 Yangting Zhao1
Yangting Zhao1 Jingfang Liu
Jingfang Liu- 1The First Clinical Medical College, Lanzhou University, Lanzhou, Gansu, China
- 2Department of Endocrinology, The First Hospital of Lanzhou University, Lanzhou, Gansu, China
Diabetic cardiomyopathy (DCM) is a serious complication of type 1 and type 2 diabetes, which leads to the aggravation of myocardial fibrosis, disorders involving systolic and diastolic functions, and increased mortality of patients with diabetes through mechanisms such as glycolipid toxicity, inflammatory response, and oxidative stress. Ferroptosis is a form of iron-dependent regulatory cell death that is attributed to the accumulation of lipid peroxides and an imbalance in redox regulation. Increased production of lipid reactive oxygen species (ROS) during ferroptosis promotes oxidative stress and damages myocardial cells, leading to myocardial systolic and diastolic dysfunction. Overproduction of ROS is an important bridge between ferroptosis and DCM, and ferroptosis inhibitors may provide new targets for the treatment of patients with DCM.
1 Introduction
Diabetes mellitus is a chronic metabolic disorder caused by a combination of genetic and environmental factors, and its prevalence is increasing annually. The global prevalence of diabetes in people aged 20-79 years is currently estimated to be 10.5% (536.6 million people) and is expected to rise to 12.2% (783.2 million people) by 2045. There are currently 141 million adults with diabetes in China, and it is expected that by 2045, more than 174 million people will have diabetes (1).
Patients with diabetes are at a significantly higher risk of cardiovascular comorbidities owing to long-term metabolic disorders, persistent high glucose status, and insulin resistance (IR). Diabetic cardiomyopathy (DCM), a specific form of heart disease, has garnered considerable attention. DCM, also known as non-vascular myocardial dysfunction, refers to the development of abnormal myocardial structures and clinical manifestations in patients with diabetes in the absence of other cardiac risk factors, such as coronary artery disease, hypertension, and severe valvular disease (2).
There is still a lack of clear expert consensus on the definition of DCM, and its prevalence and incidence remain unclear. It has been estimated that early features of DCM can be observed in one-quarter to one-third of asymptomatic patients with diabetes. Among them, 15.5% with type 1 diabetes mellitus (T1DM) have abnormal myocardial systolic or diastolic function, whereas patients with type 2 diabetes mellitus (T2DM) are mostly middle-aged and elderly, with a relatively higher risk of cardiovascular system complications (3). A prospective study showed that the prevalence of DCM in subjects diagnosed with clinical or preclinical stages of DCM was approximately 38%, with a prevalence of 48% in female patients, mostly older than 65 years (4). DCM is also a major cause of heart failure (HF) in diabetic patients, and the risk of HF in patients with diabetes increases by 74% (5). The incidence of myocardial ischemia is 2.45 to 2.99 times that of non-diabetic patients (6). DCM is the leading cause of increased mortality among patients with diabetes in both developed and developing countries, and its prevalence increases with the incidence of obesity and T2DM, thereby placing a serious burden on the global economy and health management systems.
Ferroptosis is a new type of programmed cell death that is often accompanied by massive iron accumulation and lipid peroxidation, as well as a deficiency in oxidoreductases, especially glutathione peroxidase 4 (GPX4) (6, 7). In 2012, Dixon et al. named this death pattern of iron-dependent non-apoptotic cell death as “ferroptosis” (8). Ferroptosis can affect GPX4 directly or indirectly through different pathways, causing a decrease in cellular antioxidant capacity and accumulation of lipid reactive oxygen species (ROS), ultimately leading to cell death (7).
In recent years, studies have shown that ferroptosis is strongly associated with various diseases including ischemic heart disease, kidney disease, liver damage, degenerative diseases, and diabetes (6, 9). Ferroptosis inhibitors can improve the progression of DCM-related pathologies and may be used as novel therapeutic modalities for DCM treatment (10). This paper reviews the relationship and molecular mechanisms of ferroptosis and DCM to provide new ideas and targets for the treatment of DCM.
2 Roles and regulatory mechanisms of ferroptosis
2.1 Ferroptosis and iron metabolism
Iron is an important nutrient in the human body and is involved in a variety of biological processes, such as oxygen transport, lipid metabolism, DNA and protein synthesis, and cellular respiration. Abnormalities in the content or distribution of iron in the body may lead to the occurrence and progression of certain diseases (11). Intracellular iron metabolism mainly involves iron absorption, export, utilization, and storage, and an imbalance between these processes may affect the susceptibility of cells to ferroptosis (12). On the one hand, the redox properties of iron make free Fe2+ prone to undergo Fenton reactions with lipid peroxides, which produces a highly toxic hydroxyl radical and induces a strong oxidative stress response, which is a core mechanism for ferroptosis. On the other hand, Fe2+ and Fe3+ are also cofactors that can enhance the activities of various metabolic enzymes, resulting in the formation of free radicals such as alkoxy (RO) and peroxy (RO2), promoting lipid peroxidation, and inducing ferroptosis (13, 14). In addition, the degradation of ferritin and subsequent autophagy can increase the intracellular unstable iron content and enhance sensitivity to ferroptosis. Iron regulatory protein 1 (IRP1) and iron regulatory protein 2 (IRP2) regulate intracellular iron storage, release, entry, and exit, thereby maintaining cellular iron homeostasis. Changes in their levels may affect the amount of unstable iron in cells, thereby altering the sensitivity of cells to ferroptosis (15, 16). Therefore, an abnormal iron metabolism is necessary for ferroptosis.
2.2 Characteristics of ferroptosis
Ferroptosis is mainly characterized by iron overload, which causes substantial lipid peroxide production and, ultimately, cell death (9). Ferroptosis is morphologically, biochemically, and genetically different from cell death by apoptosis, necrosis, and autophagy. Ferroptosis is morphologically characterized by the contraction of mitochondria, reduction in volume, increased membrane density, and the reduction or disappearance of mitochondrial cristae. However, the cell nuclei are normal in size, and there is no chromatin condensation. This is an essential distinction from other cell death modalities such as apoptosis and necrosis. Ferroptosis is biochemically predominantly iron-dependent and is mainly characterized by an increase in Fe2+ concentration and lipids. Excessive production of ROS by Fe2+ through Fenton reactions causes intracellular lipid peroxidation, whereas iron enhances lipoxygenase (LOX) activity to promote ferroptosis (17). Genetically, ferroptosis is a biological process regulated by multiple genes, and a variety of genes involved in iron metabolism, lipid synthesis, and oxidative stress regulation can modulate ferroptosis (7).
2.3 Mechanism of ferroptosis
In addition to iron-dependent cell death, ferroptosis is caused by an imbalance between the production and degradation of intracellular lipid ROS and is regulated by multiple metabolic and redox systems. ROS are mainly produced by the iron-dependent Fenton reaction and mitochondrial nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) family enzymes. Mitochondria are the metabolic centers of most mammalian cells, and are important sources of ROS. Excessive ROS may result in oxidative damage to mitochondrial proteins and DNA, which impairs mitochondrial function, eventually leading to ferroptosis (18).
The system Xc- -glutathione (GSH)-GPX4 axis plays a central role in limiting lipid peroxidation. Ferroptosis is associated with the inactivation of cellular antioxidant systems, especially system Xc-, which causes the accumulation of lipid peroxides (17). System Xc- is an amino acid counter-transport protein widely distributed in phospholipid bilayers and consists of two independent proteins: solute carrier family 7 member 11 (SLC7A11) and solute carrier family 3 member 2 (SLC3A2) (19). GPX is the main endogenous mechanism that prevents peroxidation, among which GPX4 is a key regulator of ferroptosis and inhibits ferroptosis by inhibiting the production of lipid peroxides. GSH is an essential cofactor for GPX4, which converts reduced GSH to oxidized GSH (GSSG) and reduces toxic lipid peroxides (L-OOH) to non-toxic alcohols (L-OH), thereby decreasing oxidative stress damage and inhibiting the onset of ferroptosis (7, 20).
Cystine and glutamate are exchanged in and out of the cell via system Xc- at a ratio of 1:1. System Xc- transports extracellular cystine into the cell and converts it to cysteine, which is then used for GSH synthesis. GSH reduces ROS and reactive nitrogen species through the action of GPX (21). Thus, the inhibition of system Xc- activity can affect GSH synthesis by inhibiting cystine uptake, contributing to a reduction in GPX activity, a decrease in cellular antioxidant capacity, accumulation of lipid ROS, and ultimately the occurrence of oxidative damage and ferroptosis.
