 Stuti Khadka
Stuti Khadka Benjamin C. Duncan
Benjamin C. Duncan Jonathan T. Busada
Jonathan T. Busada- Department of Microbiology, Immunology and Cell Biology, West Virginia University School of Medicine, Morgantown, WV, United States
Glucocorticoids are steroid hormones that regulate a host of cellular and physiological functions. However, they are arguably best known for their potent anti-inflammatory properties. Chronic inflammation is well-known to promote the development and progression of numerous types of cancer, and emerging evidence suggests that glucocorticoid regulation of inflammation affects cancer development. However, the timing, intensity, and duration of glucocorticoid signaling have important but often contradictory effects on cancer development. Moreover, glucocorticoids are widely used in parallel with radiation and chemotherapy to control pain, dyspnea, and swelling, but their use may compromise anti-tumor immunity. This review will explore the effects of glucocorticoids on cancer development and progression with particular focus on pro and anti-tumor immunity.
Background
In 1938, Compound E was synthesized from the adrenal cortex of beef cattle (1). Within the next decade, adrenal cortical hormones (glucocorticoids or corticosteroids) were used to successfully treat inflammatory diseases such as rheumatoid arthritis and rheumatic fever. Since then, glucocorticoids have risen to be one of the most commonly prescribed drugs in the world. Within the clinic, glucocorticoids are used to treat a host of conditions and diseases, ranging from improving lung development in premature infants, treating allergic reactions, autoimmune diseases, and chronic inflammatory diseases, to anti-cancer therapies. However, despite their widespread clinical use, the mechanisms whereby glucocorticoids regulate diverse cellular and physiological functions have yet to be fully elucidated.
The glucocorticoid receptor (NR3C1 or GR) is expressed by most nucleated cell types in humans. Glucocorticoids are reported to regulate up to 20% of the total transcriptome (2). However, the response to glucocorticoids varies between different cells and tissues. Similar functional diversity of glucocorticoid signaling impacts cancer, and glucocorticoids may inhibit or promote carcinogenesis depending on the cancer type. In addition, the effect of glucocorticoid signaling changes as cancers develop and progress. In some cases, glucocorticoid signaling may suppress damaging inflammation thereby protecting from neoplasia development, but these protective roles are reduced or may reverse during chronic inflammation. Moreover, despite the unclear roles of glucocorticoid signaling in solid tumors, these hormones are commonly co-administered with anti-cancer therapeutic regimens to relieve adverse side effects such as pain, nausea, and dyspepsia and to improve appetite. This review will provide an update on the current knowledge of how glucocorticoid signaling affects cancer development in the pre-tumor microenvironment and how their roles shift in mature tumors. Moreover, we will discuss how glucocorticoids interact with estrogens and androgens in breast and prostate cancer, respectively.
Steroidogenesis
Glucocorticoids are steroid hormones produced as the end products of the hypothalamic-pituitary-adrenal (HPA) axis. They are produced in a circadian rhythm and in response to psychological and physical stress (Figure 1). Glucocorticoid production is initiated in the hypothalamus with the stimulation of neurons within the paraventricular nucleus. This signals the release of corticotropin-releasing hormone (CRH), which acts on the anterior pituitary to promote the synthesis of proopiomelanocortin by corticotropic cells (3, 4). POMC is then cleaved into adrenocorticotropic hormone (ACTH) and released into circulation, acting upon steroidogenic cells within the zona fasciculata of the adrenal cortex. ACTH activates an enzymatic cascade whereby cholesterol is converted into corticosterone (mouse) or cortisol (human) and released into circulation (5, 6). While the HPA axis is well-known to be activated in a diurnal pattern and in response to stress, proinflammatory cytokines and pathogen activated-pattern recognition receptors also trigger glucocorticoid synthesis (7, 8). Upon their release into circulation, glucocorticoids inhibit further activation of the HPA axis. Acute activation of the HPA axis is necessary to suppress immune hyperactivation. However, chronic glucocorticoid signaling is associated with numerous negative health outcomes, such as elevated circulating cytokine levels, chronic inflammation, impaired wound healing, and many other maladies (9). Prolonged glucocorticoid signaling is especially relevant in cancer, where chronic pro-neoplastic infections, cancer-associated psychological stress, corticosteroid therapies, or intra-tumor steroidogenesis inhibit anti-tumor immune responses and promote tumor cell survival (10).
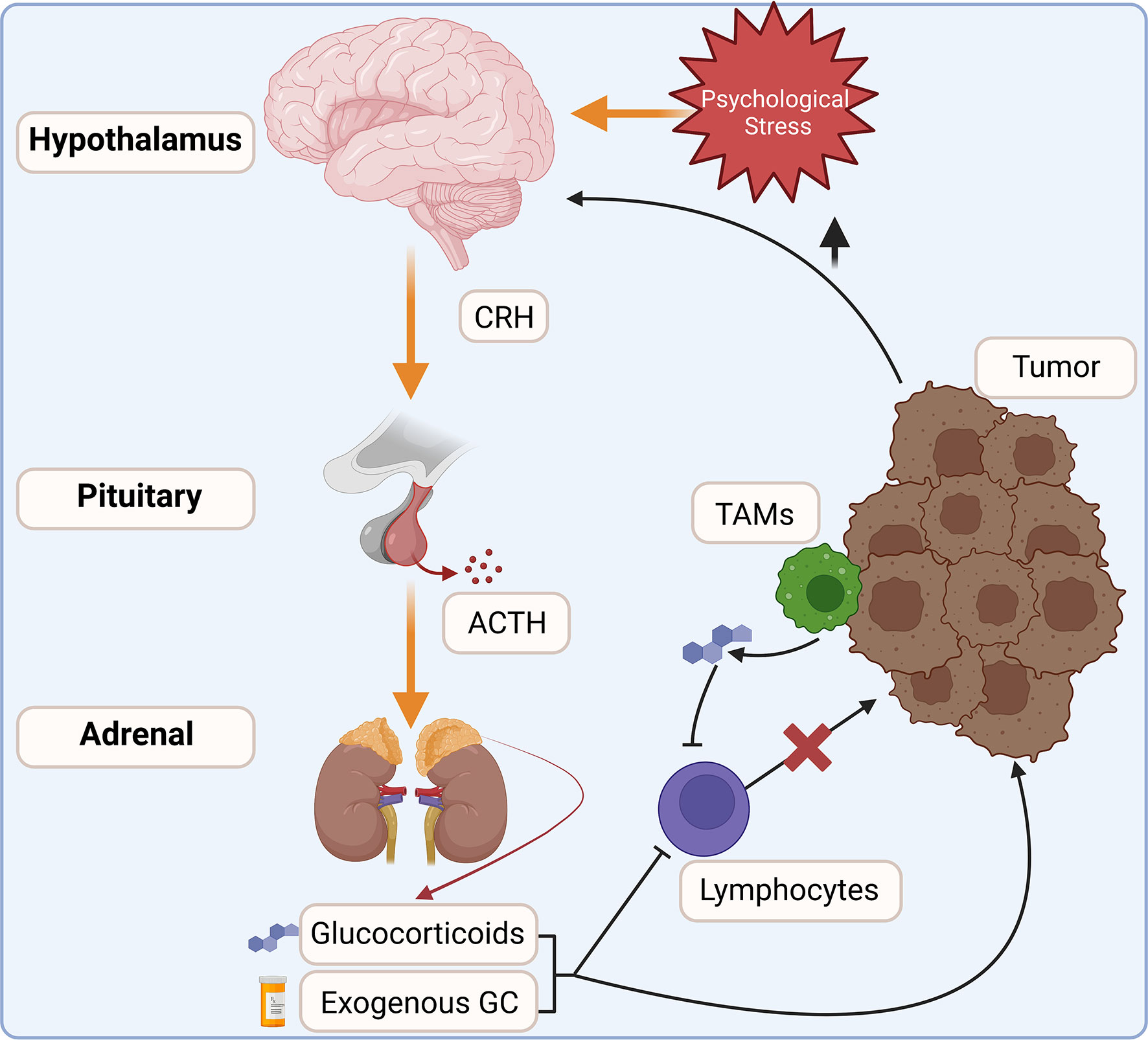
Figure 1 The HPA axis integrates internal and external cues to modulate glucocorticoid production. Increased glucocorticoid signaling is commonly associated with cancers. Increased stress and tissue damage act on the hypothalamus to stimulate the release of corticotropin-releasing hormone (CRH), which in turn stimulates adrenocorticotropic hormone (ACTH) release into circulation. ACTH acts on steroidogenic cells within the adrenal cortex to increase endogenous glucocorticoid production. Exogenous corticosteroids are also commonly co-administered with anti-cancer therapies. Within the tumor, infiltrating leukocytes, such as tumor-associated macrophages (TAMs), activate steroidogenesis and may increase glucocorticoid concentrations within the tumor. Elevated glucocorticoids inhibit anti-tumor immune responses and are linked to increased tumor cell survival.
