 Nina Makretskaya1*†
Nina Makretskaya1*† Natalia Kalinchenko2†Inna Tebieva3†Sofya Ionova1†Rena Zinchenko1
Natalia Kalinchenko2†Inna Tebieva3†Sofya Ionova1†Rena Zinchenko1 Andrey Marakhonov1†
Andrey Marakhonov1† Anatoly Tiulpakov1
Anatoly Tiulpakov1- 1Department of Genetics of Endocrine Diseases, Research Centre for Medical Genetics, Moscow, Russia
- 2Institute of Pediatric Endocrinology, Endocrinology Research Centre, Moscow, Russia
- 3Consulting and Diagnostic Department, Republic of North Ossetia-Alania (RNOA) “Republican Children’s Clinical Hospital”, Vladikavkaz, Russia
Background: Congenital adrenal hyperplasia (CAH) caused by 3β-HSD deficiency is a rare form of congenital adrenal deficiency with an autosomal recessive type of inheritance. Previously we have demonstrated that a single nucleotide variant (SNV) p.Trp230* in the homozygous state is a frequent cause of CAH among the indigenous population of North Ossetia-Alania represented by Ossetians.
Methods: Genotyping of the NM_000198.3:c.690G>A p.Trp230* variant was performed by Real-time PCR. 339 healthy individuals of Ossetian origin were included in the study. Allele frequencies, Fisher’s confidence intervals (CI) were calculated using the WinPepi v. 11.65 software. Comparison of allele frequencies was performed with the z-score test for two proportions.
Results: Eight heterozygous carriers of c.690G>A variant in HSD3B2 gene were detected in 339 samples investigated. The total allele frequency of p.Trp230* variant was 0.0118 (n=8/678, 95% CI=0.0051–0.0231). Accordingly, the heterozygous carrier rate was 0.0236 (n=8/339). The frequency of CAH caused by p.Trp230* variant in HSD3B2 in Ossetian population was 1:7183 or 13.9 per 100,000 (95% CI: 1:1874–1:38447 or 3–53 per 100,000).
Conclusion: The results demonstrate high frequency of p.Trp230* variant in Ossetians, which is most likely attributed to a founder effect.
1 Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders caused by impairment at one of the steps of cortisol biosynthesis in adrenal cortex. To date, 7 distinct clinical forms of CAH and the corresponding genes are recognized, namely: lipoid adrenal hyperplasia, STAR (OMIM #201710); deficiency of cholesterol side-chain cleavage enzyme, CYP11A1 (OMIM #613743); deficiency of 3β-hydroxysteroid dehydrogenase, HSD3B2 (OMIM #201810); deficiency of 17α-hydroxylase/17,20-lyase, CYP17A1 (OMIM #202110); deficiency of P450 oxidoreductase, POR (OMIM #613571); deficiency of 21-hydroxylase, CYP21A2 (OMIM #201910); and deficiency of 11β-hydroxylase, CYP11B1 (OMIM #202010) (1). The two most frequent enzymatic defects, deficiencies of 21-hydroxylase and 11β-hydroxylase, account for 90-99% (2, 3) and 5-8% (4) of all CAH cases, respectively, whereas the other 5 forms are exceptionally rare.
HSD3B2 is expressed in adrenal cortex and gonads where it converts Δ5-3β-hydroxysteroids into the corresponding Δ4-3-keto isomers: namely, pregnenolone to progesterone, 17α-hydroxypregnenolone (17OHPreg) to 17α-hydroxyprogesterone (17OHP) and dehydroepiandrosterone (DHEA) to androstenedione, respectively. Biallelic loss-of-function variants in HSD3B2 present with salt wasting, genital ambiguity, and hypogonadism in both sexes (5). The disorder is estimated to have an overall incidence of less than 1/1 000 000 live births (6), however the prevalence may be substantially higher in inbred populations (7).
