mp.2009.116
41. Kim H, Chen L, Lim G, Sung B, Wang S, McCabe MF, et al. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J Clin Invest (2012) 122(8):2940–54. doi: 10.1172/JCI61884
42. Fuertig R, Azzinnari D, Bergamini G, Cathomas F, Sigrist H, Seifritz E, et al. Mouse chronic social stress increases blood and brain kynurenine pathway activity and fear behaviour: both effects are reversed by inhibition of indoleamine 2,3-dioxygenase. Brain Behav Immun (2016) 54:59–72. doi: 10.1016/j.bbi.2015.12.020
43. Felger JC, Alagbe O, Hu F, Mook D, Freeman AA, Sanchez MM, et al. Effects of interferon-alpha on rhesus monkeys: a nonhuman primate model of cytokine-induced depression. Biol Psychiatry (2007) 62(11):1324–33. doi: 10.1016/j.biopsych.2007.05.026
44. Felger JC, Mun J, Kimmel HL, Nye JA, Drake DF, Hernandez CR, et al. Chronic interferon-α decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology (2013) 38(11):2179–87. doi: 10.1038/npp.2013.115
45. Vancassel S, Fanet H, Castanon N, Monchaux De Oliveira C, Cussotto S, Capuron L. Tetrahydrobiopterin modulates the behavioral neuroinflammatory response to an LPS challenge in mice. Brain Behav Immun (2022) 105:139–48. doi: 10.1016/j.bbi.2022.06.016
46. Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong J-S, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia (2007) 55(5):453–62. doi: 10.1002/glia.20467
47. Wakabayashi C, Kiyama Y, Kunugi H, Manabe T, Iwakura Y. Age-dependent regulation of depression-like behaviors through modulation of adrenergic receptor α1 A subtype expression revealed by the analysis of interleukin-1 receptor antagonist knockout mice. Neuroscience (2011) 192:475–84. doi: 10.1016/j.neuroscience.2011.06.031
48. Roman M, Irwin MR. Novel neuroimmunologic therapeutics in depression: a clinical perspective on what we know so far. Brain Behav Immun (2020) 83:7–21. doi: 10.1016/j.bbi.2019.09.016
49. Bottiglieri T, Laundy M, Crellin R, Toone BK, Carney MW, Reynolds EH. Homocysteine, folate, methylation, and monoamine metabolism in depression. J Neurol Neurosurg Psychiatry (2000) 69(2):228–32. doi: 10.1136/jnnp.69.2.228
50. Remy P, Doder M, Lees A, Turjanski N, Brooks D. Depression in parkinson's disease: loss of dopamine and noradrenaline innervation in the limbic system. Brain (2005) 128(Pt 6):1314–22. doi: 10.1093/brain/awh445
51. Murrough JW, Abdallah CG, Mathew SJ. Targeting glutamate signalling in depression: progress and prospects. Nat Rev Drug Discov (2017) 16(7):472–86. doi: 10.1038/nrd.2017.16
52. Thompson SM, Kallarackal AJ, Kvarta MD, Van Dyke AM, LeGates TA, Cai X. An excitatory synapse hypothesis of depression. Trends Neurosci (2015) 38(5):279–94. doi: 10.1016/j.tins.2015.03.003
53. Norris GT, Kipnis J. Immune cells and cns physiology: microglia and beyond. J Exp Med (2019) 216(1):60–70. doi: 10.1084/jem.20180199
54. Lawrence T. The nuclear factor nf-kappab pathway in inflammation. Cold Spring Harb Perspect Biol (2009) 1(6):a001651. doi: 10.1101/cshperspect.a001651
55. Shih R-H, Wang C-Y, Yang C-M. Nf-kappab signaling pathways in neurological inflammation: a mini review. Front Mol Neurosci (2015) 8:77. doi: 10.3389/fnmol.2015.00077
56. Brás JP, Guillot de Suduiraut I, Zanoletti O, Monari S, Meijer M, Grosse J, et al. Stress-induced depressive-like behavior in male rats is associated with microglial activation and inflammation dysregulation in the hippocampus in adulthood. Brain Behav Immun (2022) 99:397–408. doi: 10.1016/j.bbi.2021.10.018
57. Park C, Brietzke E, Rosenblat JD, Musial N, Zuckerman H, Ragguett R-M, et al. Probiotics for the treatment of depressive symptoms: an anti-inflammatory mechanism? Brain Behav Immun (2018) 73:115–24. doi: 10.1016/j.bbi.2018.07.006
58. Obermeier B, Daneman R, Ransohoff RM. Development, maintenance and disruption of the blood-brain barrier. Nat Med (2013) 19(12):1584–96. doi: 10.1038/nm.3407
59. Ren X, Chen H. Changes in Th9 and Th17 lymphocytes and functional cytokines and their relationship with thyroid-stimulating hormone receptor antibodies at different stages of graves' disease. Front Immunol (2022) 13:919681. doi: 10.3389/fimmu.2022.919681
60. Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, et al. Cellular mechanisms of il-17-induced blood-brain barrier disruption. FASEB J (2010) 24(4):1023–34. doi: 10.1096/fj.09-141978
61. Menard C, Pfau ML, Hodes GE, Kana V, Wang VX, Bouchard S, et al. Social stress induces neurovascular pathology promoting depression. Nat Neurosci (2017) 20(12):1752–60. doi: 10.1038/s41593-017-0010-3
62. Louveau A, Kipnis J. Sex, gut, and microglia. Dev Cell (2018) 44(2):137–8. doi: 10.1016/j.devcel.2018.01.003
63. Kim J, Suh YH, Chang KA. Interleukin-17 induced by cumulative mild stress promoted depression-like behaviors in young adult mice. Mol Brain (2021) 14(1):11. doi: 10.1186/s13041-020-00726-x
64. Weber MD, Godbout JP, Sheridan JF. Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology (2017) 42(1):46–61. doi: 10.1038/npp.2016.102
65. Liu Y, Hu P, Zheng Z, Zhong D, Xie W, Tang Z, et al. Photoresponsive vaccine-like car-m system with high-efficiency central immune regulation for inflammation-related depression. Adv Mater (2022) 34(11):e2108525. doi: 10.1002/adma.202108525
66. Antonelli A, Rotondi M, Fallahi P, Grosso M, Boni G, Ferrari SM, et al. Iodine-131 given for therapeutic purposes modulates differently interferon-gamma-inducible alpha-chemokine CXCL10 serum levels in patients with active graves' disease or toxic nodular goiter. J Clin Endocrinol Metab (2007) 92(4):1485–90. doi: 10.1210/jc.2006-1571
67. Leighton SP, Nerurkar L, Krishnadas R, Johnman C, Graham GJ, Cavanagh J. Chemokines in depression in health and in inflammatory illness: a systematic review and meta-analysis. Mol Psychiatry (2018) 23(1):48–58. doi: 10.1038/mp.2017.205
68. Beurel E, Lowell JA. Th17 cells in depression. Brain Behav Immun (2018) 69:28–34. doi: 10.1016/j.bbi.2017.08.001
69. Gold PW. Endocrine factors in key structural and intracellular changes in depression. Trends Endocrinol Metab (2021) 32(4):212–23. doi: 10.1016/j.tem.2021.01.003
70. Maes M, Song C, Yirmiya R. Targeting il-1 in depression. Expert Opin Ther Targets (2012) 16(11):1097–112. doi: 10.1517/14728222.2012.718331
71. Fox RI. Sjögren's syndrome. controversies progress. Clin Lab Med (1997) 17(3):431–44. doi: 10.1016/S0272-2712(18)30204-X
72. Leonard B, Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev (2012) 36(2):764–85. doi: 10.1016/j.neubiorev.2011.12.005
73. Galvis D, Zavala E, Walker JJ, Upton T, Lightman SL, Angelini GD, et al. Modelling the dynamic interaction of systemic inflammation and the hypothalamic-pituitary-adrenal (HPA) axis during and after cardiac surgery. J R Soc Interface (2022) 19(189):20210925. doi: 10.1098/rsif.2021.0925
74. Hofland J, Bakker J, Feelders RA. What's new on the hpa axis? Intensive Care Med (2015) 41(8):1477–9. doi: 10.1007/s00134-015-3771-8
75. Gold PW. The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry (2015) 20(1):32–47. doi: 10.1038/mp.2014.163
76. Ben-Skowronek I, Szewczyk L, Kulik-Rechberger B, Korobowicz E. The differences in T and b cell subsets in thyroid of children with graves' disease and hashimoto's thyroiditis. World J Pediatr (2013) 9(3):245–50. doi: 10.1007/s12519-013-0398-0
77. Cui X, Huang M, Wang S, Zhao N, Huang T, Wang Z, et al. Circulating exosomes from patients with graves' disease induce an inflammatory immune response. Endocrinology (2021) 162(3):bqaa236. doi: 10.1210/endocr/bqaa236
78. Tsatsoulis A, Johnson EO, Kalogera CH, Seferiadis K, Tsolas O. The effect of thyrotoxicosis on adrenocortical reserve. Eur J Endocrinol (2000) 142(3):231–5. doi: 10.1530/eje.0.1420231
79. Van Dam AM, Malinowsky D, Lenczowski MJ, Bartfai T, Tilders FJ. Interleukin 1 (IL-1) type I receptors mediate activation of rat hypothalamus-pituitary-adrenal axis and interleukin 6 production as shown by receptor type selective deletion mutants of IL-1beta. Cytokine (1998) 10(6):413–7. doi: 10.1006/cyto.1997.0315
80. Oprica M, Zhu S, Goiny M, Pham TM, Mohammed AH, Winblad B, et al. Transgenic overexpression of interleukin-1 receptor antagonist in the CNS influences behaviour, serum corticosterone and brain monoamines. Brain Behav Immun (2005) 19(3):223–34. doi: 10.1016/j.bbi.2004.07.006
