 Zhe Liu
Zhe Liu Jiahui Liu
Jiahui Liu Wanning Wang
Wanning Wang Xingna An
Xingna An Ling Luo
Ling Luo Dehai Yu
Dehai Yu Weixia Sun
Weixia Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 30 June 2023
Sec. Clinical Diabetes
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1133970
This article is part of the Research TopicClinical and Genetic Determinants of Diabetes and ComplicationsView all 31 articles
Diabetic kidney disease (DKD) is a common microangiopathy in diabetic patients and the main cause of death in diabetic patients. The main manifestations of DKD are proteinuria and decreased renal filtration capacity. The glomerular filtration rate and urinary albumin level are two of the most important hallmarks of the progression of DKD. The classical treatment of DKD is controlling blood glucose and blood pressure. However, the commonly used clinical therapeutic strategies and the existing biomarkers only partially slow the progression of DKD and roughly predict disease progression. Therefore, novel therapeutic methods, targets and biomarkers are urgently needed to meet clinical requirements. In recent years, increasing attention has been given to the role of epigenetic modification in the pathogenesis of DKD. Epigenetic variation mainly includes DNA methylation, histone modification and changes in the noncoding RNA expression profile, which are deeply involved in DKD-related inflammation, oxidative stress, hemodynamics, and the activation of abnormal signaling pathways. Since DKD is reversible at certain disease stages, it is valuable to identify abnormal epigenetic modifications as early diagnosis and treatment targets to prevent the progression of end-stage renal disease (ESRD). Because the current understanding of the epigenetic mechanism of DKD is not comprehensive, the purpose of this review is to summarize the role of epigenetic modification in the occurrence and development of DKD and evaluate the value of epigenetic therapies in DKD.
Diabetic kidney disease (DKD) is a common complication of diabetes and a major cause of end-stage renal disease (ESRD), which seriously affects the quality of life of patients (1–4). The main pathological features of DKD are glomerular sclerosis, podocyte detachment, epithelial-mesenchymal transition (EMT)/endothelial-to-mesenchymal transition (EndMT)/macrophage-myofibroblast transition (MMT), excessive extracellular matrix (ECM) and renal tubular fibrosis. These pathological changes affect glomerular and tubular function, leading to the progression of proteinuria and decreased glomerular filtration capacity. A long-term hyperglycemic environment in diabetics induces metabolic disorders, oxidative stress and hemodynamic changes. Although these symptoms occur with genetic mutations, they are, to a greater extent, related to epigenetic variations (5). For instance, studies have found that even after a long period of strict glycemic control, patients with diabetes may still develop complications due to early high glucose (HG) exposure (4, 6, 7). This metabolic memory phenomenon has been shown to be related to DNA methylation and histone acetylation at the promoter, which suggests that epigenetic modifications are subsumed in the pathological process of diabetes and affect patients’ conditions over a long period of time (8, 9). Therefore, a deeper understanding of the epigenetic modifications in DKD can help to better understand the pathogenesis of the disease and provide potential predictive and therapeutic targets for DKD treatment. In the current study, we conducted a comprehensive analysis and introduction of DKD-related epigenetic mechanisms and epigenetic therapies based on searching the published literature from PubMed (https://pubmed.ncbi.nlm.nih.gov) and Web of Science (http://www.webofknowledge.com/databases ). Our aim is to encourage more clinicians and researchers to pay attention to the function of epigenetic modifications in the occurrence and development of DKD and conduct laboratory, preclinical and clinical studies on the development of epigenetic drugs and therapeutic strategies for DKD.
Diabetic patients often have high blood pressure, high blood lipids, high uric acid and obesity, all of which may lead to kidney damage (10, 11). The pathogenesis of DKD is complex. The main pathological characteristics of DKD are glomerulosclerosis and renal fibrosis (12, 13). An impaired glomerular filtration barrier is the primary cause of albuminuria. Renal fibrosis and albuminuria are important causes of renal function loss, which is the consequence of multiple factors and mechanisms. DKD-associated renal fibrosis is defined by the excessive deposition of ECM caused by various adverse stimuli (14–17). Understanding the pathogenesis of DKD may help to prevent, slow down, or even reverse DKD. Figure 1 briefly summarizes the pathogenesis of DKD.
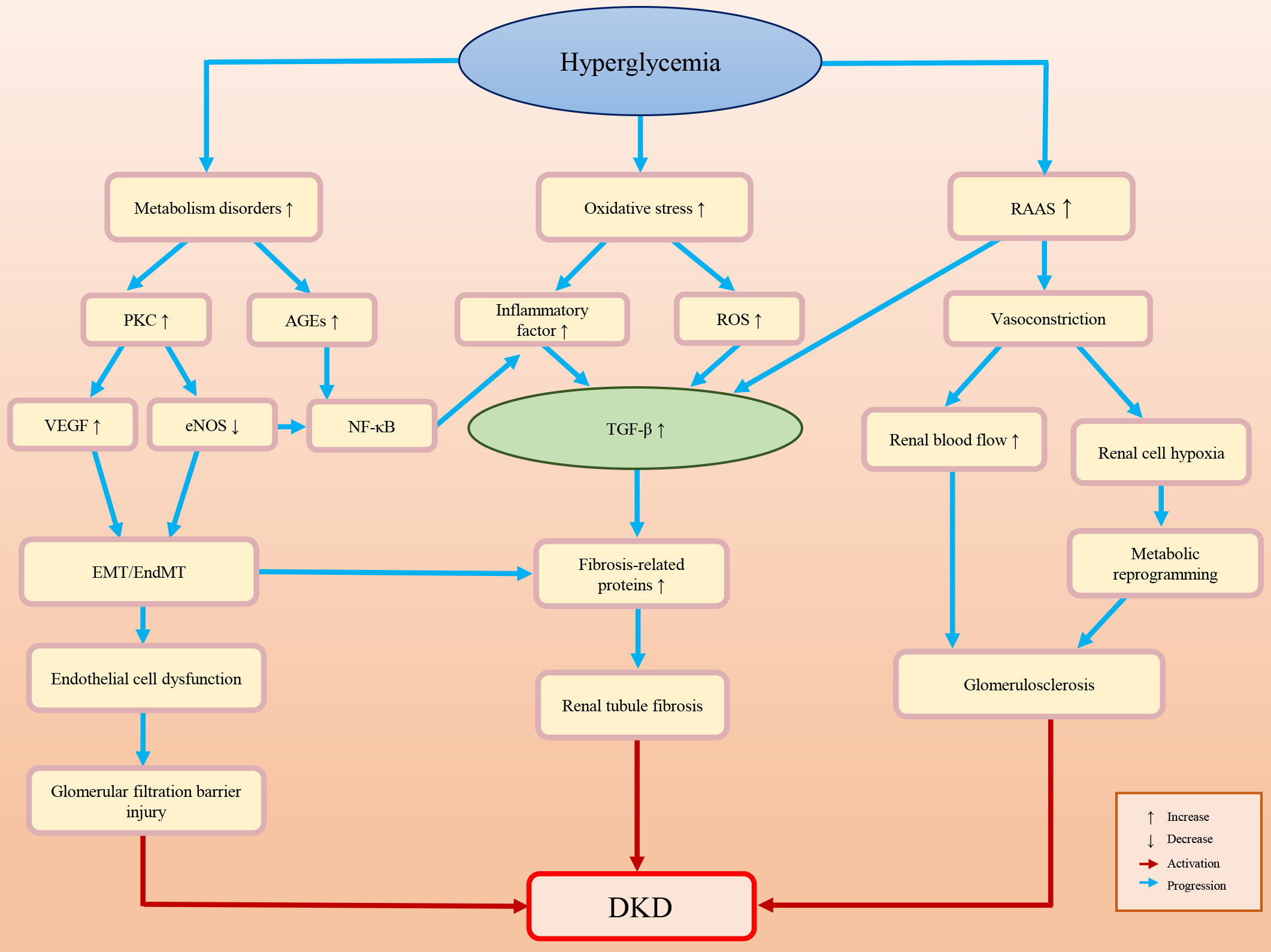
Figure 1 Briefly summarizes and illustration of the pathogenesis of DKD.
Glucose, lipid and hormone metabolism disorders caused by HG exposure may lead to the accumulation of advanced glycation end products (AGEs) and the activation of protein kinase C (PKC) (18–22). AGEs can activate related signaling pathways, such as the nuclear factor kappa-B (NF-κB) and transforming growth factor β (TGF-β) pathways, promote EMT/EndMT, and result in glomerular podocyte loss and progressive glomerulosclerosis (23–27). Activated PKC may decrease endothelial nitric oxide synthase (eNOS) production, which not only activates NF-κB-mediated inflammatory pathways but also stimulates the production of vascular endothelial growth factor (VEGF), inducing endothelial dysfunction and further (26, 28–30).
The HG environment activates polyols, PKC, hexosamine and other pathways, leading to an increase in the oxidative stress response and reactive oxygen species (ROS) (31–33). ROS mediate various signaling pathways, such as TGF-β, adenosine 5′ monophosphate-activated protein kinase (AMPK) and nuclear factor-erythroid 2-related factor 2 (Nrf2), which pertain to the cell cycle, cell proliferation, autophagy, inflammation and oxidative stress (33–35). In DKD, the activation of ROS can promote podocyte apoptosis and inflammatory factor release, and activate the renal fibrosis signaling pathways, which results in renal fibrosis and the decline of glomerular filtration function (23, 25, 27).
Diabetes is often accompanied by chronic inflammation. The expression levels of inflammatory factors (e.g. tumor necrosis factor-α (TNF-α), interleukin-6, interferon γ (IFN-γ) and interleukin-17) are elevated in DKD patients (14–16, 36–41). Abnormal expression of these cytokines may activate renal fibrosis-related signaling pathways, induce EndMT/EMT/MMT, and promote the accumulation of ECM, which ultimately stimulates the expression of fibrosis-related proteins (e.g. α-smooth muscle actin (α-SMA) and connective tissue growth factor (CTGF)) and glomerulosclerosis (42–47).
Diabetic patients’ kidneys are always exposed to an HG environment for a long time. The long-term high level filtration load may induce glomerular feedback dysfunction and aggravate glomerular sclerosis (48). The renin-angiotensin-aldosterone system (RAAS) can also be activated by HG exposure (e.g., the products of HG-induced metabolic disorders and oxidative stress-induced ROS) (49). The RAAS not only induces the constriction of blood vessels in the kidney, but also upregulates TGF-β fibrosis-associated pathways and inflammation (49–53). The decline in blood flow and oxygen delivery at the glomerular filtration barrier after renal vasoconstriction may promote glycolysis and metabolic reprogramming and produce metabolites (e.g., lactate and L-serine) (54–58). These metabolites are associated with multiple cellular behavior variations, such as mitochondrial damage, histone modification, and the activation of the renal cell fibrosis-related signaling pathway, which may affect cell senescence and survival, increase inflammation reflection, induce podocyte damage, endothelial cell dysfunction, and renal tubular cell fibrosis, and further aggravate kidney damage (59–64).
DNA methylation is a significant epigenetic regulatory mechanism. DNA methylation is catalyzed by a family of DNA methyltransferases that transfer a methyl group from S-adenyl methionine to the carbon of a cytosine residue (65). DNA methylation can change chromosome structure, conformation, stability and the interaction mode between DNA and protein, thereby participating in a variety of regulatory mechanisms (e.g., gene transcription and imprinting, cell differentiation and fibrosis) (66–70).
DNA methylation is associated with DKD. VanderJagt et al. found that many methylation modifications occur from prediabetes to diabetes. Among these methylation modifications, six genes are associated with DKD, which may induce inflammation and immunity and break urate homeostasis (71). By comparing the DNA methylation of kidney proximal tubule cells in 10-week-old db/db mice with that in normal mice, Marumo et al. considered that at the early stage of DKD, several potentially functional genes were significantly methylated, e.g., angiotensinogen (Agt) and claudin 18 (Cldn18), which may alter the progression of DKD (72). Park et al. indicated that there are extensive methylation differences in DKD kidneys, among which the change in TNF-α methylation has a close connection with kidney function decline (73). In addition, the application of reversed-phase high performance liquid chromatography (RP-HPLC) to determine DNA methylation levels in peripheral blood mononuclear cells also revealed differences in genomic methylation levels between patients with renal dysfunction and patients with simple diabetes (74, 75). These abnormal changes may be a response to a chronically hyperglycemic environment. Furthermore, the degree of methylation in DKD varies from stage to stage. Lecamwasam et al. collected blood samples from diabetic patients with chronic kidney disease (CKD) and indicated that differential methylation patterns of 5’-C-phosphate-G-3’ (CpG) sites are associated with different stages of CKD. Of note, relative to the early CKD group, the cysteine-rich secretory protein 2 (CRISP2) gene promoter carried 12 hypermethylated CpG sites in the late CKD group, which may lead to oxidative stress in inflammatory pathways (76).
Histones are an important component of nucleosomes and a general term for alkaline proteins that bind to DNA (77–79). The free N-terminus at the end of histones can undergo various modifications, including acetylation, methylation, phosphorylation, and ubiquitination (80). Once histones are modified, the function of chromatin will be changed: first, the charge of amino acids will be changed, and the affinity between histones and DNA will be decreased; second, binding to specific surfaces and regulating transcriptional activity will also be changed (79). Figure 2 briefly summarizes the histone epigenetic modifications and their regulatory roles in DKD.
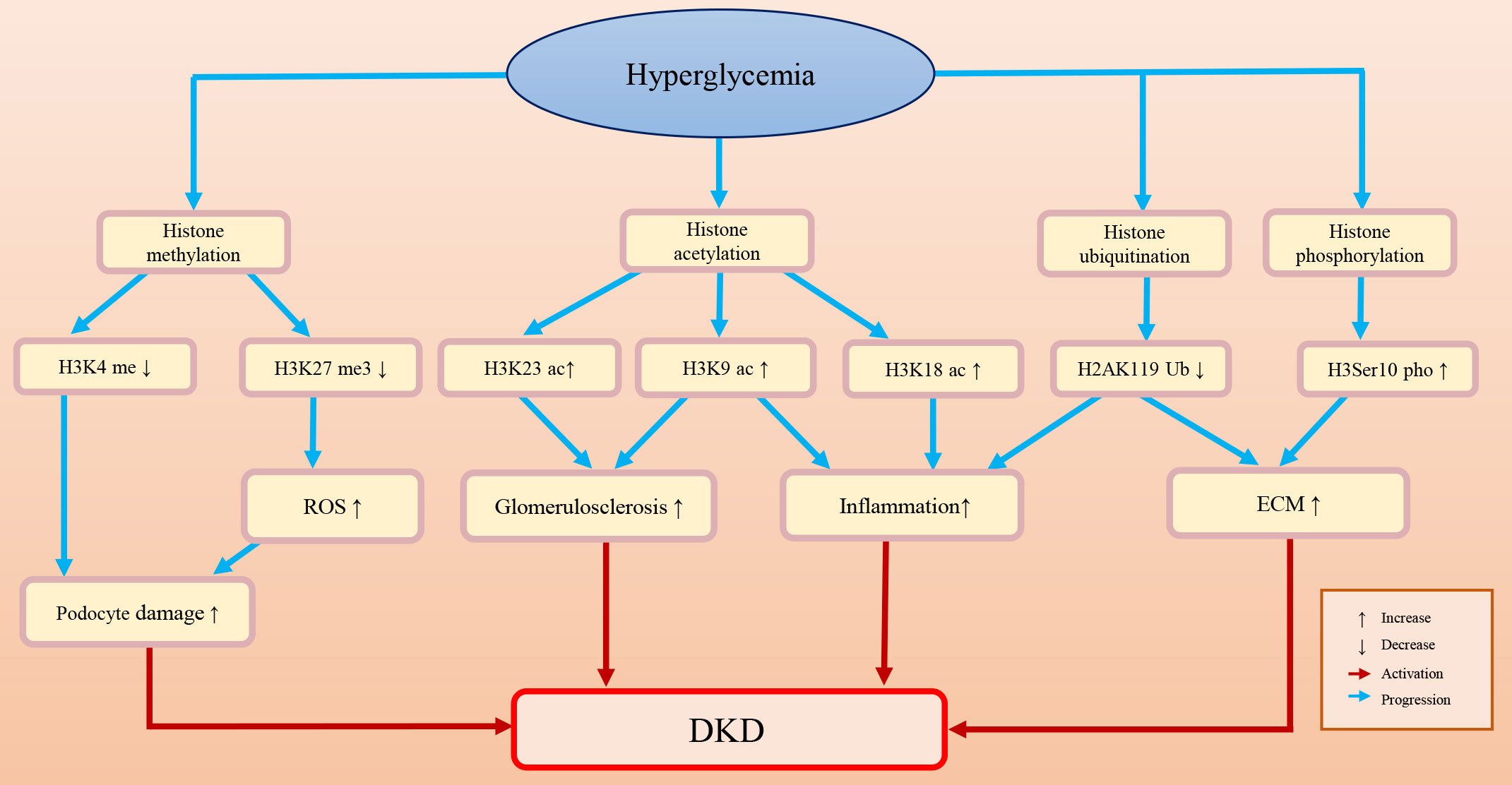
Figure 2 The histone epigenetic modifications and their roles in DKD.