Previously we have reported 2 siblings with 3β-hydroxysteroid dehydrogenase (3β-HSD) deficiency caused by homozygous p.Trp230* variant in HSD3B2 (8). Subsequently additional patients with the same variant have been described, all of which, like the first two, were of Ossetian origin (9). Based on the number of the diagnosed cases in North Ossetia–Alania (RNOA) in 2007–2017 the incidence of 3β-HSD deficiency was estimated to be 1:22470, close to that calculated during the same period for 21-hydroxylase deficiency (1:18750) (9). The above findings prompted us to study carrier frequency of the p.Trp230* variant in Ossetians.
There are 5 subethnic groups within Ossetian origin: Ironians, Digorians, Kudar, Iasi, Dvals. In this study, a comparative analysis was carried out of two of them: Ironians and Digorians.
2 Method
2.1 Ethical consideration
A written informed consent for genetic studies was obtained from the subjects. The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Review Board of the Research Centre for Medical Genetics, Moscow, Russia (protocol no. 7 of December 20, 2017).
2.2 Variant nomenclature, accession, and pathogenic status
GenBank entry NM_000198.3 was used as reference for the HSD3B2 gene nucleotide and amino acid numbering. All genome coordinates are given according to the GRCh37/hg19 assembly. Assessment of pathogenic status of the variant was performed using ACMG/AMP recommendations (10). Prediction whether variant is a subject to degradation by nonsense-mediated decay (NMD) was made using NMDEscPredictor (11).
2.3 Genetic testing and autozygosity mapping
DNA samples were analyzed using chromosomal microarray analysis (CMA). The analysis was performed using the GeneChip™ 3000 system (Thermo Fisher Scientific, USA) according to the manufacturer’s protocol on a CytoScan HD microarray containing 2.67 million markers.
Regions of autozygosity were analyzed using Chromosome Analysis Suite v. 4.2.1 software (Thermo Fisher Scientific, USA) with the subsequent visual inspection of the regions’ borders. The age of the mutation event was estimated using the method described in detail elsewhere (12). For the interpolation of hg19 physical positions into the sex-averaged map positions in Kosambi cM, Rutgers Maps v.3 were used (13).
Allele frequencies, Fisher’s confidence intervals (CI) were calculated using the WinPepi v. 11.65 software (14). Comparison of allele frequencies was performed with the z-score test for two proportions.
2.4 Subjects
The study included 339 healthy Ossetians, with a distribution by subethnic groups: 283 Ironians and 56 Digorians. Sex ratio is 32.5:67.5%, median age is 37, interquartile range 26–52 years, range 3–86.
2.5 Real-time PCR
Genomic DNA was isolated from peripheral blood leukocytes using the PureLink® Genomic DNA Mini Kit (Thermo Scientific, Waltham, MA, USA) according to the method recommended by the manufacturer.
Genotyping of the nucleotide variant NM_000198.3:c.690G>A (p.Trp230*) was performed by real-time PCR on a StepOne Plus amplifier (Thermo Scientific, Waltham, MA, USA) using the following primers and TaqMan probes:
HSD_4F CCTTGTACACTTGTGCGTTAAGACCCACA,
HSD_4R CCATCTGGAATCAAGGCGGAGGTCA,
HSD_wt (VIC)-CGGCCTGGGCCCACATTCTGGCCG-(BHQ2),
HSD_mut (FAM)-CGGCCTGAGCCCACATTCTGGCCG-(BHQ1).
3 Results
Previously, it was shown that a single nucleotide variant NM_000198.3:c.690G>A (chr1:119964814G>A, GRCh37/hg19), which produces a premature translation termination codon at position 230 of HSD3B2 protein (p.Trp230*) in the homozygous state is the major causes of CAH in the indigenous population of RNOA represented by Ossetians (9).
In order to determine the population frequency of the identified causative variant in HSD3B2 gene the population study was performed.
Among the 339 samples investigated, the variant c.690G>A was detected in 8 cases of heterozygous carriage. In Ironians, this substitution was detected in 7 cases, with allele frequency of 0.0124 (n=7/566, 95% CI=0.0036–0.0300). In the Digorian subethnic group, the variant was found in 1 case, the frequency is 0.0089 (n=1/112, 95% CI=0.0002–0.0487). There was no statistically significant difference in the allelic frequency of c.690G>A variant between the subpopulations of Ironians and Digorians (z-score = 0.3079, p = 0.7566), which indicates a single origin of the variant before the division of the Ossetian people into sub-ethnic groups and allows us to combine the data to determine the overall frequency of the disease.
