
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 28 February 2023
Sec. Cardiovascular Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1150171
This article is part of the Research Topic Insights in Cardiovascular Endocrinology: 2023 View all 8 articles
 Milan Obradovic1
Milan Obradovic1 Emina Sudar-Milovanovic1
Emina Sudar-Milovanovic1 Zoran Gluvic2Katarina Banjac1
Zoran Gluvic2Katarina Banjac1 Manfredi Rizzo3*Esma R. Isenovic1
Manfredi Rizzo3*Esma R. Isenovic1Cardiometabolic diseases (CMD) are a direct consequence of modern living and contribute to the development of multisystem diseases such as cardiovascular diseases and diabetes mellitus (DM). CMD has reached epidemic proportions worldwide. A sodium pump (Na+/K+-ATPase) is found in most eukaryotic cells’ membrane and controls many essential cellular functions directly or indirectly. This ion transporter and its isoforms are important in the pathogenesis of some pathological processes, including CMD. The structure and function of Na+/K+-ATPase, its expression and distribution in tissues, and its interactions with known ligands such as cardiotonic steroids and other suspected endogenous regulators are discussed in this review. In addition, we reviewed recent literature data related to the involvement of Na+/K+-ATPase activity dysfunction in CMD, focusing on the Na+/K+-ATPase as a potential therapeutic target in CMD.
Cardiometabolic diseases (CMD) are a direct consequence of the modern lifestyle and represent a step forward in the development of multisystem diseases such as cardiovascular diseases (CVD) and diabetes mellitus (DM) (1, 2). The prevalence of CMD achieves epidemic proportion, estimated at approximately 25% at the global level (3, 4). An unhealthy diet combined with sedentary behaviour, smoking, alcohol use and socioeconomic aspects is a substantial risk factor for the development of cluster metabolic disorders, including obesity, hypertension, dyslipidaemia and impaired glucose regulation (5, 6). Aside from prevention, there are numerous therapeutics for CMD treatment on the market, most of which are designed to improve insulin action and lipid-lowering. However, the dramatic increase in the prevalence of CMD and the inadequacy of current therapy point to the need for new therapeutic targets.
The sodium/potassium adenosine-triphosphatase (Na+/K+-ATPase) is an essential plasma membrane enzyme that maintains ion homeostasis, cell volume and contractility, electrical signaling, membrane trafficking and vascular tone (7). The Na+/K+-ATPase is the target of several controlling mechanisms. Hormones up-regulate and downregulate Na+/K+-ATPase activity/expression, which primarily comes to the fore in different CMD (8–12). Also, Na+/K+-ATPase functions as a receptor for cardiotonic steroids (CTS), with downstream molecular response affected by CTS concentration. Higher concentrations of CTS (mM range) lead to reverse the inhibition of Na+/K+-ATPase activity, causing a transient cytotoxic effect and, most importantly positive inotropic effect (13). Precisely for this reason, cardiac glycosides have been used for a long time as a drug to strengthen the force of the heartbeat in numerous heart disorders (14). In addition, CTS were among the 200 most frequently prescribed drugs in 2018 year in the USA (15). Particular mechanisms of Na+/K+-ATPase regulation arise after CTS binding to the specific site at α subunit of Na+/K+-ATPase, but at low CTS concentrations (≤ nM) which is insufficient for ion transport inhibition (16). Cell signaling, intracellular Ca2+ oscillations, gene transcription, growth, and proliferation are all activated as a result (17, 18). Since its discovery, Na+/K+-ATPase has been the subject of numerous studies, but the regulation mechanism remains unknown.
Given that CMD alters Na+/K+-ATPase activity and/or subunit expression (8, 9, 13, 19), it represents a promising therapeutic target (20, 21). Furthermore, basic and clinical studies show that improving Na+/K+-ATPase function is directly related to improving various pathological conditions of the cardiovascular system (22). In this review, we discussed recent literature data on Na+/K+-ATPase regulation in CMD as a potential target for new approaches to treating these pathologies.
The transmembrane protein, Na+/K+-ATPase transports K+ ions into the cell and Na+ ions out of the cell, and since the process requires transporting ions against their concentration gradients, Na+/K+-ATPase uses the energy derived from hydrolysis of ATP. It is composed of a ∼100 kDa catalytic α subunit, a heavily glycosylated ∼45 kDa β subunit, and a regulatory subunit, often referred to as γ-subunit (∼10 kDa), that belongs to an FXYD group of proteins (Figure 1) (13, 23). The subunits display multiple isoforms, four α subunit isoforms and three β subunit isoforms, which can assemble in 12 different Na+/K+-ATPase isozymes with tissue-specific different functional activities. Seven tissue and Na+/K+-ATPase isozymes are also specific γ-subunit isoforms (23). α-subunit has a large intracellular domain with ATP-binding and phosphorylation site, a transmembrane domain composed of ten segments responsible for ion transport, and an extracellular domain with binding sites for cardiac steroids (24). β-subunit has an essential role in the α-subunit assembly, and additionally, it increases α-subunit stability and modulates its affinity of ions (24). On the other hand, γ-subunits are tissue-specific and act as Na+/K+-ATPase modulatory proteins. Whereas heart tissue specific γ-subunit is phosholemman (PLM), which disinhibits Na+/K+-ATPase in its phosphorylated form, increasing Na+ efflux (25). The γ-subunit influences the affinity of the Na+/K+-ATPase for ions and ATP, in addition to the transport and stabilization properties (26).
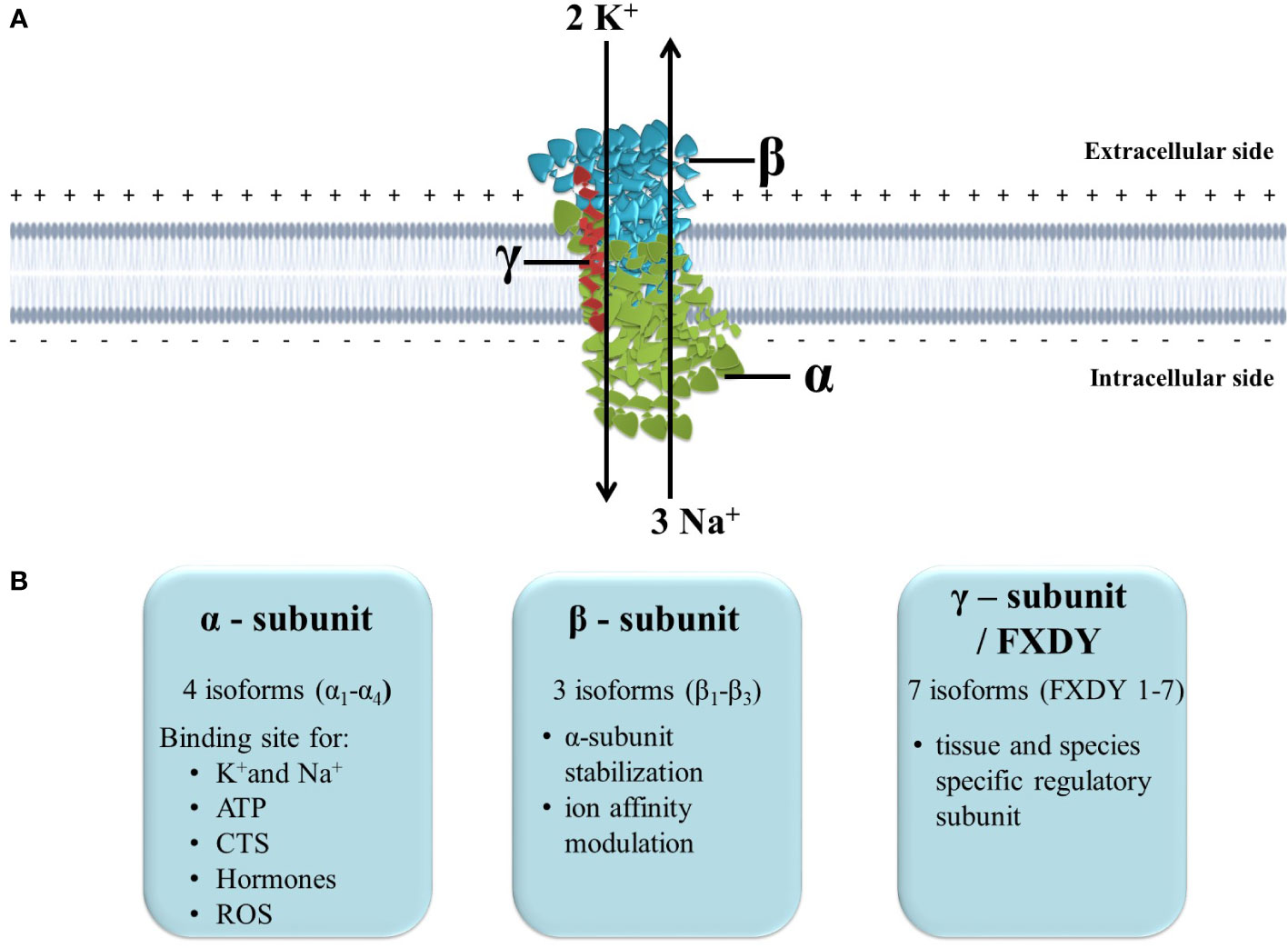
Figure 1 The structure and function of Na+/K+-ATPase. (A) Position of Na+/K+-ATPase in the plasma membrane and maintenance of ion homeostasis; (B) Subunits isoforms and specific function. ATP, adenosine triphosphate; CTS, cardiotonic steroids; ROS, reactive oxygen species.
Normal functioning Na+/K+-ATPase in the plasma membrane is vital for mammalian cells since it maintains Na+ and K+ electrochemical gradients across the plasma membrane. Many ion transporters and channels utilize these chemical gradients to transport ions, minerals, sugar and amino acids (23). Therefore it is involved in ion homeostasis regulation, intracellular pH regulation, Ca2+ signaling, fluid and volume homeostasis regulation and renal salt reabsorption (13). Additionally, the ion gradient Na+/K+-ATPase creates across the plasma membrane is essential for generating action potential that sustains cardiac muscle contraction and neuronal communication. Finally, novel studies confirm that Na+/K+-ATPase also acts as a signal transducer since it is a cardiotonic steroids receptor and can activate intracellular protein kinases (23, 27).
In the cardiovascular system, Na+/K+-ATPase is important in regulating vascular tone and cardiac remodelling (28). Animal hearts express the α1 isoform dominantly or in combination with the α2 and/or α3 isoform. Considering rodent adult cardiomyocytes, they mainly express the α1 isoform and α2 isoform (<25%) (29), while human adult cardiomyocytes have all three isoforms expressed (13). Since α1 and α2 isoforms are present in different ratios and with differential distribution in cardiac cells, it has been suggested that they have different functions. Both α1 and α2 isoforms in the heart have physical and functional associations with Na+/Ca2+ exchangers, thus favouring Ca2+ influx rather than Ca2+ efflux, which leads to increased contractility (29–31). Moreover, the α2 isoform is approximately five times more present in the T-tubules, where the Na+/Ca2+ exchanger is located (32). At the same time, data indicate that the α1 isoform in the heart regulates cell growth and survival via maintaining a global pool of Na+ throughout the cell. On the other hand, the α2 isoform regulates Ca2+ concentration in cells via regulating local Na+ and Ca2+ concentrations in sarcolemma/sarcoplasmic reticulum microdomains, thereby regulating contractility and hypertrophy (29–31). Furthermore, α2 isoform overexpression has a protective effect from pressure overload caused by cardiac dysfunction; thus, this isoform probably regulates cardiac pathological hypertrophy (29, 30). Numerous studies have also implicated aberrant Na+/K+-ATPase and PLM expression, reduction in Na+/K+-ATPase activity and subsequent increase in intracellular Na+ and Ca2+ concentrations in diseased heart (31, 33). Chronic increase in intracellular Na+ and Ca2+ concentrations lead to maladaptive cardiac hypertrophy and arrhytmogenesis (31). Additionally, several pathophysiological conditions such as insulin resistance (IR), obesity and hypertension are associated with defects in normal Na+/K+-ATPase function (8, 9, 13, 19).
