
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 21 April 2023
Sec. Pediatric Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1135768
This article is part of the Research TopicEndocrine Dysfunction in Patients with Down SyndromeView all 5 articles
 David Shaki1,2
David Shaki1,2 Eli Hershkovitz1,2*
Eli Hershkovitz1,2* Shai Tamam3
Shai Tamam3 Arkadi Bollotin2
Arkadi Bollotin2 Odeya David1,2
Odeya David1,2 Guy Yalovitsky2
Guy Yalovitsky2 Neta Loewenthal1,2Lior Carmon1,2Dganit Walker1Raphael Nowak2Alon Haim1,2
Neta Loewenthal1,2Lior Carmon1,2Dganit Walker1Raphael Nowak2Alon Haim1,2Objective: To analyze and determine the safety and efficacy of growth hormone (GH) treatment in Down syndrome (DS) pediatric patients and to weigh ethical aspects involved.
Design: Systematic review and mini meta-analysis of the literature.
Methods: A search was performed in PubMed, Embase, Scopus, and PsycINFO through August 2022. Eligible studies included those who answered at least one of the following two questions: 1) What is the effect of growth hormone treatment in children with Down syndrome? 2) What are the ethical arguments in favor and against growth hormone treatment for children with Down syndrome? Multiple reviewers independently screened each article for eligibility.
Results: In total sixteen reports detailed medical effects of GH treatment in pediatric DS patients and eight studies dealt with ethical aspects of GH treatment. Treatment with GH resulted in significantly higher growth velocity in patients with DS. The ethical complexity is great but does not present insurmountable difficulties to the therapeutic option.
Conclusions: As GH treatment is safe and effective for short-term height growth, GH therapy should be considered in long-term treatment of DS children.
Down syndrome (DS) is the most common chromosomal disorder with an incidence of one in 700 live births in the United States (1) and 1–10 in 1,000 live births worldwide, according to the WHO (2). Linear growth retardation is a cardinal characteristic of DS. Pathologic low height velocity is mainly marked in infancy and adolescence (3). A significant portion of short-stature pathogenesis in children with DS is associated with impaired GHRH-GH-IGF1 axis function, mainly due to an abnormal quantitative and qualitative capacity of the hypothalamus–pituitary axis and reduced bioactivity of endogenous growth hormone (GH) (4).
The effect of GH therapy in children with DS on height, head circumference, and cognitive and motor function has been tested in research studies since the 1950s. Various and even conflicting findings have been reported, and in the absence of a systematic review, it is difficult to establish well-founded conclusions on its true, overall effect.
Life expectancy for people with DS continues to rise – the median lifespan is now 58 years with many living into their sixties and seventies (5). Heart conditions, which can accompany DS, have been routinely corrected by surgery since the early 1980s. Consequently, longer life expectancy also raises the importance for DS patients to have a good quality of life.
This study reviews for the first time all the research reported data with references to the effect of GH therapy for children with DS on longitudinal growth, head circumference, cognitive and motor function, insulin-like growth factor 1 (IGF1) levels, bone age, as well as short and long-term treatment side effects. In addition, we review all data with references to the ethical aspects of GH therapy in DS patients.
The present systematic review was performed in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (6).
No formal ethical approval was required. An extensive literature search of four electronic databases: PubMed, Cochrane Library, Scopus, and PsycINFO via EBSCO was undertaken for studies regarding DS and growth hormone, published until January 2021. The general keywords were ‘Down syndrome’ and ‘growth hormone’, while the search strategy was updated and adapted for each database. Vocabulary supplemented with keywords were used for searches that were conducted separately for each topic and combined later. The search was restricted to humans, and no other restriction was made. Studies in all languages were included. Full-text articles of potentially relevant studies not available through the university library were requested from the authors. We repeated the search on September 2022 and received 27 additional records. A review of the title or abstract was enough to determine that they are not suitable for inclusion in this review.
The scope of data reporting in much of the original works did not allow for a full quality and risk of bias assessment to be carried out on the individual original studies; therefore, no individual quality assessment was carried out.
We used ROBIS, a tool for assessing quality and risk of bias in systematic reviews. The tool is completed in three phases: assess relevance (optional), identify concerns with the review process by 21 questions divided to 4 domains, and judge risk of bias in the review. It is the first rigorously developed tool designed specifically to assess the risk of bias in systematic reviews (7).
Eligible studies enrolled pediatric DS patients who were treated with GH during childhood and examined the effect of treatment on longitudinal growth and other aspects or dealt with the ethical aspects of such treatment. Studies that answered one of the following two questions were included:
1) What is the effect of growth hormone treatment in children with Down syndrome? In relation to this question, studies that examined the effect of such treatment on at least one of the following six outcomes: height, head circumference, cognition and motor skills, side effects, bone age, and IGF1 level, were included.
2) What are the ethical arguments in favor and against growth hormone treatment for children with Down syndrome? In relation to this question, studies that made claims in at least one of the following three categories: safety of GH treatment, necessity for GH treatment, and agreement and autonomy, were included. Two reviewers (D.S. and G.Y.) worked in duplicate independently and extracted data on study characteristics and outcomes. Studies were eligible for inclusion regardless of design, language, year, or sample size.
Data extraction included a full description of participants if available. Foreign language articles were translated by multilingual reviewers. The main outcome extracted from studies for mini meta-analysis were GH treatment characteristics, during and post-treatment height effects, head circumference, cognitive and motor function, additional therapeutic effects, adverse effects, IGF1, bone age response, and ethical arguments in favor and against GH therapy in DS patients. Additionally, we collected the number and age of the participants in the research groups as well as in the control groups, inclusion criteria, type of dose, and duration of growth hormone treatment.
