
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 15 August 2023
Sec. Endocrinology of Aging
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1139692
This article is part of the Research TopicA Year in Review: Discussions in Endocrinology of AgingView all 5 articles
 Nazanin Aghamohamadi1Faezeh Shahba1
Nazanin Aghamohamadi1Faezeh Shahba1 Ali Zarezadeh Mehrabadi1
Ali Zarezadeh Mehrabadi1 Hossein Khorramdelazad1,2Milad Karimi1
Hossein Khorramdelazad1,2Milad Karimi1 Reza Falak1*
Reza Falak1* Reza Zolfaghari Emameh3*
Reza Zolfaghari Emameh3*Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is potentially pathogenic and causes severe symptoms; in addition to respiratory syndromes, patients might experience other severe conditions such as digestive complications and liver complications injury. The abnormality in the liver is manifested by hepatobiliary dysfunction and enzymatic elevation, which is associated with morbidity and mortality. The direct cytopathic effect, immune dysfunction, cytokine storm, and adverse effects of therapeutic regimens have a crucial role in the severity of liver injury. According to aging and immune system alterations, cytokine patterns may also change in the elderly. Moreover, hyperproduction of cytokines in the inflammatory response to SARS-CoV-2 can lead to multi-organ dysfunction. The mortality rate in elderly patients, particularly those with other comorbidities, is also higher than in adults. Although the pathogenic effect of SARS-CoV-2 on the liver has been widely studied, the impact of age and immune-mediated responses at different ages remain unclear. This review discusses the association between immune system responses in coronavirus disease 2019 (COVID-19) patients of different ages and liver injury, focusing on cytokine alterations.
The coronaviridae family comprises enveloped viruses infecting amphibians, birds, and mammals. The family consists of the torovirinae and orthocoronavirinae; the subfamily orthocoronavirinae includes alpha, beta, gamma, and deltacoronaviruses (1–4). Coronaviruses have crown-like structures due to spike glycoproteins. These viruses have an outer envelope and contain positive-stranded RNA as their genomic material (5).
Some species of coronaviruses may not lead to severe disease and are typically responsible for common colds. However, severe acute respiratory syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and SARS-CoV-2 are potentially pathogenic to humans. These viruses can cause severe respiratory complications, multiple organ failure, and even death in severe stages (6, 7). SARS-CoV-2, the last known species of the coronavirus family, is a beta-coronavirus and was identified in Wuhan Hubei Province, China, in late 2019 (8). The COVID-19 clinical manifestations vary from sore throat, cough, fever, and loss of smell or taste to acute respiratory distress syndrome (ARDS), leading to patient hospitalization (9). Moreover, due to SARS-CoV-2-induced thrombotic disorders, various organ-specific complications have been observed, including heart injury, acute kidney injury, liver dysfunction, central nervous system (CNS) complications, and gastroenteritis (9, 10).
According to the lessons from patients with SARS-CoV-1, various levels of hepatopathy and elevated liver enzymes suggest that coronaviruses are responsible for inducing systemic inflammation (11–13). In a similar behavior, SARS-CoV-2 can also increase aminotransferase levels (14).
Several hepatotoxic medications, particularly those used to treat COVID-19, have been related to drug-induced liver damage (15). However, liver injury is defined as any damage to the liver that occurs during disease or treatment (16). As a result, the proportion of hospitalized COVID-19 patients with altered liver biomarkers ranges from 14% to 53%. Evidence has revealed that the levels of aminotransferase, bilirubin, alkaline phosphatase (ALP), and gamma-glutamyl transferase (GGT) are remarkably increased in patients with COVID-19 (17, 18). In contrast, serum albumin levels decrease in SARS-CoV-2-induced liver injury (19).
Cytokine storm is characterized as an abnormal, excessive, and uncontrolled systemic inflammatory response associated with the hyperproduction of pro-inflammatory cytokines. This inflammatory phenomenon is caused by infection, drugs, or an allogeneic hypersensitivity to foreign tissue, resulting in multi-organ functional impairment (20). Among the affected body organs, the liver is more susceptible to disruptions in systemic homeostasis and is influenced by various mechanisms, including vascular and immune-mediated pathways (21). The pathophysiology of liver dysfunction caused by the cytokine storm is characterized by increased vascular permeability due to cytokine release, hypoperfusion, endothelial dysfunction, reactive oxygen species (ROS) production, and nitric oxide (NO) deficiency (22). Furthermore, dysregulated cytokine release can cause an acute-phase response in hepatocytes (23). Multiple studies have demonstrated that immune responses vary with age, and this is also true for COVID-19 patients and their related cytokine responses (24, 25). Additionally, aging can affect cytokine patterns and exacerbate inflammation (26). Since the tissue damage caused by the cytokine storm is critical, aging and increased inflammation caused by SARS-CoV-2 infection may lead to more severe liver damage in the elderly than in younger patients (27).
Therefore, this review summarizes the association of age-related immune responses and cytokine patterns with liver injuries during SARS-CoV-2 infection or following treatment and vaccination.
Aging is an intricate biological process associated with numerous alterations that can affect several organs’ functions and represents the leading risk factor for geriatric diseases (28). Age-related changes occur in every immune system component, called “immunosenescence,” and affect innate and adaptive immune responses (29, 30). These alterations in the immune system consist of persistent low-grade inflammation (called inflammaging), impaired ability to respond to new antigens effectively, and increased susceptibility to infections, autoimmunity, as well as cancer (Figure 1) (31, 32).
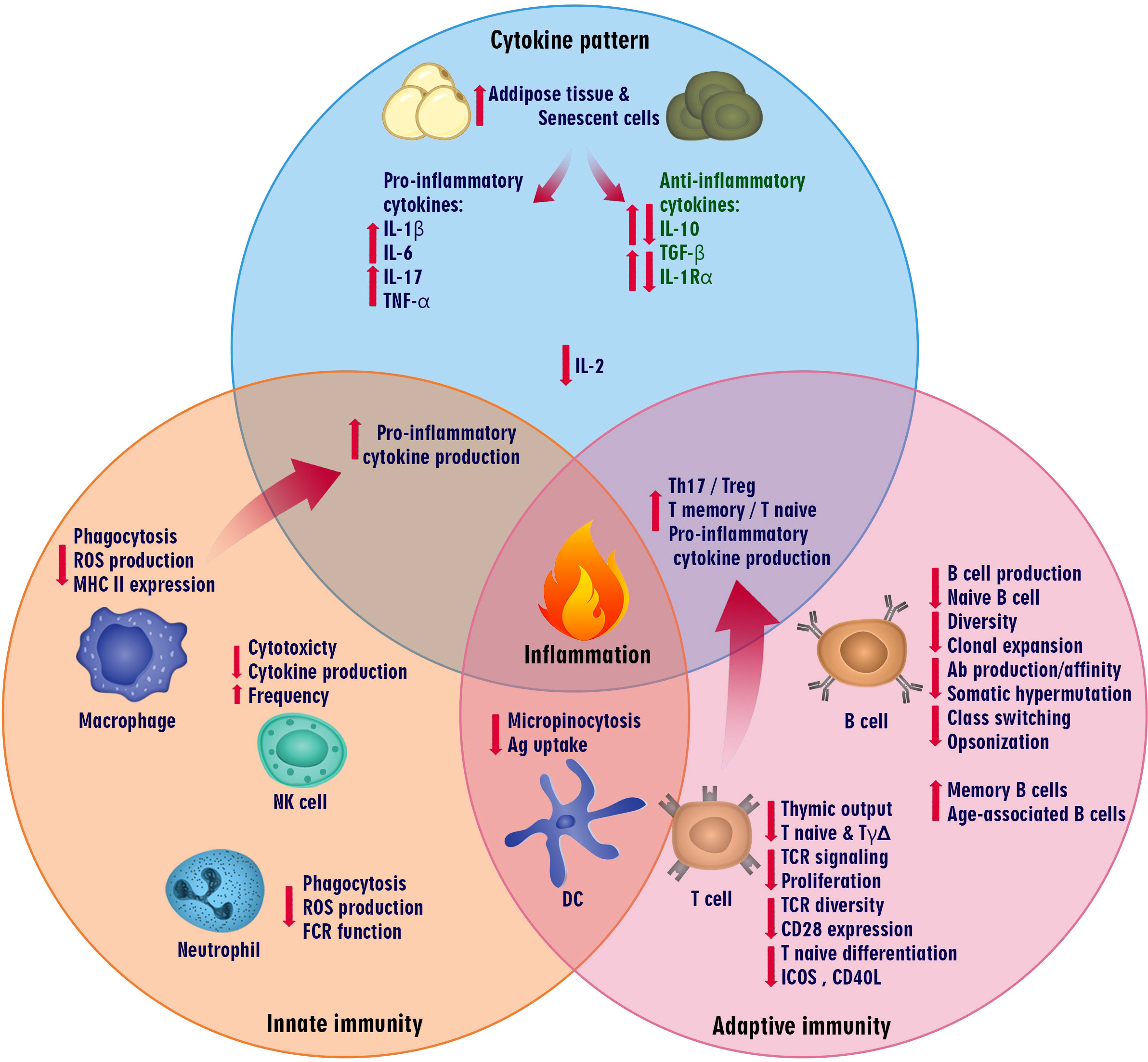
Figure 1 Immune system fluctuations in aging. The components of the innate and adaptive immune systems change in aging, affecting immune responses. These changes can also alter the cytokine pattern so that inflammatory conditions prevail in these people due to the increased expression of pro-inflammatory cytokines such as IL-1β, IL-6, IL-17, and TNF-α.
Innate immunity is the first line of defense against pathogens and is vital for protecting against several infections (33). Previous studies have shown that aging impairs epithelial barriers in the skin, lung, and gastrointestinal tract, allowing pathogenic organisms to accumulate in mucosal tissues and challenge the innate immune system (34, 35). Immunosenescence affects innate immune cells such as neutrophils, macrophages, dendritic cells, and NK cells (36).
Studies evaluating the neutrophil phagocytosis of opsonized bacteria have revealed a considerable reduction in neutrophil phagocytic ability among the elderly population (37). Furthermore, macrophages exert their function by eliminating invasive pathogens and cancer cells by releasing pro-inflammatory cytokines, which can activate signals to other immune cells (38). Previous evidence has confirmed the harmful impact of aging on macrophage activity. The macrophage population expresses lower major histocompatibility complex (MHC) II in the elderly, decreasing the CD4+ T cell-mediated immune responses (39–41).
Moreover, it has been reported that macrophages could not produce ROS in aged rats after the interferon-gamma (IFN-γ) treatment (42). Reduced superoxide anions in macrophages may cause persistent infections in old people (42). In this way, studies on the micropinocytosis capacity of dendritic cells (DCs) have shown that aged DCs in the elderly are less able to uptake fluorescein isothiocyanate–dextran than young adult Mo-DCs (43). Unlike the other innate immune cells, the NK cell absolute count is elevated in the aged population (44). However, NK cell cytotoxicity and cytokine production can be reduced through aging (45). Aging also affects phagocytic and NK cells, soluble components, cytokines, and chemokines (46). Elevated serum concentrations of IL-1β, IL-6, and TNFα have been observed among elderly populations, leading to systemic inflammation via the continuous activation of various immune cells. Therefore, pro-inflammatory cytokines can be a prognostic indicator for functional impairment, weakness, morbidity, and mortality among the aged population (47, 48).
The adaptive immune system is based on generating a diverse repertoire of T and B lymphocyte receptors (TCRs and BCRs), lymphocyte activation, and clonal expansion. The de novo generation of T and B cells has declined due to age-related alterations in the human hematopoietic system (29, 49). Hematopoietic stem cells (HSCs) are responsible for the continuous supply of myeloid and lymphoid progenitors; however, studies have shown that bone marrow (BM) cellularity and adaptive immune system functions decreased, whereas the frequency of HSC changes through aging (50–52). According to aging, the HSCs’ proliferative potency can decrease and shift toward myeloid progenitors (53, 54).
In accordance with HSC diminished capacity, B lymphocyte quantitation and qualification have changed through aging. These age-related alterations include the decreased total number of B cells, diminished diversity of B cell repertoire, especially IgVh CDR3 diversity (55), declined naïve B cells frequency, impaired response capacity to new antigens (56), decreased memory B cells clonal expansion and plasma cells production (31, 50, 57). Besides a decline in antibody production, antibody functionality is also impaired by decreased affinities and opsonizing properties (31, 50, 57). As a result, the humoral immune response to extracellular antigens and vaccination is impaired during aging (31, 58).
Immunosenescence is characterized by significant changes in the naive/memory B cell compartment through a progressive shift from naïve B cells to memory B cells (29, 31, 58). Furthermore, several studies have introduced a subtype of B cells called age-associated B cells (ABCs) that are enhanced with age and express T-bet transcription factors and CD11c (59–61). The ABC subset does not proliferate after BCR crosslinking but exhibits a robust proliferation response after stimulation with toll-like receptor (TLR)-7 and TLR-9 ligands (61–63). Furthermore, ABCs are a pro-inflammatory subset that secretes significantly high amounts of TNF-α, impairing young Pro-B cell generation (64).
Modifications in hematopoietic stem cells through aging play a crucial role in developing the T cell repertoire similar to B cells (31). The overall number of T cells, remarkably naïve CD8+ and γδTCR+ T cells, proliferation capacity of naïve T cells, TCR signaling, and diversity of TCRVβ repertoire are decreased in aging (49, 50, 65). In contrast, T memory/T naïve ratios, Th17/Treg ratios, and pro-inflammatory cytokine production is increased (29, 58, 66). Furthermore, thymo-suppressive cytokines such as IL-6, oncostatin M, and leukemia inhibitory factor increased, and thymo-stimulative cytokines such as IL-7 decreased, diminishing naïve T-cell output and increasing the susceptibility to infection, autoimmunity, and cancer (57, 58, 67).
Following the aging process, the co-stimulatory molecules such as CD28 are down-regulated in naïve T cells, reducing the differentiation of naïve T cells into central and effector memory T cells (50, 68). Moreover, an age-related Erk-TCR phosphorylation through the reduction in microRNA-181a of naive CD4+ T cells attenuated signaling in these cells (31, 69, 70). Besides, CD40L and inducible T-cell COStimulator (ICOS) expression decreased in CD4+ memory T cells, reducing B cell differentiation and specific antibody production (70).