Na+/K+-ATPase regulation is a crucial and highly complex process on various levels (Figure 2). Concerning tissue-specific mechanisms of Na+/K+-ATPase regulation, there are local and systemic regulatory mechanisms. Intracellular and extracellular Na+ and K+ concentrations are the most important local regulatory mechanism, along with hypoxia, purines, oxidative stress, pH, nitric oxide and ATP, that influence activity of Na+/K+-ATPase (34, 35). On the other hand, hormones are major factors in the systemic regulation of Na+/K+-ATPase. Hormones regulate Na+/K+-ATPase cell surface expression and activity, provoking protein kinase phosphorylation (35–41). Nonetheless, translocation from intracellular compartments to the plasma membrane is controlled by α-subunit phosphorylation, a type of posttranslational modification (12, 42, 43). Furthermore, the α subunit contains several serine, threonine, and tyrosine residues that can be phosphorylated by various kinases, influencing Na+/K+-ATPase activity (44). Besides phosphorylation, Na+/K+-ATPase can be modified via glutathionylation, which causes its inactivation (35). Additionally, except for regulating Na+/K+-ATPase cell surface expression, hormones can up-regulate α and β gene transcription, which determines the total cell content of Na+/K+-ATPase subunits along with the degradation rate [9]. Insulin, as one of the most potent regulators of Na+/K+-ATPase, increased its activity and translocation of subunits to the cell membrane via phosphatidylinositol 3-kinase (PI3K), protein kinase C (PKC), and extracellular signal-regulated kinases 1 and 2 (ERK1/2) (42, 43, 45). In contrast, leptin decreased Na+/K+ATPase activity in the rat kidney via the PI3K pathway (46). Angiotensin II (Ang II), insulin-like growth factor 1 (IGF-1) and estradiol stimulate Na+/K+-ATPase activity and gene expression in primary cultured rat vascular smooth muscle cells via PI3K, protein kinase B (Akt), and ERK1/2 (9, 37, 38). Estradiol also increased Na+/K+-ATPase activity and expression in the heart of rats via signaling pathways that involve stimulation of insulin receptor substrate 1 (IRS-1)/PI3K/Akt/ERK1/2 and suppression of Ang II receptor type 1, Rho A, and Rho-associated kinase cascade (8, 47). Furthermore, Ang II inhibits IGF-1-stimulated Na+/K+-ATPase activity in VSMC via PI3K/Akt signaling (37), whereas IGF-1 overexpression reduced Ang II production and oxidative stress in mouse cardiomyocytes (48). Several studies have shown that the signaling pathways that regulate IGF-1 and estradiol are crosslinked, implying that these hormones may have a combined effect on the regulation of Na+/K+-ATPase (49). This interactive effect of hormones indicates a complex mechanism of Na+/K+-ATPase regulation in vivo where tonic hormone release simultaneously influences Na+/K+-ATPase and balances its activity.
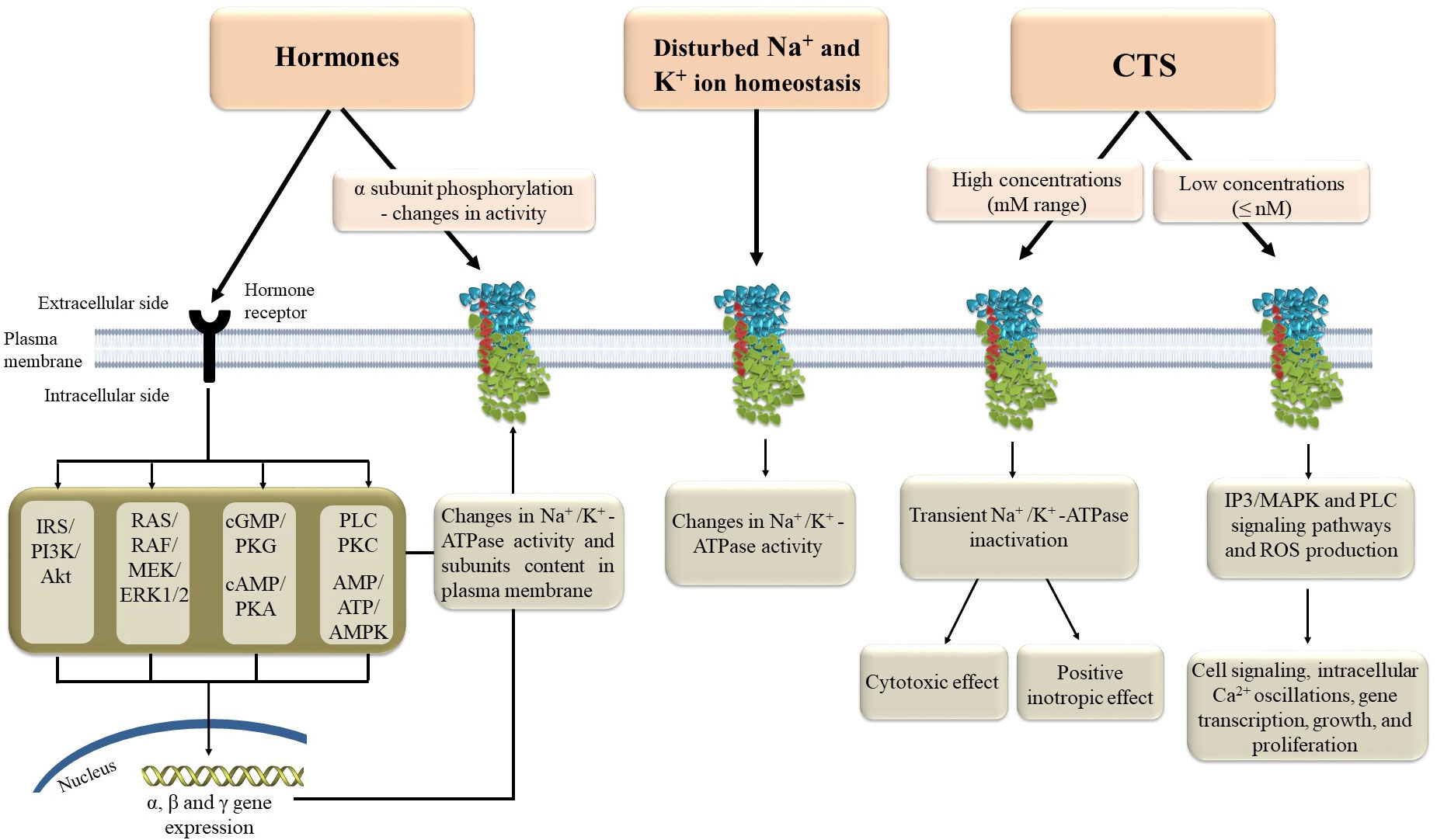
Figure 2 Molecular mechanisms of Na+/K+-ATPase regulation. AMP - adenosine monophosphate, AMPK – AMP-activated protein kinase, ATP - adenosine triphosphate, CTS – cardiotonic steroids, ERK1/2 – extracellular signal-regulated kinases 1 and 2, IP3 – inositol triphosphate, MAPK – mitogen-activated protein kinase, PKA, protein kinase A; PKC, protein kinase C; PKG, cGMP-activated protein kinase; PLC, phospholipase C; ROS, reactive oxygen species.
New researches also confirm that γ-subunits or FXYD proteins can modulate Na+/K+-ATPase binding and function via protein-protein interactions and consequent post-translational modifications (50). These effects, along with the FXYD proteins expression, are also regulated by hormones (35). Since FXYD proteins are tissue-specific, post-translational modifications fine-tune Na+/K+-ATPase binding and function according to tissue-specific needs (32). In addition, FXYD proteins can also be substrates for post-translational modulation, which modifies their regulatory function (32). Finally, cardiac steroids bind to the extracellular domain of α- subunit and stabilize and modify Na+/K+-ATPase to support its different functions. Therefore, cardiac steroids can have a positive inotropic effect through Na+/K+-ATPase inhibition and the effect of activation of different signal transduction pathways mediated by Na+/K+-ATPase. In comparison, higher cardiac steroids concentrations induce Na+/K+-ATPase inhibition, while sub-inhibitory concentrations induce activation of mitogen-activated protein kinase signal cascades, mitochondrial reactive oxygen species (ROS) production, and the phospholipase C signaling pathway (23). In addition, cardiac steroids can modulate Na+/K+-ATPase sensitivity to different regulatory proteins (51).
The emergence of CMD risk factors is unpredictable and dynamic. Cardiovascular and metabolic disruptions most often cause CMD, and a long-lasting CMD, including DM and different cardiovascular pathologies, are the main causes of death worldwide. The incidence and prevalence of CMD have increased in parallel with the rise in obesity, DM and hypertension (52). Since mortality has reduced during the past few years in high-income nations but increased in low- and middle-income countries, increases in the prevalence of CMD, such as hypertension, obesity, dyslipidaemia and DM, and their major risk factors have not been uniform (53). Cardiometabolic disorders can occur substantially before the clinical appearance of diseases. CMD-related complications are complex and multifactorial disorders, but in most cases, preventable. Many factors, such as changes in living environments, unhealthy diets, specific lifestyles, physical inactivity, and genetic and epigenetic factors, may be involved in CMD development (52). Early and accurate predictors of CMD are of great importance since the delay or prevention of morbidity is achievable via pharmacological treatments and lifestyle modulation (54–56). Timely treatment of these detrimental factors is important in their progressive and ultimate transformation into more complicated CMD. Novel mechanisms implicated in the development of CMD may open up new prognostic and therapeutic avenues.
Over the last decade, it has been generally recognized that genetic mutations are engaged in different CMD (52), including hypertension (57), impaired lipid metabolism and lipotoxicity (58). In addition to defective genes, frequent inflammation is also one of the pathological driving forces involved in various CMD (59). Pathophysiological factors, such as proinflammatory cytokines: resistin, interleukin (IL)-6, tumour necrosis, factor-alpha (TNF-α), and IL1β, as well as interactions among them and also with the molecules of the insulin signaling cascade, are involved in IR occurrence (52, 59). In addition to dysfunctional insulin signaling, proinflammatory cytokines are implicated in impaired endothelial function and dyslipidemia (52), both involved in CMD.
