
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 13 March 2023
Sec. Endocrinology of Aging
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1128622
This article is part of the Research Topic Molecular Crosstalk Between Endocrine Factors, Paracrine Signals, and the Immune System During Aging View all 6 articles
 Priya Makhijani1,2†‡
Priya Makhijani1,2†‡ Paulo José Basso3†‡
Paulo José Basso3†‡ Yi Tao Chan4
Yi Tao Chan4 Nan Chen5,6
Nan Chen5,6 Jordan Baechle2,7
Jordan Baechle2,7 Saad Khan1,5
Saad Khan1,5 David Furman2,7,8,9†‡
David Furman2,7,8,9†‡ Sue Tsai3*†‡
Sue Tsai3*†‡ Daniel A. Winer1,2,5,6,7,10*†‡
Daniel A. Winer1,2,5,6,7,10*†‡The signaling pathways downstream of the insulin receptor (InsR) are some of the most evolutionarily conserved pathways that regulate organism longevity and metabolism. InsR signaling is well characterized in metabolic tissues, such as liver, muscle, and fat, actively orchestrating cellular processes, including growth, survival, and nutrient metabolism. However, cells of the immune system also express the InsR and downstream signaling machinery, and there is increasing appreciation for the involvement of InsR signaling in shaping the immune response. Here, we summarize current understanding of InsR signaling pathways in different immune cell subsets and their impact on cellular metabolism, differentiation, and effector versus regulatory function. We also discuss mechanistic links between altered InsR signaling and immune dysfunction in various disease settings and conditions, with a focus on age related conditions, such as type 2 diabetes, cancer and infection vulnerability.
Insulin is a vital hormone that controls the availability of glucose, required for nearly all cell processes (1). The cellular response to insulin has been heavily studied in liver, skeletal muscle, and fat cells due to their role in governing systemic nutrient availability. However, immune cells also rely on insulin for their metabolic needs and function, with activated and effector immune phenotypes exhibiting increased energetic demands (2). Insulin binding receptors, including the insulin receptor (InsR) and insulin-like growth factor 1 receptor (IGF1R), are expressed on cells of both branches of the immune system. Innate immune cells (e.g., monocytes, macrophages, neutrophils, and dendritic cells), as well as adaptive immune cells (e.g., T cells and B cells) express InsR/IGF1R at various stages of activation (3–5). In this review, we will discuss how InsR signaling modulates downstream immune cell processes impacting the overall immune response. Because insulin-mediated effects on immune cells, including insulin resistance, can drive both pro- and anti-inflammatory effects, we will also discuss proposed mechanisms behind insulin’s pleiotropic effects and highlight their implications in disease.
Insulin signal transduction pathways downstream of the phosphorylated InsR/IGF1R have been studied in some immune cells. Insulin binding to the InsR and to the IGF1R promotes the uptake of glucose, its storage in the form of glycogen and lipids as well as its biosynthetic use (1). Both receptors dimerize and auto-phosphorylate at tyrosine residues upon insulin binding with differential affinities to the insulin ligand. Signal transduction events downstream of insulin binding have been extensively summarized previously (6). In brief, InsR/IGF1R signaling begins with the recruitment of insulin receptor substrates (IRS; IRS-1 through 6), with different isoforms enriched in different cell types. An IRS scaffold is generated leading to the formation of Src-homology 2 (SH2) domains capable of activating two branches of signal transduction: the Phosphatidyl inositol-kinase (PI3K)/Protein kinase B (PKB), or Akt pathway, and the Mitogen activated protein kinase (MAPK) pathway. Activation of P13K leads to phosphorylation of Akt at T308 via phosphoinositide-dependent protein kinase-1 (PDK1) and at S473 via mammalian (or mechanistic) target of rapamycin complex 2 (mTORC2). The complete phosphorylation of Akt drives activation of mTORC1 via the inactivation of Tuberous Sclerosis-1/2 (TSC1/2) (7). The activated mTORC1, in turn, activates ribosomal protein S6 kinase (S6K) and eukaryotic translation initiation factor 4E (eif4E) to exert the multiple cellular effects assigned to the complex.
Importantly, mTORC1 and Akt driven signaling cascades control downstream pattern-recognition receptor (PRR) expression, epigenetics, autophagy, and metabolic programs like glycolysis, pentose phosphate pathway (PPP), oxidative phosphorylation (OXPHOS), and glutaminolysis, in immune cells (8). Because mTORC1 and Akt also respond to nutrient availability, growth factors and stress signals, their potentiation by insulin signaling serves to couple energetic demands with immune cell function.
The response to insulin can take on distinct forms, for example, due to the context dependent use of IRS and Akt isoforms in each immune cell subtype (9, 10). Also, insulin can boost existing immune responses due to crosstalk with other pathways. For instance, in addition to mTORC1 mediated metabolic flux, InsR/IGF1R mediated Akt activation drives phosphorylation with numerous other target proteins, including inflammatory factors. For example, Akt activates the nuclear factor kappa B (NF-kB) pathway via degradation of inhibitor of kB (IkB) (11). In immune cells, NF-kB is a cardinal factor for activation, controlling maturation, proliferation and production of chemokines and cytokines such as CCL2 and TNFα. These molecules recruit and activate both innate and adaptive immune cells, orchestrating local and systemic inflammatory responses.
The second branch of InsR/IGF1R signal transduction triggered at the receptor/IRS complex is the MAPK-ERK pathway. The SH2 domains at the IRS scaffold also serve as docking sites for Shc adaptor proteins. Shc proteins activate the Ras-GTPase which then turns on the MAPK-ERK pathway. Activated ERK1/2 also has been recognized as a major pathway responsible for important consequences on immunity driving immune cell proliferation, metabolic re-programming, differentiation, inflammasome activation and cytokine production (12–15). Importantly, both MAPK and PI3K pathways promote glucose uptake via several glucose transporters (GLUTs), including GLUT3, affecting overall energy metabolism in immune cells (16). The cascade of insulin signal transduction via the PI3K and ERK pathways serves to create points of crosstalk and regulation of the insulin response. Insulin signaling thus potentiates other immune activation signals. However, it can also be dampened by several important negative feedback mechanisms. The activated S6K, IKKB and JNK proteins can block and degrade IRS-1 through serine phosphorylation of IRS-1/2 (17), modulating the InsR/IGF1R cascade at the earliest stage. Many of these proteins are also activated within inflammatory signaling pathways (e.g. TNFR-JNK) and contribute to insulin resistance (IR) (6). Together, this network of signaling events can both activate and attenuate inflammation in a context-dependent manner.
A third route of InsR signaling is through the recently described mechanism where InsR translocates into the nucleus and binds to promoter regions of key genes associated with immune cell recruitment (18). In this pathway, activated InsR colocalizes with host cell factor-1 (HCF-1) to mediate direct transcriptional activity (18). This phenomenon was primarily observed in cultured liver cells, and it remains to be seen whether a similar mechanism occurs in immune cells.
Insulin signaling is intricately tied to metabolic responses in immune cells as alluded to earlier. Thus, we will next discuss in-depth how insulin action interacts with these nutrient sensing pathways and metabolic responses to alter inflammatory states in innate and adaptive immune cells.
Macrophages and monocytes are best recognized for their ability to sense the tissue environment and polarize to orchestrate an appropriate immune response at the site of infection or damage. Protein expression of both InsR and IGF1R has been reported on monocytes and macrophages (19, 20). In vitro, insulin treatment of mouse macrophages potentiated the LPS-mediated production of pro-inflammatory cytokines, IL-6 and TNFα, in a PI3K dependent manner (21), with a similar response in human monocytes (22). Several studies using myeloid lineage knockouts support that insulin signals are required for pro-inflammatory polarization. Indeed, myeloid lineage specific genetic ablation of InsR, IGF1R or IRS leads to the anti-inflammatory (or M2-like) polarization of macrophages (3, 23, 24), consistent with an overall pro-inflammatory (or M1-like) biasing by InsR/IGF1R signaling. Key studies on the direct effect of insulin on innate and adaptive cell types are summarized in Table 1.
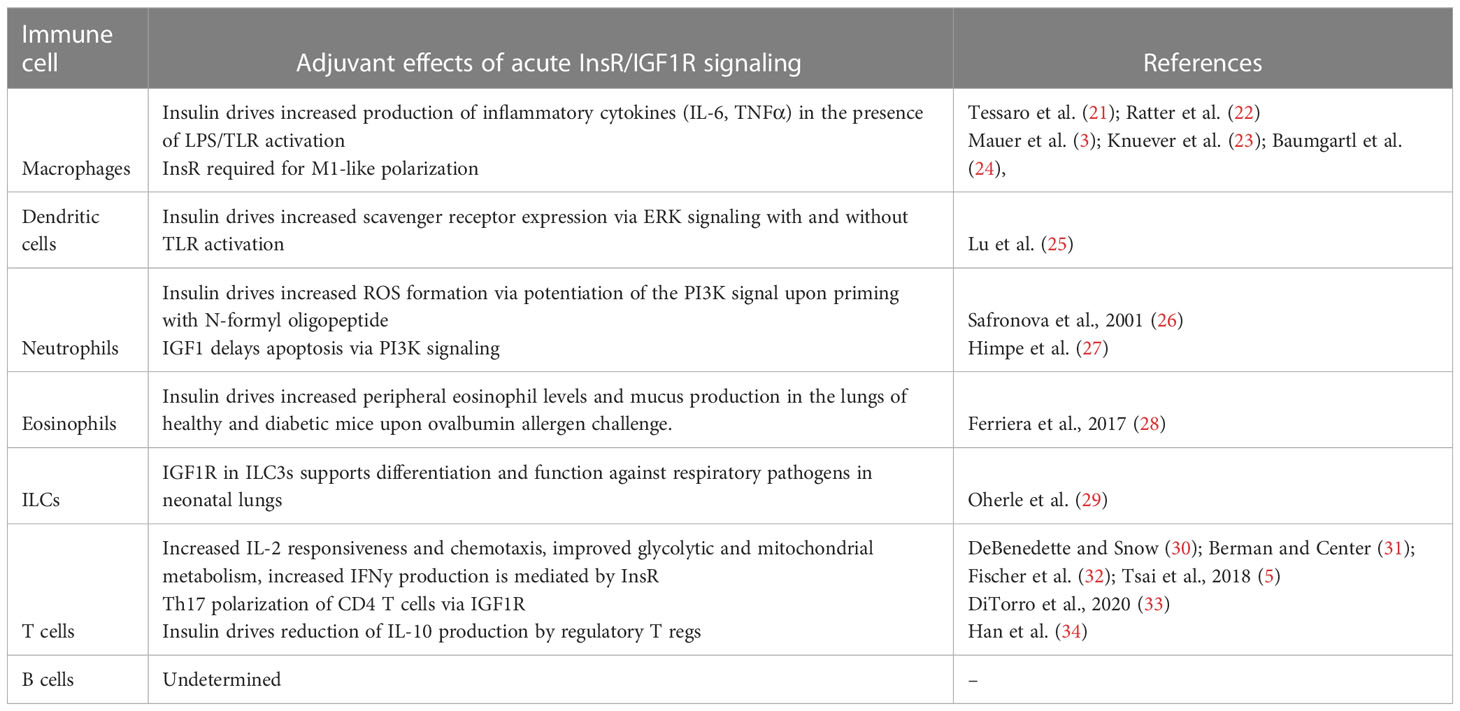
Table 1 The acute effects of insulin and IGF1R signaling on immune cells by cell type.