Histone methylation is a process in which methyl groups are transferred to lysine and arginine residues in the histone tail by histone methyltransferase (81). Histone methylation is a dynamic and reversible process because the methylation of histones can be erased by histone demethylases (82). Histone methylation plays a regulatory role similar to that of DNA methylation. Whether it functions in transcriptional repression or activation depends on the methylation degree and the modification site.
Histone methylation is an important epigenetic modification in DKD. In diabetic mice, upregulation of TGF-β may promote the recruitment of the histone H3 lysine 4 (H3K4) methylation methyltransferase SET7/9 and upregulate the expression levels of H3K4me and p21. This may lead to glomerular cell injury, severe glomerular sclerosis, albuminuria, and a decreased glomerular filtration rate (83). Histone methylation also affects podocyte survival and function. The foot processes of podocytes are attached to the basement membrane of the glomeruli. Foot process effacement and simplification can lead to proteinuria, which is a sign of podocyte injury (84, 85). Adjacent podocytes connect through the slit diaphragm and form an important barrier for glomerular filtration proteins (86). Therefore, the structure and arrangement of podocytes are very important to kidney function (85). PAX transcription activation domain interacting protein (PTIP) is a part of the H3K4 methyltransferase complex (87). Lefevre et al. found that the H3K4 trimethylation (H3K4me3) level declined in PTIP knockout mouse podocytes, which may affect the transcription of the neurotrophic tyrosine kinase receptor type 3 (Ntrk3) gene, resulting in podocyte development disorder and abnormal podocyte arrangement and eventually leading to tubulointerstitial fibrosis and glomerulosclerosis (88, 89). Furthermore, PTIP can interact with dachshund homolog 1 (DACH1) and be recruited by DACH1 to its promoter-binding sites. In podocytes, DACH1-PTIP recruitment can repress transcription, limit promoter H3K4me3, and affect the transcription of downstream genes (89–92). Cao et al. found that DACH1 played a safeguard role in podocytes. DACH1 expression is dramatically decreased in DKD patients, which may result in proteinuria. In DACH1 knockdown podocytes combined with hyperglycemia, DACH1-PTIP promoter binding was reduced, transcriptional repression was lost, and the H3K4me3 expression level was increased (88).
Decreased expression of H3K27me3 in DKD may aggravate podocyte injury and fibrosis. Enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2), a methyltransferase, can regulate podocyte oxidative stress and renal injury in diabetes (93, 94). DKD patients often have metabolic disorders and high levels of AGEs. Liebisch et al. found that in podocytes of diabetic mice, high levels of AGEs can downregulate EZH2 expression levels, decrease H3K27me3 levels, and induce podocyte injury (95). Siddiqi et al. found that in diabetic rats, depletion of EZH2 may decrease H3K27me3 levels and increase glomerular thioredoxin interacting protein (TxnIP) expression levels, which may promote ROS accumulation, increase matrix production, and lead to podocyte injury and proteinuria (96). Similarly, Ye et al. studied the safeguard role of H3K27me3 and EZH2 in a rat DKD model and indicated that in rat renal mesangial cells, TGF-β downregulated the expression of EZH2, decreased the enrichment of the epigenetic repressive mark H3K27me3 at the fibrotic gene promoter (e.g., Serpin family E member 1 (Serpine1) and C-C motif chemokine ligand 2 (Ccl2), and increased fibrosis protein expression and renal fibrosis (97). Ubiquitously transcribed tetratricopeptide repeat on chromosome X (UTX) is a demethylase that removes dimethyl and trimethyl groups from H3K27 (98). UTX expression is increased in the renal tubules of diabetic mice and DKD patients (99). Increased UTX may promote the transcription of inflammatory factor genes and DNA damage. However, administration of the H3K27 demethylase inhibitor GSK-J4 alleviated inflammatory damage to renal tubules in diabetic mice (99).
Glucocorticoid receptor (GR) and mineralocorticoid receptor (MR) play pivotal roles in DKD-associated fibrosis and inflammation. GR and MR are expressed in a variety of renal cells (e.g., podocytes, endothelial cells and fibroblasts). The absence of GR may induce renal fibrosis and albuminuria (100, 101). Overactivation of MR may lead to endothelial dysfunction, renal fibrosis, and renal water and salt metabolism dysfunction (102, 103). Water and salt metabolism disorder is a common metabolic abnormality in DKD patients (104). Disturbance of renal water and salt metabolism may lead to sodium retention, blood pressure elevation, glomerular sclerosis, and tubulointerstitial fibrosis (102). The expression levels of GR and MR are closely related to epigenetic modification. Disruptor of telomeric silencing-1 (Dot1) is a histone lysine methyltransferase whose function and activity are regulated by GR (104). When MR is deficient, GR can modify Dot1 methyltransferase activity through the serum/glucocorticoid-regulated kinase 1 (Sgk-1) and aldosterone (a corticosteroid)-dependent signaling pathways, thereby relaxing chromatin in relevant locations and promoting transcription to compensatively increase epithelial sodium channel expression (104–107). In this way, kidney salt retention can be regulated and the filtration function of the kidney can be ensured.
Histone acetylation usually occurs on lysine residues. Lysine is positively charged, and DNA is negatively charged. Under normal conditions, histone proteins and DNA are tightly bound by interaction. When histone acetylation occurs, acetyl-coenzyme A is transferred to the lysine side chain, which disrupts the interaction between histones and DNA and leads to nucleosome structure relaxation and a subsequent increase in accessibility to transcription factors (108, 109).
Histone acetylation plays an important role in the onset of DKD. Chen et al. found increased acetylation of H3K9 and H3K18 in the renal cortex of diabetic mice, which is related to inflammatory responses and glomerulosclerosis (110). Sufyan et al. found that the increased acetylation of H3K9 and H3K23 is associated with albuminuria and glomerulosclerosis in a mouse model (83). Lizotte et al. found that H3K9/14 acetylation was associated with insulin resistance, podocyte apoptosis and kidney injury (111).
Histone deacetylases (HDACs) are epigenetic regulatory factors that can reverse the histone acetylation process. HDACs can be divided into four groups according to their homology: class I includes HDAC1/2/3/8; class II includes HDAC4/5/6/7/9/10; class III includes sirtuin (SIRT)1-7; and class IV includes HDAC11 (112). Wang et al. found that the expression of HDAC2/4/5 was increased in streptozotocin (STZ)-induced diabetic rats and db/db mice, and the increased expression of HDAC4 exacerbated inflammation and led to podocyte injury (113).
HDAC3, as a profibrotic factor, plays a pivotal role during the genesis of DKD (114). The expression level of HDAC3 is upregulated in renal tubular epithelial cells of DKD mice (115). Klotho protein protects the kidney by regulating the expression of fibrinogen and prevents renal fibrosis by inhibiting profibrotic signaling pathways (e.g., TGF-β/small mothers against decapentaplegic (Smad) and wingless/integrated (Wnt)/β-catenin (115, 116). HDAC3 may modulate the expression of Klotho. Chen et al. found that HDAC3 promotes renal fibrosis by inhibiting the transcription of the antifibrotic protein Klotho (115). HDAC3 also regulates macrophage function, promotes macrophage M2 polarization activation and leads to MMT, which is a marker of renal fibrosis (117, 118). HDAC3 inhibitors can reverse M2 polarization and the phagocytic activity of macrophages and alleviate renal fibrosis (115, 118).
SIRT3 plays a protective role in DKD-related kidney injury. In DKD patients, HG can downregulate SIRT3, which inhibits the activity of antioxidant enzymes, aggravates oxidative stress, induces mitochondrial dysfunction and leads to the accumulation of metabolic substances such as ROS (60, 61). These variations cause a series of changes in kidney cells, including metabolic reprogramming and immunoreaction fibrosis, and eventually induce kidney damage (61, 119–121). Protein Kinase B (AKT) is involved in apoptosis and proliferation by regulating the phosphorylation of forkhead box O (FoxO) (122). High levels of SIRT3 may inhibit the activity of the Akt/FoxO signaling pathway and reduce oxidative stress and renal tubular epithelial cell apoptosis (123). SIRT3 also plays a role in maintaining endothelial cell homeostasis (124). Srivastava et al. reported that SIRT3 is involved in the regulation of EndMT, and SIRT3 deficiency in mouse endothelial cells may induce/aggravate renal fibrosis. However, renal fibrosis can be relieved by the overexpression of SIRT3 (124).
Ubiquitin is a protein with a highly conserved sequence (125). Histone ubiquitination often occurs at specific lysine residues in the C-terminal tails of histone H2A and histone H2B (126). Three enzymes are involved in the process of histone ubiquitination. First, the ubiquitin molecule is activated by E1 (ubiquitin-activating enzyme) in an ATP-dependent manner; then, the activated ubiquitin moiety forms a complex with E2 (ubiquitin-conjugating enzyme) with the assistance of E1, and the complex is transferred to the target protein with the assistance of specific E3 (ubiquitin ligase) (127). The process of ubiquitination is dynamic and reversible, and deubiquitination enzymes can reverse this process (128). The ubiquitin proteasome system is involved in the degradation of many types of proteins, which is associated with the regulation of a series of cell behaviors and the occurrence of diseases (129, 130).
In diabetic patients, ubiquitin A-52 residue ribosomal protein fusion product 1 gene (UbA52), which is associated with renal tubular injury, and the UbA52 expression level can be upregulated in response to increasing concentrations of glucose (131, 132). Abnormal ubiquitination modifications have also been observed in DKD models. Increased H2A ubiquitination and decreased H2B ubiquitination levels have been observed in HG-treated mesangial cells. In addition, these histone ubiquitination changes may enhance the activation of TGF-β and influence the pathogenesis of DKD (126, 133). Histone ubiquitination can regulate the expression of downstream proteins by changing their occupancy in the promoter region and thus promote renal fibrosis. For example, decreased occupancy of H2AK119 monoubiquitination (H2AK119Ub) at the TGF-β and monocyte chemoattractant protein-1 (MCP-1) promoters may upregulate TGF-β pathway-related factors in diabetic kidneys, activate fibrosis-related signals, and accelerate renal fibrosis (134). Intriguingly, histone methylation has been shown to be cross-regulated by histone ubiquitination. Goru et al. found that in diabetic kidneys, decreased occupancies of H2AK119Ub may increase occupancies of histone H3K36 dimethylation (H3K36me2) marks on the promoter of SET7/9 and upregulate the protein SET7/9 expression. Of note, the increased expression level of SET7/9 can increase the promoter occupancies of H3K4me2 on the promoter of collagen type I alpha 1 (COL1A1), which may lead to ECM deposition in the kidney and renal fibrosis (135).
Currently, ubiquitin proteasome system-related proteasome inhibitors have been approved for cancer therapy with good efficacy. However, studies on histone ubiquitination modification in DKD are few, and related drug development remains in the experimental stage. Aspirin and Carbobenzoxyl-L-leucyl-L-leucyl-L-leucine (MG132) are potential proteasome inhibitors. Aspirin can prevent and alleviate renal fibrosis in diabetic animals by increasing histone H2AK119Ub and reducing SET7 deposition-induced ECM (136). MG132 alleviates oxidative stress-induced damage to the kidney by inhibiting diabetes-increased proteasome activity and upregulating Nrf2 (137). Although these drugs are in the preclinical stage, proteasome inhibitors have shown promising therapeutic potential in DKD treatment.
Histone phosphorylation is a central step in chromosome coagulation, transcriptional regulation, and DNA damage repair during cell division (78, 138, 139). In DKD mice and patients, the increase in histone H3 serine 10 (H3Ser10) phosphorylation may upregulate vascular cell adhesion molecule 1 (VCAM-1), promote glomerular endothelial activation, and activate DKD fibrosis and inflammation progression (26, 140). Histone phosphorylation is associated with albuminuria. Sayyed et al. found that glomerulosclerosis and albuminuria were associated with increased H3Ser10 phosphorylation, and the process of histone phosphorylation could be reversed. Ccl2 blockade can prevent the progression of DKD by blocking H3Ser10 phosphorylation (83). Moreover, Tikoo et al. found that resveratrol (a kind of polyphenol) can prevent kidney disease progression by reducing H3 dephosphorylation in diabetic rats (141, 142).
LncRNAs are a class of RNA molecules whose transcript length exceeds 200 nt (143). Instead of encoding proteins, lncRNAs regulate cell behaviors by influencing gene transcription and protein translation (144). LncRNAs are associated with the occurrence and development of a variety of diseases. In DKD, lncRNAs are involved in renal fibrosis, inflammation, podocyte injury, albuminuria and other pathological processes in direct or indirect ways (145).
LncRNAs are crucial during the genesis of DKD. LncRNAs can affect protein expression by targeting microRNAs (miRNAs) and related signaling pathways. miR-96-5P regulates the expression of fibronectin, which is involved in renal fibrosis. It has been observed that the expression level of miR-96-5P is downregulated in HG-treated renal tubular epithelial cells and DKD mouse models (146). LncRNA GAS5 can bind to miR-96-5p and inhibit its expression, thereby promoting renal fibrosis (146). HG may stimulate the expression of lncRNA NR_038323. In STZ-induced DKD mice, lncRNA NR_038323 may interact with miR-324-3p, which upregulates the expression of dual-specificity protein phosphatase-1 (DUSP1), downregulates the expression of collagen I, collagen IV and fibronectin, and significantly improves renal fibrosis and glomerular hypertrophy (147). In the early stage of DKD, the expression level of lncRNA CYP4B1-PS1-001 is significantly downregulated. However, the enforced expression of lncRNA CYP4B1-PS1-001 can inhibit the proliferation and fibrosis of murine mesangial cells by interacting with nucleolin (148, 149). LncRNA SOX2OT can exert renal protective effects by inhibiting renal fibrosis (150, 151). In DKD mice, overexpression of lncRNA SOX2OT may alleviate hyperglycemia, decrease the expression of fibronectin, suppress collagen-related interstitial fibrosis, enhance the autophagy of mesentery cells, and significantly inhibit the proliferation and fibrosis of mesentery cells (150).
LncRNAs are also associated with proteinuria. The expression of lncENST00000436340 is increased in DKD patients. It has been demonstrated that lncENST00000436340 may promote the degradation of polypyrimidine tract binding protein 1 (PTBP1) by enhancing its binding to mRNA, which regulates cytoskeletal rearrangement, and leads to podocyte injury and urine protein (152). LncRNA DLX6-AS1 is highly expressed in DKD patients and podocytes cultured in HG. cAMP-response element binding protein (CREB) can target DLX6-AS1, and overexpression of CREB may increase the level of DLX6-AS1. High levels of DLX6-AS1 may disrupt the podocyte structure, increase kidney inflammation, and induce albuminuria (153).
miRNAs are a class of small and highly conserved noncoding RNAs that regulate protein expression at the posttranscriptional level by interacting directly with the 3’UTR of target genes (154). miRNAs also participate in the pathogenesis of DKD. It has been demonstrated that the expression of miRNA-5b-181p is decreased in a DKD mouse model, and supplementation with miRNA-5b-181p-mimic may reduce albuminuria and alleviate abnormal mesentery expansion (155). Since miRNA can be stably present in urine in the form of exosomes, it can be used as a biomarker to predict the progression of DKD. It has been reported that the expression of miR-342b, miR-30 and miR-2a is significantly increased in the urinary exosomes of DKD patients (156).
miR-33 and miR-21 play significant roles in renal fibrosis. miR-33 can promote fibrosis by activating the TGF-β/Smad inflammatory pathway (157, 158). In a folate-treated mouse model, miR-33 deletion enhanced fatty acid oxidation, reduced lipid accumulation, and protected mouse kidneys from fibrosis (159). miR-21 expression is upregulated in DKD patients. It has been demonstrated that miR-21 in the exosomes of renal tubular cells can target the phosphatase and tensin homolog (PTEN)/AKT pathway and promote renal fibrosis (160). TGF-β/Smad3 mediates the upregulation of miR-21 in renal tubular epithelial cells, which in turn positively regulates the expression of ECM and α-SMA in TECs and fibrotic kidneys (161). The TGF-β/Smad3 pathway also induces the expression of renal tubule collagen I, promotes ECM accumulation and accelerates renal fibrosis by promoting the expression of miR-192 (162, 163).