The total allelic frequency of the studied variant is 0.0118 (n=8/678, 95% CI=0.0051–0.0231). Accordingly, the heterozygous carrier rate is 0.0236 (n=8/339). Based on the Hardy-Weinberg equilibrium, the proportion of homozygotes for the mutant allele is 0.000139, which gives the prevalence of the disease 1:7183 subjects or 13.9 subjects per 100,000 population (95% CI=1:1874–1:38447 or 3–53 subjects per 100,000 population). The frequency of heterozygous carriers of c.690G>A variant is 1:43 subjects (95% CI: 1:22–1:99).
Since the proportion of closely related marriages among Ossetians is extremely low (15) such high estimated frequency of 3β-HSD deficiency in the studied population suggests a founder effect in the distribution of the c.690G>A variant in the indigenous population.
To test this hypothesis we performed a haplotype study using genotyping with Affymetrix high-density SNP Array in two patients with homozygous c.690G>A variant. As a result, we detected autozygosity regions at chr1:119425396_152538358 and chr1:117767918_145829474 encompassing the HSD3B2 locus and overlapping the centromere of the chromosome 1 (shown in Figure 1).
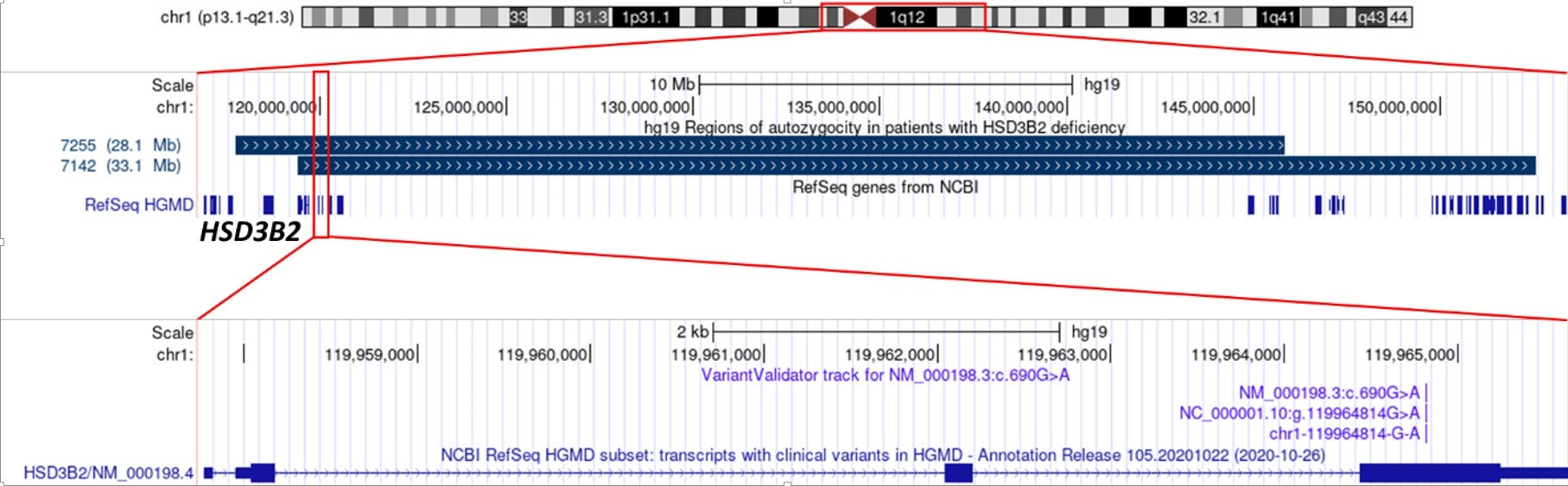
Figure 1 The position of extended autozygosity regions on chromosome 1 (upper panel), including the locus of the HSD3B2 gene (middle panel); variant c.690G>A p.Trp230* in exon 4 (lower panel).