81. Maes M, Scharpé S, Meltzer HY, Bosmans E, Suy E, Calabrese J, et al. Relationships between interleukin-6 activity, acute phase proteins, and function of the hypothalamic-pituitary-adrenal axis in severe depression. Psychiatry Res (1993) 49(1):11–27. doi: 10.1016/0165-1781(93)90027-e
82. Alesci S, Martinez PE, Kelkar S, Ilias I, Ronsaville DS, Listwak SJ, et al. Major depression is associated with significant diurnal elevations in plasma interleukin-6 levels, a shift of its circadian rhythm, and loss of physiological complexity in its secretion: clinical implications. J Clin Endocrinol Metab (2005) 90(5):2522–30. doi: 10.1210/jc.2004-1667
83. Chang FC, Opp MR. IL-1 is a mediator of increases in slow-wave sleep induced by CRH receptor blockade. Am J Physiol Regul Integr Comp Physiol (2000) 279(3):R793–802. doi: 10.1152/ajpregu.2000.279.3.R793
84. Tapp ZM, Godbout JP, Kokiko-Cochran ON. A tilted axis: maladaptive inflammation and hpa axis dysfunction contribute to consequences of tbi. Front Neurol (2019) 10:345. doi: 10.3389/fneur.2019.00345
85. Keller J, Gomez R, Williams G, Lembke A, Lazzeroni L, Murphy GM, et al. HPA axis in major depression: cortisol, clinical symptomatology and genetic variation predict cognition. Mol Psychiatry (2017) 22(4):527–36. doi: 10.1038/mp.2016.120
86. Melhem NM, Munroe S, Marsland A, Gray K, Brent D, Porta G, et al. Blunted HPA axis activity prior to suicide attempt and increased inflammation in attempters. Psychoneuroendocrinology (2017) 77:284–94. doi: 10.1016/j.psyneuen.2017.01.001
87. Haleem DJ. Glucocorticoids in the physiological and transcriptional regulation of 5-Ht1a receptor and the pathogenesis of depression. Neuroscientist (2022) 28(1):59–68. doi: 10.1177/1073858420975711
88. Kanner AM. Depression and epilepsy: do glucocorticoids and glutamate explain their relationship? Curr Neurol Neurosci Rep (2009) 9(4):307–12. doi: 10.1007/s11910-009-0046-1
89. Wakabayashi C, Numakawa T, Odaka H, Ooshima Y, Kiyama Y, Manabe T, et al. IL-1 receptor-antagonist (IL-1Ra) knockout mice show anxiety-like behavior by aging. Neurosci Lett (2015) 599:20–5. doi: 10.1016/j.neulet.2015.05.019
90. Menke A, Nitschke F, Hellmuth A, Helmel J, Wurst C, Stonawski S, et al. Stress impairs response to antidepressants via HPA axis and immune system activation. Brain Behav Immun (2021) 93:132–40. doi: 10.1016/j.bbi.2020.12.033
91. Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J (2002) 16(1):61–71. doi: 10.1096/fj.01-0245com
92. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol (2017) 17(4):233–47. doi: 10.1038/nri.2017.1
93. Munck A, Náray-Fejes-Tóth A. The ups and downs of glucocorticoid physiology. Permissive suppressive effects revisited. Mol Cell Endocrinol (1992) 90(1):C1–4. doi: 10.1016/0303-7207(92)90091-j
94. Meyer T, Stanske B, Kochen MM, Cordes A, Yüksel I, Wachter R, et al. Serum levels of interleukin-6 and interleukin-10 in relation to depression scores in patients with cardiovascular risk factors. Behav Med (2011) 37(3):105–12. doi: 10.1080/08964289.2011.609192
95. Grygiel-Górniak B, Limphaibool N, Puszczewicz M. Cytokine secretion and the risk of depression development in patients with connective tissue diseases. Psychiatry Clin Neurosci (2019) 73(6):302–16. doi: 10.1111/pcn.12826
96. Chiang JJ, Cole SW, Bower JE, Irwin MR, Taylor SE, Arevalo J, et al. Depressive symptoms and immune transcriptional profiles in late adolescents. Brain Behav Immun (2019) 80:163–9. doi: 10.1016/j.bbi.2019.03.004
97. Lane LC, Cheetham TD, Perros P, Pearce SHS. New therapeutic horizons for graves' hyperthyroidism. Endocr Rev (2020) 41(6):873–84. doi: 10.1210/endrev/bnaa022
98. Johnson EO, Kamilaris TC, Calogero AE, Gold PW, Chrousos GP. Experimentally-induced hyperthyroidism is associated with activation of the rat hypothalamic-pituitary-adrenal axis. Eur J Endocrinol (2005) 153(1):177–85. doi: 10.1530/eje.1.01923
99. Lakshmana Perumal N, Selvi J, Sridharan K, Sahoo J, Kamalanathan S. Insulin sensitivity and beta-cell function in graves'disease and their changes with the carbimazole-induced euthyroid state. Eur Thyroid J (2019) 8(2):59–63. doi: 10.1159/000496924