Ubiquinone, also known as coenzyme Q10 (CoQ10), is an electron carrier in the mitochondrial respiratory chain and a potent lipophilic antioxidant (22). Ubiquinol (CoQ10H2) is the reduced form of coenzyme Q10 that reduces oxidative stress, inhibits adipocyte differentiation, and suppresses lipid ROS accumulation (20). Mitochondrial CoQ10 inhibits apoptosis, whereas non-mitochondrial CoQ10 prevents ferroptosis. Ferroptosis suppressor protein 1 (FSP1), a flavoprotein oxidoreductase, may determine the role of CoQ10 in apoptosis and ferroptosis, and its pro-apoptotic function may be achieved through the inhibition of redox activity (18). FSP1, originally named apoptosis-inducing factor mitochondrial associated 2 (AIFM2), is also a mitochondrial pro-apoptotic protein (23). It has been found that FSP1 is able to use NADPH to regenerate lipophilic radicals to trap antioxidant; that is, FSP1 is responsible for regenerating CoQ10. FSP1 shuttles the reducing equivalents from NADPH to the lipid bilayer, allowing CoQ10 to interconvert with CoQ10H2, thereby inhibiting lipid peroxidation and maintaining the effective concentration of CoQ10H2. The FSP1-CoQ10-NADPH pathway is an independent parallel system, also known as the non-dependent GSH/GPX4 ferroptosis inhibition pathway, which is the main pathway of endogenous antioxidant enzyme regeneration, and synergistically inhibits lipid peroxidation and ferroptosis with GPX4 and GSH (24). Tetrahydrobiopterin (BH4) is an endogenous antioxidant that protects cells from ferroptosis by reducing lipid peroxidation. Dihydrobiopterin (BH2) can be reduced to BH4 by dihydrofolate reductase (DHFR) (25); therefore, DHFR is also an important regulator of ferroptosis.
Lipid peroxidation is a prerequisite for ferroptosis and is closely associated with lipid metabolism. Fatty acids (FA) are important components of cellular lipid metabolism and have a variety of cellular functions, including energy supply, cell membrane formation, and function as precursors to a variety of signaling molecules (26). The accumulation of polyunsaturated fatty acids (PUFA), such as arachidonic acid (AA), and the reduction in lipid peroxide scavenging capacity lead to ferroptosis (6). It has been shown that phosphatidyl ethanolamine (PE) containing AA or its derivative, adrenic acid (ADA), is a key phospholipid in the induction of cellular ferroptosis. Acyl CoA synthase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are two key enzymes involved in the synthesis of PE, activating PUFA and affecting their transmembrane properties. PUFA can be acylated by ACSL4 to form polyunsaturated fatty acid acyl-coenzyme A (PUFA-CoA), which is then esterified by LPCAT3 and finally reacts with PE to form PUFA- phosphatidyl ethanolamines (PUFA-PEs). PUFA-PEs can be oxidized by LOX to L-OOH products such as PE-AA-OOH or PE-ADA-OOH. Therefore, reducing the expression of ACSL4 and LPCAT3 can decrease the accumulation of intracellular lipid peroxides and inhibit ferroptosis (27).
Autophagy is a fundamental cellular homeostatic program, and excessive autophagy may trigger cell death, also known as autophagy-dependent cell death (18). Recent studies have shown that ferritinophagy is a unique form of selective autophagy mediated by nuclear receptor coactivator 4 (NCOA4) (10). Ferritinophagy promotes ferroptosis, which may be attributed to the iron overload caused by increased NCOA4 expression (28). NCOA4 mediates the degradation of ferritin after combining with ferritin heavy chain 1 (FTH1) as a selective autophagic receptor and converts ferritin-bound iron into free iron, thereby inducing ferroptosis (29) (Figure 1).
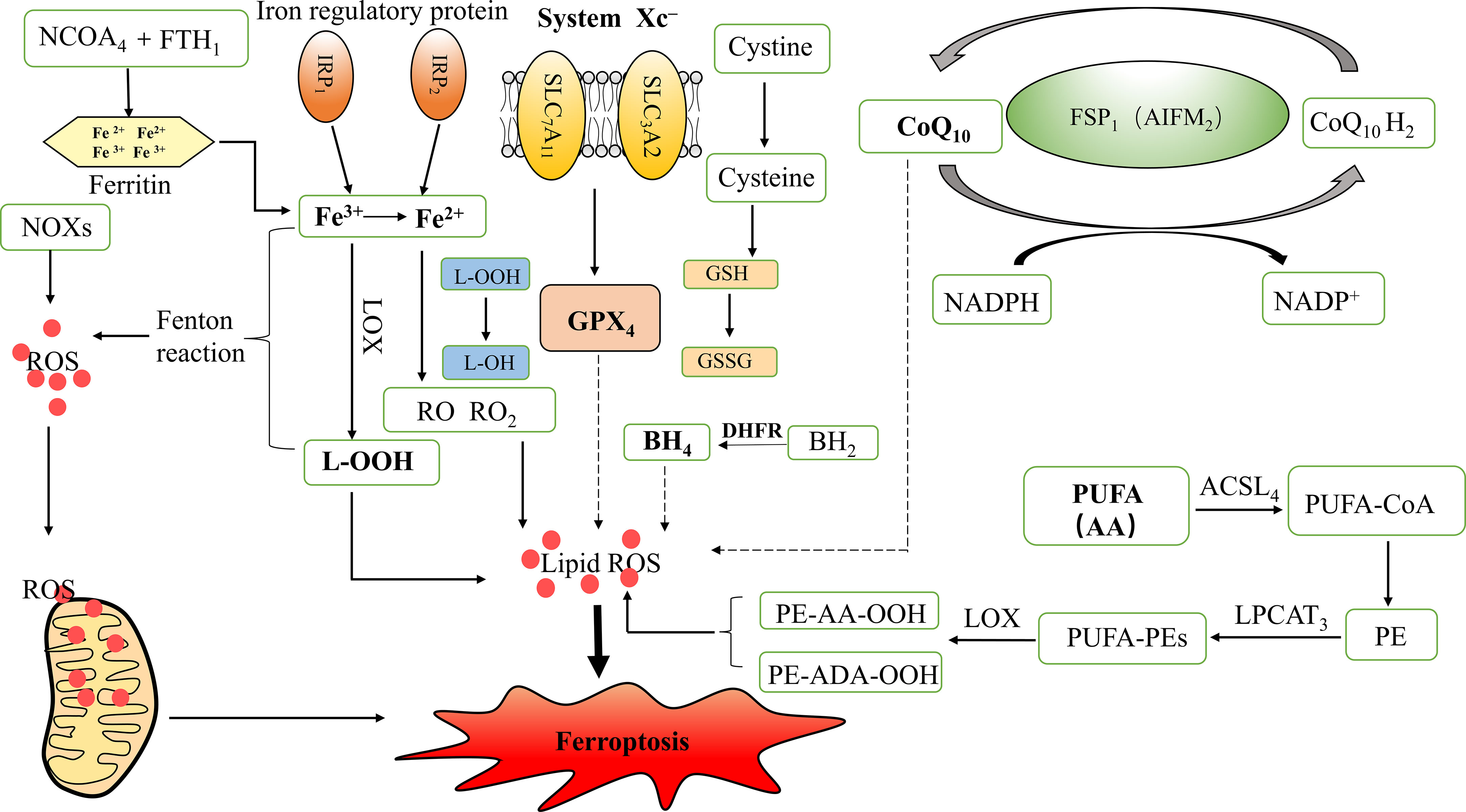
Figure 1 Mechanism of ferroptosis and iron metabolism. ROS, Reactive oxygen species; GPX4, Glutathione peroxidase 4; L-OOH, Lipid hydroperoxides; L-OH, Alcohol; RO, Alkoxy; RO2, Peroxy; IRP1, Iron regulatory protein 1; IRP2, Iron regulatory protein 2; LOX, Lipoxygenase; NADPH, Nicotinamide adenine dinucleotide phosphate; NOXs, NADPH Oxidases; GSH, Glutathione; GSSG, Oxidized GSH; SLC7A11, Solute carrier family 7 members 11; SLC3A2, Solute carrier family 3 member 2; CoQ10, Coenzyme Q10/Ubiquinone; CoQ10H2, Ubiquinol; FSP1, Ferroptosis suppressor protein 1; AIFM2, Apoptosis inducing factor mitochondrial associated 2; BH4, Tetrahydrobiopterin; BH2, Dihydrobiopterin; DHFR, Dihydrofolate reductase; PUFA, Polyunsaturated fatty acids; AA, Arachidonic acid; ADA, Adrenic acid; PE, Phosphatidyl ethanolamine; ACSL4, Acyl CoA synthase long-chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; PUFA-CoA, Polyunsaturated fatty acid acyl-coenzyme A; PUFA-PEs, PUFA- phosphatidyl ethanolamines; NCOA4, Nuclear receptor co-activator 4; FTH1, Ferritin heavy chain1.
3 Typing and diagnosis of diabetic cardiomyopathy and its pathogenesis
3.1 Typing and diagnostic criteria of diabetic cardiomyopathy
The histological features of DCM include myocardial collagen deposition, myocardial hypertrophy, and fibrosis (30). It is conventionally believed that the early stages of DCM are characterized by left ventricular (LV) hypertrophy and diastolic dysfunction, whereas the later stages progress to cardiac fibrosis and systolic dysfunction, eventually leading to HF (31). In 1954, Lundbaek first described DCM as a cardiomyopathy that affects two-thirds of elderly patients with diabetes mellitus (32). Two DCM phenotypes, HF with preserved LV ejection fraction (HFpEF) and HF with reduced LV ejection fraction (HFrEF), were identified based on the predominance of cardiomyocyte hypertrophy or apoptosis, the most common of which is the HFpEF phenotype (30).