Adrenal steroidogenesis controls systemic glucocorticoid production, but several factors contribute to local glucocorticoid availability. Approximately 80% of endogenous glucocorticoids in the blood are bound by corticosteroid-binding globulin (SERPINA6), which aids transport and regulates bioavailability (11). Proteases such as neutrophil elastase can degrade corticosteroid-binding globulin increasing glucocorticoid concentrations at sites of inflammation (12). Myeloid-derived suppressor cells commonly found within the tumor microenvironment express abundant neutrophil elastase and may utilize this mechanism to increase the glucocorticoid concentration within the tumor. At the tissue level, glucocorticoid concentrations are regulated by hydroxysteroid 11-beta dehydrogenases (HSD11B1 and HSD11B2). HSD11B1 catalyzes the conversion of inactive cortisone into cortisol, thereby increasing local glucocorticoid concentrations. HSD11B1 is widely expressed in the liver, lungs, brain, stomach, adipose tissue, adrenal cortex, uterus, ovaries, and testes (13–15). HSD11B2 catalyzes the reverse reaction by converting cortisol to cortisone and is expressed in the kidneys, colon, and salivary glands (13, 16). These tissues are critical sites of mineralocorticoid receptor signaling, which has a higher binding affinity for glucocorticoids than for its endogenous ligand aldosterone. Thus, HSD11B2 protects the mineralocorticoid receptor from activation by glucocorticoids. Tumor cells are reported to alter the expression of these enzymes, thereby modifying glucocorticoid signaling within the tumor microenvironment (17). For instance, glucocorticoid signaling opposes the development of estrogen-dependent breast cancers, and breast cancers downregulate the expression of HSD11B1 reducing glucocorticoid availability within the tumor (18). Similarly, expression of HSD11B2 is suppressed in skin cancers, increasing intra-tumor glucocorticoid concentrations and promoting invasive and metastatic phenotypes (19).
While the adrenal glands are the primary source of circulating glucocorticoids, increasing evidence suggests that local glucocorticoid production is also important for modulating the functions of healthy tissues and the tumor microenvironment. Extra-adrenal glucocorticoid production primarily exerts paracrine effects (20). Within the intestines, glucocorticoid production by cells within the crypts affects intestinal barrier function and inflammation (21). Within the thymus, nurse cell derived-glucocorticoids raise the threshold for T cell negative selection by suppressing the transcription factors NUR77 and HELIOS (21, 22). Within the tumor microenvironment, glucocorticoids produced by tumor-associated macrophages and tumor-infiltrating T cells promote the expression of T cell checkpoint proteins and inhibit anti-tumor immunity (23–25). Numerous other tissues and cell types are reported to express steroidogenesis enzymes. However, more work is needed to understand the full extent of extra-adrenal glucocorticoid synthesis and its effects on cellular and tissue function.
Glucocorticoid signaling
Glucocorticoids signal through the GR, a member of the ligand-dependent transcription factor superfamily. Structurally, the GR is similar to the other steroid hormone receptors. The GR protein is comprised of three functional domains: an N-terminal transactivation domain, a central DNA binding domain, and a C-terminal ligand binding domain (26). The transactivation domain interacts with coregulators and the transcriptional machinery. The DNA binding domain contains two zinc-finger motifs involved in binding to glucocorticoid response elements (GREs). The DNA binding domain and ligand binding domain are separated by a hinge region that is involved in receptor dimerization and contains two nuclear localization signals, NL1 and NL2. The ligand binding domain contains a hydrophobic pocket for glucocorticoid binding, which induces a conformational change (27). The GR protein also contains several sites for post-translational modifications including, phosphorylation, sumoylation, acetylation, and ubiquitination that regulate receptor activity and turn-over (28). In the absence of ligand, the GR is sequestered in the cytoplasm by a large protein complex comprised of chaperones PTGES3, HSP90, and HSPA1B, and the immunophilins FKBP5 and FKBP4 (Figure 2) (27). While bound in this complex, the ligand binding domain of the GR is readily accessible, but the nuclear localization signals are masked. Lipophilic glucocorticoids diffuse freely through the plasma membrane to bind cytosolic GRs. Upon ligand binding, the GR undergoes a conformational change, disassociating the protein complex, and exposing the nuclear localization signals. The cellular effects of this complex dissociation are two-fold; ligand-bound GR is rapidly transported into the nucleus, activating the classical genomic actions of glucocorticoid signaling. Unbound chaperone proteins engage with various other cellular pathways affiliated with some of the non-genomic effects of glucocorticoid signaling (29).
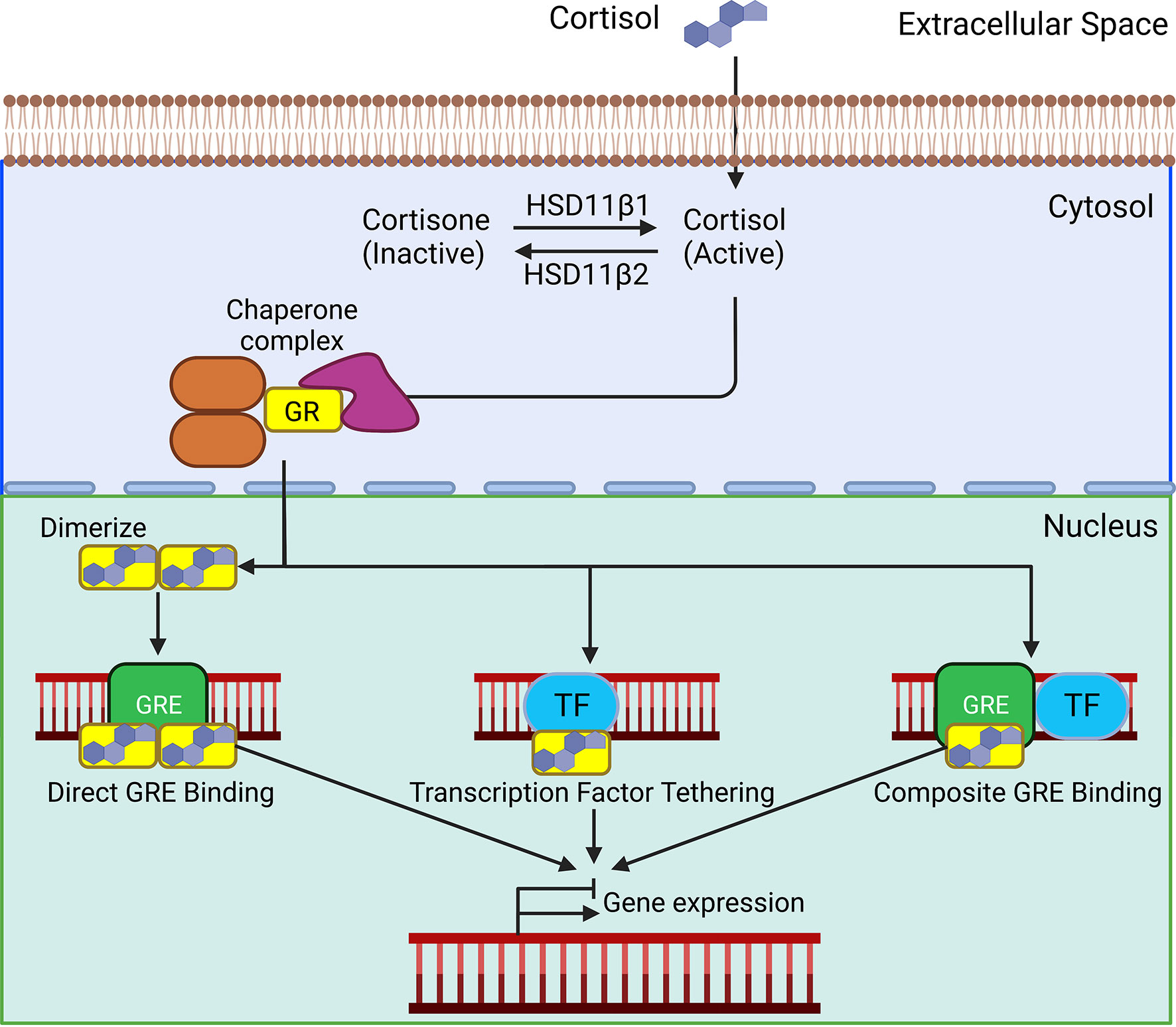
Figure 2 In the absence of ligands, the glucocorticoid receptor (GR) is bound in the cytoplasm by a complex of chaperone proteins. Lipophilic glucocorticoids freely defuse through the plasma membrane and bind to the GR. Upon ligand binding, the GR translocate to the nucleus. Homodimerized GRs bind to glucocorticoid response elements (GREs). Direct binding of the GR to transcription factors (TF) and composite binding to GREs and TF affect their transcriptional activity. These interactions ultimately act to activate or suppress gene transcription.
Once in the nucleus, the classical mechanism through which GR affects gene transcription is by binding to GREs, distinct palindromic consensus sequences separated by a 3-5 nucleotide spacer. Binding occurs through the central zinc finger DNA-binding domain, which induces a conformational change within the receptor. Subsequent allosteric interactions promote the recruitment of coactivator complexes necessary to remodel chromatin. These complexes are the cAMP response element binding protein (CREB)/p300 and pCAF, response element-binding protein (CREB)-binding protein/p300 and p/CAF, both of which contain intrinsic histone acetylase activity (30). These proteins acetylate lysine residues to induce nucleosome rearrangement and DNA unwinding, which leads to promoter accessibility and the recruitment of the basal transcription machinery, resulting in target gene transcription. GR monomers can also directly bind to other transcription factors, modifying their activity, or undergo composite interactions requiring both GRE binding and protein interactions with another transcription factor (Figure 2) (29). GR can also repress gene activation by binding as a monomer to negative GREs (31). The net effect of these interactions is to induce or suppress gene transcription.