It has been recognized that pro-inflammatory innate immune cells rely on glycolysis and the PPP to meet biosynthetic demands, while immature and regulatory immune cells rely more heavily on OXPHOS and fatty acid oxidation (FAO) for obtaining energy and to inhibit metabolic drivers of inflammation (36, 37). Consistently, inflammatory M1-like macrophages show increased glycolysis, PPP, inducible nitric oxide synthase (iNOS) activity, and fatty acid synthesis, required to meet the biosynthetic demands of activation and corresponding with pro-inflammatory cytokine burst (38). Also, anti-inflammatory M2-like macrophages show increased glutamine metabolism, associated OXPHOS, arginase-1 activity, and FAO (39). A major mechanistic mediator of insulin driven pro-inflammatory responses is the mTOR complex, which supports glycolytic metabolism (40). Therefore, we speculate that insulin driven mTORC1 activation provides metabolic support for M1-like polarization. In myeloid cells, lineage-specific deletion of Raptor, a specific component of mTORC1, elevated the M2 macrophage signature, consistent with the effect of TSC1 deletion (mTOR activating), which results in sustained mTORC1 activation and a corresponding decrease in the M2 signature (41). Insulin-mTORC1 axis is also expected to promote pro-inflammatory outcomes via the inhibition of autophagy. Autophagy contributes to OXPHOS via lipid catabolism, which is inhibited upon mTORC1 activation (42). The above studies suggest that insulin signaling may have dominant effects on preferential inflammatory pathways via boosting glycolysis, PPP, and fatty acid synthesis (Figure 1).
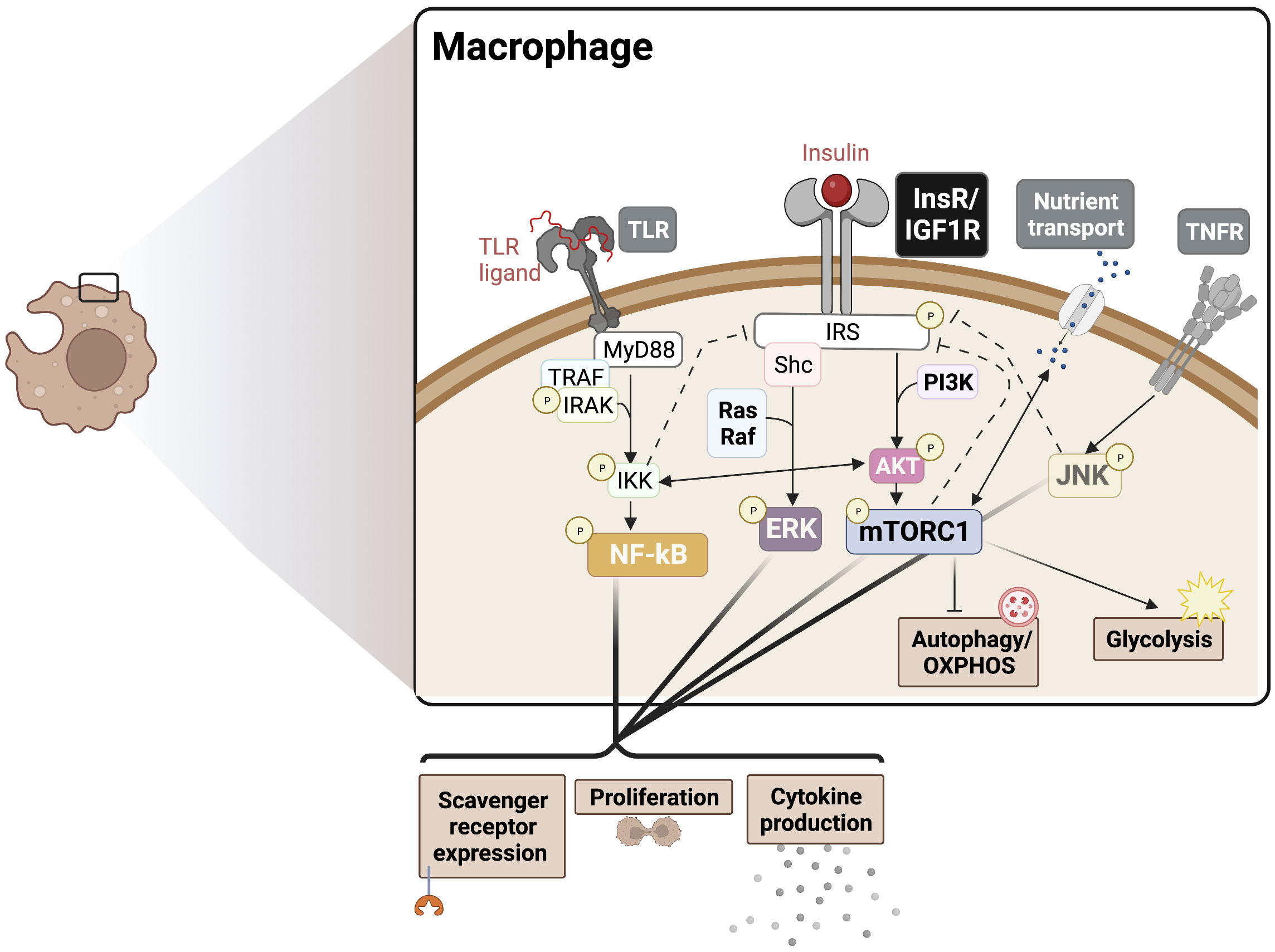
Figure 1 Insulin crosstalk with immune signalling pathways in macrophages. PAMPs/DAMPs trigger surface and endosomal TLRs, signalling largely via MyD88 to NF-kB to drive cytokine production, proliferation, and scavenger receptor expression. InsR/IGF1R signals through MAPK and Akt/mTORC1 to boost cytokine production and proliferation, supporting the TLR response. These signalling events also drive downstream metabolic programming to support the redox balance and energetic demands, promoting an inflammatory phenotype. Activation of mTORC1 also blocks autophagy, which provides substrates for OXPHOS. Various points of crosstalk exist between InsR, PRRs, cytokine receptor, and nutrient sensing responses can also drive insulin resistance via negative feedback mechanisms (dotted lines), for example in cases of chronic hyperinsulinemia. The degree to which each intersecting pathway is stimulated likely determines the nature of the pleiotropic phenotypes seen in IR macrophages. PAMPs, Pathogen-associated molecular patters; DAMPs, Damage-associated molecular patterns; OXPHOS, Oxidative phosphorylation; PRR, pattern recognition receptor; TLR, Toll-like receptor; MyD88, Myeloid differentiation primary response 88; NF-kB, Nuclear factor kappa B; InsR, Insulin receptor; IGF1R, Insulin-like growth factor 1 receptor; ERK, extracellular regulated kinase; mTORC1, mammalian target of rapamycin complex 1.
However, M2-like macrophages also require glycolysis up to a threshold level to meet energetic demands (43). Therefore, insulin signaling may also participate in immune tolerance in certain situations. A new understanding of macrophage polarization states across both pro-inflammatory and suppressive disease conditions supports the adaptation of a spectrum polarization model (44), especially in cases of chronic unresolving inflammation. This model allows us to better appreciate heterogeneous macrophage responses to insulin depending on disease context.
Neutrophils perform the important function of trapping and clearing pathogens as well as damaged tissue by producing reactive oxygen species (ROS) and releasing neutrophil extracellular traps (NETs). In vitro priming of neutrophils with insulin prior to activation with N-formyl oligopeptide leads to increased formation of ROS suggesting crosstalk with formyl peptide receptor mediated PI3K signaling (26). Furthermore, PI3K signaling was required for other neutrophil functions like cytokine production, chemotaxis, and degranulation (45), which maybe similarly potentiated by insulin treatment. Additionally, IGFR1-PI3K signaling was shown to delay apoptosis of the normally short-lived neutrophils (27). The mTORC1 pathway also promotes the release of NETs via HIF-1α (46). This suggests that insulin-mediated mTORC1 activation may also drive NET formation, influencing the clearance of infection. The context-dependent activation and metabolic reprogramming of neutrophils via InsR/IGF1R requires further study, especially given the key role neutrophils play in wound healing and infection.