The minimum diagnostic criteria for DCM include LV diastolic dysfunction and/or reduced LV ejection fraction (LVEF), LV hypertrophy, and interstitial fibrosis and can be divided into three stages: early, advanced, and end. DCM progresses from an initial subclinical phase, characterized by mild structural and functional abnormalities, then to severe diastolic HF with normal ejection fraction (EF), and eventually to HF with systolic dysfunction accompanied by a reduced EF value (33). One of the earliest features of DCM is abnormal LV wall stiffness, mainly secondary to extracellular matrix and myocardial cell remodeling. Cardiac magnetic resonance imaging (MRI) shows weakened ventricular wall motion in all components of myocardial deformation (longitudinal, radial, and circumferential strain). Reduced myocardial strain can also be observed in asymptomatic patients and in T2DM patients without other signs of etiologic heart disease. Therefore, reduced myocardial strain can be considered as a marker of preclinical DCM (30).
3.2 Pathophysiology of diabetic cardiomyopathy
Glyco-lipotoxicity, IR, inflammation, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress (ERS), and impaired intracellular Ca2+ handling may represent the molecular bases for DCM development. In addition, hyperglycemia-related epigenetic modifications may also play an important role in the development of DCM. Among them, histone modifications, especially histone deacetylases, are key regulators of cardiac fibrosis and myocardial hypertrophy (34). The synthesis of microRNA (miRNA) molecules may also be influenced by high glucose levels, and miRNA can affect inflammatory processes and cardiomyocyte survival by regulating oxidative stress response (35).
Oxidative stress can interfere with the balance between antioxidant capacity and ROS production, which is a core mechanism in the development of DCM, and a key regulator of myocardial fibrosis. Myocardial fibrosis is an important feature of cardiac remodeling in DCM (36). Wang et al. reported the induction of the pre- and early stages of DCM using a high-fat diet combined with T2DM rats induced by different doses of streptozotocin (STZ). The results showed increased ROS production in the low- and high-dose STZ treatment groups, with significantly increased cardiac weight index and myocardial collagen deposition in the high-dose group compared to the untreated group. In other words, cardiac structural remodeling, ROS production, and cell death are dysregulated in the pre- and early stages of DCM (37). Excessive ROS in cardiomyocytes leads to mitochondrial DNA damage, lipid peroxidation, post-translational modification of proteins, and eventually cell death and HF (38). ROS production activates redox-sensitive transcription factors and signal transduction pathways, such as protein kinase C (PKC), p38 mitogen-activated protein kinase (p38 MAPK), NH2-terminal Jun kinase (JNK), Janus kinase, and signal transducer and activator of transcription (JAK-STAT) pathways, further leading to impaired cardiac structure and function (39). ROS are mainly derived from NOXs, which are oxidative enzymes in the heart, particularly NOX2 and NOX4. NOX2-dependent signaling promotes several deleterious processes in cardiac pathology, including cardiomyocyte hypertrophy, contractile dysfunction, interstitial fibrosis, and cell death. In contrast, NOX4 plays important roles in endogenous detoxification reactions, angiogenesis, ERS, substrate utilization, and stress responses (40).
Increased oxidative stress in the heart is also due to cytoplasmic ROS-induced exacerbation of mitochondrial ROS production, and oxidative damage to the mitochondria further causes mitochondrial dysfunction (36). When mitochondrial ROS production exceeds endogenous clearance capacity, it leads to oxidative stress and inflammation. In addition, increased mitochondrial FA uptake and FA β-oxidation in DCM may induce the accumulation of toxic lipid metabolites, resulting in cardiac lipotoxicity and mitochondrial dysfunction (41). It has been recently shown that upregulation of A-kinase anchoring protein 1 (Akap1) may have the potential to treat myocardial injury in patients with DCM, where Akap1 expression is reduced in a diabetic mouse heart model. Thus, Akap1 deficiency may exacerbate DCM deterioration, increase mitochondrial ROS levels, and impair mitochondrial function. In contrast, cardiac-specific overexpression of Akap1 restores mitochondrial function and alleviates diabetes-induced cardiac dysfunction and myocardial fibrosis by ectopically regulating NADPH-CoQ in the mitochondria and decreasing mitochondrial ROS (42).
ERS and abnormal Ca2+ handling processes are associated with diastolic dysfunction in DCM. In patients with diabetes, persistent hyperglycemia and IR induce ERS and impair Ca2+ handling. However, impaired Ca2+ handling leads to an increased action potential duration, which consequently causes diastolic dysfunction (41). ERS causes increased myocardial ROS and impaired insulin signaling through activation of the JNK pathway, which is dependent on cellular redox status (43). Cardiomyocyte contractility is affected by impaired insulin signaling and reduced glucose uptake by cardiomyocytes. Additionally, Ca2+ is released from cardiomyocytes via the ryanodine receptor (RyR), which improves the susceptibility to oxidative stress injury induced by abnormal insulin metabolism and ROS production. Thus, it impairs Ca2+ efflux through L-type calcium channels, resulting in a reduction in efflux Ca2+ and an increase in intracellular Ca2+, which affects the systolic and diastolic functions of cardiac myocytes (44).
Altered cardiac substrate metabolic pathways and impaired energy metabolism are important factors in the pathogenesis of DCM. Under physiological conditions, the heart can employ FA and glucose as energy substrates. In contrast, in diabetic patients, the decrease in glucose uptake due to systemic and cardiac IR promotes a shift of substrates toward increased oxidation of free FA (FFA), resulting in reduced efficiency of cardiac metabolism. The heart loses the ability to utilize glucose, resulting in glucose overload in cardiomyocytes, which promotes the formation of advanced glycation end-products (AGEs) (30, 31). AGEs may play a key role in the development of DCM by stimulating collagen expression and accumulation and promoting collagen cross-linking, which causes increased myocardial fibrosis, reduced myocardial compliance, and impaired myocardial diastolic function (2). In addition, AGEs can also lead to increased intracytoplasmic ROS through activation of the AGE receptor (RAGE), thereby resulting in cardiac diastolic dysfunction (45). Activation of RAGE involves nuclear factor-κB (NF-κB) and its target genes, causing a shift from the α to the β isoform of myosin heavy chain in cardiac myocytes, which reduces myocardial contractility (46).
Alterations in the energy matrix of the heart enable increased FA uptake by cardiomyocytes to exceed the oxidative capacity of mitochondria, leading to excessive lipid storage and production of lipotoxic metabolites that impede cardiomyocyte metabolism and contractility, thereby promoting cardiomyocyte death (47). Excessive accumulation of FA in cardiac tissue and lipotoxicity can also impair insulin signaling, causing reduced cardiac metabolic capacity, increased myocardial oxygen consumption, and abnormal cardiac morphology and structure, ultimately resulting in a significant reduction in cardiac diastolic function (31). In cardiac tissues, the accumulation of lipid metabolites results in the reduced expression of glucose transporter 4 (GLUT4) and reduced translocation of GLUT4 from the cytoplasm to the cell membrane. As a result, glucose uptake by cardiomyocytes is significantly reduced. On the one hand, it may promote cardiac remodeling; on the other hand, it may inhibit cardioprotective pathways (48). Hyperglycemia and IR activate the PKC signaling pathway, allowing for an increase in myocardial endothelial cell permeability, resulting in endothelial dysfunction. In the pathogenesis of DCM, there is an imbalance in the release of vasoconstrictors [for example, prostanoids, endothelin, and angiotensin-II(Ang-II)] and diastolic agents [for example, nitric oxide (NO), prostacyclin (PGI2), bradykinin, and endothelium-derived hyperpolarizing factor (EDHF)] (49). NO, PGI2, and EDHF are released from coronary artery endothelial cells, allowing vasodilation. In the early stages of DCM and IR, NO-induced vasodilation is impaired and EDHF-mediated vasodilation is usually maintained or even enhanced to maintain a normal vascular tone. However, in later stages, both NO- and EDHF-induced vasodilation may eventually be impaired, resulting in microvascular dysfunction (41).
Diabetes mellitus is an inflammatory disease, and increased ROS levels induce an increase in inflammatory factors. Cardiac inflammation in DCM is mostly caused or exacerbated by increased ROS, and this pro-inflammatory state is largely caused by activation of ROS-triggered inflammatory vesicle nucleotide-binding oligomerization domain-like receptor pyrin domain containing 3 (NLRP3) (39). Furthermore, in obesity and IR, M1 macrophages polarize and secrete inflammatory cytokines, causing reduced cardiac insulin signaling and promoting DCM development. Conversely, M2 macrophages secrete IL-10, which inhibits cardiomyocyte hypertrophy and cardiac fibrosis (33). T-lymphocyte inducers contribute to an increase in pro-fibrotic cytokine levels in mouse heart tissue. In addition, T-helper lymphocytes secrete pro-inflammatory cytokines, which can bring about cardiac oxidative stress and coronary artery dysfunction, ultimately leading to cardiac remodeling, fibrosis, and diastolic dysfunction (50). C-reactive protein (CRP) is a cardiovascular pathogenic factor and well-known indicator of inflammation. Mano et al. showed that diabetic mice with CRP overexpression have increased levels of pro-inflammatory cytokines (IL-6 and TNF-α), increased type I collagen, increased expression of brain natriuretic peptide (BNP), and reduced LVEF on echocardiography compared to non-diabetic mice. This means that CRP overexpression may exacerbate LV function, cardiac remodeling, and myocardial fibrosis in DCM patients, possibly through inflammation and oxidative stress (51). Thus, systemic and local adaptive immune responses and inflammation may bring about alterations involving myocardial structure and metabolism, which in turn may cause diastolic dysfunction and eventually HF (Figure 2).
