 Huimin Li
Huimin Li Jing Ren
Jing Ren Yusi Li
Yusi Li Qian Wu
Qian Wu Junping Wei
Junping Wei
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 03 April 2023
Sec. Clinical Diabetes
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1134025
This article is part of the Research Topic Clinical and Genetic Determinants of Diabetes and Complications View all 31 articles
Obesity has been associated with oxidative stress. Obese patients are at increased risk for diabetic cognitive dysfunction, indicating a pathological link between obesity, oxidative stress, and diabetic cognitive dysfunction. Obesity can induce the biological process of oxidative stress by disrupting the adipose microenvironment (adipocytes, macrophages), mediating low-grade chronic inflammation, and mitochondrial dysfunction (mitochondrial division, fusion). Furthermore, oxidative stress can be implicated in insulin resistance, inflammation in neural tissues, and lipid metabolism disorders, affecting cognitive dysfunction in diabetics.
The prevalence of obesity has been on the rise globally for the last half century (1). Obesity prevalence has doubled since 1980 in more than 70 countries. Furthermore, women of all ages had a higher prevalence of obesity than men (2). Obesity causes many twenty-first-century chronic diseases worldwide and imposes enormous socioeconomic burdens (1). Numerous risk factors for chronic diseases, including cardiovascular disease (CVD) (3), type 2 diabetes (T2DM), and cognitive impairment, are influenced by obesity (4). Diabetes prevalence has been increasing, especially with T2DM, due to changes in lifestyle factors such as diet, obesity, and lack of exercise (5). The IDF Diabetes Atlas indicates that prevalence in 20–79-year-olds in 2021 was estimated to be 10.5% (536.6 million people), rising to 12.2% (783.2 million) in 2045 (6). Patients with diabetes are also at risk for complications as they age (7). Diabetes patients have been found to have an increased risk for dementia (8). In a US study, people with diabetes had an overall prevalence of dementia and cognitive impairment of 13.1% for people aged 65–74 and 24.2% for those aged 75 and over (9). Those who suffer from cognitive impairment in diabetes experience cognitive dysfunction, delayed executive, function, and impeded information processing speed, and pathology may include neuro amyloid plaques and tau protein tangles (10). There is a correlation between diabetes and cognitive impairment, which negatively affects patient quality of life (11). A cross-sectional analysis of baseline data shows that high BMI and low mood are associated with worse cognitive function among overweight/obese elderly with metabolic syndrome (12). In older people, BMI has been associated with a higher risk of developing type 2 diabetes (13). This review aims to explore how oxidative stress processes could contribute to obesity-related cognitive dysfunction in diabetics.
Oxidative stress (OS) regulates biological components, and it has been proposed to be a mediator of the relationship between obesity and cognitive impairment in diabetes. In 1985, “oxidative stress” was introduced as a concept in redox biology and medicine (14); the concept of biological oxidative stress was defined as “an imbalance between oxidants and antioxidants in favor of oxidants, leading to a disruption of redox signaling and control and molecular damage” (15). Redox reactions contribute to regulation, where endogenous and exogenous regulatory factors, such as the number of biochemical components (oxygen, nitrogen, and sulfur), can contribute to the oxidative stress. Furthermore, these reactive species, called reactive substances, mainly reactive oxygen species (ROS), reactive nitrogen species (RNS), and reactive sulfur species (RSS), stimulate the metabolic processes of cells (16). Reactive species participate in several oxidative signaling pathways, such as NF-κB, JAK-STAT, Nrf-2, and HIF-1; they are also involved in the development of several diseases, including cardiovascular diseases, cancer, and diabetes (17). The production of ROS contributes to the inflammatory response process, which leads to an increase in adipocyte size, promotes adipogenesis and lipogenesis, and adipocyte differentiation (18). Studies have demonstrated that fat accumulation is an early trigger and a fundamental cause of obesity-associated metabolic syndrome resulting in increased oxidative stress (19). Prolonged exposure of adipocytes to ROS leads to insulin-induced activation of PI3-kinase and Akt, resulting in impaired islet function and facilitated glucose transporter member 4 (GLUT4) translocation(20). Due to their sensitivity to oxidative damage, neuronal cells are especially susceptible to neurodegenerative diseases, such as diabetes-related cognitive impairment (21). Mitochondrial homeostasis plays a crucial role in maintaining neuronal and axonal energetic homeostasis. Bioenergetic deficits contribute significantly to the cognitive decline observed in aging and neurodegenerative diseases. Neurons are particularly susceptible to mitochondrial dysfunction due to their intrinsic properties (22). ROS synthesis is derived from mitochondria, and when mitochondria become dysfunctional, ROS production of ROS and oxidative stress increase, and mitochondrial maldistribution disrupts neuronal axonal energy homeostasis. Oxidative stress disrupts neurological metabolism resulting in hypoglucose metabolism in the brain (23). The brains exhibit structural changes due to an accumulation of disease-specific protein aggregates (24). These structural changes may contribute to neuronal and synaptic dysfunction, resulting in cognitive impairment (25).
Oxidative stress is produced by reactive oxygen/nitrogen species (ROS/RNS) (26). Furthermore, oxidative stress alters the balance between the production of ROS and antioxidant defenses. By-products of aerobic metabolism, ROS, can pose a health risk when exposed to stressful environments (27). ROS are primarily derived from mitochondria and electron transport chain (ETC), in which mitochondria produce adenosine triphosphate (ATP) through a series of oxidative phosphorylation processes. However, ROS also contain a variety of chemical entities, including nitric oxide, peroxynitrite, hypochlorous acid, singlet oxygen and hydroxyl radicals (28).
Several studies have shown that obesity induces the formation of oxidative stress. The high-fat diet induces oxidative stress in the white adipose tissue of rats (29). When there is a high intake of nutrients, oxidative stress increases, and inflammation is induced through signaling pathways mediated by the nuclear factor-kappa B (30). High consumption of fat-rich diets promotes mitochondrial β-oxidation of free fatty acids (FFAs), and subsequent use of cytochrome-c oxidase leading to excess electron flow increases the accumulation of ROS, ROS, and lipid peroxidation deplete vitamins and antioxidant enzymes (31). We summarise the link between obesity and oxidative stress in terms of disruption of the adipose microenvironment, chronic inflammation in obesity, and mitochondrial dysfunction (Figure 1).
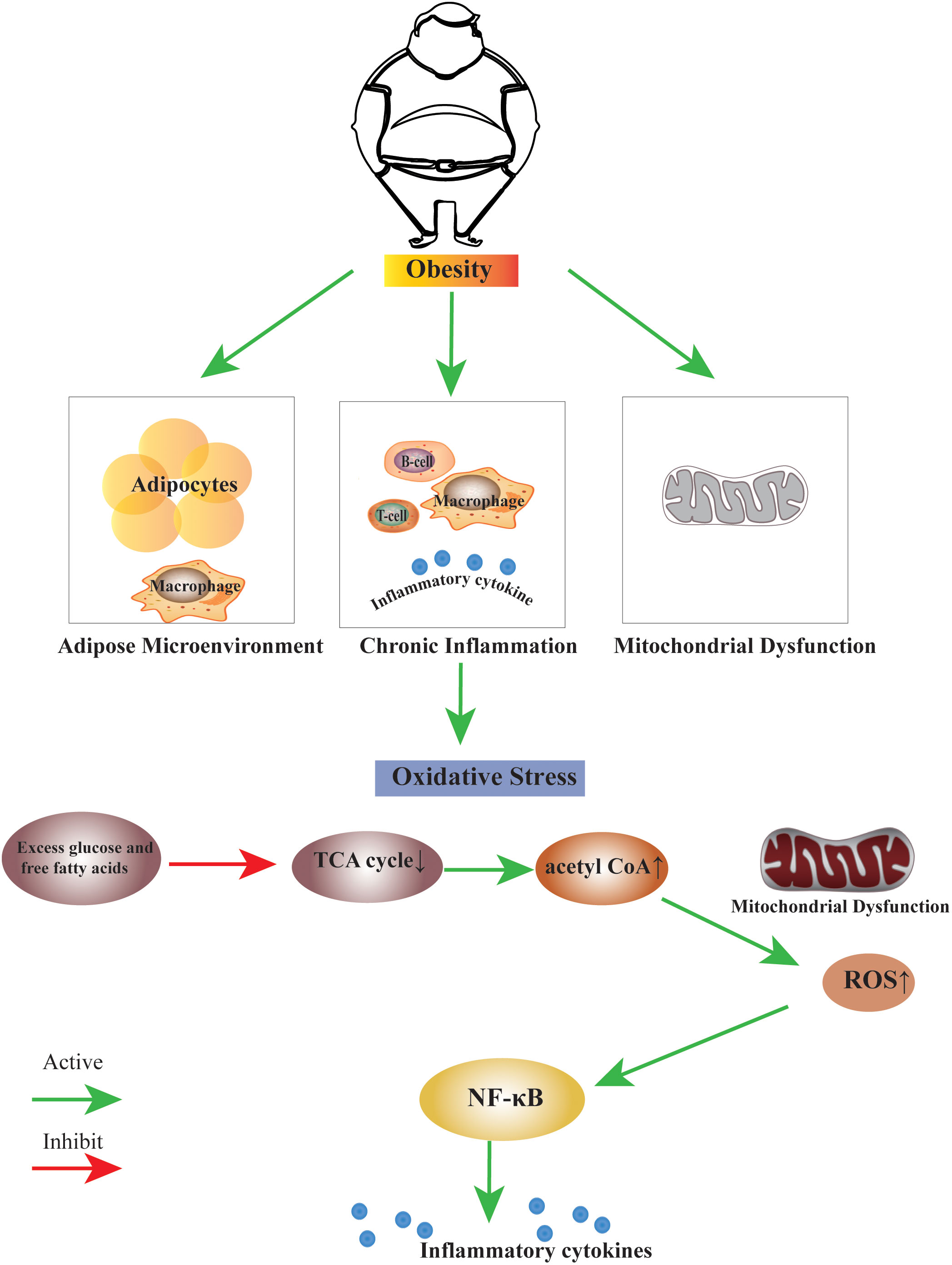
Figure 1 The link between obesity and oxidative stress. Obesity can induce the biological process of oxidative stress by disrupting the adipose microenvironment, mediating chronic inflammation, and mitochondrial dysfunction. The process is probably that excess glucose and free fatty acids suppress the TCA cycle, leading to an increase in the production of acetyl CoA. Excess acetyl CoA stimulates mitochondrial dysfunction, resulting in an increase of ROS within the cell, this change may activate many factors, the nuclear factor κB is the main inflammatory factor.TCA cycle, Tricarboxylic Acid cycle; acetyl CoA, Acetoacetyl coenzyme A; ROS, Reactive Oxygen Species; NF-κB, nuclear factor κB.
Obesity is an increase in lipid content in adipose tissue, manifested by an increase in the size and number of adipose cells (32). Adipose tissue can be divided into three categories: white adipose tissue (WAT), brown adipose tissue (BAT) and beige adipocytes.