Many cardiometabolic complications, including obesity, DMT2, hyperlipidemia, dyslipidemia, nephropathy, hypertension, and nonalcoholic fatty liver disease, are closely interrelated (52). Among the major causes of CMD and related complications is DM. Obesity usually predisposes to DM, especially central obesity per se, and is associated with severe comorbidities, influencing every system of organs, particularly affecting cardiometabolic comorbidities (60). Moreover, obesity is associated with an increased risk for CVD independently from other CVD risk factors and is also considered a modulator of other CVD risk factors. Thus, treating obesity should be the most important management strategy to reduce cardiometabolic risk (60, 61). Furthermore, it is considered a chronic metabolic disorder associated with chronic low-grade inflammation and results in marked alterations of proinflammatory cytokines, adipokines, and other molecules affecting CVS function and CMD development. In an observational cohort study in which 1.3 million overweight or obese adults participated, four commonly observed cardiac risk factors were found: the prevalence of hypertension, prediabetes, decreased HDL and elevated TG. An earlier study by National Health and Nutrition Examination Survey (NHANES), which included individuals with diabetes, showed that 52% of adults overweight and 32% of adults with obesity had no cardiac risk factors or only one, suggesting that different phenotypes of obesity, such as subcutaneous versus abdominal fat, may pose various health risks (62, 63). The authors concluded that being overweight or obese increases cardiometabolic risk, but the quantity and developed cardiac risk factors differed substantially by age, even among participants with morbid obesity (62, 63).
Furthermore, vitamin D effects on insulin sensitivity may be compromised in obese individuals (64), and in these individuals, hyperinsulinemia and/or IR may be responsible for reduced vitamin D concentration, which underscores this paradigm (52). In addition, alterations at a hormonal, inflammatory and endothelial level associated with obesity induce stimulation of several factors contributing to the hypertensive state and development of CVD and cardiovascular morbidity. The most recognized factors connecting obesity and hypertension are impaired sodium homeostasis, endocrine alterations, altered hemodynamics, autonomic nervous system imbalance, renal dysfunction, oxidative stress and inflammation, and vascular injury (65).
The development of complications and increased mortality influenced by obesity indirectly affect other risk factors such as IR, dyslipidemia, and hypertension (66). In addition, an important link between obesity and CVD development is dyslipidemia (67). Dyslipidemia occurs when the levels of triglyceride (TG), small dense LDL (sdLDL) particles, very low-density lipoprotein (VLDL) cholesterol and total cholesterol are increased, while high-density lipoprotein (HDL) cholesterol levels are decreased (68, 69). Persons with visceral adiposity usually have indicators for CVD development, such as an increased ratio of apolipoprotein (Apo) B to Apo A1 (70), a rise in sdLDL particles (71), and low HDL cholesterol level (72). In the last decade, dyslipidemia occurring due to IR and obesity has been recognized as “metabolic dyslipidemia” (73). Its main features are increased levels of TG accompanied by decreased HDL cholesterol level, while LDL cholesterol level could be mildly increased or optimal, even though the number of LDL particles (LDL-P) can also be elevated. Also, atherogenic lipoproteins, such as lipoprotein(a) (Lp (a)), are critical in the development of various CVD (74), leading to CMD (52). In addition, endothelial and vascular dysfunction caused by obesity leads to CVD (75). Furthermore, obesity predisposes to heart disease through various mechanisms, including causing structural and functional changes in the heart, affecting heart morphology and leading to pathological heart hypertrophy, characterized by cardiomyocyte enhancement and increased protein synthesis (76, 77). However, it is not accompanied by a rise in capillaries supplying the myocardium, finally leading to ischemic changes in the myocardium (78).
Among others, in patients with CMD, response to ischemic insults may also be impaired. Patients with cardiovascular risk, especially patients with hypertension and diabetes, exhibited an abnormal reactive hyperemic response to ischemic insults, which are associated with myocardial infarction (52, 59).
Many difficult problems must be solved to improve CMD diagnosis, prognosis, therapy, and management. Cardiometabolic risks are a complex group of disease entities, and risk assessment, prediction, and management are also difficult because the underlying causes that promote or precipitate cardiac risk factors in these metabolic diseases are unknown.
Altered Na+/K+-ATPase activity/expression is the basis for vascular complication and cardiac dysfunction in different CMD (Figure 3) (8, 9, 19, 79–82). Decreased Na+/K+-ATPase activity and high concentrations of Na+ in cytosol lead to impaired myocardial contractility in advanced heart failure (83). The link between CMD and altered Na+/K+-ATPase activity is somewhat predictable given that CMD causes changes in hormone levels, most notably insulin, insulin-like growth factor 1, angiotensin II (Ang II), estradiol, and leptin, all of which are potent regulators of the Na+/K+-ATPase (12, 49). The function of Na+/K+-ATPase is impaired at different levels of regulation in hearts, aorta and erythrocytes in human and animal models of induced obesity, insulin resistance and hypertension (Table 1) (8, 19, 84–92, 94–101, 104–107). It has been demonstrated that leptin reduces Na+/K+-ATPase activity in fibroblasts (108), which may be important in the obese state frequently associated with hyperleptinemia. Evidence suggests that long-term activation of Na+/K+-ATPase signaling may promote cardiac fibrosis and the development of heart dysfunction (109–113). Furthermore, using Na+/K+-ATPase signaling antagonists, such as pNaKtide, has shown promise in reducing organ fibrosis (109, 113). We also found that a high-fat diet induces obesity and IR in rats, resulting in decreased activity and α1 and α2 subunits of expression of Na+/K+-ATPase in cardiac tissue, which is accompanied by heart hypertrophy (8, 76). In addition, decreased Na+/K+-ATPase activity is detected in erythrocytes of obese, IR and DM patients (19, 102, 103, 114). Increased Ang II and (ROS) inhibit Na+/K+-ATPase by glutathionylation of β1 subunit that may have pathophysiological effects in the cardiovascular system of obese and DM patients (115–117). The activity of cardiac Na+/K+-ATPase is decreased in hypertensive male rats (91). Also, earlier studies reported altered expression of α1 and α2 subunits of Na+/K+-ATPase in the aorta and heart of hypertensive rats (118, 119). Mice with an ouabain-resistant α2 subunit of Na+/K+-ATPase are protected from hypertension development after treatment with adrenocorticotropic hormone (120, 121). Genetic silencing of the α2 subunit of the Na+/K+-ATPase decreased pathological heart hypertrophy and cardiac remodeling (93, 122). The Na+/K+-ATPase signaling is activated with ROS and CTS (33, 123). However, in pathophysiological conditions such as obesity and related disorders, increased ROS and CTS promote Na+/K+-ATPase signaling, leading to the overproduction of ROS and inflammatory markers creating an oxidant amplification loop that consequently alters the metabolic profile (124). Recent research reveals an important role of Na+/K+-ATPase in autosis, that is characterized as an autophagy-dependent non-apoptotic form of cell death in different (125, 126). The increased interaction of Na+/K+-ATPase with the autophagy protein Beclin 1 was detected in ischemic conditions of hearts (127). Further studies are needed to enhance our knowledge of Na+/K+-ATPase in oxidant amplification and autosis, which may be a target option for CMD treatment.
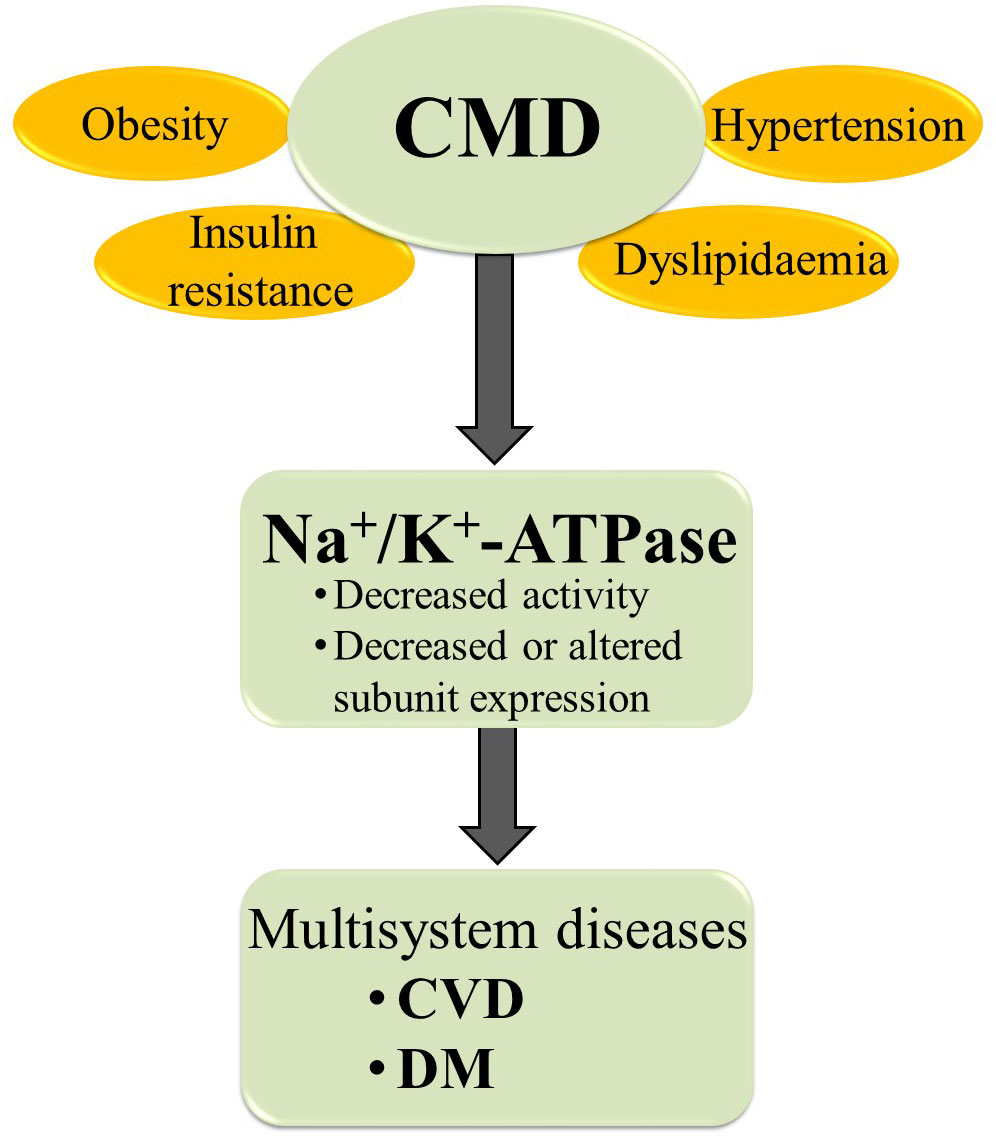
Figure 3 The relationships between CMD and Na+/K+-ATPase. CMD, cardiometabolic diseases; CVD, cardiovascular diseases; DM, diabetes mellitus.