Fibroblast growth factor receptor 1 (FGFR1) plays a key role in the anti-EndMT process and in reducing kidney fibrosis (164, 165). Koya et al. performed a series of studies on DKD-related EndMT and found that there was EndMT-related crosstalk between miR-29, miR-let-7 family members and FGFR1 (166–168). Overactivation of the TGF-β/Smad signaling pathway may decrease the expression of miR-29, which promotes the transcription of the inflammatory factor IFN-γ and inhibits FGFR1, leading to a downregulation of FGFR1-dependent miR-let-7 (166, 169, 170). The decreased expression of miR-let-7a enhances glycolysis, increases lactic acid and ROS accumulation, turns on metabolic reprogramming and leads to EndMT (54, 55, 167). Furthermore, N-acetyl-seryl-aspartyl-lysyl-proline (AcSDKP) can maintain endothelial homeostasis and protect the kidney from fibrosis by activating FGFR1 and miR-let-7 (167, 171–173).
CircRNAs are a class of single-stranded closed-loop RNAs that mainly exist in the cytoplasm or exosomes. Functionally, circRNAs can interact with proteins and other RNAs by acting as microRNA sponges and regulate transcription in either a competitive or noncompetitive fashion; in some cases, circRNAs can also be translated into polypeptides and perform regulatory functions (174–176).
CircRNA profiles vary with different physiological states, so they can be used as biomarkers and therapeutic targets of diseases. The most common function of circRNAs in DKD is serving as molecular sponges through targeting miRNA and functional proteins, such as SIRT6, SRY-Box Transcription Factor 6 (SOX6), TGF-β1 and NF-κB. CircRNAs are widely involved in DKD-related oxidative stress, inflammation, ECM accumulation and renal fibrosis (177). Qin et al. found that the HG environment can increase the expression levels of circ_0123996 and SOX6 and decrease the expression of miR-203a-3p in mesenchymal cells. Silencing circ_0123996 can suppress cell proliferation and alleviate inflammation and fibrosis (178). Ge et al. found that after exposing mesangial cells to a similar HG environment as in DKD patients, the expression of circ_0000064 was increased (179). Knockdown of circ_0000064 may inhibit the expression of fibrosis-related proteins, such as type I collagen, type IV collagen, and fibronectin (25, 179). Table 1 summarizes the DKD-related circRNAs. Studies of the function of circRNAs in DKD remain at the animal and cell experimental stages, and to date, no circRNA drug has been approved for the clinical treatment of DKD.
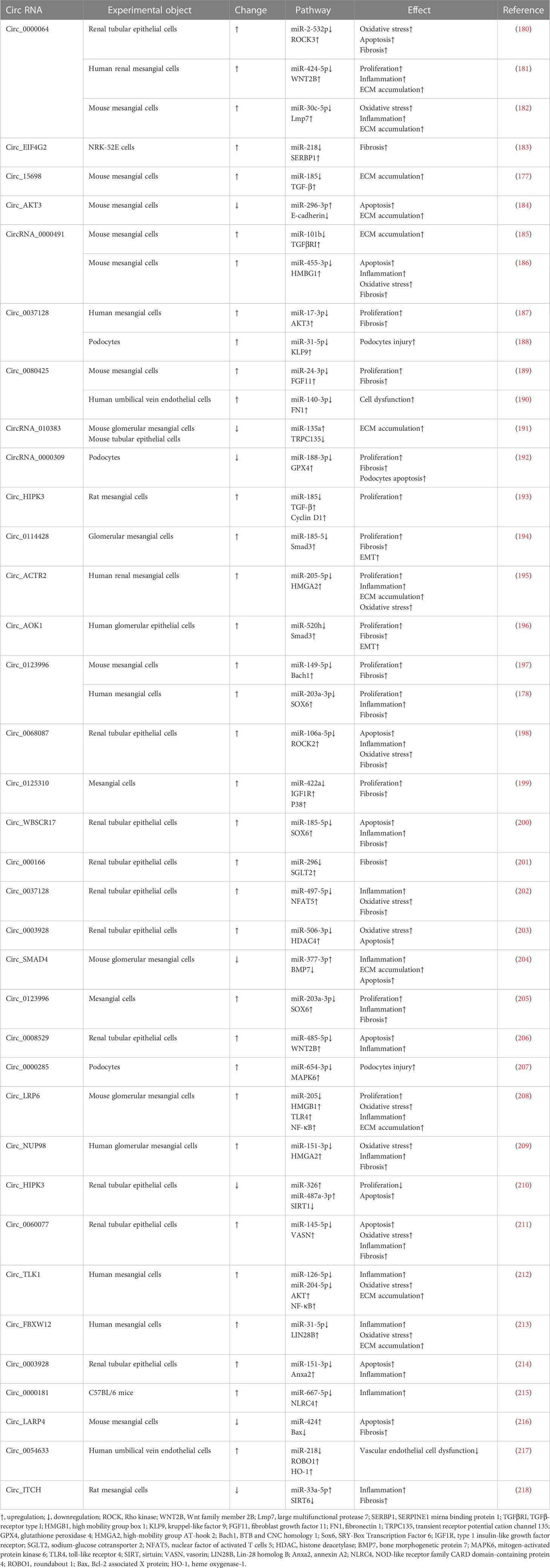
Table 1 DKD related circRNA.
Currently, the main therapeutic strategies for DKD are to alleviate or avoid proteinuria by controlling blood glucose and blood pressure and enhancing renal filtration capacity. Since the direct cause of DKD in diabetic patients is high blood glucose, lowering blood glucose is the priority for controlling the progression of DKD. Some hypoglycemic drugs also have therapeutic effects on renal disorders. For example, SGLT2 inhibitors (e.g., empagliflozin) not only reduce the tubule reabsorption of glucose but also improve the kidney filtration capacity and delay the progression of kidney disease by reducing glomerular pressure and albuminuria (219). Overactivation of the RAAS may trigger glomerular hypertension, which in turn promotes the constriction of bulbar arterioles, damages endothelial cells, and leads to albuminuria. Therefore, the use of antihypertensive drugs can significantly prevent renal dysfunction while maintaining normal blood pressure (220). RAAS inhibitors are widely used drugs for the treatment of DKD and have been proven to be effective in all stages of DKD (220–222). Table 2 summarizes the main regular drugs for DKD treatment.
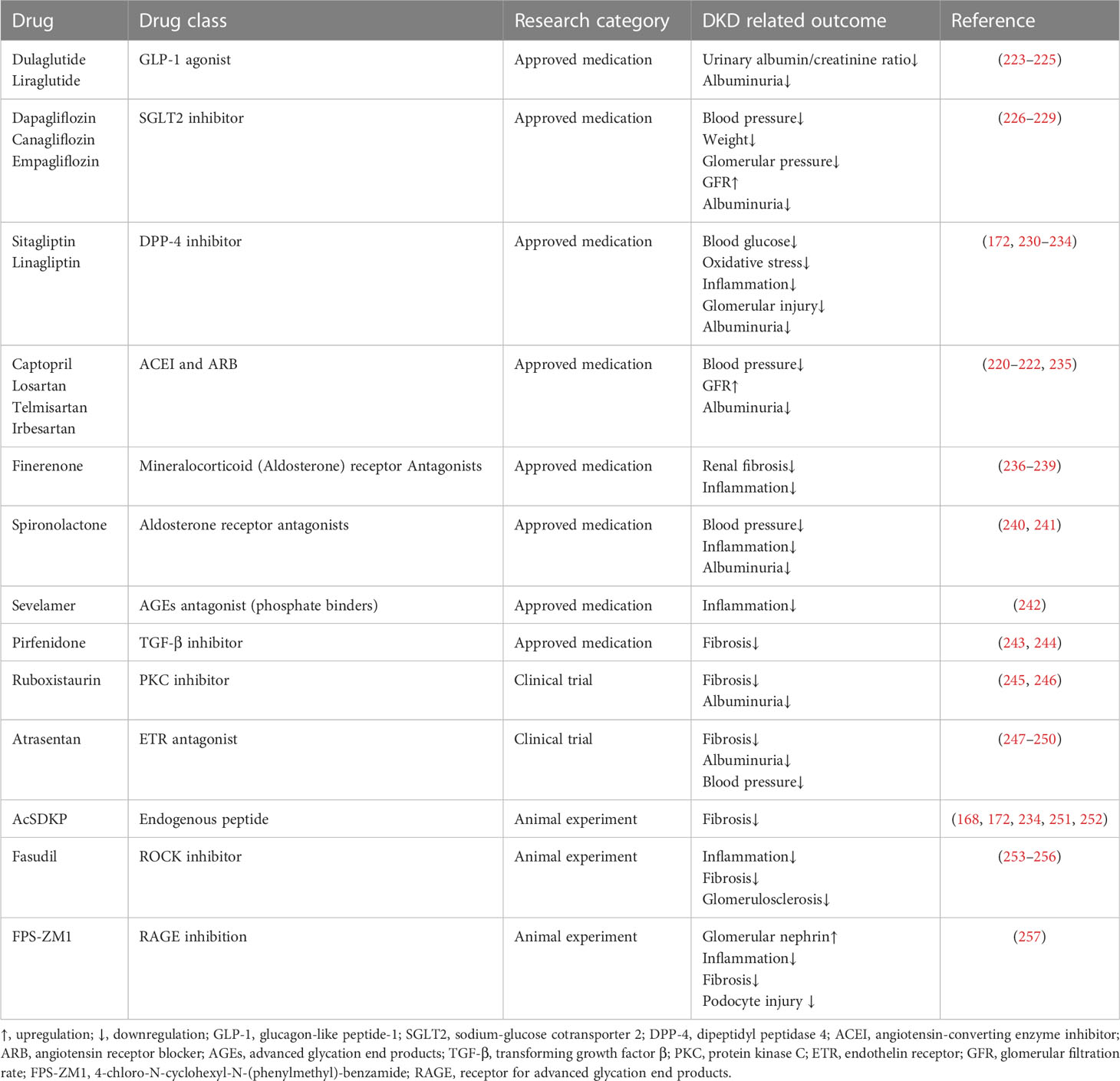
Table 2 Regular therapies of DKD.
Presently, studies of epigenetic drugs for DKD mostly remain at the animal experimental stage, and histone acetylation inhibitors are a research hotspot. We summarized the potential epigenetic therapies for DKD in Table 3. HDACIs have been widely studied in tumors and approved for the treatment of cutaneous T-cell lymphoma and multiple myeloma. HDACIs also have a protective effect against diabetic kidney damage. For example, HDAC2 expression is increased in diabetic rats, and administration of trichostatin A (TSA) may decrease ECM-related protein and mRNA expression and prevent (262). TSA also inhibits the activity of the class II type of HDAC, which plays a similar role in blocking EMT. Xu et al. found that the expression of HDAC5 was increased in the renal tubules of diabetic mice. After TSA administration, the expression of HDAC5 was decreased and the accumulation of ECM was alleviated (264). Valproate (VPA), sodium butyrate (NaB), and vorinostat are all HDACIs that inhibit class I and II HDACs (265). VPA is a branched short-chain fatty acid that can alleviate the damage to renal tubules in STZ-induced diabetic rats, reduce autophagy and stress, reduce proteinuria, and prevent kidney fibrosis (258, 259, 266). NaB is another branched short-chain fatty acid that can reduce inflammation and oxidative damage and relieve albuminuria in diabetic rats (260, 267). Vorinostat can relieve oxidative stress in STZ-induced diabetic rats, and decrease renal tubular cell proliferation and glomerular matrix production (261, 268).
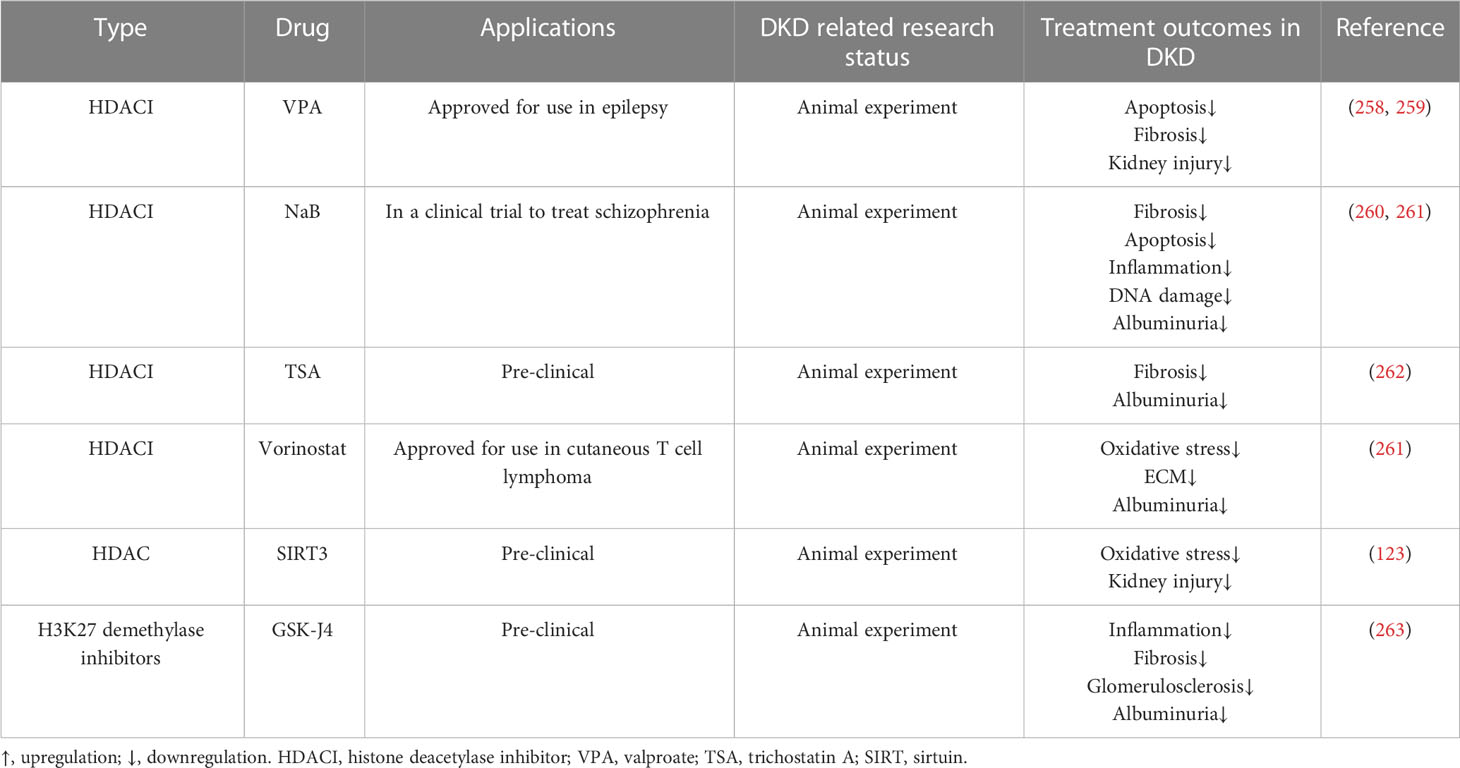
Table 3 Epigenetic therapies of DKD.
Although HDACIs have great potential in the treatment of DKD, their drawbacks, such as adverse effects and poor tolerance, should not be ignored (265, 269, 270). For life-threatening diseases such as cancer, side effects such as nausea, vomiting, and liver toxicity are acceptable. However, whether the application of HDACIs is a good choice for chronic diseases such as DKD should be discussed with great deliberation. In addition, the specificity of HDACIs is poor. Because class I, II and IV HDACs are all dependent on zinc for enzymatic reactions, and most HDACIs target the zinc domain, HDACIs have broad spectrum effects (commonly called pan-HDACIs) (270, 271).