We used a random effects meta-analysis in order to assess heterogeneity. We assessed the degree of inconsistency in the results between studies using the I² statistic; this statistic explains the proportion of inconsistency between studies that cannot be explained by chance alone, and is likely due to real differences in the population or the conduct of the studies (8).
Height standard deviation scores (SDS) were estimated in each study and pooled using a random effects meta-analysis (9). SDS expressed height differences in terms of the SD for the height of the reference population. Most studies reported SDS in reference to their national databases. To compare the change in height SDS along time between GH treated and untreated DS patients and also between DS patients with and without proven growth hormone deficiency under GH treatment, we extrapolated data using studies in which we could extract mean and standard deviations for all children in the different time points. We used two-way repeated-measures ANOVA to assess the difference between groups over time. R statistical programming language was used for extrapolation and statistical analysis. Publication selection bias could not be calculated due to missing relevant data.
An initial search of the literature yielded 281 publications and 24 eligible studies (Figure 1). Sixteen reports detailed medical effects of treatment with GH. Some are follow-up studies reporting late treatment outcomes or completing statistical analyses that did not appear in the initial report. Eight studies dealt with the ethical aspects of GH treatment in DS patients. The main characteristics and findings of the included studies are presented in Supplementary Table 1 and Table 1.
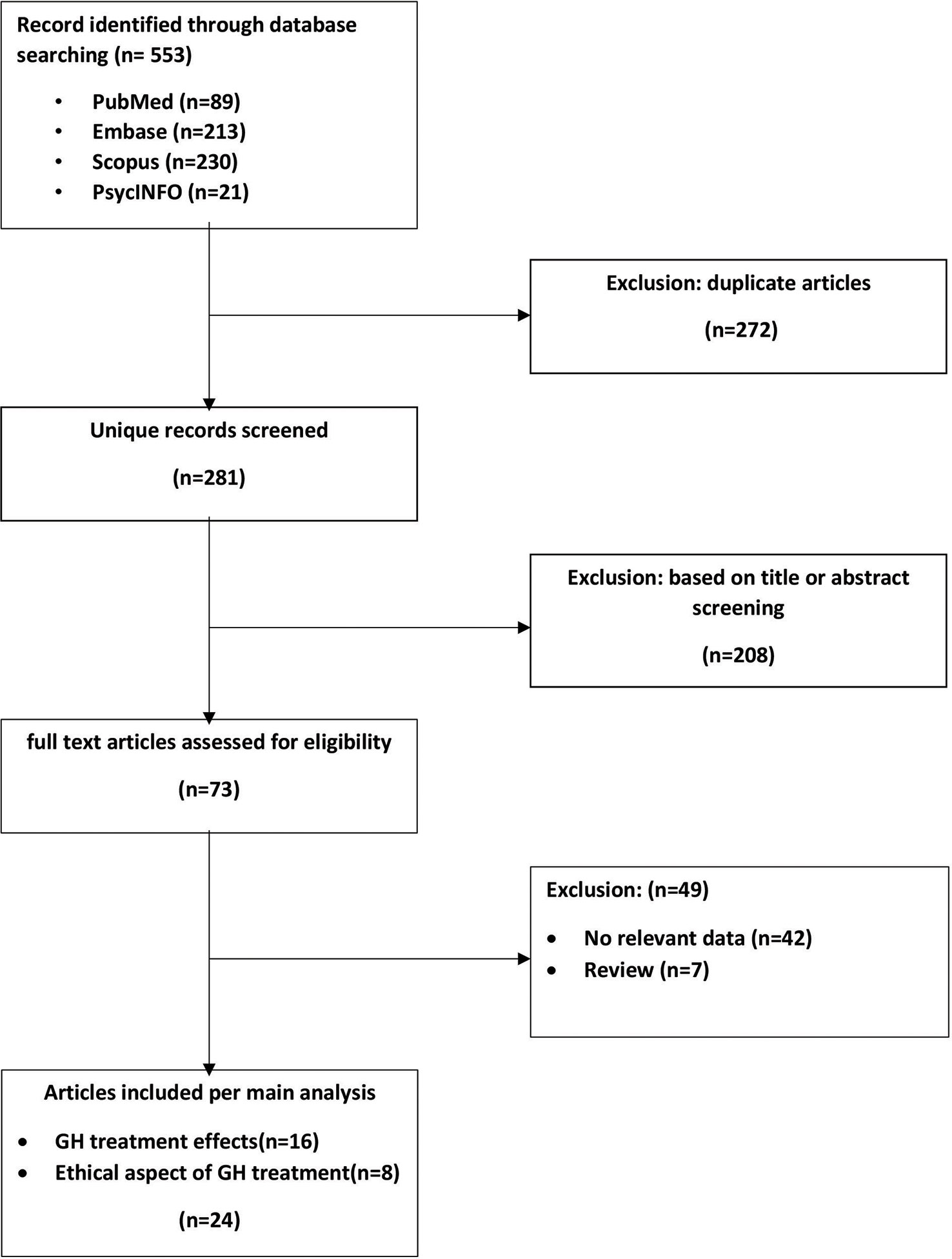
Figure 1 Flowchart of article screening and inclusion.