According to aging and the alterations of the immune system, it seems that cytokine patterns also change with aging. Inflammaging is a physiological process involving various organs and tissues, such as adipose tissue, and the accumulation of senescent cells in tissues (30, 57). It has been reported that inflammaging is a persistent low-grade inflammation associated with several age-related disorders. This phenomenon is characterized by increased levels of pro-inflammatory mediators (31, 50, 58). Evidence has revealed that adipose tissue could increase throughout the body in aging and act as an endocrine source of mediators such as acute-phase proteins, hormones, pro-inflammatory cytokines, and growth factors (58, 71, 72). In this regard, the levels of circulating pro-inflammatory mediators such as IL-1β, IL-6, TNF-α, and IFN-γ increase in both homeostatic situations and in response to infections in the elderly compared with adults (57, 73, 74). Although the cytokine pattern in aging is variable, the results of clinical studies in this field are contradictory. In contrast, experimental studies showed an age-related shift from type 1 cytokines (IL-2, IL-12, IFN-γ) to type 2 cytokines (IL-4, IL-6, IL-10) in animal models (75–77).
In a physiologic condition, the expression of IL-6 is undetectable in the serum of young individuals. However, IL-6 levels can be increased by aging, which has been called “a cytokine for gerontologists” (73, 78, 79). Another age-associated pro-inflammatory cytokine is TNF-α, which can be increased in elderly individuals compared with young people, leading to the progression of age-related diseases (80–82). Increased TNF-α and IL-6 are also associated with weakness, a significant decline in muscle strength, an increased risk of cardiovascular and cerebrovascular conditions, and cognitive disorders among the elderly (73, 79, 83). Controversial findings have also been reported on the role of IL-1 in the aging process. Some studies have shown an age-associated increase in the IL-1R antagonist (IL-1Rα) and no significant age-associated changes in IL-1 (73, 79, 84). Additionally, IFN-γ is essential in cell-mediated immune response and defense against intracellular pathogens and viruses. The expression of IFN-γ in CD8+ naïve, effector, and memory T cells increased in the elderly compared with the young population. However, the expression of IFN-γ in naïve, effector, and memory CD4+ T cells decreased in old comparison with young individuals (73, 79, 85). Generally, findings are contradictory about the role and levels of IFN-γ in aging (50, 75, 79). IL-2, the “T-cell growth factor (TCGF),” is critical in different immunological responses. Most studies reported an age-related decline in the production of IL-2 by peripheral blood mononuclear cells (PBMC) in the elderly compared to young people (50, 73, 75). In contrast, previous studies indicated that IL-2 intracellular expression might be upregulated in CD8+ T cells among elderly individuals (75, 85). Additionally, it was reported that IL-2 serum levels were not altered in the healthy-aged population compared to the young controls (86).
As an anti-inflammatory cytokine, IL-10 can repress pro-inflammatory cytokine production and cell proliferation (87). Furthermore, conflicting results have been observed for IL-10 in the elderly population. Most studies have revealed that IL-10 production decreases or might be unchanged through aging (75, 88, 89). However, some studies have reported that serum IL-10 levels and its production is increased during aging due to the attempt of the immune system to suppress the pro-inflammatory response and return the immune homeostasis (73, 79, 90). Transforming growth factor beta (TGF-β) is another anti-inflammatory cytokine that can be elevated in the serum levels of octogenarians and centenarians (73, 91, 92). However, it was reported that there are no significant differences in TGF-β levels between the elderly and young women (93). These findings indicate that the cytokine pattern can be sex-dependent in the elderly.
Understanding the immunopathogenesis of infectious diseases is essential because investigating its various aspects and monitoring the immune system’s responses can lead to discovering diagnostic biomarkers and therapeutic targets. This section discusses the immunopathogenesis of SARS-CoV-2 infection and the destructive role of pro-inflammatory cytokines.
Following the SARS-CoV-2 infection and virus entrance, angiotensin-converting enzyme 2 (ACE2)-expressing type II alveolar epithelial cells are the main target of the virus (94, 95). However, a wide range of other cells in different organs, such as cholangiocytes in the liver, can express ACE2 and become infected (96).
The pathogen-associated molecular pattern (PAMP) and danger-associated molecular pattern (DAMP) are identified by immune sensors such as TLR-3, TLR-7, retinoic acid-inducible gene I (RIG-I)-like receptors such as RIG-I, and melanoma differentiation-associated protein 5 (MDA5), initiating IFN-I downstream signals (97, 98). The ligation of IFNs-I to their receptors (IFNRs) activates Janus kinases (JAKs), signal transducer and activator of transcription 1 (STAT1) and STAT2, expressing interferon-stimulated genes (ISGs) and stimulating antiviral immune responses (99).
After releasing IFNs and other pro-inflammatory cytokines and chemokines, immune cells, including neutrophils and monocytes, are recruited to the site of infection. Moreover, the epithelial and fibroblast cells can release pro-inflammatory cytokines, including TNF-α, IL-1β, IL-6, monocyte infiltration factor (MIF), IL-12, TGF-β, IL-21, IL-23, and IL-27, contributing to cytokine storm as well as neutrophil/monocyte migration and activation. The infiltration of these immune cells is the cause of neutrophilia and monocytosis in patients with COVID-19 (13, 100). Cytokine storm is a hallmark of COVID-19 pathogenesis resulting from the secretion of excessive proinflammatory cytokines and chemokines, and it induces SARS-CoV-2-related complications, particularly ARDS and multi-organ failures.
Considering the increase of neutrophils and the decrease of lymphocytes in COVID-19 patients, the ratio of neutrophils to lymphocytes (N/L ratio) is a suitable biomarker for predicting the severity of the disease (101).
The first line of defense against SARS-CoV-2 is phagocytosis. Neutrophils try to prevent the virus from spreading via neutrophil extracellular trap (NET) formation and releasing neutrophilic elastase and myeloperoxidase (MPO) into the extracellular space. Accordingly, NETosis can damage the lung and other tissues. Therefore, neutrophils act as a double-edged sword in SARS-CoV-2 infection, and neutrophilia is usually associated with poor prognosis, especially in hospitalized patients (102).
Monocytes and macrophages are other immune cells that control viral infections. They sense and capture viral antigens and present them to CD8+ T cells. These activated T cells highly express IFN-γ and IL-2 to eradicate viral pathogens and recruit other anti-viral immune cells (103). However, in SARS-CoV-2 pathogenesis, monocytes alter their phenotype from CD14++CD16– to CD14++CD16++ and their functionality from anti-inflammatory to inflammatory (104). Studies have shown that T and B cells cooperate in shifting monocyte phenotype. In this scenario, T cells can activate monocytes via expressing colony-stimulating factors 1 and 2 (CSF1 and CSF2) as ligands to their receptors on the monocytes and thus cause hyperinflammation (105).
Additionally, B cells secrete a large amount of IL-6, lymphotoxin beta (LTB), and lymphotoxin alpha (LTA), which bind to monocytes’ receptors. High concentrations of IL-6 can induce T cells to produce inflammatory chemokines and cytokines such as IFN-γ and IL-1β (106). Moreover, these interactions activate monocytes to induce cytokine hyperproduction and promote cytokine-induced tissue damage in patients infected with SARS-CoV-2 (107). Therefore, similar to neutrophils, monocyte, and macrophage inflammatory phenotype in SARS-CoV-2 pathogenesis raises the question of whether monocytes are friend or foe.
Lymphopenia as an immune abnormality is detected in 96.1% of severe COVID-19 patients, and its degree correlates with clinical outcomes (108). An investigation reported that recovered patients exhibited a slow increase in CD4+ and CD8+ T cell frequency compared to healthy individuals despite a decline in disease onset (109). Several reasons have been speculated for COVID-19 lymphopenia. SARS-CoV-2 infects lymphocytes through the ACE2 receptor and CD147 (ACE2-dependent and ACE2-independent routes), reducing the number of lymphocytes (110). Additionally, lymphoid organs such as the thymus and spleen are involved in lymphopenia. Coronavirus animal models have demonstrated alterations in the weight and cellularity of the thymus and reduction of the CD4+/CD8+ thymocytes (111).
Moreover, considering the rapid clinical recovery of blood lymphocytes in COVID-19 patients, lymphocyte sequestration in the lungs and the gastrointestinal tract has been proposed as a simple reason for lymphopenia (112). It has been suggested that T cells from COVID-19 patients undergo apoptosis due to the macrophage-derived TNF-α (113). Additionally, in COVID-19 patients, serum levels of IL-6, TNF-α, IL-8, and IL-10 were negatively correlated with lymphocyte counts (114).
Collectively, neutrophilia, lymphopenia, dysregulated monocyte and macrophages, cytokine storm, and coagulation disorders lead to organ failure and other COVID-19-associated disorders, which may be exacerbated in the elderly due to the inflammatory conditions of these disorders following SARS-CoV-2 infection.
In accordance with the knowledge of coronaviruses, it appears that cytokines and chemokines play a pivotal role in the immunopathogenesis of SARS-CoV, MERS, and SARS-CoV-2. An imbalanced production of these mediators causes hyperinflammation and damage to various organs, including the lungs, liver, kidneys, and heart (113). There is a possibility that some COVID-19-infected patients develop severe symptoms, resulting in hyperinflammation caused by cytokines/chemokines overexpression, a pathologic condition known as cytokine release syndrome (CRS), which can lead to pneumonia and ARDS (115). A series of immune responses trigger a systemic CRS after the virus invades the respiratory mucosa and infects various immune and non-immune cells, such as alveolar type II (116). CRS usually manifests as a systemic inflammatory immune response associated with the elevation of inflammatory biomarkers and multiple organ failure due to an intensified release of pro-inflammatory cytokines (117, 118). Tissue damage by SARS-CoV-2 infection or an overactivated immune system can cause CRS. A sign of this overactive immune system is the infiltration of macrophages and neutrophils in the affected tissues. These cells migrate to the infected tissue to provide protection, but their uncontrolled functions cause hyperinflammation and tissue damage (119). Although numerous cytokines and chemokines are involved in the pathogenesis of SARS-CoV-2, the most significant inflammatory cytokines, including TNF-α, IL-1β, IL-6, IL-8, and IL-17 that cause tissue damage and multi-organ failure, have been briefly discussed here (Table 1).
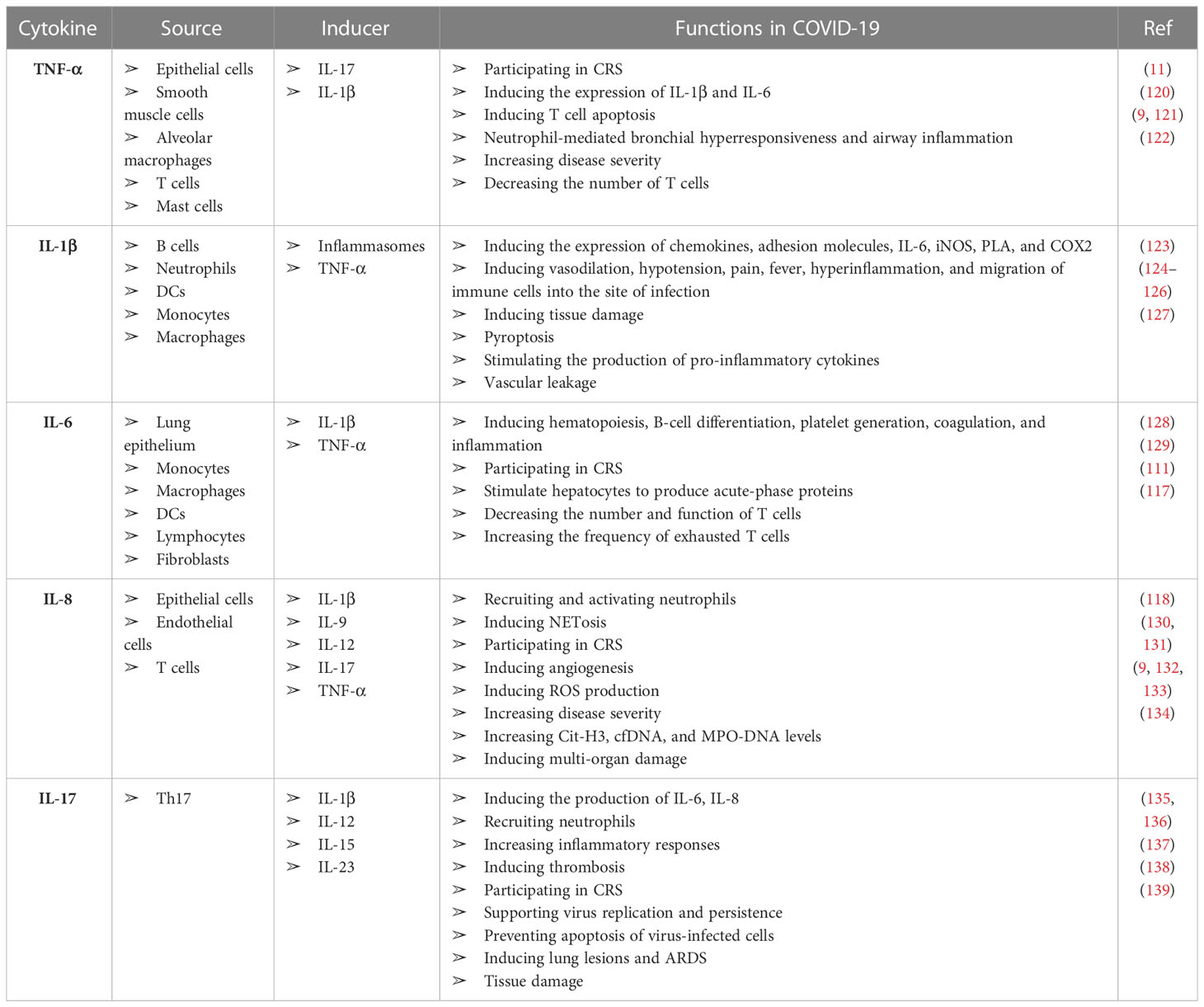
Table 1 Role of the most studied pro-inflammatory cytokines in the pathogenesis of COVID-19.
It has been demonstrated that TNF-α is a crucial factor in the pathophysiology of CRS in patients with SARS-CoV-2 infection. Several types of cells in the airways are responsible for TNF-α production, including epithelial cells, smooth muscle cells, alveolar macrophages, T cells, and mast cells. The synthesis of this cytokine is primarily triggered by PAMPs and IL-1β following NF-κB activation. IL-17 also induces the expression and release of TNF-α (140). Interestingly, TNF-α induces the expression of IL-1β and IL-6 as well as T cell apoptosis (12). It has been reported that inhaling TNF-α leads to neutrophil-mediated bronchial hyperresponsiveness and airway inflammation in healthy individuals (120). Recent studies have shown that serum levels of TNF-α have increased significantly in patients with COVID-19, and there is a positive and significant association between elevated serum TNF-α levels and disease severity (10, 130). In contrast, increasing TNF-α levels are negatively associated with the number of T cells because, as discussed, this cytokine can induce T-cell apoptosis (131).