100. Mitrou P, Boutati E, Lambadiari V, Tsegka A, Raptis AE, Tountas N, et al. Insulin resistance in hyperthyroidism: the role of IL6 and TNF alpha. Eur J Endocrinol (2010) 162(1):121–6. doi: 10.1530/EJE-09-0622
101. Atis G, Dalkilinc A, Altuntas Y, Atis A, Gurbuz C, Ofluoglu Y, et al. Hyperthyroidism: a risk factor for female sexual dysfunction. J Sex Med (2011) 8(8):2327–33. doi: 10.1111/j.1743-6109.2011.02354.x
102. Skjöldebrand Sparre L, Kollind M, Carlström K. Ovarian ultrasound and ovarian and adrenal hormones before and after treatment for hyperthyroidism. Gynecol Obstet Invest (2002) 54(1):50–5. doi: 10.1159/000065573
103. Rasgon NL, Kenna HA, Wong M-L, Whybrow PC, Bauer M. Hypothalamic-pituitary-end organ function in women with bipolar depression. Psychoneuroendocrinology (2007) 32(3):279–86. doi: 10.1016/j.psyneuen.2006.12.014
104. Feldt-Rasmussen U, Effraimidis G, Klose M. The hypothalamus-pituitary-thyroid (HPT)-axis and its role in physiology and pathophysiology of other hypothalamus-pituitary functions. Mol Cell Endocrinol (2021) 525:111173. doi: 10.1016/j.mce.2021.111173
105. Smith TJ, Hegedüs L. Graves' disease. N Engl J Med (2016) 375(16):1552–65. doi: 10.1056/NEJMra1510030
106. Fountoulakis KN, Kantartzis S, Siamouli M, Panagiotidis P, Kaprinis S, Iacovides A, et al. Peripheral thyroid dysfunction in depression. World J Biol Psychiatry (2006) 7(3):131–7. doi: 10.1080/15622970500474739
107. Mano T, Sakamoto H, Fujita K, Makino M, Kakizawa H, Nagata M, et al. Effects of thyroid hormone on catecholamine and its metabolite concentrations in rat cardiac muscle and cerebral cortex. Thyroid (1998) 8(4):353–8. doi: 10.1089/thy.1998.8.353
108. Eshel N, Roiser JP. Reward and punishment processing in depression. Biol Psychiatry (2010) 68(2):118–24. doi: 10.1016/j.biopsych.2010.01.027
109. Kristensen AS, Andersen J, Jørgensen TN, Sørensen L, Eriksen J, Loland CJ, et al. SLC6 neurotransmitter transporters: structure, function, and regulation. Pharmacol Rev (2011) 63(3):585–640. doi: 10.1124/pr.108.000869
110. Villanueva I, Alva-Sánchez C, Pacheco-Rosado J. The role of thyroid hormones as inductors of oxidative stress and neurodegeneration. Oxid Med Cell Longev (2013) 2013:218145. doi: 10.1155/2013/218145
111. Bruscolini A, Sacchetti M, La Cava M, Nebbioso M, Iannitelli A. Quality of life and neuropsychiatric disorders in patients with graves' orbitopathy: current concepts. Autoimmun Rev (2018) 17(7):639–43. doi: 10.1016/j.autrev.2017.12.012
112. Medici M, Direk N, Visser WE, Korevaar TIM, Hofman A, Visser TJ, et al. Thyroid function within the normal range and the risk of depression: a population-based cohort study. J Clin Endocrinol Metab (2014) 99(4):1213–9. doi: 10.1210/jc.2013-3589
113. Cappola AR, Desai AS, Medici M, Cooper LS, Egan D, Sopko G, et al. Thyroid and cardiovascular disease research agenda for enhancing knowledge, prevention, and treatment. Circulation (2019) 13:10. doi: 10.1161/CIRCULATIONAHA.118.036859
114. Tribulova N, Kurahara LH, Hlivak P, Hirano K, Szeiffova Bacova B. Pro-arrhythmic signaling of thyroid hormones and its relevance in subclinical hyperthyroidism. Int J Mol Sci (2020) 21(8):2844. doi: 10.3390/ijms21082844
115. Thrall G, Lip GYH, Carroll D, Lane D. Depression, anxiety, and quality of life in patients with atrial fibrillation. Chest (2007) 132(4):1259–64. doi: 10.1378/chest.07-0036
116. van Sloten TT, Sedaghat S, Carnethon MR, Launer LJ, Stehouwer CDA. Cerebral microvascular complications of type 2 diabetes: stroke, cognitive dysfunction, and depression. Lancet Diabetes Endocrinol (2020) 8(4):325–36. doi: 10.1016/S2213-8587(19)30405-X
117. van Sloten TT, Sigurdsson S, van Buchem MA, Phillips CL, Jonsson PV, Ding J, et al. Cerebral small vessel disease and association with higher incidence of depressive symptoms in a general elderly population: the ages-reykjavik study. Am J Psychiatry (2015) 172(6):570–8. doi: 10.1176/appi.ajp.2014.14050578