White adipocytes are the primary cell type found in human adipose tissue. Energy-yielding triglycerides and cholesterol esters are stored within the sizeable intracellular lipid droplets. Leptin, adiponectin, and other adipokines are among the proteins secreted by white adipocytes.
Brown adipocytes: BAT is widely present throughout the body (33); BAT is rich in multiple lipid droplets and contains uncoupling protein 1– containing mitochondria; these adipocytes mediate thermogenic respiration (34).
Beige adipocytes: Beige adipocytes are derived from white adipocytes tissue, and browning of white adipocytes tissue can be induced by cold stimulation, exercise, and some endocrine hormones; beige adipocytes have thermogenic effects because of rich uncoupling protein 1 (35).
The different adipocytes and macrophages of the adipose tissue constitute the adipose microenvironment. Dysfunction at the WAT level may influence the development of obesity-associated metabolic complications (36). An obesity-induced immune response occurs when metabolic cells (including adipocytes) are involved (e.g., adipocytes). Overnutrition leads to adipotoxicity, which produces inflammatory factors (37). Studies have shown that macrophages infiltrate adipocytes in obese individuals and promote an inflammatory response (38). Kinase inhibitors (IKK), c-jun n-terminal kinase (JNK), and protein kinase r (PKR) can transmit nutrient signals from metabolic tissues to inflammatory cells. This process is accompanied by oxidative stress, and these kinases and their downstream pro-inflammatory targeting factors can be significantly upregulated in obese subjects (39). Due to the accumulation of oxidative biomolecules in adipocytes, the homeostatic system that regulates oxidative stress and the antioxidant regulatory system are suppressed mainly in obese adipocytes. Excess ROS irreversibly damages DNA, lipids, and proteins and adversely affects cellular function (28).
Obesity is primarily caused by an energy imbalance between excessive calories consumed and insufficient calories expended (40). Adipose tissue is regarded as an energy storage for calories and an essential endocrine organ. It produces many bioactive molecules, including chemokines and cytokines, called adipokines (or adipocytokines), when secreted by adipose tissue. They are not only regulators of systemic metabolism, but also have immunomodulatory properties (41). Adipose tissue is responsible for the production and secretion of many biologically active adipokines, including leptin, adiponectin, resistin, visfatin, and schelatin, that can lead to chronic complications (42). Obesity leads to an increase in adipocytes and enlargement of adipose tissue. The ensuing decrease in oxygen tension leads to hypoxia and massive accumulation of hypoxia-inducible factor (HIF-1) in adipocytes. Furthermore, hypoxia has been linked to adipose inflammation and macrophage infiltration (43). Studies have shown that macrophages accumulate in adipose tissue of obese people as well as in the obese B6.V Lepob/ob mouse model, and macrophages promote the secretion and expression of adipokines, including tumor necrosis factor-alpha (TNF-α), iNOS and interleukin-6 (IL-6) (38).
Obesity is a chronic low-grade inflammatory condition, with adipose tissue infiltrated by macrophages and elevated inflammatory markers and cytokines. This low-grade chronic inflammation in adipose tissue may contribute to developing related metabolic diseases, such as insulin resistance and T2DM (44). Adipocytes produce large amounts of adipokines with inflammatory functions, such as IL-6, IL-1, and TNF-α, which induce ROS production and mediate oxidative stress (45). TNF-α is produced mainly by macrophages and is also a critical adipokine. Fat accumulation leads to adipocyte damage, leading to high production of cytokines such as TNF-α, which produces ROS in tissues and increases the rate of lipid peroxidation (46). TNF-α also activates the NF-kB signaling pathway to aggravate the inflammatory response (47). During oxidative stress, adipokines, including leptin, IL-6, and lipocalin, resist all functions (45).
Oxidative stress impairs islet beta-cell function in several ways; it significantly reduces insulin production, impairs the ability of insulinogenic vesicles to enter the plasma membrane, and reduces the response to hyperglycemia. Oxidative stress can induce islet β-cell apoptosis, and excess free radicals interfere with β-cell neogenesis (48). Oxidative stress leads to reduced GLUT4 expression and ultimately reduces insulin sensitivity by disrupting the binding of nuclear proteins to the insulin response element in the GLUT4 promoter (49). Oxidative stress was also involved in the development of diabetic encephalopathy. Oxidative stress inhibits the islet signaling system. HFD/STZ induced a significant increase in relevant oxidative stress parameters such as TBARS, NO levels, and XO activity in the brain tissue of rats compared with controls, and serum peripheral TNF-α and IL-6 inflammatory cytokine levels were significantly increased in the diabetic rats, and a similar brain AD-related miRNA expression profile was observed in the diabetic rats (50).
Mitochondria are intracellular organelles that play an important role in the cell by metabolizing nutrients and producing adenosine triphosphate (ATP). Mitochondria regulate energy, maintenance of cellular calcium homeostasis, production and removal of reactive oxygen species, and regulation of cell death (51). Mitochondria produce energy in the form of ATP through the oxidative metabolism of nutrients, consisting of two main steps: 1) oxidation of NADH or FADH2 produced during glycolysis, TCA or β-oxidation of fatty acids, with most of the ATP being produced through the TCA cycle through the ETC; 2) oxidative phosphorylation (OXPHOS) to produce ATP. Mitochondria continuously metabolize oxygen and produce ROS during the combination of electron transport and protons in the ETC, which is the primary source of ROS (52).
Mitochondrial dysfunction can manifest itself by loss of mitochondrial membrane potential, altered ETC function, increased ROS production, and decreased oxygen consumption. There is a reduction in the efficiency of mitochondrial ATP production (53). Mitochondrial dysfunction can also occur when mitochondrial molecular dynamics is impaired. Mitochondria is a dynamic energy organelle that responds to energy demands and environmental stimuli through fusion, fission, and movement to maintain cellular homeostasis (54). Fission can also promote mitochondrial autophagy and biogenesis, two events that can occur because of mitochondrial fission (55, 56). Mitochondria generate several stress response pathways, including the mitochondrial unfolded protein response and degrading mislocalized proteins in mitochondria dysfunction (57, 58). Severely damaged mitochondria can be identified and degraded through the process of mitochondrial autophagy (59). When mitochondrial autophagy is dysregulated, ROS produced by mitochondria can activate inflammatory vesicles composed of NLRP3, the bridging protein ASC, and caspase-1, triggering inflammation. It has been reported that defects in the autophagy gene PINK1 increase NLRP3 expression and lead to brown fat dysfunction in mice (60, 61).
Studies have shown that excessive nutrient intake leads to hyperglycemia, increases ROS production, and causes mitochondrial dysfunction in adipocytes, suggesting that obesity triggers oxidative stress and mitochondrial dysfunction (57). When mitochondrial function is impaired, major adipocyte pathways are altered, resulting in decreased adipogenesis, increased lipolysis, and decreased fatty acid esterification; these alterations promote changes in insulin sensitivity (62). A study showed that high fat-induced obese (DIO) mice exhibit insulin resistance, mitochondrial dysfunction, hepatic lipid deposition, and oxidative stress (63). A study indicated that the expression of mitofusin-2 (Mfn2, a mitochondrial fusion protein) was decreased in the muscles of obese subjects or type 2 diabetics, leading to an imbalance between mitochondrial fusion and fission events and mitochondrial dysfunction, which may be involved in insulin resistance (64). Significantly elevated levels of acylcarnitine in patients with nonalcoholic fatty liver mark mitochondrial dysfunction and impaired fatty acid oxidation (65). A study indicated a decrease in mitochondrial biosynthesis in a rodent model of obesity (66). Down-regulation of mitochondrial biogenesis in obesity is associated with metabolic alterations, insulin resistance, and low-grade inflammation (67). Chronic high-fat diet feeding promotes excessive apoptosis in mouse HK-2 cells by inducing oxidative stress and mitochondrial disorders in kidney cells (68). It has been shown that a high-fat diet induces oxidative damage in the brain of obese (DIO) rats and that a high-fat diet increases lipid oxidation in the brain tissue of DIO rats as well as the level of mitochondrial ROS (69). Mitochondrial dysfunction in the brain due to obesity may lead to insulin resistance and cognitive dysfunction(70, 71). Jheng found smaller and shorter mitochondria and increased mitochondrial fission in the skeletal muscle of obese mice, suggesting that altered mitochondrial fission is associated with mitochondrial dysfunction in the skeletal muscle and insulin resistance (72).
A high-fat diet induces mitochondrial expansion in rodent brown fat, and excess leads to inhibition of mitochondrial fusion, resulting in fragmentation and autophagy, leading to mitochondrial dysfunction (73). The excess leads to cellular oxidative stress, which subsequently induces an inflammatory cascade response. This process is likely caused by excess glucose and free fatty acids suppressing the TCA cycle, increasing acetyl CoA. Excess acetyl CoA stimulates the mitochondria to produce excess superoxide in the electron transport chain, which increases ROS within the cell. This change may activate many factors; the nuclear factor κB is the primary inflammatory factor (74).
Oxidative stress is an imbalance between the production of oxidants and antioxidant defenses that may result in damage to biological systems. Oxidative stress is considered one of the crucial factors in the development and progression of cognitive impairment in diabetes mellitus (75). In cognitive impairment, there are interconnections between oxidative stress, insulin resistance, neuroinflammation, and abnormal lipid metabolism (76–78) (Figure 2).