IL-1β is a pro-inflammatory pleiotropic cytokine that plays a pivotal role in inflammatory-based disorders. B cells, neutrophils, DCs, monocytes, macrophages, and synovial fibroblasts can produce IL-1β (122). Moreover, IL-1β induces the expression of chemokines, adhesion molecules, IL-6, inducible nitric oxide synthase (iNOS), phospholipase A (PLA), and type 2 cyclooxygenase (COX2). The release of the mentioned mediators leads to vasodilation, hypotension, pain, fever, hyperinflammation, and migration of immune cells into the site of infection, inducing tissue damage (122–124). Recent studies have reported a significant increase in the expression of IL-1β at the mRNA and protein levels in patients with COVID-19 (10, 141).
Moreover, inflammasomes play a significant role in the production of IL-1β. For example, the activation of NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) activates mechanisms related to pyroptosis, inflammatory-based programmed cell death, resulting in IL-1β release by the pyroptotic cell, which is a common phenomenon in cytopathic viral infections (138). A crucial role of pyroptosis in SARS-CoV-2 infection is stimulating the production of pro-inflammatory cytokines, which lead to destructive inflammatory responses and tissue damage. It has previously been observed in patients with SARS-CoV infection that viral infection and replication could cause high levels of virus-induced pyroptosis and vascular leakage (142). Therefore, IL-1β should be considered a critical immune mediator in the pathogenesis of COVID-19.
Another pro-inflammatory and pleiotropic cytokine is IL-6, expressed by several cells, including monocytes, macrophages, DCs, lymphocytes, and fibroblasts (125). The circulatory levels of IL-6 increase in several inflammatory states, such as viral infections, septic shock, burns, and trauma (125). IL-6 is an immune system regulator involved in hematopoiesis, B-cell differentiation, platelet generation, coagulation, and inflammation. This cytokine also significantly participates in CRS (117). Another critical function of IL-6 is stimulating hepatocytes to produce acute phase proteins such as C-reactive protein (CRP). A significant increase in this protein has been reported in COVID-19 patients (141). In patients with moderate or severe COVID-19, serum levels of IL-6 are significantly elevated. However, some studies reported that circulatory concentrations of IL-6 were associated with disease severity (143–145). Although the origin of this increase in serum level is probably not circulating blood cells like peripheral blood mononuclear cells (PBMCs) and is more related to the lung epithelium. Studies have shown that IL-6 transcript levels in COVID-19 patients did not change significantly compared to controls that confirm this theory (146). Like TNF-α, an increased level of IL-6 is associated with a decrease in the number and function of T cells. Furthermore, in COVID-19 patients with increased levels of IL-6, the frequency of exhausted T cells is elevated (147).
IL-8, or CXCL8, is a chemoattractant and priming factor for neutrophils and is also involved in inflammatory responses, NETosis (neutrophil extracellular trap [NET]), and angiogenesis (132). In addition, IL-8 activates neutrophils to produce inflammatory mediators and clearance of bacterial infections by inducing reactive oxygen species (ROS) production, as well as NET formation (128, 129). IL-8 levels significantly increase in patients with SARS-CoV-2 infection and are directly related to the disease severity (10, 134, 148). Following the formation of NETosis in patients with COVID-19, citrullinated histone H3 (Cit-H3), cell-free DNA (cfDNA), and myeloperoxidase (MPO)-DNA levels are increased, inducing multi-organ damage and death from severe SARS-CoV-2 infection (149). Since neutrophils are one of the main factors in the pathogenesis of COVID-19, IL-8, as a chemotactic factor of these cells, can play an essential role in increasing neutrophil-mediated destructive inflammatory responses.
The IL-17 family comprises six members (A-F), which IL-17A and IL-17F types are involved in inflammation (150). This cytokine is secreted from Th17 cells and can induce the production of IL-6, IL-8, and several chemokines in response to viral infections, recruiting neutrophils and increasing inflammatory responses (151, 152). The systemic effects of IL-17 can be explained by inducing coagulation pathways and coagulopathy, as well as CRS (153, 154). Evidence demonstrated that IL-17 and TNF-α could create a synergistic effect in initiating these processes. Microthrombosis can damage various organs during and after SARS-CoV-2 infection (135).
Moreover, IL-17, along with IL-6, can support virus replication and persistence, as well as prevent apoptosis of virus-infected cells (136). IL-17 concentrations significantly increased in COVID-19 patients, possibly associated with lung lesions and ARDS (155). The analysis of biopsy samples taken from the liver, lung, and heart tissue of patients who died of severe SARS-CoV-2 infection has shown that the infiltration of Th17 and CD8+ T cells in the lung tissue of these people has increased significantly (156). These findings indicate that the dysregulated increase in the activity of Th17 and cytotoxic CD8+ T cells has led to severe tissue damage and, finally, the death of patients.
The reviewed studies indicate that the excessive increase in pro-inflammatory cytokines during SARS-CoV-2 infection can cause tissue damage through different mechanisms. However, the patterns of these cytokines at different ages are probably different. Therefore, targeting these cytokines or their receptors can be a potential treatment strategy. However, more studies are required because inhibiting cytokines can act like a double-edged sword and increase viral load or co-infections.
Although SARS-CoV mainly affects the respiratory system, it can also lead to systemic and organ-specific disorders. During the earlier SARS-CoV-2 outbreak, about 60% of patients experienced varying degrees of liver impairment. According to evolutionary similarities, it is conceivable that SARS-CoV-2 also damages the liver with hepatobiliary manifestation and enzyme elevation. Patients with COVID-19 may have pre-existing liver disease or not (157, 158). A recent systematic review and meta-analysis reported that 14-53% of COVID-19 patients suffer from varying degrees (mild to severe conditions) of liver injury (159). However, the mechanism of liver injury has not been fully discovered. It might be due to several underlying mechanisms, including severe inflammatory responses, direct cytopathic effects, aggravation of the pre-existing liver disease, and cytokine storm. Moreover, drug-induced liver injury can occur in undertreatment patients, as explained in the next part (Figure 2) (137, 160).
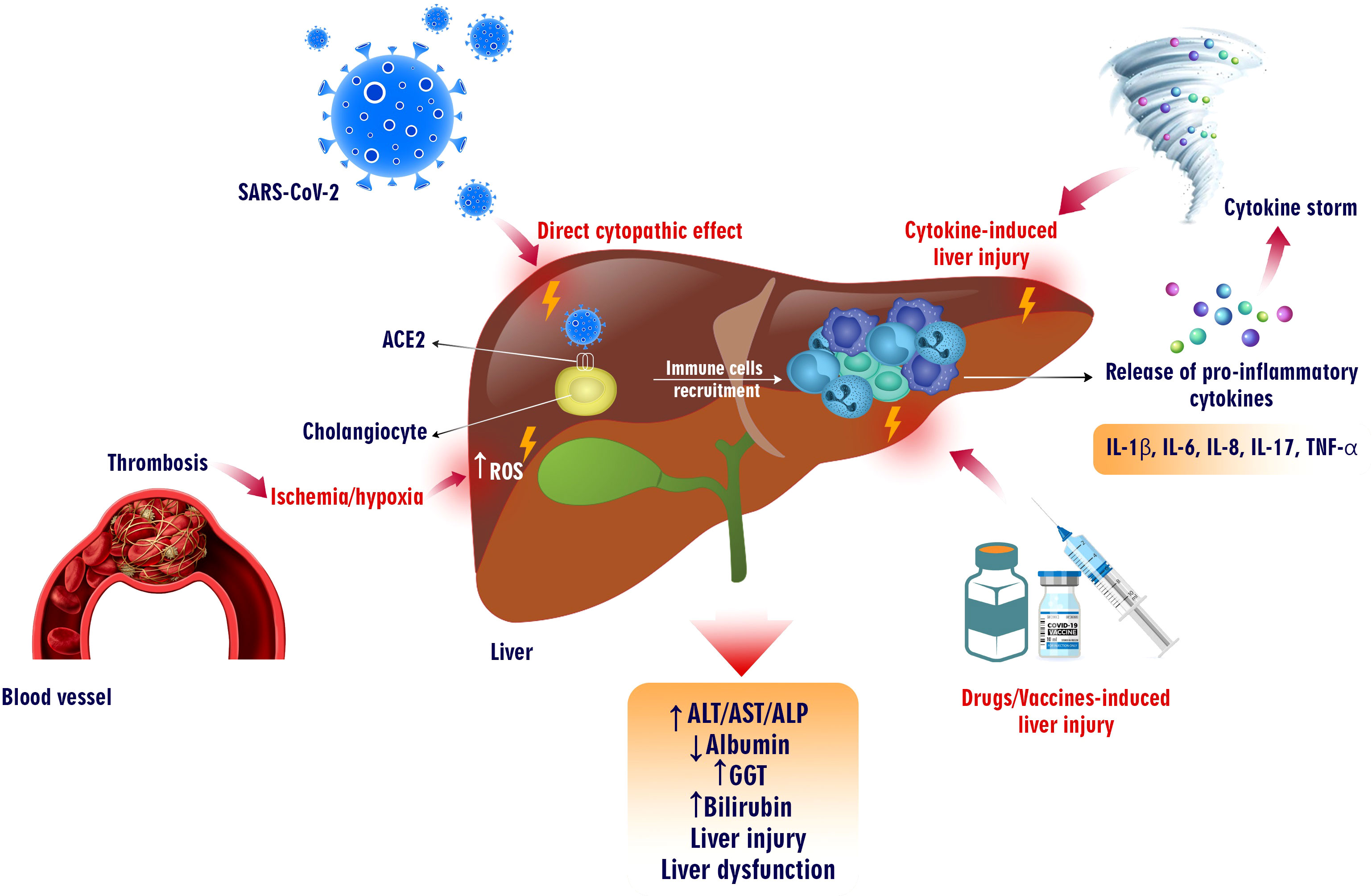
Figure 2 COVID-19-induced liver injury. SARS-CoV-2 infection can cause liver failure in different ways. This virus can directly infect liver cells expressing ACE2 and exert cytopathic effects. Additionally, the increase in the production of pro-inflammatory cytokines from virus-infected cells has led to the recruiting of immune cells into the liver, which can cause a cytokine storm and tissue injury. Moreover, thrombosis causing hypoxia and ischemia induces ROS overproduction, which harms liver tissue. On the other hand, vaccinations and drugs prescribed for COVID-19 can also increase liver enzymes, bilirubin, GGT, and various degrees of liver damage.
SARS-CoV-2 can damage the liver directly through ACE2, an entry receptor located on bile duct cells (cholangiocytes) (59.7%) and less on hepatocytes (2.6%), leading to hepatocyte apoptosis (161). However, recent data revealed that other causative factors, such as drug-induced injury, precede direct cytotoxicity (162). Liver biopsies of COVID-19 patients showed lobular inflammation, acidophilic bodies, mild activity of the portal vein, and moderate microvascular steatosis. Moreover, autopsies from dead patients revealed hepatomegaly, lobular necrosis, slight lymphocyte infiltration, and sinusoidal dilation of the central lobule in at least 50% of patients. It is unknown if the viral infection or the medications are responsible for these histopathological changes (158, 163, 164).
The severity of COVID-19 elevated IL-6 or ferritin levels and younger age are considered strong risk factors for liver injury during hospitalization and are associated with mortality (160, 165). The mitochondrial alterations and metabolic acidosis aggravation are related to SARS-CoV-2 infection and result in ischemic/hypoxic liver injury (166). Several studies revealed that patients had abnormal liver function tests, including a moderate increase in bilirubin, aspartate aminotransferase (AST), and alanine aminotransferase (ALT), which are frequently observed in patients with severe COVID-19 compared with non-severe patients (167–169). The alteration of these biomarkers correlates with liver function, and hepatocyte integrity is involved in acute liver infection and disease severity. Besides, the transaminase increase could be associated with morbidity and mortality (170).
However, hypoalbuminemia has been reported due to increased capillary permeability and decreased hepatic synthesis in severe patients. Elevated alkaline phosphatase (ALP) and GGT levels are observed in acute inflammatory oxidative stress and are less common in COVID-19 (26, 171). Interestingly, skin darkening and hyperpigmentation caused by increased estrogen and alteration of tyrosine into melanin in patients after recovery are recognized because of liver injury (172).
It has been revealed that patients with pre-existing liver disease and systematic inflammation are more susceptible to COVID-19-mediated liver injury (173). Comorbidities such as metabolic-associated fatty liver disease (MAFLD), alcohol-related liver disease, autoimmune hepatitis (AIH), and chronic liver disease (CLD) aggravate COVID-19 severity (171). In patients with fatty liver disease, alcohol use disorder (AUD), and non-alcoholic fatty liver disease (NAFLD), GGT levels were elevated compared with those without pre-existing liver disease, indicating that these patients are more prone to severe disease phenotype (162, 170).
Several studies have investigated the association between age and severity of liver injury in COVID-19 patients. A retrospective study comprising 900 patients classified for ages 18-39, 40-69, and more than 70 years old demonstrated that comorbidity and C-reactive peptide (CRP) are positively associated with aging. Moreover, abnormalities in liver function tests and liver-related mortality were observed, especially in 40-69 years old patients. Additionally, they had higher ALT levels than the other groups (174).
Another study reported that hepatic symptoms are more frequent in young and obese patients with pre-existing liver diseases like MAFLD. These symptoms are aggressive in adult patients having severe COVID-19 (175). With advanced age and increased comorbidities, such as arterial hypertension, diabetes mellitus, chronic liver, cardiovascular disease, and malignancy, the risk of liver injury and mortality increases in elderly patients (176). A retrospective cohort study revealed that COVID-19-associated liver injury is more common in patients aged ≥ 65 years, especially with other comorbidities. Elevated levels of AST and total bilirubin were indicators of increased risk of mortality (177).
Some studies agree that COVID-19 patients under 18 suffer less from liver injury than adults (17, 178). A retrospective case series study reported that two COVID-19 patients under one year without previous liver disease presented with acute liver failure, and three patients aged 2, 8, and 13 exhibited hepatitis with cholestasis in liver biopsies (179). Indeed, liver injury in children with COVID-19 is mild, with low changes in laboratory and radiological evidence (180). Taken together, there seems to be a positive association between aging and liver injuries, particularly regarding pre-existing liver involvement.