Epigenetic modifications are common in diseases and some epigenetic variations are highly specific in a certain disease or a certain stage of disease, which provides us with potential therapeutic targets in clinical treatments (272, 273). Presently, many studies have confirmed the role of epigenetics in DKD. In this review, we concluded the evidence for epigenetic modifications associated with DKD by summarizing the relevant literature, and we found that epigenetic modifications are involved in the inhibition/activation of a variety of pathogenic signaling pathways. Epigenetic variations affect multiple renal cell functions, such as the activity of GR and glucose metabolism (274, 275). In particular, epigenetic variation-induced EndMT/EMT processes are pivotal in the genesis of DKD, which are the core events in kidney fibrosis (Figure 3). Epigenetic modifications are a consequence of exposure to HG and contribute to the progression of DKD. Since DKD is the result of multiple factors and their complex interactions, different epigenetic modifications may contribute to the same outcome through different signaling pathways and mechanisms. However, most of the existing epigenetic studies have focused on the effect of a single variation on the changes in the signaling pathway to promote or mitigate the occurrence of DKD processes. Therefore, drugs or biomarkers designed for a single target are probably not accurate, and the joint use of multiple epigenetic drugs targeting different epigenetic variations should be considered in future DKD treatment. In addition, most of these studies were conducted in diabetic animals or cell models under HG conditions, but we believe that the human body environment is more complex and that more influential factors and mechanisms should be involved in DKD than animals and cells. Therefore, additional solid experimental and clinical trial data from clinical specimens and patients are eagerly anticipated.
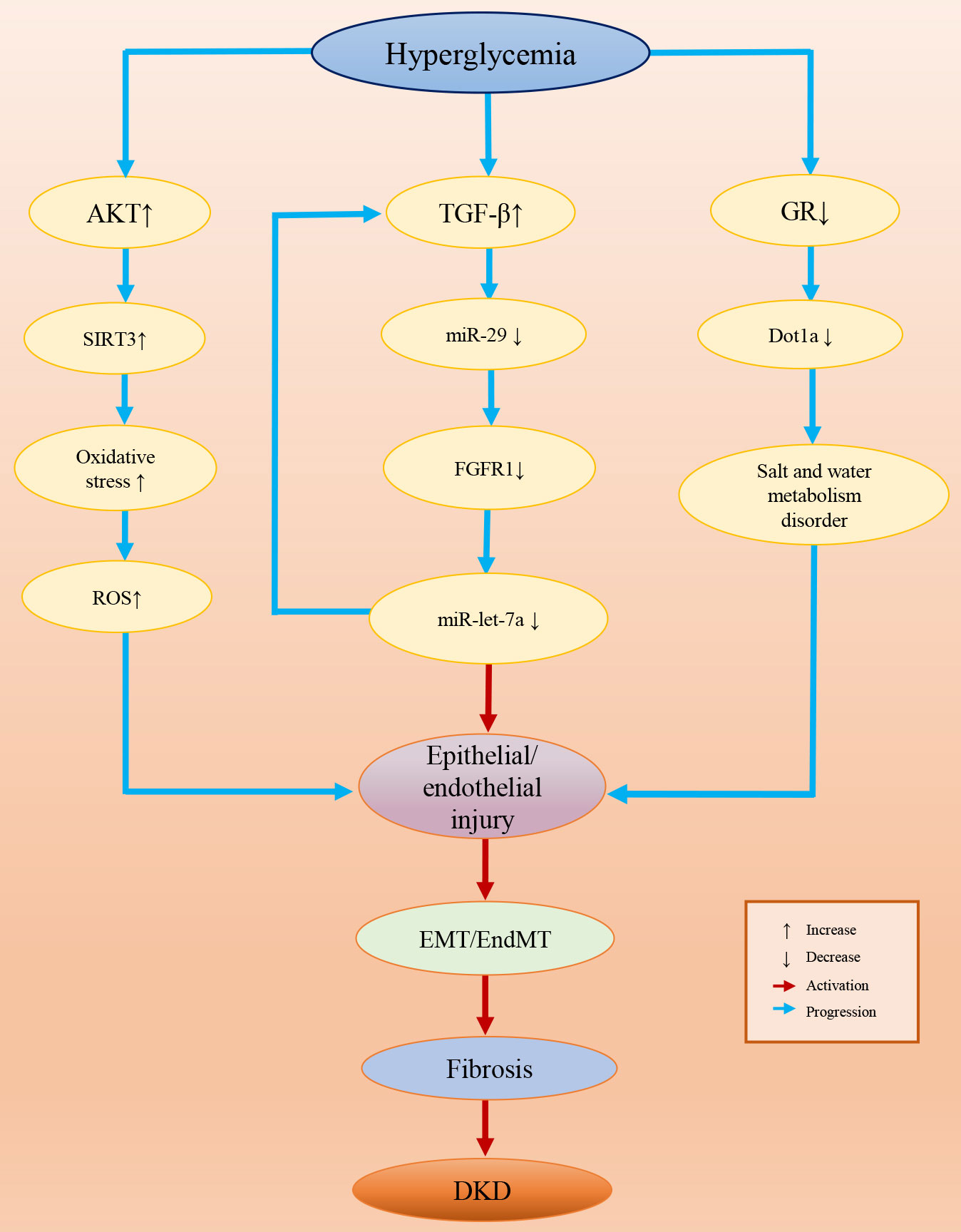
Figure 3 Epigenetic variation-induced EndMT/EMT processes in DKD.
In recent years, epigenetic detection technology has developed rapidly. With the wide application of high-throughput sequencing technology in the clinic, the detection of epigenetic changes (mainly DNA methylation and noncoding RNA profiles) in kidney tissues or the peripheral blood of DKD patients has become easier, faster and cheaper to implement (276, 277). These sequencing results are of great value for the precision diagnosis and drug development of DKD. Moreover, the CRISPR−Cas9 system is being tried as a novel tool for editing a specific epigenetic variation, which is a potential approach for the prevention and treatment of DKD (276, 278, 279).
WS and DY conceived the manuscript. ZL and DY drafted the manuscript. ZL drew the figures. ZL and WW proofread the manuscript and made revisions. LL and XA collected the references. All authors contributed to the article and approved the submitted version.
This work was supported in part by Natural Science Foundation of Jilin Province (20200201428JC to WS and 20210101339JC to WW), Jilin International Collaboration Grant (20220402066GH to DY), the Subject Arrangement Program from Science and Technology Department of Jilin Province (20200201123JC to DY), National Natural Science Foundation of China (82000688 to WW), and Science and technology research project of Jilin Provincial Department of Education (JJKH20211185KJ to WW).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Retnakaran R, Cull CA, Thorne KI, Adler AI, Holman RR, Group US. Risk factors for renal dysfunction in type 2 diabetes: U.K. prospective diabetes study 74. Diabetes (2006) 55(6):1832–9. doi: 10.2337/db05-1620
2. L'Heveder R, Nolan T. International diabetes federation. Diabetes Res Clin Pract (2013) 101(3):349–51. doi: 10.1016/j.diabres.2013.08.003
3. Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, et al. Diabetic kidney disease. Nat Rev Dis Primers (2015) 1:15018. doi: 10.1038/nrdp.2015.18
4. Thomas MC. Epigenetic mechanisms in diabetic kidney disease. Curr Diabetes Rep (2016) 16(3):31. doi: 10.1007/s11892-016-0723-9
5. Simeone CA, Wilkerson JL, Poss AM, Banks JA, Varre JV, Guevara JL, et al. A dominant negative adipoq mutation in a diabetic family with renal disease, hypoadiponectinemia, and hyperceramidemia. NPJ Genom Med (2022) 7(1):43. doi: 10.1038/s41525-022-00314-z
6. Chalmers J, Cooper ME. Ukpds and the legacy effect. N Engl J Med (2008) 359(15):1618–20. doi: 10.1056/NEJMe0807625
7. Reddy MA, Zhang E, Natarajan R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia (2015) 58(3):443–55. doi: 10.1007/s00125-014-3462-y
8. Miao F, Chen Z, Genuth S, Paterson A, Zhang L, Wu X, et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes (2014) 63(5):1748–62. doi: 10.2337/db13-1251
9. Chen Z, Miao F, Paterson AD, Lachin JM, Zhang L, Schones DE, et al. Epigenomic profiling reveals an association between persistence of DNA methylation and metabolic memory in the Dcct/Edic type 1 diabetes cohort. Proc Natl Acad Sci USA (2016) 113(21):E3002–11. doi: 10.1073/pnas.1603712113
10. Theofilis P, Vordoni A, Tsimihodimos V, Kalaitzidis RG. Metabolic dysfunction- associated fatty liver disease in newly diagnosed, treatment-naive hypertensive patients and its association with cardiorenal risk markers. High Blood Press Cardiovasc Prev (2023) 30(1):63–72. doi: 10.1007/s40292-023-00558-5
11. Zhang X, Liu J, Shao S, Yang Y, Qi D, Wang C, et al. Sex differences in the prevalence of and risk factors for abnormal glucose regulation in adults aged 50 years or older with normal fasting plasma glucose levels. Front Endocrinol (Lausanne) (2020) 11:531796. doi: 10.3389/fendo.2020.531796
12. Stenvinkel P, Chertow GM, Devarajan P, Levin A, Andreoli SP, Bangalore S, et al. Chronic inflammation in chronic kidney disease progression: Role of Nrf2. Kidney Int Rep (2021) 6(7):1775–87. doi: 10.1016/j.ekir.2021.04.023
13. Chen Y, Zou H, Lu H, Xiang H, Chen S. Research progress of endothelial-mesenchymal transition in diabetic kidney disease. J Cell Mol Med (2022) 26(12):3313–22. doi: 10.1111/jcmm.17356
14. Duran-Salgado MB, Rubio-Guerra AF. Diabetic nephropathy and inflammation. World J Diabetes (2014) 5(3):393–8. doi: 10.4239/wjd.v5.i3.393
15. Smith MJ, Simmons KM, Cambier JC. B cells in type 1 diabetes mellitus and diabetic kidney disease. Nat Rev Nephrol (2017) 13(11):712–20. doi: 10.1038/nrneph.2017.138
16. Navarro-González JF, Mora-Fernández C. The role of inflammatory cytokines in diabetic nephropathy. J Am Soc Nephrol (2008) 19(3):433–42. doi: 10.1681/asn.2007091048
17. Zheng W, Guo J, Liu ZS. Effects of metabolic memory on inflammation and fibrosis associated with diabetic kidney disease: An epigenetic perspective. Clin Epigenet (2021) 13(1):87. doi: 10.1186/s13148-021-01079-5
18. Moorhead JF, Chan MK, El-Nahas M, Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet (1982) 2(8311):1309–11. doi: 10.1016/s0140-6736(82)91513-6
19. Lin PH, Duann P. Dyslipidemia in kidney disorders: Perspectives on mitochondria homeostasis and therapeutic opportunities. Front Physiol (2020) 11:1050. doi: 10.3389/fphys.2020.01050
20. Jang HS, Noh MR, Kim J, Padanilam BJ. Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front Med (Lausanne) (2020) 7:65. doi: 10.3389/fmed.2020.00065
21. Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest (2011) 121(11):4210–21. doi: 10.1172/JCI45161
22. Wang H, Zhang S, Guo J. Lipotoxic proximal tubular injury: A primary event in diabetic kidney disease. Front Med (Lausanne) (2021) 8:751529. doi: 10.3389/fmed.2021.751529
23. Tan AL, Forbes JM, Cooper ME. Age, rage, and ros in diabetic nephropathy. Semin Nephrol (2007) 27(2):130–43. doi: 10.1016/j.semnephrol.2007.01.006
24. Li JH, Wang W, Huang XR, Oldfield M, Schmidt AM, Cooper ME, et al. Advanced glycation end products induce tubular epithelial-myofibroblast transition through the rage- Erk1/2 map kinase signaling pathway. Am J Pathol (2004) 164(4):1389–97. doi: 10.1016/S0002-9440(10)63225-7
25. Xu Y, Nie L, Yin YG, Tang JL, Zhou JY, Li DD, et al. Resveratrol protects against hyperglycemia-induced oxidative damage to mitochondria by activating Sirt1 in rat mesangial cells. Toxicol Appl Pharmacol (2012) 259(3):395–401. doi: 10.1016/j.taap.2011.09.028
26. Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, Garcia-Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol (2011) 7(6):327–40. doi: 10.1038/nrneph.2011.51
27. Rabbani N, Thornalley PJ. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int (2018) 93(4):803–13. doi: 10.1016/j.kint.2017.11.034
28. Nilsson S, Makela S, Treuter E, Tujague M, Thomsen J, Andersson G, et al. Mechanisms of estrogen action. Physiol Rev (2001) 81(4):1535–65. doi: 10.1152/physrev.2001.81.4.1535
29. Dhindsa S, Reddy A, Karam JS, Bilkis S, Chaurasia A, Mehta A, et al. Prevalence of subnormal testosterone concentrations in men with type 2 diabetes and chronic kidney disease. Eur J Endocrinol (2015) 173(3):359–66. doi: 10.1530/EJE-15-0359
30. Vikan T, Schirmer H, Njolstad I, Svartberg J. Low testosterone and sex hormone- binding globulin levels and high estradiol levels are independent predictors of type 2 diabetes in men. Eur J Endocrinol (2010) 162(4):747–54. doi: 10.1530/EJE-09-0943
31. Zhou X, Liu Z, Ying K, Wang H, Liu P, Ji X, et al. Wj-39, an aldose reductase inhibitor, ameliorates renal lesions in diabetic nephropathy by activating Nrf2 signaling. Oxid Med Cell Longev (2020) 2020:7950457. doi: 10.1155/2020/7950457
32. Kang Q, Yang C. Oxidative stress and diabetic retinopathy: Molecular mechanisms, pathogenetic role and therapeutic implications. Redox Biol (2020) 37:101799. doi: 10.1016/j.redox.2020.101799
33. Ma X, Ma J, Leng T, Yuan Z, Hu T, Liu Q, et al. Advances in oxidative stress in pathogenesis of diabetic kidney disease and efficacy of tcm intervention. Ren Fail (2023) 45(1):2146512. doi: 10.1080/0886022X.2022.2146512
34. Wu L, Liu C, Chang DY, Zhan R, Zhao M, Man Lam S, et al. The attenuation of diabetic nephropathy by annexin A1 Via regulation of lipid metabolism through the Ampk/Pparalpha/Cpt1b pathway. Diabetes (2021) 70(10):2192–203. doi: 10.2337/db21-0050
35. Alshehri AS. Kaempferol attenuates diabetic nephropathy in streptozotocin-induced diabetic rats by a hypoglycaemic effect and concomitant activation of the nrf-2/Ho- 1/Antioxidants axis. Arch Physiol Biochem (2021), 1–14. doi: 10.1080/13813455.2021.1890129
36. Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The orphan nuclear receptor rorgammat directs the differentiation program of proinflammatory il-17+ t helper cells. Cell (2006) 126(6):1121–33. doi: 10.1016/j.cell.2006.07.035
37. Moon JY, Jeong KH, Lee TW, Ihm CG, Lim SJ, Lee SH. Aberrant recruitment and activation of t cells in diabetic nephropathy. Am J Nephrol (2012) 35(2):164–74. doi: 10.1159/000334928
38. Lei L, Mao Y, Meng D, Zhang X, Cui L, Huo Y, et al. Percentage of circulating Cd8+ t lymphocytes is associated with albuminuria in type 2 diabetes mellitus. Exp Clin Endocrinol Diabetes (2014) 122(1):27–30. doi: 10.1055/s-0033-1358666
39. Eller K, Kirsch A, Wolf AM, Sopper S, Tagwerker A, Stanzl U, et al. Potential role of regulatory t cells in reversing obesity-linked insulin resistance and diabetic nephropathy. Diabetes (2011) 60(11):2954–62. doi: 10.2337/db11-0358
40. Liu G, Ma H, Qiu L, Li L, Cao Y, Ma J, et al. Phenotypic and functional switch of macrophages induced by regulatory Cd4+Cd25+ t cells in mice. Immunol Cell Biol (2011) 89(1):130–42. doi: 10.1038/icb.2010.70
41. Zhang C, Xiao C, Wang P, Xu W, Zhang A, Li Q, et al. The alteration of Th1/Th2/Th17/Treg paradigm in patients with type 2 diabetes mellitus: Relationship with diabetic nephropathy. Hum Immunol (2014) 75(4):289–96. doi: 10.1016/j.humimm.2014.02.007
42. Theodorou K, Boon RA. Endothelial cell metabolism in atherosclerosis. Front Cell Dev Biol (2018) 6:82. doi: 10.3389/fcell.2018.00082
43. Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, et al. Epithelial notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest (2010) 120(11):4040–54. doi: 10.1172/JCI43025
44. Zhang Q, Xiao X, Zheng J, Li M, Yu M, Ping F, et al. Qishen yiqi dripping pill protects against diabetic nephropathy by inhibiting the Wnt/Beta-catenin and transforming growth factor-Beta/Smad signaling pathways in rats. Front Physiol (2020) 11:613324. doi: 10.3389/fphys.2020.613324
45. Wilmer WA, Dixon CL, Hebert C. Chronic exposure of human mesangial cells to high glucose environments activates the P38 mapk pathway. Kidney Int (2001) 60(3):858–71. doi: 10.1046/j.1523-1755.2001.060003858.x
46. Cargnello M, Roux PP. Activation and function of the mapks and their substrates, the mapk-activated protein kinases. Microbiol Mol Biol Rev (2011) 75(1):50–83. doi: 10.1128/MMBR.00031-10
47. Ying Q, Wu G. Molecular mechanisms involved in podocyte emt and concomitant diabetic kidney diseases: An update. Ren Fail (2017) 39(1):474–83. doi: 10.1080/0886022X.2017.1313164
48. Tuttle KR. Back to the future: Glomerular hyperfiltration and the diabetic kidney. Diabetes (2016) 66(1):14–6. doi: 10.2337/dbi16-0056
49. Kang AK, Miller JA. Effects of gender on the renin-angiotensin system, blood pressure, and renal function. Curr Hypertens Rep (2002) 4(2):143–51. doi: 10.1007/s11906-002-0039-9
50. Sparks MA, Crowley SD, Gurley SB, Mirotsou M, Coffman TM. Classical renin- angiotensin system in kidney physiology. Compr Physiol (2014) 4(3):1201–28. doi: 10.1002/cphy.c130040
51. Labandeira-Garcia JL, Rodriguez-Pallares J, Villar-Cheda B, Rodriguez-Perez AI, Garrido-Gil P, Guerra MJ. Aging, angiotensin system and dopaminergic degeneration in the substantia nigra. Aging Dis (2011) 2(3):257–74.