Glucocorticoids are pleiotropic steroid hormones that exert a wide array of effects, such as embryological development, metabolism, cellular proliferation, apoptosis, inflammation, and cancer development (27, 29, 32). Some reports estimate glucocorticoids regulate up to 20% of transcribed genes (2). However, the effects of glucocorticoid signaling are cell-type specific, owing to their interactions with numerous transcription factors, differential chromatin accessibility, and non-genomic effects. The GR mRNA also contains 7 internal translation start sites, and each GR translational isoform has unique transcriptomes (33, 34). Moreover, interactions between the GR and other transcription factors can provide cell-specific effects on transcription. These interactions and cell-type specific effects are important within cancer, where glucocorticoids have significantly divergent roles depending on the cancer type. More work is needed to uncover the mechanisms regulating cell-type specific actions of glucocorticoids both in healthy tissues and in cancer.
Glucocorticoid regulation of inflammation within the pre-tumor and tumor microenvironments
The relationship between the immune system and cancer is complex. The immune system is a frontline defense against cancer development. Natural killer cells and CD8+ cytotoxic T cells remove nascent cancer cells with altered MHC I expression or displaying mutated proteins (35, 36). Anti-cancer therapies such as immune checkpoint blockade (ICB) rely on increasing anti-tumor immune activity by stimulating T cells. However, inflammation is a double-edged sword that can also promote cancer development and progression (37). Immune responses to infections and or exposure to irritants, such as inhaled particles, inevitably cause damage to the surrounding tissue. During a “normal” inflammatory response, the pathogens are quickly cleared, the inflammation resolves, and the damaged tissue is healed (Figure 3). However, chronic “smoldering” inflammation and the resultant prolonged tissue damage leads to metaplasia development and may promote carcinogenesis. Inflammation associated with chronic infections such as Helicobacter pylori (gastric cancer) and Human Papillomavirus (HPV, cervical cancers), chronic inflammatory diseases (chronic obstructive pulmonary disorder (COPD) and Crohn's Disease), and through chronic exposure to irritants such as tobacco use (oral and lung cancers) promote cancer development (38–40). At therapeutic doses, glucocorticoids exert powerful anti-inflammatory effects and have become one of the most prescribed drug classes worldwide. However, severe side effects associated with long-term use limit their effectiveness in treating chronic inflammatory diseases. Glucocorticoids suppress the recruitment of additional leukocytes to the site of inflammation by blocking the production of chemokines, histamine, and prostaglandins and by reducing the expression of adhesion molecules such as integrins and selectins (32). Glucocorticoids also limit the expression of proinflammatory cytokines such as IL1B, TNF, INFG, and IL6 while promoting the expression of anti-inflammatory cytokines such as IL10 and TGFB. Dysregulation of these inflammatory processes is associated with cancer development (41).
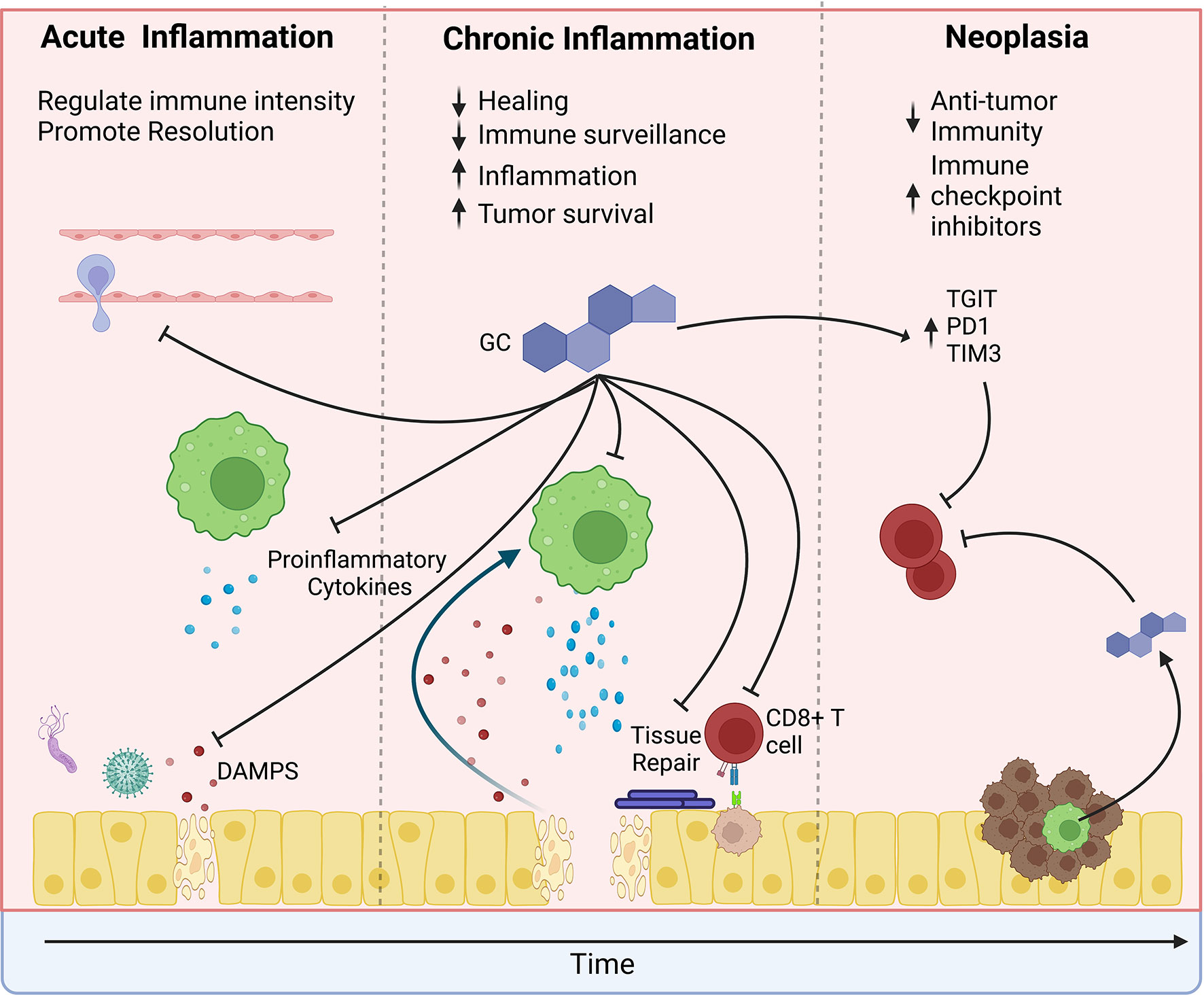
Figure 3 The effects of glucocorticoids change over the course of tumor development. In the pre-tumor microenvironment, glucocorticoids may protect from cancer by limiting the intensity of inflammation. Glucocorticoids suppress immune cell recruitment to sites of damage or infection and suppress proinflammatory cytokine production. However, during chronic inflammation, glucocorticoids may promote tumor development. Prolonged glucocorticoid signaling inhibits wound healing, thereby causing a feedforward loop of increased release of damage-associated molecular patterns (DAMPs), which increase inflammation and further promote tissue damage. Simultaneously, glucocorticoids suppress anti-tumor surveillance by CD8 T cells. Similarly, glucocorticoids suppress anti-tumor immunity within the tumor microenvironment and promote the expression of immune checkpoint proteins. However, their effects are compounded by increased psychological stress, exogenous steroid treatment, and local steroidogenesis by tumor-infiltrating immune cells. Furthermore, glucocorticoids may directly promote tumor cell survival by suppressing apoptosis and promoting tumor cell growth.
Approximately 15-20% of all cancers are preceded by infection (37, 42, 43). Reduced GR expression and polymorphisms that decrease GR activity have long been associated with an increased risk of various cancers. However, the exact stage of cancer development affected by glucocorticoid signaling is unclear. Gastric cancer is the most common pathogen-associated cancer (44). Approximately 90% of gastric cancers are associated with long-term Helicobacter pylori infection. Chronic inflammation is a requisite driver of H. pylori-associated gastric cancer. Immune-deficient mice are resistant to Helicobacter-associated gastric pathologies (45). Moreover, overexpression of the proinflammatory cytokines IL1β or INFγ is sufficient to drive gastric cancer development even in the absence of Helicobacter infection (46, 47), underscoring that inflammation is both necessary and sufficient to drive gastric carcinogenesis. Disruption of glucocorticoid signaling is associated with an increased risk of gastric cancer (48). Given their anti-inflammatory effects, glucocorticoids likely suppress pro-neoplastic gastric inflammation. Recently, we reported that adrenalectomized mice developed spontaneous gastric inflammation stemming from the activation of type 2 innate lymphoid cells (ILC2s) and the recruitment of monocyte-derived macrophages (49, 50). Adrenalectomy-induced gastric inflammation drove atrophic gastritis and pyloric metaplasia development, which are precursors of gastric adenocarcinoma. It is important to note that the adrenalectomy-induced gastric inflammation and pre-neoplastic lesions occurred without Helicobacter infection, suggesting that glucocorticoids are master regulators of gastric inflammation. While these studies show an essential role for glucocorticoids in protecting the stomach from pro-neoplastic inflammation, excessive glucocorticoid signaling may compromise protective gastric immunity. For instance, treatment with the synthetic glucocorticoid dexamethasone during H. pylori infection protects from pre-neoplastic epithelial changes but also increases gastric bacterial load (51). Excessive glucocorticoid signaling is also linked to peptic ulcer development (52). Thus, the magnitude and duration of glucocorticoid signaling likely contribute to how these hormones affect carcinogenesis. Further investigation is needed to uncover how glucocorticoid signaling during H. pylori infection affects gastric cancer development.