Systemic homeostasis disruptions in both vascular and immune-mediated pathways affect multiple mechanisms in the liver. Some studies have demonstrated that COVID-19 patients have minor early symptoms but might develop quickly into the final stage of multiple organ dysfunction (22). This trend is associated with a viral infection-driven rapid onset of inflammatory cytokines. As discussed, dysregulated activation of inflammatory pathways leads to the hyperproduction of cytokines and, eventually, the formation of a cytokine storm (181). The overexpression of CRP, ferritin, LDH, TNF-α, IL-1β, IL-6, IL-2, IFN-γ, and vascular endothelial growth factor (VEGF) can be associated with COVID-19 severity (160, 166, 182). It has been reported that there is no difference between cytokine profiles in mild or severe COVID-19, ARDS, and sepsis. However, the therapeutic regime in various cytokine storm-induced disorders differs (183). Several studies suggested that pro-inflammatory cytokines contribute to hepatologic abnormalities in the early and late phases (171, 184, 185). A large cohort study demonstrated a correlation between systematic inflammation and the acute phase of liver injury in which IL-6 is accompanied by CRP or ferritin-caused elevation in AST levels (186). The increase in ALP and GGT levels is related to the advanced phase of liver injury, which is mediated by cytokines and causes hepatocellular cholestasis (187). Moreover, SARS-CoV-2, by activating twenty family member 2 and C-type lectins on myeloid cells, strengthened pro-inflammatory cytokine responses (170). In inflammatory states, peripheral blood evaluation revealed hyperactivation of CD4+ T cells, particularly CCR6+ Th17 cells, and CD8+ cytotoxic T cells, inducing hepatocellular dysfunction (188).
The pathogenic effect of cytokines on liver injury caused by COVID-19 is not well known. However, according to the evidence, some pathophysiological prosses can be suggested that are associated with hepatocellular damage. Cytokine can induce hypoperfusion, vascular dysfunction, and oxidative stress. Moreover, these mediators can cause vascular permeability by increasing the adherence of immune cells to the endothelial (22). Both vascular permeability and ROS propagation result from cytokine activation (22, 189). For instance, TNF-α can impair NO-mediated vasodilation, and capillary leak syndrome and hypoxia occur dramatically with the continuation of this process. Capillary leakage causes edema in some organs, such as the liver, brain, heart, and kidneys. Upregulation of IFN-related JAK-STAT signalling in liver autopsies exhibited vascular damage, endothelial injury, and recruiting immune cells, leading to clot formation (190). Hepatocellular injury can activate Kupffer cells to produce ROS, NO, and proinflammatory cytokines. These products induce liver sinusoidal endothelial cells (LSECs) to respond to the cytokines that cause more liver damage (191). Under the influence of TNF, hepatocytes secrete IL-6, which activates caspase 3 and hepatocellular apoptosis. Moreover, the generated NO causes mitochondrial damage and hepatocellular necrosis (192).
SARS-CoV-2, through ACE2 and upregulation of angiotensin II (Ang II), plays a crucial role in RAS signalling, promoting inflammation, tissue injury, and migration of endothelial cells (193). hypercytokinemia is involved in the renin-angiotensin-aldosterone system. Therefore, the imbalance of this system by inducing the accumulation of bradykinin and plasminogen activator inhibitor-I leads to thrombosis (22). The consequence of this circumstance is inflammation in the liver and hepatocyte injury (194).
The COVID-19 pandemic has caused widespread morbidity and mortality. This virus can affect numerous organs such as the lung, kidney, heart, CNS, and liver causing multiorgan failures. As a result, there is an urgent need for an effective method to limit the spread of the COVID-19 virus, and vaccine development may be a promising option (195). Although adverse effects from vaccination have been minor, there are also reports of liver damage following vaccination with different platforms of the COVID-19 vaccine (Table 2) (217).

Table 2 Impacts of drugs/vaccines on liver injury in patients with COVID-19.
Several COVID-19 vaccine platforms have been effective in preventing SARS-CoV-2 infection. Despite mild side effects, no evidence of acute liver injury (ALI) has been reported among vaccine recipients, possibly due to the small sample size or other involved factors. Since the extensive implementation of COVID-19 immunization programs worldwide, reports of adverse events have been reported, including rare hepatic adverse effects with various characteristics in the form of ALI resembling autoimmune hepatitis (AIH). Since the widespread implementation of COVID-19 immunization programs worldwide, reports of adverse events have been made, including rare hepatic adverse effects with various characteristics in the form of ALI resembling autoimmune hepatitis (AIH) (206, 230, 236). The exact mechanism of liver injury caused by the COVID-19 vaccine is unknown. Therefore, there are no data to prove the existence of a relationship between vaccination and the incidence of AIH (237).
Vaccine-induced AIH was reported in both mRNA and non‐mRNA vaccines, suggesting that these adverse effects could be independent of the vaccine mechanisms (208). Some patients had a liver or autoimmune disease history, whereas others had neither liver nor autoimmune disease history (211).
The pattern of liver injury reported was predominantly hepatocellular, and some patients showed features of immune-mediated hepatitis (214). In some patients, liver injury was observed after the first vaccination dose, whereas in others, it occurred after the second dose (214). Elevated bilirubin levels and liver enzymes, which clinically manifest as jaundice due to hepatotoxicity, are examples of abnormal liver function tests (230, 236). The most frequently reported symptom was jaundice (211). [249] The seropositivity rate for anti-smooth muscle antibody (ASMA), anti-nuclear antibody (ANA), and anti-mitochondrial antibody (AMA) varied between studies, and the IgG level was high in most of the ¬patients (214). Liver biopsies exhibited a varied pattern, including histological features consistent with AIH, such as interface hepatitis, portal inflammation, and non-specific centrilobular necrosis (228). Additionally, after vaccination, severe thrombosis in the portal and splenic veins and immune thrombocytopenic purpura were linked with ALI (218, 220).
A case series described two patients with hepatocellular liver injury without autoimmune features on serology and histology following COVID-19 vaccination (215). However, another case series analysis showed no evidence of ALI after COVID-19 vaccination (217).
Drug-induced liver injury (DILI) is a rare and potentially life-threatening adverse effect seen with different chemicals or drugs (224). Some liver abnormalities observed in COVID-19 are associated with DILI manifested by high liver enzyme levels and, less commonly, high bilirubin levels. Patients’ histology showed moderate microvascular steatosis and minor hepatic inflammation, possibly due to DILI from treating the virus or its symptoms. DILI in patients with COVID-19 is a mild hepatic inflammation that occurs following antiviral treatments or the disease itself. Histological examination shows moderate microvascular steatosis, manifested by elevated liver enzymes and rarely bilirubin (158, 205).
Antiviral (remdesivir, favipiravir, lopinavir/ritonavir, and umifenovir), antibiotics (azithromycin), immunomodulators (dexamethasone and tocilizumab), antimalarial (hydroxychloroquine), and antipyretic medications (acetaminophen) have been administred to patients with COVID-19 (225, 227). Several of the mentioned medications have previously been linked to various degrees of hepatotoxicity when used to treat other disorders, such as viral infections. The administration of many of these drugs in similar viral infections is associated with any grade of hepatotoxicity.
Almost all medications used to treat COVID-19 are metabolized in the liver, but some degree of liver damage is unavoidable (229). However, it is unknown what causes the elevated liver enzymes in this population (the disease or DILI). Additionally, due to drug combinations, it is challenging to imagine a relationship between each drug and liver damage (232).
Remdesivir is a nucleoside analog currently approved by the United States food and drug administration (FDA) and recommended for some hospitalized patients with COVID-19 (233). This antiviral drug shortens recovery time in hospitalized patients with lower respiratory tract SARS-CoV-2 infection. Despite its efficacy in reducing recovery times, its healing properties are yet to be proven (238). Increasing ALT/AST, hyperbilirubinemia, and hypoalbuminemia are considered the most common adverse effects in patients under treatment with remdesivir (197, 234, 239).
The efficacy of Umifenovir in COVID-19 patients was assessed in a randomized clinical trial and showed significantly improved clinical and laboratory parameters and decreased hospitalization duration; however, the most common liver-associated disorder in patients using umifenovir was abnormal liver function tests (235).
LPV/r is a co-formulation of ritonavir and lopinavir structurally related protease inhibitors (203). Conflicting published data results have sparked debate about using LPV/r in COVID-19 patients. Two clinical trials showed that a combination of LPV and ritonavir made no difference to the standard of care. However, some studies have reported that LPV/r could decrease the SARS-CoV-2 shedding and the median time of clinical improvement (240–242). Abnormal liver function has been reported in patients under treatment with lopinavir/ritonavir, and the most prevalent increase in test findings was seen in GGT and total bilirubin (205, 243).
As an RNA polymerase inhibitor, favipiravir reduces viral clearance time and improves chest imaging. However, its administration could be associated with adverse effects, such as increased liver enzymes (244, 245).
The liver is the primary site of azithromycin metabolism, and prior studies found that azithromycin had a modest risk of acute, temporary, and asymptomatic increases in serum aminotransferases, occurring in up to 2% of individuals with incomplete treatment. Thus, when given to COVID-19 patients, especially in small doses and for a brief time, this medication can be excluded from further evaluation associated with liver injury (231, 246).
Patients with severe COVID-19 have a high level of circulating IL-2, IL-6, IL-7, IL-10, and IFN-γ (10). Therefore, immunomodulator drugs could help regulate inflammatory responses, enhancing COVID-19 prognosis (224). Dexamethasone is the first drug shown to reduce mortality in patients with severe COVID-19 (247). This drug is metabolized in the liver and has little effect on hepatic dysfunction in COVID-19 patients because of the low prescribed dosage and short treatment duration (248). Additionally, tocilizumab is a recombinant humanized monoclonal antibody that inhibits the IL-6 receptor and has been used to reduce IL-6-mediated inflammatory responses in patients with severe COVID-19. Patients receiving tocilizumab had a better median overall survival than hospitalization time. A retrospective study of patients treated with tocilizumab showed no evidence of adverse liver effects (249). Other studies reported DILI after using tocilizumab and mild to moderate elevations in liver enzyme levels (219, 221).
SARS-CoV-2 infects ACE2-expressing cells, and chloroquine may inhibit the ligation of the virus to the ACE2 by inhibiting terminal glycosylation (250). The effectiveness of hydroxychloroquine/chloroquine is controversial in patients with COVID-19 (251, 252). It has been reported that hydroxychloroquine could be a probable but uncommon cause of DILI (246). A clinical study demonstrated that patients taking hydroxychloroquine alone or along with azithromycin had higher liver enzymes (213). Furthermore, a case study showed hepatotoxicity and transaminase elevation after using hydroxychloroquine (253). Acetaminophen has been the medication for treating fever and myalgia associated with COVID-19 (254), and using acetaminophen in supratherapeutic amounts for several days might lead to hepatitis, cholestasis, or other nonspecific elevations of liver enzymes (255).
JAK inhibitors, such as baricitinib, imatinib, and tofacitinib, help reduce mortality and intubation rates. These inhibitors have been approved in several countries to treat COVID-19. However, administering JAK inhibitors could be associated with increased liver enzymes and bilirubin in less than 1% of patients with COVID-19. Furthermore, no severe DILI cases have been reported following treatment with JAK inhibitors (256).
According to the available studies on COVID-19, liver dysfunction generally develops in male, obese, and elderly patients. Several mechanisms are involved in liver injury, including severe inflammatory response, a direct cytopathic effect, and the idiosyncratic effect of therapeutic regimens and cytokine storm. Mild to severe liver injury is frequent and occurs in approximately 50% of patients with COVID-19 and is associated with the development of the disease, particularly in the elderly with pre-existing liver disease and other comorbidities. The patients hospitalized due to the worsening of the disease have a significant elevation in serum levels of AST, ALT, and ALP and reduced albumin levels. Elevations in liver enzymes are transient and decrease after recovery. Therefore, due to the changed conditions of the immune system and chronic inflammation caused by aging, the elderly should be under more care for managing tissue damage, especially the lung, liver and heart, because the increased levels of pro-inflammatory cytokines or the prescribed drugs worsen the condition of these patients. Furthermore, post-vaccination liver complications should be monitored in the elderly.
All authors participated in drafting the manuscript. RF and RE supervised the study and reviewed the final version of the manuscript. All authors contributed to the article and approved the submitted version.
This study has been performed through the collaboration agreement between the National Institute of Genetic Engineering and Biotechnology (NIGEB, Tehran, Iran) and the Iran University of Medical Sciences (Tehran, Iran) (Agreement No.: 1401S/ZH/566).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
3. Gorbalenya A, Lauber C, Siddell S. Taxonomy of viruses. (2019). doi: 10.1016/B978-0-12-801238-3.99237-7
4. Franks TJ, Galvin JR. Coronavirus. In: Fraire A., Woda B., Welsh R., Kradin R. Eds. Viruses and the Lung. Springer, Berlin, Heidelberg. (2014), 109–16. doi: 10.1007/978-3-642-40605-8_13
5. De Wit E, Van Doremalen N, Falzarano D, Munster VJ. SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Microbiol (2016) 14(8):523–34. doi: 10.1038/nrmicro.2016.81
6. Khan S, Siddique R, Shereen MA. The emergence of a novel coronavirus (SARS-CoV-2), their biology and therapeutic options. J Clin Microbiol (2020) 58(5):00187–20. doi: 10.1128/jcm.00187-20
7. Li X, Chang J, Chen S, Wang L, Yau TO, Zhao Q, et al. Genomic feature analysis of betacoronavirus provides insights into SARS and COVID-19 pandemics. Front Microbiol (2021) 12:614494. doi: 10.3389/fmicb.2021.614494
8. Guan W-j, Ni Z-y, Hu Y, Liang W-h, Ou C-q, He J-x, et al. Clinical characteristics of coronavirus disease 2019 in China. New Engl J Med (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
9. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet (2020) 395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5
10. Chau TN, Lee KC, Yao H, Tsang TY, Chow TC, Yeung YC, et al. SARS-associated viral hepatitis caused by a novel coronavirus: report of three cases. Hepatology (2004) 39(2):302–10. doi: 10.1002/hep.20111
11. Peiris J, Lai S, Poon L, Guan Y, Yam L, Lim W, et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet (2003) 361(9366):1319–25. doi: 10.1016/S0140-6736(03)13077-2
12. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus–infected pneumonia in Wuhan, China. jama (2020) 323(11):1061–9. doi: 10.1001/jama.2020.1585
13. Sun J, Aghemo A, Forner A, Valenti L. COVID-19 and liver disease. Liver Int (2020) 40(6):1278–81. doi: 10.1111/liv.14470
14. Kukla M, Skonieczna-Żydecka K, Kotfis K, Maciejewska D, Łoniewski I, Lara LF, et al. COVID-19, MERS and SARS with concomitant liver injury—systematic review of the existing literature. J Clin Med (2020) 9(5):1420. doi: 10.3390/jcm9051420
15. Sodeifian F, Seyedalhosseini ZS, Kian N, Eftekhari M, Najari S, Mirsaeidi M, et al. Drug-induced liver injury in COVID-19 patients: a systematic review. Front Med (2021) 8:731436. doi: 10.3389/fmed.2021.731436
16. Saha L, Vij S, Rawat K. COVID-19 drug-induced liver injury: A recent update of the literature. World J Gastroenterol (2022) 28(45):6314–27. doi: 10.3748/wjg.v28.i45.6314
17. Yadav DK, Singh A, Zhang Q, Bai X, Zhang W, Yadav RK, et al. Involvement of liver in COVID-19: systematic review and meta-analysis. Gut (2021) 70(4):807–9. doi: 10.1136/gutjnl-2020-322072