52. Rianto F, Hoang T, Revoori R, Sparks MA. Angiotensin receptors in the kidney and vasculature in hypertension and kidney disease. Mol Cell Endocrinol (2021) 529:111259. doi: 10.1016/j.mce.2021.111259
53. Miller JA, Anacta LA, Cattran DC. Impact of gender on the renal response to angiotensin ii. Kidney Int (1999) 55(1):278–85. doi: 10.1046/j.1523-1755.1999.00260.x
54. Bao MH, Zhang YW, Lou XY, Cheng Y, Zhou HH. Protective effects of let-7a and let- 7b on oxidized low-density lipoprotein induced endothelial cell injuries. PloS One (2014) 9(9):e106540. doi: 10.1371/journal.pone.0106540
55. Chae U, Kim HS, Lee HS, Lee SR, Lee DS. Drp1-dependent mitochondrial fission regulates P62-mediated autophagy in lps-induced activated microglial cells. Biosci Biotechnol Biochem (2019) 83(3):409–16. doi: 10.1080/09168451.2018.1549933
56. Gaur P, Prasad S, Kumar B, Sharma SK, Vats P. High-altitude hypoxia induced reactive oxygen species generation, signaling, and mitigation approaches. Int J Biometeorol (2021) 65(4):601–15. doi: 10.1007/s00484-020-02037-1
57. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, et al. Metabolic regulation of gene expression by histone lactylation. Nature (2019) 574(7779):575–80. doi: 10.1038/s41586-019-1678-1
58. Lee DY, Kim JY, Ahn E, Hyeon JS, Kim GH, Park KJ, et al. Associations between local acidosis induced by renal ldha and renal fibrosis and mitochondrial abnormalities in patients with diabetic kidney disease. Transl Res (2022) 249:88–109. doi: 10.1016/j.trsl.2022.06.015
59. Wu Y, Tang L, Huang H, Yu Q, Hu B, Wang G, et al. Phosphoglycerate dehydrogenase activates Pkm2 to phosphorylate histone H3t11 and attenuate cellular senescence. Nat Commun (2023) 14(1):1323. doi: 10.1038/s41467-023-37094-8
60. Wang Z, Zhai J, Zhang T, He L, Ma S, Zuo Q, et al. Canagliflozin ameliorates epithelial- mesenchymal transition in high-salt diet-induced hypertensive renal injury through restoration of sirtuin 3 expression and the reduction of oxidative stress. Biochem Biophys Res Commun (2023) 653:53–61. doi: 10.1016/j.bbrc.2023.01.084
61. Morita M, Kanasaki K. Sodium-glucose cotransporter-2 inhibitors for diabetic kidney disease: Targeting warburg effects in proximal tubular cells. Diabetes Metab (2020) 46(5):353–61. doi: 10.1016/j.diabet.2020.06.005
62. Zhang Z, Liang W, Luo Q, Hu H, Yang K, Hu J, et al. Pfkp activation ameliorates foot process fusion in podocytes in diabetic kidney disease. Front Endocrinol (Lausanne) (2021) 12:797025. doi: 10.3389/fendo.2021.797025
63. Wang Z, Hao D, Fang D, Yu J, Wang X, Qin G. Transcriptome analysis reveal candidate genes and pathways responses to lactate dehydrogenase inhibition (Oxamate) in hyperglycemic human renal proximal epithelial tubular cells. Front Endocrinol (Lausanne) (2022) 13:785605. doi: 10.3389/fendo.2022.785605
64. Wang Z, Nielsen PM, Laustsen C, Bertelsen LB. Metabolic consequences of lactate dehydrogenase inhibition by oxamate in hyperglycemic proximal tubular cells. Exp Cell Res (2019) 378(1):51–6. doi: 10.1016/j.yexcr.2019.03.001
65. Moore LD, Le T, Fan G. DNA Methylation and its basic function. Neuropsychopharmacology (2013) 38(1):23–38. doi: 10.1038/npp.2012.112
66. Liu Y, Wen D, Ho C, Yu L, Zheng D, O'Reilly S, et al. Epigenetics as a versatile regulator of fibrosis. J Transl Med (2023) 21(1):164. doi: 10.1186/s12967-023-04018-5
67. Mattei AL, Bailly N, Meissner A. DNA methylation: A historical perspective. Trends Genet (2022) 38(7):676–707. doi: 10.1016/j.tig.2022.03.010
68. Lassén E, Daehn IS. Molecular mechanisms in early diabetic kidney disease: Glomerular endothelial cell dysfunction. Int J Mol Sci (2020) 21(24):9456. doi: 10.3390/ijms21249456
69. Lu Z, Liu N, Wang F. Epigenetic regulations in diabetic nephropathy. J Diabetes Res (2017) 2017:7805058.
70. Prattichizzo F, Giuliani A, Ceka A, Rippo MR, Bonfigli AR, Testa R, et al. Epigenetic mechanisms of endothelial dysfunction in type 2 diabetes. Clin Epigenet (2015) 7(1):56. doi: 10.1186/s13148-015-0090-4
71. VanderJagt TA, Neugebauer MH, Morgan M, Bowden DW, Shah VO. Epigenetic profiles of pre-diabetes transitioning to type 2 diabetes and nephropathy. World J Diabetes (2015) 6(9):1113–21. doi: 10.4239/wjd.v6.i9.1113
72. Marumo T, Yagi S, Kawarazaki W, Nishimoto M, Ayuzawa N, Watanabe A, et al. Diabetes induces aberrant DNA methylation in the proximal tubules of the kidney. J Am Soc Nephrol (2015) 26(10):2388–97. doi: 10.1681/ASN.2014070665
73. Park J, Guan Y, Sheng X, Gluck C, Seasock MJ, Hakimi AA, et al. Functional methylome analysis of human diabetic kidney disease. JCI Insight (2019) 4(11):e128886. doi: 10.1172/jci.insight.128886
74. Maghbooli Z, Larijani B, Emamgholipour S, Amini M, Keshtkar A, Pasalar P. Aberrant DNA methylation patterns in diabetic nephropathy. J Diabetes Metab Disord (2014) 13:69. doi: 10.1186/2251-6581-13-69
75. Das P, Veazey KJ, Van HT, Kaushik S, Lin K, Lu Y, et al. Histone methylation regulator ptip is required to maintain normal and leukemic bone marrow niches. Proc Natl Acad Sci USA (2018) 115(43):E10137–E46. doi: 10.1073/pnas.1806019115
76. Lecamwasam A, Novakovic B, Meyer B, Ekinci EI, Dwyer KM, Saffery R. DNA methylation profiling identifies epigenetic differences between early versus late stages of diabetic chronic kidney disease. Nephrol Dial Transplant (2021) 36(11):2027–38. doi: 10.1093/ndt/gfaa226
77. Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature (1997) 389(6648):251–60. doi: 10.1038/38444
78. Wang S, Zhang X, Wang Q, Wang R. Histone modification in podocyte injury of diabetic nephropathy. J Mol Med (Berl) (2022) 100(10):1373–86. doi: 10.1007/s00109-022-02247-7
79. Strahl BD, Allis CD. The language of covalent histone modifications. Nature (2000) 403(6765):41–5. doi: 10.1038/47412
80. Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell (1999) 98(3):285–94. doi: 10.1016/s0092-8674(00)81958-3
81. Chen Y, Ren B, Yang J, Wang H, Yang G, Xu R, et al. The role of histone methylation in the development of digestive cancers: A potential direction for cancer management. Signal Transduct Target Ther (2020) 5(1):143. doi: 10.1038/s41392-020-00252-1
82. Greer EL, Shi Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat Rev Genet (2012) 13(5):343–57. doi: 10.1038/nrg3173
83. Sayyed SG, Gaikwad AB, Lichtnekert J, Kulkarni O, Eulberg D, Klussmann S, et al. Progressive glomerulosclerosis in type 2 diabetes is associated with renal histone H3k9 and H3k23 acetylation, H3k4 dimethylation and phosphorylation at serine 10. Nephrol Dial Transplant (2010) 25(6):1811–7. doi: 10.1093/ndt/gfp730
84. Lee JH, Jang SH, Cho NJ, Heo NH, Gil HW, Lee EY, et al. Severity of foot process effacement is associated with proteinuria in patients with iga nephropathy. Kidney Res Clin Pract (2020) 39(3):295–304. doi: 10.23876/j.krcp.20.017
85. Hu YF, Tan Y, Yu XJ, Wang H, Wang SX, Yu F, et al. Podocyte involvement in renal thrombotic microangiopathy: A clinicopathological study. Am J Nephrol (2020) 51(9):752–60. doi: 10.1159/000510141
86. Kawachi H, Fukusumi Y. New insight into podocyte slit diaphragm, a therapeutic target of proteinuria. Clin Exp Nephrol (2020) 24(3):193–204. doi: 10.1007/s10157-020-01854-3
87. Kim D, Patel SR, Xiao H, Dressler GR. The role of ptip in maintaining embryonic stem cell pluripotency. Stem Cells (2009) 27(7):1516–23. doi: 10.1002/stem.79
88. Cao A, Li J, Asadi M, Basgen JM, Zhu B, Yi Z, et al. Dach1 protects podocytes from experimental diabetic injury and modulates ptip-H3k4me3 activity. J Clin Invest (2021) 131(10):e141279. doi: 10.1172/JCI141279
89. Lefevre GM, Patel SR, Kim D, Tessarollo L, Dressler GR. Altering a histone H3k4 methylation pathway in glomerular podocytes promotes a chronic disease phenotype. PloS Genet (2010) 6(10):e1001142. doi: 10.1371/journal.pgen.1001142
90. Takeshita T, Yamamoto-Ibusuki M, Yamamoto Y, Omoto Y, Honda Y, Iyama K, et al. Ptip associated protein 1, Pa1, is an independent prognostic factor for lymphnode negative breast cancer. PloS One (2013) 8(11):e80552. doi: 10.1371/journal.pone.0080552
91. Patel SR, Kim D, Levitan I, Dressler GR. The brct-domain containing protein ptip links Pax2 to a histone H3, lysine 4 methyltransferase complex. Dev Cell (2007) 13(4):580–92. doi: 10.1016/j.devcel.2007.09.004
92. Cho EA, Prindle MJ, Dressler GR. Brct domain-containing protein ptip is essential for progression through mitosis. Mol Cell Biol (2003) 23(5):1666–73. doi: 10.1128/MCB.23.5.1666-1673.2003
93. Zhou X, Zang X, Ponnusamy M, Masucci MV, Tolbert E, Gong R, et al. Enhancer of zeste homolog 2 inhibition attenuates renal fibrosis by maintaining Smad7 and phosphatase and tensin homolog expression. J Am Soc Nephrol (2016) 27(7):2092–108. doi: 10.1681/ASN.2015040457
94. Lin P, Qiu F, Wu M, Xu L, Huang D, Wang C, et al. Salvianolic acid b attenuates tubulointerstitial fibrosis by inhibiting Ezh2 to regulate the Pten/Akt pathway. Pharm Biol (2023) 61(1):23–9. doi: 10.1080/13880209.2022.2148169
95. Liebisch M, Wolf G. Age-induced suppression of Ezh2 mediates injury of podocytes by reducing H3k27me3. Am J Nephrol (2020) 51(9):676–92. doi: 10.1159/000510140
96. Siddiqi FS, Majumder S, Thai K, Abdalla M, Hu P, Advani SL, et al. The histone methyltransferase enzyme enhancer of zeste homolog 2 protects against podocyte oxidative stress and renal injury in diabetes. J Am Soc Nephrol (2016) 27(7):2021–34. doi: 10.1681/ASN.2014090898
97. Jia Y, Reddy MA, Das S, Oh HJ, Abdollahi M, Yuan H, et al. Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-Beta1-Induced gene expression in mesangial cells and diabetic kidney. J Biol Chem (2019) 294(34):12695–707. doi: 10.1074/jbc.RA119.007575
98. Hong S, Cho YW, Yu LR, Yu H, Veenstra TD, Ge K. Identification of jmjc domain- containing utx and Jmjd3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA (2007) 104(47):18439–44. doi: 10.1073/pnas.0707292104
99. Chen H, Huang Y, Zhu X, Liu C, Yuan Y, Su H, et al. Histone demethylase utx is a therapeutic target for diabetic kidney disease. J Physiol (2019) 597(6):1643–60. doi: 10.1113/JP277367
100. Srivastava SP, Zhou H, Setia O, Liu B, Kanasaki K, Koya D, et al. Loss of endothelial glucocorticoid receptor accelerates diabetic nephropathy. Nat Commun (2021) 12(1):2368. doi: 10.1038/s41467-021-22617-y
101. Srivastava SP, Zhou H, Setia O, Dardik A, Fernandez-Hernando C, Goodwin J. Podocyte glucocorticoid receptors are essential for glomerular endothelial cell homeostasis in diabetes mellitus. J Am Heart Assoc (2021) 10(15):e019437. doi: 10.1161/JAHA.120.019437
102. Gonzalez-Juanatey JR, Gorriz JL, Ortiz A, Valle A, Soler MJ, Facila L. Cardiorenal benefits of finerenone: Protecting kidney and heart. Ann Med (2023) 55(1):502–13. doi: 10.1080/07853890.2023.2171110
103. DuPont JJ, Jaffe IZ. 30 years of the mineralocorticoid receptor: The role of the mineralocorticoid receptor in the vasculature. J Endocrinol (2017) 234(1):T67–82. doi: 10.1530/JOE-17-0009
104. Bartlett AA, Lapp HE, Hunter RG. Epigenetic mechanisms of the glucocorticoid receptor. Trends Endocrinol Metab (2019) 30(11):807–18. doi: 10.1016/j.tem.2019.07.003
105. Kone BC. Epigenetics and the control of the collecting duct epithelial sodium channel. Semin Nephrol (2013) 33(4):383–91. doi: 10.1016/j.semnephrol.2013.05.010
106. Zhang D, Yu ZY, Cruz P, Kong Q, Li S, Kone BC. Epigenetics and the control of epithelial sodium channel expression in collecting duct. Kidney Int (2009) 75(3):260–7. doi: 10.1038/ki.2008.475
107. Zhang X, Zhou Q, Chen L, Berger S, Wu H, Xiao Z, et al. Mineralocorticoid receptor antagonizes Dot1a-Af9 complex to increase alphaenac transcription. Am J Physiol Renal Physiol (2013) 305(10):F1436–44. doi: 10.1152/ajprenal.00202.2013
108. Jenuwein T, Allis CD. Translating the histone code. Science (2001) 293(5532):1074–80. doi: 10.1126/science.1063127
109. Millan-Zambrano G, Burton A, Bannister AJ, Schneider R. Histone post-translational modifications - cause and consequence of genome function. Nat Rev Genet (2022) 23(9):563–80. doi: 10.1038/s41576-022-00468-7
110. Chen H, Li J, Jiao L, Petersen RB, Li J, Peng A, et al. Apelin inhibits the development of diabetic nephropathy by regulating histone acetylation in akita mouse. J Physiol (2014) 592(3):505–21. doi: 10.1113/jphysiol.2013.266411
111. Lizotte F, Denhez B, Guay A, Gevry N, Cote AM, Geraldes P. Persistent insulin resistance in podocytes caused by epigenetic changes of shp-1 in diabetes. Diabetes (2016) 65(12):3705–17. doi: 10.2337/db16-0254
112. Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science (1996) 272(5260):408–11. doi: 10.1126/science.272.5260.408