The lung is also highly susceptible to inflammation-associated neoplasia. Lung inflammation caused by tobacco smoking, particle inhalation, or exposure to gases such as ozone is linked to lung cancer development (53). While tobacco smoking is the top risk factor for lung cancers, smokers with chronic lung inflammatory diseases such as COPD are 3-10 fold more likely to develop lung cancer than smokers without COPD (54). In mouse mutant KRAS lung cancer models, lung inflammation accelerates tumor formation. However, depleting discrete inflammatory cell populations or reducing the concentration of proinflammatory cytokines protects from tumor development (55–57). Similarly, in urethane lung tumor models, glucocorticoid treatment suppressed lung tumor development while adrenalectomy significantly increased tumor number (58, 59). Glucocorticoid treatment also reduced cancer risk in mice exposed to cigarette smoke (60). Therefore, glucocorticoid treatments that reduce lung inflammation protect from cancer development. Within the clinic, glucocorticoids are a frontline therapy for treating lung inflammatory diseases (61). Epidemiological studies have found that inhaled steroid use dramatically reduces the risk of lung cancer in individuals with lung inflammation, such as COPD (62, 63) and asthma (64). Thus, glucocorticoids may be critical in suppressing pro-neoplastic lung inflammation.
Glucocorticoid signaling within the pre-tumor microenvironment is a double-edged sword. While glucocorticoid signaling suppresses pro-neoplastic inflammation and limits tissue damage, they may also activate processes that promote neoplasia development (Figure 3). Glucocorticoids inhibit wound healing by suppressing collagen deposition, growth factor expression, and vascularization (65). These delays in wound healing may increase inflammation, epithelial proliferation, and metaplasia, promoting carcinogenesis. Tang et al. reported that while deletion of the GR from the intestinal epithelium increased acute inflammation, intestinal GR knockout mice exhibited accelerated tissue healing and fewer intestinal tumors than GR wild type mice (66). Similarly, treatment with the GR agonist betamethasone suppressed acuteintestinal inflammation but also prevented epithelial healing promoting tumor development. Therefore, sustained glucocorticoid suppression of tissue repair promotes chronic inflammation and may promote cancer development. In addition to preventing wound healing, glucocorticoid signaling also suppresses anti-cancer immune surveillance by CD8+ cytotoxic T cells (67). Thus, as mutations occur, glucocorticoids may suppress the removal of developing cancer cells.
Cancer cell escape from immune surveillance is critical for tumor formation, survival, and growth. Glucocorticoids potently suppress anti-tumor immunity. Within the myeloid compartment, glucocorticoids alter the phenotype of tumor-associated macrophages, suppressing their proinflammatory functions while promoting anti-inflammatory roles and driving the production of the anti-inflammatory cytokines TGFB and IL10 (68). Glucocorticoids also curtail the anti-tumor effects of dendritic cells by inducing the expression of TSC23D3 (GILZ), thereby reducing proinflammatory cytokine expression and suppressing antigen presentation to CD8+ T cells (69). Within the lymphoid compartment, glucocorticoids directly suppress CD8+ T cell activation by driving the expression of the immune checkpoint inhibitors TGIT, PD1, TIM3, and LAG3 and through promoting the expression of the anti-inflammatory cytokine IL10 (23). CD8+ T cells are key mediators of anti-tumor immunity, and suppression of these cells through the induction of checkpoint proteins significantly promotes tumor growth and metastasis. Immune-activating therapies such as ICB are designed to suppress these checkpoint proteins and promote anti-tumor immunity. However, elevated expression of GR-regulated genes is associated with reduced ICB responsiveness (23), and high systemic glucocorticoid levels are linked to ICB failure (70, 71). Consistent with their ability to antagonize ICB efficacy, glucocorticoids are the standard treatment for immune-related adverse events associated with ICB (72).
Glucocorticoid effects on cancer cells and tumor progression
In contrast to their heterogeneous effects on tumor development, glucocorticoid signaling within established solid tumors is typically linked to poor clinical outcomes. Elevated glucocorticoid signaling during cancer occurs through the activation of the HPA axis and treatment with exogenous steroids. In preclinical models, chronic psychological stress suppresses anti-tumor immunity and increases tumor growth (73). Similar outcomes are reported in clinical studies where depression and poor patient mood increase systemic glucocorticoid production and are linked to suppression of anti-tumor immunity and increased mortality (69, 74). Glucocorticoids are also commonly co-administered with chemotherapy to reduce negative side effects such as nausea and edema and to improve appetite and energy (75). Similar to the negative effects of elevated endogenous glucocorticoids, corticosteroid treatment is associated with reduced anti-tumor immune responses, tumor treatment resistance, and increased metastasis. In addition to a systemic increase of adrenal-derived glucocorticoids, tumor-infiltrating immune cells also may produce glucocorticoids within the tumor (10, 23–25). Local steroidogenesis may be an important paracrine mechanism by which tumor cells can evade the immune system, but the overall impact of extra-adrenal glucocorticoid production on tumor progression and patient outcomes remains unknown.
Glucocorticoid signaling in cancer cells is heterogenous, and their impacts depend on the cancer type and stage. Within tumor cells, glucocorticoids promote proliferation, survival, and migration. These pro-tumor effects are manifested through a variety of mechanisms. In some cases, direct GR transcriptional targets are critical for promoting tumor cell survival. For instance, treatment with dexamethasone protects thyroid cancer cells from apoptosis by induction of BCL-XL (76). However, consistent with its high-level role as a transcriptional regulator, many cellular effects are manifested through the induction of gene expression or signaling cascades that ultimately promote tumor progression. This is illustrated by the GR-target glucocorticoid-regulated kinase 1 (SGK1), which promotes tumor cell survival through interacting with the PI3K pathway and through the activation of the Forkhead box family of transcription factors such as FKHRL1 (77). Furthermore, SGK1 phosphorylation of N-MYC downstream regulated gene 1 (NDRG1) promotes tumor cell growth and tissue extravasation. NDRG1 expression is elevated in esophageal squamous cell carcinoma, wherein expression is correlated with local tumor invasion and poor prognosis (78). Additionally, NDRG1 dysregulation in esophageal cancer has pro-oncogenic functions by activating the WNT signaling pathway (79). Similarly, betamethasone promotes increased expression of cytoskeletal proteins such as fibronectin, promoting the activation and nuclear accumulation of the oncoprotein YAP and the expansion of chemotherapy-resistant cancer stem cells (80). Therefore, the effects of glucocorticoids on cancer cells may often be masked by downstream effectors, and there are likely numerous effects of glucocorticoid signaling that have not yet been described.
In addition to their classical transcriptional effects, the GR impacts cancer through rapid non-genomic actions. Within the A549 lung carcinoma cell line, the GR regulates the activation of both WT and mutated RAS through non-genomic actions (81). In the absence of glucocorticoids, cytoplasmic GR directly binds to RAS, inhibiting its activation. GR suppression of RAS blocks tumor cell proliferation, and GR-deficient A549 cell xenografts developed into larger tumors than WT cells which express endogenous GR (81). Importantly, GR suppression of RAS is blocked by treatment with dexamethasone. Thus, increased glucocorticoid levels within the tumor microenvironment would block GR binding to RAS and promote tumor growth.
While glucocorticoid signaling in solid tumors is often linked to poor clinical outcomes, this is not universally true. Patient hepatocellular carcinomas actively suppress glucocorticoid signaling by upregulating HSD11B2. Bypassing the HSD11B1/B2 axis through long-term administration of dexamethasone protects from liver carcinogenesis in mice by suppressing glycolysis and promoting gluconeogenesis (82). Similarly, short-term administration of hydrocortisone attenuates the progression of hepatocellular carcinoma in rats (83). The GR transcriptional target TSC22D3 suppresses RAS and RAF signaling by directly binding these proteins and preventing the activation of the MAPK pathway and RB phosphorylation (84). Similarly, GR promotes the expression of the phosphatase DUSP1, which inhibits multiple components of the MAPK pathway including, p38 kinase, JNK, and ERK (85, 86). These seemingly conflicting results highlight the cell-type-specific effects of glucocorticoid signaling and the need for further research on the impact of glucocorticoids on cancer.
Glucocorticoid interactions with other steroid hormone receptors
All steroid hormone receptors primarily function as ligand-dependent transcription factors. Many of the mechanisms utilized to regulate transcription are shared between the steroid hormone receptors. Estrogen and androgen signaling are central players in breast and prostate cancer development and progression, respectively. Glucocorticoids can modulate the activity of these steroid hormones through multiple mechanisms, including heterodimerizing with the estrogen receptor (ER) and androgen receptor (AR) and competing for co-factors (87–89). Moreover, the DNA response elements recognized by each steroid hormone receptor are highly similar, often only differing by a few nucleotides (90). Therefore, certain ER and AR-regulated genes can also be targeted by the GR.