A cell type closely related to neutrophils is the myeloid derived suppressor cell (MDSC). Mainly studied in the context of cancer and wound healing, MDSCs are similar to alternatively activated neutrophils and function to drive immune tolerance (47). While MDSCs rely on Akt-mediated STAT3 and HIF-1α signaling to produce regulatory effectors like ROS, IL-10 and Arg-1 (48, 49), they also require autophagy for MHC downregulation and rely on OXPHOS metabolism (50). The impact of insulin on MDSCs biology is a new consideration, with very little known about the expression of InsR/IGF1R on MDSCs or the basic in vitro effect insulin has on this cell type.
Eosinophils have been well-recognized for their response to allergens and helminth infection with accumulating evidence for their role in tissue regeneration upon injury (51). Activated eosinophils upregulate IGF1R and therapeutic targeting of IGF1R can attenuate eosinophil mediated inflammation (52), suggesting a possible role for insulin/IGF signals in eosinophil function. Similarly to other immune cells, glycolysis is increased upon activation in eosinophils (53). Correspondingly, insulin treatment leads to increased peripheral eosinophil levels and mucus production in the lungs of healthy mice upon ovalbumin allergen challenge (28). Furthermore, eosinophils play a key role in immune cell crosstalk. For example, activated eosinophils have been shown to produce IL-4 and IL-13 to sustain the M2-like suppressive macrophage phenotype (54), thereby promoting wound healing and fibrosis. More work is needed to understand the impact of insulin in these processes, such as whether insulin exposed eosinophils can feedback onto inflammatory macrophages to regulate tissue homeostasis.
Dendritic cells (DCs) perform the key functions of antigen capture and presentation to lymphocytes required for adaptive immunity. In vitro insulin treatment drives maturation and increased scavenger receptor expression in human monocyte-derived DCs in an ERK-dependent manner (25). Additionally, TLR activation in DCs and subsequent Akt-phosphorylation directly boosts hexokinase 2 (HK2) function, an enzyme which catalyzes the first step in glucose metabolism, leading to increased glycolysis and PPP-derived NADPH to fuel lipid synthesis (55). Consistent with its adjuvant functions in other immune cells, insulin likely intersects with TLR signaling at boosting phosphorylated Akt signals upstream of mTORC1, potentially contributing to overlapping inflammatory mediator induction in DCs. In plasmacytoid DCs (pDCs), a subset of DCs shown to be important in anti-viral responses and autoimmune pathophysiology, type I IFN production upon TLR activation requires mTOR signaling (56). This interaction suggests that insulin-mediated mTORC1 activation may also augment IFN-I production in pDCs, aggravating autoimmunity or boosting anti-viral responses.
Although the direct role of the InsR-mTOR pathway on DCs has not been studied, the role of mTORC1 on DC activation and maturation has been highlighted in various studies (57). The cDC1 subset is described to cross-present antigen to CD8+ T cells, while cDC2s present antigen to CD4+ T cells. Studies using mice with conditional knockout of Raptor in the DC lineage showed that mTORC1 supports glycolysis while impairing CD8+ T cell priming by cDC1s, with no effects on CD4+ Th2 priming by cDC2s (58). This effect suggests a likely role for insulin-mediated mTORC1 activation in inflammatory metabolic reprograming in DCs, while inhibiting autophagy-dependent functions, such as antigen presentation, in DCs in a subtype-specific manner. Fully understanding the complex effect of insulin on antigen presentation in DCs will provide critical insights into their role in orchestrating immune dysregulation in diseases with disrupted insulin homeostasis.
ILCs are a family of lymphocytes with the capacity to rapidly release cytokines and perform killing functions in response to injury or infection but lacking adaptive rearranging antigen receptors like traditional lymphocytes but with the capacity to rapidly release cytokines and perform killing functions in response to injury or infection. Three widely accepted groups of ILCs have been documented: ILC1, ILC2 and ILC3, which are classified into further subtypes depending on the tissue (59). Whether and how ILCs respond to insulin/IGF1 as seen in other innate immune cell studies with a key role for mTORC1 and PI3K pathways is mostly unknown. Natural cytotoxicity receptors, found on all ILC subtypes, signal via the PI3K pathway (60) and could potentially cross-talk with InsR to aggravate ILC-mediated cytotoxicity and inflammation. Group 1 ILCs include NK cells and mediate crosstalk between the tissue and the immune response with the capacity to produce inflammatory cytokines. In NK cells, mTORC1 driven glycolytic metabolism was shown to be required for activation, cytotoxicity, and maturation (61). Interestingly, autophagy was required for catabolic processes and inhibition of apoptosis in ILC1s upon cell stress (62). Thus, we speculate, that insulin may boost acute ILC1/NK cell activation through mTORC1, while insulin-mediated inhibition of autophagy, may be detrimental for ILC1 survival in the face of stress.
In contrast to ILC1s, ILC2s are producers of type 2 cytokines like IL-4, IL-13, and IL-5 and can also drive tolerogenic activation of other immune cells. ILC2s were found to require glycolytic metabolism for cytokine production mediated by mTORC1 upon activation (63). Interestingly, the baseline level of OXPHOS required to the sustain naïve ILC2s, was found to be greater compared to NK cells (64). Speculating on the pro-inflammatory role of insulin in other immune cells, we expect differential effects of insulin on the suppressive ILC2s versus on pro-inflammatory ILC1/NK cells due to their differential thresholds of OXPHOS at baseline.
ILC3s are a distinct population of ILCs that produce IL-22 and IL-17 and play a role in mucosal homeostasis and Th17 responses. In ILC3s, mTORC1 was required for sustained HIF-1α and RORγt expression and production of cytokines (29). Further, conditional Igf1r ablation in ILC precursors driven by the RORγt promoter, lead to aberrant ILC3 differentiation and poor defense against respiratory pathogens in neonatal lungs, implying crosstalk with resident IGF1 producing fibroblasts and a role for ILC3s in early-onset asthma (30). The role for insulin/IGF1 on ILC3s in other mucosal tissues like the gut remains to be studied considering its critical role in gut healthy and barrier function in sustaining metabolic homeostasis.
In contrast to the large body of evidence supporting the pro-inflammatory role of insulin in innate immune cells, some studies suggest that insulin may also dampen inflammation in certain ways. For example, the response to insulin in obese subjects or purified immune cells led to reduced TLR activation and transcription of TLRs (65, 66). Further, PI3K-Akt also inhibits the transcription factor, forkhead box O1 (FOXO1), a downstream target of Akt which is involved in cell proliferation, energy metabolism and oxidative stress (67). In macrophages, deficiency of FOXO1 has been shown to promote alternative polarizaion via the inhibition of glycolysis (68) and downregulation of target gene tlr4 (65). The reduction in FOXO1 and TLR transcription may serve to dampen insulin mediated inflammation.
Insulin is also expected to stimulate the anti-inflammatory factor Nrf2 via the ERK pathway (69). Among many other functions, Nrf2 blocks production of inflammatory effectors such as ROS and IL-1β (70), which may support the anti-inflammatory polarization of macrophages and neutrophils. As alluded to earlier, insulin-mTORC1 axis also inhibits autophagy required for antigen presentation by macrophages and DCs and thereby, the activation of the adaptive immune response (57). Outside of these cell intrinsic effects, insulin also drives a systemic reduction in glucose levels. Low glucose levels result in fewer advanced glycation end products (AGEs) leading to dampened RAGE-mediated NF-kB activation and reduced inflammation (71). To better understand the action of insulin on innate immunity, we will need to delineate the short- and long-term as well as dose-dependent effects of insulin exposure, as the immune response to insulin may be subject to negative feedback mechanisms, which may have evolved to prevent tissue damage and unresolving inflammation.
The adaptive immune system is highly specialized and orchestrates tumor surveillance, anti-microbial responses, and generates immunological memory. Notably, cells of the adaptive immune system also express the InsR, which rises in expression upon activation (72–74). This increase in InsR expression likely reflects the T cells’ increasing energetic and biosynthetic demands. While a limited number of studies ascribed an anti-inflammatory role to insulin (73), emerging data also support an important pro-inflammatory role of InsR signaling in optimal T cell-mediated immunity (5, 33, 75). Addition of insulin to cultured T cells in vitro promoted IL-2 responsiveness (31), and chemotactic activity (32), which was accompanied by increased nutrient uptake and glycolytic reprogramming necessary for optimal T effector function (5). Thus, T cells lacking InsR expression exhibited an impaired glycolytic and mitochondrial metabolism, and reduced both IFNγ production and antigen-specific expansion upon influenza infection (5, 33). Consistent with the role of InsR in promoting inflammation and immunity, InsR signaling has been shown to dampen regulatory T cell (Treg) function in the setting of obesity and aging (35, 76). Interestingly, mTORC1 inhibition during the contraction phase promoted CD8+ T cell memory formation and function (77, 78). A recent study showed that InsR is dispensable for an effective memory CD8+ T cell response (79), suggesting that the immunostimulatory effects of insulin may selectively act during T cell activation and effector differentiation stages where glycolytic metabolism is critical, but not during memory differentiation which is predominantly supported by mitochondrial fatty acid metabolism.
T cells also express IGF1R, which signals through the Akt-mTORC1 axis (80) to regulate Th17/Treg differentiation both in vitro and in vivo (34). In vitro, stimulation with natural ligands of IGF1R favored the polarization of naïve CD4+ T cells towards Th17 phenotype and away from Foxp3+ Tregs. This change was accompanied by upregulated HK2 transcripts and glycolytic and mitochondrial metabolism within in vitro-differentiated Th17 cells. Furthermore, igfr1 deletion in CD4+ T cells reduced IL-17A-expressing CD4+ T cell levels, increased central nervous systems-infiltrating Tregs, and protected against experimental autoimmune encephalomyelitis (34). Of note, these findings are in contrast to earlier reports, where IGF-1 was shown to expand Tregs and protect against autoimmune disease models of T1D and EAE (81). Here, Treg-specific genetic deletion of igfr1 led to the loss of IGF-1’s protective effects. The discrepancies could arise from differences in the differentiation stage at which IGF-1 signals are perceived, during the activation and differentiation of naïve T cells (34, 82), or in pre-activated/differentiated T cell subsets (80, 83). The type and dosage of IGF-1R ligands, and signaling through IGF1R vs IGF1R/InsR heterodimers, could also contribute to the observed inconsistencies.