118. Najjar S, Pearlman DM, Devinsky O, Najjar A, Zagzag D. Neurovascular unit dysfunction with blood-brain barrier hyperpermeability contributes to major depressive disorder: a review of clinical and experimental evidence. J Neuroinflamm (2013) 10:142. doi: 10.1186/1742-2094-10-142
119. Aragão CN, Souza LL, Cabanelas A, Oliveira KJ, Pazos-Moura CC. Effect of experimental hypo- and hyperthyroidism on serum adiponectin. Metabolism (2007) 56(1):6–11. doi: 10.1016/j.metabol.2006.08.015
120. Fasciolo G, Napolitano G, Aprile M, Cataldi S, Costa V, Ciccodicola A, et al. Hepatic insulin resistance in hyperthyroid rat liver: vitamin e supplementation highlights a possible role of ros. Antioxidants (Basel) (2022) 11(7):1295. doi: 10.3390/antiox11071295
121. Armitage M, Franklyn J, Scott-Morgan L, Parr J, Borsey DQ, Sheppard M, et al. Insulin autoantibodies in graves' disease–before and after carbimazole therapy. Diabetes Res Clin Pract (1990) 8(3):169–76. doi: 10.1016/0168-8227(90)90113-8
122. Underwood EL, Thompson LT. High-fat diet impairs spatial memory and hippocampal intrinsic excitability and sex-dependently alters circulating insulin and hippocampal insulin sensitivity. Biol Sex Differ (2016) 7:9. doi: 10.1186/s13293-016-0060-3
123. Sen ZD, Danyeli LV, Woelfer M, Lamers F, Wagner G, Sobanski T, et al. Linking atypical depression and insulin resistance-related disorders via low-grade chronic inflammation: integrating the phenotypic, molecular and neuroanatomical dimensions. Brain Behav Immun (2021) 93:335–52. doi: 10.1016/j.bbi.2020.12.020
124. Barrett EJ, Liu Z, Khamaisi M, King GL, Klein R, Klein BEK, et al. Diabetic microvascular disease: an endocrine society scientific statement. J Clin Endocrinol Metab (2017) 102(12):4343–410. doi: 10.1210/jc.2017-01922
125. Bogush M, Heldt NA, Persidsky Y. Blood brain barrier injury in diabetes: unrecognized effects on brain and cognition. J Neuroimmune Pharmacol (2017) 12(4):593–601. doi: 10.1007/s11481-017-9752-7
126. Cline BH, Costa-Nunes JP, Cespuglio R, Markova N, Santos AI, Bukhman YV, et al. Dicholine succinate, the neuronal insulin sensitizer, normalizes behavior, REM sleep, hippocampal pGSK3 beta and mRNAs of NMDA receptor subunits in mouse models of depression. Front Behav Neurosci (2015) 9:37. doi: 10.3389/fnbeh.2015.00037
127. Maratou E, Hadjidakis DJ, Peppa M, Alevizaki M, Tsegka K, Lambadiari V, et al. Studies of insulin resistance in patients with clinical and subclinical hyperthyroidism. Eur J Endocrinol (2010) 163(4):625–30. doi: 10.1530/EJE-10-0246
128. Noble RE. Depression in women. Metabolism (2005) 54(5 Suppl 1):49–52. doi: 10.1016/j.metabol.2005.01.014
129. Burch HB, Cooper DS. Management of graves disease: a review. JAMA (2015) 314(23):2544–54. doi: 10.1001/jama.2015.16535
130. Kundakovic M, Rocks D. Sex hormone fluctuation and increased female risk for depression and anxiety disorders: from clinical evidence to molecular mechanisms. Front Neuroendocrinol (2022) 66:101010. doi: 10.1016/j.yfrne.2022.101010
131. Möckel T, Basta F, Weinmann-Menke J, Schwarting A. B cell activating factor (BAFF): structure, functions, autoimmunity and clinical implications in systemic lupus erythematosus (SLE). Autoimmun Rev (2021) 20(2):102736. doi: 10.1016/j.autrev.2020.102736
132. Zhang J, Roschke V, Baker KP, Wang Z, Alarcón GS, Fessler BJ, et al. Cutting edge: a role for b lymphocyte stimulator in systemic lupus erythematosus. J Immunol (2001) 166(1):6–10. doi: 10.4049/jimmunol.166.1.6
133. Mariette X, Roux S, Zhang J, Bengoufa D, Lavie F, Zhou T, et al. The level of BLyS (BAFF) correlates with the titre of autoantibodies in human sjögren's syndrome. Ann Rheum Dis (2003) 62(2):168–71. doi: 10.1136/ard.62.2.168
134. Migita K, Ilyassova B, Kovzel EF, Nersesov A, Abiru S, Maeda Y, et al. Serum BAFF and APRIL levels in patients with PBC. Clin Immunol (2010) 134(2):217–25. doi: 10.1016/j.clim.2009.09.007
135. Cheng CW, Fang WF, Tang KT, Lin JD. Possible interplay between estrogen and the BAFF may modify thyroid activity in graves' disease. Sci Rep (2021) 11(1):21350. doi: 10.1038/s41598-021-00903-5