In ER-positive breast cancer, GR expression is correlated to a favorable prognosis (91, 92). Glucocorticoids may inhibit the development of ER-positive tumors by antagonizing the activity of ER alpha. GR is reported to antagonize ER signaling through several distinct mechanisms, such as displacing ER alpha binding to estrogen response elements and by heterodimerizing with the ER to recruit co-repressors (87, 93). GR-ER binding is also associated with increased expression of differentiation markers, inhibition of ER-induced growth factors, and increased survival of patients with ER-positive breast cancer (92). Thus, glucocorticoid signaling may be protective in estrogen-dependent breast cancer. Further supporting this idea, a recent epidemiological study found that systemic glucocorticoid therapies were associated with reduced incidence of ER-positive but not ER-negative invasive breast cancers (94). However, protection was only associated with grade I and II tumors, while glucocorticoid use was associated with an increased risk of metastasis for higher-grade tumors, potentially due to reduced estrogen dependence in more advanced tumors. In contrast to their putative protective roles in early-stage ER-positive breast cancers, glucocorticoids are linked to poor outcomes in patients with ER-negative tumors (91, 92, 95). In these tumors, glucocorticoids are reported to assume a role similar to the ER by inducing the expression of ER-regulated genes, promoting growth and metastasis (80, 96). Further information about the role of glucocorticoids in breast cancer can be found in this cited review article (97).
In prostate cancer, glucocorticoids play important roles in promoting resistance to anti-androgen therapies. Various reports disagree on whether the GR is expressed by healthy human prostate epithelial cells. However, GR expression increases as the tumors acquire resistance to anti-androgen therapies (98, 99). Treatment with AR antagonists is also associated with increased circulating cortisol levels and decreased expression of the cortisol-degrading enzyme HSD11B2 by prostate cancer cells, increasing local cortisol concentrations (100, 101). Exogenous corticosteroids are also commonly co-administered with anti-androgen therapies to alleviate side effects (102). Collectively, these changes promote increased glucocorticoid signaling within prostate cancer cells.
The GR and AR are structurally similar and share nearly identical DNA response elements (90). Cells that co-express the AR and GR have significant overlap in their regulated transcriptomes (98, 103). During anti-androgen therapies, when only the GR is transcriptionally active, the GR drives the expression of typically AR-regulated genes related to apoptotic resistance and tumor cell survival (104, 105). It is important to note that while there is significant overlap between AR and GR-regulated transcriptomes, they are not identical, and each steroid hormone receptor regulates a host of specific genes and cellular functions. However, the similarity between these receptors allows for some functional overlap. Indeed, numerous studies have reported the immunomodulatory roles of androgens (106, 107). We recently reported that glucocorticoids and androgens regulate the transcription of proinflammatory cytokines within ILC2s, suppressing gastric inflammation and metaplasia (50). Therefore, many of the effects of glucocorticoids on anti-cancer immunity could also be influenced by androgens. Interactions between glucocorticoids and androgens likely play critical roles in the development of numerous types of cancers and warrant further investigation.
Clinical implications
Glucocorticoids are used extensively as cancer therapy adjuvants for the treatment of solid tumors. Synthetic glucocorticoids such as dexamethasone, prednisolone, and betamethasone have long been utilized for improving adverse symptoms associated with chemotherapy and radiation therapy (108–110). Dexamethasone ameliorates chemotherapy-induced cytotoxicity especially in breast and prostate cancer (104, 111). In brain tumors, dexamethasone is used to reduce intracranial pressure and edema (112). Despite their significant use for managing symptoms in solid tumors, corticosteroid use is linked to the failure of anti-tumor therapies and poor patient outcomes. Glucocorticoid treatments are linked to suppression of anti-tumor immunity, ICB failure, cancer cell resistance to chemotherapy, and increased tumor metastases (113–115). In a retrospective study of 181 glioblastoma patients receiving anti-PDL1 therapy, dexamethasone treatment was the strongest predictor for reduced patient survival (71, 116). Corticosteroid treatment as an adjuvant to radiotherapy for glioblastoma has consistently shown a reduction in overall patient survival and promotes tumor recurrence (117, 118). Similarly, in non-small cell lung cancer and urothelial carcinoma, glucocorticoid treatment is associated with reduced patient survival (119).
Despite these host of negative effects, glucocorticoid treatments offer considerable positive benefits. As a chemotherapy adjuvant, glucocorticoids suppress nausea, improve appetite and energy, and reduce pain (75). Suppression of these negative side effects improves patient quality of life and increases patient adherence to oral chemotherapy regimens (120). Corticosteroids are often utilized during tumor resections to reduce anesthesia-related nausea and vomiting and suppress inflammation within the surgical site. Glucocorticoid treatments after resection of Barret’s esophagus or esophageal carcinoma protect from the development of esophageal strictures (121). Retrospective studies have found that intra- and post-operative treatments with corticosteroids have no impact on patient survival or cancer recurrence, demonstrating that carefully constructed therapeutic regimens may be able to deliver the benefits while avoiding the negative complications of glucocorticoid therapies (122, 123). However, the exact effects of glucocorticoids likely vary depending on tumor type, and much more work is needed to fully elucidate how glucocorticoid adjuvant therapies affect various cancer types.
Conclusions
Glucocorticoid signaling is highly divergent across different cancer types, and their effects significantly change as cancers develop. Despite their widespread clinical use to combat the side effects of chemotherapy, there is a significant gap in our knowledge concerning how glucocorticoid signaling affects specific cancer types. However, even less is known about how glucocorticoids affect early tumor development. Our summary of the literature suggests that glucocorticoids may be a double-edged sword in the pre-tumor microenvironment where they protect from pro-neoplastic inflammation early, but sustained activation may promote carcinogenesis by promoting chronic inflammation and inhibiting wound repair. Significantly more work is needed to improve our knowledge of how these hormones affect cancer development and how the clinical use of glucocorticoids affects anti-cancer treatment efficacy. Finally, interactions between glucocorticoids and other steroid hormones clearly affect breast and prostate cancer, but these interactions likely affect numerous other cancer types. Glucocorticoid and androgen signaling, in particular, shape the anti-tumor T cell response. More work is needed to uncover how interactions between stress and sex hormones affect anti-tumor immunity.
Author contributions
JB conceived the topic and constructed the original outline. SK, SD, BD, and JB wrote the original manuscript. BD and JB constructed the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by West Virginia University start-up funds (JB) and a grant from National Institutes of Health P20GM121322 (JB).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
GR, Glucocorticoid Receptor; ER, Estrogen Receptor; AR, Androgen Receptor; HPA axis, Hypothalamic-Pituitary-Adrenal axis; CRH, Corticotropin-Releasing Hormone; ACTH, Adrenocorticotropic Hormone; GRE, Glucocorticoid Response Elements; ICB, Immune Checkpoint Blockade; HPV, Human Papillomavirus; COPD, Chronic Obstructive Pulmonary Disorder; ILC2, Type 2 Innate Lymphoid Cell.