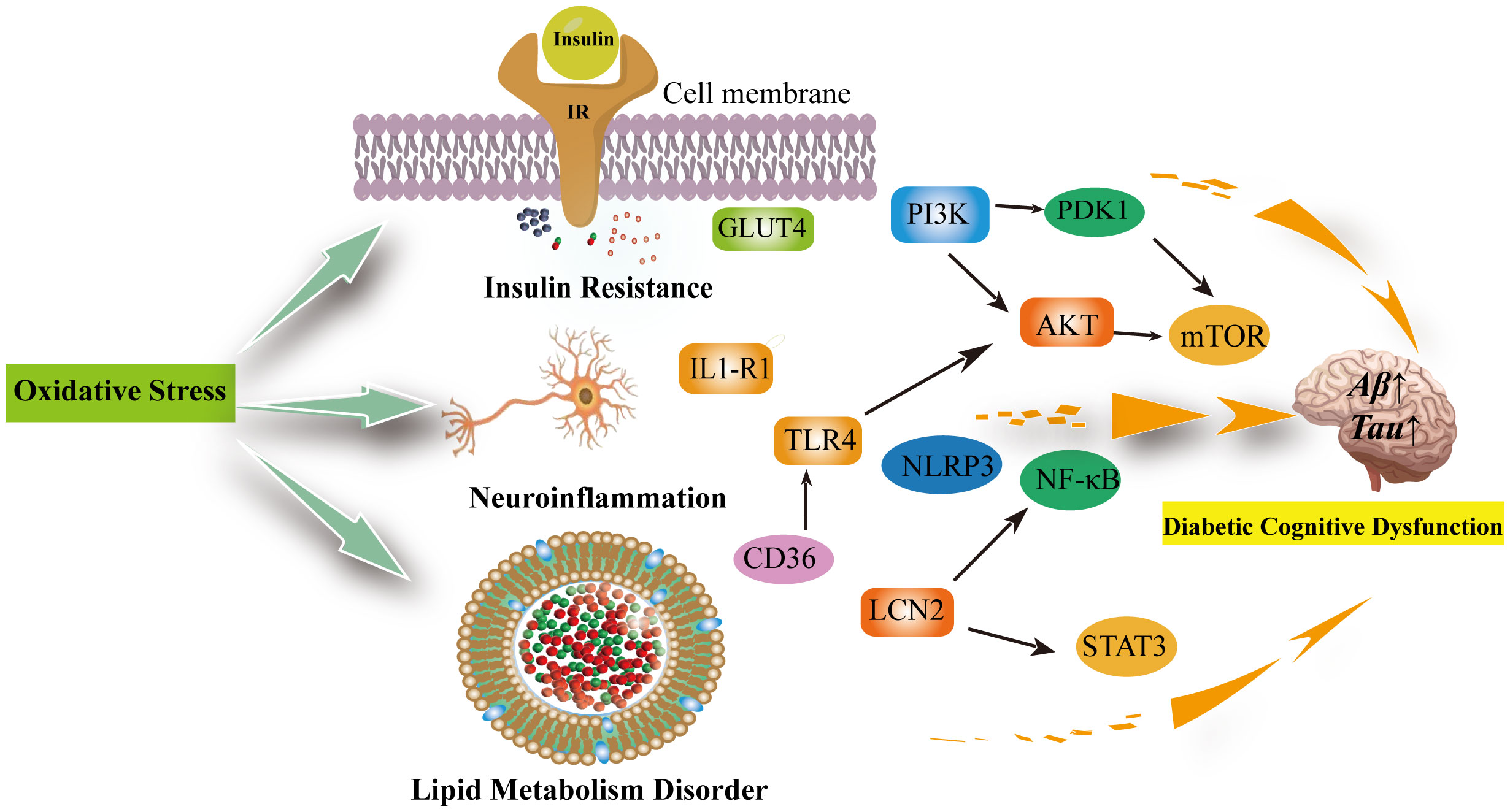
Figure 2 Oxidative stress can be involved in insulin resistance, neuroinflammation, lipid metabolism disorders leading to diabetic cognitive dysfunction. Oxidative stress reduces GLUT-4 expression and the translocation of GLUT-4 to the cell membrane, decreasing insulin sensitivity; In the brain, insulin activates the PI3K/PDK1/AKT and the PI3K/AKT/mTOR signaling pathway to inhibit apoptosis and promote neuronal development and survival, which are inhibited when insulin is resistant and increase the production of inflammatory factors. Neuroinflammation is associated with excessive microglia activation. activation of IL1-R1 signaling pathway, NLRP3/IL-1β signaling pathway, NK-κB signaling pathway and release of pro-inflammatory factors exacerbate neuroinflammation and neuronal damage in the brain. The inhibition of TLR4/AKT/mTOR signaling pathway inhibits cellular autophagy as well as promotes neuroinflammation and microglia apoptosis. Diabetic cognitive dysfunction is also exacerbated by the presence of impaired lipid metabolism in the brain. CD36 recognizes oxidized low-density lipoprotein receptors (TLRS) and triggers a toll-like response to stimulate sterile inflammation. Meanwhile, LCN2 is mainly produced in glial cells of the brain under oxidative stress. It promotes cellular neuroinflammation by activating the NF-kB pathway as well as the STAT3 signalling pathway to promote microglia activation. IR, Insluin resistance; GLUT4, facilitated glucose transporter member 4; PI3K, phosphatidylinositol 3' -kinase; PDK1, pyruvate dehydrogenase kinase isoform 1; AKT, Protein Kinase B; mTOR, mammalian target of rapamycin; TLR4, toll-like receptor 4; IL1-R1, interleukin 1 receptor type I; NLRP3,NLR family pyrin domain containing 3; NF-κB, Nuclear factor kappa B; LCN2, Lipocalin 2; STAT3, signal transduction and transcription 3; CD36, Platelet glycoprotein 4.
Insulin resistance means that systemic target tissues such as fat, muscle, and liver are less sensitive to insulin and cannot properly regulate the pathological state of glucose homeostasis (79). Inflammation, dysfunction, and elevated OS levels lead to insulin signaling cascade disorder and are important triggers of insulin resistance (80–82). In pro-inflammatory conditions, activation of glial cells can lead to progressive neuronal damage (83). Additionally, insulin regulates metabolic pathways that maintain learning and memory at the brain level and glucose transport/metabolism (83). In diabetic cognitive dysfunction, insulin resistance weakens the metabolic raw material of dysfunctional neurons and affects memory function (84).Currently, the molecular mechanism of the development of insulin resistance has not been fully elucidated. Insulin receptor substrates (IRSs) work as scaffold protein driving activation of two primary insulin signaling pathways: 1) PI3K/PDK1/Akt pathway; And 2) MAPK pathway (85). The former is closely related to insulin metabolism, while the latter is mainly involved in cell growth differentiation (86).
ROS can act as both a signaling agent and a damaging agent. Low levels of endogenous reactive oxygen species play an essential role in the signaling pathway and have crucial physiological significance (87). In insulin signaling, there are redox initiation steps in which some oxidizing agents, such as hydrogen peroxide (H2O2), promote the phosphorylation of insulin receptors (83). In addition, it can inhibit protein tyrosine phosphatase PTP1B, which deactivates IR by dephosphorylating A-ring phosphotyrosine (88).Thus, insulin-induced H2O2 acts as a net positive regulator in acting insulin receptors. Furthermore, as age increases, OS levels increase and glutathione (GSH) levels decrease, which has been verified in aging models (89). In insulin resistance or T2DM mice, oxidative stress markers increased, and glutathione levels decreased in the brain(90). Previous studies have shown that brain plasticity, the ability of the brain to undergo structural and functional changes due to environmental stimulation, is carefully regulated by dietary and nutritional-dependent hormones, including insulin (91). Therefore, it can be shown that OS is closely related to the development of insulin resistance and cognitive dysfunction. Changes in insulin signals in the central nervous system can accelerate brain aging, affect brain plasticity, and promote synaptic loss and nerve degradation (92).
Oxidative stress can cause β-cell dysfunction (93). Because the antioxidant defense system of β-cell is low, OS is widely found in diabetes mellitus and plays an essential role in β-cell dysfunction (48, 93). OS can reduce the production of insulin, impair the contents of the original insulin vesicles into plasma membrane, and reduce the exocytosis of glucose into circulation (93, 94). Since apoptotic agents are highly sensitive to OS, OS can induce pancreatic cell apoptosis and lead to β-cell apoptosis (94, 95). An overload of free radicals can affect the normal metabolic pathway in β-cells, damage the KATP channel, and lead to decreased insulin secretion (93, 96). Previous studies have shown that OS activates Nf-κ B, JNK/SAPK, p38 MAPK, hexosamine pathway, and toll-like receptor (TLRs), thereby impairing β-cell function (48, 93). In addition, β-cell mitochondrial dysfunction induced by oxidative stress may be an important mechanism leading to β-cell dysfunction (48, 93). β-cell dysfunction, which results from oxidative stress, can lead to insulin resistance and in turn diabetes cognitive dysfunction.
Oxidative stress can reduce insulin sensitivity in insulin-dependent cells such as adipocytes and myocytes (48). Normal GLUT-4 expression and localization are necessary to maintain these tissues’ insulin sensitivity (97). Reduction of GLUT-4 expression/localization is one of the main molecular mechanisms by which oxidative stress induces insulin resistance and promotes the development of cognitive dysfunction in diabetes mellitus (48). Studies have found lower expression and localization of GLUT-4 in patients with insulin resistance and T2DM (98–100). Oxidative stress can reduce the translocation of GLUT-4 to the cell membrane (101). Long-term oxidative stress inhibits transcription factors and microscopic RNAs involved in GLUT-4 expression (101–103).
Oxidative stress can impair normal insulin signal transduction (IST) at different levels (104). Hyperglycemic-induced OS activates different stress-sensitive serine/threonine (Ser/Thr) kinases such as IKK-β, which phosphorylates multiple targets such as IR, IRS-1 and IRS-2, leading to adverse downstream effects, including decreased PI3K activation and insulin resistance (48, 105). Oxidative stress can damage insulin sensitivity and lead to insulin resistance and cognitive dysfunction of diabetes by downregulating the proteins involved in normal IST, such as Insulin-degrading enzyme (IDE), Biliverdin reductase-A (BVR-A), Akt, IRS, IRS-1 and GSK-3 (48, 83, 50). Therefore, IST abnormality is one of the important mechanisms of insulin resistance caused by oxidative stress.
Insulin resistance can occur in both obesity and diabetes and is manifested in peripheral and central insulin resistance (106). In the brain, insulin acts as a neuromodulator to regulate activity-dependent synaptic plasticity by activating PI3K/PDK1/Akt signaling pathways (107). Insulin can inhibit apoptosis by activating the Akt signaling pathway to promote neuronal cell survival (108). Insulin resistance is characterized by the down-regulation of insulin receptor expression and impaired IRS proteins (86). IRs are localized in both presynaptic and postsynaptic neurons (109, 110). IRs recruit and activate PI3K complexes, which subsequently activate AKT (111), AKT downstream of GLUT4 and mTOR complexes, AKT-mediated stimulation of mTOR and its downstream targets regulates protein and lipid synthesis and promotes dendritic spine formation, as well as many aspects of neuronal development, survival, autophagy, and long-term synaptic plasticity (84, 111). When oxidative stress processes activate kinases such as JNK and IKK in neurons, insulin signaling pathways become abnormal, such as the P13K/AKT pathway, thereby the downstream of the pathway is inhibited (112). Failure of insulin signaling causes tau protein hyperphosphorylation (113), in addition to increased neurotoxic Aβ deposition at specific levels of hyperinsulinemia (114), all of which can lead to decreased cognitive function in diabetes. In addition, chronic elevated blood glucose can induce inflammation and can cause insulin resistance. Pro-inflammatory mediators such as TNF-α, IL1-β and IL-6 further exacerbate the inflammatory state through feedback inhibition of insulin receptors and through feedforward mechanisms that disrupt mitochondrial function to stimulate the production of reactive oxygen species, thereby producing an inflammatory environment with reduced insulin sensitivity. This chronic inflammatory environment increases NF-κB inducible kinase (NIK), which independently impairs mitochondrial function to further promote insulin resistance (86). Pro-inflammatory cytokines secreted into the bloodstream (across the blood-brain barrier) during chronic hyperglycemia and inflammatory cytokines within the brain’s innate immune system, soluble misfolded Aβ can induce inflammatory cytokines (e.g., TNF-α) through a NIK-dependent pathway that can lead to neuroinflammation and exacerbate cognitive deficits (115).