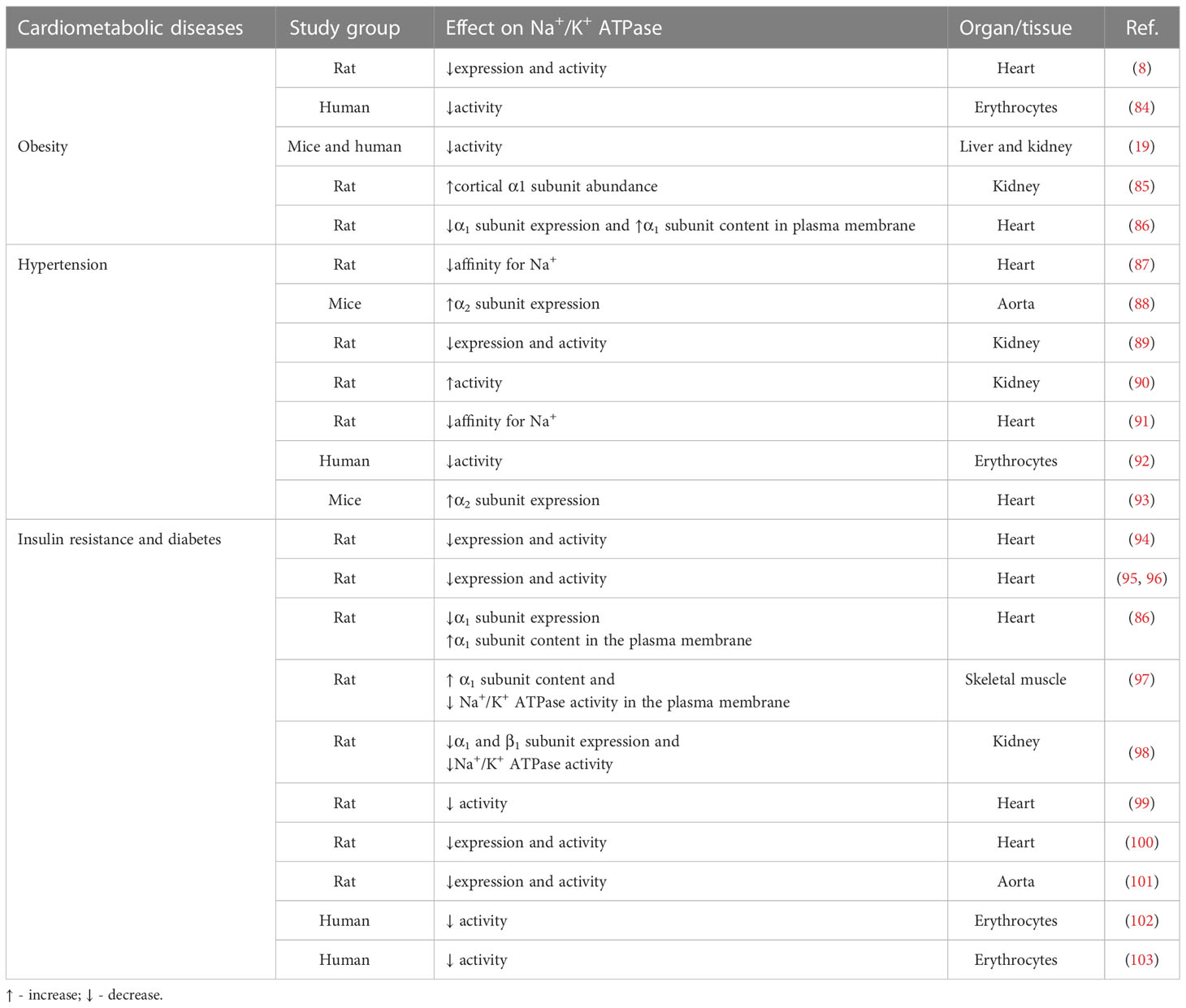
Table 1 The link between cardiometabolic diseases and Na+/K+-ATPase activity/expression.
Because of its critical role in numerous cellular processes that extrapolate to overall body function, the Na+/K+-ATPase is a promising drug target. Even though Na+/K+-ATPase was studied decades ago, the mechanism of Na+/K+-ATPase regulation is very complex and is still not fully understood. An important issue in Na+/K+-ATPase regulation is balancing its activity and its function as a receptor through which signaling pathways are activated.
Since the discovery of Na+-K+-ATPase, it has been evident that modulation of its activity could serve as a pharmacology and therapeutic target (128). According to this, Na+/K+-ATPase activity inhibition can be promoted by various agents (i.e. endogenous and exogenous cardiac steroids), but it can also be seen in different pathologies, such as heart failure, with a significant decrease (40%) in myocardial Na+/K+-ATPase concentration (14, 129–131). Insulin and β adrenergic agonists are important in increasing the amount of Na+/K+-ATPase, which promotes K+ transport into cells (131, 132). Because Na+/K+-ATPase is a non-specific receptor for ROS, the Na+/K+-ATPase-Src oxidant amplification loop is important in the ageing process, obesity, and atherosclerotic CVD (123, 133).
The pharmacological mechanism of CTS cardiovascular effects is based on the inhibition of Na+/K+-ATPase, followed by an increase in intracellular Ca2+ concentration, and then the promotion of positive inotropic and negative chronotropic effects (134, 135). CTS are classified as endogenous or exogenous (cardiac glycoside) based on their source. Endogenous CTS functions in mammals as endogenous digitalis-like factors (135). Among the most extensively studied endogenous CTS are cardenolides (ouabain and digoxin) and bufadienolides (marinobufagenin, telocinobufagin, and 19-Norbufalin) (14, 136). Marinobufagenin and its reduced form, telocinobufagin, were found in the bodily fluids of patients suffering from myocardial infarction, acute renal failure, end-stage renal disease, and heart failure (137–141). Patients with hypertension and pregnant women with preeclampsia had higher ouabain levels (142, 143). Digitalis lanata and Digitalis purpurea are the primary sources of cardiac glycosides (134). Digitoxin, digoxin, lantoside C, and strophanthin K are clinical preparations in use (135).
A disturbed transarcolemmal Na+ gradient characterizes ventricular wall hypertrophy and dilation (144, 145). Increased intracellular Na+ content inhibits Ca2+ mitochondrial uniporter/exchanger function, causing the mitochondria to become metabolically exhausted due to an ATP supply-demand mismatch (146). Furthermore, mitochondrial dysfunction promotes the production of ROS (147). Ouabain, a cardiotonic glycoside, binds to the subunit and inhibits Na+/K+-ATPase (148). Digoxin and digitoxin inhibit the Na+/K+-ATPase directly (149). Such Na+/K+-ATPase inhibition in the myocardium causes an increase in K+ efflux at the same time as intracellular Na+ accumulation, resulting in decreased Na+/Ca2+ channel exchanger activity and an increase in the sarcoplasmic reticulum and cytosolic Ca2+ content in cardiomyocytes (150). Furthermore, digitalis glycosides attenuate Ca2+ influx in cells (13). The net effect is increased intracellular Ca2+, which strengthen heart contractility (129). Furthermore, cardiac glycosides favour a longer atrioventricular node refractory period and sinoatrial depression (both beneficial in atrial fibrillation), an increase in cardiomyocyte automatism (which promotes ventricular arrhythmogenic foci), and a decrease in atrioventricular impulse conduction (151). Digitalis additionally slows heart rate through vagal activation (152). Digoxin is now used to treat persistent heart failure symptoms in patients already receiving modern therapy and control heart rate in patients with atrial fibrillation and heart failure, but it does not affect mortality rates (151–153). Other pharmacological agents, in addition to cardiotonic glycosides, influence Na+/K+-ATPase activity. Diuretic-induced K+ loss and secondary hyperaldosteronism associated with heart failure reduce myocardial Na+/K+-ATPase activity (154), whereas angiotensin-converting enzyme inhibitors (ACEi) and spironolactone may stimulate myocardial Na+/K+-ATPase activity (130, 155). Aside from plant-derived cardiotonic glycosides, endogenous vertebrate-derived aglycones such as bufalin and marinobufagenin, whose production in the adrenals and possibly hypothalamus is primarily under humoral control (ACTH, Ang II) are also detected as Na+/K+-ATPase inhibitors (156–158). Subnanomolar concentrations of plant- and vertebrate-derived glycosides have been found in various diseases such as hypertension (142), renal failure (159), and atherosclerotic CVD (160, 161). In addition to the beneficial roles of endogenous cardiotonic steroids in heart contractility, heart rate control, natriuresis, and blood pressure regulation, chronic exposure causes deleterious effects such as ventricular and vascular wall remodelling, myocardial fibrosis, and arrhythmia risks (14). Exogenous CTS (digoxin, digitoxin) is intended for patients with heart failure with reduced ejection fraction and AF with rapid ventricular rate, especially if previously approved therapy (diuretics, angiotensin-converting enzyme inhibitors (ACEI)/angiotensin receptor blockers, -blockers, and aldosterone receptor antagonists) fails (162–167). Digoxin improves cardiac function and prognosis, and lowers hospitalization rates in HF patients but has no effect on all-cause mortality (168, 169). In contrast, the cardiovascular remodeling caused by long-term CTS exposure promotes the development of cardiac fibrosis pro-arrhythmic foci (170). Because of digoxin’s narrow therapeutic range should be used with caution in elderly, malnourished, and hypokalemic patients (167). The possibility of a deleterious effect of concomitantly administered digoxin, the most common type of CTS in clinical use, arises in patients with already elevated levels of endogenous CTS (14, 135). Digitoxin could be used in patients with impaired renal function (167, 171).
Reduced Na+/K+-ATPase activity and expression are detected in chronic kidney-related heart injury (172). Zheng et al. show that targeting the DR extracellular region (897DVEDSYGQQWTYEQR911) of α1 subunit’s Na+/K+-ATPase with DRm217 antibody stimulates Na+/K+-ATPase activities and protects ischemic injury and cardiac remodeling injury in rats (20). β3 adrenoceptor agonist increased Na+/K+-ATPase activity and reduced indices of organ congestion in a rabbit model, suggesting that decreased Na+/K+-ATPase activity may serve as a treatment target in a state of congestive heart failure (21).
The modulation of myocardial Na+/K+-ATPase activity and expression by different exogenous and endogenous cardiac steroids in animal models helps unravel all the molecular mechanisms in which Na+/K+-ATPase are involved. The interventional and dose-tapering studies in humans are necessary to elucidate the beneficial effects and mechanisms of selected cardiac steroids on human hearts.
Because of the specific modulation of Na+/K+-ATPase activity, Na+/K+-ATPase is a very intriguing drug target. The site of Na+/K+-ATPase modulation could be either Na+/K+-ATPase itself or downstream cascade pathways. The identification of pNaKtide as an antagonist of Na+/K+-ATPase signalling was the first step in this direction (123). Further cardiovascular damage could be avoided by inhibiting the Na+/K+-ATPase-Src oxidant amplification cascade (173). Additionally, it is important to assess the activity and expression of Na+/K+-ATPase and post-receptor cascades in distinctive specific and conjoint diseases, such as CMD, and further, evaluate the effects of different associated molecular targets’ inhibition or stimulation in such patients. The relationship between endogenous and exogenous CTS must be thoroughly investigated. Despite the restricted use of glycosides according to current guidelines recommended by specific cardiology associations, detecting some new CTS or elucidating some unknown effects recognizes the CTS as the focus of translational medicine trials.
MO designed and wrote the paper, ESM, KB, and ZG wrote the paper, MR critically revised the paper, and EI designed and critically revised the paper. All authors contributed to the article and approved the submitted version.
This work was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Contract No# 451-03-47/2023-01/200017).