18. Teijaro JR. Cytokine storms in infectious diseases. In: Seminars in immunopathology. (Springer) (2017).
19. Edwards L, Wanless IR. Mechanisms of liver involvement in systemic disease. Best Pract Res Clin gastroenterol (2013) 27(4):471–83. doi: 10.1016/j.bpg.2013.08.002
20. Anirvan P, Narain S, Hajizadeh N, Aloor FZ, Singh SP, Satapathy SK. Cytokine-induced liver injury in coronavirus disease-2019 (COVID-19): untangling the knots. Eur J Gastroenterol Hepatology. (2022) 33(1):e42–e9. doi: 10.1097/MEG.0000000000002034
21. Moshage H. Cytokines and the hepatic acute phase response. J Pathology: A J Pathological Soc Great Britain Ireland. (1997) 181(3):257–66. doi: 10.1002/(SICI)1096-9896(199703)181:3<257::AID-PATH756>3.0.CO;2-U
22. Ciarambino T, Para O, Giordano M. Immune system and COVID-19 by sex differences and age. Women’s Health (2021) 17:17455065211022262. doi: 10.1177/17455065211022262
23. Bajaj V, Gadi N, Spihlman AP, Wu SC, Choi CH, Moulton VR. Aging, immunity, and COVID-19: how age influences the host immune response to coronavirus infections? Front Physiol (2021) 11:571416. doi: 10.3389/fphys.2020.571416
24. Cichoż-Lach H, Michalak A. Liver injury in the era of COVID-19. World J gastroenterol (2021) 27(5):377. doi: 10.3748/wjg.v27.i5.377
25. Pirabe A, Heber S, Schrottmaier WC, Schmuckenschlager A, Treiber S, Pereyra D, et al. Age related differences in monocyte subsets and cytokine pattern during acute COVID-19—A prospective observational longitudinal study. Cells (2021) 10(12):3373. doi: 10.3390/cells10123373
26. Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell (2014) 159(4):709–13. doi: 10.1016/j.cell.2014.10.039
27. Fuentes E, Fuentes M, Alarcón M, Palomo I. Immune system dysfunction in the elderly. Anais da Academia Bras Ciências. (2017) 89:285–99. doi: 10.1590/0001-3765201720160487
28. Nikolich-Žugich J. The twilight of immunity: emerging concepts in aging of the immune system. Nat Immunol (2018) 19(1):10–9. doi: 10.1038/s41590-017-0006-x
29. Akha AAS. Aging and the immune system: An overview. J Immunol Methods (2018) 463:21–6. doi: 10.1016/j.jim.2018.08.005
30. Goronzy JJ, Weyand CM. Understanding immunosenescence to improve responses to vaccines. Nat Immunol (2013) 14(5):428–36. doi: 10.1038/ni.2588
31. Medzhitov R, Janeway C. Innate immunity. New Engl J Med (2000) 343(5):338–44. doi: 10.1056/NEJM200008033430506
32. Gomez CR, Boehmer ED, Kovacs EJ. The aging innate immune system. Curr Opin Immunol (2005) 17(5):457–62. doi: 10.1016/j.coi.2005.07.013
33. Nomellini V, Gomez CR, Kovacs EJ. Aging and impairment of innate immunity. Trends Innate Immunity. (2008) 15:188–205. doi: 10.1159/000136358
34. Weiskopf D, Weinberger B, Grubeck-Loebenstein B. The aging of the immune system. Transplant Int (2009) 22(11):1041–50. doi: 10.1111/j.1432-2277.2009.00927.x
35. Wenisch C, Patruta S, Daxböck F, Krause R, Hörl W. Effect of age on human neutrophil function. J leukocyte Biol (2000) 67(1):40–5. doi: 10.1002/jlb.67.1.40
36. Emanuelli G, Lanzio M, Anfossi T, ROmano S, Anfossi G, Calcamuggi G. Influence of age on polymorphonuclear leukocytes in vitro: phagocytic activity in healthy human subjects. Gerontology (1986) 32(6):308–16. doi: 10.1159/000212809
37. Butcher S, Chahal H, Nayak L, Sinclair A, Henriquez N, Sapey E, et al. Senescence in innate immune responses: reduced neutrophil phagocytic capacity and CD16 expression in elderly humans. J leukocyte Biol (2001) 70(6):881–6. doi: 10.1189/jlb.70.6.881
38. Plowden J, Renshaw-Hoelscher M, Engleman C, Katz J, Sambhara S. Innate immunity in aging: impact on macrophage function. Aging Cell (2004) 3(4):161–7. doi: 10.1111/j.1474-9728.2004.00102.x
39. Herrero C, Sebastián C, Marqués L, Comalada M, Xaus J, Valledor AF, et al. Immunosenescence of macrophages: reduced MHC class II gene expression. Exp gerontology (2002) 37(2-3):389–94. doi: 10.1016/S0531-5565(01)00205-4
40. Plackett TP, Boehmer ED, Faunce DE, Kovacs EJ. Aging and innate immune cells. J leukocyte Biol (2004) 76(2):291–9. doi: 10.1189/jlb.1103592
41. Agrawal A, Agrawal S, Gupta S. Dendritic cells in human aging. Exp gerontol (2007) 42(5):421–6. doi: 10.1016/j.exger.2006.11.007
42. Miyaji C, Watanabe H, Toma H, Akisaka M, Tomiyama K, Sato Y, et al. Functional alteration of granulocytes, NK cells, and natural killer T cells in centenarians. Hum Immunol (2000) 61(9):908–16. doi: 10.1016/S0198-8859(00)00153-1
43. Borrego F, Alonso M, Galiani M, Carracedo J, Ramirez R, Ostos B, et al. NK phenotypic markers and IL2 response in NK cells from elderly people. Exp gerontol (1999) 34(2):253–65. doi: 10.1016/S0531-5565(98)00076-X
44. Ershler WB, Keller ET. Age-associated increased interleukin-6 gene expression, late-life diseases, and frailty. Annu Rev Med (2000) 51(1):245–70. doi: 10.1146/annurev.med.51.1.245
45. Bruunsgaard H, Andersen-Ranberg K, vB Hjelmborg J, Pedersen BK, Jeune B. Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am J Med (2003) 115(4):278–83. doi: 10.1016/S0002-9343(03)00329-2
46. O’Mahony L, Holland J, Jackson J, Feighery C, Hennessy T, Mealy K. Quantitative intracellular cytokine measurement: age-related changes in proinflammatory cytokine production. Clin Exp Immunol (1998) 113(2):213–9. doi: 10.1046/j.1365-2249.1998.00641.x
47. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin science. (2018) 132(12):1243–52. doi: 10.1042/CS20180306
48. Gao H-M, Hong J-S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol (2008) 29(8):357–65. doi: 10.1016/j.it.2008.05.002
49. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci (2011) 108(50):20012–7. doi: 10.1073/pnas.1116110108
50. Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci (2005) 102(26):9194–9. doi: 10.1073/pnas.0503280102
51. Beerman I, Bhattacharya D, Zandi S, Sigvardsson M, Weissman IL, Bryder D, et al. Functionally distinct hematopoietic stem cells modulate hematopoietic lineage potential during aging by a mechanism of clonal expansion. Proc Natl Acad Sci (2010) 107(12):5465–70. doi: 10.1073/pnas.1000834107
52. Lansdorp PM, Dragowska W, Thomas T, Little M, Mayani H. Age-related decline in proliferative potential of purified stem cell candidates. Blood Cells (1994) 20(2-3):376–80.
53. Gibson KL, Wu YC, Barnett Y, Duggan O, Vaughan R, Kondeatis E, et al. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell (2009) 8(1):18–25. doi: 10.1111/j.1474-9726.2008.00443.x
54. Roukens AH, Soonawala D, Joosten SA, de Visser AW, Jiang X, Dirksen K, et al. Elderly subjects have a delayed antibody response and prolonged viraemia following yellow fever vaccination: a prospective controlled cohort study. PloS One (2011) 6(12):e27753. doi: 10.1371/journal.pone.0027753
55. Müller L, Di Benedetto S, Pawelec G. The immune system and its dysregulation with aging.In: Harris J., Korolchuk V. (eds) Biochemistry and Cell Biology of Ageing: Part II Clinical Science. Subcellular Biochemistry 91. Springer, Singapore. doi: 10.1007/978-981-13-3681-2_2
56. Frasca D, Diaz A, Romero M, Garcia D, Blomberg BB. B cell immunosenescence. Annu Rev Cell Dev Biol (2020) 36:551–74. doi: 10.1146/annurev-cellbio-011620-034148
57. Frasca D, van der Put E, Riley RL, Blomberg BB. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J Immunol (2004) 172(4):2155–62. doi: 10.4049/jimmunol.172.4.2155
58. Castelo-Branco C, Soveral I. The immune system and aging: a review. Gynecological Endocrinol (2014) 30(1):16–22. doi: 10.3109/09513590.2013.852531
59. Kogut I, Scholz JL, Cancro MP, Cambier JC. B cell maintenance and function in aging. Seminars in immunology. Amsterdam: Elsevier (2012).
60. Hao Y, O’Neill P, Naradikian MS, Scholz JL, Cancro MP. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood J Am Soc Hematology. (2011) 118(5):1294–304. doi: 10.1182/blood-2011-01-330530
61. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)–driven accumulation of a novel CD11c+ B-cell population is important for the development of autoimmunity. Blood J Am Soc Hematology. (2011) 118(5):1305–15. doi: 10.1182/blood-2011-01-331462
62. Blomberg BB, Ryan JG, Schwartz R, Riley RL, Frasca D, Landin AM, et al. Aging down-regulates the transcription. J Immunol (2008) 180:5283–90. doi: 10.1111/imr.12380
63. Naradikian MS, Hao Y, Cancro MP. Age-associated B cells: key mediators of both protective and autoreactive humoral responses. Immunol Rev (2016) 269(1):118–29. doi: 10.1111/imr.12380
64. Ratliff M, Alter S, Frasca D, Blomberg BB, Riley RL. In senescence, age-associated B cells secrete TNF α and inhibit survival of B-cell precursors. Aging Cell (2013) 12(2):303–11. doi: 10.1111/acel.12055
65. Britanova OV, Putintseva EV, Shugay M, Merzlyak EM, Turchaninova MA, Staroverov DB, et al. Age-related decrease in TCR repertoire diversity measured with deep and norMalized sequence profiling. J Immunol (2014) 192(6):2689–98. doi: 10.4049/jimmunol.1302064
66. Schmitt V, Rink L, Uciechowski P. The Th17/Treg balance is disturbed during aging. Exp gerontol (2013) 48(12):1379–86. doi: 10.1016/j.exger.2013.09.003
67. Sempowski GD, Hale LP, Sundy JS, Massey JM, Koup RA, Douek DC, et al. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J Immunol (2000) 164(4):2180–7. doi: 10.4049/jimmunol.164.4.2180
68. Vallejo AN, Brandes JC, Weyand CM, Goronzy J. Modulation of CD28 expression: distinct regulatory pathways during activation and replicative senescence. J Immunol (1999) 162(11):6572–9. doi: 10.4049/jimmunol.162.11.6572
69. Li G, Yu M, Lee W-W, Tsang M, Krishnan E, Weyand CM, et al. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat Med (2012) 18(10):1518–24. doi: 10.1038/nm.2963
70. Yu M, Li G, Lee W-W, Yuan M, Cui D, Weyand CM, et al. Signal inhibition by the dual-specific phosphatase 4 impairs T cell-dependent B-cell responses with age. Proc Natl Acad Sci (2012) 109(15):E879–E88.
71. Franceschi C. Obesity in geroscience—is cellular senescence the culprit? Nat Rev Endocrinol (2017) 13(2):76–8. doi: 10.1038/nrendo.2016.213
72. Ponti F, Santoro A, Mercatelli D, Gasperini C, Conte M, Martucci M, et al. Aging and imaging assessment of body composition: from fat to facts. Front endocrinol (2020) 10:861. doi: 10.3389/fendo.2019.00861
73. Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol (2018) 586. doi: 10.3389/fimmu.2018.00586
74. Franceschi C, Garagnani P, Vitale G, Capri M, Salvioli S. Inflammaging and ‘Garb-aging’. Trends Endocrinol Metab (2017) 28(3):199–212. doi: 10.1016/j.tem.2016.09.005
75. Gardner EM, Murasko DM. Age-related changes in Type 1 and Type 2 cytokine production in humans. Biogerontology (2002) 3:271–90. doi: 10.1023/A:1020151401826
76. Albright JW, Zúñiga-Pflücker JC, Albright JF. Transcriptional control of IL-2 and IL-4 in T cells of young and old mice. Cell Immunol (1995) 164(2):170–5. doi: 10.1006/cimm.1995.1158
77. Pelletier I, Sato S. Specific recognition and cleavage of galectin-3 by Leishmania major through species-specific polygalactose epitope. J Biol Chem (2002) 277(20):17663–70. doi: 10.1074/jbc.M201562200
78. Hobbs M, Weigle W, Noonan D, Torbett B, McEvilly R, Koch R, et al. Patterns of cytokine gene expression by CD4+ T cells from young and old mice. J Immunol (Baltimore Md: 1950). (1993) 150(8):3602–14. doi: 10.4049/jimmunol.150.8.3602
79. Ershler W, Sun W, Binkley N, Gravenstein S, Volk M, Kamoske G, et al. Interleukin-6 and aging: blood levels and mononuclear cell production increase with advancing age and in vitro production is modifiable by dietary restriction. Lymphokine Cytokine Res (1993) 12(4):225–30.