References
1. Miller WL, White PC. History of adrenal research: From ancient anatomy to contemporary molecular biology. Endocr Rev (2023) 44(1):70–116. doi: 10.1210/endrev/bnac019
2. Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J (2002) 16(1):61–71. doi: 10.1096/fj.01-0245com
3. Vettorazzi S, Nalbantoglu D, Gebhardt JCM, Tuckermann J. A guide to changing paradigms of glucocorticoid receptor function–a model system for genome regulation and physiology. FEBS J (2022) 289(19):5718–43. doi: 10.1111/febs.16100
4. Sheng JA, Bales NJ, Myers SA, Bautista AI, Roueinfar M, Hale TM, et al. The hypothalamic-Pituitary-Adrenal axis: Development, programming actions of hormones, and maternal-fetal interactions. Front Behav Neurosci (2020) 14:601939. doi: 10.3389/fnbeh.2020.601939
5. Kraemer F, Shen WJ, Natu V, Patel S, Osuga J, Ishibashi S. Adrenal neutral cholesteryl ester hydrolase: identification, subcellular distribution, and sex differences. Endocrinology (2002) 143:801–6. doi: 10.1210/endo.143.3.8693
6. Miller WL. Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta (BBA)-Mol Cell Biol Lipids (2007) 1771(6):663–76. doi: 10.1016/j.bbalip.2007.02.012
7. Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci (2000) 917:608–17. doi: 10.1111/j.1749-6632.2000.tb05426.xæ
8. Bornstein SR, Ziegler CG, Krug AW, Kanczkowski W, Rettori V, McCann SM, et al. The role of toll-like receptors in the immune-adrenal crosstalk. Ann N Y Acad Sci (2006) 1088:307–18. doi: 10.1196/annals.1366.027
9. Wirtz PH, von Känel R. Psychological stress, inflammation, and coronary heart disease. Curr Cardiol Rep (2017) 19(11):111. doi: 10.1007/s11886-017-0919-x
10. Tang R, Acharya N, Subramanian A, Purohit V, Tabaka M, Hou Y, et al. Tim-3 adapter protein Bat3 acts as an endogenous regulator of tolerogenic dendritic cell function. Sci Immunol (2022) 7(69):eabm0631. doi: 10.1126/sciimmunol.abm0631
11. Hammond GL. Plasma steroid-binding proteins: primary gatekeepers of steroid hormone action. J Endocrinol (2016) 230(1):R13–25. doi: 10.1530/JOE-16-0070
12. Hammond GL, Smith CL, Paterson NA, Sibbald WJ. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab (1990) 71(1):34–9. doi: 10.1210/jcem-71-1-34
13. Monder C, Lakshmi V. Corticosteroid llβ-dehydrogenase of rat tissues: Immunological studies. Endocrinology (1990) 126(5):2435–43. doi: 10.1210/endo-126-5-2435
14. Paulsen SK, Pedersen SB, Fisker S, Richelsen B. 11β-HSD type 1 expression in human adipose tissue: Impact of gender, obesity, and fat localization. Obesity (2007) 15(8):1954–60. doi: 10.1038/oby.2007.233
15. Ricketts ML, Verhaeg JM, Bujalska I, Howie AJ, Rainey WE, Stewart PM. Immunohistochemical localization of type 1 11β-hydroxysteroid dehydrogenase in human tissues. J Clin Endocrinol Metab (1998) 83(4):1325–35. doi: 10.1210/jcem.83.4.4706
16. Smith RE, Maguire JA, Stein-Oakley AN, Sasano H, Takahashi K, Fukushima K, et al. Localization of 11 beta-hydroxysteroid dehydrogenase type II in human epithelial tissues. J Clin Endocrinol Metab (1996) 81(9):3244–8. doi: 10.1210/jcem.81.9.8784076
17. Voisin M, de Medina P, Mallinger A, Dalenc F, Huc-Claustre E, Leignadier J, et al. Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc Natl Acad Sci U S A (2017) 114(44):E9346–e55. doi: 10.1073/pnas.1707965114
18. Lu L, Zhao G, Luu-The V, Ouellet J, Fan Z, Labrie F, et al. Expression of 11β-hydroxysteroid dehydrogenase type 1 in breast cancer and adjacent non-malignant tissue. Immunocytochem Study Pathol Oncol Res (2011) 17(3):627–32. doi: 10.1007/s12253-011-9361-3
19. Cirillo N, Morgan DJ, Pedicillo MC, Celentano A, Lo Muzio L, McCullough MJ, et al. Characterisation of the cancer-associated glucocorticoid system: key role of 11β-hydroxysteroid dehydrogenase type 2. Br J Cancer (2017) 117(7):984–93. doi: 10.1038/bjc.2017.243
20. Taves MD, Gomez-Sanchez CE, Soma KK. Extra-adrenal glucocorticoid and mineralocorticoid biosynthesis: evidence for local synthesis, regulation, and function. Am J Physiol Endocrinol Metab (2011) 301(1):11–24. doi: 10.1152/ajpendo.00100.2011
21. Phan TS, Merk VM, Brunner T. Extra-adrenal glucocorticoid synthesis at epithelial barriers. Genes Immun (2019) 20(8):627–40. doi: 10.1038/s41435-019-0058-z
22. Mittelstadt PR, Taves MD, Ashwell JD. Glucocorticoids oppose thymocyte negative selection by inhibiting Helios and Nur77. J Immunol (2019) 203(8):2163–70. doi: 10.4049/jimmunol.1900559
23. Acharya N, Madi A, Zhang H, Klapholz M, Escobar G, Dulberg S, et al. Endogenous glucocorticoid signaling regulates CD8(+) T cell differentiation and development of dysfunction in the tumor microenvironment. Immunity (2020) 53(3):658–71.e6. doi: 10.1016/j.immuni.2020.08.005
24. Mahata B, Pramanik J, van der Weyden L, Polanski K, Kar G, Riedel A, et al. Tumors induce de novo steroid biosynthesis in T cells to evade immunity. Nat Commun (2020) 11(1):3588. doi: 10.1038/s41467-020-17339-6
25. Mahata B, Zhang X, Kolodziejczyk AA, Proserpio V, Haim-Vilmovsky L, Taylor AE, et al. Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep (2014) 7(4):1130–42. doi: 10.1016/j.celrep.2014.04.011
26. Giguère V, Hollenberg SM, Rosenfeld MG, Evans RM. Functional domains of the human glucocorticoid receptor. Cell (1986) 46(5):645–52. doi: 10.1016/0092-8674(86)90339-9
27. Cruz-Topete D, Oakley RH, Cidlowski JA. Glucocorticoid signaling and the aging heart. Front Endocrinol (Lausanne) (2020) 11:347. doi: 10.3389/fendo.2020.00347
28. Scheschowitsch K, Leite JA, Assreuy J. New insights in glucocorticoid receptor signaling-more than just a ligand-binding receptor. Front Endocrinol (Lausanne) (2017) 8:16. doi: 10.3389/fendo.2017.00016
29. Busada JT, Cidlowski JA. Mechanisms of glucocorticoid action during development. Curr Top Dev Biol (2017) 125:147–70. doi: 10.1016/bs.ctdb.2016.12.004
30. Deroo BJ, Archer TK. Glucocorticoid receptor-mediated chromatin remodeling in vivo. Oncogene (2001) 20(24):3039–46. doi: 10.1038/sj.onc.1204328
31. Surjit M, Ganti KP, Mukherji A, Ye T, Hua G, Metzger D, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell (2011) 145(2):224–41. doi: 10.1016/j.cell.2011.03.027
32. Cain DW, Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol (2017) 17(4):233–47. doi: 10.1038/nri.2017.1
33. Lu NZ, Cidlowski JA. Translational regulatory mechanisms generate n-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell (2005) 18(3):331–42. doi: 10.1016/j.molcel.2005.03.025
34. Oakley RH, Ramamoorthy S, Foley JF, Busada JT, Lu NZ, Cidlowski JA. Glucocorticoid receptor isoform-specific regulation of development, circadian rhythm, and inflammation in mice. FASEB J (2018) 32(10):5258–71. doi: 10.1096/fj.201701153R
35. Huntington ND, Cursons J, Rautela J. The cancer-natural killer cell immunity cycle. Nat Rev Cancer (2020) 20(8):437–54. doi: 10.1038/s41568-020-0272-z
36. van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer (2020) 20(4):218–32. doi: 10.1038/s41568-019-0235-4
37. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell (2010) 140(6):883–99. doi: 10.1016/j.cell.2010.01.025
38. Amieva M, Peek RM Jr. Pathobiology of helicobacter pylori-induced gastric cancer. Gastroenterology (2016) 150(1):64–78. doi: 10.1053/j.gastro.2015.09.004
39. Araldi RP, Sant'Ana TA, Módolo DG, de Melo TC, Spadacci-Morena DD, de Cassia Stocco R, et al. The human papillomavirus (HPV)-related cancer biology: An overview. BioMed Pharmacother (2018) 106:1537–56. doi: 10.1016/j.biopha.2018.06.149
40. Faye AS, Holmer AK, Axelrad JE. Cancer in inflammatory bowel disease. Gastroenterol Clin North Am (2022) 51(3):649–66. doi: 10.1016/j.gtc.2022.05.003
41. Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol (2013) 33 Suppl 1:S79–84. doi: 10.1007/s10875-012-9847-0
42. Greten FR, Grivennikov SI. Inflammation and cancer: Triggers, mechanisms, and consequences. Immunity (2019) 51(1):27–41. doi: 10.1016/j.immuni.2019.06.025
43. Sakowska J, Arcimowicz Ł, Jankowiak M, Papak I, Markiewicz A, Dziubek K, et al. Autoimmunity and cancer-two sides of the same coin. Front Immunol (2022) 13:793234. doi: 10.3389/fimmu.2022.793234
44. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71(3):209–49. doi: 10.3322/caac.21660
45. Roth KA, Kapadia SB, Martin SM, Lorenz RG. Cellular immune responses are essential for the development of helicobacter felis-associated gastric pathology. J Immunol (1999) 163(3):1490–7. doi: 10.4049/jimmunol.163.3.1490
46. Syu LJ, El-Zaatari M, Eaton KA, Liu Z, Tetarbe M, Keeley TM, et al. Transgenic expression of interferon-gamma in mouse stomach leads to inflammation, metaplasia, and dysplasia. Am J Pathol (2012) 181(6):2114–25. doi: 10.1016/j.ajpath.2012.08.017