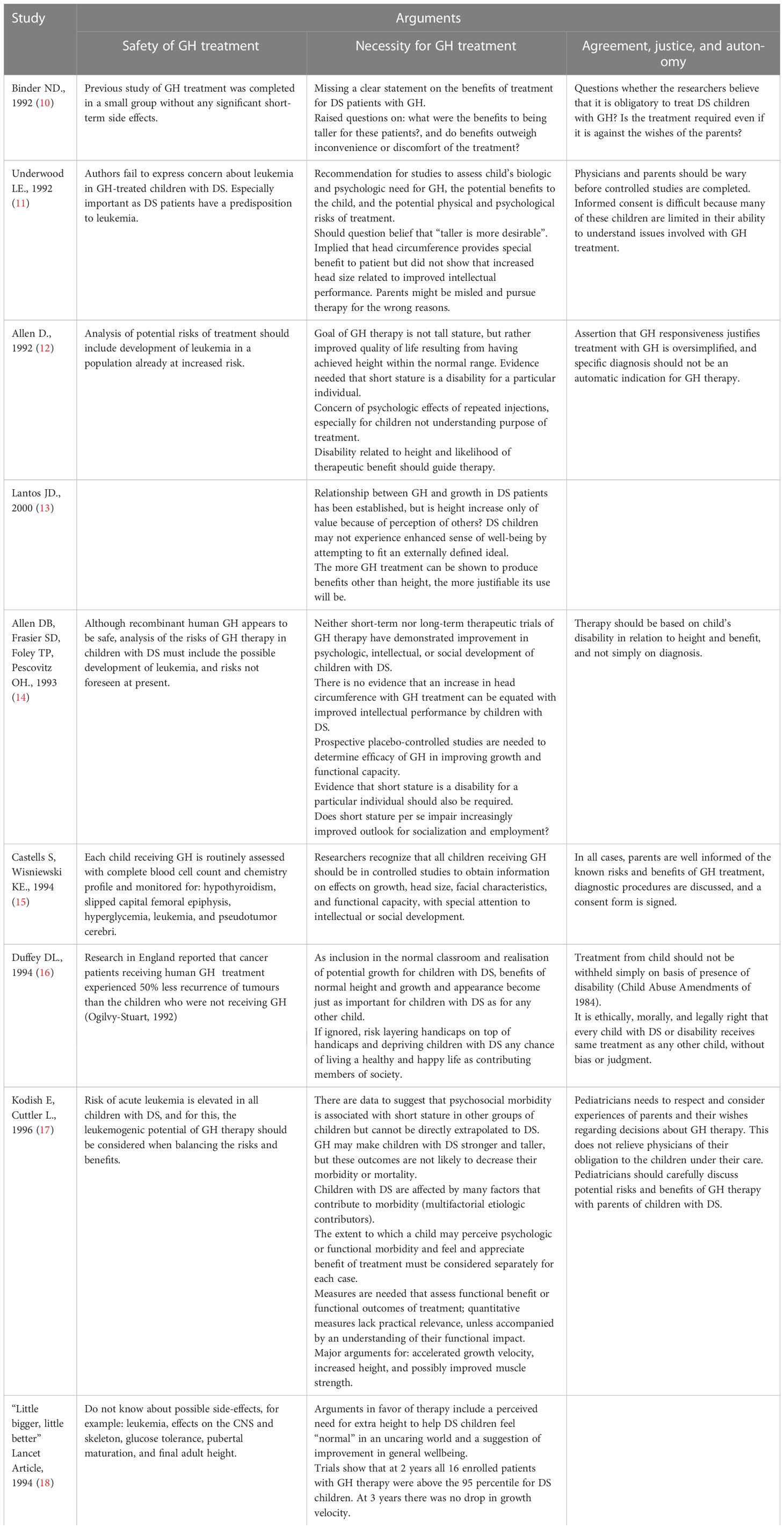
Table 1 Arguments regarding the ethical aspect of growth hormone treatment in children with Down syndrome.
Most studies present height data by using average height SDS and growth curves of a normal population, while some also present the average growth velocity. The problem with using growth velocity is the normal variation in this parameter is dependent on the age, gender, and pubertal status of the patient.
One important study presented height data by using a percentile range for each patient at the start and end points of a GH treatment period (19). Another study presented height data by using growth velocity alone (20). Unfortunately, it was impossible to utilize both studies’ essential height data within the mini meta-analysis. Nonetheless, both studies presented an impressive longitudinal growth response to GH therapy over three years of treatment in children with DS. Only one study showed continuation of GH treatment in a small proportion of children until they reached their final height.
A control group was recruited in two studies (21, 22) However, detailed control group height data were not presented and, therefore, one was not included in the statistical analysis (21). No GH deficiency diagnosis was required in three of the four studies included in the mini meta-analysis (21–23). A diagnosis of GH deficiency was required in one study included in the meta-analysis (24), as well as in two other studies that could not be included (19, 20).