113. Wang X, Liu J, Zhen J, Zhang C, Wan Q, Liu G, et al. Histone deacetylase 4 selectively contributes to podocyte injury in diabetic nephropathy. Kidney Int (2014) 86(4):712–25. doi: 10.1038/ki.2014.111
114. Zhang L, Cao W. Histone deacetylase 3 (Hdac3) as an important epigenetic regulator of kidney diseases. J Mol Med (Berl) (2022) 100(1):43–51. doi: 10.1007/s00109-021-02141-8
115. Chen F, Gao Q, Wei A, Chen X, Shi Y, Wang H, et al. Histone deacetylase 3 aberration inhibits klotho transcription and promotes renal fibrosis. Cell Death Differ (2021) 28(3):1001–12. doi: 10.1038/s41418-020-00631-9
116. Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, et al. Klotho inhibits transforming growth factor-Beta1 (Tgf-Beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J Biol Chem (2011) 286(10):8655–65. doi: 10.1074/jbc.M110.174037
117. Casalena G, Daehn I, Bottinger E. Transforming growth factor-beta, bioenergetics, and mitochondria in renal disease. Semin Nephrol (2012) 32(3):295–303. doi: 10.1016/j.semnephrol.2012.04.009
118. Yang J, Yin S, Bi F, Liu L, Qin T, Wang H, et al. Timap repression by tgfbeta and Hdac3- associated smad signaling regulates macrophage M2 phenotypic phagocytosis. J Mol Med (Berl) (2017) 95(3):273–85. doi: 10.1007/s00109-016-1479-z
119. Murugasamy K, Munjal A, Sundaresan NR. Emerging roles of Sirt3 in cardiac metabolism. Front Cardiovasc Med (2022) 9:850340. doi: 10.3389/fcvm.2022.850340
120. Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, et al. Sirt3 opposes reprogramming of cancer cell metabolism through Hif1alpha destabilization. Cancer Cell (2011) 19(3):416–28. doi: 10.1016/j.ccr.2011.02.014
121. Srivastava SP, Li J, Kitada M, Fujita H, Yamada Y, Goodwin JE, et al. Sirt3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis (2018) 9(10):997. doi: 10.1038/s41419-018-1057-0
122. Zhang X, Tang N, Hadden TJ, Rishi AK. Akt, foxo and regulation of apoptosis. Biochim Biophys Acta (2011) 1813(11):1978–86. doi: 10.1016/j.bbamcr.2011.03.010
123. Jiao XC, Li Y, Zhang T, Liu MD, Chi YQ. Role of Sirtuin3 in high glucose-induced apoptosis in renal tubular epithelial cells. Biochem Bioph Res Co (2016) 480(3):387–93. doi: 10.1016/j.bbrc.2016.10.060
124. Srivastava SP, Li J, Takagaki Y, Kitada M, Goodwin JE, Kanasaki K, et al. Endothelial Sirt3 regulates myofibroblast metabolic shifts in diabetic kidneys. iScience (2021) 24(5):102390. doi: 10.1016/j.isci.2021.102390
125. Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem (2001) 70:503–33. doi: 10.1146/annurev.biochem.70.1.503
126. Gao CL, Chen G, Liu L, Li X, He JH, Jiang L, et al. Impact of high glucose and proteasome inhibitor Mg132 on histone H2a and H2b ubiquitination in rat glomerular mesangial cells. J Diabetes Res (2013) 2013:589474. doi: 10.1155/2013/589474
127. Hershko A, Ciechanover A, Varshavsky A. The ubiquitin system. Nat Med (2000) 6(10):1073–81. doi: 10.1038/80384
128. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med (2014) 20(11):1242–53. doi: 10.1038/nm.3739
129. Rajan V, Mitch WE. Ubiquitin, proteasomes and proteolytic mechanisms activated by kidney disease. Biochim Biophys Acta (2008) 1782(12):795–9. doi: 10.1016/j.bbadis.2008.07.007
130. Asmamaw MD, Liu Y, Zheng YC, Shi XJ, Liu HM. Skp2 in the ubiquitin-proteasome system: A comprehensive review. Med Res Rev (2020) 40(5):1920–49. doi: 10.1002/med.21675
131. Dihazi H, Muller GA, Lindner S, Meyer M, Asif AR, Oellerich M, et al. Characterization of diabetic nephropathy by urinary proteomic analysis: Identification of a processed ubiquitin form as a differentially excreted protein in diabetic nephropathy patients. Clin Chem (2007) 53(9):1636–45. doi: 10.1373/clinchem.2007.088260
132. Sun L, Pan XM, Wada J, Haas CS, Wuthrich RP, Danesh FR, et al. Isolation and functional analysis of mouse Uba52 gene and its relevance to diabetic nephropathy (Retraction of vol 277, pg 29953, 2002). J Biol Chem (2019) 294(26):10382–. doi: 10.1074/jbc.W119.009588
133. Asano Y, Ihn H, Yamane K, Kubo M, Tamaki K. Impaired Smad7-Smurf-Mediated negative regulation of tgf-beta signaling in scleroderma fibroblasts. J Clin Invest (2004) 113(2):253–64. doi: 10.1172/JCI16269
134. Pandey A, Goru SK, Kadakol A, Malek V, Sharma N, Gaikwad AB. H2ak119 monoubiquitination regulates angiotensin ii receptor mediated macrophage infiltration and renal fibrosis in type 2 diabetic rats. Biochimie (2016) 131:68–76. doi: 10.1016/j.biochi.2016.09.016
135. Goru SK, Kadakol A, Pandey A, Malek V, Sharma N, Gaikwad AB. Histone H2ak119 and H2bk120 mono-ubiquitination modulate Set7/9 and Suv39h1 in type 1 diabetes-induced renal fibrosis. Biochem J (2016) 473(21):3937–49. doi: 10.1042/BCJ20160595
136. Goru SK, Gaikwad AB. Novel reno-protective mechanism of aspirin involves H2ak119 monoubiquitination and Set7 in preventing type 1 diabetic nephropathy. Pharmacol Rep (2018) 70(3):497–502. doi: 10.1016/j.pharep.2017.11.018
137. Cui W, Li B, Bai Y, Miao X, Chen Q, Sun W, et al. Potential role for Nrf2 activation in the therapeutic effect of Mg132 on diabetic nephropathy in Ove26 diabetic mice. Am J Physiol Endocrinol Metab (2013) 304(1):E87–99. doi: 10.1152/ajpendo.00430.2012
138. Rossetto D, Avvakumov N, Côté J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics (2012) 7(10):1098–108. doi: 10.4161/epi.21975
139. Bahl S, Seto E. Regulation of histone deacetylase activities and functions by phosphorylation and its physiological relevance. Cell Mol Life Sci (2021) 78(2):427–45. doi: 10.1007/s00018-020-03599-4
140. Alghamdi TA, Batchu SN, Hadden MJ, Yerra VG, Liu Y, Bowskill BB, et al. Histone H3 serine 10 phosphorylation facilitates endothelial activation in diabetic kidney disease. Diabetes (2018) 67(12):2668–81. doi: 10.2337/db18-0124
141. Tikoo K, Singh K, Kabra D, Sharma V, Gaikwad A. Change in histone H3 phosphorylation, map kinase P38, sir 2 and P53 expression by resveratrol in preventing streptozotocin induced type i diabetic nephropathy. Free Radic Res (2008) 42(4):397–404. doi: 10.1080/10715760801998646
142. Berman AY, Motechin RA, Wiesenfeld MY, Holz MK. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis Oncol (2017) 1:35. doi: 10.1038/s41698-017-0038-6
143. Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The gencode V7 catalog of human long noncoding rnas: Analysis of their gene structure, evolution, and expression. Genome Res (2012) 22(9):1775–89. doi: 10.1101/gr.132159.111
144. St Laurent G, Wahlestedt C, Kapranov P. The landscape of long noncoding rna classification. Trends Genet (2015) 31(5):239–51. doi: 10.1016/j.tig.2015.03.007
145. Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding rna function in development and disease. Cell Mol Life Sci (2016) 73(13):2491–509. doi: 10.1007/s00018-016-2174-5
146. Wang W, Jia YJ, Yang YL, Xue M, Zheng ZJ, Wang L, et al. Lncrna Gas5 exacerbates renal tubular epithelial fibrosis by acting as a competing endogenous rna of mir-96-5p. BioMed Pharmacother (2020) 121:109411. doi: 10.1016/j.biopha.2019.109411
147. Ge Y, Wang J, Wu D, Zhou Y, Qiu S, Chen J, et al. Lncrna Nr_038323 suppresses renal fibrosis in diabetic nephropathy by targeting the mir-324-3p/Dusp1 axis. Mol Ther Nucleic Acids (2019) 17:741–53. doi: 10.1016/j.omtn.2019.07.007
148. Wang S, Chen X, Wang M, Yao D, Chen T, Yan Q, et al. Long non-coding rna Cyp4b1- Ps1-001 inhibits proliferation and fibrosis in diabetic nephropathy by interacting with nucleolin. Cell Physiol Biochem (2018) 49(6):2174–87. doi: 10.1159/000493821
149. Wang M, Wang S, Yao D, Yan Q, Lu W. A novel long non-coding rna Cyp4b1-Ps1- 001 regulates proliferation and fibrosis in diabetic nephropathy. Mol Cell Endocrinol (2016) 426:136–45. doi: 10.1016/j.mce.2016.02.020
150. Chen K, Yu B, Liao J. Lncrna Sox2ot alleviates mesangial cell proliferation and fibrosis in diabetic nephropathy Via Akt/Mtor-mediated autophagy. Mol Med (2021) 27(1):71. doi: 10.1186/s10020-021-00310-6
151. Zhang X, Shang J, Wang X, Cheng G, Jiang Y, Liu D, et al. Microarray analysis reveals long Non-Coding rna Sox2ot as a novel candidate regulator in diabetic nephropathy. Mol Med Rep (2018) 18(6):5058–68. doi: 10.3892/mmr.2018.9534
152. Hu J, Wang Q, Fan X, Zhen J, Wang C, Chen H, et al. Long noncoding rna Enst00000436340 promotes podocyte injury in diabetic kidney disease by facilitating the association of Ptbp1 with Rab3b. Cell Death Dis (2023) 14(2):130. doi: 10.1038/s41419-023-05658-7
153. Zheng W, Guo J, Lu X, Qiao Y, Liu D, Pan S, et al. Camp-response element binding protein mediates podocyte injury in diabetic nephropathy by targeting lncrna Dlx6-As1. Metabolism (2022) 129:155155. doi: 10.1016/j.metabol.2022.155155
154. Srivastava SP, Hedayat AF, Kanasaki K, Goodwin JE. Microrna crosstalk influences epithelial-to-Mesenchymal, endothelial-to-Mesenchymal, and macrophage-to-Mesenchymal transitions in the kidney. Front Pharmacol (2019) 10:904. doi: 10.3389/fphar.2019.00904
155. Ishii H, Kaneko S, Yanai K, Aomatsu A, Hirai K, Ookawara S, et al. Microrna expression profiling in diabetic kidney disease. Transl Res (2021) 237:31–52. doi: 10.1016/j.trsl.2021.05.008
156. Eissa S, Matboli M, Bekhet MM. Clinical verification of a novel urinary microrna panal: 133b, -342 and -30 as biomarkers for diabetic nephropathy identified by bioinformatics analysis. BioMed Pharmacother (2016) 83:92–9. doi: 10.1016/j.biopha.2016.06.018
157. Yu B, Li W, Al F, Chen Z. Microrna-33a deficiency inhibits proliferation and fibrosis through inactivation of tgf-Beta/Smad pathway in human cardiac fibroblasts. Pharmazie (2017) 72(8):456–60. doi: 10.1691/ph.2017.7561
158. Pezzolesi MG, Satake E, McDonnell KP, Major M, Smiles AM, Krolewski AS. Circulating tgf-Beta1-Regulated mirnas and the risk of rapid progression to esrd in type 1 diabetes. Diabetes (2015) 64(9):3285–93. doi: 10.2337/db15-0116
159. Price NL, Miguel V, Ding W, Singh AK, Malik S, Rotllan N, et al. Genetic deficiency or pharmacological inhibition of mir-33 protects from kidney fibrosis. JCI Insight (2019) 4(22):e131102. doi: 10.1172/jci.insight.131102
160. Zhao S, Li W, Yu W, Rao T, Li H, Ruan Y, et al. Exosomal mir-21 from tubular cells contributes to renal fibrosis by activating fibroblasts Via targeting pten in obstructed kidneys. Theranostics (2021) 11(18):8660–73. doi: 10.7150/thno.62820
161. Zhong X, Chung AC, Chen HY, Meng XM, Lan HY. Smad3-mediated upregulation of mir-21 promotes renal fibrosis. J Am Soc Nephrol (2011) 22(9):1668–81. doi: 10.1681/ASN.2010111168
162. Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi JJ, et al. Microrna-192 in diabetic kidney glomeruli and its function in tgf-Beta-Induced collagen expression Via inhibition of e-box repressors. Proc Natl Acad Sci USA (2007) 104(9):3432–7. doi: 10.1073/pnas.0611192104
163. Chung AC, Huang XR, Meng X, Lan HY. Mir-192 mediates tgf-Beta/Smad3-Driven renal fibrosis. J Am Soc Nephrol (2010) 21(8):1317–25. doi: 10.1681/ASN.2010020134
164. Li J, Shi S, Srivastava SP, Kitada M, Nagai T, Nitta K, et al. Fgfr1 is critical for the anti- endothelial mesenchymal transition effect of n-Acetyl-Seryl-Aspartyl-Lysyl-Proline Via induction of the Map4k4 pathway. Cell Death Dis (2017) 8(8):e2965. doi: 10.1038/cddis.2017.353
165. Pan JA, Lin H, Yu JY, Zhang HL, Zhang JF, Wang CQ, et al. Mir-21-3p inhibits adipose browning by targeting Fgfr1 and aggravates atrial fibrosis in diabetes. Oxid Med Cell Longev (2021) 2021:9987219. doi: 10.1155/2021/9987219
166. Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The microrna mir-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon- gamma. Nat Immunol (2011) 12(9):861–9. doi: 10.1038/ni.2073
167. Srivastava SP, Shi S, Kanasaki M, Nagai T, Kitada M, He J, et al. Effect of antifibrotic micrornas crosstalk on the action of n-Acetyl-Seryl-Aspartyl-Lysyl-Proline in diabetes- related kidney fibrosis. Sci Rep (2016) 6:29884. doi: 10.1038/srep29884
168. Srivastava SP, Goodwin JE, Kanasaki K, Koya D. Metabolic reprogramming by n- acetyl-Seryl-Aspartyl-Lysyl-Proline protects against diabetic kidney disease. Br J Pharmacol (2020) 177(16):3691–711. doi: 10.1111/bph.15087
169. Chen PY, Qin L, Barnes C, Charisse K, Yi T, Zhang X, et al. Fgf regulates tgf-beta signaling and endothelial-to-Mesenchymal transition Via control of let-7 mirna expression. Cell Rep (2012) 2(6):1684–96. doi: 10.1016/j.celrep.2012.10.021
170. Wang B, Komers R, Carew R, Winbanks CE, Xu B, Herman-Edelstein M, et al. Suppression of microrna-29 expression by tgf-Beta1 promotes collagen expression and renal fibrosis. J Am Soc Nephrol (2012) 23(2):252–65. doi: 10.1681/ASN.2011010055