47. Tu S, Bhagat G, Cui G, Takaishi S, Kurt-Jones EA, Rickman B, et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell (2008) 14(5):408–19. doi: 10.1016/j.ccr.2008.10.011
48. Gu Y, Deng B, Kong J, Yan C, Huang T, Yang J, et al. Functional polymorphisms in NR3C1 are associated with gastric cancer risk in Chinese population. Oncotarget (2017) 8(62):105312–9. doi: 10.18632/oncotarget.22172
49. Busada JT, Ramamoorthy S, Cain DW, Xu X, Cook DN, Cidlowski JA. Endogenous glucocorticoids prevent gastric metaplasia by suppressing spontaneous inflammation. J Clin Invest (2019) 129(3):1345–58. doi: 10.1172/JCI123233
50. Busada JT, Peterson KN, Khadka S, Xu X, Oakley RH, Cook DN, et al. Glucocorticoids and androgens protect from gastric metaplasia by suppressing group 2 innate lymphoid cell activation. Gastroenterology (2021) 161(2):637–52.e4. doi: 10.1053/j.gastro.2021.04.075
51. Naka Y, Aihara T, Keto Y, Okabe S. Effects of dexamethasone and FK506 on helicobacter pylori-induced gastritis and bacterial viability in Mongolian gerbils. J Physiol Paris (2001) 95(1-6):443–51. doi: 10.1016/S0928-4257(01)00061-4
52. Bandyopadhyay U, Biswas K, Bandyopadhyay D, Ganguly CK, Banerjee RK. Dexamethasone makes the gastric mucosa susceptible to ulceration by inhibiting prostaglandin synthetase and peroxidase–two important gastroprotective enzymes. Mol Cell Biochem (1999) 202(1-2):31–6. doi: 10.1023/A:1007018212822
53. Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int J Environ Res Public Health (2013) 10(9):3886–907. doi: 10.3390/ijerph10093886
54. Young RP, Hopkins RJ, Christmas T, Black PN, Metcalf P, Gamble GD. COPD prevalence is increased in lung cancer, independent of age, sex and smoking history. Eur Respir J (2009) 34(2):380–6. doi: 10.1183/09031936.00144208
55. Moghaddam SJ, Li H, Cho SN, Dishop MK, Wistuba II, Ji L, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol (2009) 40(4):443–53. doi: 10.1165/rcmb.2008-0198OC
56. Caetano MS, Zhang H, Cumpian AM, Gong L, Unver N, Ostrin EJ, et al. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras-Mutant lung cancer. Cancer Res (2016) 76(11):3189–99. doi: 10.1158/0008-5472.CAN-15-2840
57. Gong L, Cumpian AM, Caetano MS, Ochoa CE, de la Garza MM, Lapid DJ, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer (2013) 12(1):154. doi: 10.1186/1476-4598-12-154
58. Stathopoulos GT, Sherrill TP, Cheng DS, Scoggins RM, Han W, Polosukhin VV, et al. Epithelial NF-kappaB activation promotes urethane-induced lung carcinogenesis. Proc Natl Acad Sci U S A (2007) 104(47):18514–9. doi: 10.1073/pnas.0705316104
59. Droms KA, Fernandez CA, Thaete LG, Malkinson AM. Effects of adrenalectomy and corticosterone administration on mouse lung tumor susceptibility and histogenesis. J Natl Cancer Inst (1988) 80(5):365–9. doi: 10.1093/jnci/80.5.365
60. Balansky R, Ganchev G, Iltcheva M, Steele VE, De Flora S. Prevention of cigarette smoke-induced lung tumors in mice by budesonide, phenethyl isothiocyanate, and n-acetylcysteine. Int J Cancer (2010) 126(5):1047–54. 10.1002/ijc.24942
61. Samransamruajkit R, Jitchaiwat S, Deerojanawong J, Sritippayawan S, Praphal N. Adrenal insufficiency in early phase of pediatric acute lung injury/acute respiratory distress syndrome. J Crit Care (2007) 22(4):314–8. doi: 10.1016/j.jcrc.2007.03.003
62. Raymakers AJN, Sadatsafavi M, Sin DD, FitzGerald JM, Marra CA, Lynd LD. Inhaled corticosteroids and the risk of lung cancer in COPD: a population-based cohort study. Eur Respir J (2019) 53(6). doi: 10.1183/13993003.01257-2018
63. Parimon T, Chien JW, Bryson CL, McDonell MB, Udris EM, Au DH. Inhaled corticosteroids and risk of lung cancer among patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2007) 175(7):712–9. doi: 10.1164/rccm.200608-1125OC
64. Lee CH, Hyun MK, Jang EJ, Lee NR, Kim K, Yim JJ. Inhaled corticosteroid use and risks of lung cancer and laryngeal cancer. Respir Med (2013) 107(8):1222–33. doi: 10.1016/j.rmed.2012.12.002
65. Christian LM, Graham JE, Padgett DA, Glaser R, Kiecolt-Glaser JK. Stress and wound healing. Neuroimmunomodulation (2006) 13(5-6):337–46. doi: 10.1159/000104862
66. Tang S, Zhang Z, Oakley RH, Li W, He W, Xu X, et al. Intestinal epithelial glucocorticoid receptor promotes chronic inflammation-associated colorectal cancer. JCI Insight (2021) 6(24):e151815. doi: 10.1172/jci.insight
67. Shimba A, Ikuta K. Control of immunity by glucocorticoids in health and disease. Semin Immunopathol (2020) 42(6):669–80. doi: 10.1007/s00281-020-00827-8
68. Skytthe MK, Graversen JH, Moestrup SK. Targeting of CD163(+) macrophages in inflammatory and malignant diseases. Int J Mol Sci (2020) 21(15):5497. doi: 10.3390/ijms21155497
69. Yang H, Xia L, Chen J, Zhang S, Martin V, Li Q, et al. Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat Med (2019) 25(9):1428–41. doi: 10.1038/s41591-019-0566-4
70. Tokunaga A, Sugiyama D, Maeda Y, Warner AB, Panageas KS, Ito S, et al. Selective inhibition of low-affinity memory CD8(+) T cells by corticosteroids. J Exp Med (2019) 216(12):2701–13. doi: 10.1084/jem.20190738
71. Iorgulescu JB, Gokhale PC, Speranza MC, Eschle BK, Poitras MJ, Wilkens MK, et al. Concurrent dexamethasone limits the clinical benefit of immune checkpoint blockade in glioblastoma. Clin Cancer Res (2021) 27(1):276–87. doi: 10.1158/1078-0432.CCR-20-2291
72. Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB. Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol (2017) 8:49. doi: 10.3389/fphar.2017.00049
73. Colon-Echevarria CB, Ortiz T, Maldonado L, Hidalgo-Vargas MJ, Pérez-Morales J, Aquino-Acevedo AN, et al. Zoledronic acid abrogates restraint stress-induced macrophage infiltration, PDGF-AA expression, and ovarian cancer growth. Cancers (Basel) (2020) 12(9):2671. doi: 10.3390/cancers12092671
74. Batty GD, Russ TC, Stamatakis E, Kivimäki M. Psychological distress in relation to site specific cancer mortality: pooling of unpublished data from 16 prospective cohort studies. Bmj (2017) 356:j108. doi: 10.1136/bmj.j108
75. Lossignol D. A little help from steroids in oncology. J Transl Int Med (2016) 4(1):52–4. doi: 10.1515/jtim-2016-0011
76. Petrella A, Ercolino SF, Festa M, Gentilella A, Tosco A, Conzen SD, et al. Dexamethasone inhibits TRAIL-induced apoptosis of thyroid cancer cells via bcl-xL induction. Eur J Cancer (2006) 42(18):3287–93. doi: 10.1016/j.ejca.2006.07.018
77. Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol Cell Biol (2001) 21(3):952–65. doi: 10.1128/MCB.21.3.952-965.2001
78. Gokon Y, Fujishima F, Taniyama Y, Ishida H, Yamagata T, Sawai T, et al. Glucocorticoid receptor and serum-and glucocorticoid-induced kinase-1 in esophageal adenocarcinoma and adjacent barrett's esophagus. Pathol Int (2020) 70(6):355–63. doi: 10.1111/pin.12922
79. Ai R, Sun Y, Guo Z, Wei W, Zhou L, Liu F, et al. NDRG1 overexpression promotes the progression of esophageal squamous cell carcinoma through modulating wnt signaling pathway. Cancer Biol Ther (2016) 17(9):943–54. doi: 10.1080/15384047.2016.1210734
80. Sorrentino G, Ruggeri N, Zannini A, Ingallina E, Bertolio R, Marotta C, et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat Commun (2017) 8:14073. doi: 10.1038/ncomms14073
81. Caratti B, Fidan M, Caratti G, Breitenecker K, Engler M, Kazemitash N, et al. The glucocorticoid receptor associates with RAS complexes to inhibit cell proliferation and tumor growth. Sci Signal (2022) 15(726):eabm4452. doi: 10.1126/scisignal.abm4452
82. Ma R, Zhang W, Tang K, Zhang H, Zhang Y, Li D, et al. Switch of glycolysis to gluconeogenesis by dexamethasone for treatment of hepatocarcinoma. Nat Commun (2013) 4(1):2508. doi: 10.1038/ncomms3508
83. Liu X, Cui H, Niu H, Wang L, Li X, Sun J, et al. Hydrocortisone suppresses early paraneoplastic inflammation and angiogenesis to attenuate early hepatocellular carcinoma progression in rats. OncoTargets Ther (2019) 12:9481. doi: 10.2147/OTT.S224618
84. Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, et al. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of ras signaling. J Clin Invest (2007) 117(6):1605–15. doi: 10.1172/JCI30724
85. Hoppstädter J, Ammit AJ. Role of dual-specificity phosphatase 1 in glucocorticoid-driven anti-inflammatory responses. Front Immunol (2019) 10:1446. doi: 10.3389/fimmu.2019.01446
86. Cirstea IC, Moll HP, Tuckermann J. Glucocorticoid receptor and RAS: an unexpected couple in cancer. Trends Cell Biol (2022) 8924(22)00253-7. doi: 10.1016/j.tcb.2022.11.002
87. Karmakar S, Jin Y, Nagaich AK. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) α and activator protein 1 (AP1) in dexamethasone-mediated interference of ERα activity. J Biol Chem (2013) 288(33):24020–34. doi: 10.1074/jbc.M113.473819