Significant difference in the change in height SDS over time was found between GH treated and untreated DS patients. (Means of -1.22 and 0.81, p-value<0.0001) (Figure 2). No statistically significant difference in the change in height SDS over time was found between children with and without proven GH deficiency (means and standard deviations of 0.86 +/- 1.2 and 0.78 +/- 1.4, p-value=0.73)
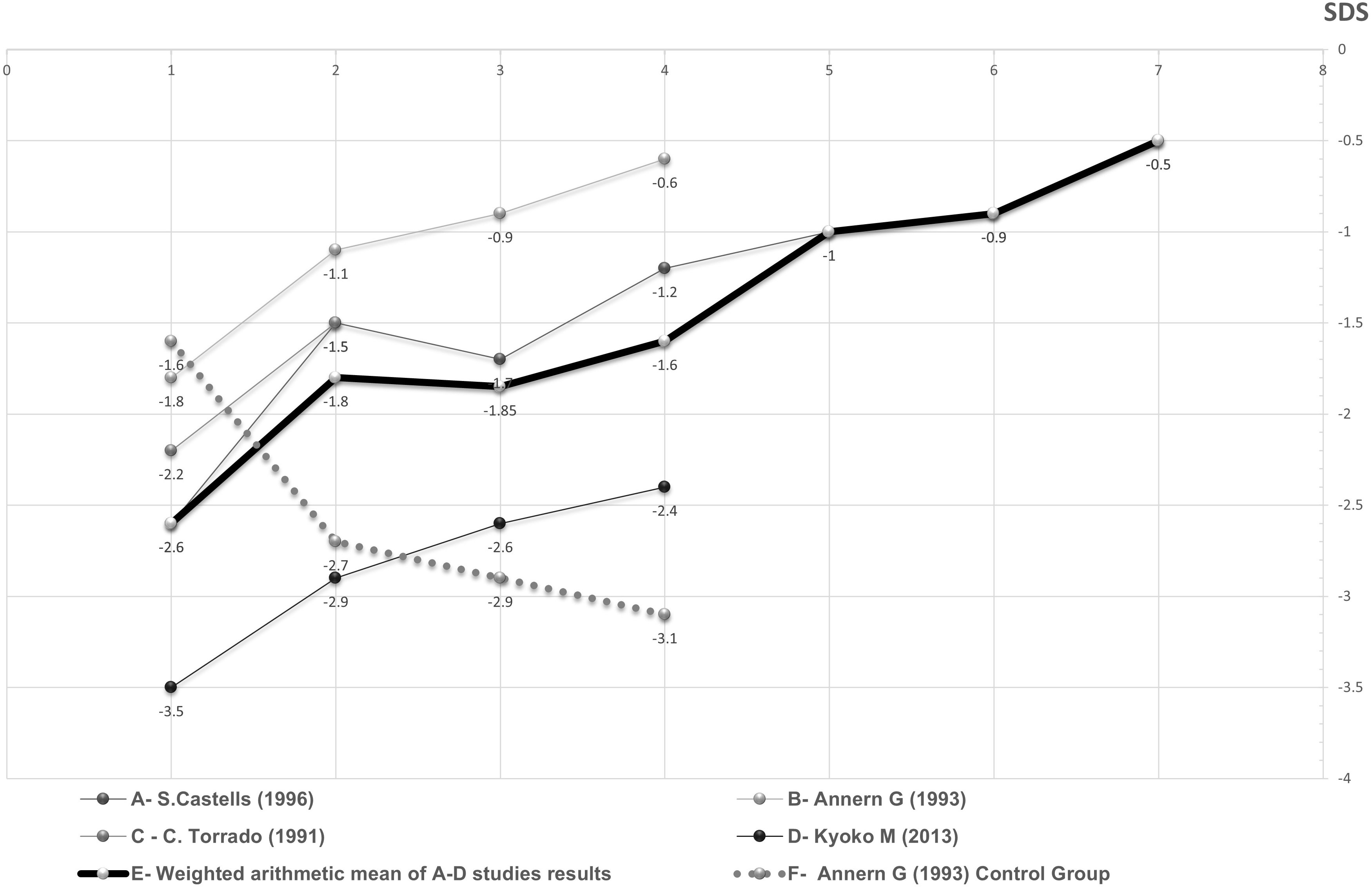
Figure 2 Periodic average height SDS of DS patients during GH treatment as a function of duration of treatment in years. The fragmented and continuous lines represent different DS patients groups whose source data appear in the studies mentioned in the figure itself. The thickened line represents the weighted arithmetic mean of GH treated groups. The dotted line represents the control group.
The description of height response to GH therapy in all studies is described in Supplementary Table 1. Height SDS data of DS children treated with GH in the four studies providing this parameter, as well as their weighted average and control group data, are presented in Figure 2.
Risk of bias of systematic review was evaluated according to ROBIS. Phase 2 includes 3 essential domains: identification and selection of studies, data collection and study appraisal, and synthesis and findings. 14 signaling questions in three domains were corresponded to “low risk of bias” while 2 signaling questions, one in domain 3 and one in domain 4, were classified as “no information,” and hence corresponded to “unclear risk of bias”.
Heterogeneity index I-squared was calculated to be 78.2%. The estimate of between-study variance tau-squared was 0.3897. A forest plot of the mini meta-analysis is presented in Figure 3.