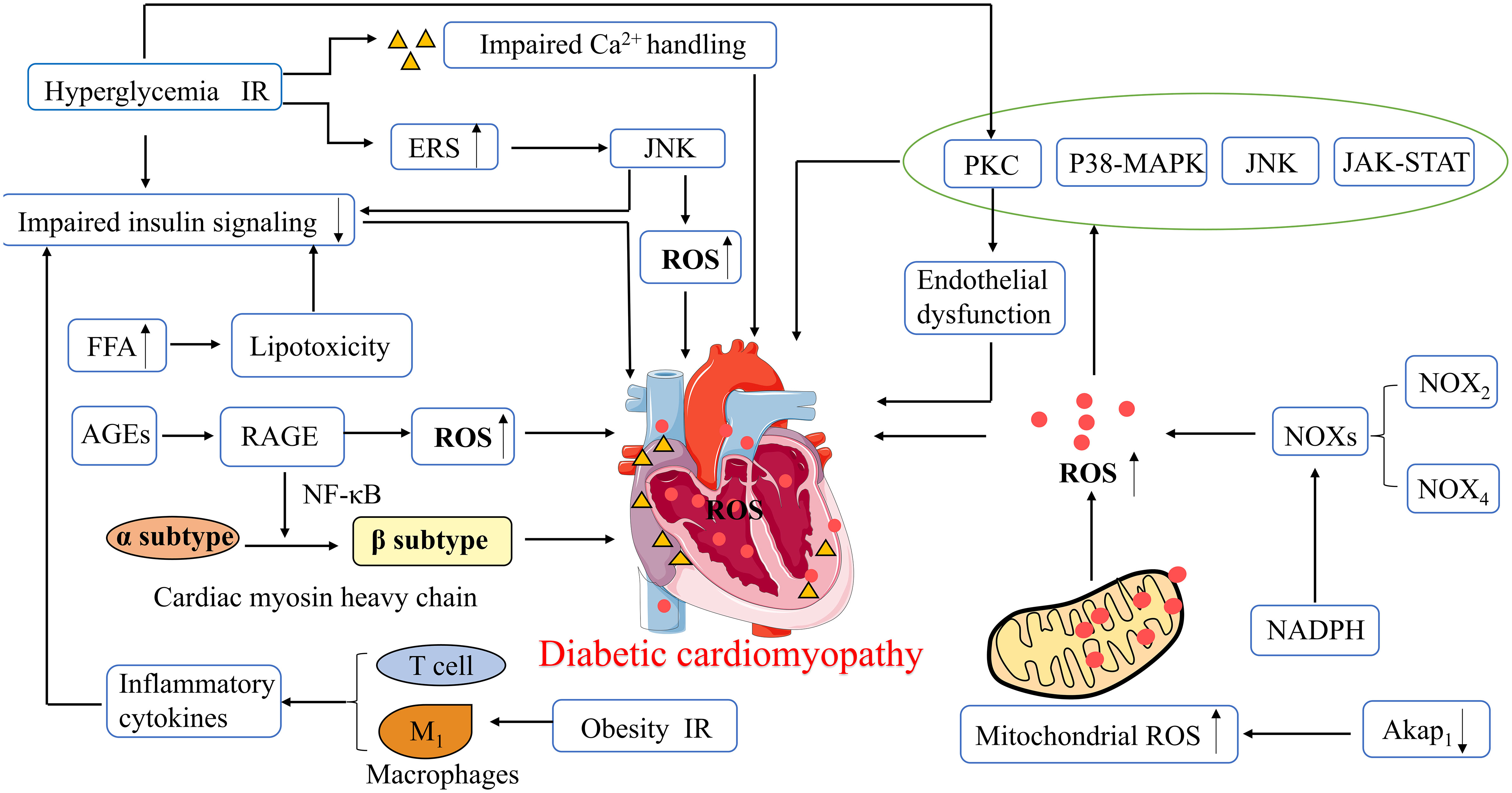
Figure 2 Pathophysiological mechanisms of diabetic cardiomyopathy. ROS, Reactive oxygen species; ERS, Endoplasmic reticulum stress; PKC, protein kinase C; p38 MAPK, p38 Mitogen activated protein kinase; JNK, Jun kinase; JAK -STAT, Janus kinase and signal transducer and activator of transcription; NADPH, Nicotinamide adenine dinucleotide phosphate; NOXs, NADPH Oxidases; Akap1, A-kinase anchoring protein 1; FFA, Free fatty acids; AGEs, Advanced glycation end-products; RAGE, AGE receptor; NF-κB, nuclear factor-κB; IR, Insulin resistance.
4 Role of ferroptosis in DCM
It has been shown that cardiomyocytes in diabetic patients are extremely sensitive to cell death, and their cardiomyocyte mortality is 85 times higher than that of non-diabetic patients. Consequently, cell death significantly affects the development of diabetes and its complications. Apoptosis is the main cause of cell death in DCM and promotes DCM cardiomyocyte injury through multiple upstream signaling pathways (52). However, the role of autophagy in DCM pathogenesis of DCM is unclear. Numerous studies have shown that autophagy protects cardiomyocytes in DCM; however, it has also been shown that autophagy can trigger cardiomyocyte injury. In a fructose-induced T2DM diabetic mouse model, upregulated autophagy was associated with elevated cardiac superoxide production and fibrosis, suggesting that autophagy may contribute to DCM (53). In addition, inhibition of various signaling pathways associated with necrosis may reduce myocardial damage in patients with diabetes (52). Ferroptosis is a specific type of cell death characterized by iron dependence and lipid peroxidation, which allows for increased levels of ROS and may promote the occurrence and development of DCM.
Dysregulation of internal environmental homeostasis and iron metabolism is closely related to the development of cardiac diseases, and iron overload is significantly detrimental to the development of ferroptosis and HF in cardiomyocytes (54). Studies have shown that ferroptosis may cause mitochondrial damage in the heart through iron overload, including abnormal mitochondrial structure, altered mitochondrial membrane potential, and increased mitochondrial ROS, which are also considered important features of ferroptosis (55). Iron overload in diabetic patients not only increases the risk of IR and diabetes progression but may also exacerbate cardiovascular complications through the Fenton response (56). DCM development is primarily associated with excessive ROS production and impaired antioxidant capacity. However, the high correlation between ferroptosis and lipid ROS production suggests that the inhibition of ferroptosis may be an important target for DCM prevention and treatment. Abnormal mitochondrial ferroptosis is observed in the hearts of diabetic mice, mainly manifested by decreased mitochondrial membrane potential, downregulated expression of key enzymes involved in antioxidant defence (superoxide dismutase [SOD2] and glutathione peroxidase 1 [GPX1]) in the mitochondria, and significantly increased mitochondrial ROS levels (57). Another study showed that mice fed a high-iron diet can cause severe myocardial damage and have a typical molecular profile of ferroptosis, including increased lipid peroxidation and reduced GSH levels (58). Thus, iron overload has a detrimental effect on cardiomyocyte function.
In the hyperglycemic state, oxidative stress and impaired antioxidant systems underlie DCM pathogenesis. Ferroptosis, which causes an imbalance between oxidation and antioxidation, typically causes the excessive production of ROS. This is clinically important because cardiomyocytes are highly susceptible to oxidative damage, and lipid peroxidation is involved in ROS-induced cardiac injury. Sampaio et al. reported increased expression of oxidative stress-carbonyl protein markers and myocardial fibrosis (type III collagen) in iron-overloaded T1DM diabetic rats (59). Ghosh et al. showed that GSH was reduced and ROS levels were increased in cardiomyocytes in an STZ diabetic rat model (60).
Glucose-induced persistent cardiomyocyte peroxide accumulation triggers ferroptosis and results in cellular damage at the whole-organism and cellular levels (61). In addition, during DCM onset, the energy metabolism of cardiomyocytes shifts from glycogenolysis to FA oxidation, resulting in increased intracellular lipid accumulation and lipotoxicity (62). However, ferroptosis involves multiple metabolic processes including iron metabolism, lipid metabolism, and in particular lipid peroxide production (63). Therefore, removal of lipid peroxide may attenuate cardiomyocyte injury.
A recent study suggested that the inhibition of ferroptosis by the activation of nuclear factor-erythroid 2-related factor 2 (NRF2) may represent a potential therapeutic target in DCM. NRF2 regulates multiple antioxidants and plays a key role in maintaining cellular redox reactions (64). NRF2 and its target genes have antioxidant, anti-inflammatory, anti-apoptotic, anti-ferroptosis, and anti-fibrotic functions that protect islet β-cells from high-glucose-induced oxidative damage in DCM (65).