171. Hu Q, Li J, Nitta K, Kitada M, Nagai T, Kanasaki K, et al. Fgfr1 is essential for n-acetyl- seryl-Aspartyl-Lysyl-Proline regulation of mitochondrial dynamics by upregulating microrna let-7b-5p. Biochem Biophys Res Commun (2018) 495(3):2214–20. doi: 10.1016/j.bbrc.2017.12.089
172. Kanasaki K, Nagai T, Nitta K, Kitada M, Koya D. N-Acetyl-Seryl-Aspartyl-Lysyl-Proline: A valuable endogenous anti-fibrotic peptide for combating kidney fibrosis in diabetes. Front Pharmacol (2014) 5:70. doi: 10.3389/fphar.2014.00070
173. Nagai T, Kanasaki M, Srivastava SP, Nakamura Y, Ishigaki Y, Kitada M, et al. N-acetyl- seryl-Aspartyl-Lysyl-Proline inhibits diabetes-associated kidney fibrosis and endothelial- mesenchymal transition. BioMed Res Int (2014) 2014:696475. doi: 10.1155/2014/696475
174. Jin J, Sun H, Shi C, Yang H, Wu Y, Li W, et al. Circular rna in renal diseases. J Cell Mol Med (2020) 24(12):6523–33. doi: 10.1111/jcmm.15295
175. Ivanov A, Memczak S, Wyler E, Torti F, Porath HT, Orejuela MR, et al. Analysis of intron sequences reveals hallmarks of circular rna biogenesis in animals. Cell Rep (2015) 10(2):170–7. doi: 10.1016/j.celrep.2014.12.019
176. Tu C, Wang L, Wei L, Jiang Z. The role of circular rna in diabetic nephropathy. Int J Med Sci (2022) 19(5):916–23. doi: 10.7150/ijms.71648
177. Hu W, Han Q, Zhao L, Wang L. Circular rna Circrna_15698 aggravates the extracellular matrix of diabetic nephropathy mesangial cells Via mir-185/Tgf-Beta1. J Cell Physiol (2019) 234(2):1469–76. doi: 10.1002/jcp.26959
178. Qin Y, Xu Y, Peng H, Cao M, Zhao K, Zhu Y. Circ_0123996 promotes the proliferation, inflammation, and fibrosis of mesangial cells by sponging mir-203a-3p to upregulate Sox6 in diabetic nephropathy. J Biochem Mol Toxicol (2022) 36(11):e23139. doi: 10.1002/jbt.23139
179. Ge X, Xi L, Wang Q, Li H, Xia L, Cang Z, et al. Circular rna Circ_0000064 promotes the proliferation and fibrosis of mesangial cells Via mir-143 in diabetic nephropathy. Gene (2020) 758:144952. doi: 10.1016/j.gene.2020.144952
180. Wang H, Huang S, Hu T, Fei S, Zhang H. Circ_0000064 promotes high glucose-induced renal tubular epithelial cells injury to facilitate diabetic nephropathy progression through mir-532-3p/Rock1 axis. BMC Endocr Disord (2022) 22(1):67. doi: 10.1186/s12902-022-00968-x
181. Li J, Min Y, Zhao Q. Circ_0000064 knockdown attenuates high glucose-induced proliferation, inflammation and extracellular matrix deposition of mesangial cells through mir-424-5p-Mediated Wnt2b inhibition in cell models of diabetic nephropathy. Clin Exp Nephrol (2022) 26(10):943–54. doi: 10.1007/s10157-022-02241-w
182. Sun L, Han Y, Shen C, Luo H, Wang Z. Emodin alleviates high glucose-induced oxidative stress, inflammation and extracellular matrix accumulation of mesangial cells by the Circ_0000064/Mir-30c-5p/Lmp7 axis. J Receptors Signal Transduction (2022) 42(3):302–12. doi: 10.1080/10799893.2021.1933028
183. Xu B, Wang Q, Li W, Xia L, Ge X, Shen L, et al. Circular rna Circeif4g2 aggravates renal fibrosis in diabetic nephropathy by sponging mir-218. J Cell Mol Med (2022) 26(6):1799–805. doi: 10.1111/jcmm.16129
184. Tang B, Li W, Ji T-T, Li X-Y, Qu X, Feng L, et al. Circ-Akt3 inhibits the accumulation of extracellular matrix of mesangial cells in diabetic nephropathy Via modulating mir-296- 3p/E-cadherin signals. J Cell Mol Med (2020) 24(15):8779–88. doi: 10.1111/jcmm.15513
185. Mou X, Chen JW, Zhou DY, Liu K, Chen LJ, Zhou D, et al. A novel identified circular rna, Circ_0000491, aggravates the extracellular matrix of diabetic nephropathy glomerular mesangial cells through suppressing mir-101b by targeting tgf beta ri. Mol Med Rep (2020) 22(5):3785–94. doi: 10.3892/mmr.2020.11486
186. Wang J, Yang S, Li W, Zhao M, Li K. Circ_0000491 promotes apoptosis, inflammation, oxidative stress, and fibrosis in high glucose-induced mesangial cells by regulating mir- 455-3p/Hmgb1 axis. Nephron (2022) 146(1):72–83. doi: 10.1159/000516870
187. Wang Q, Cang Z, Shen L, Peng W, Xi L, Jiang X, et al. Circ_0037128/Mir-17-3p/Akt3 axis promotes the development of diabetic nephropathy. Gene (2021) 765:145076. doi: 10.1016/j.gene.2020.145076
188. Fang R, Cao X, Zhu Y, Chen Q. Hsa_Circ_0037128 aggravates high glucose-induced podocytes injury in diabetic nephropathy through mediating mir-31-5p/Klf9. Autoimmunity (2022) 55(4):254–63. doi: 10.1080/08916934.2022.2037128
189. Liu H, Wang X, Wang Z-Y, Li L. Circ_0080425 inhibits cell proliferation and fibrosis in diabetic nephropathy Via sponging mir-24-3p and targeting fibroblast growth factor 11. J Cell Physiol (2020) 235(5):4520–9. doi: 10.1002/jcp.29329
190. Zhang L, Jin G, Zhang W, Wang X, Li Z, Dong Q. Silencing Circ_0080425 alleviates high-Glucose-Induced endothelial cell dysfunction in diabetic nephropathy by targeting mir-140-3p/Fn1 axis. Clin Exp Nephrol (2023) 27(1):12–23. doi: 10.1007/s10157-022-02273-2
191. Peng F, Gong W, Li S, Yin B, Zhao C, Liu W, et al. Circrna_010383 acts as a sponge for mir-135a, and its downregulated expression contributes to renal fibrosis in diabetic nephropathy. Diabetes (2021) 70(2):603–15. doi: 10.2337/db20-0203
192. Jin J, Wang Y, Zheng D, Liang M. He q. a novel identified circular rna, Mmu_Mmu_Circrna_0000309, involves in germacrone-mediated improvement of diabetic nephropathy through regulating ferroptosis by targeting mir-188-3p/Gpx4 signaling axis. Antioxidants Redox Signaling (2022) 36(10-12):740–59. doi: 10.1089/ars.2021.0063
193. Liu R, Zhang M, Ge Y. Circular rna Hipk3 exacerbates diabetic nephropathy and promotes proliferation by sponging mir-185. Gene (2021) 765:145065. doi: 10.1016/j.gene.2020.145065
194. Li B, Sun G, Yu H, Meng J, Wei F. Circ_0114428 promotes proliferation, fibrosis and emt process of high glucose-induced glomerular mesangial cells through regulating the mir-185-5p/Smad3 axis. Autoimmunity (2022) 55(7):462–72. doi: 10.1080/08916934.2022.2103797
195. Yun J, Ren J, Liu Y, Dai L, Song L, Ma X, et al. Circ-Actr2 aggravates the high glucose- induced cell dysfunction of human renal mesangial cells through mediating the mir-205- 5p/Hmga2 axis in diabetic nephropathy. Diabetol Metab Syndrome (2021) 13(1):72. doi: 10.1186/s13098-021-00692-x
196. Li B, Sun G, Yu H, Meng J, Wei F. Exosomal Circtaok1 contributes to diabetic kidney disease progression through regulating Smad3 expression by sponging mir-520h. Int Urol Nephrol (2022) 54(9):2343–54. doi: 10.1007/s11255-022-03139-y
197. Wang W, Feng J, Zhou H, Li Q. Circ_0123996 promotes cell proliferation and fibrosis in mouse mesangial cells through sponging mir-149-5p and inducing Bach1 expression. Gene (2020) 761:144971. doi: 10.1016/j.gene.2020.144971
198. Feng F, Yang J, Wang G, Huang P, Li Y, Zhou B. Circ_0068087 promotes high glucose- induced human renal tubular cell injury through regulating mir-106a-5p/Rock2 pathway. Nephron (2022). doi: 10.1159/000525440
199. Zhu Y, Zha F, Tang B, Ji TT, Li XY, Feng L, et al. Exosomal Hsa_Circ_0125310 promotes cell proliferation and fibrosis in diabetic nephropathy Via sponging mir-422a and targeting the Igf1r/P38 axis. J Cell Mol Med (2022) 26(1):151–62. doi: 10.1111/jcmm.17065
200. Li G, Qin Y, Qin S, Zhou X, Zhao W, Zhang D. Circ_Wbscr17 aggravates inflammatory responses and fibrosis by targeting mir-185-5p/Sox6 regulatory axis in high glucose- induced human kidney tubular cells. Life Sci (2020) 259:118269. doi: 10.1016/j.lfs.2020.118269
201. Chen S. Circ_000166/Mir-296 aggravates the process of diabetic renal fibrosis by regulating the Sglt2 signaling pathway in renal tubular epithelial cells. Dis Markers (2022) 2022:6103086. doi: 10.1155/2022/6103086
202. Feng T, Li W, Li T, Jiao W, Chen S. Circular Rna_0037128 aggravates high glucose- induced damage in hk-2 cells Via regulation of microrna-497-5p/Nuclear factor of activated t cells 5 axis. Bioengineered (2021) 12(2):10959–70. doi: 10.1080/21655979.2021.2001912
203. Liu Q, Cui Y, Ding N, Zhou C. Knockdown of Circ_0003928 ameliorates high glucose- induced dysfunction of human tubular epithelial cells through the mir-506-3p/Hdac4 pathway in diabetic nephropathy. Eur J Med Res (2022) 27(1):55. doi: 10.1186/s40001-022-00679-y
204. Wu R, Niu Z, Ren G, Ruan L, Sun L. Circsmad4 alleviates high glucose-induced inflammation, extracellular matrix deposition and apoptosis in mouse glomerulus mesangial cells by relieving mir-377-3p-Mediated Bmp7 inhibition. Diabetol Metab Syndr (2021) 13(1):137. doi: 10.1186/s13098-021-00753-1
205. Qin Y, Xu Y, Peng H, Cao M, Zhao K, Zhu Y. Circ_0123996 promotes the proliferation, inflammation, and fibrosis of mesangial cells by sponging mir-203a-3p to upregulate Sox6 in diabetic nephropathy. J Biochem Mol Toxicol (2022). doi: 10.1002/jbt.23139
206. Wang W, Lu H. High glucose-induced human kidney cell apoptosis and inflammatory injury are alleviated by Circ_0008529 knockdown Via Circ_0008529-mediated mir-485-5p/Wnt2b signaling. Appl Biochem Biotechnol (2022). doi: 10.1007/s12010-022-04088-z
207. Yao T, Zha D, Hu C, Wu X. Circ_0000285 promotes podocyte injury through sponging mir-654-3p and activating Mapk6 in diabetic nephropathy. Gene (2020) 747:144661. doi: 10.1016/j.gene.2020.144661
208. Chen B, Li Y, Liu Y, Xu Z. Circlrp6 regulates high glucose-induced proliferation, oxidative stress, ecm accumulation, and inflammation in mesangial cells. J Cell Physiol (2019) 234(11):21249–59. doi: 10.1002/jcp.28730
209. Dong Q, Dong L, Zhu Y, Wang X, Li Z, Zhang L. Circular ribonucleic acid nucleoporin 98 knockdown alleviates high glucose-induced proliferation, fibrosis, inflammation and oxidative stress in human glomerular mesangial cells by regulating the microribonucleic acid-151-3p-High mobility group at-hook 2 axis. J Diabetes Investig (2022) 13(8):1303–15. doi: 10.1111/jdi.13821
210. Zhuang L, Wang Z, Hu X, Yang Q, Pei X, Jin G. Circhipk3 alleviates high glucose toxicity to human renal tubular epithelial hk-2 cells through regulation of mir-326/Mir- 487a-3p/Sirt1. Diabetes Metab Syndrome Obesity-Targets Ther (2021) 14:729–40. doi: 10.2147/dmso.S289624
211. Zhou J, Peng X, Ru Y, Xu J. Circ_0060077 knockdown alleviates high-Glucose-Induced cell apoptosis, oxidative stress, inflammation and fibrosis in hk-2 cells Via mir-145- 5p/Vasn pathway. Inflammation (2022). doi: 10.1007/s10753-022-01649-6
212. Qiu B, Qi X, Wang J. Circtlk1 downregulation attenuates high glucose-induced human mesangial cell injury by blocking the Akt/Nf-kappa b pathway through sponging mir-126- 5p/Mir-204-5p. Biochem Genet (2022) 60(5):1471–87. doi: 10.1007/s10528-021-10146-8
213. Sun A, Sun N, Liang X, Hou Z. Circ-Fbxw12 aggravates the development of diabetic nephropathy by binding to mir-31-5p to induce Lin28b. Diabetol Metab Syndrome (2021) 13(1):141. doi: 10.1186/s13098-021-00757-x
214. An L, Ji D, Hu W, Wang J, Jin X, Qu Y, et al. Interference of Hsa_Circ_0003928 alleviates high glucose-induced cell apoptosis and inflammation in hk-2 cells Via mir-151- 3p/Anxa2. Diabetes Metab Syndrome Obesity-Targets Ther (2020) 13:3157– 68. doi: 10.2147/dmso.S265543
215. Li Y, Yu W, Xiong H, Yuan F. Circ_0000181 regulates mir-667-5p/Nlrc4 axis to promote pyroptosis progression in diabetic nephropathy. Sci Rep (2022) 12(1):11994. doi: 10.1038/s41598-022-15607-7
216. Wang Y, Qi Y, Ji T, Tang B, Li X, Zheng P, et al. Circ_Larp4 regulates high glucose- induced cell proliferation, apoptosis, and fibrosis in mouse mesangial cells. Gene (2021) 765:145114. doi: 10.1016/j.gene.2020.145114
217. Pan L, Lian W, Zhang X, Han S, Cao C, Li X, et al. Human circular rna-0054633 regulates high glucose-induced vascular endothelial cell dysfunction through the microrna-218/Roundabout 1 and microrna-218/Heme oxygenase-1 axes. Int J Mol Med (2018) 42(1):597–606. doi: 10.3892/ijmm.2018.3625
218. Liu J, Duan P, Xu C, Xu D, Liu Y, Jiang J. Circrna circ-itch improves renal inflammation and fibrosis in streptozotocin-induced diabetic mice by regulating the mir-33a-5p/Sirt6 axis. Inflamm Res (2021) 70(7):835–46. doi: 10.1007/s00011-021-01485-8
219. Colbert GB, Madariaga HM, Gaddy A, Elrggal ME, Lerma EV. Empagliflozin in adults with chronic kidney disease (Ckd): Current evidence and place in therapy. Ther Clin Risk Manag (2023) 19:133–42. doi: 10.2147/TCRM.S398163
220. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting- enzyme inhibition on diabetic nephropathy. the collaborative study group. N Engl J Med (1993) 329(20):1456–62. doi: 10.1056/NEJM199311113292004
221. Parving HH, Lehnert H, Brochner-Mortensen J, Gomis R, Andersen S, Arner P. [Effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes]. Ugeskr Laeger (2001) 163(40):5519–24.