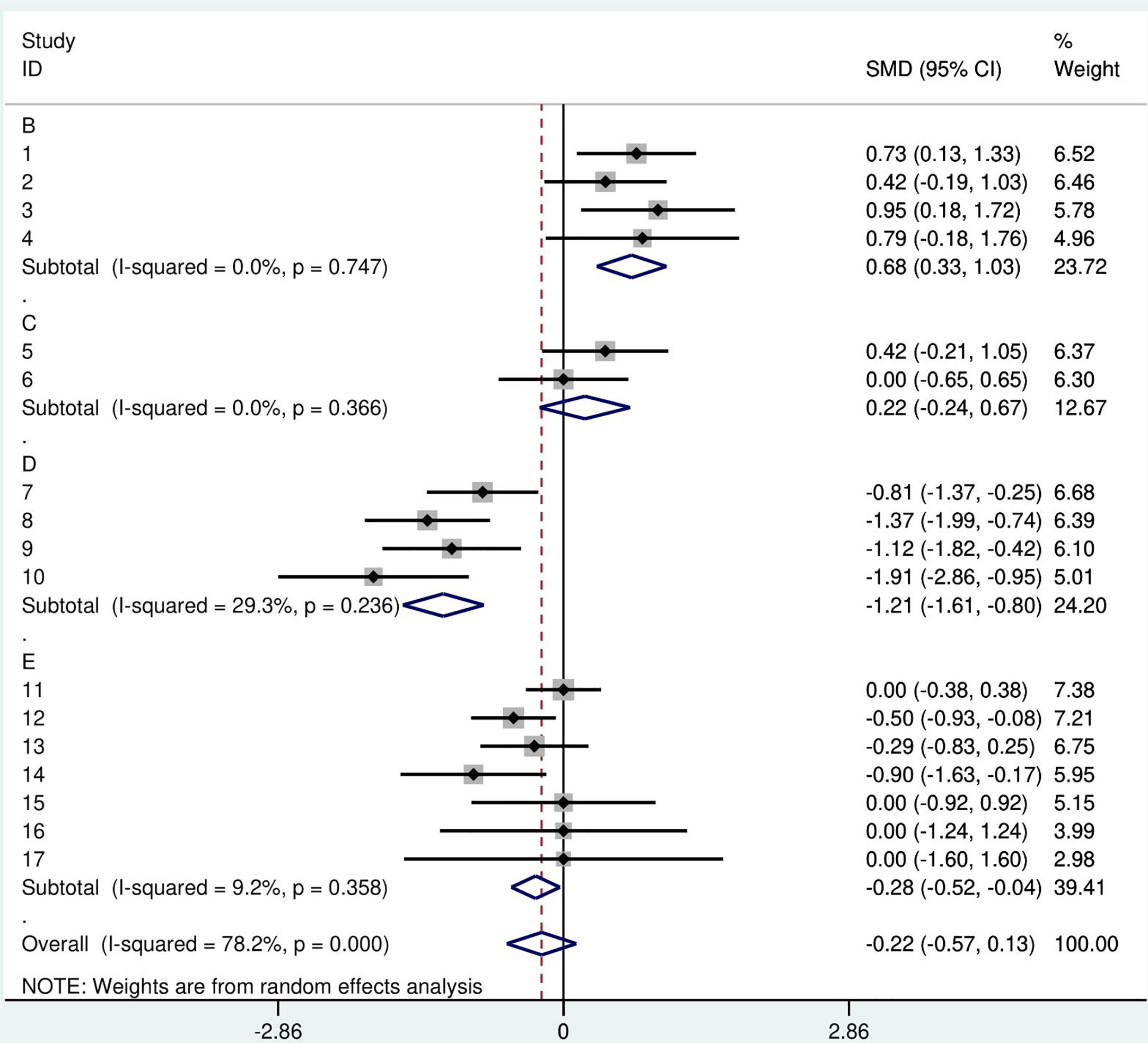
Figure 3 A forest plot of the GH therapy mini meta-analysis by study. Random effects meta-analysis of height SDS was performed in comparison to the reference group. Squares- point represent the result of each study, each year of GH treatment, or follow-up. A- Castells S. 1996. B- Annerén G.1993. C- Torrado C. 1991. D- Meguri K 2013. E- weighted arithmetic mean of GH treated groups, F- Annerén G. 1993 control group.
One study reported that growth velocity returned to baseline after stopping treatment (25). In one study, the predicted adult height (PAH) was calculated during therapy, and was found to be 170.7 ± 10.8 cm in 19 males and 167.1 ± 5.2 cm in 16 females. PAH was normalized in 91% of the children receiving treatment (21). A series of consecutive studies showed that the accelerated growth benefit was maintained for the short term, three years from the end of treatment (26), but was not maintained for the long term, ten years from the end of the treatment. Final height in those patients did not differ between the GH-treated subjects and the extended group of controls (27). Another study showed that the benefit of growth is maintained even 10–15 years from the end of treatment, with an improvement in the final height on average of 5.16 cm in males and 7.35 cm in females, compared to C. Cronk DS growth charts (20).
All studies investigating the effect of recombinant human GH treatment in the early years of life on head circumference showed a positive effect (21–23, 27, 28). The degree of impact reported ranges from very slight improvement to dramatic improvement and normalization of the head circumference.
There was a significant change in cognitive function compared to the control group in one study alone. Approximately 15 years after GH therapy termination, there was no statistical preference to the GH therapy group in brief IQ scores, but significantly higher scores in all subtests of cognitive tests Leiter-R and WISC-III and in all but one subtest of the motor BOT-2 test (27). The other studies that tested cognitive function found no change (25, 29–31).
One study showed an improvement in fine motor performance in the short term (30) and in all but one subtest of the motor BOT-2 test in the long term (27). One case report showed a dramatic improvement in both fine and gross motor function (32).
One study showed possible craniofacial growth and dental maturation that may be associated to the treatment (31).
Although the length of menstrual cycles in DS is within the normal range, a reduction in reproductive function is generally observed. A study examining the function of HPG axis showed reduced sensitivity of ovaries to FSH in patients with DS compared to controls. A treatment with GH normalized the ovarian response to FSH (33).
Almost all studies examined the response of IGF1 level to growth hormone treatment and showed that IGF1 levels in plasma were below normal range before treatment and increased to normal range shortly after starting treatment and remained normal throughout treatment.
Bone age was delayed before treatment began. Some studies showed advances in bone age during treatment (21, 25, 26, 29), and others showed no significant change (22–24).
Two studies reported an increase in mean neutrophil lobe count (25, 34). One study also showed an increase in neutrophil count during treatment and a fall thereafter (25). One study (three articles) reported on a single child with slightly elevated liver enzymes (22, 25, 29). Another study reported on mild subclinical hypothyroidism (19). Precocious puberty was observed in one boy 3.4 yrs after GH treatment initiation (24). In no study was the development of leukemia reported.
The ethical legitimacy of GH therapy in children with DS has been widely questioned over the years. The claim put forth in Torrado et al. (23), which argues that responsiveness to GH therapy by DS children justifies prescribing a course of treatment, was challenged by several respondents.
Various arguments have been presented for and against the treatment. These arguments can be divided into 3 categories: safety of GH treatment, necessity for GH treatment, and agreement, justice, and autonomy. A summary of all arguments that have appeared in the various studies on this subject appear in Table 1.