Rutin, an NRF2 activator and phytochemical, has multiple pharmacological activities, including hypoglycemic and antioxidant activities. In a diabetic mouse model, rutin improved glucolipid metabolism in diabetic mice, attenuated myocardial damage caused by oxidative stress, including ventricular hypertrophy, ventricular remodeling, and ventricular dysfunction, and prevented the progression of myocardial fibrosis, thereby effectively alleviating DCM (66). Additionally, some key regulators of ferroptosis are also the downstream targets of NRF2, such as SLC7A11 and ferritin. SLC7A11 transports the precursor of GSH, cystine, into the cell matrix, and ferritin plays an important role in iron metabolism by storing excess cellular iron and alleviating the Fenton reaction (67). Almost all genes associated with ferroptosis are transcriptionally regulated by NRF2, including GSH-regulated and NADPH-regenerating genes that are essential for GPX4 activity, lipid peroxidation, and iron metabolism. The NRF2/Kelch-like-epichlorohydrin (ECH)-associated protein 1 (KEAP1)/antioxidant response element (ARE) pathway is the main mechanism of myocardial defence against oxidative damage in diabetes and hyperglycemia. It regulates the expression of several genes, most of which are associated with the reduction of oxidative stress and cell death (68). In addition, it has also been shown that the NRF2/ferroportin 1 (FPN1) signaling pathway is a key mechanism in diabetic myocardial injury, inhibiting ferroptosis by regulating iron metabolic homeostasis, and its activation alleviates diabetic myocardial injury to some extent (64). Furthermore, most patients with diabetes have abnormal lipid metabolism and produce excess saturated FA such as palmitic acid (PA), which plays a role in cardiomyocyte death and the development of DCM. Ferroptosis is associated with PA-induced myocardial injury, and ferroptosis inhibitors significantly reduce cell death in H9c2 and rat cardiomyocytes exposed to PA (10).
BH4 inhibits ferroptosis by inhibiting lipid peroxidation. BH4 is synthesized by GTP (guanosine triphosphate) cyclohydrolase 1 (GCH1) and acts as a cofactor in a variety of biosynthetic pathways, such as the synthesis of aromatic amino acids, neurotransmitters, and NO (69). Overexpression of GCH1 protects the heart from DCM and improves cardiac remodeling and dysfunction in a T1DM mouse heart model; therefore, GCH1 may serve as a new target for DCM therapy (70).
The increase in oxidative stress generated by iron overload through the Fenton reaction promotes the formation of AGEs, leading to lipid peroxidation, which is an important mechanism in DCM pathogenesis. In an ACE-treated diabetic rat model, ferroptosis inhibitors prevented the diastolic dysfunction of DCM, indicating an important role of ferroptosis in DCM. AGEs induce ferroptosis in engineered cardiac tissues (ECT), as evidenced by increased levels of unstable iron and lipid peroxides and decreased levels of GSH and SLC7A11 (67).
Endosomal sorting complexes required for transport (ESCRT) play multiple roles in membrane bending or outgrowth as multi-subunit machinery in the material transport process. Various cellular processes, including cell death, are affected by this membrane-remodeling mechanism. In particular, ESCRT-III initiates membrane repair to prevent various types of cell death, including necrosis, apoptosis, and ferroptosis. Activation of ESCRT-III on cell membranes requires Ca2+, confirming the role of calcium homeostasis and ERS in the control of ferroptosis (71). Abnormal calcium homeostasis and ERS are also important mechanisms in DCM development. ERS is a cellular response to endoplasmic reticulum dysfunction that can be induced by ROS, and inhibition of ferroptosis alleviates diabetic myocardial ischemia/reperfusion injury, which may provide a new therapeutic target for DCM. ROS are produced by the interaction between Fe2+ and NADPH oxidase during ferroptosis. ERS is characterized by the activation of the transcription factor 4 (ATF4)- CCAAT-enhancer-binding protein (C/EBP) homologous protein (CHOP) pathway. It has been shown that, in a diabetic rat model (high glucose and myocardial injury), the diabetic myocardial injury group showed increased levels of ACSL4, ATF4, and CHOP, severe impairment of cellular arrangement, cell swelling, and most of the myocardial fibers were broken compared to normal control group animals. However, when ferroptosis inhibitors were added, the degree of myocardial injury was significantly reduced in rats, and the levels of ROS, intracellular Fe2+ concentration, ACSL4, ATF4 and CHOP decreased, indicating that ferroptosis inhibitors can improve diabetic myocardial injury by reducing ERS (72).
Autophagy is a fundamental intracellular homeostatic process. Appropriate autophagy may be a pro-survival response; however, excessive autophagy, especially selective autophagy, and impaired lysosomal activity may promote ferroptosis. NCOA4-dependent ferritinophagy promotes ferroptosis by releasing free iron from ferritin (73). Many autophagic vesicles have been observed in cardiomyocytes of diabetic patients (74). However, in diabetic mice, 1,25-dihydroxyvitamin-D3 [1-25(OH)2VitD3] improved myocardial fibrosis and cardiac function in DCM through an autophagy-related vitamin D receptor (VDR)-dependent mechanism and the β-catenin/T-cell factor/lymphoid enhancer factor (TCF4)/glycogen synthase kinase-3β (GSK-3β)/mammalian target of rapamycin (mTOR) pathway (75). Glycogen autophagy plays an important role in DCM pathogenesis. In cultured primary cardiomyocytes, glycogen autophagy is regulated by extracellular glucose and insulin and occurs simultaneously with glycogen accumulation (76). In addition, glycogen autophagy is associated with a higher risk of heart disease (including DCM) in female patients, possibly because glycogen autophagy is associated with selective glycogen accumulation in the female myocardium, in which estrogen may upregulate the expression of signaling intermediates that promote glycogen storage (77) (Figure 3).
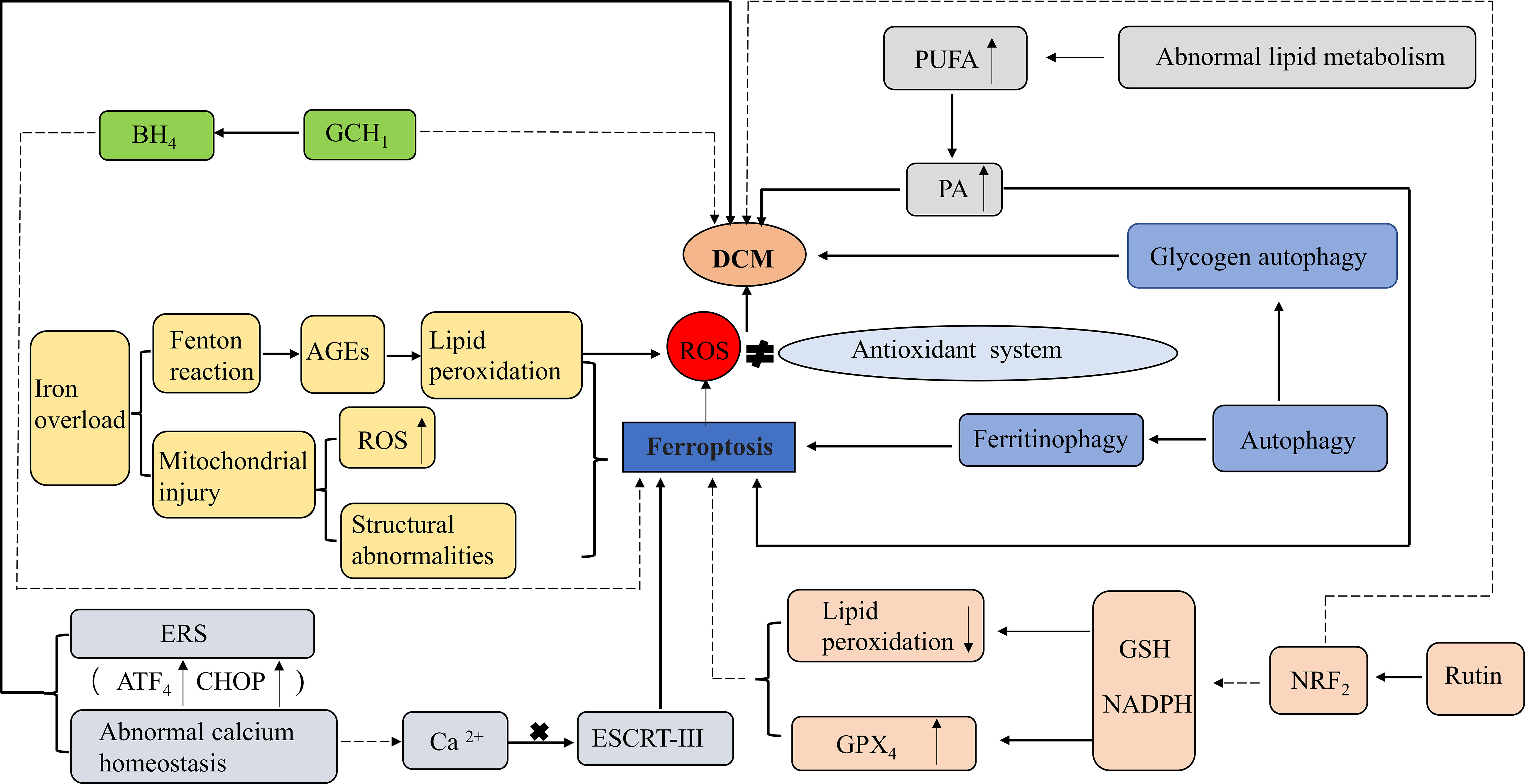
Figure 3 Role of ferroptosis in DCM. ROS, Reactive oxygen species;GPX4, Glutathione peroxidase 4; GSH, Glutathione; NADPH, Nicotinamide adenine dinucleotide phosphate; NRF2, nuclear factor-erythroid 2-related factor 2; PUFA, Polyunsaturated fatty acids; PA, palmitic acid; AGEs, Advanced glycation end-products; BH4, Tetrahydrobiopterin; GCH1GTP, (Guanosine triphosphate) cyclohydrolase 1; ERS, Endoplasmic reticulum stress; ESCRT-III, Endosomal sorting complexes required for transport III; ATF4, the activation of the transcription factor 4 (ATF4)- CCAAT-enhancer-binding protein (C/EBP) homologous protein (CHOP). Activation: Inhibition.