Using Rutgers Map v.2 (13) the physical boundaries of autozygosity regions determined by the SNP array were converted into 6.06 and 3.44 cM (Kosambi) sex-averaged genetic distances, which corresponds to a common ancestor of 8.25–14.53 generations ago. Based on the average length of generations in humans of 25 years (12), these estimates suggest the age of the mutation of 206.21–363.32 years.
4 Discussion
To our knowledge, apart from the observations in Amish (7), the previously reported cases of Ossetian origin (8, 9) represent the largest cohort of patients with 3β-HSD deficiency caused by the same variant.
The variant NM_000198.3:c.690G>A p.Trp230* (chr1:119964814G>A) is not present in gnomAD (16) and Human Gene Mutation Database (HGMD) (www.hgmd.cf.ac.uk). Nevertheless, in addition to our cases (8, 9) the variant was described in a compound-heterozygote state (p.[Ala82Asp];[Trp230*]) in a patient “of Russian origin” reported by Nordenström et al. (17). It cannot be ruled out that that heterozygous variant has the same origin as the cases detected by our group.
Analysis of the probable molecular consequences of the detected p.Trp230* variant suggests that it most likely does not activate the mechanism of nonsense-mediated mRNA decay according to the NMDEscPredictor (11), since it is located in the last exon (shown in Figure 1), but leads to the synthesis of a shortened by 143 (out of 372) amino acid residues of HSD3B2 protein. The lost residues include a 21-amino acid sequence (positions 287-307) that is essential for fixation of the enzyme at the inner surfaces of the membranes of cellular organelles (18). According to the ACMG criteria, the variant can be classified as pathogenic with a significance level of PM2, PVS1, PP1-M (10).
We have demonstrated that the estimated prevalence of 3β-HSD deficiency due to this founder mutation (1:7183) is higher than the prevalence of the most common form of CAH, 21-hydroxylase deficiency, in the majority of studied populations (1). To date, the sensitivity of 17-OHP-based neonatal screening for 3β-HSD deficiency is not known. Taking into account that in the simple virilizing and nonclassic forms of 21OH-deficiency the sensitivity may be as low 79.7% and 32.4%, respectively (19), missed cases of 3β-HSD deficiency cannot be ruled out. The regional screening data of 2007–2017 showed the incidence that was lower than theoretically expected (1:22470 vs 1:7183), although the difference was not significant (z-score = 0.8509, p = 0.3953). In the literature, several reports document detection of 3β-HSD deficiency by the 17-OHP-based neonatal screening (9, 20, 21), however both negative (20) and borderline (21) results also exist. Further studies in North Ossetia-Alania will show whether modifications for the national screening program are needed to improve the diagnostics of 3β-HSD deficiency for this region. Direct steroid hormone quantification in dried blood spots using ultra-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) (22) or a lower 17-OHP immunoassay cut-off combined with LC-MS/MS as the second-tier test (23, 24) may be considered as the options.
The high estimated allele frequency of the p.Trp230* variant in HSD3B2 gene as well as identification of extended regions of autozigosity in two unrelated patients with 3β-HSD deficiency of Ossetian origin could suggest the possible “founder effect” as well as the “bottleneck” effect in Ossetians. These observations are supported by the fact that before the 1930s, most of the population of Russia lived in rural areas, practicing assorted marriages based on geographical proximity and ethnicity. In addition, at the end of XVIII – early XIX centuries, the population of the RNOA decreased 10 times (from 200,000 people to 20,000) due to the spread of plague in this area, which also corresponds to the identified age of the mutation (25).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
Ethics statement
The studies involving human participants were reviewed and approved by the Institutional Review Board of the Research Centre for Medical Genetics, Moscow, Russia (protocol no. 7 of December 20, 2017). The patients/participants provided their written informed consent to participate in this study.