Consistent with the results of this meta-analysis, it can be concluded that there is a significant short-term beneficial effect of GH therapy on longitudinal growth in children with DS. The growth velocity of patients with DS treated with GH was found to be significantly higher than in non-treated DS patients.
Except for a single study that reported on the maintenance of the therapeutic effect even after stopping treatment, all the other studies show that the effect of short-term treatment gradually fades after the end of treatment. Importantly, there is almost no data on the effect of long-term treatment. It is possible that long-term treatment until the end of growth may preserve and even increase the therapeutic effect, even if the final height does not ultimately reach the forecasted height.
In addition, we have shown that there is a probable effect on head circumference and a possible effect on motor and even cognitive function. A discrepancy between longitudinal and head circumference results may indicate that the reduced head circumference in DS is not only a result of growth retardation but is mainly due to limited brain growth.
Despite the great importance of the treatments’ effect on quality of life, no study has formally examined this aspect by accepted and valid questionnaires.
No significant adverse effect of GH treatment in DS patients was ever reported even in the long- term. Certainly, more research is needed to describe the effect of longer-term treatment, but the results of this review provide a strong basis for considering that the risks of GH therapy in children with DS are not significantly different from other children.
The literature on height outcome in DS pediatric patients, who were treated with recombinant human GH, is heterogeneous, with variable age at diagnosis, years of follow-up, and variable criteria and methods of height measures. Although the number of studies included in this mini meta-analysis is not large, it is important to note that to the best of our knowledge this review included all studies that examined the effect of GH treatment in children with DS, from whom a reliable and comparable measurement tool, like height SDS, could be derived. It is also noteworthy that even studies that could not be included in the analysis reached the same conclusions about the beneficial effect of GH therapy on the longitudinal growth of children with DS.
Recently, an original study assessed whether anthropometric measurements of children with DS correlate with their IQ. The results showed that full-scale, verbal, and nonverbal IQ correlated with height percentile in a multiple linear regression analysis. The results of this study suggest an association between growth and IQ in children with DS, and this finding may be valuable for this population in light of increasing access to GH therapy in various genetic syndromes associated with short stature (35). DS shares many common clinical features with other genetic syndromes such as Prader-Willi syndrome (PWS). Children with PWS are also treated by GH treatment starting from the first year of life. Early GH treatment has been shown to promote mental and motor development as well as adaptive functioning in the PWS population in addition to improving growth velocity and metabolism (36).
Heterogeneity indices were calculated and were found to be relatively high. It is reasonable to conclude that one study (23) is responsible for the most heterogeneity from looking at the forest plot graph and from other calculations. In this study, the initial height index was significantly lower compared to the other studies. However, the growth rate through the treatment period was very similar to the growth rate observed in the other studies, as can be seen from Figure 2.
GH treatment was found to be effective equally in DS children who were diagnosed with GH deficiency and in DS children who were not diagnosed with GH deficiency. This finding corresponds to the conclusion that despite the increased prevalence of GH deficiency in children with DS, the quantitative component of the growth hormone is only one of the possible damaged elements in the GHRH-GH-IGF1 axis in DS patients, and apart from that, an impairment was also detected in GH neurosecretory function and in the bio-activity of GH, both of which are likely to be missed if relying solely on growth hormone stimulation tests (4).
It should be noted that all three of the above components negatively affect the level of IGF1 in patients with DS. It was recently found that biomarkers of neurodegeneration are associated with IGF1 deficiency in DS and that short stature is associated with lower IGF1 and with higher biomarkers (37).
Exogenous growth hormone treatment is expected to provide a response to these three issues.
In contrast to more distal disturbances such as growth hormone receptor resistance and IGF-1 receptor deficiency, the administration of proper recombinant GH circumvents both the quantitative and qualitative disturbances in the production and secretion of GH, as well as any defects in the endogenous protein structure. It seems that a diagnosis of growth hormone deficiency in children with DS should not be a criterion for GH treatment.
The main ethical arguments that have been raised over the years concerning GH treatment of DS patients consider the safety and necessity of GH treatment, while also raising the issue of patient autonomy and agreement (Table 1).
In the “safety” category, the main health concern that has been raised is the fear of developing leukemia, importantly as DS patients have a predisposition to developing leukemia. The fear of developing leukemia arose at a time when there was a similar general concern about growth hormone treatment in the general population. Since then, large studies have already shown that this fear is unwarranted in the general population. Correspondingly, we have shown herein that there is no evidence of the development of leukemia in small studies, most of which have documented short-term follow-up but also in the minority that documented long-term follow-up.
In the “necessity” category, it has been argued that short stature does not constitute a significant limitation to DS patients necessitating the need for potentially harmful and expensive GH treatment because of their developmental intellectual disability. Also, growth hormone treatment is given every day by subcutaneous injection which involves discomfort for the patient. Identifying with the purpose of the treatment makes it easier for the patient to cope with the pain. In the absence of such identification, as in the case of a child with Down syndrome due to developmental delay, the child’s difficulty in dealing with the pain and the parent’s difficulty in providing the treatment may increase.
In the “agreement, justice, and autonomy” category, it has been argued that informed consent is difficult because many of these children are limited in their ability to understand issues involved with GH treatment. Further, there is concern of the psychologic effects of repeated GH injections to children who may not understand the purpose of the treatment and may view the treatment as an additional “punishment” to their condition.
The degree of developmental intellectual disability in DS patients is highly variable. Average IQ of standard DS is around 50 and that of mosaic DS is around 65, showing that most DS patients have a mild degree of developmental intellectual disability. A large survey in the US reported that 57 percent of adult DS people were working a paid job (38).