This work is part of the collaboration between the Department of Radiobiology and Molecular Genetics, “VINČA” Institute of Nuclear Sciences - National Institute of the Republic of Serbia, University of Belgrade, Belgrade, Serbia, Clinic for Internal Medicine, Department of Endocrinology and Diabetes, Zemun Clinical Hospital, School of Medicine, University of Belgrade, Belgrade, Serbia, and School of Medicine, Promise Department, University of Palermo, Palermo, Italy.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Powell-Wiley TM, Poirier P, Burke LE, Després JP, Gordon-Larsen P, Lavie CJ, et al. Obesity and cardiovascular disease: A scientific statement from the American heart association. Circulation (2021) 143:e984–e1010. doi: 10.1161/CIR.0000000000000973
2. Stefan N. Causes, consequences, and treatment of metabolically unhealthy fat distribution. Lancet Diabetes Endocrinol (2020) 8:616–27. doi: 10.1016/S2213-8587(20)30110-8
3. Kones R, Rumana U. Cardiometabolic diseases of civilization: history and maturation of an evolving global threat. An update and call to action. Ann Med (2017) 49:260–74. doi: 10.1080/07853890.2016.1271957
4. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 study. J Am Coll Cardiol (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
5. Eduard MS, Julio PF, Alejandra RF. Co-Occurrence of cardiometabolic disease risk factors: Unhealthy eating, tobacco, alcohol, sedentary lifestyle and socioeconomic aspects. Arq Bras Cardiol (2019) 113:710–1. doi: 10.5935/abc.20190213
6. Marques MDC, Pires R, Perdigão M, Sousa L. Patient-centered care for patients with cardiometabolic diseases: An integrative review. J Pers Med (2021) 11:1289. doi: 10.3390/jpm11121289
7. Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem (2002) 71:511–35. doi: 10.1146/annurev.biochem.71.102201.141218
8. Obradovic M, Zafirovic S, Jovanovic A, Milovanovic ES, Mousa SA, Labudovic-Borovic M, et al. Effects of 17beta-estradiol on cardiac Na/K-ATPase in high fat diet fed rats. Mol Cell Endocrinol (2015) 15:46–56. doi: 10.1016/j.mce.2015.08.020
9. Sudar E, Velebit J, Gluvic Z, Zakula Z, Lazic E, Vuksanovic-Topic L, et al. Hypothetical mechanism of sodium pump regulation by estradiol under primary hypertension. J Theor Biol (2008) 251:584–92. doi: 10.1016/j.jtbi.2007.12.023
10. McDonough AA, Brown TA, Horowitz B, Chiu R, Schlotterbeck J, Bowen J, et al. Thyroid hormone coordinately regulates na+-K+-ATPase alpha- and beta-subunit mRNA levels in kidney. Am J Physiol (1988) 254:C323–329. doi: 10.1152/ajpcell.1988.254.2.C323
11. Ewart HS, Klip A. Hormonal regulation of the na(+)-K(+)-ATPase: mechanisms underlying rapid and sustained changes in pump activity. Am J Physiol (1995) 269:C295–311. doi: 10.1152/ajpcell.1995.269.2.C295
12. Al-Khalili L, Yu M, Chibalin AV. Na+,K+-ATPase trafficking in skeletal muscle: insulin stimulates translocation of both alpha 1- and alpha 2-subunit isoforms. FEBS Lett (2003) 536:198–202. doi: 10.1016/S0014-5793(03)00047-4
13. Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front Physiol (2017) 8:371. doi: 10.3389/fphys.2017.00371
14. Pavlovic D. Endogenous cardiotonic steroids and cardiovascular disease, where to next? Cell Calcium (2020) 86:102156. doi: 10.1016/j.ceca.2019.102156
15. Bejček J, Jurášek M, Spiwok V, Rimpelová S. Quo vadis cardiac glycoside research? Toxins (2021) 13:344. doi: 10.3390/toxins13050344
16. Hamlyn JM, Blaustein MP. Endogenous ouabain: Recent advances and controversies. Hypertension (2016) 68:526–32. doi: 10.1161/HYPERTENSIONAHA.116.06599
17. Chomczynski PW, Vires KM. A real-time PCR method to genotype mutant mouse models with altered affinity for cardiotonic steroids on the Na,K-ATPase. PLoS One (2022) 17:. doi: 10.1371/journal.pone.0267348
18. Lan YL, Wang X, Lou JC, Xing JS, Zou S, Yu ZL, et al. Marinobufagenin inhibits glioma growth through sodium pump α1 subunit and ERK signaling-mediated mitochondrial apoptotic pathway. Cancer Med (2018) 7:2034–47. doi: 10.1002/cam4.1469
19. Iannello S, Milazzo P, Belfiore F. Animal and human tissue Na,K-ATPase in obesity and diabetes: A new proposed enzyme regulation. Am J Med Sci (2007) 333:1–9. doi: 10.1097/00000441-200701000-00001
20. Zheng J, Lan P, Meng X, Kang MC, Huang X, Yan X. Na(+)/K(+)-ATPase DR region antibody ameliorated cardiac hypertrophy and fibrosis in rats with 5/6 nephrectomy. Exp Biol Med (Maywood) (2022) 247:1785–94. doi: 10.1177/15353702221108910
21. Fry NAS, Liu C-C, Garcia A, Hamilton EJ, Galougahi KK, Kim YJ, et al. Targeting cardiac myocyte na+-k+ pump function with β3 adrenergic agonist in rabbit model of severe congestive heart failure. Circulation: Heart Failure (2020) 13:e006753. doi: 10.1161/CIRCHEARTFAILURE.119.006753
22. Reiter-Brennan C, Dzaye O, Davis D, Blaha M, Eckel RH. Comprehensive care models for cardiometabolic disease. Curr Cardiol Rep (2021) 23:22. doi: 10.1007/s11886-021-01450-1
23. Gagnon KB, Delpire E. Sodium transporters in human health and disease. Front Physiol (2021) 11:588664. doi: 10.3389/fphys.2020.588664
24. Li Z, Langhans SA. Transcriptional regulators of Na,K-ATPase subunits. Front Cell Dev Biol 26 (2015) 3:66. doi: 10.3389/fcell.2015.00066
25. Boguslavskyi A, Tokar S, Prysyazhna O, Rudyk O, Sanchez-Tatay D, Lemmey HAL, et al. Phospholemman phosphorylation regulates vascular tone, blood pressure, and hypertension in mice and humans. Circulation (2021) 143:1123–38. doi: 10.1161/CIRCULATIONAHA.119.040557
26. Gao C, Li T, Liu J, Guo Q, Tian L. Endothelial functioning and hemodynamic parameters in rats with subclinical hypothyroid and the effects of thyroxine replacement. PloS One (2015) 10:e0131776. doi: 10.1371/journal.pone.0131776
27. Aperia A, Akkuratov EE, Fontana JM, Brismar H. Na+-K+-ATPase, a new class of plasma membrane receptors. Am J Physiol Cell Physiol (2016) 310:C491–5. doi: 10.1152/ajpcell.00359.2015
28. Yan Y, Shapiro JI. The physiological and clinical importance of sodium potassium ATPase in cardiovascular diseases. Curr Opin Pharmacol (2016) 27:43–9. doi: 10.1016/j.coph.2016.01.009
29. Liu L, Wu J, Kennedy DJ. Regulation of cardiac remodeling by cardiac Na(+)/K(+)-ATPase isoforms. Front Physiol (2016) 7:382. doi: 10.3389/fphys.2016.00382
30. Matchkov VV, Krivoi II. Specialized functional diversity and interactions of the Na,K-ATPase. Front Physiol (2016) 7:179. doi: 10.3389/fphys.2016.00179
31. Pavlovic D, Fuller W, Shattock MJ. Novel regulation of cardiac Na pump via phospholemman. J Mol Cell Cardiol (2013) 61:83–93. doi: 10.1016/j.yjmcc.2013.05.002
32. Yap JQ, Seflova J, Sweazey R, Artigas P, Robia SL. FXYD proteins and sodium pump regulatory mechanisms. J Gen Physiol (2021) 153:e202012633. doi: 10.1085/jgp.202012633
33. Srikanthan K, Shapiro JI, Sodhi K. The role of Na/K-ATPase signaling in oxidative stress related to obesity and cardiovascular disease. Molecules (2016) 21:1172. doi: 10.3390/molecules21091172
34. Geering K. FXYD proteins: new regulators of Na-K-ATPase. Am J Physiol Renal Physiol (2006) 290:F241–50. doi: 10.1152/ajprenal.00126.2005
35. Pirkmajer S, Chibalin AV. Chapter ten - hormonal regulation of na+-K+-ATPase from the evolutionary perspective. In: Orlov SN, editor. Current topics in membranes, vol. 83 .Cambridge, MA 02139, United States: Academic Press (2019). p. 315–51.
36. Pirkmajer S, Chibalin AV. Na,K-ATPase regulation in skeletal muscle. Am J Physiol Endocrinol Metab (2016) 311:E1–E31. doi: 10.1152/ajpendo.00539.2015
37. Isenovic ER, Meng Y, Jamali N, Milivojevic N, Sowers JR. Ang II attenuates IGF-1-stimulated na+, k(+)-ATPase activity via PI3K/Akt pathway in vascular smooth muscle cells. Int J Mol Med (2004) 13:915–22. doi: 10.3892/ijmm.13.6.915
38. Isenovic ER, Jacobs DB, Kedees MH, Sha Q, Milivojevic N, Kawakami K, et al. Angiotensin II regulation of the na+ pump involves the phosphatidylinositol-3 kinase and p42/44 mitogen-activated protein kinase signaling pathways in vascular smooth muscle cells. Endocrinology (2004) 145:1151–60. doi: 10.1210/en.2003-0100
39. Dzurba A, Ziegelhoffer A, Vrbjar N, Styk J, Slezak J. Estradiol modulates the sodium pump in the heart sarcolemma. Mol Cell Biochem (1997) 176:113–8. doi: 10.1023/A:1006835214312
40. Liu CG, Xu KQ, Xu X, Huang JJ, Xiao JC, Zhang JP, et al. 17Beta-oestradiol regulates the expression of Na+/K+-ATPase beta1-subunit, sarcoplasmic reticulum Ca2+-ATPase and carbonic anhydrase iv in H9C2 cells. Clin Exp Pharmacol Physiol (2007) 34:998–1004. doi: 10.1111/j.1440-1681.2007.04675.x
41. Palacios J, Marusic ET, Lopez NC, Gonzalez M, Michea L. Estradiol-induced expression of n(+)-K(+)-ATPase catalytic isoforms in rat arteries: gender differences in activity mediated by nitric oxide donors. Am J Physiol Heart Circ Physiol (2004) 286:H1793–1800. doi: 10.1152/ajpheart.00990.2003
42. Al-Khalili L, Kotova O, Tsuchida H, Ehrén I, Féraille E, Krook A, et al. ERK1/2 mediates insulin stimulation of Na(+),K(+)-ATPase by phosphorylation of the alpha-subunit in human skeletal muscle cells. J Biol Chem (2004) 279:25211–8. doi: 10.1074/jbc.M402152200
43. Comellas AP, Kelly AM, Trejo HE, Briva A, Lee J, Sznajder JI, et al. Insulin regulates alveolar epithelial function by inducing Na+/K+-ATPase translocation to the plasma membrane in a process mediated by the action of akt. J Cell Sci (2010) 123:1343–51. doi: 10.1242/jcs.066464
44. Mohan S, Tiwari MN, Biala Y, Yaari Y. Regulation of neuronal Na(+)/K(+)-ATPase by specific protein kinases and protein phosphatases. J Neurosci (2019) 39:5440–51. doi: 10.1523/JNEUROSCI.0265-19.2019
45. Serhan MF, Kreydiyyeh SI. Insulin targets the Na+/K+ ATPase in enterocytes via PI3K, PKC, and MAPKS. J Recept Signal Transduct Res (2011) 31:299–306. doi: 10.3109/10799893.2011.587821
46. Beltowski J, Marciniak A, Wojcicka G. Leptin decreases renal medullary na(+), k(+)-ATPase activity through phosphatidylinositol 3-kinase dependent mechanism. J Physiol Pharmacol (2004) 55:391–407.