Neuroinflammation is the inflammatory response of factors of the central nervous system (CNS) acting on homeostasis in the body (116). This response includes distinct types of cells in the central nervous system, such as microglia and astrocytes (116, 117). Neuroinflammation aims to restore neuronal homeostasis and protect neuronal integrity (118). During the acute phase, neuroinflammation can protect neurological homeostasis, promote nerve growth, repair damaged cells, and remove protein plaques (119). During the chronic phase, neuroinflammatory-induced maladaptive results will cause neuronal damage to worsen (119). Neuroinflammation is a common pathogenic factor in neurological disorders, including Alzheimer’s disease (AD), diabetic cognitive dysfunction, and depression (120). Neuroinflammation includes various inflammatory events in the central nervous system under pathological conditions. Brain alterations in AD and diabetic cognitive dysfunction can manifest amyloid-β plaques and are associated with neuroinflammation (112). Abnormal activation of glial cells (microglia and astrocytes) can mediate neuroinflammation leading to neurodegenerative disease, and dysfunctional neurons alter the clearance of amyloid-β plaques in AD, promoting neuroinflammation and cognitive impairment (121). Obesity-induced chronic inflammation also affects the central nervous system (122), with obesity-promoting peripheral inflammation and increasing the permeability of the blood-brain barrier (BBB), and elevated levels of inflammatory mediators in diabetic patients promote neuroinflammation by triggering harmful neutrophil/microglia activation in the diabetic brain (123). Elevated levels of related proteins in inflammatory cells (e.g., lipid transport protein 2, LCN2 and tension enhancer binding protein, TonEBP) can adversely affect diabetic encephalopathy by leaking through the BBB (124).
Neuroinflammation and oxidative stress are essential in the onset and development of neurodegenerative lesions and are closely linked in their pathogenesis. ROS and RNS can further enhance intracellular signaling cascades and increase the expression of pro-inflammatory factors. On the other hand, inflammatory cells secrete active substances that produce ROS (116, 125). Therefore, neuroinflammation and oxidative stress can stimulate and interact with each other. An imbalance in redox and insufficient inflammatory response in the central nervous system causes neuroinflammation (116).
The blood-brain barrier (BBB) is a protective barrier for the CNS that prevents harmful substances from entering the brain and maintains intracerebral homeostasis by regulating the transport of essential molecules, including glucose. Pericytes and astrocytes contribute to the formation of the basal membrane of the blood-brain barrier (126, 127). Hyperglycemia increases the rate at which pericytes and astrocytes respire, producing ROS production and oxidative stress in people with diabetes (127, 128). Increased ROS further stimulates the upregulation of inflammatory cytokines and activates the NF-κB signaling pathway, leading to leakage of the blood-brain barrier (129). Neuroinflammation resulting from these injuries promotes the opening of the BBB and the influx of high blood sugar into the CNS (127). Long-term high glucose levels can cause disturbances in glucose metabolism pathways, decrease essential cofactors in redox reactions, including NADPH and NAD +, and produce advanced glycosylated end products (AGEs) (130–132). AGEs bind to AGE receptors (RAGEs) on the cell surface, producing excessive ROS (127, 133). High concentrations of ROS can initiate misfolding of proteins in neurons mitochondria, causing dysfunction of mitochondria, causing neuroinflammation, exacerbating tissue damage, and destroying neuronal regeneration (127, 134).
Mitochondria generate the energy required for almost all biological functions of the body and are an essential organelles. Neurons have high energy requirements, and neuronal mitochondria provide constant energy to neuronal cells (127, 135). Mitochondrial dysfunction causes intracellular energy, leading to inflammation and cell death (119). Previous studies have found that brain neurons are more susceptible to oxidative stress (136). Recent studies have shown that mitochondrial dysfunction plays vital role in hyperglycemic-induced neuronal damage (127, 136, 137). Oxidative stress is one of the leading causes of mitochondrial dysfunction (119). Oxidative stress can interrupt one or more mitochondrial functions, increasing membrane permeability (138). Furthermore, oxidative stress can increase neurotoxic glutamate levels, affecting mitochondrial phagocytosis (119, 139).Microglia are one of the important cells involved in neuroinflammation. They play an important role in maintaining neuronal homeostasis, neuron growth, building extra synapses, removing fragments of cells, removing protein aggregates and neuroplasticity (119, 140). Microglia can identify pathogens, protein aggregates, or fragments through pattern recognition receptors (PRRs) in pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMP), and activate phagocytosis pathogens, release cytokines, chemokines, ROS/RNS until an immune response is eliminated (141). Due to mitochondrial dysfunction, mitochondrial membrane damage releases DAMP, which initiates multiple inflammatory cascades leading to neuroinflammation. The DAMP released by mitochondria (TLR, TNF receptor, inflammasome) can be identified by PRRs of microglia, activate the TLR/NF-κ B inflammatory pathway, and promote the release of pro-inflammatory factor cytokines and chemokines (119). Inflammation caused by the DAMP released by mitochondria can lead to mitochondrial dysfunction, increase ROS, and exacerbate inflammatory circulation (119).
Neuroinflammation is closely associated with microglia hyperactivation. Studies have shown that NLRP3/IL-1β signaling may underlie the correlation between visceral obesity and cognitive impairment in humans, with high-fat diets feeding WT and NLRP3-KO mice, WT mice activating IL1R1 signaling in microglia, leading to hippocampal IL1β accumulation and neuroinflammation, and consequently cognitive impairment, while NLRP3-KO mice are protective against obesity-induced peripheral inflammation (142). Study indicated that neuroinflammation in diabetic cognitive dysfunction is associated with autophagy, continuous hyperglycemia under diabetes can trigger activation of the NF-κ B pathway and release of pro-inflammatory factors, leading to the inflammatory response, and neuronal damage (143). Pharmacological administration of mTOR inhibitors and autophagy stimulators improves inflammation in vivo by inhibiting NF-κ B signaling (144). In a study of Cui, they found melatonin (MLT) could improve learning and memory in diabetes-associated cognitive dysfunction mice by activating autophagy via the TLR4/Akt/mTOR pathway, thereby inhibiting neuroinflammation and microglial apoptosis (145). A study showed that lncRNA MEG3 overexpression significantly improved diabetic cognitive impairments by regulating the Rac1/ROS axis, and by inhibiting mitochondria-related apoptosis. In addition, MEG3 overexpression or Rac1 inhibition promoted FUNDC1 dephosphorylation and inhibited oxidative stress and neuroinflammation (146).
Lipids are a class of organic compounds that act as structural components of cell membranes, chemical energy sources and cell signaling molecules, involving many biological processes (147). Lipids can be divided into the following categories according to their structure: fatty acids, triglycerides, sphingolipids, phospholipids, glycolipids, sterol lipids, isopropylene enols and polyketides (148). Lipid metabolism can be defined as the synthesis, storage and breakdown of lipids (149). These processes are necessary to maintain complex homeostasis and lipid diversity and produce products involved in multiple cellular processes. The liver and adipose tissue play a key role in lipid metabolism. The liver helps digest, uptake, storage, and biosynthesis of dietary lipids, which can be exported as lipoproteins to provide energy or structural components (139). Adipose tissue can be used for long-term energy storage (150). Adipose tissue can be used for long-term energy storage (150). Insulin regulates fat storage by inhibiting or stimulating fat mobilization (151). Moreover, most body cells can synthesize cholesterol, but the amount depends on the cell’s needs (152).
Lipids are abundant in the brain, accounting for about 50–60% of the dry weight, especially fatty acids, glycerophospholipids, sphingolipids, and cholesterol (153, 154). A study has shown that “adipose inclusions” or “lipid particles” can be found in AD brains (155). In neurons, oligomeric Aβ peptides can alter cellular cholesterol metabolism (156). Obesity and abnormal blood lipids are the main risk factors for cognitive dysfunction in diabetes mellitus (157). Physiological studies have found that cholesterol metabolism, inflammation, and innate immunity are closely related to neurodegenerative lesions (158). Previous studies have found that several risk factors for Alzheimer’s disease (AD) involve genes for lipid metabolism and transport, such as APOϵ4, CLU, and ABCA7(159–161). Therefore, abnormal lipid metabolism may be important in diabetes-associated cognitive dysfunction.
Lipid types and levels in the brain are vital determinants of brain function. Studies found that human liposomes change with age and aging can cause damage to the distribution of brain lipids and cause brain dysfunction (162, 163). The increase in oxidative stress is one of the signs of aging. Redox imbalance in the body damages to the cellular mechanism (164). Increased levels of ROS and RNS in patients with DM and AD (48, 165). Significant increase in oxidized proteins and lipids in patients with the brain in AD (166, 167). Studies such as Cutler found that changes in sphingolipid and cholesterol metabolism caused by membrane-related oxidative stress can cause neurodegenerative cascades (168). Increased oxidative stress and lipid peroxidation are associated with cognitive dysfunction in diabetes mellitus. One of the most significant hypotheses for neurodegenerative lesions is the amyloid hypothesis, which holds that excessive amounts of insoluble A subtypes cause tau to be over phosphorylated, resulting in free radical generation, inflammation, and oxidative damage (169). Low-density lipid lipoprotein receptor-related protein (LRP1) is involved in the clearance of Aβ peptide. Oxidation of LRP1 will inhibit its ability to remove Aβ peptide, resulting in the accumulation of Aβ peptide in the brain (170). High concentrations of ROS can lead to increased lipid peroxidation in the brain and change membrane permeability and membrane receptor and associated enzyme activity (171). Lipid peroxidation produces active aldehydes, such as malonaldehyde (MDA) and 4-hydroxynonenaldehyde (4-HNE), which combine and modify proteins involved in metabolism, antioxidant defense systems, and axon growth. The tau protein can be modified by 4-HNE, which indirectly leads to increased entanglement of neuronal fibers (172). In addition, LRP1 is also a covalently modified protein that further leads to the production of lipid peroxidation products. These products can cause normal initiation cascade dysregulation in neurons (173).