80. Palmeri M, Misiano G, Malaguarnera M, Forte GI, Vaccarino L, Milano S, et al. Cytokine serum profile in a group of Sicilian nonagenarians. J Immunoassay Immunochemistry. (2012) 33(1):82–90. doi: 10.1080/15321819.2011.601781
81. Pedersen B, Bruunsgaard H, Ostrowski K, Krabbe K, Hansen H, Krzywkowski K, et al. Cytokines in aging and exercise. Int J sports Med (2000) 21(Sup. 1):4–9. doi: 10.1055/s-2000-1444
82. Minciullo PL, Catalano A, Mandraffino G, Casciaro M, Crucitti A, Maltese G, et al. Inflammaging and anti-inflammaging: the role of cytokines in extreme longevity. Archivum immunologiae therapiae experimentalis. (2016) 64:111–26. doi: 10.1007/s00005-015-0377-3
83. Huang H, Patel DD, Manton KG. The immune system in aging: roles of cytokines, T cells and NK cells. Front Biosci (2005) 10(4):192–215. doi: 10.2741/1521
84. Bruunsgaard H, Andersen-Ranberg K, Jeune B, Pedersen AN, Skinhøj P, Pedersen BK. A high plasma concentration of TNF-α is associated with dementia in centenarians. Journals Gerontology Ser A: Biomed Sci Med Sci (1999) 54(7):M357–M64. doi: 10.1093/gerona/54.7.M357
85. Michaud M, Balardy L, Moulis G, Gaudin C, Peyrot C, Vellas B, et al. Proinflammatory cytokines, aging, and age-related diseases. J Am Med Directors Assoc (2013) 14(12):877–82. doi: 10.1016/j.jamda.2013.05.009
86. Jylhä M, Paavilainen P, Lehtimäki T, Goebeler S, Karhunen PJ, Hervonen A, et al. Interleukin-1 receptor antagonist, interleukin-6, and C-reactive protein as predictors of mortality in nonagenarians: the vitality 90+ study. Journals Gerontology Ser A: Biol Sci Med Sci (2007) 62(9):1016–21. doi: 10.1093/gerona/62.9.1016
87. Zanni F, Vescovini R, Biasini C, Fagnoni F, Zanlari L, Telera A, et al. Marked increase with age of type 1 cytokines within memory and effector/cytotoxic CD8+ T cells in humans: a contribution to understand the relationship between inflammation and immunosenescence. Exp gerontol (2003) 38(9):981–7. doi: 10.1016/S0531-5565(03)00160-8
88. Alberti S, Cevenini E, Ostan R, Capri M, Salvioli S, Bucci L, et al. Age-dependent modifications of Type 1 and Type 2 cytokines within virgin and memory CD4+ T cells in humans. Mech Ageing Dev (2006) 127(6):560–6. doi: 10.1016/j.mad.2006.01.014
89. Rea I, Stewart M, Campbell P, Alexander H, Crockard A, Morris T. Changes in lymphocyte subsets, interleukin 2, and soluble interleukin 2 receptor in old and very old age. Gerontology (1996) 42(2):69–78. doi: 10.1159/000213775
90. Caruso C, Candore G, Cigna D, Di Lorenzo G, Sireci G, Dieli F, et al. Cytokine production pathway in the elderly. Immunologic Res (1996) 15:84–90. doi: 10.1007/BF02918286
91. Myśliwska J, Bryl E, Foerster J, Myśliwski A. Increase of interleukin 6 and decrease of interleukin 2 production during the ageing process are influenced by the health status. Mech Ageing Dev (1998) 100(3):313–28. doi: 10.1016/S0047-6374(97)00154-1
92. Candore G, Di Lorenzo G, Caruso C, Modica MA, Colucci AT, Crescimanno G, et al. The effect of age on mitogen responsive T cell precursors in human beings is completely restored by interleukin-2. Mech Ageing Dev (1992) 63(3):297–307. doi: 10.1016/0047-6374(92)90007-Z
93. Saxena A, Khosraviani S, Noel S, Mohan D, Donner T, Hamad ARA. Interleukin-10 paradox: A potent immunoregulatory cytokine that has been difficult to harness for immunotherapy. Cytokine (2015) 74(1):27–34. doi: 10.1016/j.cyto.2014.10.031
94. Bruunsgaard H, Pedersen AN, Schroll M, Skinhøj P, Pedersen B. Proliferative responses of blood mononuclear cells (BMNC) in a cohort of elderly humans: role of lymphocyte phenotype and cytokine production. Clin Exp Immunol (2000) 119(3):433–40. doi: 10.1046/j.1365-2249.2000.01146.x
95. Llorente L, Richaud-Patin Y, Alvarado C, Vidaller A, Jakez-Ocampo J. Autoantibody production in healthy elderly people is not promoted by interleukin-10 although this cytokine is expressed in them by a peculiar CD8+ CD3+ large granular cell subpopulation. Scandinavian J Immunol (1997) 45(4):401–7. doi: 10.1046/j.1365-3083.1997.d01-409.x
96. Hirokawa K, Utsuyama M, Hayashi Y, Kitagawa M, Makinodan T, Fulop T. Slower immune system aging in women versus men in the Japanese population. Immun Ageing. (2013) 10(1):1–9. doi: 10.1186/1742-4933-10-19
97. Sansoni P, Vescovini R, Fagnoni F, Biasini C, Zanni F, Zanlari L, et al. The immune system in extreme longevity. Exp gerontol (2008) 43(2):61–5. doi: 10.1016/j.exger.2007.06.008
98. Rea IM, Maxwell LD, McNerlan SE, Alexander HD, Curran MD, Middleton D, et al. Killer immunoglobulin-like receptors (KIR) haplogroups A and B track with natural killer cells and cytokine profile in aged subjects: observations from octo/nonagenarians in the Belfast Elderly Longitudinal Free-living Aging Study (BELFAST). Immun Ageing. (2013) 10:1–12. doi: 10.1186/1742-4933-10-35
99. Forsey R, Thompson J, Ernerudh J, Hurst T, Strindhall J, Johansson B, et al. Plasma cytokine profiles in elderly humans. Mech Ageing Dev (2003) 124(4):487–93. doi: 10.1016/S0047-6374(03)00025-3
100. Carrieri G, Marzi E, Olivieri F, Marchegiani F, Cavallone L, Cardelli M, et al. The G/C915 polymorphism of transforming growth factor β1 is associated with human longevity: a study in Italian centenarians. Aging Cell (2004) 3(6):443–8. doi: 10.1111/j.1474-9728.2004.00129.x
101. Halper B, Hofmann M, Oesen S, Franzke B, Stuparits P, Vidotto C, et al. Influence of age and physical fitness on miRNA-21, TGF-β and its receptors in leukocytes of healthy women. Exerc Immunol Rev (2015) 21(154):63.
102. Hamming I, Timens W, Bulthuis M, Lely A, Gv N, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol (2004) 203(2):631–7. doi: 10.1002/path.1570
103. Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, et al. Specific ACE2 expression in cholangiocytes may cause liver damage after 2019-nCoV infection. biorxiv (2020). doi: 10.1101/2020.02.03.931766
104. Merad M, Blish CA, Sallusto F, Iwasaki A. The immunology and immunopathology of COVID-19. Science (2022) 375(6585):1122–7. doi: 10.1126/science.abm8108
105. Kouwaki T, Nishimura T, Wang G, Oshiumi H. RIG-I-like receptor-mediated recognition of viral genomic RNA of severe acute respiratory syndrome coronavirus-2 and viral escape from the host innate immune responses. Front Immunol (2021) 12:700926. doi: 10.3389/fimmu.2021.700926
106. Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol (2020) 20(9):537–51. doi: 10.1038/s41577-020-0288-3
107. Costela-Ruiz VJ, Illescas-Montes R, Puerta-Puerta JM, Ruiz C, Melguizo-Rodríguez L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth factor Rev (2020) 54:62–75. doi: 10.1016/j.cytogfr.2020.06.001
108. Wang Y, Zhao J, Yang L, Hu J, Yao Y. Value of the neutrophil-lymphocyte ratio in predicting COVID-19 severity: a meta-analysis. Dis Markers. (2021) 2021:2571912. doi: 10.1155/2021/2571912
109. Ma Y, Zhang Y, Zhu L. Role of neutrophils in acute viral infection. Immunity Inflammation disease. (2021) 9(4):1186–96. doi: 10.1002/iid3.500
110. Campos-Valdez M, Monroy-Ramírez HC, Armendáriz-Borunda J, Sánchez-Orozco LV. Molecular mechanisms during hepatitis B infection and the effects of the virus variability. Viruses (2021) 13(6):1167. doi: 10.3390/v13061167
111. Gómez-Rial J, Rivero-Calle I, Salas A, Martinon-Torres F. Role of monocytes/macrophages in Covid-19 pathogenesis: implications for therapy. Infection Drug Resistance (2020) 13:2485–93. doi: 10.2147/IDR.S258639
112. Meidaninikjeh S, Sabouni N, Marzouni HZ, Bengar S, Khalili A, Jafari R. Monocytes and macrophages in COVID-19: Friends and foes. Life Sci (2021) 269:119010. doi: 10.1016/j.lfs.2020.119010
113. Wen W, Su W, Tang H, Le W, Zhang X, Zheng Y, et al. Immune cell profiling of COVID-19 patients in the recovery stage by single-cell sequencing. Cell discovery. (2020) 6(1):31. doi: 10.1038/s41421-020-0168-9
114. Copaescu A, Smibert O, Gibson A, Phillips EJ, Trubiano JA. The role of IL-6 and other mediators in the cytokine storm associated with SARS-CoV-2 infection. J Allergy Clin Immunol (2020) 146(3):518–34.e1. doi: 10.1016/j.jaci.2020.07.001
115. Deng Z, Zhang M, Zhu T, Zhili N, Liu Z, Xiang R, et al. Dynamic changes in peripheral blood lymphocyte subsets in adult patients with COVID-19. Int J Infect Diseases. (2020) 98:353–8. doi: 10.1016/j.ijid.2020.07.003
116. Chai X, Hu L, Zhang Y, Han W, Lu Z, Ke A, et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. bioRxiv (2020) 2020.02.03.93176. doi: 10.1101/2020.02.03.931766
117. Godfraind C, Holmes KV, Coutelier J-P. Thymus involution induced by mouse hepatitis virus A59 in BALB/c mice. J Virology. (1995) 69(10):6541–7. doi: 10.1128/jvi.69.10.6541-6547.1995
118. Li T, Qiu Z, Zhang L, Han Y, He W, Liu Z, et al. Significant changes of peripheral T lymphocyte subsets in patients with severe acute respiratory syndrome. J Infect diseases. (2004) 189(4):648–51. doi: 10.1086/381535
119. Jamilloux Y, Henry T, Belot A, Viel S, Fauter M, El Jammal T, et al. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun Rev (2020) 19(7):102567. doi: 10.1016/j.autrev.2020.102567
120. Hou H, Zhang B, Huang H, Luo Y, Wu S, Tang G, et al. Using IL-2R/lymphocytes for predicting the clinical progression of patients with COVID-19. Clin Exp Immunol (2020) 201(1):76–84. doi: 10.1111/cei.13450
121. Yan D, Liu X-Y, Y-n Z, Huang L, Dan B-t, Zhang G-j, et al. Factors associated with prolonged viral shedding and impact of lopinavir/ritonavir treatment in hospitalised non-critically ill patients with SARS-CoV-2 infection. Eur Respir J (2020) 56(1). doi: 10.1183/13993003.00799-2020
122. Abbasifard M, Khorramdelazad H. The bio-mission of interleukin-6 in the pathogenesis of COVID-19: A brief look at potential therapeutic tactics. Life Sci (2020) 257:118097. doi: 10.1016/j.lfs.2020.118097
123. Farnoosh G, Ghanei M, Khorramdelazad H, Alishiri G, Farahani AJ, Shahriary A, et al. Are Iranian sulfur mustard gas-exposed survivors more vulnerable to SARS-CoV-2? Some similarity in their pathogenesis. Disaster Med Public Health preparedness. (2020) 14(6):826–32. doi: 10.1017/dmp.2020.156
124. Shaath H, Vishnubalaji R, Elkord E, Alajez NM. Single-cell transcriptome analysis highlights a role for neutrophils and inflammatory macrophages in the pathogenesis of severe COVID-19. Cells (2020) 9(11):2374. doi: 10.3390/cells9112374
125. Makwana R, Gozzard N, Spina D, Page C. TNF-α-induces airway hyperresponsiveness to cholinergic stimulation in Guinea pig airways. Br J Pharmacol (2012) 165(6):1978–91. doi: 10.1111/j.1476-5381.2011.01675.x
126. Ye X, Luo Y, Xia S, Sun Q, Ding J, Zhou Y, et al. Clinical efficacy of lopinavir/ritonavir in the treatment of Coronavirus disease 2019. Eur Rev Med Pharmacol Sci (2020) 24 - N. 6:3390–6. doi: 10.26355/eurrev_202003_20706
127. Gori A, Dondossola D, Antonelli B, Mangioni D, Alagna L, Reggiani P, et al. Coronavirus disease 2019 and transplantation: a view from the inside. Am J Transplantation. (2020) 20(7):1939. doi: 10.1111/ajt.15853
128. Sironi M, Breviario F, Proserpio P, Biondi A, Vecchi A, Van Damme J, et al. IL-1 stimulates IL-6 production in endothelial cells. J Immunol (Baltimore Md: 1950). (1989) 142(2):549–53. doi: 10.4049/jimmunol.142.2.549
129. Warke T, Fitch P, Brown V, Taylor R, Lyons J, Ennis M, et al. Exhaled nitric oxide correlates with airway eosinophils in childhood asthma. Thorax (2002) 57(5):383–7. doi: 10.1136/thorax.57.5.383
130. Darif D, Hammi I, Kihel A, Saik IEI, Guessous F, Akarid K. The pro-inflammatory cytokines in COVID-19 pathogenesis: What goes wrong? Microbial pathogenesis (2021) 153:104799. doi: 10.1016/j.micpath.2021.104799
131. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
132. Ren K, Torres R. Role of interleukin-1β during pain and inflammation. Brain Res Rev (2009) 60(1):57–64. doi: 10.1016/j.brainresrev.2008.12.020
133. Khorramdelazad H, Kazemi MH, Najafi A, Keykhaee M, Emameh RZ, Falak R. Immunopathological similarities between COVID-19 and influenza: Investigating the consequences of Co-infection. Microbial pathogenesis. (2021) 152:104554. doi: 10.1016/j.micpath.2020.104554
134. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science (2020) 369(6504):718–24. doi: 10.1126/science.abc6027
135. Wang Z, Yang B, Li Q, Wen L, Zhang R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin Infect diseases. (2020) 71(15):769–77. doi: 10.1093/cid/ciaa272
136. Chen X, Zhao B, Qu Y, Chen Y, Xiong J, Feng Y, et al. Detectable serum SARS-CoV-2 viral load (RNAaemia) is closely associated with drastically elevated interleukin 6 (IL-6) level in critically ill COVID-19 patients. MedRxiv (2020) 2020.02.29.2002952. doi: 10.1101/2020.02.29.20029520
137. Hot A, Lenief V, Miossec P. Combination of IL-17 and TNFα induces a pro-inflammatory, pro-coagulant and pro-thrombotic phenotype in human endothelial cells. Ann rheumatic diseases. (2012) 71(5):768–76. doi: 10.1136/annrheumdis-2011-200468
138. Matsushima K, Yang D, Oppenheim JJ. Interleukin-8: An evolving chemokine. Cytokine (2022) 153:155828. doi: 10.1016/j.cyto.2022.155828
139. Dong L, Hu S, Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug discoveries Ther (2020) 14(1):58–60. doi: 10.5582/ddt.2020.01012
140. Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerging Microbes infections. (2020) 9(1):761–70. doi: 10.1080/22221751.2020.1747363
141. Chen N, Zhou M, Dong X, Qu J, Gong F, Han Y, et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: a descriptive study. Lancet (2020) 395(10223):507–13. doi: 10.1016/S0140-6736(20)30211-7
142. Muñoz-Carrillo JL, Contreras-Cordero JF, Gutiérrez-Coronado O, Villalobos-Gutiérrez PT, Ramos-Gracia LG, Hernández-Reyes VE. Cytokine profiling plays a crucial role in activating immune system to clear infectious pathogens. Immune response activation immunomodulation (2018).