88. Miranda TB, Voss TC, Sung MH, Baek S, John S, Hawkins M, et al. Reprogramming the chromatin landscape: interplay of the estrogen and glucocorticoid receptors at the genomic level. Cancer Res (2013) 73(16):5130–9. doi: 10.1158/0008-5472.CAN-13-0742
89. Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, et al. Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell (2016) 165(3):593–605. doi: 10.1016/j.cell.2016.02.067
90. Claessens F, Joniau S, Helsen C. Comparing the rules of engagement of androgen and glucocorticoid receptors. Cell Mol Life Sci (2017) 74(12):2217–28. doi: 10.1007/s00018-017-2467-3
91. Abduljabbar R, Negm OH, Lai CF, Jerjees DA, Al-Kaabi M, Hamed MR, et al. Clinical and biological significance of glucocorticoid receptor (GR) expression in breast cancer. Breast Cancer Res Treat (2015) 150(2):335–46. doi: 10.1007/s10549-015-3335-1
92. West DC, Pan D, Tonsing-Carter EY, Hernandez KM, Pierce CF, Styke SC, et al. GR and ER coactivation alters the expression of differentiation genes and associates with improved ER+ breast cancer outcome. Mol Cancer Res (2016) 14(8):707–19. doi: 10.1158/1541-7786.MCR-15-0433
93. Severson TM, Kim Y, Joosten SEP, Schuurman K, van der Groep P, Moelans CB, et al. Characterizing steroid hormone receptor chromatin binding landscapes in male and female breast cancer. Nat Commun (2018) 9(1):482. doi: 10.1038/s41467-018-02856-2
94. Cairat M, Al Rahmoun M, Gunter MJ, Heudel PE, Severi G, Dossus L, et al. Use of systemic glucocorticoids and risk of breast cancer in a prospective cohort of postmenopausal women. BMC Med (2021) 19(1):186. doi: 10.1186/s12916-021-02004-6
95. Pan D, Kocherginsky M, Conzen SD. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res (2011) 71(20):6360–70. doi: 10.1158/0008-5472.CAN-11-0362
96. Chen Z, Lan X, Wu D, Sunkel B, Ye Z, Huang J, et al. Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat Commun (2015) 6:8323. doi: 10.1038/ncomms9323
97. Noureddine LM, Trédan O, Hussein N, Badran B, Le Romancer M, Poulard C. Glucocorticoid receptor: A multifaceted actor in breast cancer. Int J Mol Sci (2021) 22(9):4446. doi: 10.3390/ijms22094446
98. Isikbay M, Otto K, Kregel S, Kach J, Cai Y, Vander Griend DJ, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer (2014) 5(2):72–89. doi: 10.1007/s12672-014-0173-2
99. Shah N, Wang P, Wongvipat J, Karthaus WR, Abida W, Armenia J, et al. Regulation of the glucocorticoid receptor via a BET-dependent enhancer drives antiandrogen resistance in prostate cancer. Elife (2017) 6:e27861. doi: 10.7554/eLife.27861
100. Alyamani M, Li J, Patel M, Taylor S, Nakamura F, Berk M, et al. Deep androgen receptor suppression in prostate cancer exploits sexually dimorphic renal expression for systemic glucocorticoid exposure. Ann Oncol (2020) 31(3):369–76. doi: 10.1016/j.annonc.2019.12.002
101. Li J, Alyamani M, Zhang A, Chang KH, Berk M, Li Z, et al. Aberrant corticosteroid metabolism in tumor cells enables GR takeover in enzalutamide resistant prostate cancer. Elife (2017) 6:e20183. doi: 10.7554/eLife.20183
102. Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab (2010) 21(5):315–24. doi: 10.1016/j.tem.2010.01.002
103. Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell (2013) 155(6):1309–22. doi: 10.1016/j.cell.2013.11.012
104. Sahu B, Laakso M, Pihlajamaa P, Ovaska K, Sinielnikov I, Hautaniemi S, et al. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res (2013) 73(5):1570–80. doi: 10.1158/0008-5472.CAN-12-2350
105. Cleutjens CB, Steketee K, van Eekelen CC, van der Korput JA, Brinkmann AO, Trapman J. Both androgen receptor and glucocorticoid receptor are able to induce prostate-specific antigen expression, but differ in their growth-stimulating properties of LNCaP cells. Endocrinology (1997) 138(12):5293–300. doi: 10.1210/endo.138.12.5564
106. Cephus JY, Stier MT, Fuseini H, Yung JA, Toki S, Bloodworth MH, et al. Testosterone attenuates group 2 innate lymphoid cell-mediated airway inflammation. Cell Rep (2017) 21(9):2487–99. doi: 10.1016/j.celrep.2017.10.110
107. Laffont S, Blanquart E, Savignac M, Cenac C, Laverny G, Metzger D, et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J Exp Med (2017) 214(6):1581–92. doi: 10.1084/jem.20161807
108. Lundholm K, Gelin J, Hyltander A, Lönnroth C, Sandström R, Svaninger G, et al. Anti-inflammatory treatment may prolong survival in undernourished patients with metastatic solid tumors. Cancer Res (1994) 54(21):5602–6.
109. Rutz HP. Effects of corticosteroid use on treatment of solid tumours. Lancet (2002) 360(9349):1969–70. doi: 10.1016/S0140-6736(02)11922-2
110. Hughes MA, Parisi M, Grossman S, Kleinberg L. Primary brain tumors treated with steroids and radiotherapy: low CD4 counts and risk of infection. Int J Radiat Oncol Biol Phys (2005) 62(5):1423–6. doi: 10.1016/j.ijrobp.2004.12.085
111. Karmakar S, Jin Y, Nagaich AK. Interaction of glucocorticoid receptor (GR) with estrogen receptor (ER) alpha and activator protein 1 (AP1) in dexamethasone-mediated interference of ERalpha activity. J Biol Chem (2013) 288(33):24020–34. doi: 10.1074/jbc.M113.473819
112. Dietrich J, Rao K, Pastorino S, Kesari S. Corticosteroids in brain cancer patients: benefits and pitfalls. Expert Rev Clin Pharmacol (2011) 4(2):233–42. doi: 10.1586/ecp.11.1
113. Herr I, Pfitzenmaier J. Glucocorticoid use in prostate cancer and other solid tumours: implications for effectiveness of cytotoxic treatment and metastases. Lancet Oncol (2006) 7(5):425–30. doi: 10.1016/S1470-2045(06)70694-5
114. Zhang C, Wenger T, Mattern J, Ilea S, Frey C, Gutwein P, et al. Clinical and mechanistic aspects of glucocorticoid-induced chemotherapy resistance in the majority of solid tumors. Cancer Biol Ther (2007) 6(2):278–87. doi: 10.4161/cbt.6.2.3652
115. Cook AM, McDonnell AM, Lake RA, Nowak AK. Dexamethasone co-medication in cancer patients undergoing chemotherapy causes substantial immunomodulatory effects with implications for chemo-immunotherapy strategies. Oncoimmunology (2016) 5(3):e1066062. doi: 10.1080/2162402X.2015.1066062
116. Swildens KX, Sillevis Smitt PAE, van den Bent MJ, French PJ, Geurts M. The effect of dexamethasone on the microenvironment and efficacy of checkpoint inhibitors in glioblastoma: a systematic review. Neurooncol Adv (2022) 4(1):vdac087. doi: 10.1093/noajnl/vdac087
117. Pitter KL, Tamagno I, Alikhanyan K, Hosni-Ahmed A, Pattwell SS, Donnola S, et al. Corticosteroids compromise survival in glioblastoma. Brain (2016) 139(Pt 5):1458–71. doi: 10.1093/brain/aww046
118. Goh JJ, See SJ, Ang E, Ng WH. Vanishing glioblastoma after corticosteroid therapy. J Clin Neurosci (2009) 16(9):1226–8. doi: 10.1016/j.jocn.2008.10.029
119. Drakaki A, Dhillon PK, Wakelee H, Chui SY, Shim J, Kent M, et al. Association of baseline systemic corticosteroid use with overall survival and time to next treatment in patients receiving immune checkpoint inhibitor therapy in real-world US oncology practice for advanced non-small cell lung cancer, melanoma, or urothelial carcinoma. Oncoimmunology (2020) 9(1):1824645. doi: 10.1080/2162402X.2020.1824645
120. Djedovic V, Lee YY, Kollara A, May T, Brown TJ. The two faces of adjuvant glucocorticoid treatment in ovarian cancer. Horm Cancer (2018) 9(2):95–107. doi: 10.1007/s12672-017-0319-0
121. Ishihara R, Arima M, Iizuka T, Oyama T, Katada C, Kato M, et al. Endoscopic submucosal dissection/endoscopic mucosal resection guidelines for esophageal cancer. Dig Endosc (2020) 32(4):452–93. doi: 10.1111/den.13654
122. Cata JP, Jones J, Sepesi B, Mehran RJ, Rodriguez-Restrepo A, Lasala J, et al. Lack of association between dexamethasone and long-term survival after non-small cell lung cancer surgery. J Cardiothorac Vasc Anesth (2016) 30(4):930–5. doi: 10.1053/j.jvca.2016.01.004
Keywords: glucocorticoids, inflammation, steroid hormones, cancer development, corticosteroids, endocrine
Citation: Khadka S, Druffner SR, Duncan BC and Busada JT (2023) Glucocorticoid regulation of cancer development and progression. Front. Endocrinol. 14:1161768. doi: 10.3389/fendo.2023.1161768
Received: 08 February 2023; Accepted: 28 March 2023;
Published: 18 April 2023.
Edited by:
Mauricio Rodriguez-Dorantes, National Institute of Genomic Medicine (INMEGEN), MexicoReviewed by:
Jonathan Wolf Mueller, University of Birmingham, United KingdomKarla Vazquez Santillan, National Institute of Genomic Medicine (INMEGEN), Mexico
Isabel Ben-Batalla, German Cancer Research Center (DKFZ), Germany
Copyright © 2023 Khadka, Druffner, Duncan and Busada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan T. Busada, am9uYXRoYW4uYnVzYWRhQGhzYy53dnUuZWR1