222. Makino H, Haneda M, Babazono T, Moriya T, Ito S, Iwamoto Y, et al. Prevention of transition from incipient to overt nephropathy with telmisartan in patients with type 2 diabetes. Diabetes Care (2007) 30(6):1577–8. doi: 10.2337/dc06-1998
223. Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. Dulaglutide and renal outcomes in type 2 diabetes: An exploratory analysis of the rewind randomised, placebo-controlled trial. Lancet (2019) 394(10193):131–8. doi: 10.1016/S0140-6736(19)31150-X
224. de Boer IH. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med (2017) 377(22):2198. doi: 10.1056/NEJMc1713042
225. Hanefeld M, Arteaga JM, Leiter LA, Marchesini G, Nikonova E, Shestakova M, et al. Efficacy and safety of lixisenatide in patients with type 2 diabetes and renal impairment. Diabetes Obes Metab (2017) 19(11):1594–601. doi: 10.1111/dom.12986
226. Laffel LM, Danne T, Klingensmith GJ, Tamborlane WV, Willi S, Zeitler P, et al. Efficacy and safety of the Sglt2 inhibitor empagliflozin versus placebo and the dpp-4 inhibitor linagliptin versus placebo in young people with type 2 diabetes (Dinamo): A multicentre, randomised, double-blind, parallel group, phase 3 trial. Lancet Diabetes Endocrinol (2023) 11(3):169–81. doi: 10.1016/S2213-8587(22)00387-4
227. Nincevic V, Omanovic Kolaric T, Roguljic H, Kizivat T, Smolic M, Bilic Curcic I. Renal benefits of sglt 2 inhibitors and glp-1 receptor agonists: Evidence supporting a paradigm shift in the medical management of type 2 diabetes. Int J Mol Sci (2019) 20(23):5831. doi: 10.3390/ijms20235831
228. Norgaard SA, Briand F, Sand FW, Galsgaard ED, Sondergaard H, Sorensen DB, et al. Nephropathy in diabetic Db/Db mice is accelerated by high protein diet and improved by the Sglt2 inhibitor dapagliflozin. Eur J Pharmacol (2019) 860:172537. doi: 10.1016/j.ejphar.2019.172537
229. Garofalo C, Borrelli S, Liberti ME, Andreucci M, Conte G, Minutolo R, et al. Sglt2 inhibitors: Nephroprotective efficacy and side effects. Medicina (Kaunas) (2019) 55(6):268. doi: 10.3390/medicina55060268
230. Gupta S, Sen U. More than just an enzyme: Dipeptidyl peptidase-4 (Dpp-4) and its association with diabetic kidney remodelling. Pharmacol Res (2019) 147:104391. doi: 10.1016/j.phrs.2019.104391
231. Schernthaner G, Mogensen CE, Schernthaner GH. The effects of glp-1 analogues, dpp- 4 inhibitors and Sglt2 inhibitors on the renal system. Diabetes Vasc Dis Res (2014) 11(5):306–23. doi: 10.1177/1479164114542802
232. Marques C, Goncalves A, Pereira PMR, Almeida D, Martins B, Fontes-Ribeiro C, et al. The dipeptidyl peptidase 4 inhibitor sitagliptin improves oxidative stress and ameliorates glomerular lesions in a rat model of type 1 diabetes. Life Sci (2019) 234:116738. doi: 10.1016/j.lfs.2019.116738
233. Kanasaki K. The pathological significance of dipeptidyl peptidase-4 in endothelial cell homeostasis and kidney fibrosis. Diabetol Int (2016) 7(3):212–20. doi: 10.1007/s13340-016-0281-z
234. Kanasaki K. N-Acetyl-Seryl-Aspartyl-Lysyl-Proline is a valuable endogenous antifibrotic peptide for kidney fibrosis in diabetes: An update and translational aspects. J Diabetes Investig (2020) 11(3):516–26. doi: 10.1111/jdi.13219
235. Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med (2001) 345(12):861–9. doi: 10.1056/NEJMoa011161
236. Bakris GL, Agarwal R, Chan JC, Cooper ME, Gansevoort RT, Haller H, et al. Effect of finerenone on albuminuria in patients with diabetic nephropathy: A randomized clinical trial. JAMA (2015) 314(9):884–94. doi: 10.1001/jama.2015.10081
237. Dey S, Garg J, Wang A, Holzner E, Frishman WH, Aronow WS. Finerenone: Efficacy of a new nonsteroidal mineralocorticoid receptor antagonist in treatment of patients with chronic kidney disease and type 2 diabetes. Cardiol Rev (2023). doi: 10.1097/CRD.0000000000000548
238. Kim DL, Lee SE, Kim NH. Renal protection of mineralocorticoid receptor antagonist, finerenone, in diabetic kidney disease. Endocrinol Metab (Seoul) (2023) 38(1):43–55. doi: 10.3803/EnM.2022.1629
239. Filippatos G, Anker SD, Pitt B, Rossing P, Joseph A, Kolkhof P, et al. Finerenone and heart failure outcomes by kidney Function/Albuminuria in chronic kidney disease and diabetes. JACC Heart Fail (2022) 10(11):860–70. doi: 10.1016/j.jchf.2022.07.013
240. Esteghamati A, Noshad S, Jarrah S, Mousavizadeh M, Khoee SH, Nakhjavani M. Long- term effects of addition of mineralocorticoid receptor antagonist to angiotensin ii receptor blocker in patients with diabetic nephropathy: A randomized clinical trial. Nephrol Dial Transplant (2013) 28(11):2823–33. doi: 10.1093/ndt/gft281
241. El Mokadem M, Abd El Hady Y, Aziz A. A prospective single-blind randomized trial of ramipril, eplerenone and their combination in type 2 diabetic nephropathy. Cardiorenal Med (2020) 10(6):392–401. doi: 10.1159/000508670
242. Bravo-Soto GA, Madrid T. Sevelamer versus calcium-based phosphate binders for chronic kidney disease. Medwave (2017) 17(Suppl2):e6942. doi: 10.5867/medwave.2017.6942
243. Jin J, Togo S, Kadoya K, Tulafu M, Namba Y, Iwai M, et al. Pirfenidone attenuates lung fibrotic fibroblast responses to transforming growth factor-Beta1. Respir Res (2019) 20(1):119. doi: 10.1186/s12931-019-1093-z
244. Ruwanpura SM, Thomas BJ, Bardin PG. Pirfenidone: Molecular mechanisms and potential clinical applications in lung disease. Am J Respir Cell Mol Biol (2020) 62(4):413–22. doi: 10.1165/rcmb.2019-0328TR
245. Tuttle KR, McGill JB, Bastyr EJ 3rd, Poi KK, Shahri N, Anderson PW. Effect of ruboxistaurin on albuminuria and estimated gfr in people with diabetic peripheral neuropathy: Results from a randomized trial. Am J Kidney Dis (2015) 65(4):634–6. doi: 10.1053/j.ajkd.2014.11.024
246. Al-Onazi AS, Al-Rasheed NM, Attia HA, Al-Rasheed NM, Ahmed RM, Al-Amin MA, et al. Ruboxistaurin attenuates diabetic nephropathy Via modulation of tgf-Beta1/Smad and grap pathways. J Pharm Pharmacol (2016) 68(2):219–32. doi: 10.1111/jphp.12504
247. Heerspink HJL, Parving HH, Andress DL, Bakris G, Correa-Rotter R, Hou FF, et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (Sonar): A double-blind, randomised, placebo-controlled trial. Lancet (2019) 393(10184):1937–47. doi: 10.1016/S0140-6736(19)30772-X
248. Hudkins KL, Wietecha TA, Steegh F, Alpers CE. Beneficial effect on podocyte number in experimental diabetic nephropathy resulting from combined atrasentan and raas inhibition therapy. Am J Physiol Renal Physiol (2020) 318(5):F1295–F305. doi: 10.1152/ajprenal.00498.2019
249. Rafnsson A, Bohm F, Settergren M, Gonon A, Brismar K, Pernow J. The endothelin receptor antagonist bosentan improves peripheral endothelial function in patients with type 2 diabetes mellitus and microalbuminuria: A randomised trial. Diabetologia (2012) 55(3):600–7. doi: 10.1007/s00125-011-2415-y
250. Kose D, Kose A, Halici Z, Cadirci E, Tavaci T, Gurbuz MA, et al. Bosentan, a drug used in the treatment of pulmonary hypertension, can prevent development of osteoporosis. Iran J Basic Med Sci (2021) 24(7):922–7. doi: 10.22038/ijbms.2021.54152.12172
251. Nitta K, Shi S, Nagai T, Kanasaki M, Kitada M, Srivastava SP, et al. Oral administration of n-Acetyl-Seryl-Aspartyl-Lysyl-Proline ameliorates kidney disease in both type 1 and type 2 diabetic mice Via a therapeutic regimen. BioMed Res Int (2016) 2016:9172157. doi: 10.1155/2016/9172157
252. Nitta K, Nagai T, Mizunuma Y, Kitada M, Nakagawa A, Sakurai M, et al. N-Acetyl-Seryl- aspartyl-Lysyl-Proline is a potential biomarker of renal function in normoalbuminuric diabetic patients with egfr >/= 30 Ml/Min/1.73 M(2). Clin Exp Nephrol (2019) 23(8):1004–12. doi: 10.1007/s10157-019-01733-6
253. Komers R, Oyama TT, Beard DR, Tikellis C, Xu B, Lotspeich DF, et al. Rho kinase inhibition protects kidneys from diabetic nephropathy without reducing blood pressure. Kidney Int (2011) 79(4):432–42. doi: 10.1038/ki.2010.428
254. Qi XJ, Ning W, Xu F, Dang HX, Fang F, Li J. Fasudil, an inhibitor of rho-associated coiled-coil kinase, attenuates hyperoxia-induced pulmonary fibrosis in neonatal rats. Int J Clin Exp Pathol (2015) 8(10):12140–50.
255. Nagai Y, Matoba K, Yako H, Ohashi S, Sekiguchi K, Mitsuyoshi E, et al. Rho-kinase inhibitor restores glomerular fatty acid metabolism in diabetic kidney disease. Biochem Biophys Res Commun (2023) 649:32–8. doi: 10.1016/j.bbrc.2023.01.088
256. Matoba K, Takeda Y, Nagai Y, Kanazawa Y, Kawanami D, Yokota T, et al. Rock inhibition may stop diabetic kidney disease. JMA J (2020) 3(3):154–63. doi: 10.31662/jmaj.2020-0014
257. Sanajou D, Ghorbani Haghjo A, Argani H, Roshangar L, Ahmad SNS, Jigheh ZA, et al. Fps-Zm1 and valsartan combination protects better against glomerular filtration barrier damage in streptozotocin-induced diabetic rats. J Physiol Biochem (2018) 74(3):467–78. doi: 10.1007/s13105-018-0640-2
258. Sun XY, Qin HJ, Zhang Z, Xu Y, Yang XC, Zhao DM, et al. Valproate attenuates diabetic nephropathy through inhibition of endoplasmic reticulum Stress-Induced apoptosis. Mol Med Rep (2016) 13(1):661–8. doi: 10.3892/mmr.2015.4580
259. Khan S, Jena G, Tikoo K. Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the inhibition of histone deacetylases in diabetic rat. Exp Mol Pathol (2015) 98(2):230–9. doi: 10.1016/j.yexmp.2015.01.003
260. Khan S, Jena G. Sodium butyrate, a hdac inhibitor ameliorates enos, inos and tgf-Beta1- induced fibrogenesis, apoptosis and DNA damage in the kidney of juvenile diabetic rats. Food Chem Toxicol (2014) 73:127–39. doi: 10.1016/j.fct.2014.08.010
261. Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, et al. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol (2011) 178(5):2205–14. doi: 10.1016/j.ajpath.2011.01.044
262. Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-Beta1-Induced renal injury. Am J Physiol Renal Physiol (2009) 297(3):F729–39. doi: 10.1152/ajprenal.00086.2009
263. Hung PH, Hsu YC, Chen TH, Ho C, Lin CL. The histone demethylase inhibitor gsk-J4 is a therapeutic target for the kidney fibrosis of diabetic kidney disease Via Dkk1 modulation. Int J Mol Sci (2022) 23(16):9407. doi: 10.3390/ijms23169407
264. Xu Z, Jia K, Wang H, Gao F, Zhao S, Li F, et al. Mettl14-regulated Pi3k/Akt signaling pathway Via pten affects Hdac5-mediated epithelial-mesenchymal transition of renal tubular cells in diabetic kidney disease. Cell Death Dis (2021) 12(1):32. doi: 10.1038/s41419-020-03312-0
265. Hadden MJ, Advani A. Histone deacetylase inhibitors and diabetic kidney disease. Int J Mol Sci (2018) 19(9):2630. doi: 10.3390/ijms19092630
266. Khan S, Jena G, Tikoo K, Kumar V. Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of nf-Kappab/Inos signaling in diabetic rat. Biochimie (2015) 110:1–16. doi: 10.1016/j.biochi.2014.12.015
267. Dong W, Jia Y, Liu X, Zhang H, Li T, Huang W, et al. Sodium butyrate activates Nrf2 to ameliorate diabetic nephropathy possibly Via inhibition of hdac. J Endocrinol (2017) 232(1):71–83. doi: 10.1530/JOE-16-0322
268. Gilbert RE, Huang Q, Thai K, Advani SL, Lee K, Yuen DA, et al. Histone deacetylase inhibition attenuates diabetes-associated kidney growth: Potential role for epigenetic modification of the epidermal growth factor receptor. Kidney Int (2011) 79(12):1312–21. doi: 10.1038/ki.2011.39
269. Foss F, Advani R, Duvic M, Hymes KB, Intragumtornchai T, Lekhakula A, et al. A phase ii trial of belinostat (Pxd101) in patients with relapsed or refractory peripheral or cutaneous t-cell lymphoma. Br J Haematol (2015) 168(6):811–9. doi: 10.1111/bjh.13222
270. Zhao C, Dong H, Xu Q, Zhang Y. Histone deacetylase (Hdac) inhibitors in cancer: A patent review (2017-present). Expert Opin Ther Pat (2020) 30(4):263–74. doi: 10.1080/13543776.2020.1725470
271. Liu N, Zhuang S. Treatment of chronic kidney diseases with histone deacetylase inhibitors. Front Physiol (2015) 6:121. doi: 10.3389/fphys.2015.00121
272. Miranda Furtado CL, Dos Santos Luciano MC, Silva Santos RD, Furtado GP, Moraes MO, Pessoa C. Epidrugs: Targeting epigenetic marks in cancer treatment. Epigenetics (2019) 14(12):1164–76. doi: 10.1080/15592294.2019.1640546
273. He X, Hui Z, Xu L, Bai R, Gao Y, Wang Z, et al. Medicinal chemistry updates of novel hdacs inhibitors (2020 to present). Eur J Med Chem (2022) 227:113946. doi: 10.1016/j.ejmech.2021.113946
274. Dewanjee S, Bhattacharjee N. Microrna: A new generation therapeutic target in diabetic nephropathy. Biochem Pharmacol (2018) 155:32–47. doi: 10.1016/j.bcp.2018.06.017
275. Liu R, Lee K, He JC. Genetics and epigenetics of diabetic nephropathy. Kidney Dis (Basel) (2015) 1(1):42–51. doi: 10.1159/000381796
276. Kato M, Natarajan R. Epigenetics and epigenomics in diabetic kidney disease and metabolic memory. Nat Rev Nephrol (2019) 15(6):327–45. doi: 10.1038/s41581-019-0135-6
277. Dirks RA, Stunnenberg HG, Marks H. Genome-wide epigenomic profiling for biomarker discovery. Clin Epigenet (2016) 8:122. doi: 10.1186/s13148-016-0284-4
278. Doudna JA, Charpentier E. Genome editing. the new frontier of genome engineering with crispr-Cas9. Science (2014) 346(6213):1258096. doi: 10.1126/science.1258096
Keywords: epigenetic modification, diabetic kidney disease, metabolic disorder, biomarker, noncoding RNA
Citation: Liu Z, Liu J, Wang W, An X, Luo L, Yu D and Sun W (2023) Epigenetic modification in diabetic kidney disease. Front. Endocrinol. 14:1133970. doi: 10.3389/fendo.2023.1133970
Received: 29 December 2022; Accepted: 30 May 2023;
Published: 30 June 2023.
Edited by:
Sen Li, Beijing University of Chinese Medicine, ChinaReviewed by:
Yang Danyi, Central South University, ChinaCopyright © 2023 Liu, Liu, Wang, An, Luo, Yu and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dehai Yu, eXVkZWhhaUBqbHUuZWR1LmNu; Weixia Sun, c3Vud3hAamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.