143. Capucetti A, Albano F, Bonecchi R. Multiple roles for chemokines in neutrophil biology. Front Immunol (2020) 11:1259. doi: 10.3389/fimmu.2020.01259
144. Teijeira A, Garasa S, MdC O, Cirella A, Olivera I, Glez-Vaz J, et al. Differential Interleukin-8 thresholds for chemotaxis and netosis in human neutrophils. Eur J Immunol (2021) 51(9):2274–80. doi: 10.1002/eji.202049029
145. Kesmez Can F, Özkurt Z, Öztürk N, Sezen S. Effect of IL-6, IL-8/CXCL8, IP-10/CXCL 10 levels on the severity in COVID 19 infection. Int J Clin Practice. (2021) 75(12):e14970. doi: 10.1111/ijcp.14970
146. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol (2020) 827. doi: 10.3389/fimmu.2020.00827
147. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
148. Freeman TL, Swartz TH. Targeting the NLRP3 inflammasome in severe COVID-19. Front Immunol (2020) 1518. doi: 10.3389/fimmu.2020.01518
149. da Costa LS, Outlioua A, Anginot A, Akarid K, Arnoult D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death disease. (2019) 10(5):346. doi: 10.1038/s41419-019-1579-0
150. Ma A, Zhang L, Ye X, Chen J, Yu J, Zhuang L, et al. High levels of circulating IL-8 and soluble IL-2R are associated with prolonged illness in patients with severe COVID-19. Front Immunol (2021) 12:626235. doi: 10.3389/fimmu.2021.626235
151. Jones SA, Jenkins BJ. Recent insights into targeting the IL-6 cytokine family in inflammatory diseases and cancer. Nat Rev Immunol (2018) 18(12):773–89. doi: 10.1038/s41577-018-0066-7
152. Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harbor Perspect Biol (2014) 6(10):a016295. doi: 10.1101/cshperspect.a016295
153. Zuo Y, Yalavarthi S, Shi H, Gockman K, Zuo M, Madison JA, et al. Neutrophil extracellular traps in COVID-19. JCI Insight (2020) 5(11):e138999. doi: 10.1172/jci.insight.138999
154. Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine (2013) 64(2):477–85. doi: 10.1016/j.cyto.2013.07.022
155. Herold T, Jurinovic V, Arnreich C, Lipworth BJ, Hellmuth JC, von Bergwelt-Baildon M, et al. Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J Allergy Clin Immunol (2020) 146(1):128–36.e4. doi: 10.1016/j.jaci.2020.05.008
156. Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med (1996) 183(6):2593–603. doi: 10.1084/jem.183.6.2593
157. Ryzhakov G, CC-k L, Blazek K, To K-w, Hussell T, Udalova I. IL-17 boosts proinflammatory outcome of antiviral response in human cells. J Immunol (2011) 187(10):5357–62. doi: 10.4049/jimmunol.1100917
158. Bulat V, Situm M, Azdajic MD, Likic R. Potential role of IL-17 blocking agents in the treatment of severe COVID-19? Br J Clin Pharmacol (2021) 87(3):1578. doi: 10.1111/bcp.14437
159. Raucci F, Mansour AA, Casillo GM, Saviano A, Caso F, Scarpa R, et al. Interleukin-17A (IL-17A), a key molecule of innate and adaptive immunity, and its potential involvement in COVID-19-related thrombotic and vascular mechanisms. Autoimmun Rev (2020) 19(7):102572. doi: 10.1016/j.autrev.2020.102572
160. Hou W, Jin Y-H, Kang HS, Kim BS. Interleukin-6 (IL-6) and IL-17 synergistically promote viral persistence by inhibiting cellular apoptosis and cytotoxic T cell function. J virology. (2014) 88(15):8479–89. doi: 10.1128/JVI.00724-14
161. Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci (2020) 63:364–74. doi: 10.1007/s11427-020-1643-8
162. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
163. Jothimani D, Venugopal R, Abedin MF, Kaliamoorthy I, Rela M. COVID-19 and the liver. J hepatology. (2020) 73(5):1231–40. doi: 10.1016/j.jhep.2020.06.006
164. Li J, Fan J-G. Characteristics and mechanism of liver injury in 2019 coronavirus disease. J Clin Trans hepatology. (2020) 8(1):13. doi: 10.14218/JCTH.2020.00019
165. Abdulla S, Hussain A, Azim D, Abduallah EH, Elawamy H, Nasim S, et al. COVID-19-induced hepatic injury: a systematic review and meta-analysis. Cureus (2020) 12(10). doi: 10.7759/cureus.10923
166. Mohammed SA, Eid KM, Anyiam FE, Wadaaallah H, Muhamed MAM, Morsi MH, et al. Liver injury with COVID-19: laboratory and histopathological outcome—systematic review and meta-analysis. Egyptian Liver J (2022) 12(1):9. doi: 10.1186/s43066-022-00171-6
167. Sivandzadeh GR, Askari H, Safarpour AR, Ejtehadi F, Raeis-Abdollahi E, Lari AV, et al. COVID-19 infection and liver injury: Clinical features, biomarkers, potential mechanisms, treatment, and management challenges. World J Clin cases. (2021) 9(22):6178. doi: 10.12998/wjcc.v9.i22.6178
168. Wu J, Song S, Cao H-C, Li L-J. Liver diseases in COVID-19: Etiology, treatment and prognosis. World J gastroenterol (2020) 26(19):2286. doi: 10.3748/wjg.v26.i19.2286
169. Russo FP, Burra P, Zanetto A. COVID-19 and liver disease: where are we now? Nat Rev Gastroenterol Hepatol (2022) 19(5):277–8. doi: 10.1038/s41575-022-00607-9
170. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Modern Pathology. (2020) 33(6):1007–14. doi: 10.1038/s41379-020-0536-x
171. Zhong P, Xu J, Yang D, Shen Y, Wang L, Feng Y, et al. COVID-19-associated gastrointestinal and liver injury: clinical features and potential mechanisms. Signal transduction targeted Ther (2020) 5(1):256. doi: 10.1038/s41392-020-00373-7
172. Gracia-Ramos AE, Jaquez-Quintana JO, Contreras-Omana R, Auron M. Liver dysfunction and SARS-CoV-2 infection. World J Gastroenterol (2021) 27(26):3951. doi: 10.3748/wjg.v27.i26.3951
173. Cai Y, Ye L-P, Song Y-Q, Mao X-L, Wang L, Jiang Y-Z, et al. Liver injury in COVID-19: Detection, pathogenesis, and treatment. World J Gastroenterol (2021) 27(22):3022. doi: 10.3748/wjg.v27.i22.3022
174. Ahmed J, Rizwan T, Malik F, Akhter R, Malik M, Ahmad J, et al. COVID-19 and liver injury: a systematic review and meta-analysis. Cureus (2020) 12(7). doi: 10.7759/cureus.9424
175. Li D, Ding X, Xie M, Tian D, Xia L. COVID-19-associated liver injury: from bedside to bench. J Gastroenterol (2021) 56:218–30. doi: 10.1007/s00535-021-01760-9
176. Omrani-Nava V, Maleki I, Ahmadi A, Moosazadeh M, Hedayatizadeh-Omran A, Roozbeh F, et al. Evaluation of hepatic enzymes changes and association with prognosis in COVID-19 patients. Hepatitis Monthly (2020) 20(4):e103179. doi: 10.5812/hepatmon.103179
177. Yu D, Du Q, Yan S, Guo X-G, He Y, Zhu G, et al. Liver injury in COVID-19: clinical features and treatment management. Virol J (2021) 18(1):1–9. doi: 10.1186/s12985-021-01593-1
178. Dufour J-F, Marjot T, Becchetti C, Tilg H. COVID-19 and liver disease. Gut (2022) 71(11):2350–62. doi: 10.1136/gutjnl-2021-326792
179. Burra P. Liver abnorMalities and endocrine diseases. Best Pract Res Clin Gastroenterol (2013) 27(4):553–63. doi: 10.1016/j.bpg.2013.06.014
180. Verma HK, Bhaskar L. SARS-CoV-2 infection in people with pre-existing liver disease: Further research is warranted. World J Gastroenterol (2021) 27(45):7855. doi: 10.3748/wjg.v27.i45.7855
181. Hartl L, Haslinger K, Angerer M, Jachs M, Simbrunner B, Bauer DJ, et al. Age-adjusted mortality and predictive value of liver chemistries in a Viennese cohort of COVID-19 patients. Liver Int (2022) 42(6):1297–307. doi: 10.1111/liv.15274
182. Marjot T, Webb GJ, Barritt AS IV, Moon AM, Stamataki Z, Wong VW, et al. COVID-19 and liver disease: mechanistic and clinical perspectives. Nat Rev Gastroenterol hepatology. (2021) 18(5):348–64. doi: 10.1038/s41575-021-00426-4
183. Xu J, Xiao W, Liang X, Shi L, Zhang P, Wang Y, et al. A meta-analysis on the risk factors adjusted association between cardiovascular disease and COVID-19 severity. BMC Public Health (2021) 21(1):1–49. doi: 10.1186/s12889-021-11051-w
184. Chuan Q, Luoqi Z, Ziwei H, Shuoqi Z, Sheng Y, Yu T, et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin Infect Dis (2020) 10.
185. Zhang S-S, Dong L, Wang G-M, Tian Y, Ye X-F, Zhao Y, et al. Progressive liver injury and increased mortality risk in COVID-19 patients: a retrospective cohort study in China. World J gastroenterol (2021) 27(9):835. doi: 10.3748/wjg.v27.i9.835
186. Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. Jama (2020) 323(20):2052–9. doi: 10.1001/jama.2020.6775
187. Cooper S, Tobar A, Konen O, Orenstein N, Kropach Gilad N, Landau YE, et al. Long COVID-19 liver manifestation in children. J Pediatr Gastroenterol Nutr (2022) 75(3):244–51. doi: 10.1097/MPG.0000000000003521
188. Xu Y, Li X, Zhu B, Liang H, Fang C, Gong Y, et al. Characteristics of pediatric SARS-CoV-2 infection and potential evidence for persistent fecal viral shedding. Nat Med (2020) 26(4):502–5. doi: 10.1038/s41591-020-0817-4
189. Yang R-X, Zheng R-D, Fan J-G. Etiology and management of liver injury in patients with COVID-19. World J gastroenterol (2020) 26(32):4753. doi: 10.3748/wjg.v26.i32.4753
190. McConnell MJ, Kondo R, Kawaguchi N, Iwakiri Y. Covid-19 and liver injury: role of inflammatory endotheliopathy, platelet dysfunction, and thrombosis. Hepatol Commun (2022) 6(2):255–69. doi: 10.1002/hep4.1843
191. Wilson JG, Simpson LJ, Ferreira A-M, Rustagi A, Roque J, Asuni A, et al. Cytokine profile in plasma of severe COVID-19 does not differ from ARDS and sepsis. JCI Insight (2020) 5(17):e140289. doi: 10.1172/jci.insight.140289
192. Wijarnpreecha K, Ungprasert P, Panjawatanan P, Harnois DM, Zaver HB, Ahmed A, et al. COVID-19 and liver injury: a meta-analysis. Eur J Gastroenterol Hepatol (2021) 33(7):990–5. doi: 10.1097/MEG.0000000000001817
193. Effenberger M, Grander C, Grabherr F, Griesmacher A, Ploner T, Hartig F, et al. Systemic inflammation as fuel for acute liver injury in COVID-19. Digestive Liver Disease. (2021) 53(2):158–65. doi: 10.1016/j.dld.2020.08.004
194. Shafran N, Issachar A, Shochat T, Shafran IH, Bursztyn M, Shlomai A. Abnormal liver tests in patients with SARS-CoV-2 or influenza–prognostic similarities and temporal disparities. JHEP Rep (2021) 3(3):100258. doi: 10.1016/j.jhepr.2021.100258
195. Dawood RM, Salum GM, Abd El-Meguid M. The impact of COVID-19 on liver injury. Am J Med Sci (2022) 363(2):94–103. doi: 10.1016/j.amjms.2021.11.001
196. Moore N, Bosco-Levy P, Thurin N, Blin P, Droz-Perroteau C. NSAIDs and COVID-19: a systematic review and meta-analysis. Drug safety. (2021) 44(9):929–38. doi: 10.1007/s40264-021-01089-5
197. Teijaro JR, Farber DL. COVID-19 vaccines: modes of immune activation and future challenges. Nat Rev Immunol (2021) 21(4):195–7. doi: 10.1038/s41577-021-00526-x
198. Her M, Lee Y, Jung E, Kim T, Kim D. Liver enzyme abnorMalities in systemic lupus erythematosus: a focus on toxic hepatitis. Rheumatol Int (2011) 31:79–84. doi: 10.1007/s00296-009-1237-4
199. Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med virology. (2021) 93(1):250–6. doi: 10.1002/jmv.26232
200. Ledford H. Coronavirus breakthrough: dexamethasone is first drug shown to save lives. Nature (2020) 582(7813):469–70. doi: 10.1038/d41586-020-01824-5
201. Marra F, Smolders EJ, El-Sherif O, Boyle A, Davidson K, Sommerville AJ, et al. Recommendations for dosing of repurposed COVID-19 medications in patients with renal and hepatic impairment. Drugs R&D. (2021) 21:9–27. doi: 10.1007/s40268-020-00333-0
202. Bril F, Al Diffalha S, Dean M, Fettig DM. Autoimmune hepatitis developing after coronavirus disease 2019 (COVID-19) vaccine: causality or casualty? J Hepatol (2021) 75(1):222–4. doi: 10.1016/j.jhep.2021.04.003
203. Dubert M, Visseaux B, Isernia V, Bouadma L, Deconinck L, Patrier J, et al. Case report study of the first five COVID-19 patients treated with remdesivir in France. Int J Infect diseases. (2020) 98:290–3. doi: 10.1016/j.ijid.2020.06.093
204. Mann R, Sekhon S, Sekhon S. Drug-induced liver injury after COVID-19 vaccine. Cureus (2021) 13(7). doi: 10.7759/cureus.16491
205. Fimiano F, D’Amato D, Gambella A, Marzano A, Saracco GM, Morgando A. Autoimmune hepatitis or drug-induced autoimmune hepatitis following Covid-19 vaccination? Liver Int (2022) 42(5):1204. doi: 10.1111/liv.15224