Several studies have confirmed the presence of disorders of lipid metabolism, including SP metabolism, Trp metabolism, and GP metabolism, in both patients with cognitive impairment in diabetes or in rat models (174, 175). Lipid metabolism can regulate numerous cellular signaling pathways involved in inflammatory responses (e.g., fatty acids, diacylglycerol (DAG), sphingolipids, CD36, Lipocalin 2). Microglia lipid metabolism is specifically involved in the control of microglia activation and effector functions such as migration, phagocytosis and inflammatory signaling, and minor disturbances in microglia lipid processing are associated with altered brain function in diseases characterized by neuroinflammation (176). Furthermore, peroxides produced by lipid peroxidation interfere with the structure of the cell membrane and protein function and stimulate intracellular signaling and other pathways leading to cell death. As a type B scavenger receptor, CD36 recognizes low-density lipoprotein (LDL), oxidized phospholipids, and beta-amyloid and is also an FA transporter. By activating the innate immune response, phagocytosis, and oxidant production in microglia, cd36g orchestrates transcriptional and metabolic remodeling. By recognizing oxidized parenchymal LDL receptors (TLRs) and internalizing receptor-ligand complexes, cd36 triggers Toll-like responses to stimulate sterile inflammation, which is closely related to neuroinflammation in cognitive impairment and mild inflammation in obesity (176). Lipocalin 2 (LCN2) functions in the regulation of the immune system and inflammatory processes. LCN 2 is mainly produced in the glia of the brain under oxidative stress and can disrupt the blood-brain barrier by promoting astrocyte and brain endothelial cell damage. Studies have shown that LCN2 regulates cellular activity in the central nervous system, controlling iron accumulation, and modulating neuroinflammation by activating glial cells (177). It may regulate neuroinflammation by activating the NF-κB signaling, activating signal transduction and transcription 3 (STAT 3) pathway to activate microglia, promoting astrocyte activation, further activation of microglia, and inhibiting neuroprotective cell pathways in the brain by regulating cytokines such as IL-1β, TNF-α, and IL-6 (178). LCN 2 is associated with inflammatory responses in metabolic disorders (including obesity and insulin resistance), where the pathway involves NF-κB, C/EBP, and estrogen response elements (179, 180). Moreover, insulin induces LCN-2 expression, thought to be via the phosphatidylinositol 3-kinase and mitogen-activated protein kinase signaling pathways (180). Excess ROS, RNS are highly susceptible to oxidation of lipids containing carbon-carbon double bonds, especially polyunsaturated fatty acid (PUFA), peroxides of PUFA and their reactive aldehydes, their end products-reactive aldehydes such as 4-HNE-lead to protein carbonylation, and 4-HNE and other lipophiles mediate cytokinesis through protein adduct toxicity (181). Furthermore, 4-HNE processing activates pathways including DNA damage, antioxidant, ER stress, and heat shock responses associated with neuroinflammation, insulin resistance, and other diseases (182).
Recent epidemiological and experimental data provide evidence of a bidirectional interaction between obesity and cognitive dysfunction in diabetes. Obesity and diabetes are both risk factors for cognitive dysfunction, and cognitive dysfunction in diabetes is one of the complications of diabetes (78). Therefore, hyperglycemia in diabetic patients is a cause of cognitive dysfunction, while obesity is the primary cause. Insulin resistance, neuroinflammation, and lipid metabolism disorders are exacerbated by obesity in diabetic patients, resulting in cognitive dysfunction in diabetic patients. The physiological processes of obesity and diabetic cognitive impairment are mediated by biological processes of oxidative stress. Oxidative stress in obese tissues is mainly caused by the disruption of the adipose microenvironment, chronic low-grade inflammation, and mitochondrial dysfunction. Furthermore, oxidative stress affects insulin resistance, neuroinflammation, and lipid metabolism in diabetic brain tissue, affecting physiological and pathological processes. In addition to accelerating the destruction of neurons, aging, and tau protein deposition in the brain, diabetic brains are prone to cognitive dysfunction. The obese diabetic population should be regularly evaluated for cognitive function because obesity is a risk factor for diabetic cognitive dysfunction. Managing diabetic cognitive dysfunction disease in obese and metabolically impaired individuals requires meticulous management, including antioxidants if necessary.
HL and JR designed the work of review; HL, JR, and YL reviewed the literature available on this topic and wrote the paper; HL and JR contributed to the scientific writing of the manuscript; HL, JR, QW, and JW revised the manuscript. HL, JR, YL, QW, and JW contributed equally to this work. All authors contributed to the article and approved the submitted version.
The study was funded by Scientific and technological innovation project of China Academy of Chinese Medical Sciences”, Chinese medicine prevention and treatment of type 2 diabetes evidence-based capacity-building projects——Huashi Jiangzhuo Decoction in the treatment of newly diagnosed type 2 diabetes damp heat syndrome of multi-center randomized controlled clinical study, the national natural science foundation of China with funding number 82074412.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hanson P, Weickert MO, Barber TM. Obesity: Novel and unusual predisposing factors. Ther Adv Endocrinol Metab (2020) 11:2042018820922018. doi: 10.1177/2042018820922018
2. Collaborators, G. B. D. O, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med (2017) 377:13–27. doi: 10.1056/NEJMoa1614362
3. Piche ME, Poirier P, Lemieux I, Despres JP. Overview of epidemiology and contribution of obesity and body fat distribution to cardiovascular disease: An update. Prog Cardiovasc Dis (2018) 61:103–13. doi: 10.1016/j.pcad.2018.06.004
4. Li Y, Shang S, Fei Y, Chen C, Jiang Y, Dang L, et al. Interactive relations of type 2 diabetes and abdominal obesity to cognitive impairment: A cross-sectional study in rural area of Xi'an in China. J Diabetes Complicat (2018) 32:48–55. doi: 10.1016/j.jdiacomp.2017.09.006
5. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet (2014) 383:1068–83. doi: 10.1016/S0140-6736(13)62154-6
6. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF diabetes atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
7. Papatheodorou K, Banach M, Edmonds M, Papanas N, Papazoglou D. Complications of diabetes. J Diabetes Res (2015) 2015:189525. doi: 10.1155/2015/189525
8. Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: A systematic review. Lancet Neurol (2006) 5:64–74. doi: 10.1016/S1474-4422(05)70284-2
9. Feil DG, Rajan M, Soroka O, Tseng CL, Miller DR, Pogach LM. Risk of hypoglycemia in older veterans with dementia and cognitive impairment: Implications for practice and policy. J Am Geriatr Soc (2011) 59:2263–72. doi: 10.1111/j.1532-5415.2011.03726.x
10. Luo A, Xie Z, Wang Y, Wang X, Li S, Yan J, et al. Type 2 diabetes mellitus-associated cognitive dysfunction: Advances in potential mechanisms and therapies. Neurosci Biobehav Rev (2022) 137:104642. doi: 10.1016/j.neubiorev.2022.104642
11. You Y, Liu Z, Chen Y, Xu Y, Qin J, Guo S, et al. The prevalence of mild cognitive impairment in type 2 diabetes mellitus patients: A systematic review and meta-analysis. Acta Diabetol (2021) 58:671–85. doi: 10.1007/s00592-020-01648-9
12. Mallorqui-Bague N, Lozano-Madrid M, Toledo E, Corella D, Salas-Salvado J, Cuenca-Royo A, et al. Type 2 diabetes and cognitive impairment in an older population with overweight or obesity and metabolic syndrome: Baseline cross-sectional analysis of the PREDIMED-plus study. Sci Rep (2018) 8:16128. doi: 10.1038/s41598-018-33843-8
13. Ye M, Robson PJ, Eurich DT, Vena JE, Xu JY, Johnson JA. Changes in body mass index and incidence of diabetes: A longitudinal study of alberta's tomorrow project cohort. Prev Med (2018) 106:157–63. doi: 10.1016/j.ypmed.2017.10.036
14. Sies H. Oxidative stress: A concept in redox biology and medicine. Redox Biol (2015) 4:180–3. doi: 10.1016/j.redox.2015.01.002
15. Sies H. Oxidative stress: Concept and some practical aspects. Antioxidants (Basel) (2020) 9:852. doi: 10.3390/antiox9090852
16. Bourgonje AR, Feelisch M, Faber KN, Pasch A, Dijkstra G, Van Goor H. Oxidative stress and redox-modulating therapeutics in inflammatory bowel disease. Trends Mol Med (2020) 26:1034–46. doi: 10.1016/j.molmed.2020.06.006
17. Sies H, Jones DP. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol (2020) 21:363–83. doi: 10.1038/s41580-020-0230-3
18. Perez-Torres I, Castrejon-Tellez V, Soto ME, Rubio-Ruiz ME, Manzano-Pech L, Guarner-Lans V. Oxidative stress, plant natural antioxidants, and obesity. Int J Mol Sci (2021) 22:1786. doi: 10.3390/ijms22041786
19. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest (2004) 114:1752–61. doi: 10.1172/JCI21625
20. Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes (1998) 47:1562–9. doi: 10.2337/diabetes.47.10.1562
21. Muriach M, Flores-Bellver M, Romero FJ, Barcia JM. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid Med Cell Longev (2014) 2014:102158. doi: 10.1155/2014/102158
22. Trigo D, Avelar C, Fernandes M, Sa J, Da Cruz ESO. Mitochondria, energy, and metabolism in neuronal health and disease. FEBS Lett (2022) 596:1095–110. doi: 10.1002/1873-3468.14298
23. Yin F, Sancheti H, Patil I, Cadenas E. Energy metabolism and inflammation in brain aging and alzheimer's disease. Free Radic Biol Med (2016) 100:108–22. doi: 10.1016/j.freeradbiomed.2016.04.200
24. Morimoto RI. Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev (2008) 22:1427–38. doi: 10.1101/gad.1657108
25. Jellinger KA. Basic mechanisms of neurodegeneration: a critical update. J Cell Mol Med (2010) 14:457–87. doi: 10.1111/j.1582-4934.2010.01010.x
26. Georgiou CD, Margaritis LH. Oxidative stress and NADPH oxidase: Connecting electromagnetic fields, cation channels and biological effects. Int J Mol Sci (2021) 22:10041. doi: 10.3390/ijms221810041
27. Rahal A, Kumar A, Singh V, Yadav B, Tiwari R, Chakraborty S, et al. Oxidative stress, prooxidants, and antioxidants: The interplay. BioMed Res Int (2014) 2014:761264. doi: 10.1155/2014/761264
28. Masschelin PM, Cox AR, Chernis N, Hartig SM. The impact of oxidative stress on adipose tissue energy balance. Front Physiol (2019) 10:1638. doi: 10.3389/fphys.2019.01638
29. Charradi K, Elkahoui S, Limam F, Aouani E. High-fat diet induced an oxidative stress in white adipose tissue and disturbed plasma transition metals in rat: Prevention by grape seed and skin extract. J Physiol Sci (2013) 63:445–55. doi: 10.1007/s12576-013-0283-6
30. Jia QQ, Wang JC, Long J, Zhao Y, Chen SJ, Zhai JD, et al. Sesquiterpene lactones and their derivatives inhibit high glucose-induced NF-kappaB activation and MCP-1 and TGF-beta1 expression in rat mesangial cells. Molecules (2013) 18:13061–77. doi: 10.3390/molecules181013061
31. Tan BL, Norhaizan ME, Liew WP. Nutrients and oxidative stress: Friend or foe? Oxid Med Cell Longev (2018) 2018:9719584. doi: 10.1155/2018/9719584
32. Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. N Engl J Med (2017) 376:254–66. doi: 10.1056/NEJMra1514009
33. Alkhawaldeh K, Alavi A. Quantitative assessment of FDG uptake in brown fat using standardized uptake value and dual-time-point scanning. Clin Nucl Med (2008) 33:663–7. doi: 10.1097/RLU.0b013e318184b3de
34. Saito S, Saito CT, Shingai R. Adaptive evolution of the uncoupling protein 1 gene contributed to the acquisition of novel nonshivering thermogenesis in ancestral eutherian mammals. Gene (2008) 408:37–44. doi: 10.1016/j.gene.2007.10.018
35. Lizcano F. The beige adipocyte as a therapy for metabolic diseases. Int J Mol Sci (2019) 20:5058. doi: 10.3390/ijms20205058
36. Kusminski CM, Bickel PE, Scherer PE. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat Rev Drug Discov (2016) 15:639–60. doi: 10.1038/nrd.2016.75
37. Singh M, Benencia F. Inflammatory processes in obesity: Focus on endothelial dysfunction and the role of adipokines as inflammatory mediators. Int Rev Immunol (2019) 38:157–71. doi: 10.1080/08830185.2019.1638921
38. Weisberg SP, Mccann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest (2003) 112:1796–808. doi: 10.1172/JCI200319246
39. Solinas G, Karin M. JNK1 and IKKbeta: molecular links between obesity and metabolic dysfunction. FASEB J (2010) 24:2596–611. doi: 10.1096/fj.09-151340
40. Bluher M. Obesity: Global epidemiology and pathogenesis. Nat Rev Endocrinol (2019) 15:288–98. doi: 10.1038/s41574-019-0176-8
41. Mcardle MA, Finucane OM, Connaughton RM, Mcmorrow AM, Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: Insights into the emerging role of nutritional strategies. Front Endocrinol (Lausanne) (2013) 4:52. doi: 10.3389/fendo.2013.00052
42. Zorena K, Jachimowicz-Duda O, Slezak D, Robakowska M, Mrugacz M. Adipokines and obesity. potential link to metabolic disorders and chronic complications. Int J Mol Sci (2020) 21:3570. doi: 10.3390/ijms21103570
43. Johnson AR, Milner JJ, Makowski L. The inflammation highway: Metabolism accelerates inflammatory traffic in obesity. Immunol Rev (2012) 249:218–38. doi: 10.1111/j.1600-065X.2012.01151.x
44. Keane KN, Calton EK, Carlessi R, Hart PH, Newsholme P. The bioenergetics of inflammation: Insights into obesity and type 2 diabetes. Eur J Clin Nutr (2017) 71:904–12. doi: 10.1038/ejcn.2017.45
45. Fernandez-Sanchez A, Madrigal-Santillan E, Bautista M, Esquivel-Soto J, Morales-Gonzalez A, Esquivel-Chirino C, et al. Inflammation, oxidative stress, and obesity. Int J Mol Sci (2011) 12:3117–32. doi: 10.3390/ijms12053117
46. Khan NI, Naz L, Yasmeen G. Obesity: An independent risk factor for systemic oxidative stress. Pak J Pharm Sci (2006) 19:62–5.