47. Obradovic M, Stewart AJ, Pitt SJ, Labudovic-Borovic M, Sudar E, Petrovic V, et al. In vivo effects of 17β-estradiol on cardiac Na(+)/K(+)-ATPase expression and activity in rat heart. Mol Cell Endocrinol (2014) 388:58–68. doi: 10.1016/j.mce.2014.03.005
48. Kajstura J, Fiordaliso F, Andreoli AM, Li B, Chimenti S, Medow MS, et al. IGF-1 overexpression inhibits the development of diabetic cardiomyopathy and angiotensin II-mediated oxidative stress. Diabetes (2001) 50:1414–24. doi: 10.2337/diabetes.50.6.1414
49. Obradovic M, Stanimirovic J, Panic A, Bogdanovic N, Sudar-Milovanovic E, Cenic-Milosevic D, et al. Regulation of Na+/K+-ATPase by estradiol and IGF-1 in cardio-metabolic diseases. Curr Pharm Des (2017) 23:1551–61. doi: 10.2174/1381612823666170203113455
50. Mishra NK, Peleg Y, Cirri E, Belogus T, Lifshitz Y, Voelker DR, et al. FXYD proteins stabilize Na,K-ATPase: amplification of specific phosphatidylserine-protein interactions. J Biol Chem (2011) 286:9699–712. doi: 10.1074/jbc.M110.184234
51. Klimanova EA, Petrushanko IY, Mitkevich VA, Anashkina AA, Orlov SN, Makarov AA, et al. Binding of ouabain and marinobufagenin leads to different structural changes in Na,K-ATPase and depends on the enzyme conformation. FEBS Lett (2015) 589:2668–74. doi: 10.1016/j.febslet.2015.08.011
52. Ndisang JF, Rastogi S. Cardiometabolic diseases and related complications: current status and future perspective. BioMed Res Int (2013) 2013:467682. doi: 10.1155/2013/467682
53. Miranda JJ, Barrientos-Gutierrez T, Corvalan C, Hyder AA, Lazo-Porras M, Oni T, et al. Understanding the rise of cardiometabolic diseases in low- and middle-income countries. Nat Med (2019) 25:1667–79. doi: 10.1038/s41591-019-0644-7
54. Kones R. Primary prevention of coronary heart disease: integration of new data, evolving views, revised goals, and role of rosuvastatin in management. a comprehensive survey. Drug Des Devel Ther (2011) 5:325–80. doi: 10.2147/DDDT.S14934
55. Okovityi SV, Kulikov AN, Shulenin KS. [Pharmacological prevention of cardiovascular diseases]. Eksp Klin Farmakol (2011) 74:43–7.
56. Stoner L, Stoner KR, Young JM, Fryer S. Preventing a cardiovascular disease epidemic among indigenous populations through lifestyle changes. Int J Prev Med (2012) 3:230–40.
57. Kataoka M, Aimi Y, Yanagisawa R, Ono M, Oka A, Fukuda K, et al. Alu-mediated nonallelic homologous and nonhomologous recombination in the BMPR2 gene in heritable pulmonary arterial hypertension. Genet Med (2013) 15:941–7. doi: 10.1038/gim.2013.41
58. Fiorillo C, Brisca G, Cassandrini D, Scapolan S, Astrea G, Valle M, et al. Subclinical myopathy in a child with neutral lipid storage disease and mutations in the PNPLA2 gene. Biochem Biophys Res Commun (2013) 430:241–4. doi: 10.1016/j.bbrc.2012.10.127
59. Ndisang JF. Role of heme oxygenase in inflammation, insulin-signalling, diabetes and obesity. Mediators Inflamm (2010) 2010:359732. doi: 10.1155/2010/359732
60. Iwamoto SJ, Abushamat LA, Zaman A, Millard AJ, Cornier MA. Obesity management in cardiometabolic disease: State of the art. Curr Atheroscler Rep (2021) 23:59. doi: 10.1007/s11883-021-00953-0
61. Twig G, Yaniv G, Levine H, Leiba A, Goldberger N, Derazne E, et al. Body-mass index in 2.3 million adolescents and cardiovascular death in adulthood. N Engl J Med (2016) 374:2430–40. doi: 10.1056/NEJMoa1503840
62. Wildman RP, Muntner P, Reynolds K, McGinn AP, Rajpathak S, Wylie-Rosett J, et al. The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999-2004). Arch Intern Med (2008) 168:1617–24. doi: 10.1001/archinte.168.15.1617
63. Cardiol J, Ther C, Rao G. Cardiometabolic diseases: A global perspective. J Cardiol Cardiovasc Ther (2018) 12:1–5. doi: 10.19080/JOCCT.2018.12.555834
64. Reyman M, Verrijn Stuart AA, van Summeren M, Rakhshandehroo M, Nuboer R, de Boer FK, et al. Vitamin d deficiency in childhood obesity is associated with high levels of circulating inflammatory mediators, and low insulin sensitivity. Int J Obes (Lond) (2014) 38:46–52. doi: 10.1038/ijo.2013.75
65. Susic D, Varagic J. Obesity: A perspective from hypertension. Med Clin North Am (2017) 101:139–57. doi: 10.1016/j.mcna.2016.08.008
66. Saguy AC, Riley KW. Weighing both sides: morality, mortality, and framing contests over obesity. J Health Polit Policy Law. (2005) 30:869–921. doi: 10.1215/03616878-30-5-869
67. Vekic J, Zeljkovic A, Stefanovic A, Jelic-Ivanovic Z, Spasojevic-Kalimanovska V. Obesity and dyslipidemia. Metabolism (2019) 92:71–81. doi: 10.1016/j.metabol.2018.11.005
68. Howard BV, Ruotolo G, Robbins DC. Obesity and dyslipidemia. Endocrinol Metab Clin North Am (2003) 32:855–67. doi: 10.1016/S0889-8529(03)00073-2
69. Zaric B, Obradovic M, Trpkovic A, Banach M, Mikhailidis DP, Isenovic ER. Endothelial dysfunction in dyslipidaemia: Molecular mechanisms and clinical implications. Curr Med Chem (2020) 27:1021–40. doi: 10.2174/0929867326666190903112146
70. Tian M, Li R, Shan Z, Wang DW, Jiang J, Cui G. Comparison of apolipoprotein B/A1 ratio, framingham risk score and TC/HDL-c for predicting clinical outcomes in patients undergoing percutaneous coronary intervention. Lipids Health Dis (2019) 18:202. doi: 10.1186/s12944-019-1144-y
71. Jin X, Yang S, Lu J, Wu M. Small, dense low-density lipoprotein-cholesterol and atherosclerosis: Relationship and therapeutic strategies. Front Cardiovasc Med (2021) 8:804214. doi: 10.3389/fcvm.2021.804214
72. Caselli C, De Caterina R, Smit JM, Campolo J, El Mahdiui M, Ragusa R, et al. Triglycerides and low HDL cholesterol predict coronary heart disease risk in patients with stable angina. Sci Rep (2021) 11:20714. doi: 10.1038/s41598-021-00020-3
73. Kaze AD, Santhanam P, Musani SK, Ahima R, Echouffo-Tcheugui JB. Metabolic dyslipidemia and cardiovascular outcomes in type 2 diabetes mellitus: Findings from the look AHEAD study. J Am Heart Assoc (2021) 10:e016947. doi: 10.1161/JAHA.120.016947
74. Wei DH, Zhang XL, Wang R, Zeng JF, Zhang K, Yang J, et al. Oxidized lipoprotein(a) increases endothelial cell monolayer permeability via ROS generation. Lipids (2013) 48:579–86. doi: 10.1007/s11745-013-3795-1
75. Wang Z, Nakayama T. Inflammation, a link between obesity and cardiovascular disease. Mediators Inflamm (2010) 2010:535918. doi: 10.1155/2010/535918
76. Obradovic M, Sudar E, Zafirovic S, Stanimirovic J, Labudovic-Borovic M, Isenovic ER. Estradiol in vivo induces changes in cardiomyocytes size in obese rats. Angiology (2015) 66:25–35. doi: 10.1177/0003319713514477
77. Maximilian Buja L, Vela D. Cardiomyocyte death and renewal in the normal and diseased heart. Cardiovasc Pathol (2008) 17:349–74. doi: 10.1016/j.carpath.2008.02.004
78. Artham SM, Lavie CJ, Milani RV, Ventura HO. Obesity and hypertension, heart failure, and coronary heart disease-risk factor, paradox, and recommendations for weight loss. Ochsner J (2009) 9:124–32.
79. Obradovic MM, Bjelogrlic P, Rizzo M, Katsiki N, Haidara M, Stewart AJ, et al. Effects of obesity and estradiol on Na+/K+-ATPase and their relevance to cardiovascular disease. J Endocrinol (2013) 218:R13–23. doi: 10.1530/JOE-13-0144
80. Vrbjar N, Bernatova I, Pechanova O. Functional alterations of cardiac (Na,K)-ATPase in l-NAME induced hypertension. Gen Physiol Biophys (1999) 18(Suppl 1):10–2.
81. Iannello S, Milazzo P, Belfiore F. Animal and human tissue Na,K-ATPase in normal and insulin-resistant states: regulation, behaviour and interpretative hypothesis on NEFA effects. Obes Rev (2007) 8:231–51. doi: 10.1111/j.1467-789X.2006.00276.x
82. Liu C, Bai Y, Chen Y, Wang Y, Sottejeau Y, Liu L, et al. Reduction of Na/K-ATPase potentiates marinobufagenin-induced cardiac dysfunction and myocyte apoptosis. J Biol Chem (2012) 287:16390–8. doi: 10.1074/jbc.M111.304451
83. Bay J, Kohlhaas M, Maack C. Intracellular na+ and cardiac metabolism. J Mol Cell Cardiol (2013) 61:20–7. doi: 10.1016/j.yjmcc.2013.05.010
84. Luise MD, Blackburn GL, Flier JS. Reduced activity of the red-cell sodium-potassium pump in human obesity. N Engl J Med (1980) 303:1017–22. doi: 10.1056/NEJM198010303031801
85. Bickel CA, Verbalis JG, Knepper MA, Ecelbarger CA. Increased renal Na-K-ATPase, NCC, and β-ENaC abundance in obese zucker rats. Am J Physiol Renal Physiol (2001) 281:F639–48. doi: 10.1152/ajprenal.2001.281.4.F639
86. Koricanac G, Tepavcevic S, Romic S, Milosavljevic T, Stojiljkovic M, Zakula Z. Expression and cellular distribution of glucose transporters and alpha subunits of Na+/K+-ATPase in the heart of fructose-fed female rats: the role of estradiol. Horm Metab Res (2014) 46:109–15. doi: 10.1055/s-0033-1354401
87. Vlkovičová J, Javorkova V, Mézešová L, Pecháňová O, Vrbjar N. Regulatory role of nitric oxide on the cardiac Na, K-ATPase in hypertension. Physiol Res (2008) 57:S15–22. doi: 10.33549/physiolres.931547
88. Dostanic I, Paul RJ, Lorenz JN, Theriault S, Huysse JWV, Lingrel JB. The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol (2005) 288:H477–85. doi: 10.1152/ajpheart.00083.2004
89. Liu J, Yan Y, Liu L, Xie Z, Malhotra D, Joe B, et al. Impairment of Na/K-ATPase signaling in renal proximal tubule contributes to Dahl salt-sensitive hypertension. J Biol Chem (2011) 286:22806–13. doi: 10.1074/jbc.M111.246249
90. Beltowski J, Jamroz-Wisniewska A, Borkowska E, Wójcicka G. Up-regulation of renal Na, K ATPase: the possible novel mechanism of leptin-induced hypertension. Pol J Pharmacol (2004) 56:213–22.