136. Estienne V, Duthoit C, Reichert M, Praetor A, Carayon P, Hunziker W, et al. Androgen-dependent expression of FcgammaRIIB2 by thyrocytes from patients with autoimmune graves' disease: a possible molecular clue for sex dependence of autoimmune disease. FASEB J (2002) 16(9):1087–92. doi: 10.1096/fj.01-0998hyp
137. Pinar C, Fontaine CJ, Triviño-Paredes J, Lottenberg CP, Gil-Mohapel J, Christie BR. Revisiting the flip side: long-term depression of synaptic efficacy in the hippocampus. Neurosci Biobehav Rev (2017) 80:394–413. doi: 10.1016/j.neubiorev.2017.06.001
138. Duman RS, Aghajanian GK, Sanacora G, Krystal JH. Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med (2016) 22(3):238–49. doi: 10.1038/nm.4050
139. Docimo G, Cangiano A, Romano RM, Pignatelli MF, Offi C, Paglionico VA, et al. The human microbiota in endocrinology: implications for pathophysiology, treatment, and prognosis in thyroid diseases. Front Endocrinol (Lausanne) (2020) 11:586529. doi: 10.3389/fendo.2020.586529
140. Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell (2014) 157(1):121–41. doi: 10.1016/j.cell.2014.03.011
141. Khan R, Petersen FC, Shekhar S. Commensal bacteria: an emerging player in defense against respiratory pathogens. Front Immunol (2019) 10:1203. doi: 10.3389/fimmu.2019.01203
142. Frank DN, Pace NR. Gastrointestinal microbiology enters the metagenomics era. Curr Opin Gastroenterol (2008) 24(1):4–10. doi: 10.1097/MOG.0b013e3282f2b0e8
143. Evrensel A, Ceylan ME. The gut-brain axis: the missing link in depression. Clin Psychopharmacol Neurosci (2015) 13(3):239–44. doi: 10.9758/cpn.2015.13.3.239
144. Dash S, Clarke G, Berk M, Jacka FN. The gut microbiome and diet in psychiatry: focus on depression. Curr Opin Psychiatry (2015) 28(1):1–6. doi: 10.1097/yco.0000000000000117
145. Forsythe P, Sudo N, Dinan T, Taylor VH, Bienenstock J. Mood and gut feelings. Brain behavior Immun (2010) 24(1):9–16. doi: 10.1016/j.bbi.2009.05.058
146. Bastiaanssen TFS, Cowan CSM, Claesson MJ, Dinan TG, Cryan JF. Making sense of … the microbiome in psychiatry. Int J Neuropsychopharmacol (2019) 22(1):37–52. doi: 10.1093/ijnp/pyy067
147. Li Y, Luo B, Tong B, Xie Z, Cao J, Bai X, et al. The role and molecular mechanism of gut microbiota in graves' orbitopathy. J Endocrinol Invest (2023) 46(2):305–17. doi: 10.1007/s40618-022-01902-7
148. Knezevic J, Starchl C, Tmava Berisha A, Amrein K. Thyroid-Gut-Axis: how does the microbiota influence thyroid function? Nutrients (2020) 12(6):1769. doi: 10.3390/nu12061769
149. Cuan-Baltazar Y, Soto-Vega E. Microorganisms associated to thyroid autoimmunity. Autoimmun Rev (2020) 19(9):102614. doi: 10.1016/j.autrev.2020.102614
150. Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, et al. Altered fecal microbiota composition in patients with major depressive disorder. Brain behavior Immun (2015) 48:186–94. doi: 10.1016/j.bbi.2015.03.016
151. Cornejo-Pareja I, Ruiz-Limón P, Gómez-Pérez AM, Molina-Vega M, Moreno-Indias I, Tinahones FJ. Differential microbial pattern description in subjects with autoimmune-based thyroid diseases: a pilot study. J Pers Med (2020) 10(4):192. doi: 10.3390/jpm10040192
152. Ishaq HM, Mohammad IS, Shahzad M, Ma C, Raza MA, Wu X, et al. Molecular alteration analysis of human gut microbial composition in graves' disease patients. Int J Biol Sci (2018) 14(11):1558–70. doi: 10.7150/ijbs.24151
153. Su X, Yin X, Liu Y, Yan X, Zhang S, Wang X, et al. Gut dysbiosis contributes to the imbalance of treg and Th17 cells in graves' disease patients by propionic acid. J Clin Endocrinol Metab (2020) 105(11):dgaa511. doi: 10.1210/clinem/dgaa511
154. Yan HX, An WC, Chen F, An B, Pan Y, Jin J, et al. Intestinal microbiota changes in graves' disease: a prospective clinical study. Biosci Rep (2020) 40(9):BSR20191242. doi: 10.1042/bsr20191242
155. Hou J, Tang Y, Chen Y, Chen D. The role of the microbiota in graves' disease and graves' orbitopathy. Front Cell Infect Microbiol (2021) 11:739707. doi: 10.3389/fcimb.2021.739707
156. Kelly JR, Borre Y, O' Brien C, Patterson E, El Aidy S, Deane J, et al. Transferring the blues: depression-sssociated gut microbiota induces neurobehavioural changes in the rat. J Psychiatr Res (2016) 82:109–18. doi: 10.1016/j.jpsychires.2016.07.019