47. Lastra G, Manrique CM, Hayden MR. The role of beta-cell dysfunction in the cardiometabolic syndrome. J Cardiometab Syndr (2006) 1:41–6. doi: 10.1111/j.0197-3118.2006.05458.x
48. Yaribeygi H, Sathyapalan T, Atkin SL, Sahebkar A. Molecular mechanisms linking oxidative stress and diabetes mellitus. Oxid Med Cell Longev (2020) 2020:8609213. doi: 10.1155/2020/8609213
49. Pessler D, Rudich A, Bashan N. Oxidative stress impairs nuclear proteins binding to the insulin responsive element in the GLUT4 promoter. Diabetologia (2001) 44:2156–64. doi: 10.1007/s001250100024
50. Balbaa M, Abdulmalek SA, Khalil S. Oxidative stress and expression of insulin signaling proteins in the brain of diabetic rats: Role of nigella sativa oil and antidiabetic drugs. PloS One (2017) 12:e0172429. doi: 10.1371/journal.pone.0172429
51. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis (2017) 1863:1066–77. doi: 10.1016/j.bbadis.2016.11.010
52. Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochem (Mosc) (2005) 70:200–14. doi: 10.1007/s10541-005-0102-7
53. Videla LA, Mariman A, Ramos B, Jose Silva M, Del Campo A. Standpoints in mitochondrial dysfunction: Underlying mechanisms in search of therapeutic strategies. Mitochondrion (2022) 63:9–22. doi: 10.1016/j.mito.2021.12.006
54. Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet (2009) 18:R169–76. doi: 10.1093/hmg/ddp326
55. Tilokani L, Nagashima S, Paupe V, Prudent J. Mitochondrial dynamics: overview of molecular mechanisms. Essays Biochem (2018) 62:341–60. doi: 10.1042/EBC20170104
56. Senos Demarco R, Jones DL. Mitochondrial fission regulates germ cell differentiation by suppressing ROS-mediated activation of epidermal growth factor signaling in the drosophila larval testis. Sci Rep (2019) 9:19695. doi: 10.1038/s41598-019-55728-0
57. Wrobel L, Topf U, Bragoszewski P, Wiese S, Sztolsztener ME, Oeljeklaus S, et al. Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol. Nature (2015) 524:485–8. doi: 10.1038/nature14951
58. Melber A, Haynes CM. UPR(mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res (2018) 28:281–95. doi: 10.1038/cr.2018.16
59. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ (2013) 20:31–42. doi: 10.1038/cdd.2012.81
60. Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science (2011) 333:1109–12. doi: 10.1126/science.1201940
61. Ko MS, Yun JY, Baek IJ, Jang JE, Hwang JJ, Lee SE, et al. Mitophagy deficiency increases NLRP3 to induce brown fat dysfunction in mice. Autophagy (2021) 17:1205–21. doi: 10.1080/15548627.2020.1753002
62. Kusminski CM, Scherer PE. Mitochondrial dysfunction in white adipose tissue. Trends Endocrinol Metab (2012) 23:435–43. doi: 10.1016/j.tem.2012.06.004
63. Wang Z, Hou L, Huang L, Guo J, Zhou X. Exenatide improves liver mitochondrial dysfunction and insulin resistance by reducing oxidative stress in high fat diet-induced obese mice. Biochem Biophys Res Commun (2017) 486:116–23. doi: 10.1016/j.bbrc.2017.03.010
64. Zorzano A, Liesa M, Palacin M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch Physiol Biochem (2009) 115:1–12. doi: 10.1080/13813450802676335
65. Peng KY, Watt MJ, Rensen S, Greve JW, Huynh K, Jayawardana KS, et al. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J Lipid Res (2018) 59:1977–86. doi: 10.1194/jlr.M085613
66. Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol (2010) 52:727–36. doi: 10.1016/j.jhep.2009.11.030
67. Heinonen S, Buzkova J, Muniandy M, Kaksonen R, Ollikainen M, Ismail K, et al. Impaired mitochondrial biogenesis in adipose tissue in acquired obesity. Diabetes (2015) 64:3135–45. doi: 10.2337/db14-1937
68. Sun Y, Ge X, Li X, He J, Wei X, Du J, et al. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis (2020) 11:914. doi: 10.1038/s41419-020-03122-4
69. Ma W, Yuan L, Yu H, Xi Y, Xiao R. Mitochondrial dysfunction and oxidative damage in the brain of diet-induced obese rats but not in diet-resistant rats. Life Sci (2014) 110:53–60. doi: 10.1016/j.lfs.2014.07.018
70. Sripetchwandee J, Chattipakorn N, Chattipakorn SC. Links between obesity-induced brain insulin resistance, brain mitochondrial dysfunction, and dementia. Front Endocrinol (Lausanne) (2018) 9:496. doi: 10.3389/fendo.2018.00496
71. Terzo S, Amato A, Mule F. From obesity to alzheimer's disease through insulin resistance. J Diabetes Complicat (2021) 35:108026. doi: 10.1016/j.jdiacomp.2021.108026
72. Jheng HF, Tsai PJ, Guo SM, Kuo LH, Chang CS, Su IJ, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol (2012) 32:309–19. doi: 10.1128/MCB.05603-11
73. Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab (2013) 17:491–506. doi: 10.1016/j.cmet.2013.03.002
74. Munoz A, Costa M. Nutritionally mediated oxidative stress and inflammation. Oxid Med Cell Longev (2013) 2013:610950. doi: 10.1155/2013/610950
75. He L, Sun Y. The potential role of Keap1-Nrf2 pathway in the pathogenesis of alzheimer's disease, type 2 diabetes, and type 2 diabetes-related alzheimer's disease. Metab Brain Dis (2021) 36:1469–79. doi: 10.1007/s11011-021-00762-z
76. Mittal K, Katare DP. Shared links between type 2 diabetes mellitus and alzheimer's disease: A review. Diabetes Metab Syndr (2016) 10:S144–9. doi: 10.1016/j.dsx.2016.01.021
77. Solov'eva EY, Dzhutova ED, Knyazeva VS. [Pathogenetic approaches to treatment of cognitive disorders in patients with diabetes mellitus]. Zh Nevrol Psikhiatr Im S S Korsakova (2016) 116:85–93. doi: 10.17116/jnevro20161167185-93
78. Pugazhenthi S, Qin L, Reddy PH. Common neurodegenerative pathways in obesity, diabetes, and alzheimer's disease. Biochim Biophys Acta Mol Basis Dis (2017) 1863:1037–45. doi: 10.1016/j.bbadis.2016.04.017
79. Yaribeygi H, Farrokhi FR, Butler AE, Sahebkar A. Insulin resistance: Review of the underlying molecular mechanisms. J Cell Physiol (2019) 234:8152–61. doi: 10.1002/jcp.27603
80. Butterfield DA, Di Domenico F, Barone E. Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim Et Biophys Acta (2014) 1842:1693–706. doi: 10.1016/j.bbadis.2014.06.010
81. Ferreira ST, Clarke JR, Bomfim TR, De Felice FG. Inflammation, defective insulin signaling, and neuronal dysfunction in alzheimer's disease. Alzheimer's Dementia: J Alzheimer's Assoc (2014) 10:S76–83. doi: 10.1016/j.jalz.2013.12.010
82. Lin D, Chun T-H, Kang L. Adipose extracellular matrix remodelling in obesity and insulin resistance. Biochem Pharmacol (2016) 119:8–16. doi: 10.1016/j.bcp.2016.05.005
83. Barone E, Di Domenico F, Perluigi M, Butterfield DA. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic Biol Med (2021) 176:16–33. doi: 10.1016/j.freeradbiomed.2021.09.006
84. Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang H-Y, Ahima RS, et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat Rev Neurol (2018) 14:168–81. doi: 10.1038/nrneurol.2017.185
85. Saltiel AR. Insulin signaling in health and disease. J Clin Invest (2021) 131:e142241. doi: 10.1172/JCI142241
86. Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev (2018) 98:2133–223. doi: 10.1152/physrev.00063.2017
87. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radical Biol Med (2016) 100:14–31. doi: 10.1016/j.freeradbiomed.2016.04.001
88. Goldstein BJ, Mahadev K, Wu X. Redox paradox. Diabetes (2005) 54:311–21. doi: 10.2337/diabetes.54.2.311
89. Sultana R, Perluigi M, Butterfield DA. Oxidatively modified proteins in alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: role of abeta in pathogenesis. Acta Neuropathologica (2009) 118:131–50. doi: 10.1007/s00401-009-0517-0
90. García-Fernández M, Delgado G, Puche JE, González-Barón S, Castilla Cortázar I. Low doses of insulin-like growth factor I improve insulin resistance, lipid metabolism, and oxidative damage in aging rats. Endocrinology (2008) 149:2433–42. doi: 10.1210/en.2007-1190
91. Mainardi M, Fusco S, Grassi C. Modulation of hippocampal neural plasticity by glucose-related signaling. Neural Plast (2015) 2015:657928. doi: 10.1155/2015/657928
92. Kullmann S, Heni M, Hallschmid M, Fritsche A, Preissl H, Häring H-U. Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol Rev (2016) 96:1169–209. doi: 10.1152/physrev.00032.2015
93. Drews G, Krippeit-Drews P, Düfer M. Oxidative stress and beta-cell dysfunction. Pflugers Archiv: Eur J Physiol (2010) 460:703–18. doi: 10.1007/s00424-010-0862-9
94. Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxidants Redox Signaling (2017) 26:501–18. doi: 10.1089/ars.2016.6755
95. Kang B, Wang X, Xu Q, Wu Y, Si X, Jiang D. Effect of 3-nitropropionic acid inducing oxidative stress and apoptosis of granulosa cells in geese. Bioscience Rep (2018) 38:BSR20180274. doi: 10.1042/BSR20180274