206. Guaraldi G, Meschiari M, Cozzi-Lepri A, Milic J, Tonelli R, Menozzi M, et al. Tocilizumab in patients with severe COVID-19: a retrospective cohort study. Lancet Rheumatol (2020) 2(8):e474–e84. doi: 10.1016/S2665-9913(20)30173-9
207. Chow KW, Pham NV, Ibrahim BM, Hong K, Saab S. Autoimmune hepatitis-like syndrome following COVID-19 vaccination: a systematic review of the literature. Digestive Dis Sci (2022) 67(9):4574–80. doi: 10.1007/s10620-022-07504-w
208. Muhović D, Bojović J, Bulatović A, Vukčević B, Ratković M, Lazović R, et al. First case of drug-induced liver injury associated with the use of tocilizumab in a patient with COVID-19. Liver Int (2020) 40(8):1901–5. doi: 10.1111/liv.14516
209. Efe C, Kulkarni AV, Terziroli Beretta-Piccoli B, Magro B, Stättermayer A, Cengiz M, et al. Liver injury after SARS-CoV-2 vaccination: Features of immune-mediated hepatitis, role of corticosteroid therapy and outcome. Hepatology (2022) 76(6):1576–86. doi: 10.1002/hep.32572
210. Fan Z, Chen L, Li J, Cheng X, Yang J, Tian C, et al. Clinical features of COVID-19-related liver functional abnorMality. Clin Gastroenterol Hepatology. (2020) 18(7):1561–6. doi: 10.1016/j.cgh.2020.04.002
211. Serviddio G, Villani R, Stallone G, Scioscia G, Foschino-Barbaro MP, Lacedonia D. Tocilizumab and liver injury in patients with COVID-19. Ther Adv Gastroenterol (2020) 13:1756284820959183. doi: 10.1177/1756284820959183
212. Hines A, Shen JG, Olazagasti C, Shams S. Immune thrombocytopenic purpura and acute liver injury after COVID-19 vaccine. BMJ Case Rep CP. (2021) 14(7):e242678. doi: 10.1136/bcr-2021-242678
213. Schultz NH, Sørvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT, et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. New Engl J Med (2021) 384(22):2124–30. doi: 10.1056/NEJMoa2104882
214. Vincent MJ, Bergeron E, Benjannet S, Erickson BR, Rollin PE, Ksiazek TG, et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol J (2005) 2(1):1–10. doi: 10.1186/1743-422X-2-69
215. Cavalcanti AB, Zampieri FG, Rosa RG, Azevedo LC, Veiga VC, Avezum A, et al. Hydroxychloroquine with or without azithromycin in mild-to-moderate covid-19. New Engl J Med (2020) 383(21):2041–52. doi: 10.1056/NEJMoa2019014
216. Andrade RJ, Chalasani N, Björnsson ES, Suzuki A, Kullak-Ublick GA, Watkins PB, et al. Drug-induced liver injury. Nat Rev Dis Primers. (2019) 5(1):58. doi: 10.1038/s41572-019-0105-0
217. Premkumar M, Kedarisetty CK. Cytokine storm of COVID-19 and its impact on patients with and without chronic liver disease. J Clin Trans Hepatology. (2021) 9(2):256. doi: 10.14218/JCTH.2021.00055
218. Gautret P, Lagier J-C, Parola P, Meddeb L, Mailhe M, Doudier B, et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int J antimicrobial agents. (2020) 56(1):105949. doi: 10.1016/j.ijantimicag.2020.105949
219. Abubakar AR, Sani IH, Godman B, Kumar S, Islam S, Jahan I, et al. Systematic review on the therapeutic options for COVID-19: clinical evidence of drug efficacy and implications. Infection Drug resistance (2020) 13:4673–95. doi: 10.2147/IDR.S289037
220. Boulware DR, Pullen MF, Bangdiwala AS, Pastick KA, Lofgren SM, Okafor EC, et al. A randomized trial of hydroxychloroquine as postexposure prophylaxis for Covid-19. New Engl J Med (2020) 383(6):517–25. doi: 10.1056/NEJMoa2016638
221. Guan G-W, Gao L, Wang J-W, Wen X-J, Mao T-H, Peng S-W, et al. Exploring the mechanism of liver enzyme abnorMalities in patients with novel coronavirus-infected pneumonia. Zhonghua gan zang bing za zhi= Zhonghua ganzangbing zazhi= Chin J hepatology. (2020) 28(2):100–6. doi: 10.3760/cma.j.issn.1007-3418.2020.02.002
222. Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of Covid-19—preliminary report. New Engl J Med (2020) 383(19):1813–36. doi: 10.1056/NEJMoa2007764
223. Antinori S, Cossu MV, Ridolfo AL, Rech R, Bonazzetti C, Pagani G, et al. Compassionate remdesivir treatment of severe Covid-19 pneumonia in intensive care unit (ICU) and Non-ICU patients: Clinical outcome and differences in post-treatment hospitalisation status. Pharmacol Res (2020) 158:104899. doi: 10.1016/j.phrs.2020.104899
224. Falcão MB, de Goes Cavalcanti LP, Filgueiras Filho NM, de Brito CAA. Case report: hepatotoxicity associated with the use of hydroxychloroquine in a patient with COVID-19. Am J Trop Med hygiene. (2020) 102(6):1214. doi: 10.4269/ajtmh.20-0276
225. Boeckmans J, Rodrigues RM, Demuyser T, Pierard D, Vanhaecke T, Rogiers V. COVID-19 and drug-induced liver injury: a problem of plenty or a petty point? Arch Toxicol (2020) 94:1367–9. doi: 10.1007/s00204-020-02734-1
226. Wang D, Yin Y, Yao Y. Advances in sepsis-associated liver dysfunction. Burns trauma. (2014) 2(3):2321–3868.132689. doi: 10.4103/2321-3868.132689
227. Naeem A, Khamuani MK, Kumar P, Pooja F, Raj D, Lal K, et al. Impact of coronavirus diseases on liver enzymes. Cureus (2021) 13(9):e17650. doi: 10.7759/cureus.17650
228. Nojomi M, Yasin Z, Keyvani H, Makiani MJ, Roham M, Laali A, et al. Effect of arbidol on COVID-19: a randomized controlled trial. BMC Infect Dis (2020) 20, 954. doi: 10.21203/rs.3.rs-78316/v1
229. Limen RY, Sedono R, Sugiarto A, Hariyanto TI. Janus kinase (JAK)-inhibitors and coronavirus disease 2019 (Covid-19) outcomes: a systematic review and meta-analysis. Expert Rev Anti-Infective Ther (2022) 20(3):425–34. doi: 10.1080/14787210.2021.1982695
230. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A trial of lopinavir–ritonavir in adults hospitalized with severe Covid-19. New Engl J Med (2020) 382(19):1787–99. doi: 10.1056/NEJMoa2001282
231. Chen C, Huang J, Yin P, Zhang Y, Cheng Z, Wu J, et al. Favipiravir versus arbidol for COVID-19: a randomized clinical trial. MedRxiv (2020) 2020.03.17.20037432. doi: 10.1101/2020.03.17.20037432
232. Wang Y, Zhang D, Du G, Du R, Zhao J, Jin Y, et al. Remdesivir in adults with severe COVID-19: a randomised, double-blind, placebo-controlled, multicentre trial. Lancet (2020) 395(10236):1569–78. doi: 10.1016/S0140-6736(20)31022-9
233. Spinner CD, Gottlieb RL, Criner GJ, López JRA, Cattelan AM, Viladomiu AS, et al. Effect of remdesivir vs standard care on clinical status at 11 days in patients with moderate COVID-19: a randomized clinical trial. Jama (2020) 324(11):1048–57. doi: 10.1001/jama.2020.16349
234. Durante-Mangoni E, Andini R, Bertolino L, Mele F, Florio LL, Murino P, et al. Early experience with remdesivir in SARS-CoV-2 pneumonia. Infection (2020) 48:779–82. doi: 10.1007/s15010-020-01448-x
235. Leegwater E, Strik A, Wilms E, Bosma L, Burger D, Ottens T. Drug-induced liver injury in a COVID-19 patient: potential interaction of remdesivir with P-glycoprotein inhibitors. Clin Infect Dis (2020) 72 (7), 1256–1258. doi: 10.1093/cid/ciaa883
236. Alqahtani SA, Schattenberg JM. Liver injury in COVID-19: The current evidence. United Eur Gastroenterol J (2020) 8(5):509–19. doi: 10.1177/2050640620924157
237. Cvetkovic RS, Goa KL. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs (2003) 63(8):769–802. doi: 10.2165/00003495-200363080-00004
238. Puccioni-Sohler M, Poton AR, Franklin M, SJd S, Brindeiro R, Tanuri A. Current evidence of neurological features, diagnosis, and neuropathogenesis associated with COVID-19. Rev da Sociedade Bras Medicina Trop (2020) 53:e20200477. doi: 10.1590/0037-8682-0477-2020
239. Wang X, Lei J, Li Z, Yan L. Potential effects of coronaviruses on the liver: An update. Front Med (2021) 8:651658. doi: 10.3389/fmed.2021.651658
240. Lee C, Ahn MY, Byeon K, Choi J-P, Hahm C, Kim H, et al. Clinical experience with use of remdesivir in the treatment of severe acute respiratory syndrome coronavirus 2: a case series. Infection chemotheR (2020) 52(3):369. doi: 10.3947/ic.2020.52.3.369
241. Zampino R, Mele F, Florio LL, Bertolino L, Andini R, Galdo M, et al. Liver injury in remdesivir-treated COVID-19 patients. Hepatol Int (2020) 14:881–3. doi: 10.1007/s12072-020-10077-3
242. Carothers C, Birrer K, Vo M. Acetylcysteine for the treatment of suspected remdesivir-associated acute liver failure in COVID-19: A case series. Pharmacotherapy (2020) 40(11):1166–71. doi: 10.1002/phar.2464
243. Li Y, Xie Z, Lin W, Cai W, Wen C, Guan Y, et al. Efficacy and safety of lopinavir/ritonavir or arbidol in adult patients with mild/moderate COVID-19: an exploratory randomized controlled trial. Med (2020) 1(1):105–13.e4. doi: 10.1016/j.medj.2020.04.001
244. Cai Q, Huang D, Yu H, Zhu Z, Xia Z, Su Y, et al. COVID-19: Abnormal liver function tests. J hepatology. (2020) 73(3):566–74. doi: 10.1016/j.jhep.2020.04.006
245. Sun J, Deng X, Chen X, Huang J, Huang S, Li Y, et al. Incidence of adverse drug reactions in COVID-19 patients in China: an active monitoring study by hospital pharmacovigilance system. Clin Pharmacol Ther (2020) 108(4):791–7. doi: 10.1002/cpt.1866
246. Yamazaki S, Suzuki T, Sayama M, T-a N, Igari H, Ishii I. Suspected cholestatic liver injury induced by favipiravir in a patient with COVID-19. J Infection Chemother (2021) 27(2):390–2. doi: 10.1016/j.jiac.2020.12.021
247. Kelly M, O’Connor R, Townsend L, Coghlan M, Relihan E, Moriarty M, et al. Clinical outcomes and adverse events in patients hospitalised with COVID-19, treated with off-label hydroxychloroquine and azithromycin. Br J Clin Pharmacol (2021) 87(3):1150–4. doi: 10.1111/bcp.14482
248. Stone JH, Frigault MJ, Serling-Boyd NJ, Fernandes AD, Harvey L, Foulkes AS, et al. Efficacy of tocilizumab in patients hospitalized with Covid-19. New Engl J Med (2020) 383(24):2333–44. doi: 10.1056/NEJMoa2028836
249. Rocco A, Sgamato C, Compare D, Nardone G. Autoimmune hepatitis following SARS-CoV-2 vaccine: may not be a casuality. J Hepatology. (2021) 75(3):728–9. doi: 10.1016/j.jhep.2021.05.038
250. Shroff H, Satapathy SK, Crawford JM, Todd NJ, VanWagner LB. Liver injury following SARS-CoV-2 vaccination: a multicenter case series. J hepatology. (2022) 76(1):211–4. doi: 10.1016/j.jhep.2021.07.024
251. Izagirre A, Arzallus T, Garmendia M, Torrente S, Castiella A, Zapata EM. Autoimmune hepatitis following COVID-19 vaccination. J Autoimmun (2022) 132:102874. doi: 10.1016/j.jaut.2022.102874
252. Palla P, Vergadis C, Sakellariou S, Androutsakos T. Autoimmune hepatitis after COVID-19 vaccination: A rare adverse effect? Hepatology (2022) 75(2):489–90. doi: 10.1002/hep.32156
253. Rela M, Jothimani D, Vij M, Rajakumar A, Rammohan A. Auto-immune hepatitis following COVID vaccination. J Autoimmunity. (2021) 123:102688. doi: 10.1016/j.jaut.2021.102688
254. Camacho-Domínguez L, Rodríguez Y, Polo F, Gutierrez JCR, Zapata E, Rojas M, et al. COVID-19 vaccine and autoimmunity. A new case of autoimmune hepatitis and review of the literature. J Trans autoimmunity. (2022) 5:100140. doi: 10.1016/j.jtauto.2022.100140
255. Londoño M-C, Gratacós-Ginès J, Sáez-Peñataro J. Another case of autoimmune hepatitis after SARS-CoV-2 vaccination–still casualty? J Hepatol (2021) 75(5):1248–9. doi: 10.1016/j.jhep.2021.06.004
Keywords: COVID-19, SARS-CoV-2, cytokine storm, interleukin, liver injury, aging
Citation: Aghamohamadi N, Shahba F, Zarezadeh Mehrabadi A, Khorramdelazad H, Karimi M, Falak R and Emameh RZ (2023) Age-dependent immune responses in COVID-19-mediated liver injury: focus on cytokines. Front. Endocrinol. 14:1139692. doi: 10.3389/fendo.2023.1139692
Received: 07 January 2023; Accepted: 21 July 2023;
Published: 15 August 2023.
Edited by:
Marc R Blackman, United States Department of Veterans Affairs, United StatesReviewed by:
Arup Acharjee, Allahabad University, IndiaCopyright © 2023 Aghamohamadi, Shahba, Zarezadeh Mehrabadi, Khorramdelazad, Karimi, Falak and Emameh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Reza Zolfaghari Emameh, em9sZmFnaGFyaUBuaWdlYi5hYy5pcg==; Reza Falak, RmFsYWsuckBpdW1zLmFjLmly
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.