91. Vlkovicová J, Javorková V, Pechánová O, Vrbjar N. Gender difference in functional properties of Na,K-ATPase in the heart of spontaneously hypertensive rats. Life Sci (2005) 76:971–82. doi: 10.1016/j.lfs.2004.10.013
92. Rodrigo R, Miranda-Merchak A, Valenzuela Grau R, Bachler JP, Vergara L. Modulation of (Na,K)-ATPase activity by membrane fatty acid composition: therapeutic implications in human hypertension. Clin Experiment Hyperte (2014) 36:17–26. doi: 10.3109/10641963.2013.783048
93. Correll RN, Eder P, Burr AR, Despa S, Davis J, Bers DM, et al. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res (2014) 114:249–56. doi: 10.1161/CIRCRESAHA.114.302293
94. Rosta K, Tulassay E, Enzsoly A, Ronai K, Szantho A, Pandics T, et al. Insulin induced translocation of Na+/K+ -ATPase is decreased in the heart of streptozotocin diabetic rats. Acta Pharmacol Sin (2009) 30:1616–24. doi: 10.1038/aps.2009.162
95. Kato K, Lukas A, Chapman DC, Rupp H, Dhalla NS. Differential effects of etomoxir treatment on cardiac na+-k+ ATPase subunits in diabetic rats. Mol Cell Biochem (2002) 232:57–62. doi: 10.1023/A:1014841216418
96. Vlkovicová J, Javorková V, Stefek M, Kysel’ová Z, Gajdosíková A, Vrbjar N. Effect of the pyridoindole antioxidant stobadine on the cardiac Na(+),K(+)-ATPase in rats with streptozotocin-induced diabetes. Gen Physiol Biophys (2006) 25:111–24.
97. Galuska D, Kotova O, Barrès R, Chibalina D, Benziane B, Chibalin AV. Altered expression and insulin-induced trafficking of na+-K+-ATPase in rat skeletal muscle: effects of high-fat diet and exercise. Am J Physiol Endocrinol Metab (2009) 297:E38–49. doi: 10.1152/ajpendo.90990.2008
98. Tsimaratos M, Coste TC, Djemli-Shipkolye A, Daniel L, Shipkolye F, Vague P, et al. Evidence of time-dependent changes in renal medullary Na, K-ATPase activity and expression in diabetic rats. Cell Mol Biol (Noisy-le-grand (2001) 47:239–46.
99. Babu PVA, Sabitha KE, Shyamaladevi CS. Green tea impedes dyslipidemia, lipid peroxidation, protein glycation and ameliorates Ca2+-ATPase and Na+/K+-ATPase activity in the heart of streptozotocin-diabetic rats. Chem Biol Interact (2006) 162:157–64. doi: 10.1016/j.cbi.2006.05.020
100. Kowluru RA, Engerman RL, Kern TS. Diabetes-induced metabolic abnormalities in myocardium: effect of antioxidant therapy. Free Radic Res (2000) 32:67–74. doi: 10.1080/10715760000300071
101. Michea L, Irribarra V, Goecke IA, Marusic ET. Reduced Na-K pump but increased Na-K-2Cl cotransporter in aorta of streptozotocin-induced diabetic rat. Am J Physiol Heart Circ Physiol (2001) 280:H851–858. doi: 10.1152/ajpheart.2001.280.2.H851
102. Mimura M, Makino H, Kanatsuka A, Asai T, Yoshida S. Reduction of erythrocyte (Na(+)-K+)ATPase activity in type 2 (non-insulin-dependent) diabetic patients with microalbuminuria. Horm Metab Res (1994) 26:33–8. doi: 10.1055/s-2007-1000768
103. Baldini P, Incerpi S, Lambert-Gardini S, Spinedi A, Luly P. Membrane lipid alterations and na+-pumping activity in erythrocytes from IDDM and NIDDM subjects. Diabetes (1989) 38:825–31. doi: 10.2337/diab.38.7.825
104. Gupta D, Ahmad F, Suhail M. In vivo alloxan induced alterations in the rat erythrocyte membrane bound adenosine triphosphatases. Bioved (1991) 2:141–6.
105. Ver A, Szanto I, Banyasz T, Csermely P, Vegh E, Somogyi J. Changes in the expression of Na+/K+-ATPase isoenzymes in the left ventricle of diabetic rat hearts: effect of insulin treatment. Diabetologia (1997) 40:1255–62. doi: 10.1007/s001250050818
106. Kjeldsen K, Braendgaard H, Sidenius P, Larsen JS, Norgaard A. Diabetes decreases na+-k+ pump concentration in skeletal muscles, heart ventricular muscle, and peripheral nerves of rat. Diabetes (1987) 36:842–8. doi: 10.2337/diab.36.7.842
107. Gerbi A, Barbey O, Raccah D, Coste T, Jamme I, Nouvelot A, et al. Alteration of Na,K-ATPase isoenzymes in diabetic cardiomyopathy: effect of dietary supplementation with fish oil (n-3 fatty acids) in rats. Diabetologia (1997) 40:496–505. doi: 10.1007/s001250050707
108. Sweeney G, Klip A. Mechanisms and consequences of Na+,K+-pump regulation by insulin and leptin. Cell Mol Biol (2001) 47:363–72.
109. Fan X, Xie J, Tian J. Reducing cardiac fibrosis: Na/K-ATPase signaling complex as a novel target. Cardiovasc Pharm Open Access (2017) 6:204. doi: 10.4172/2329-6607.1000204
110. Shapiro JI, Tian J. Signaling through the Na/K-ATPase: implications for cardiac fibrosis. Am J Physiol Heart Circ Physiol (2011) 300:H29–30. doi: 10.1152/ajpheart.01038.2010
111. Elkareh J, Kennedy DJ, Yashaswi B, Vetteth S, Shidyak A, Kim EG, et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension (2007) 49:215–24. doi: 10.1161/01.HYP.0000252409.36927.05
112. Simonini M, Pozzoli S, Bignami E, Casamassima N, Messaggio E, Lanzani C, et al. Endogenous ouabain: An old cardiotonic steroid as a new biomarker of heart failure and a predictor of mortality after cardiac surgery. BioMed Res Int (2015) 2015:714793. doi: 10.1155/2015/714793
113. Drummond CA, Fan X, Haller ST, Kennedy DJ, Liu J, Tian J. Na/K-ATPase signaling mediates miR-29b-3p regulation and cardiac fibrosis formation in mice with chronic kidney disease. PloS One (2018) 13:e0197688. doi: 10.1371/journal.pone.0197688
114. Chibalin AV. Regulation of the Na,K-ATPase: Special implications for cardiovascular complications of metabolic syndrome. Pathophysiology (2007) 14:153–8. doi: 10.1016/j.pathophys.2007.09.004
115. Bibert S, Liu CC, Figtree GA, Garcia A, Hamilton EJ, Marassi FM, et al. FXYD proteins reverse inhibition of the na+-k+ pump mediated by glutathionylation of its beta1 subunit. J Biol Chem (2011) 286:18562–72. doi: 10.1074/jbc.M110.184101
116. Figtree GA, Liu CC, Bibert S, Hamilton EJ, Garcia A, White CN, et al. Reversible oxidative modification: a key mechanism of na+-k+ pump regulation. Circ Res (2009) 105:185–93. doi: 10.1161/CIRCRESAHA.109.199547
117. White CN, Figtree GA, Liu CC, Garcia A, Hamilton EJ, Chia KK, et al. Angiotensin II inhibits the na+-k+ pump via PKC-dependent activation of NADPH oxidase. Am J Physiol Cell Physiol (2009) 296:4. doi: 10.1152/ajpcell.00648.2008
118. Herrera VL, Chobanian AV, Ruiz-Opazo N. Isoform-specific modulation of na+, k+-ATPase alpha-subunit gene expression in hypertension. Science (1988) 241:221–3. doi: 10.1126/science.2838907
119. Sahin-Erdemli I, Medford RM, Songu-Mize E. Regulation of Na+,K(+)-ATPase alpha-subunit isoforms in rat tissues during hypertension. Eur J Pharmacol (1995) 292:163–71. doi: 10.1016/0926-6917(95)90009-8
120. Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci U S A (2005) 102:15845–50. doi: 10.1073/pnas.0507358102
121. Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ, et al. ACTH-induced hypertension is dependent on the ouabain-binding site of the alpha2-Na+-K+-ATPase subunit. Am J Physiol Heart Circ Physiol (2008) 295:16. doi: 10.1152/ajpheart.00183.2008
122. Cellini A, Höfler D, Arias-Loza PA. The α2-isoform of the Na(+)/K(+)-ATPase protects against pathological remodeling and β-adrenergic desensitization after myocardial infarction. Am J Physiol Heart Circ Physiol (2021) 321:H650–h662. doi: 10.1152/ajpheart.00808.2020
123. Bartlett DE, Miller RB, Thiesfeldt S, Lakhani HV, Shapiro JI, Sodhi K. The role of Na/K-ATPase signaling in oxidative stress related to aging: Implications in obesity and cardiovascular disease. Int J Mol Sci (2018) 19:2139. doi: 10.3390/ijms19072139
124. Pratt R, Lakhani HV, Zehra M, Desauguste R, Pillai SS, Sodhi K. Mechanistic insight of Na/K-ATPase signaling and HO-1 into models of obesity and nonalcoholic steatohepatitis. Int J Mol Sci (2019) 21:87. doi: 10.3390/ijms21010087
125. Liu Y, Shoji-Kawata S, Sumpter RM Jr., Wei Y, Ginet V, Zhang L, et al. Autosis is a Na+,K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A (2013) 110:20364–71. doi: 10.1073/pnas.1319661110
126. Zhang G, Luk BT, Hamidy M, Zhang L, Spector SA. Induction of a Na(+)/K(+)-ATPase-dependent form of autophagy triggers preferential cell death of human immunodeficiency virus type-1-infected macrophages. Autophagy (2018) 14:1359–75. doi: 10.1080/15548627.2018.1476014
127. Fernández ÁF, Liu Y, Ginet V, Shi M, Nah J, Zou Z, et al. Interaction between the autophagy protein beclin 1 and Na+,K+-ATPase during starvation, exercise, and ischemia. JCI Insight (2020) 5:133282. doi: 10.1172/jci.insight.133282
128. Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta (1957) 23:394–401. doi: 10.1016/0006-3002(57)90343-8
129. Fuller W, Tulloch LB, Shattock MJ, Calaghan SC, Howie J, Wypijewski KJ. Regulation of the cardiac sodium pump. Cell Mol Life Sci (2013) 70:1357–80. doi: 10.1007/s00018-012-1134-y
131. Nørgaard A, Bagger JP, Bjerregaard P, Baandrup U, Kjeldsen K, Thomsen PE. Relation of left ventricular function and Na,K-pump concentration in suspected idiopathic dilated cardiomyopathy. Am J Cardiol (1988) 61:1312–5. doi: 10.1016/0002-9149(88)91175-7
132. Minakata Y, Suzuki S, Grygorczyk C, Dagenais A, Berthiaume Y. Impact of beta-adrenergic agonist on na+ channel and na+-K+-ATPase expression in alveolar type II cells. Am J Physiol (1998) 275:L414–422. doi: 10.1152/ajplung.1998.275.2.L414
133. Chen Y, Kennedy DJ, Ramakrishnan DP, Yang M, Huang W, Li Z, et al. Oxidized LDL-bound CD36 recruits an Na+/K+-ATPase-Lyn complex in macrophages that promotes atherosclerosis. Sci Signal (2015) 8:ra91. doi: 10.1126/scisignal.aaa9623