96. Gier B, Krippeit-Drews P, Sheiko T, Aguilar-Bryan L, Bryan J, Düfer M, et al. Suppression of KATP channel activity protects murine pancreatic beta cells against oxidative stress. J Clin Invest (2009) 119:3246–56. doi: 10.1172/JCI38817
97. Hurrle S, Hsu WH. The etiology of oxidative stress in insulin resistance. Biomed J (2017) 40:257–62. doi: 10.1016/j.bj.2017.06.007
98. O'gorman DJ, Karlsson HKR, Mcquaid S, Yousif O, Rahman Y, Gasparro D, et al. Exercise training increases insulin-stimulated glucose disposal and GLUT4 (SLC2A4) protein content in patients with type 2 diabetes. Diabetologia (2006) 49:2983–92. doi: 10.1007/s00125-006-0457-3
99. Hussey SE, Mcgee SL, Garnham A, Mcconell GK, Hargreaves M. Exercise increases skeletal muscle GLUT4 gene expression in patients with type 2 diabetes. Diabetes Obes Metab (2012) 14:768–71. doi: 10.1111/j.1463-1326.2012.01585.x
100. Reno CM, Puente EC, Sheng Z, Daphna-Iken D, Bree AJ, Routh VH, et al. Brain GLUT4 knockout mice have impaired glucose tolerance, decreased insulin sensitivity, and impaired hypoglycemic counterregulation. Diabetes (2017) 66:587–97. doi: 10.2337/db16-0917
101. Fazakerley DJ, Minard AY, Krycer JR, Thomas KC, Stöckli J, Harney DJ, et al. Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J Biol Chem (2018) 293:7315–28. doi: 10.1074/jbc.RA117.001254
102. She H, Mao Z. Regulation of myocyte enhancer factor-2 transcription factors by neurotoxins. Neurotoxicology (2011) 32:563–6. doi: 10.1016/j.neuro.2011.05.019
103. Esteves JV, Enguita FJ, Machado UF. MicroRNAs-mediated regulation of skeletal muscle GLUT4 expression and translocation in insulin resistance. J Diabetes Res (2017) 2017:7267910. doi: 10.1155/2017/7267910
104. Eriksson JW. Metabolic stress in insulin's target cells leads to ROS accumulation - a hypothetical common pathway causing insulin resistance. FEBS Lett (2007) 581:3734–42. doi: 10.1016/j.febslet.2007.06.044
105. Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes (2003) 52:1–8. doi: 10.2337/diabetes.52.1.1
106. Erichsen JM, Fadel JR, Reagan LP. Peripheral versus central insulin and leptin resistance: Role in metabolic disorders, cognition, and neuropsychiatric diseases. Neuropharmacology (2022) 203:108877. doi: 10.1016/j.neuropharm.2021.108877
107. Van Der Heide LP, Kamal A, Artola A, Gispen WH, Ramakers GMJ. Insulin modulates hippocampal activity-dependent synaptic plasticity in a n-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J Neurochem (2005) 94:1158–66. doi: 10.1111/j.1471-4159.2005.03269.x
108. Kim SJ, Han Y. Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase b (Akt). J Neural Transm (Vienna Austria: 1996) (2005) 112:179–91. doi: 10.1007/s00702-004-0163-6
109. Bockmann J, Kreutz MR, Gundelfinger ED, Bockers TM. ProSAP/Shank postsynaptic density proteins interact with insulin receptor tyrosine kinase substrate IRSp53. J Neurochem (2002) 83:1013–7. doi: 10.1046/j.1471-4159.2002.01204.x
110. Gralle M. The neuronal insulin receptor in its environment. J Neurochem (2017) 140:359–67. doi: 10.1111/jnc.13909
111. Spinelli M, Fusco S, Grassi C. Brain insulin resistance and hippocampal plasticity: Mechanisms and biomarkers of cognitive decline. Front Neurosci (2019) 13:788. doi: 10.3389/fnins.2019.00788
112. Dutta BJ, Singh S, Seksaria S, Das Gupta G, Singh A. Inside the diabetic brain: Insulin resistance and molecular mechanism associated with cognitive impairment and its possible therapeutic strategies. Pharmacol Res (2022) 182:106358. doi: 10.1016/j.phrs.2022.106358
113. Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology (2009) 150:5294–301. doi: 10.1210/en.2009-0695
114. Yuan XY, Wang XG. Mild cognitive impairment in type 2 diabetes mellitus and related risk factors: A review. Rev Neurosci (2017) 28:715–23. doi: 10.1515/revneuro-2017-0016
115. Sędzikowska A, Szablewski L. Insulin and insulin resistance in alzheimer's disease. Int J Mol Sci (2021) 22(18):9987. doi: 10.3390/ijms22189987
116. Teleanu DM, Niculescu A-G, Lungu II, Radu CI, Vladâcenco O, Roza E, et al. An overview of oxidative stress, neuroinflammation, and neurodegenerative diseases. Int J Mol Sci (2022) 23:5938. doi: 10.3390/ijms23115938
117. Solleiro-Villavicencio H, Rivas-Arancibia S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4T cells in neurodegenerative diseases. Front Cell Neurosci (2018) 12:114. doi: 10.3389/fncel.2018.00114
118. Degan D, Ornello R, Tiseo C, Carolei A, Sacco S, Pistoia F. The role of inflammation in neurological disorders. Curr Pharm Design (2018) 24:1485–501. doi: 10.2174/1381612824666180327170632
119. Mishra A, Bandopadhyay R, Singh PK, Mishra PS, Sharma N, Khurana N. Neuroinflammation in neurological disorders: Pharmacotherapeutic targets from bench to bedside. Metab Brain Dis (2021) 36:1591–626. doi: 10.1007/s11011-021-00806-4
120. Kölliker-Frers R, Udovin L, Otero-Losada M, Kobiec T, Herrera MI, Palacios J, et al. Neuroinflammation: An integrating overview of reactive-neuroimmune cell interactions in health and disease. Mediators Inflammation (2021) 2021:1–20. doi: 10.1155/2021/9999146
121. Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in alzheimer's disease. J Neuroinflammation (2022) 19:206. doi: 10.1186/s12974-022-02565-0
122. Kanneganti TD, Dixit VD. Immunological complications of obesity. Nat Immunol (2012) 13:707–12. doi: 10.1038/ni.2343
123. Stranahan AM, Hao S, Dey A, Yu X, Baban B. Blood-brain barrier breakdown promotes macrophage infiltration and cognitive impairment in leptin receptor-deficient mice. J Cereb Blood Flow Metab (2016) 36:2108–21. doi: 10.1177/0271678X16642233
124. Jeong EA, Lee J, Shin HJ, Lee JY, Kim KE, An HS, et al. Tonicity-responsive enhancer-binding protein promotes diabetic neuroinflammation and cognitive impairment via upregulation of lipocalin-2. J Neuroinflammation (2021) 18:278. doi: 10.1186/s12974-021-02331-8
125. Obrador E, Salvador R, López-Blanch R, Jihad-Jebbar A, Vallés SL, Estrela JM. Oxidative stress, neuroinflammation and mitochondria in the pathophysiology of amyotrophic lateral sclerosis. Antioxidants (2020) 9:901. doi: 10.3390/antiox9090901
126. Haddad-Tóvolli R, Dragano NRV, Ramalho AFS, Velloso LA. Development and function of the blood-brain barrier in the context of metabolic control. Front Neurosci (2017) 11:224. doi: 10.3389/fnins.2017.00224
127. Liyanagamage D, Martinus RD. Role of mitochondrial stress protein HSP60 in diabetes-induced neuroinflammation. Mediators Inflamm (2020) 2020:8073516. doi: 10.1155/2020/8073516
128. Li W, Roy Choudhury G, Winters A, Prah J, Lin W, Liu R, et al. Hyperglycemia alters astrocyte metabolism and inhibits astrocyte proliferation. Aging Dis (2018) 9:674–84. doi: 10.14336/AD.2017.1208
129. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
130. Ruud J, Steculorum SM, Brüning JC. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat Commun (2017) 8:15259. doi: 10.1038/ncomms15259
131. Fournet M, Bonté F, Desmoulière A. Glycation damage: A possible hub for major pathophysiological disorders and aging. Aging Dis (2018) 9:880–900. doi: 10.14336/AD.2017.1121
132. Yan L-J. Redox imbalance stress in diabetes mellitus: Role of the polyol pathway. Anim Models Exp Med (2018) 1:7–13. doi: 10.1002/ame2.12001
133. Haspula D, Clark MA. Molecular basis of the brain renin angiotensin system in cardiovascular and neurologic disorders: Uncovering a key role for the astroglial angiotensin type 1 receptor AT1R. J Pharmacol Exp Ther (2018) 366:251–64. doi: 10.1124/jpet.118.248831
134. Krämer-Albers E-M. Exosomes deliver ROS for regeneration. Nat Cell Biol (2018) 20:225–6. doi: 10.1038/s41556-018-0048-9
135. Sheng Z-H. The interplay of axonal energy homeostasis and mitochondrial trafficking and anchoring. Trends In Cell Biol (2017) 27:403–16. doi: 10.1016/j.tcb.2017.01.005
136. Cenini G, Lloret A, Cascella R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid Med Cell Longev (2019) 2019:1–18. doi: 10.1155/2019/2105607
137. Golpich M, Amini E, Mohamed Z, Azman Ali R, Mohamed Ibrahim N, Ahmadiani A. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci Ther (2017) 23:5–22. doi: 10.1111/cns.12655
138. Jaiswal P, Mandal M, Mishra A. Effect of hesperidin on fluoride-induced neurobehavioral and biochemical changes in rats. J Biochem Mol Toxicol (2020) 34:e22575. doi: 10.1002/jbt.22575
139. Lin W-J, Kuang H-Y. Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy (2014) 10:1692–701. doi: 10.4161/auto.36076
140. Katsumoto A, Lu H, Miranda AS, Ransohoff RM. Ontogeny and functions of central nervous system macrophages. J Immunol (2014) 193:2615–21. doi: 10.4049/jimmunol.1400716
141. Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol (2021) 17:157–72. doi: 10.1038/s41582-020-00435-y