Gradual but dramatic changes over time in life expectancy, quality of life, social involvement, and functional level highlight some of the important values associated with GH treatment that should not be ignored. While GH treatment may not improve intellectual performance, by improving stature, it may affect a variety of factors which are beyond mere centimeters. It is well established that short stature impairs outlook for socialization, employment, general well-being, and happiness (39). Some parents of DS children have stressed the fact that restricting DS children of GH therapy risks layering handicaps on handicaps, ultimately depriving them of any chance of living a healthy and happy life as contributing members of society, since attaining a “normal” height and growth becomes just as important for children with DS as for any other child (16).
It seems ethically, morally, and legally right that children with DS receive the same treatment as any other child without bias or judgement, and pediatricians need to respect and consider the experiences of parents and their wishes regarding decisions about GH therapy. At the same time, GH responsiveness and DS diagnosis should not be an automatic indication for GH therapy, rather the decision should be made based on well-informed consultations with caregivers and their wishes after discussing benefits and potential risks of GH therapy and clarifying potential misconceptions.
Inferences presented in this review are weakened by the risk of potential bias caused by the design of included studies. Any attempt at pooling the data should be done with caution to derive a clinically meaningful result. The relatively small total number of participants, especially the control groups, limits our ability to draw unambiguous conclusions.
On the other hand, the systematic nature of this review, which is not limited by language or a year limit, gives it its strength.
There is a significant short-term beneficial effect of GH therapy on longitudinal growth in children with DS. The presented findings may be valuable for improving access to GH therapy for pediatric DS patients. However, these findings should be confirmed by further research with a longitudinal sample of children with DS.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
DS, EH, and AH initiated the study and devised its main idea. AB and ST were invaluable in performing the meta-analysis. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1135768/full#supplementary-material
SUPPLEMENTARY TABLE 1 | GH Treatment mini meta-analysis - Main characteristics and findings of included studies.
1. Presson AP, Partyka G, Jensen KM, Devine OJ, Rasmussen SA, McCabe LL, et al. Current estimate of down syndrome population prevalence in the united states. J Pediatr (2013) 163:1163–8. doi: 10.1016/j.jpeds.2013.06.013
2. Al-Biltagi MA. Epidemiology and prevalence of down syndrome. In: Al-Biltagi MA, editor. Down syndrome children - an update. Bentham Science Publishers (2015). p. 3–44. doi: 10.2174/97816810813421150101
3. Cronk C, Crocker AC, Pueschel SM, Shea AM, Zackai E, Pickens G, et al. Growth charts for children with down syndrome: 1 month to 18 years of age. Pediatrics (1988) 81:102–10.
4. Shaki D, Hershkovitz E, Tamam S, Bollotin A, David O, Yalovitsky G, et al. GHRH-GH-IGF1 axis in pediatric down syndrome: a systematic review and mini meta-analysis. Front Pediatr (2023) 11:1132296. doi: 10.3389/fped.2023.1132296
5. Baird PA, Sadovnick AD. Life expectancy in down syndrome. J Pediatr (1987) 110:849–54. doi: 10.1016/s0022-3476(87)80395-5
6. Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med (2009) 6:e1000097. doi: 10.1371/journal.pmed.1000097
7. Whiting P, Savović J, Higgins JPT, Caldwell DM, Reeves BC, Shea B, et al. ROBIS: a new tool to assess risk of bias in systematic reviews was developed. J Clin Epidemiol (2016) 69:225–34. doi: 10.1016/j.jclinepi.2015.06.005
8. Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta-analyses. BMJ (2003) 327:557–60. doi: 10.1136/bmj.327.7414.557
9. DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials (1986) 7:177–88. doi: 10.1016/0197-2456(86)90046-2
10. Binder ND. Growth hormone therapy in children with down syndrome. J Pediatr (1992) 120:832–3. doi: 10.1016/s0022-3476(05)80266-5
11. Underwood LE. Growth hormone therapy in children with down syndrome. J Pediatr (1992) 120:833. doi: 10.1016/s0022-3476(05)80267-7
12. Allen DB, Frasier SD, Foley TP, Pescovitz OH. Growth hormone for children with down syndrome. J Pediatr (1993) 123:742–3. doi: 10.1016/s0022-3476(05)80849-2
13. Lantos JD. Growth hormone therapy for prader-Willie and down syndromes: a post-modern medical dilemma. Growth Horm IGF Res (2000) 10:S93–4. doi: 10.1016/S1096-6374(00)80017-9
14. Allen DB. Growth hormone therapy for children with down syndrome [6]. J Pediatr (1992) 120:332–3. doi: 10.1016/s0022-3476(05)80463-9
15. Castells S, Wisniewski KE. Growth hormone treatment in down syndrome [1]. J Pediatr (1994) 124:158–9. doi: 10.1016/s0022-3476(94)70277-2
16. Duffey DL. Treatment of poor growth in children with down syndrome. J Pediatr (1994) 125:508. doi: 10.1016/S0022-3476(05)83316-5
17. Kodish E, Cuttler L. Ethical issues in emerging new treatments such as growth hormone therapy for children with down syndrome and prader-willi syndrome. Curr Opin Pediatr (1996) 8:401–5. doi: 10.1097/00008480-199608000-00018
19. Ragusa L, Alberti A, Proto C, Romano C, Colabucci F. Recombinant human growth hormone treatment in down syndrome: the troina experience. Dev Brain Dysfunction (1996) 9:158–64.
20. Pallotti S, Giuliano S, Giambi C. Growth disorders in down’s syndrome: growth hormone treatment. Minerva endocrinologica (2002) 27:59–64.