In STZ-induced T1DM mice, the heart showed significant features of DCM, such as myocardial systolic and diastolic dysfunction, impaired Ca2+ handling, and myocardial fibrosis (78). The inhibition of autophagy may allow an imbalance in NRF2-mediated metabolism as well as redox regulation, which increases iron deposition and lipid peroxidation, promoting cardiomyocyte ferroptosis and the progression of T1DM cardiomyopathy (79). However, in high-fat diet + STZ-induced T2DM mice, the heart is mainly characterized by myocardial steatosis, impaired systolic function, and mitochondrial dysfunction (78). Abnormal glucose and lipid metabolism causes the accumulation of AGEs in the extracellular matrix of the heart, leading to iron overload and lipid peroxidation, which ultimately induce ferroptosis in cardiomyocytes (67). The performance of ferroptosis in different animal models of diabetes is summarised in Table 1. Although ferroptosis has been shown to significantly improve cardiac function in animal models and cultured cells, no clinical trials have been performed to date; therefore, population-based studies are needed to determine whether selective blockade of ferroptosis improves the prognosis and/or outcomes of DCM.
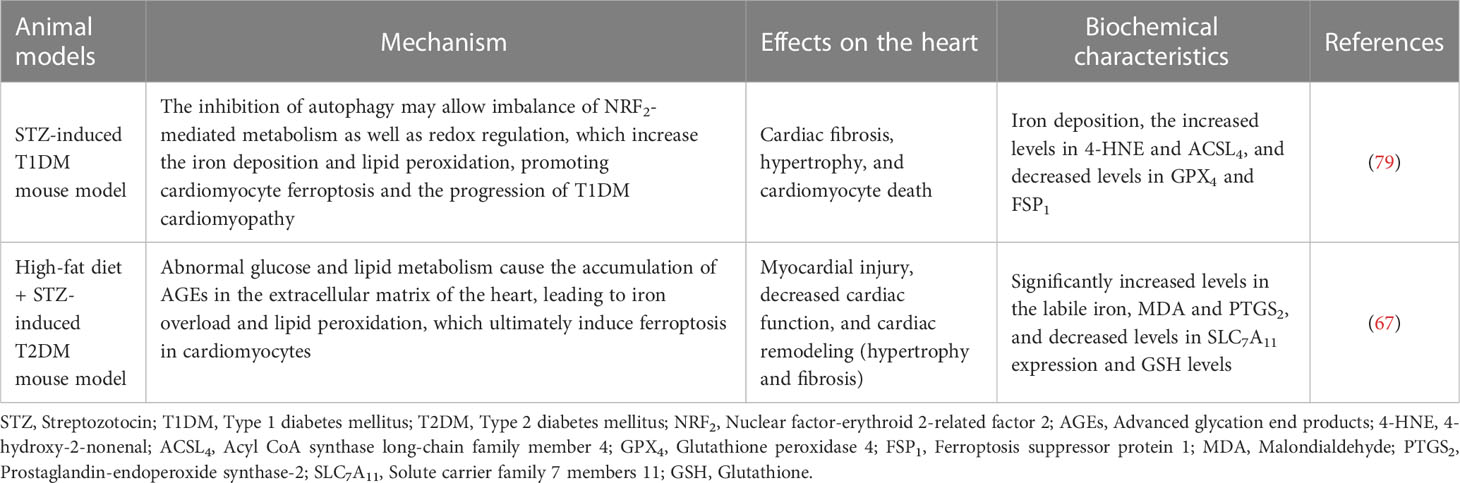
Table 1 The performance of ferroptosis in different types of animal models of diabetes.
5 Potential applications of ferroptosis inducers and inhibitors in DCM
Ferroptosis is promoted by class I (e.g., erastin) and class II (e.g., Ras-selective lethal 3 [RSL-3]) inducers, which indirectly and directly inhibit GPX4 activity, respectively (80). Erastin induces ferroptosis by inhibiting system Xc-, indirectly decreasing GPX4 activity, and directly targeting voltage-dependent anion channels 2 and 3 (VDAC2/3). In contrast, class II RSL-3 promotes ferroptosis primarily by inhibiting the endogenous lipid ROS inhibitor GPX4 (81).
Ferroptosis inhibitors act by inhibiting iron accumulation and reducing iron overload (e.g., deferoxamine [DFO]) or by inhibiting lipid peroxidation and reducing lipid ROS production (e.g., liproxstatin-1, vitamin E, and ferrostatin‐1 [Fer-1]) (7).
DFO has been shown to ameliorate cardiomyocyte injury in an in vitro DCM model of ECT exposed to AGE, reducing the expression of the lipid peroxidation marker malondialdehyde (MDA) and the ferroptosis marker prostaglandin-endoperoxide synthase-2 (PTGS2), improving ECT cardiomyocyte function. Liproxstatin-1 alleviated the decreased diastolic function 3 months after the onset of diabetes, further demonstrating the importance of ferroptosis in the pathogenesis of DCM (67).
Vitamin E is not only a ferroptosis inhibitor but also an endogenous antioxidant defence factor that plays an important role in DCM. Hamblin et al. found that the expression of two myocardial markers of oxidative stress, 8-iso-prostaglandin F2α (8-iso PGF2α) and GSSG, was increased, whereas that of LVEF was decreased in STZ-induced type I diabetic rats. However, after vitamin E supplementation, myocardial oxidative stress decreased and hemodynamic function was enhanced, further demonstrating the role of myocardial oxidative stress in DCM (82).
Fer-1, a potent inhibitor of ferroptosis, acts via lipid peroxidation. Treatment with moderate to high doses of Fer-1 reduced ACSL4 levels and enhanced GPX4 levels, thus reducing mitochondrial ROS production, alleviating mitochondrial dysfunction, and improving LV function in rats with cardiac injury (83). Herceptin (trastuzumab), a human epidermal growth factor receptor 2 (Her-2) gene-related targeted therapeutic agent for the treatment of breast cancer, also exerts toxic effects on the heart. H9c2 rat cardiomyocytes treated with herceptin exhibited decreased GPX4 and SLC7A11 expression with increasing doses of herceptin, inducing H9c2 cardiomyocyte injury, oxidative stress, mitochondrial dysfunction, and ferroptosis. However, the addition of Fer-1 restored GPX4 and SLC7A11 expression levels, which were otherwise inhibited by herceptin, and reversed the herceptin-induced increase in ACSL4 expression and increased mitochondrial ROS and iron levels, protecting H9c2 cardiomyocytes from herceptin-induced cardiomyocyte injury and ferroptosis (84).
Heat shock factor 1 (HSF1) is a stress-inducible transcription and defence factor against ferroptosis in cardiomyocytes that acts through the transcriptional activation of various heat shock proteins (HSP). A recent study found that PA induced cell death in cardiomyocytes in a dose- and time-dependent manner. Excess unoxidized PA in cardiomyocytes induces oxidative stress, mitochondrial dysfunction, and ceramide accumulation, whereas HSF1 significantly inhibits the death of H9c2 and rat cardiomyocytes exposed to PA by regulating the expression of GPX4 (10). The potential applications of ferroptosis inducers and inhibitors in DCM are summarised in Table 2.

Table 2 Potential applicationsof ferroptosis inducers and inhibitors in DCM.
6 Conclusions
An increasing number of studies have confirmed the relationship between ferroptosis and metabolic diseases, one of the more serious complications of diabetes mellitus, mainly through mechanisms such as glucolipotoxicity, inflammatory responses, and oxidative stress, resulting in increased myocardial tissue fibrosis and impaired systolic and diastolic functions. Among these, ROS overproduction may be considered an important bridge between the two. Large amounts of ROS promote oxidative stress and damage cardiac myocytes, which in turn leads to myocardial systolic and diastolic dysfunction. Thus, ferroptosis may be a new therapeutic target for diabetic cardiomyopathy. However, the role of ferroptosis in DCM is still poorly understood, and in-depth clinical studies are lacking. In addition, clinically reliable and sensitive markers for ferroptosis in early DCM are needed. Finally, objective DCM diagnostic criteria are still lacking, making it difficult to determine whether myocardial injury, hemodynamic changes, and decreased cardiac function caused by ferroptosis should be considered DCM or possibly other cardiovascular diseases, such as coronary atherosclerosis or ischemic cardiomyopathy.