B cells are primary mediators of the adaptive and humoral immune response, achieving their functions via cytokine production, antibody secretion, and modulation of other immune cells (84). The exact role of InsR signaling in B cells has not been elucidated. However, the InsR has been shown to be expressed on B cells (74, 85) and is speculated to activate downstream PI3K/Akt and MAPK pathways much like in other immune cells.
During B cell development, PI3K signaling mediated by IL-7R and pre-BCR are required for the transition from the pro-B to pre-B and beyond the pre-B stages (86). In the peripheral B cell subsets, PI3K and Akt play a significant role in the specification of marginal zone (MZ) and B-1 cells (87–90). PI3K/Akt signaling is also crucial to B cell survival, activation, differentiation, and effector functions (Figure 2) (86, 91, 92). Cumulative PI3K signaling through tonic BCR signaling and BAFF-R is required for the growth and survival of peripheral B cells (93–96). Interestingly, the anti-diabetic drug metformin was found to attenuate BAFF-induced B cell proliferation and survival by inhibiting the mTORC1-PTEN/Akt-Erk1/2 signaling pathway (97). In the germinal center (GC) of peripheral lymphoid organs, PI3K plays a dual role. It is required for BCL6 expression, the transcription factor regulator of GC formation and differentiation of B cells into long-lived plasma cells (98), and maintenance of the GC reaction in B cells (86, 98–101). In contrast, PI3K also represses B cell identity, favoring plasma cell fate decision through IRF4 and Blimp-1 expression (101–104). These features are consistent with PI3K’s role in maintenance of the GC reaction especially in the light zone (105). Given this important role for PI3K in orchestrating GC reactions, it will be interesting to dissect how insulin and its receptor impacts such reactions in B cells.
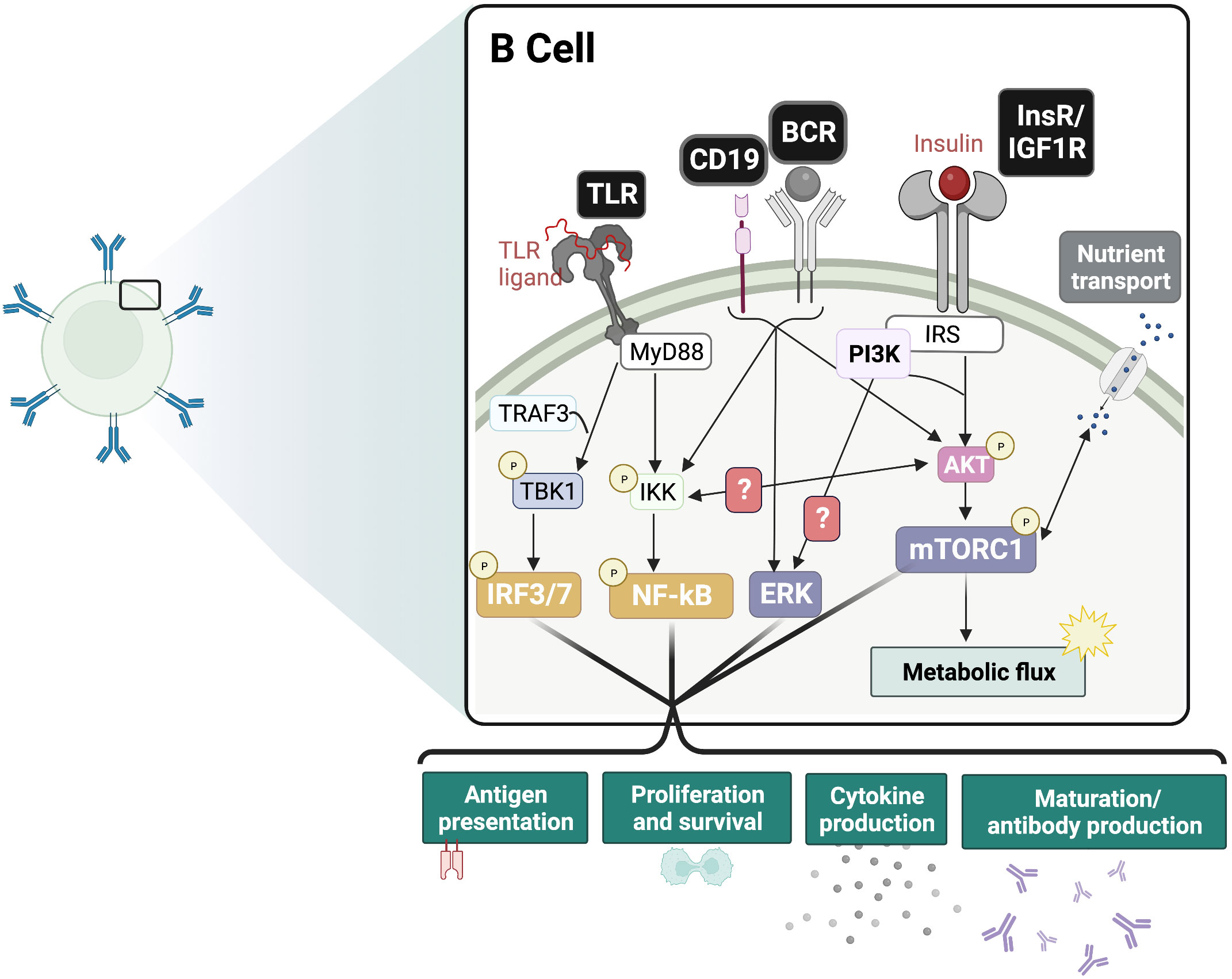
Figure 2 Insulin signaling functions as an adjuvant of B cell responses. Proposed crosstalk between InsR and BCR/CD19 and PRR signaling pathways. In the presence of antigen-BCR mediated ERK signaling and TLR-ligand mediated IRF and NF-kB signaling, the additive effect of InsR/IGF1 signaling is expected to drive inflammatory responses. This includes boosting glycolysis and nutrient transport, as well as maturation and antibody production, cytokine production, antigen presentation and proliferation. BCR, B cell receptor; TLR, Toll-like receptor; InsR, Insulin receptor; IGF1R, Insulin-like growth factor 1 receptor; mTORC1, mammalian target of rapamycin complex 1; ERK, extracellular regulated kinase; IRF, interferon regulatory factor; NF-kB, Nuclear factor kappa B.
Interestingly, PI3K signaling has also been shown to modulate class switch recombination (CSR), as pharmacological inhibition or B cell-specific genetic deletion of p110δ/α enhanced both IgG1 and IgE CSR in mice (106–108). Pten deletion, a negative regulator of PI3K signaling, also demonstrated significant reductions in IgG1 and IgE CSR (106). Together, IgG1 and IgE are mostly involved in type 2 immune responses, such as in the development of allergic Th2-mediated airway inflammation (109), and pharmacological blockade of p110δ PI3K has been suggested to be an effective treatment for allergies and atopic disease (108). More work is needed to determine if these pathways are similarly impacted by insulin signaling.
The role of Ras/MAPK signaling in B cells is well documented (110, 111). Erk signaling is necessary for the tonic BCR signaling required for immature B cells to differentiate into transitional and mature B cells (112). Erk was also shown to play a role in survival and maintenance of mature, antigen-specific IgG1+ B cells in the periphery (113). Erk is also necessary for the expression of Blimp-1 in plasma cells (114) and degradation of BCL6 (115).
In B cells, IGF1R expression is high in the pre-pro- and non-B cell bone marrow cells and decreases during differentiation, whereas InsR and IRS-2 expression increase during differentiation (74). The IGF1R was also expressed in the MZ and follicular (FO) splenic B cell populations. IGF1R deletion in B cells of chimeric mice showed no significant change in B cell development or peripheral subsets and did not affect BCR signaling. However, IGF1R deletion impaired T cell independent- but not T cell dependent-antibody response. The authors speculated that this effect was due to impaired IRS-PI3K signaling which regulates T cell independent responses (74). In patients with Grave’s disease, there was a subset of B cells with increased IGF1R expression which showed increased B cell proliferation and IgG production in response to IGF1 and CpG (TLR9 ligand) treatment (116). IGF1 has also been shown to increase IgG subtype production in human tonsillar and peripheral B cells in a CD40-dependent manner (117, 118). Interestingly, IGF2 was also shown to enhance regulatory B cells (Breg) proliferation and IL-10 production in response to specific antigen in an ovalbumin (OVA) allergic intestinal inflammation model (119), suggesting a role for IGFs in anti-inflammatory functions.
Although the effects of B cell InsR signaling have been less studied, there is likely significant metabolic overlap. Upon activation via BCR signaling, B cells rapidly increase glucose and amino acid uptake and glucose transporter GLUT1 expression in a PI3K-dependent manner (120–123). It is interesting to note that B cells rely less on aerobic glycolysis compared to T cells upon activation (120). Instead, BCR signaling diverts glucose catabolism away from glycolysis to: 1) the PPP to generate NADPH, crucial for neutralizing ROS and maintaining cellular redox status (121), 2) the de novo lipogenesis via ATP-citrate lyase to supply activated B cells with phospholipids for morphological changes (123), 3) the TCA cycle for efficient production of ATP through OXPHOS (120) and 4) the hexosamine pathway to allow for antibody glycosylation in long-lived plasma cells (124). Glycolysis appears to be dispensable for B cell activation and instead supports activation through redox maintenance through the PPP pathway, whereas OXPHOS and glutamine metabolism were shown to be indispensable for B cell growth and differentiation (125). Future studies using reductionist models will be important to tease out effects of insulin on B cell metabolism.