21. Castells S, Abdel-Khalek IA, Wisniewski KE. Long-term effects of recombinant human growth hormone on children with down syndrome and growth retardation. Dev Brain Dysfunction (1996) 9:144–57.
22. Anneren G, Gustafsson J, Sara VR, Tuvemo T. Normalized growth velocity in children with down’s syndrome during growth hormone therapy. J INTELLECT Disabil Res (1993) 37:381–7. doi: 10.1111/j.1365-2788.1993.tb00881.x
23. Torrado C, Bastian W, Wisniewski KE, Castells S. Treatment of children with down syndrome and growth retardation with recombinant human growth hormone. J Pediatr (1991) 119:478–83. doi: 10.1016/s0022-3476(05)82068-2
24. Meguri K, Inoue M, Narahara K, Sato T, Takata A, Ohki N, et al. Therapeutic efficacy and safety of GH in Japanese children with down syndrome short stature accompanied by GH deficiency. Clin Pediatr Endocrinol (2013) 22:65–72. doi: 10.1297/cpe.22.65
25. Annerén G, Sara VR, Hall K, Tuvemo T. Growth and somatomedin responses to growth hormone in down’s syndrome. Arch Dis Child (1986) 61:48–52. doi: 10.1136/adc.61.1.48
26. Annerén G, Tuvemo T, Carlsson-Skwirut C, Lönnerholm T, Bang P, Sara VR, et al. Growth hormone treatment in young children with down’s syndrome: effects on growth and psychomotor development. Arch Dis Child (1999) 80:334–8. doi: 10.1136/adc.80.4.334
27. Myrelid A, Bergman S, Elfvik Strömberg M, Jonsson B, Nyberg F, Gustafsson J, et al. Late effects of early growth hormone treatment in down syndrome. Acta Paediatrica Int J Paediatrics (2010) 99:763–9. doi: 10.1111/j.1651-2227.2009.01679.x
28. Wisniewski KE, Castells S, Mandys V. Growth hormone neuropathology in down syndrome (trisomy 21). Dev Brain DYSFUNCTION (1996) 9:100–13.
29. Annerén G, Carlsson-Skwirut C, Sara VR, Tuvemo T, Gustafsson J. The effect of growth hormone therapy on growth and mental development in children with down sydrome. Dev Brain DYSFUNCT (1996) 9:138–43.
30. Annerén G, Tuvemo T, Gustafsson J. Growth hormone therapy in young children with down syndrome and a clinical comparison of down and prader-willi syndromes. Growth Horm IGF Res (2000) 10:S87–91. doi: 10.1016/S1096-6374(00)80016-7
31. Carlstedt K, Anneren G, Huggare J, Modéer T, Dahllöf G. The effect of growth hormone therapy on craniofacial growth and dental maturity in children with down syndrome. J Craniofacial Genet Dev Biol (1999) 19:20–3.
32. Yasuhara A, Yoshida Y. Beneficial effect of growth hormone on severe delay in motor development in a child with down syndrome. Clin Pediatr Endocrinol (2001) 10:137–40. doi: 10.1297/cpe.10.137
33. Cento RM, Ragusa L, Proto C, Alberti A, Fiore G, Soranna L, et al. Growth hormone administration normalizes the ovarian responsiveness to follicle-stimulating-hormone in the early stages of the follicular maturation in women with down syndrome. J Endocrinological Invest (1998) 21:342–7. doi: 10.1007/BF03350768
34. Berg JM, Kirman BH, Stern J, Mittwoch U. Treatment of mongolism with pituitary extract. J Ment Sci (1961) 107:475–80. doi: 10.1192/bjp.107.448.475
35. Kłosowska A, Kuchta A, Ćwiklińska A, Sałaga-Zaleska K, Jankowski M, Kłosowski P, et al. Relationship between growth and intelligence quotient in children with down syndrome. Transl Pediatr (2022) 11:505–13. doi: 10.21037/tp-21-424
36. Festen D a. M, Wevers M, Lindgren AC, Böhm B, Otten BJ, Wit JM, et al. Mental and motor development before and during growth hormone treatment in infants and toddlers with prader-willi syndrome. Clin Endocrinol (Oxf) (2008) 68:919–25. doi: 10.1111/j.1365-2265.2007.03126.x
37. Araya P, Kinning KT, Coughlan C, Smith KP, Granrath RE, Enriquez-Estrada BA, et al. IGF1 deficiency integrates stunted growth and neurodegeneration in down syndrome. Cell Rep (2022) 41:111883. doi: 10.1016/j.celrep.2022.111883
38. Kumin L, Schoenbrodt L. Employment in adults with down syndrome in the united states: results from a national survey. J Appl Res Intellect Disabil (2016) 29:330–45. doi: 10.1111/jar.12182
Keywords: growth hormone, GH, rGH treatment, Down syndrome, insulin-like growth factor 1, IGF1
Citation: Shaki D, Hershkovitz E, Tamam S, Bollotin A, David O, Yalovitsky G, Loewenthal N, Carmon L, Walker D, Nowak R and Haim A (2023) GH treatment in pediatric Down syndrome: a systematic review and mini meta-analysis. Front. Endocrinol. 14:1135768. doi: 10.3389/fendo.2023.1135768
Received: 01 January 2023; Accepted: 05 April 2023;
Published: 21 April 2023.
Edited by:
Artur Mazur, University of Rzeszow, PolandReviewed by:
Rossella Gaudino, University Hospital of Verona, ItalyCopyright © 2023 Shaki, Hershkovitz, Tamam, Bollotin, David, Yalovitsky, Loewenthal, Carmon, Walker, Nowak and Haim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eli Hershkovitz, ZWxpaEBiZ3UuYWMuaWw=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.