157. Kiyohara C, Yoshimasu K. Molecular epidemiology of major depressive disorder. Environ Health Prev Med (2009) 14(2):71–87. doi: 10.1007/s12199-008-0073-6
158. Trullas R, Skolnick P. Functional antagonists at the NMDA receptor complex exhibit antidepressant actions. Eur J Pharmacol (1990) 185(1):1–10. doi: 10.1016/0014-2999(90)90204-j
159. Matsukawa N, Furuya Y, Ogura H, Ojika K. HCNP precursor protein transgenic mice display a depressive-like phenotype in old age. Brain Res (2010) 1349:153–61. doi: 10.1016/j.brainres.2010.06.041
160. Fogaça MV, Wu M, Li C, Li XY, Picciotto MR, Duman RS. Inhibition of GABA interneurons in the mPFC is sufficient and necessary for rapid antidepressant responses. Mol Psychiatry (2021) 26(7):3277–91. doi: 10.1038/s41380-020-00916-y
161. Hayakawa K, Kimura M, Kasaha K, Matsumoto K, Sansawa H, Yamori Y. Effect of a gamma-aminobutyric acid-enriched dairy product on the blood pressure of spontaneously hypertensive and normotensive wistar-Kyoto rats. Br J Nutr (2004) 92(3):411–7. doi: 10.1079/bjn20041221
162. Quandt G, Höfner G, Wanner KT. Synthesis and evaluation of n-substituted nipecotic acid derivatives with an unsymmetrical bis-aromatic residue attached to a vinyl ether spacer as potential GABA uptake inhibitors. Bioorg Med Chem (2013) 21(11):3363–78. doi: 10.1016/j.bmc.2013.02.056
163. Lener MS, Niciu MJ, Ballard ED, Park M, Park LT, Nugent AC, et al. Glutamate and gamma-aminobutyric acid systems in the pathophysiology of major depression and antidepressant response to ketamine. Biol Psychiatry (2017) 81(10):886–97. doi: 10.1016/j.biopsych.2016.05.005
164. Pytka K, Dziubina A, Młyniec K, Dziedziczak A, Żmudzka E, Furgała A, et al. TThe role of glutamatergic, GABA-ergic, and cholinergic receptors in depression and antidepressant-like effect. Pharmacol Rep (2016) 68(2):443–50. doi: 10.1016/j.pharep.2015.10.006
165. Barrett E, Ross RP, O'Toole PW, Fitzgerald GF, Stanton C. γ-aminobutyric acid production by culturable bacteria from the human intestine. J Appl Microbiol (2012) 113(2):411–7. doi: 10.1111/j.1365-2672.2012.05344.x
166. Zhou H, Zhang S, Zhang X, Zhou H, Wen T, Wang J. Depression-like symptoms due to Dcf1 deficiency are alleviated by intestinal transplantation of lactobacillus murine and lactobacillus reuteri. Biochem Biophys Res Commun (2022) 593:137–43. doi: 10.1016/j.bbrc.2022.01.026
167. Rocha-Ramírez LM, Pérez-Solano RA, Castañón-Alonso SL, Moreno Guerrero SS, Ramírez Pacheco A, García Garibay M, et al. Probiotic lactobacillus strains stimulate the inflammatory response and activate human macrophages. J Immunol Res (2017) 2017:4607491. doi: 10.1155/2017/4607491
168. Liu N, Sun S, Wang P, Sun Y, Hu Q, Wang X. The mechanism of secretion and metabolism of gut-derived 5-hydroxytryptamine. Int J Mol Sci (2021) 22(15):7931. doi: 10.3390/ijms22157931
169. Wu CH, Chang CS, Yang YK, Shen LH, Yao WJ. Comparison of brain serotonin transporter using [I-123]-ADAM between obese and non-obese young adults without an eating disorder. PloS One (2017) 12(2):e0170886. doi: 10.1371/journal.pone.0170886
170. Warma S, Lee Y, Brietzke E, McIntyre RS. Microbiome abnormalities as a possible link between diabetes mellitus and mood disorders: pathophysiology and implications for treatment. Neurosci Biobehav Rev (2022) 137:104640. doi: 10.1016/j.neubiorev.2022.104640
171. Sun J, Wang F, Hong G, Pang M, Xu H, Li H, et al. Antidepressant-like effects of sodium butyrate and its possible mechanisms of action in mice exposed to chronic unpredictable mild stress. Neurosci Lett (2016) 618:159–66. doi: 10.1016/j.neulet.2016.03.003
172. Lukić I, Getselter D, Koren O, Elliott E. Role of tryptophan in microbiota-induced depressive-like behavior: evidence from tryptophan depletion study. Front Behav Neurosci (2019) 13:123. doi: 10.3389/fnbeh.2019.00123
173. Clarke G, Grenham S, Scully P, Fitzgerald P, Moloney RD, Shanahan F, et al. The microbiome-gut-brain axis during early life regulates the hippocampal serotonergic system in a sex-dependent manner. Mol Psychiatry (2013) 18(6):666–73. doi: 10.1038/mp.2012.77