134. Deng LJ, Qi M, Li N, Lei YH, Zhang DM, Chen JX. Natural products and their derivatives: Promising modulators of tumor immunotherapy. J Leukoc Biol (2020) 108:493–508. doi: 10.1002/JLB.3MR0320-444R
135. Ren J, Gao X, Guo X, Wang N, Wang X. Research progress in pharmacological activities and applications of cardiotonic steroids. Front Pharmacol (2022) 13:902459. doi: 10.3389/fphar.2022.902459
136. Schoner W, Scheiner-Bobis G. Endogenous and exogenous cardiac glycosides: their roles in hypertension, salt metabolism, and cell growth. Am J Physiol Cell Physiol (2007) 293:C509–536. doi: 10.1152/ajpcell.00098.2007
137. Bagrov AY, Fedorova OV, Dmitrieva RI, Howald WN, Hunter AP, Kuznetsova EA, et al. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension (1998) 31:1097–103. doi: 10.1161/01.HYP.31.5.1097
138. Bignami E, Casamassima N, Frati E, Lanzani C, Corno L, Alfieri O, et al. Preoperative endogenous ouabain predicts acute kidney injury in cardiac surgery patients. Crit Care Med (2013) 41:744–55. doi: 10.1097/CCM.0b013e3182741599
139. Komiyama Y, Dong XH, Nishimura N, Masaki H, Yoshika M, Masuda M, et al. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin Biochem (2005) 38:36–45. doi: 10.1016/j.clinbiochem.2004.08.005
140. Gottlieb SS, Rogowski AC, Weinberg M, Krichten CM, Hamilton BP, Hamlyn JM. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation (1992) 86:420–5. doi: 10.1161/01.CIR.86.2.420
141. Kennedy DJ, Shrestha K, Sheehey B, Li XS, Guggilam A, Wu Y, et al. Elevated plasma marinobufagenin, an endogenous cardiotonic steroid, is associated with right ventricular dysfunction and nitrative stress in heart failure. Circ Heart Fail (2015) 8:1068–76. doi: 10.1161/CIRCHEARTFAILURE.114.001976
142. Pierdomenico SD, Bucci A, Manunta P, Rivera R, Ferrandi M, Hamlyn JM, et al. Endogenous ouabain and hemodynamic and left ventricular geometric patterns in essential hypertension. Am J Hypertens (2001) 14:44–50. doi: 10.1016/S0895-7061(00)01225-5
143. Adair CD, Buckalew VM, Graves SW, Lam GK, Johnson DD, Saade G, et al. Digoxin immune fab treatment for severe preeclampsia. Am J Perinatol (2010) 27:655–62. doi: 10.1055/s-0030-1249762
144. Pieske B, Maier LS, Piacentino V, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation (2002) 106:447–53. doi: 10.1161/01.CIR.0000023042.50192.F4
145. Verdonck F, Volders PG, Vos MA, Sipido KR. Intracellular na+ and altered na+ transport mechanisms in cardiac hypertrophy and failure. J Mol Cell Cardiol (2003) 35:5–25. doi: 10.1016/S0022-2828(02)00280-8
146. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res (2006) 99:172–82. doi: 10.1161/01.RES.0000232546.92777.05
147. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, et al. Elevated cytosolic na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation (2010) 121:1606–13. doi: 10.1161/CIRCULATIONAHA.109.914911
148. Lingrel JB. The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu Rev Physiol (2010) 72:395–412. doi: 10.1146/annurev-physiol-021909-135725
149. Wang X, Liu J, Drummond CA, Shapiro JI. Sodium potassium adenosine triphosphatase (Na/K-ATPase) as a therapeutic target for uremic cardiomyopathy. Expert Opin Ther Targets (2017) 21:531–41. doi: 10.1080/14728222.2017.1311864
150. Blaustein MP. Sodium ions, calcium ions, blood pressure regulation, and hypertension: a reassessment and a hypothesis. Am J Physiol (1977) 232:C165–173. doi: 10.1152/ajpcell.1977.232.5.C165
151. Virgadamo S, Charnigo R, Darrat Y, Morales G, Elayi CS. Digoxin: A systematic review in atrial fibrillation, congestive heart failure and post myocardial infarction. World J Cardiol (2015) 7:808–16. doi: 10.4330/wjc.v7.i11.808
152. Ziff OJ, Kotecha D. Digoxin: The good and the bad. Trends Cardiovasc Med (2016) 26:585–95. doi: 10.1016/j.tcm.2016.03.011
153. McDonagh TA, Metra M. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J (2021) 42:3599–726. doi: 10.1093/eurheartj/ehab368
154. Mihailidou AS, Bundgaard H, Mardini M, Hansen PS, Kjeldsen K, Rasmussen HH. Hyperaldosteronemia in rabbits inhibits the cardiac sarcolemmal na(+)-k(+) pump. Circ Res (2000) 86:37–42. doi: 10.1161/01.RES.86.1.37
155. Hool LC, Whalley DW, Doohan MM, Rasmussen HH. Angiotensin-converting enzyme inhibition, intracellular na+, and na(+)-k+ pumping in cardiac myocytes. Am J Physiol (1995) 268:C366–375. doi: 10.1152/ajpcell.1995.268.2.C366
156. Fedorova OV, Zernetkina VI, Shilova VY, Grigorova YN, Juhasz O, Wei W, et al. Synthesis of an endogenous steroidal Na pump inhibitor marinobufagenin, implicated in human cardiovascular diseases, is initiated by CYP27A1 via bile acid pathway. Circ Cardiovasc Genet (2015) 8:736–45. doi: 10.1161/CIRCGENETICS.115.001217
157. Słabiak-Błaż N, Piecha G. Endogenous mammalian cardiotonic steroids-a new cardiovascular risk factor?-a mini-review. Life (Basel) (2021) 11:727. doi: 10.3390/life11080727
158. Zhang XQ, Moorman JR, Ahlers BA, Carl LL, Lake DE, Song J, et al. Phospholemman overexpression inhibits na+-K+-ATPase in adult rat cardiac myocytes: relevance to decreased na+ pump activity in postinfarction myocytes. J Appl Physiol (1985) (2006) . 100:212–20. doi: 10.1152/japplphysiol.00757.2005
159. Stella P, Manunta P, Mallamaci F, Melandri M, Spotti D, Tripepi G, et al. Endogenous ouabain and cardiomyopathy in dialysis patients. J Intern Med (2008) 263:274–80. doi: 10.1111/j.1365-2796.2007.01883.x
160. Strauss M, Smith W, Fedorova OV, Schutte AE. The Na(+)K(+)-ATPase inhibitor marinobufagenin and early cardiovascular risk in humans: a review of recent evidence. Curr Hypertens Rep (2019) 21:38. doi: 10.1007/s11906-019-0942-y
161. Bagrov AY, Shapiro JI, Fedorova OV. Endogenous cardiotonic steroids: physiology, pharmacology, and novel therapeutic targets. Pharmacol Rev (2009) 61:9–38. doi: 10.1124/pr.108.000711
162. Bauersachs J, Veltmann C. Heart rate control in heart failure with reduced ejection fraction: the bright and the dark side of the moon. Eur J Heart Fail (2020) 22:539–42. doi: 10.1002/ejhf.1733
163. Vamos M, Erath JW, Hohnloser SH. Digoxin-associated mortality: a systematic review and meta-analysis of the literature. Eur Heart J (2015) 36:1831–8. doi: 10.1093/eurheartj/ehv143
164. Washam JB, Stevens SR, Lokhnygina Y, Halperin JL, Breithardt G, Singer DE, et al. Digoxin use in patients with atrial fibrillation and adverse cardiovascular outcomes: a retrospective analysis of the rivaroxaban once daily oral direct factor xa inhibition compared with vitamin K antagonism for prevention of stroke and embolism trial in atrial fibrillation (ROCKET AF). Lancet (2015) 385:2363–70. doi: 10.1016/S0140-6736(14)61836-5
165. Braunwald E. Effects of digitalis on the normal and the failing heart. J Am Coll Cardiol (1985) 5:51a–9a. doi: 10.1016/S0735-1097(85)80463-0
166. Belkin MN, Cifu AS, Pinney S. Management of heart failure. Jama (2022) 328:1346–7. doi: 10.1001/jama.2022.16667
167. McDonagh TA, Metra M. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the task force for the diagnosis and treatment of acute and chronic heart failure of the European society of cardiology (ESC). with the special contribution of the heart failure association (HFA) of the ESC. Eur Heart J (2022) 24:4–131. doi: 10.1016/j.rec.2022.05.005
168. Ziff OJ, Lane DA, Samra M, Griffith M, Kirchhof P, Lip GY, et al. Safety and efficacy of digoxin: systematic review and meta-analysis of observational and controlled trial data. BMJ (2015) 351:h4451. doi: 10.1136/bmj.h4451
169. Malik A, Masson R, Singh S, Wu WC, Packer M, Pitt B, et al. Digoxin discontinuation and outcomes in patients with heart failure with reduced ejection fraction. J Am Coll Cardiol (2019) 74:617–27. doi: 10.1016/j.jacc.2019.05.064
170. Schoner W, Scheiner-Bobis G. Role of endogenous cardiotonic steroids in sodium homeostasis. Nephrol Dial Transpl (2008) 23:2723–9. doi: 10.1093/ndt/gfn325
171. Bavendiek U, Berliner D, Dávila LA, Schwab J, Maier L, Philipp SA, et al. Rationale and design of the DIGIT-HF trial (DIGitoxin to improve ouTcomes in patients with advanced chronic heart failure): a randomized, double-blind, placebo-controlled study. Eur J Heart Fail (2019) 21:676–84. doi: 10.1002/ejhf.1452
172. Xiao J, Zhang X, Fu C, Yang Q, Xie Y, Zhang Z, et al. Impaired na+–K+-ATPase signaling in renal proximal tubule contributes to hyperuricemia-induced renal tubular injury. Exp Mol Med (2018) 50:e452–2. doi: 10.1038/emm.2017.287
Keywords: Na+/K+-ATPase, cardiometabolic diseases, cardiovascular diseases, type 2 diabetes mellitus, therapy
Citation: Obradovic M, Sudar-Milovanovic E, Gluvic Z, Banjac K, Rizzo M and Isenovic ER (2023) The Na+/K+-ATPase: A potential therapeutic target in cardiometabolic diseases. Front. Endocrinol. 14:1150171. doi: 10.3389/fendo.2023.1150171
Received: 23 January 2023; Accepted: 14 February 2023;
Published: 28 February 2023.
Edited by:
Gaetano Santulli, Albert Einstein College of Medicine, United StatesReviewed by:
Aleksandra Jovanovic, University of California, Berkeley, United StatesCopyright © 2023 Obradovic, Sudar-Milovanovic, Gluvic, Banjac, Rizzo and Isenovic. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manfredi Rizzo, bWFuZnJlZGkucml6em9AdW5pcGEuaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.