Author contributions
All authors contributed to the study conception and design. The first draft of the manuscript was written by YZ, and all authors commented on previous versions of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the National Natural Science Foundation of China (No. 81960155), the Natural Science Foundation of Gansu Province (20JR10RA690), and the Health Industry Scientific Research Project of Gansu Province (No. GSWSKY-2019-07).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Park JJ. Epidemiology, pathophysiology, diagnosis and treatment of heart failure in diabetes. Diabetes Metab J (2021) 45(2):146–57. doi: 10.4093/dmj.2020.0282
3. Jensen MT, Sogaard P, Andersen HU, Bech J, Hansen TF, Galatius S, et al. Prevalence of systolic and diastolic dysfunction in patients with type 1 diabetes without known heart disease: the thousand & 1 study. Diabetologia (2014) 57(4):672–80. doi: 10.1007/s00125-014-3164-5
4. Kiencke S, Handschin R, von Dahlen R, Muser J, Brunner-Larocca HP, Schumann J, et al. Pre-clinical diabetic cardiomyopathy: prevalence, screening, and outcome. Eur J Heart Fail (2010) 12(9):951–7. doi: 10.1093/eurjhf/hfq110
5. Gulsin GS, Athithan L, McCann GP. Diabetic cardiomyopathy: prevalence, determinants and potential treatments. Ther Adv Endocrinol Metab (2019) 10:2042018819834869. doi: 10.1177/2042018819834869
6. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res (2021) 2021:9999612. doi: 10.1155/2021/9999612
7. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis (2020) 11(2):88. doi: 10.1038/s41419-020-2298-2
8. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
9. Li S, Huang Y. Ferroptosis: an iron-dependent cell death form linking metabolism, diseases, immune cell and targeted therapy. Clin Transl Oncol (2022) 24(1):1–12. doi: 10.1007/s12094-021-02669-8
10. Wang N, Ma H, Li J, Meng C, Zou J, Wang H, et al. HSF1 functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J Mol Cell Cardiol (2021) 150:65–76. doi: 10.1016/j.yjmcc.2020.10.010
11. Ravingerová T, Kindernay L, Barteková M, Ferko M, Adameová A, Zohdi V, et al. The molecular mechanisms of iron metabolism and its role in cardiac dysfunction and cardioprotection. Int J Mol Sci (2020) 21(21):7889. doi: 10.3390/ijms21217889
12. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell (2019) 51(5):575–586.e4. doi: 10.1016/j.devcel.2019.10.007
13. Valko M, Jomova K, Rhodes CJ, Kuča K, Musílek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol (2016) 90(1):1–37. doi: 10.1007/s00204-015-1579-5
14. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer (2013) 13(5):342–55. doi: 10.1038/nrc3495
15. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy (2016) 12(8):1425–8. doi: 10.1080/15548627.2016.1187366
16. Andrews NC, Schmidt PJ. Iron homeostasis. Annu Rev Physiol (2007) 69:69–85. doi: 10.1146/annurev
17. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
19. Zheng K, Dong Y, Yang R, Liang Y, Wu H, He Z. Regulation of ferroptosis by bioactive phytochemicals: Implications for medical nutritional therapy. Pharmacol Res (2021) 168:105580. doi: 10.1016/j.phrs.2021.105580
20. Zhang Y, Xin L, Xiang M, Shang C, Wang Y, Wang Y, et al. The molecular mechanisms of ferroptosis and its role in cardiovascular disease. BioMed Pharmacother (2022) 145:112423. doi: 10.1016/j.biopha.2021.112423
21. Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal (2013) 18(5):522–55. doi: 10.1089/ars.2011.4391
22. Frei B, Kim MC, Ames BN. Ubiquinol-10 is an effective lipid-soluble antioxidant at physiological concentrations. Proc Natl Acad Sci U.S.A. (1990) 87(12):4879–83. doi: 10.1073/pnas.87.12.4879
23. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
24. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
25. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol (2020) 16(12):1351–60. doi: 10.1038/s41589-020-0613-y
26. DeBose-Boyd RA. Significance and regulation of lipid metabolism. Semin Cell Dev Biol (2018) 81:97. doi: 10.1016/j.semcdb.2017.12.003
27. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol (2017) 13(1):81–90. doi: 10.1038/nchembio.2238
28. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature (2014) 509(7498):105–9. doi: 10.1038/nature13148
29. Qin Y, Qiao Y, Wang D, Tang C, Yan G. Ferritinophagy and ferroptosis in cardiovascular disease: Mechanisms and potential applications. BioMed Pharmacother (2021) 141:111872. doi: 10.1016/j.biopha.2021.111872
30. Joubert M, Manrique A, Cariou B, Prieur X. Diabetes-related cardiomyopathy: The sweet story of glucose overload from epidemiology to cellular pathways. Diabetes Metab (2019) 45(3):238–47. doi: 10.1016/j.diabet.2018.07.003
31. Paolillo S, Marsico F, Prastaro M, Renga F, Esposito L, De Martino F, et al. Diabetic cardiomyopathy: Definition, diagnosis, and therapeutic implications. Heart Fail Clin (2019) 15(3):341–7. doi: 10.1016/j.hfc.2019.02.003
32. Lundbaek K. Diabetic angiopathy: a specific vascular disease. Lancet (1954) 266(6808):377–9. doi: 10.1016/s0140-6736(54)90924-1
33. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol (2016) 12(3):144–53. doi: 10.1038/nrendo.2015.216
34. Ke X, Lin Z, Ye Z, Leng M, Chen B, Jiang C, et al. Histone deacetylases in the pathogenesis of diabetic cardiomyopathy. Front Endocrinol (Lausanne) (2021) 12:679655. doi: 10.3389/fendo.2021.679655
35. Jakubik D, Fitas A, Eyileten C, Jarosz-Popek J, Nowak A, Czajka P, et al. MicroRNAs and long non-coding RNAs in the pathophysiological processes of diabetic cardiomyopathy: emerging biomarkers and potential therapeutics. Cardiovasc Diabetol (2021) 20(1):55. doi: 10.1186/s12933-021-01245-2
36. Sung MM, Hamza SM, Dyck JR. Myocardial metabolism in diabetic cardiomyopathy: potential therapeutic targets. Antioxid Redox Signal (2015) 22(17):1606–30. doi: 10.1089/ars.2015.6305
37. Wang D, Liu K, Zhong J, Li X, Zhang J, Wang G, et al. Molecular correlates of early onset of diabetic cardiomyopathy: Possible therapeutic targets. Oxid Med Cell Longev (2022) 2022:9014155. doi: 10.1155/2022/9014155
38. Byrne NJ, Rajasekaran NS, Abel ED, Bugger H. Therapeutic potential of targeting oxidative stress in diabetic cardiomyopathy. Free Radic Biol Med (2021) 169:317–42. doi: 10.1016/j.freeradbiomed.2021.03.046
39. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res (2020) 126(11):1501–25. doi: 10.1161/CIRCRESAHA.120.315913
40. Hansen SS, Aasum E, Hafstad AD. The role of NADPH oxidases in diabetic cardiomyopathy. Biochim Biophys Acta Mol Basis Dis (2018) 1864(5 Pt B):1908–13. doi: 10.1016/j.bbadis.2017.07.025
41. Jia G, Whaley-Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia- and insulin-resistance-induced heart disease. Diabetologia (2018) 61(1):21–8. doi: 10.1007/s00125-017-4390-4
42. Qi B, He L, Zhao Y, Zhang L, He Y, Li J, et al. Akap1 deficiency exacerbates diabetic cardiomyopathy in mice by NDUFS1-mediated mitochondrial dysfunction and apoptosis. Diabetologia (2020) 63(5):1072–87. doi: 10.1007/s00125-020-05103-w
43. Hotamisligil GS. Inflammation and endoplasmic reticulum stress in obesity and diabetes. Int J Obes (Lond) (2008) 32 Suppl 7(Suppl 7):S52–4. doi: 10.1038/ijo.2008.238
44. Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res (2018) 122(10):1460–78. doi: 10.1161/CIRCRESAHA.118.310082
45. Ma H, Li SY, Xu P, Babcock SA, Dolence EK, Brownlee M, et al. Advanced glycation endproduct (AGE) accumulation and AGE receptor (RAGE) up-regulation contribute to the onset of diabetic cardiomyopathy. J Cell Mol Med (2009) 13(8B):1751–64. doi: 10.1111/j.1582-4934.2008.00547.x
46. Aragno M, Mastrocola R, Medana C, Catalano MG, Vercellinatto I, Danni O, et al. Oxidative stress-dependent impairment of cardiac-specific transcription factors in experimental diabetes. Endocrinology (2006) 147(12):5967–74. doi: 10.1210/en.2006-0728
47. Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitus. Circ Res (2019) 124(1):121–41. doi: 10.1161/CIRCRESAHA.118.311371
48. Atkinson LL, Kozak R, Kelly SE, Onay Besikci A, Russell JC, Lopaschuk GD. Potential mechanisms and consequences of cardiac triacylglycerol accumulation in insulin-resistant rats. Am J Physiol Endocrinol Metab (2003) 284(5):E923–30. doi: 10.1152/ajpendo.00360.2002