Given the recent advances summarized above, we propose that insulin plays a critical role in regulating both innate and adaptive immune cell function through fine tuning intrinsic metabolic programming. These findings have particularly important implications in diseases where insulin signaling is dysregulated, as such alterations could promote or potentiate the immune response. In the following sections, we discuss implications of altered InsR signaling in immune cells in disease states that are accompanied by inflammatory changes and how they contribute to shaping disease outcomes. Of note, while we focus on only a few conditions, it is plausible that any condition involving an immune response, including autoimmunity, would have relevant insulin-immune interactions.
A major driver of obesity-related IR is the establishment of low-grade chronic inflammation in metabolic tissues, including liver, muscle, and adipose tissue, especially visceral adipose tissue (VAT) (126). Within these tissues, immune cells such as macrophages and lymphocytes accumulate and produce pro-inflammatory cytokines, which in turn leads to IR and hyperinsulinemia. The role for hyperinsulinemia as a driving factor in this process is poorly understood, though previous work has implicated insulin as a global driver of VAT inflammation (127). Reduction of circulating insulin levels by approximately 50% in obese mice, using a diazoxide or streptozotocin (STZ) β-cell ablation regimen, led to a decrease in VAT macrophage expansion and a reduction in expression of pro-inflammatory cytokines/chemokines, such as CCL2, TNFα, IL-6, or IL-1β in VAT (127). Diet-induced obesity leads to accumulation of resident ILC1s by IL-12 production within the VAT which in turn drove pro-inflammatory polarization of macrophages via IFNγ production (128). However, eosinophils, which produce type 2 cytokines and promote M2-like states in macrophages, have been shown to have protective effects in obesity (54). Myeloid lineage-specific knockout of InsR protected against IR and associated VAT inflammation (3). In such studies with high-fat diet (HFD) fed mice, effects of reducing pro-inflammatory parameters in fat are seen in mice as early as 4 weeks of HFD. Thus it is likely that insulin is a critical driver of VAT inflammation early in disease (3, 24), potentially before marked changes in glucose levels occur.
Immune-mediated inflammation plays an important role in driving obesity-related IR (3, 21–24). We and others have shown that obesity is associated with increased infiltration of proinflammatory macrophages and adaptive immune cells in the liver, the intestines, and in the expanding VAT (84, 129–133), as well as a reduction in Tregs (35). On the other hand, this meta-inflammatory state is associated with an inability of the immune system to respond appropriately to certain foreign or tumor antigenic cues. In the context of anti-microbial immunity, functional defects in both humoral and cellular arms of the immune response have been observed across multiple species and disease models (134–137), although mechanistic insights are limited and inconclusive.
One possible mechanism that could explain these hallmark complications of diabetes might be that the cells of the immune system are also becoming insulin resistant themselves, leading to weakened overall effector function (138). This notion would raise the question of whether such cell-intrinsic IR underlies the observed immune impairments. Indeed, some work points to insulin being a new type of “metabolic adjuvant” to acutely boost immune cell action (5). For instance, mice in which the InsR is ablated specifically in T cells, a model of near complete immune cell IR, are vulnerable to viral infections such as H1N1 influenza due to poor T cell responses (5). Consistently, as we will discuss below, viral infections induce pro-inflammatory cytokine signaling which acutely reduces systemic insulin sensitivity in order to boost available insulin to immune cells for optimal anti-viral immunity (75). However, prolonged exposure to hyperinsulinemia would likely break this system if immune cells themselves became resistant to the action of insulin. This concept may have been especially relevant during the recent COVID-19 pandemic since obesity was a risk factor for poor outcome (139), as it was associated with weaker adaptive responses to clear the virus (140). While poor glycemic control associated with obesity also likely contributes to a dysfunctional immune response (141), studies from autopsied patients dying of severe COVID-19 show that insulin signaling pathways are amongst the most downregulated in tissue/immune cell infiltrated lung samples, linking local tissue insulin resistance to poor outcome (142, 143).
Some of the mechanisms of intrinsic immune system insulin resistance at the InsR are likely related to negative feedback mechanisms induced by hyperinsulinemia. Mechanistically, the accumulation of S6K and IKK, part of the mTORC1 and NF-kB pathways respectively, can degrade IRS-1 (11, 17). Because the IRS complex is required for early events in the insulin signaling pathway, negative feedback mechanisms targeting IRS may completely reduce the insulin response with overstimulation. In addition to built-in negative feedback mechanisms, disease-associated aberrant inflammatory and metabolic signaling can also promote the serine phosphorylation of IRS and dampen insulin sensitivity. For instance, inflammatory cytokines such as TNFα directly signal to impair insulin sensitivity (reviewed in 144). Elevated free fatty acids can also impair insulin-stimulated PI3K activation (145). These additional mediators of intrinsic immune cell insulin resistance may occur during both obesity and infection.
Another possible mechanistic consequence of intrinsic immune cell IR is differential use of Akt isoforms. Akt has three leading isoforms, of which Akt2 is the major isoform mediating insulin signals involved in glucose homeostasis (146). Greater levels of mTORC1 activity and lower Akt2 activation levels were observed in IR macrophages correlating with increased glycolysis (138). Accordingly, Akt2 deficiency increased M2-like signature, while Akt1 deficiency led to increased M1-like inflammatory signatures, supporting that Akt2 silencing can phenocopy insulin resistant macrophages in macrophages (138, 147). Differential effects of Akt1 and Akt2 have also been described in neutrophils and adaptive immune cells (148, 149).
Nonetheless, whether immune cells become insulin resistant with longstanding exposure to hyperinsulinemia and inflammatory stimuli is only beginning to be addressed. Exposure to insulin at supraphysiological levels and/or extended duration can lead to IR and altered immune cell function in culture studies. Culture with high levels of insulin inhibited Treg-derived IL-10, and impaired the ability of Tregs to exert cytokine mediated immune suppression, potentially explaining reduced Treg function in obesity (35). Culture of macrophages with high insulin levels led to macrophage-intrinsic IR and altered metabolic programming (150), which could impact functional polarization. In mice, both short term (7 days) and long term (10 weeks) HFD was sufficient to turn thioglycolate-elicited peritoneal macrophages (TEPMs) into an insulin resistant state. As mentioned, in macrophages intrinsic insulin resistance was linked to an unique state characterized by defective Akt2, coupled to sustained mTORC1 activation, increased glycolysis, but with a reduced M1-like cytokine profile and induction of M2-like regulatory genes (e.g. Arg1, Fizz1, Ym1) (138).
Less is known about whether adaptive immune cells can become insulin resistant with chronic obesity. As stated earlier, B cells are potential candidates since they express the InsR to significant amounts basally and upon stimulation. In contrast, it will be interesting to examine to what extent T cells, which only upregulate InsR upon activation, can become insulin resistant, and the metabolic and immune signals that promote IR in T cells during infection and obesity. Understanding how insulin signaling pathways intersect with BCR, TCR, and TLR signaling under longstanding obesity in terms of effector functioning is an important avenue of further work, as well as how it relates to immunological memory, including trained immunity (see below). Such studies may also give insights on observed complications of obesity or aging, such as heightened innate immunity to danger, coupled to reduced adaptive immune function.
On this topic, it has been shown that simply infusing insulin to better glycemic control may not necessarily benefit all patients with viral infection. In some cases, insulin infusions during COVID-19 was linked to cytokine storm in obesity (151, 152). This model would fit an immune cell IR hypothesis if sufficient insulin is infused to overcome immune cell-intrinsic IR, or if it elicits a form of trained immunity. If this renewed insulin signal occurs in innate immune cells, then an ensuing glycolytic burst associated with cytokine storm and mortality may occur. Interestingly, other glucose lowering therapies do not show this same effect (152), and they might potentially work also in part by relieving intrinsic immune cell IR themselves. These intricacies highlight the significant, potentially life or death implications, that may be learned from studying insulin action on the immune system. Thus, “immune system IR” is an important, but developing concept that still needs further study for its validation.
Obesity fuels tumorigenic inflammation and alters cancer cell metabolism and growth (153). In addition, the growing tumor microenvironment presents challenges to the resident immune cells, such as hypoxia, nutrient deprivation, and the emergence of tumor-associated suppressive mechanisms. These challenges are further compounded by potential deleterious effects of obesity-associated hormonal and nutritional changes to dampen anti-tumor immunity.
In a mouse model of spontaneous pancreatic ductal adenocarcinoma (PDAC) with HFD-induced hyperinsulinemia, the reduction of insulin 2 gene expression (through Ins2 heterozygocity) in the Ins1 null background, led to reduced formation of early pancreatic lesions (154). The authors reported significant changes in immune cells in hyperinsulinemic PDAC-bearing mice using single cell sequencing analysis, with the greatest pathway alterations in B cells. Another reason for this protective effect, could be driven by insulin’s effect on the neutrophil-like, intratumoral MDSCs prevalent in early PDAC lesions (155). MDSCs require Akt-dependent phosphorylation of STAT3 for their suppressive activation (49), which is augmented by insulin signaling. In DCs, autophagy is required for antigen presentation (156). Autophagy inhibited by insulin-mTORC1 may also improve DC mediated cancer immune surveillance and antigen-specific responses explaining the protective effects in this model. While the immune cell intrinsic dysfunction caused by IR prior to PDAC remains to be fully understood, these studies suggest that insulin-mTORC1 driven inhibition of autophagy and Akt-STAT3 activation may drive tumor immune suppression via action on adaptive and innate immune cells.
These observations have important implications in metabolic syndrome, where loss of insulin receptiveness could differentially affect the ability of tumor infiltrating immune cell populations to compete with tumor cells for limiting nutrients and resources within the tumor microenvironment, leading to impaired anti-tumor function (157, 158). Additional investigative efforts are warranted to examine the role of insulin signaling and resistance in tumor-specific immunity.