Author contributions
NM: application of statistical study data, conducting a research and investigation process, specifically performing the experiments and data collection; preparation, creation and presentation of the published work; acquisition of the financial support for the project leading to this publication. NK: conducting a research and investigation process, specifically performing the experiments and data collection, provision of study materials. IT: conducting a research and investigation process, specifically performing the experiments and data collection, provision of study materials. SI: Application of statistical study data; conducting a research and investigation process, specifically performing the experiments and data collection. RZ: formulation or evolution of overarching research goals and aims, provision of study materials; preparation, creation and presentation of the published work; management and coordination responsibility for the research activity planning and execution. AM: application of statistical study data, conducting a research and investigation process, specifically performing the experiments and data collection; preparation, creation and presentation of the published work. AT: formulation or evolution of overarching research goals and aims, development or design of methodology; preparation, creation and presentation of the published work, management and coordination responsibility for the research activity planning and execution. All authors contributed to the article and approved the submitted version.
Funding
This work was supported grant from the President of the Russian Federation № МК-5272.2022.3. This study was also supported in part by the state assignment of the Ministry of Science and Higher Education of the Russian Federation.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Claahsen-van der Grinten HL, Speiser PW, Ahmed SF, Arlt W, Auchus RJ, Falhammar H, et al. Congenital adrenal hyperplasia-current insights in pathophysiology, diagnostics, and management. Endocr Rev (2022) 43(1):91–159. doi: 10.1210/endrev/bnab016
2. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev (2000) 21(3):245–91. doi: 10.1210/edrv.21.3.0398
3. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, et al. One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet Diabetes Endocrinol (2013) 1(1):35–42. doi: 10.1016/S2213-8587(13)70007-X
4. White PC, Dupont J, New MI, Leiberman E, Hochberg Z, Rösler A. A mutation in CYP11B1 (Arg-448–-His) associated with steroid 11 beta-hydroxylase deficiency in jews of Moroccan origin. J Clin Invest (1991) 87(5):1664–7. doi: 10.1172/JCI115182
5. Simard J, Moisan AM, Morel Y. Congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase/Delta(5)-Delta(4) isomerase deficiency. Semin Reprod Med (2002) 20(3):255–76. doi: 10.1055/s-2002-35373
6. Simard J, Ricketts ML, Gingras S, Soucy P, Feltus FA, Melner MH. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr Rev (2005) 26(4):525–82. doi: 10.1210/er.2002-0050
7. Benkert AR, Young M, Robinson D, Hendrickson C, Lee PA, Strauss KA. Severe salt-losing 3beta-hydroxysteroid dehydrogenase deficiency: treatment and outcomes of HSD3B2 c.35G>A homozygotes. J Clin Endocrinol Metab (2015) 100(8):E1105–15. doi: 10.1210/jc.2015-2098
8. Melikyan MA, Rubtsov PM, Tyulpakov AN. Congenital adrenal hyperplasia due to 3beta-hydroxysteroid dehydrogenase deficiency: molecular diagnostics and clinical presentations in two mixed-sex siblings. Probl Endokrinol (Mosk) (2008) 54(5):25–9. doi: 10.14341/probl200854526-30
9. Tebieva IS, Calinchenko NY, Getoeva ZK, Tiulpakov AN. Genetical aspects of congenital adrenal hyperplasia in the republic of north ossetia - alania. Med Genet (2019) 18(6):11–20. doi: 10.25557/2073-7998.2019.06.11-20
10. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med (2015) 17(5):405–24. doi: 10.1038/gim.2015.30
11. Coban-Akdemir Z, White JJ, Song X, Jhangiani SN, Fatih JM, Gambin T, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-Function alleles. Am J Hum Genet (2018) 103(2):171–87. doi: 10.1016/j.ajhg.2018.06.009