174. Qin J, Zhou J, Fan C, Zhao N, Liu Y, Wang S, et al. Increased circulating Th17 but decreased CD4+Foxp3+ treg and CD19+CD1dHiCD5+ breg subsets in new-onset graves' disease. BioMed Res Int (2017) 2017:8431838. doi: 10.1155/2017/8431838
175. Chen Y, Jiang T, Chen P, Ouyang J, Xu G, Zeng Z, et al. Merging tendency towards autoimmune process in major depressive patients: a novel insight from Th17 cells. Psychiatry Res (2011) 188(2):224–30. doi: 10.1016/j.psychres.2010.10.029
176. Davami MH, Baharlou R, Ahmadi Vasmehjani A, Ghanizadeh A, Keshtkar M, Dezhkam I, et al. Elevated il-17 and TGF-β serum levels: a positive correlation between T-helper 17 cell-related pro-inflammatory responses with major depressive disorder. Basic Clin Neurosci (2016) 7(2):137–42. doi: 10.15412/j.Bcn.03070207
177. Nadeem A, Ahmad SF, Al-Harbi NO, Fardan AS, El-Sherbeeny AM, Ibrahim KE, et al. IL-17A causes depression-like symptoms via NFκB and p38MAPK signaling pathways in mice: implications for psoriasis associated depression. Cytokine (2017) 97:14–24. doi: 10.1016/j.cyto.2017.05.018
178. Rudolf J, Grond M, Neveling M, Heiss WD. Catatony in graves' disease. Eur Psychiatry (1998) 13(8):419–20. doi: 10.1016/S0924-9338(99)80689-0
179. Calissendorff J, Mikulski E, Larsen EH, Möller M. A prospective investigation of graves' disease and selenium: thyroid hormones, auto-antibodies and self-rated symptoms. Eur Thyroid J (2015) 4(2):93–8. doi: 10.1159/000381768
180. Elberling TV, Rasmussen AK, Feldt-Rasmussen U, Hørding M, Perrild H, Waldemar G. Impaired health-related quality of life in graves' disease. A prospective study.Eur J Endocrinol (2004) 151(5):549–55. doi: 10.1530/eje.0.1510549
181. Stern RA, Robinson B, Thorner AR, Arruda JE, Prohaska ML, Prange AJ. A survey study of neuropsychiatric complaints in patients with graves' disease. J Neuropsychiatry Clin Neurosci (1996) 8(2):181–5. doi: 10.1176/jnp.8.2.181
182. Bunevicius R, Velickiene D, Prange AJ. Mood and anxiety disorders in women with treated hyperthyroidism and ophthalmopathy caused by graves' disease. Gen Hosp Psychiatry (2005) 27(2):133–9. doi: 10.1016/j.genhosppsych.2004.10.002
183. Alli SR, Gorbovskaya I, Liu JCW, Kolla NJ, Brown L, Müller DJ. The gut microbiome in depression and potential benefit of prebiotics, probiotics and synbiotics: a systematic review of clinical trials and observational studies. Int J Mol Sci (2022) 23(9):4494. doi: 10.3390/ijms23094494
184. Huo D, Cen C, Chang H, Ou Q, Jiang S, Pan Y, et al. Probiotic bifidobacterium longum supplied with methimazole improved the thyroid function of graves' disease patients through the gut-thyroid axis. Commun Biol (2021) 4(1):1046. doi: 10.1038/s42003-021-02587-z
185. Coulter I, Frewin S, Krassas GE, Perros P. Psychological implications of graves' orbitopathy. Eur J Endocrinol (2007) 157(2):127–31. doi: 10.1530/EJE-07-0205
186. Fukao A, Takamatsu J, Kubota S, Miyauchi A, Hanafusa T. The thyroid function of graves' disease patients is aggravated by depressive personality during antithyroid drug treatment. Biopsychosoc Med (2011) 5:9. doi: 10.1186/1751-0759-5-9
Keywords: Graves’ disease, depression, autoimmune, gut, hormone
Citation: Song Y, Wang X, Ma W, Yang Y, Yan S, Sun J, Zhu X and Tang Y (2023) Graves’ disease as a driver of depression: a mechanistic insight. Front. Endocrinol. 14:1162445. doi: 10.3389/fendo.2023.1162445
Received: 09 February 2023; Accepted: 05 April 2023;
Published: 20 April 2023.
Edited by:
Benjamin Samuel Weeks, Adelphi University, United StatesReviewed by:
Wei Peng, Chengdu University of Traditional Chinese Medicine, ChinaJun Li, Shanghai University of Traditional Chinese Medicine, China
Zhiyong Yan, Southwest Jiaotong University, China
Copyright © 2023 Song, Wang, Ma, Yang, Yan, Sun, Zhu and Tang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiapan Sun, c3VuanAzQG1haWwyLnN5c3UuZWR1LmNu; Xiaoyun Zhu, cWllYmVuYmVuQDE2My5jb20=; Yang Tang, dGFuZ3lhbmdAYnVjbS5lZHUuY24=
†These authors have contributed equally to this work and shared first authorship