Pre-eclampsia (PE) presents as high blood pressure and kidney damage and is correlated with the failure of immune homeostasis and increased inflammation at the maternal-fetal interface. Because both obesity and gestational diabetes are a pre-disposition to PE, researchers have studied the role of hyperglycemia and InsR/mTOR in the context of PE. Hyperglycemia resulted in the observed increase in AGEs in maternal blood and fetal tissues, which correlated significantly with an increased TNFα via the RAGE-NF-kB pathway (159). Moreover, as previously discussed, mTORC1 can dampen Treg responses which has also been suspected to invert the Th17/Treg balance in PE elevating responses to fetal antigens (160, 161).
In general, whole body IR increases the risk of infections, and infections also contribute to IR development and progression (75, 162). While the mechanisms underlying this association have recently begun to emerge, immunometabolism is particularly important in this context. Recent evidence suggests that T cells depend on insulin-induced signaling to respond against H1N1 influenza virus infection in mice by supporting T cell metabolic reprogramming and consequently, cell proliferation, activation, and cytokine production (5). Furthermore, since macrophages chronically exposed to insulin develop intrinsic IR and acquire a unique potentially anti-inflammatory phenotype coupled to an increased mTORC1-driven glycolytic profile (138), this undermines the resolution of infectious processes that requires pro-inflammatory macrophages. Neutrophils from diabetic rats also showed diminished phagocytosis and hydrogen peroxide production, which were recovered after insulin treatment (163). Although the impact of IR on metabolism in several immune cell subsets remains elusive, we anticipate that an impaired insulin sensitivity within immune and other cells alter cell phenotype and function, creating a favorable environmental condition for developing and progression of several infectious diseases.
The extent to which infection precedes and contributes to IR development is less clear. In general, it is plausible to consider that if the inflammatory status contributes to reduced insulin sensitivity, a sustained inflammatory response triggered by an infectious agent can also lead to IR (75, 164). In addition, specific therapies (e.g., steroids), hormones (e.g., cortisol), pro-inflammatory cytokines (e.g., TNFα, IL-1β, IL-6), nutrient status and type of pathogen may also potentiate or contribute to IR development. It is described that Staphylococcus aureus produces a protein (eLatS) with high affinity to insulin and blocks Insulin-InsR interaction, thus leading to IR (165).
The proposed bidirectional relationship between IR and infection has recently gained prominence with the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) (166). Various observational studies have found association between severe hyperglycemia and poor prognosis of COVID-19 (167). Noteworthy, COVID-19 patients without pre-existing metabolic-related diseases showed dysfunctional lipid and glucose metabolism and developed IR after discharge (168, 169). Moreover, another study described that both hyperglycemic and euglycemic patients hospitalized with COVID-19 showed impaired glucose metabolism and cytokine profile as well as developed IR at least 2 months after recovering from the disease (170). Whether reduced beta cell function as a result of SARS-CoV-2 infection is an additional contributor of these processes requires further examination (171). Induction of a transient insulin resistant, hyperinsulinemic state by a viral infection has been proposed as a physiological response to promote anti-viral CD8+ effector T cell activity (75). In the case of obesity, infection-induced persistent IR ultimately could be a maladaptive response if immune cells themselves also become IR, where deregulated glucose metabolism dampens antiviral immunity.
IR patients with uncontrolled blood glucose levels also have a greater risk of exacerbated COVID-19. Current studies showed that monocytes increase ROS-mediated HIF-1α stabilization which, in turn, enhance aerobic glycolysis during SARS-CoV-2 infection (172). These changes are associated with a glucose-enriched microenvironment, and they induce viral replication and cytokine storm, causing T cell dysfunction and lung epithelial cell death. The cytokine storm is also a hypothesized mechanism associated with COVID-19-related diabetes, since SARS-CoV-2 activates renin-angiotensin-aldosterone system (RAAS), which increases the production of IL-1α, IL-2, IL-6, TNFα, IFNγ, and MCP-1 and leads to pancreatic beta cell and systemic dysfunction (152, 173). Interestingly, insulin usage in patients in intensive care units is associated with increased COVID-19 mortality, possibly linked to increased glycolysis and associated cytokine storm, highlighting the importance and urgency of understanding pathological insulin action in the immune system (152). In the setting of chronic longstanding obesity and hyperglycemia, it is plausible that a high enough insulin dose could overcome intrinsic immune cell IR to facilitate increased glycolysis and cytokine storm, though more studies are needed to assess this possibility.
In general, blood glucose levels are reasonable predictors of disease severity in COVID-19 (174). Metformin, the most used antihyperglycemic drug, has also been prescribed as anti-inflammatory and immunomodulatory drug (175) with promising benefits to treat COVID-19. In addition to regulating glucose levels and increasing insulin sensitivity, metformin, as an activator of AMP-activated protein kinase (AMPK) and an endosomal pH modulator, could ameliorate disease through reducing the infectivity and survival of SARS-CoV-2 (176). Metformin also inhibits mitochondrial complex I and, consequently, oxidative stress and pro-inflammatory cytokine production, thereby promoting endothelial protection (173, 177). It will be interesting to determine if restoring insulin sensitivity inside IR immune cells during obesity is another mechanism of action by which metformin can boost immune cell clearance of SARS-CoV-2. Indeed, several studies have shown positive outcomes in metformin-receiving COVID-19 patients (178–180). However, the use of metformin must be avoided in patients with congestive heart failure and kidney diseases due to alterations in levels of other metabolic by products (181, 182).
More recently, the increased GP73 levels, a glucogenic hormone, were also correlated to SARS-CoV-2 infection in a mouse model, enhancing both glucogenesis and fasting blood glucose levels. The inhibition of GP73 was enough to restore the glucose levels, serving as another potential target to treat or avoid the progression to IR in COVID-19 (183). Overall, there is a bidirectional causal association between IR and infection. However, the mechanisms describing how infection leads to IR development, including the role of immune system metabolism in this context, still requires further investigation.
Emerging evidence suggests that the interplay between insulin and other pro-inflammatory signaling pathways contributes to trained immunity (reviewed in 184). Trained immunity is observed in innate immune cells, as a parallel to adaptive cell memory, and is defined as the long-term reprogramming by primary inflammatory insults that leads to an altered response to secondary challenges (185, 186). Innate cells are thought to achieve trained immunity via two interconnecting mechanisms: 1) biasing of metabolic circuits and 2) epigenetic memory established during primary exposure (185). Inflammatory stimuli like infections and vaccinations drive trained immunity via PRR driven activation of Akt-mTORC1 and NF-kB (185). The pro-inflammatory metabolic profiles these pathways drive remain active after the primary stimuli, despite complete resolution of inflammation, to drive more rapid pro-inflammatory responses to secondary stimuli (187). As previously detailed, InsR/IGF1R signaling also synergizes with Akt-mTORC1 and TLR-NF-kB activation. By boosting glycolytic cell metabolism and inflammatory cytokine production, insulin signaling also partakes in trained immunity.
In chronic obesity-associated IR, peripheral macrophages may take on an insulin resistant M2-like phenotype (138), while some adipose tissue macrophages, such as recruited types, display inflammatory M1-like polarization (188). The IR-dependent aberrant Akt signaling sustains mTORC1 activation resulting in enhanced glycolysis, which can occur acutely with insulin action or chronically during an insulin resistant immune state (138). Thus, both acute hyperinsulinemia driving Akt and mTORC1 activity, as well as an altered insulin resistant macrophage during chronic obesity with increased basal glycolysis and mTORC1 activity, might both facilitate downstream effects known to induce trained immunity (184). Some of these effects might be on mTOR-HIF-1α signaling, which alters metabolites to mediate histone modifications for trained immunity (189, 190), though more work is needed to dissect out pertinent metabolites in these settings. Further studies to test this idea are also needed to better delineate effects of insulin action and resistance in recruited vs resident macrophages, and in the setting of innate tolerance, which is considered distinct from trained immunity (186). This direction of work is especially important to reconcile observed phenotypes in immune function during chronic obesity, and whether these occur due to intrinsic immune system IR, such as discussed in the previous section on Insulin Resistance in the Immune System - an emerging paradigm? For instance, it may be possible that reduced insulin action cripples adaptive immunity but potentiates some forms of innate immunity through trained immunity.
The second key mediator of trained immunity is epigenetic memory, governing access to inflammatory gene loci. Systemic hyperglycemia was correlated with epigenetic marks like (e.g., H3K9me3 and H3K4me3) resulting in sustained NF-kB transcriptional activity (191). Elevated oxidized low density lipoprotein (OxLDL) levels observed in IR also promoted H3K4me3 methylation in monocytes driving NF-kB driven cytokine production (192). Epigenetic reprogramming at the hematopoietic stem cell level also leads to a higher myeloid to lymphoid ratio upon re-activation (193). These systemic consequences of whole body IR may also contribute to trained immunity (184).
An emerging hallmark of aging is a state of chronic low-grade inflammation, which is also a characteristic of obesity. This state is known as inflammaging, and it is coupled to overall dysfunction, termed immunosenescence (194). Mechanisms fueling dysfunction of the aged immune system and onset of immunosenescence are of broad interest. As well it is of interest to identify biomarkers and develop therapeutic targets for age-related chronic disease.
Insulin signaling is an evolutionarily conserved axis that correlates negatively with longevity across several species, including Caenorhabditis elegans (195), Drosophila melanogaster (196), Harpegnathos saltator (197) and mice (198, 199). Notably, aging in mice is associated with a declined capacity for insulin clearance resulting in hyperinsulinemia (200). In humans, low plasma insulin levels and high insulin sensitivity have been attributed to improved survival and are key features observed in centenarians, suggesting a key role for insulin in regulating human lifespan (201).
Low level chronic activation of the InsR during aging might explain inflammatory shifts within the immune system that are observed with age, including the emergence of pro-inflammatory cell types (202). In the adaptive immune compartment, InsR engagement can constitutively inhibit FOXO1, a pro-longevity transcription factor whose deletion, along with FOXO3, in T cells heightens levels of pro-inflammatory cytokines, such as IFNγ or IL-17 (203). FOXO1 is also required to suppress a state of activation in CD8+ T cells, thereby allowing antigen-activated and expanded CD8+ T cells to maintain self-renewal and preventing their senescence (204–206). In C. elegans, genetic inhibition of DAF-2 (C. elegans InsR/IGF1R homolog) is shown to enhance immunocompetence, while DAF-16 (C. elegans FOXO homolog) is mainly responsible in delaying immune aging in DAF-2 mutants (195). These observations support the notion that a dysfunctional InsR/IGF1R pathway during aging might underlie the impaired immune responses of elderly individuals.