12. Budde BS, Namavar Y, Barth PG, Poll-The BT, Nürnberg G, Becker C, et al. tRNA splicing endonuclease mutations cause pontocerebellar hypoplasia. Nat Genet (2008) 40(9):1113–8. doi: 10.1038/ng.204
13. Matise TC, Chen F, Chen W, de la Vega FM, Hansen M, He C, et al. A second-generation combined linkage physical map of the human genome. Genome Res (2007) 17(12):1783–6. doi: 10.1101/gr.7156307
14. Abramson JH. WINPEPI updated: computer programs for epidemiologists, and their teaching potential. Epidemiol Perspect Innov (2011) 8(1):1. doi: 10.1101/gr.7156307
15. El’chinova GI, Kadyshev VV, Getoeva ZK, Dzhadzhieva MYU, Vekshina AB, Lepeshinskaia AO, et al. Endogamy in population of north ossetia (Late 20th century). Russian J Genet (2020) 56(7):880–4. doi: 10.1134/s1022795420070042
16. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature (2020) 581(7809):434–43. doi: 10.1038/s41586-020-2308-7
17. Nordenström A, Forest MG, Wedell A. A case of 3beta-hydroxysteroid dehydrogenase type II (HSD3B2) deficiency picked up by neonatal screening for 21-hydroxylase deficiency: difficulties and delay in etiologic diagnosis. Horm Res (2007) 68(4):204–8. doi: 10.1159/000102593
18. Thomas JL, Bose HS. Regulation of human 3-beta-hydroxysteroid dehydrogenase type-2 (3betaHSD2) by molecular chaperones and the mitochondrial environment affects steroidogenesis. J Steroid Biochem Mol Biol (2015) 151:74–84. doi: 10.1016/j.jsbmb
19. Gidlöf S, Wedell A, Guthenberg C, von Döbeln U, Nordenström A. Nationwide neonatal screening for congenital adrenal hyperplasia in sweden: a 26-year longitudinal prospective population-based study. JAMA Pediatr (2014) 168(6):567–74. doi: 10.1001/jamapediatrics.2013.5321
20. Jeandron DD, Sahakitrungruang T. A novel homozygous Q334X mutation in the HSD3B2 gene causing classic 3beta-hydroxysteroid dehydrogenase deficiency: an unexpected diagnosis after a positive newborn screen for 21-hydroxylase deficiency. Horm Res Paediatr (2012) 77(5):334–8. doi: 10.1159/000336004
21. Alkhatib EH, Adams SD, Miller ER. Case of an unreported genetic variant of salt losing 3-beta-hydroxysteroid dehydrogenase deficiency. Oxf Med Case Rep (2021) 5, omab021. doi: 10.1093/omcr/omab021
22. Panzer K, Ekhaguere OA, Darbro B, Cook J, Shchelochkov OA. Uniparental isodisomy of chromosome 1 unmasking an autosomal recessive 3-beta hydroxysteroid dehydrogenase type II-related congenital adrenal hyperplasia. J Clin Res Pediatr Endocrinol (2017) 9(1):70–3. doi: 10.4274/jcrpe.368
23. Janzen N, Sander S, Terhardt M, Steuerwald U, Peter M, Das AM, et al. Rapid steroid hormone quantification for congenital adrenal hyperplasia (CAH) in dried blood spots using UPLC liquid chromatography-tandem mass spectrometry. Steroids (2011) 76(13):1437–42. doi: 10.1016/j.steroids.2011.07.013
24. Janzen N, Riepe FG, Peter M, Sander S, Steuerwald U, Korsch E, et al. Neonatal screening: identification of children with 11beta-hydroxylase deficiency by second-tier testing. Horm Res Paediatr (2012) 77(3):195–9. doi: 10.1159/000337974
Keywords: CAH, real-time PCR, HSD3B2, deficiency of 3β-hydroxysteroid dehydrogenase, Ossetians
Citation: Makretskaya N, Kalinchenko N, Tebieva I, Ionova S, Zinchenko R, Marakhonov A and Tiulpakov A (2023) High carrier frequency of a nonsense p.Trp230* variant in HSD3B2 gene in Ossetians. Front. Endocrinol. 14:1146768. doi: 10.3389/fendo.2023.1146768
Received: 17 January 2023; Accepted: 04 May 2023;
Published: 16 May 2023.
Edited by:
Aleksandra Gilis-Januszewska, Jagiellonian University Medical College, PolandReviewed by:
Amalia Sertedaki, National and Kapodistrian University of Athens, GreeceSichang Zheng, Shanghai Jiao Tong University, China
Copyright © 2023 Makretskaya, Kalinchenko, Tebieva, Ionova, Zinchenko, Marakhonov and Tiulpakov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nina Makretskaya, bWFrcmV0c2theWFuQGdtYWlsLmNvbQ==
†These authors have contributed equally to this work