49. Knapp M, Tu X, Wu R. Vascular endothelial dysfunction, a major mediator in diabetic cardiomyopathy. Acta Pharmacol Sin (2019) 40(1):1–8. doi: 10.1038/s41401-018-0042-6
50. Yu Q, Vazquez R, Zabadi S, Watson RR, Larson DF. T-Lymphocytes mediate left ventricular fibrillar collagen cross-linking and diastolic dysfunction in mice. Matrix Biol (2010) 29(6):511–8. doi: 10.1016/j.matbio.2010.06.003
51. Mano Y, Anzai T, Kaneko H, Nagatomo Y, Nagai T, Anzai A, et al. Overexpression of human c-reactive protein exacerbates left ventricular remodeling in diabetic cardiomyopathy. Circ J (2011) 75(7):1717–27. doi: 10.1253/circj.cj-10-1199
52. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol (2020) 11:42. doi: 10.3389/fphar.2020.00042
53. Mellor KM, Bell JR, Young MJ, Ritchie RH, Delbridge LM. Myocardial autophagy activation and suppressed survival signaling is associated with insulin resistance in fructose-fed mice. J Mol Cell Cardiol (2011) 50(6):1035–43. doi: 10.1016/j.yjmcc.2011.03.002
54. Mancardi D, Mezzanotte M, Arrigo E, Barinotti A, Roetto A. Iron overload, oxidative stress, and ferroptosis in the failing heart and liver. Antioxid (Basel) (2021) 10(12):1864. doi: 10.3390/antiox10121864
55. Sumneang N, Siri-Angkul N, Kumfu S, Chattipakorn SC, Chattipakorn N. The effects of iron overload on mitochondrial function, mitochondrial dynamics, and ferroptosis in cardiomyocytes. Arch Biochem Biophys (2020) 680:108241. doi: 10.1016/j.abb.2019.108241
56. Liu Q, Sun L, Tan Y, Wang G, Lin X, Cai L. Role of iron deficiency and overload in the pathogenesis of diabetes and diabetic complications. Curr Med Chem (2009) 16(1):113–29. doi: 10.2174/092986709787002862
57. Wang SY, Zhu S, Wu J, Zhang M, Xu Y, Xu W, et al. Exercise enhances cardiac function by improving mitochondrial dysfunction and maintaining energy homoeostasis in the development of diabetic cardiomyopathy. J Mol Med (Berl) (2020) 98(2):245–61. doi: 10.1007/s00109-019-01861-2
58. Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin h facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res (2020) 127(4):486–501. doi: 10.1161/CIRCRESAHA.120.316509
59. Sampaio AF, Silva M, Dornas WC, Costa DC, Silva ME, Dos Santos RC, et al. Iron toxicity mediated by oxidative stress enhances tissue damage in an animal model of diabetes. Biometals (2014) 27(2):349–61. doi: 10.1007/s10534-014-9717-8
60. Ghosh S, Ting S, Lau H, Pulinilkunnil T, An D, Qi D, et al. Increased efflux of glutathione conjugate in acutely diabetic cardiomyocytes. Can J Physiol Pharmacol (2004) 82(10):879–87. doi: 10.1139/y04-060
61. Wei Z, Shaohuan Q, Pinfang K, Chao S. Curcumin attenuates ferroptosis-induced myocardial injury in diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc Ther (2022) 2022:3159717. doi: 10.1155/2022/3159717
62. Avagimyan A, Popov S, Shalnova S. The pathophysiological basis of diabetic cardiomyopathy development. Curr Probl Cardiol (2022) 47(9):101156. doi: 10.1016/j.cpcardiol.2022.101156
63. Yang WS, Stockwell BR. Ferroptosis: Death by lipid peroxidation. Trends Cell Biol (2016) 26(3):165–76. doi: 10.1016/j.tcb.2015.10.014
64. Tian H, Xiong Y, Zhang Y, Leng Y, Tao J, Li L, et al. Activation of NRF2/FPN1 pathway attenuates myocardial ischemia-reperfusion injury in diabetic rats by regulating iron homeostasis and ferroptosis. Cell Stress Chaperones (2021) 27(2):149–64. doi: 10.1007/s12192-022-01257-1
65. Zhai Z, Zou P, Liu F, Xia Z, Li J. Ferroptosis is a potential novel diagnostic and therapeutic target for patients with cardiomyopathy. Front Cell Dev Biol (2021) 9:649045. doi: 10.3389/fcell.2021.649045
66. Huang R, Shi Z, Chen L, Zhang Y, Li J, An Y. Rutin alleviates diabetic cardiomyopathy and improves cardiac function in diabetic ApoEknockout mice. Eur J Pharmacol (2017) 814:151–60. doi: 10.1016/j.ejphar.2017.08.023
67. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B (2022) 12(2):708–22. doi: 10.1016/j.apsb.2021.10.005
68. Ge ZD, Lian Q, Mao X, Xia Z. Current status and challenges of NRF2 as a potential therapeutic target for diabetic cardiomyopathy. Int Heart J (2019) 60(3):512–20. doi: 10.1536/ihj.18-476
69. Nehring H, Meierjohann S, Friedmann Angeli JP. Emerging aspects in the regulation of ferroptosis. Biochem Soc Trans (2020) 48(5):2253–9. doi: 10.1042/BST20200523
70. Wu HE, Baumgardt SL, Fang J, Paterson M, Liu Y, Du J, et al. Cardiomyocyte GTP cyclohydrolase 1 protects the heart against diabetic cardiomyopathy. Sci Rep (2016) 6:27925. doi: 10.1038/srep27925
71. Dai E, Meng L, Kang R, Wang X, Tang D. ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun (2020) 522(2):415–21. doi: 10.1016/j.bbrc.2019.11.110
72. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial Ischemia/Reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol (2020) 39(2):210–25. doi: 10.1089/dna.2019.5097
73. Liu J, Kuang F, Kroemer G, Klionsky DJ, Kang R, Tang D. Autophagy-dependent ferroptosis: Machinery and regulation. Cell Chem Biol (2020) 27(4):420–35. doi: 10.1016/j.chembiol.2020.02.005
74. Kanamori H, Naruse G, Yoshida A, Minatoguchi S, Watanabe T, Kawaguchi T, et al. Morphological characteristics in diabetic cardiomyopathy associated with autophagy. J Cardiol (2021) 77(1):30–40. doi: 10.1016/j.jjcc.2020.05.009
75. Wei H, Qu H, Wang H, Ji B, Ding Y, Liu D, et al. 1,25-Dihydroxyvitamin-D3 prevents the development of diabetic cardiomyopathy in type 1 diabetic rats by enhancing autophagy via inhibiting the β-catenin/TCF4/GSK-3β/mTOR pathway. J Steroid Biochem Mol Biol (2017) 168:71–90. doi: 10.1016/j.jsbmb.2017.02.007
76. Mellor KM, Varma U, Stapleton DI, Delbridge LM. Cardiomyocyte glycophagy is regulated by insulin and exposure to high extracellular glucose. Am J Physiol Heart Circ Physiol (2014) 306(8):H1240–5. doi: 10.1152/ajpheart.00059.2014
77. Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LM. Myocardial glycophagy - a specific glycogen handling response to metabolic stress is accentuated in the female heart. J Mol Cell Cardiol (2013) 65:67–75. doi: 10.1016/j.yjmcc.2013.09.014
78. Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech (2009) 2(9-10):454–66. doi: 10.1242/dmm.001941
79. Zang H, Wu W, Qi L, Tan W, Nagarkatti P, Nagarkatti M, et al. Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice. Diabetes (2020) 69(12):2720–34. doi: 10.2337/db19-1176
80. Ju J, Song YN, Wang K. Mechanism of ferroptosis: A potential target for cardiovascular diseases treatment. Aging Dis (2021) 12(1):261–76. doi: 10.14336/AD.2020.0323
81. Baba Y, Higa JK, Shimada BK, Horiuchi KM, Suhara T, Kobayashi M, et al. Protective effects of the mechanistic target of rapamycin against excess iron and ferroptosis in cardiomyocytes. Am J Physiol Heart Circ Physiol (2018) 314(3):H659–68. doi: 10.1152/ajpheart.00452.2017
82. Hamblin M, Smith HM, Hill MF. Dietary supplementation with vitamin e ameliorates cardiac failure in type I diabetic cardiomyopathy by suppressing myocardial generation of 8-iso-prostaglandin F2alpha and oxidized glutathione. J Card Fail (2007) 13(10):884–92. doi: 10.1016/j.cardfail.2007.07.002
83. Luo Y, Apaijai N, Liao S, Maneechote C, Chunchai T, Arunsak B, et al. Therapeutic potentials of cell death inhibitors in rats with cardiac ischaemia/reperfusion injury. J Cell Mol Med (2022) 26(8):2462–76. doi: 10.1111/jcmm.17275
Keywords: diabetes mellitus, diabetic cardiomyopathy, ferroptosis, iron metabolism, oxidative stress
Citation: Zhao Y, Pan B, Lv X, Chen C, Li K, Wang Y and Liu J (2023) Ferroptosis: roles and molecular mechanisms in diabetic cardiomyopathy. Front. Endocrinol. 14:1140644. doi: 10.3389/fendo.2023.1140644
Received: 09 January 2023; Accepted: 28 March 2023;
Published: 20 April 2023.
Edited by:
Guanghong Jia, University of Missouri, United StatesReviewed by:
Rajesh Mohanraj, United Arab Emirates University, United Arab EmiratesRui He, Ningxia Medical University, China
Ramoji Kosuru, Versiti Blood Research Institute, United States
Copyright © 2023 Zhao, Pan, Lv, Chen, Li, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingfang Liu, bGpmODI0MTY4QDEyNi5jb20=