142. Guo DH, Yamamoto M, Hernandez CM, Khodadadi H, Baban B, Stranahan AM. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J Clin Invest (2020) 130:1961–76. doi: 10.1172/JCI126078
143. Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab (2011) 13:11–22. doi: 10.1016/j.cmet.2010.12.008
144. Zhou M, Xu W, Wang J, Yan J, Shi Y, Zhang C, et al. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-kappaB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine (2018) 35:345–60. doi: 10.1016/j.ebiom.2018.08.035
145. Cui Y, Yang M, Wang Y, Ren J, Lin P, Cui C, et al. Melatonin prevents diabetes-associated cognitive dysfunction from microglia-mediated neuroinflammation by activating autophagy via TLR4/Akt/mTOR pathway. FASEB J (2021) 35:e21485. doi: 10.1096/fj.202002247RR
146. Wang Z, Xia P, Hu J, Huang Y, Zhang F, Li L, et al. LncRNA MEG3 alleviates diabetic cognitive impairments by reducing mitochondrial-derived apoptosis through promotion of FUNDC1-related mitophagy via Rac1-ROS axis. ACS Chem Neurosci (2021) 12:2280–307. doi: 10.1021/acschemneuro.0c00682
147. Subramaniam S, Fahy E, Gupta S, Sud M, Byrnes RW, Cotter D, et al. Bioinformatics and systems biology of the lipidome. Chem Rev (2011) 111:6452–90. doi: 10.1021/cr200295k
148. Fahy E, Subramaniam S, Murphy RC, Nishijima M, Raetz CRH, Shimizu T, et al. Update of the LIPID MAPS comprehensive classification system for lipids. J Lipid Res (2009) 50(Suppl):S9–14. doi: 10.1194/jlr.R800095-JLR200
149. Ooi K-LM, Vacy K, Boon WC. Fatty acids and beyond: Age and alzheimer's disease related changes in lipids reveal the neuro-nutraceutical potential of lipids in cognition. Neurochem Int (2021) 149:105143. doi: 10.1016/j.neuint.2021.105143
150. Hussain MM. Intestinal lipid absorption and lipoprotein formation. Curr Opin Lipidology (2014) 25:200–6. doi: 10.1097/MOL.0000000000000084
151. Frayn KN, Arner P, Yki-Järvinen H. Fatty acid metabolism in adipose tissue, muscle and liver in health and disease. Essays Biochem (2006) 42:89–103. doi: 10.1042/bse0420089
152. Ikonen E. Cellular cholesterol trafficking and compartmentalization. nature reviews. Mol Cell Biol (2008) 9:125–38. doi: 10.1038/nrm2336
153. Woods AS, Jackson SN. Brain tissue lipidomics: Direct probing using matrix-assisted laser desorption/ionization mass spectrometry. AAPS J (2006) 8:E391–5. doi: 10.1007/BF02854910
154. Sun Y, Ma C, Sun H, Wang H, Peng W, Zhou Z, et al. Metabolism: A novel shared link between diabetes mellitus and alzheimer's disease. J Diabetes Res (2020) 2020:4981814. doi: 10.1155/2020/4981814
155. Fester L, Zhou L, Bütow A, Huber C, Von Lossow R, Prange-Kiel J, et al. Cholesterol-promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus (2009) 19:692–705. doi: 10.1002/hipo.20548
156. Grimm MOW, Grimm HS, Pätzold AJ, Zinser EG, Halonen R, Duering M, et al. Regulation of cholesterol and sphingomyelin metabolism by amyloid-beta and presenilin. Nat Cell Biol (2005) 7:1118–23. doi: 10.1038/ncb1313
157. Sunil B, Ashraf AP. Dyslipidemia in pediatric type 2 diabetes mellitus. Curr Diabetes Rep (2020) 20:1–9. doi: 10.1007/s11892-020-01336-6
158. Robinson M, Lee BY, Hanes FT. Recent progress in alzheimer's disease research, part 2: Genetics and epidemiology. J Alzheimer's Disease: JAD (2018) 61:459. doi: 10.3233/JAD-179007
159. Deary IJ, Whiteman MC, Pattie A, Starr JM, Hayward C, Wright AF, et al. Cognitive change and the APOE epsilon 4 allele. Nature (2002) 418:932. doi: 10.1038/418932a
160. Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with alzheimer's disease. Nat Genet (2011) 43:429–35. doi: 10.1038/ng.803
161. Bertram L, Tanzi RE. Alzheimer Disease risk genes: 29 and counting. nature reviews. Neurology (2019) 15:191–2. doi: 10.1038/s41582-019-0158-4
162. Kuhla A, Blei T, Jaster R, Vollmar B. Aging is associated with a shift of fatty metabolism toward lipogenesis. Journals Gerontol Ser A Biol Sci Med Sci (2011) 66:1192–200. doi: 10.1093/gerona/glr124
163. Yu Q, He Z, Zubkov D, Huang S, Kurochkin I, Yang X, et al. Lipidome alterations in human prefrontal cortex during development, aging, and cognitive disorders. Mol Psychiatry (2020) 25:2952–69. doi: 10.1038/s41380-018-0200-8
164. Lavrovsky Y, Chatterjee B, Clark RA, Roy AK. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp Gerontol (2000) 35:521–32. doi: 10.1016/S0531-5565(00)00118-2
165. Kowluru RA, Mishra M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim Et Biophys Acta (2015) 1852:2474–83. doi: 10.1016/j.bbadis.2015.08.001
166. Ceretta LB, Réus GZ, Abelaira HM, Ribeiro KF, Zappellini G, Felisbino FF, et al. Increased oxidative stress and imbalance in antioxidant enzymes in the brains of alloxan-induced diabetic rats. Exp Diabetes Res (2012) 2012:302682. doi: 10.1155/2012/302682
167. Agrawal I, Jha S. Mitochondrial dysfunction and alzheimer's disease: Role of microglia. Front In Aging Neurosci (2020) 12:252. doi: 10.3389/fnagi.2020.00252
168. Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and alzheimer's disease. Proc Natl Acad Sci USA (2004) 101:2070–5. doi: 10.1073/pnas.0305799101
169. Small SA, Duff K. Linking abeta and tau in late-onset alzheimer's disease: A dual pathway hypothesis. Neuron (2008) 60:534–42. doi: 10.1016/j.neuron.2008.11.007
170. Shinohara M, Tachibana M, Kanekiyo T, Bu G. Role of LRP1 in the pathogenesis of alzheimer's disease: Evidence from clinical and preclinical studies. J Lipid Res (2017) 58:1267–81. doi: 10.1194/jlr.R075796
171. Demirci-Çekiç S, Özkan G, Avan AN, Uzunboy S, Çapanoğlu E, Apak R. Biomarkers of oxidative stress and antioxidant defense. J Pharm Biomed Anal (2022) 209:114477. doi: 10.1016/j.jpba.2021.114477
172. Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F. Oxidative stress and the amyloid beta peptide in alzheimer's disease. Redox Biol (2018) 14:450–64. doi: 10.1016/j.redox.2017.10.014
173. Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in alzheimer's disease brain contribute to neuronal death. Neurobiol Aging (2002) 23:655–64. doi: 10.1016/S0197-4580(01)00340-2
174. Chen R, Zeng Y, Xiao W, Zhang L, Shu Y. LC-MS-Based untargeted metabolomics reveals early biomarkers in STZ-induced diabetic rats with cognitive impairment. Front Endocrinol (Lausanne) (2021) 12:665309. doi: 10.3389/fendo.2021.665309
175. Begcevic Brkovic I, Zohrer B, Scholz M, Reinicke M, Dittrich J, Kamalsada S, et al. Simultaneous mass spectrometry-based apolipoprotein profiling and apolipoprotein e phenotyping in patients with ASCVD and mild cognitive impairment. Nutrients (2022) 14:2474. doi: 10.3390/nu14122474
176. Chausse B, Kakimoto PA, Kann O. Microglia and lipids: How metabolism controls brain innate immunity. Semin Cell Dev Biol (2021) 112:137–44. doi: 10.1016/j.semcdb.2020.08.001
177. Egashira Y, Hua Y, Keep RF, Iwama T, Xi G. Lipocalin 2 and blood-brain barrier disruption in white matter after experimental subarachnoid hemorrhage. Acta Neurochirurgica. Supplement (2016) 121:131–4. doi: 10.1007/978-3-319-18497-5_23
178. Lim D, Jeong J-H, Song J. Lipocalin 2 regulates iron homeostasis, neuroinflammation, and insulin resistance in the brains of patients with dementia: Evidence from the current literature. CNS Neurosci Ther (2021) 27:883–94. doi: 10.1111/cns.13653
179. Wu G, Li H, Zhou M, Fang Q, Bao Y, Xu A, et al. Mechanism and clinical evidence of lipocalin-2 and adipocyte fatty acid-binding protein linking obesity and atherosclerosis. Diabetes/metabolism Res Rev (2014) 30:447–56. doi: 10.1002/dmrr.2493
180. Jaberi SA, Cohen A, D'souza C, Abdulrazzaq YM, Ojha S, Bastaki S, et al. Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomedicine Pharmacother (2021) 142:112002. doi: 10.1016/j.biopha.2021.112002
181. Cheng Y, Song Y, Chen H, Li Q, Gao Y, Lu G, et al. Ferroptosis mediated by lipid reactive oxygen species: A possible causal link of neuroinflammation to neurological disorders. Oxid Med Cell Longev (2021) 2021:5005136. doi: 10.1155/2021/5005136
Keywords: oxidative stress, obesity, cognitive dysfunction in diabetes, reactive oxygen species, insulin resistance, neuroinflammation, lipid metabolism disorders
Citation: Li H, Ren J, Li Y, Wu Q and Wei J (2023) Oxidative stress: The nexus of obesity and cognitive dysfunction in diabetes. Front. Endocrinol. 14:1134025. doi: 10.3389/fendo.2023.1134025
Received: 29 December 2022; Accepted: 02 March 2023;
Published: 03 April 2023.
Edited by:
Xiaodong Sun, Affiliated Hospital of Weifang Medical University, ChinaReviewed by:
Xian-pei Heng, People’s Hospital Affiliated Fujian TCM University, ChinaCopyright © 2023 Li, Ren, Li, Wu and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junping Wei, d2VpanVucGluZ0AxMjYuY29t
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.