The interplay between aging and tissue infiltrating Tregs has also gained increasing attention over recent years. During aging, FoxP3+ Tregs secreting IL-10 are sustained by IL-6, a major cytokine of age, and accumulate across different tissues in old mice. This increase in Tregs may occur possibly to compensate for the low-grade unresolving inflammation associated with aging (207, 208). Single cell analyses have unveiled that aged Tregs possess an activated phenotype and display enhanced suppressive abilities compared to young counterparts, akin to those observed within tumors of patients with solid cancers (209). Interestingly, inside VAT, FoxP3+ Tregs accumulation has been shown to be associated with aging-related insulin resistance (210). Consistently, FoxP3+ Treg-specific InsR knockout mice aged to 52 weeks under normal chow diet were protected from the metabolic effects of aging. Improved metabolism in aged Treg InsR knockout mice were linked to significantly reduced numbers of both ST2+ and ST2− Tregs in VAT, but not in brown adipose tissue (76). Interestingly, the VAT of the aged mutant mice also showed increased expression of inflammatory genes, such as Ifng, Tnf, Il1b, and Tlr2, implicating that some pro-inflammatory regulation can be favorable in restoring insulin sensitivity in age-associated glucose metabolism (76). Thus, insulin action on resident metabolic tissue immune cells is likely sufficient to dictate metabolic and tissue inflammatory outcomes with age.
Multiple drugs with anti-aging properties also link nutrient sensing to insulin action with potential ramification on immune function. Rapamycin, an anti-inflammatory drug inhibiting insulin-mTORC1 axis, has long been considered effective in extending lifespan (211), boosting immunity in the elderly (212), and delaying several age-related diseases (213–216). The mechanism of this immunostimulatory effect of rapamycin are not well characterized, but could be linked to inhibition of mTORC1-mediated S6 kinase negative feedback on age-associated chronic insulin signaling (5, 217). Similarly, metformin is another therapeutic that promotes anti-aging, pro-longevity, and improved health span outcomes by downregulating InsR-mTORC1 activity and the overall immunosenescence burden (218). The idea that rapamycin and metformin might improve insulin sensitivity within immune cells as one mechanism of immune rejuvenation remains to be validated.
Another set of anti-aging compounds, senolytic drugs, such as quercetin, dasatinib (Q+D) and fisetin, have shown protective effects against aging, and the ability to prevent adverse outcomes against infectious diseases, such as COVID-19 (219, 220). Interestingly, quercetin, may also have some capacity to block aspects of InsR action, though these effects were mostly seen in vitro, in higher doses. Nonetheless, this mechanism might be worthwhile to further investigate as an immunoregulatory agent to regulate nutrient-sensing networks during age-related metabolic disorders (221). Consistently, insulin infusion has been linked to cytokine storm, increased glycolysis and unopposed immune cell activation with COVID-19 (5, 152, 217). Senolytic drugs possess a “hit-and-run” activity, meaning that they reach maximal efficacy while minimizing side effects with only intermittent administration (222). This property might make senolytics a promising therapy over rapamycin for the aging population, considering that long-term rapamycin intake potentiates IR by inhibiting mTORC2 (223).
Recent advances highlight the existence of an endocrine-immune axis, where insulin plays a critical role in regulating immune cell function and metabolism. However, much remains to be learned regarding the molecular basis of this endocrine-immune interaction. Given that multiple arms of the InsR signaling pathway converge with those downstream of antigen and costimulatory receptors, PRRs, cytokines and nutrient sensing pathways, future efforts are needed to tease apart the signaling networks insulin uses in specific immune cell types and how this is regulated in the face of different stimuli. We propose that Akt and mTORC1 are central regulatory hubs that integrate insulin signals with immune receptors and metabolic signals, given their established role in these pathways, both as a signal transducer/integrator and an off switch for negative feedback. This notion would fit with a model where homeostatic levels of insulin signaling allow for optimal immune responses, while perturbed signaling, such as during IR states where chronic inflammation and hyperinsulinemia foster immune cell-intrinsic IR, facilitates immune dysfunction. In the context of metabolic syndrome, systemic IR and associated hyperglycemia, dyslipidemia, and inflammation can act hand in hand with immune cell-intrinsic IR, coupled to chronic basal activation of danger signals like TLRs, to further dysregulate immune cell metabolism and functional outcomes. The net result would yield immune cells with a basal inflammatory tone, unable to boost its metabolism in the face of new antigen, which are hallmarks of immune dysfunction in obesity and aging. Whether aspects of trained immunity or tolerance (e.g. sustained TLR ligation) also exist during the insulin resistant state in innate immune cells during chronic obesity is an avenue of future research.
As outlined in Figure 3, disrupted insulin-mediated metabolic homeostasis perpetuates a chronic meta-inflammatory loop that facilitates immune dysfunction. Conditions such as over-nutrition, aging, and pregnancy, as well as genetic and environmental factors could predispose individuals to developing IR, a state in which cells become impaired at perceiving insulin signals. A compensatory rise in insulin production to offset this impairment then leads to hyperinsulinemia and beta cell stress followed by functional decline. IR-associated nutrient imbalance, such as hyperglycemia and dyslipidemia, especially early in disease, together with potential intrinsic immune system IR, likely occurring later in disease progression, could thus drive dysregulation in immune cells with both pro-inflammatory and suppressive consequences, and contribute to the pleiotropic mechanisms of pathophysiology. These metabolic-immune interactions contribute to the pathogenesis of obesity, cancer, viral infection, pre-eclampsia, and age-related degeneration.
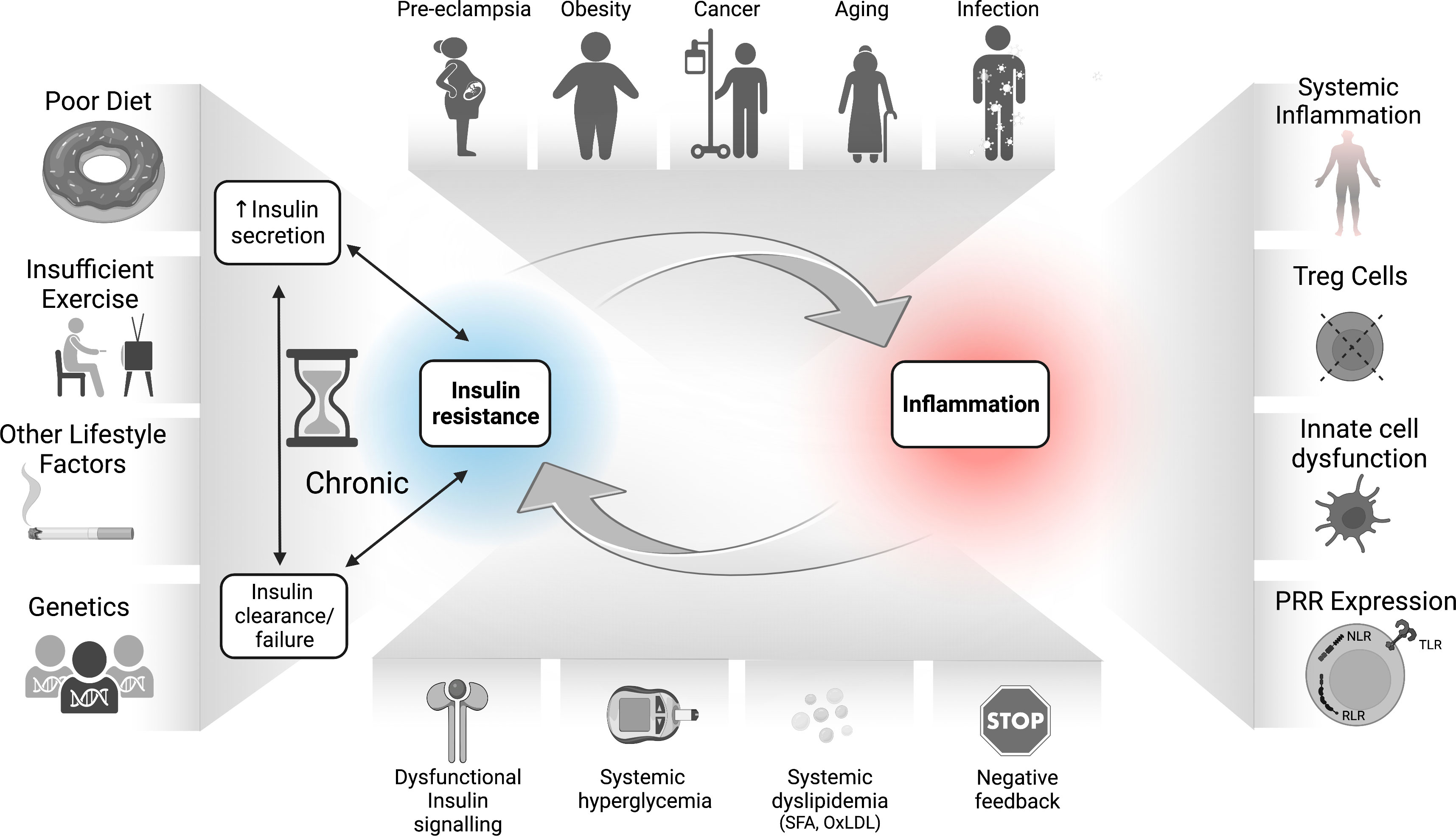
Figure 3 The bidirectional relationship between IR and inflammation in the pathogenesis of inflammatory diseases. Environmental, genetic, and dietary factors and their inflammatory responses can lead to hyperinsulinemia which drives dysfunctional responses in immune cells, including increased inflammatory tone and altered Treg function. The sustained IR-associated hyperglycemia and dyslipidemia also drive SFA and OxLDL production, increased RAGE activation, epigenetic changes, and accumulation of negative feedback signals on the InsR. Together with increased inflammation and PRR signaling, these immune/metabolic changes lead to whole-body as well as potentially immune cell-intrinsic IR. Immune cell IR in turn may undermine protective immunity and/or immune tolerance, which further exacerbates disease pathology. IR, insulin resistance; NF-kB, nuclear factor kappa B; mTORC1, mechanistic (or mammalian) target of rapamycin complex 1; NLR, nucleotide oligomerization domain (NOD)-like receptors; OxLDL, oxidized low density lipoprotein (LDL); PRR, pattern recognition receptor; RAGEs, receptor for advanced glycation end products; RLR, retinoic acid-inducible gene I-like receptors; SFA, saturated fatty acids; TLR, toll-like receptors; Treg, regulatory T cells.
From a therapy perspective, the declined protective immunity in obese and/or aged insulin resistant individuals to infections and cancer raises the important question of whether and how insulin signaling pathway can be harnessed to rejuvenate the immune system. Strategies that dampen obesity or age-related inflammation would exert systemic beneficial, insulin-sensitizing effects. Insulin sensitizing agents such as AMPK agonists, including metformin, are potential candidates as they already are commonly prescribed medications in patients with T2DM, and mediate improved insulin sensitivity by increasing InsR tyrosine kinase activity, and increasing the recruitment and activity of the glucose transporters. In immune cells, this effect is expected to reactivate cell intrinsic insulin response.
Overall, immune cell insulin receptor signaling could represent a critical missing link in understanding manifestations and complications of obesity- and age-associated immune dysfunction. Considering the recurrent emergence of obesity, diabetes and aging as leading risk factors for poor clinical outcomes, especially in the context of severe respiratory infections, vaccination, cancer, and many other inflammatory diseases, a better understanding of the molecular basis of insulin-mediated immune regulation is imperative and may contribute to the design of new targeted strategies.
DW, PM, PB, ST, YC, NC, and JB wrote parts of the manuscript, and/or generated figures/tables. SK and DF provided feedback. All authors had a chance to review the final manuscript. All authors contributed to the article and approved the submitted version.
This work was funded in part through funds derived from the Buck Institute for Research on Aging (DW) and the National Institutes of Health (R01DK128435) (DW).
Figures for this manuscript were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol (2014) 6:a009191. doi: 10.1101/cshperspect.a009191
2. van Niekerk G, Christowitz C, Conradie D, Engelbrecht A-M. Insulin as an immunomodulatory hormone. Cytokine Growth Factor Rev (2020) 52:34–44. doi: 10.1016/j.cytogfr.2019.11.006
3. Mauer J, Chaurasia B, Plum L, Quast T, Hampel B, Blüher M, et al. Myeloid cell-restricted insulin receptor deficiency protects against obesity-induced inflammation and systemic insulin resistance. PloS Genet (2010) 6:e1000938. doi: 10.1371/journal.pgen.1000938
4. Helderman JH, Strom TB. Specific insulin binding site on T and b lymphocytes as a marker of cell activation. Nature (1978) 274:62–3. doi: 10.1038/274062a0
5. Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, Chan YT, et al. Insulin receptor-mediated stimulation boosts T cell immunity during inflammation and infection. Cell Metab (2018) 28:922–934.e4. doi: 10.1016/j.cmet.2018.08.003
6. Haeusler RA, McGraw TE, Accili D. Biochemical and cellular properties of insulin receptor signalling. Nat Rev Mol Cell Biol (2018) 19:31–44. doi: 10.1038/nrm.2017.89
7. Chao LH, Avruch J. Cryo-EM insight into the structure of MTOR complex 1 and its interactions with rheb and substrates. F1000Research (2019) 8:F1000. doi: 10.12688/f1000research.16109.1
8. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol (2012) 30:39–68. doi: 10.1146/annurev-immunol-020711-075024
9. Kubota T, Inoue M, Kubota N, Takamoto I, Mineyama T, Iwayama K, et al. Downregulation of macrophage Irs2 by hyperinsulinemia impairs IL-4-indeuced M2a-subtype macrophage activation in obesity. Nat Commun (2018) 9:4863. doi: 10.1038/s41467-018-07358-9
10. Guerau-de-Arellano M, Piedra-Quintero ZL, Tsichlis PN. Akt isoforms in the immune system. Front Immunol (2022) 13:990874. doi: 10.3389/fimmu.2022.990874
11. Lamb RF. Negative feedback loops: Nutrient starvation employs a new tr(IKK) to inhibit PI3K. Mol Cell (2012) 45:705–6. doi: 10.1016/j.molcel.2012.03.015
12. D’Souza WN, Chang C-F, Fischer AM, Li M, Hedrick SM. The Erk2 MAPK regulates CD8 T cell proliferation and survival. J Immunol Baltim Md 1950 (2008) 181:7617–29. doi: 10.4049/jimmunol.181.11.7617
13. Fischer AM, Katayama CD, Pagès G, Pouysségur J, Hedrick SM. The role of Erk1 and Erk2 in multiple stages of T cell development. Immunity (2005) 23:431–43. doi: 10.1016/j.immuni.2005.08.013
14. Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES, et al. Inflammasome priming by LPS is dependent upon ERK signaling and proteasome function. J Immunol Baltim Md 1950 (2014) 192:3881–8. doi: 10.4049/jimmunol.1301974
15. Través PG, de Atauri P, Marín S, Pimentel-Santillana M, Rodríguez-Prados J-C, Marín de Mas I, et al. Relevance of the MEK/ERK signaling pathway in the metabolism of activated macrophages: A metabolomic approach. J Immunol Baltim Md 1950 (2012) 188:1402–10. doi: 10.4049/jimmunol.1101781
16. Fu Y, Maianu L, Melbert BR, Garvey WT. Facilitative glucose transporter gene expression in human lymphocytes, monocytes, and macrophages: A role for GLUT isoforms 1, 3, and 5 in the immune response and foam cell formation. Blood Cells Mol Dis (2004) 32:182–90. doi: 10.1016/j.bcmd.2003.09.002
17. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia (2012) 55:2565–82. doi: 10.1007/s00125-012-2644-8
18. Hancock ML, Meyer RC, Mistry M, Khetani RS, Wagschal A, Shin T, et al. Insulin receptor associates with promoters genome-wide and regulates gene expression. Cell (2019) 177:722–736.e22. doi: 10.1016/j.cell.2019.02.030
19. Bar RS, Gorden P, Roth J, Kahn CR, De Meyts P. Fluctuations in the affinity and concentration of insulin receptors on circulating monocytes of obese patients: Effects of starvation, refeeding, and dieting. J Clin Invest (1976) 58:1123–35. doi: 10.1172/JCI108565
20. Higashi Y, Sukhanov S, Shai S-Y, Danchuk S, Tang R, Snarski P, et al. Insulin-like growth factor-1 receptor deficiency in macrophages accelerates atherosclerosis and induces an unstable plaque phenotype in apolipoprotein e-deficient mice. Circulation (2016) 133:2263–78. doi: 10.1161/CIRCULATIONAHA.116.021805
21. Tessaro FHG, Ayala TS, Nolasco EL, Bella LM, Martins JO. Insulin influences LPS-induced TNF-α and IL-6 release through distinct pathways in mouse macrophages from different compartments. Cell Physiol Biochem (2017) 42:2093–104. doi: 10.1159/000479904
22. Ratter JM, van Heck JIP, Rooijackers HMM, Jansen HJ, van Poppel PCM, Tack CJ, et al. Insulin acutely activates metabolism of primary human monocytes and promotes a proinflammatory phenotype. J Leukoc Biol (2021) 110:885–91. doi: 10.1002/JLB.3AB0120-019RR
23. Knuever J, Willenborg S, Ding X, Akyüz MD, Partridge L, Niessen CM, et al. Myeloid cell–restricted Insulin/IGF-1 receptor deficiency protects against skin inflammation. J Immunol (2015) 195:5296–308. doi: 10.4049/jimmunol.1501237
24. Baumgartl J, Baudler S, Scherner M, Babaev V, Makowski L, Suttles J, et al. Myeloid lineage cell-restricted insulin resistance protects apolipoproteinE-deficient mice against atherosclerosis. Cell Metab (2006) 3:247–56. doi: 10.1016/j.cmet.2006.02.010
25. Lu H, Huang D, Yao K, Li C, Chang S, Dai Y, et al. Insulin enhances dendritic cell maturation and scavenger receptor-mediated uptake of oxidised low-density lipoprotein. J Diabetes Complications (2015) 29:465–71. doi: 10.1016/j.jdiacomp.2015.03.005
26. Safronova VG, Gabdoulkhakova AG, Miller AV, Kosarev IV, Vasilenko RN. Variations of the effect of insulin on neutrophil respiratory burst. the role of tyrosine kinases and phosphatases. Biochem Mosc (2001) 66:840–9. doi: 10.1023/A:1011944400908
27. Himpe E, Degaillier C, Coppens A, Kooijman R. Insulin-like growth factor-1 delays fas-mediated apoptosis in human neutrophils through the phosphatidylinositol-3 kinase pathway. J Endocrinol (2008) 199:69–80. doi: 10.1677/JOE-08-0028
28. Ferreira SS, Nunes FPB, Casagrande FB, Martins JO. Insulin modulates cytokine release, collagen and mucus secretion in lung remodeling of allergic diabetic mice. Front Immunol (2017) 8:633. doi: 10.3389/fimmu.2017.00633
29. Oherle K, Acker E, Bonfield M, Wang T, Gray J, Lang I, et al. Insulin-like growth factor 1 supports a pulmonary niche that promotes type 3 innate lymphoid cell development in newborn lungs. Immunity (2020) 52:275–294.e9. doi: 10.1016/j.immuni.2020.01.005
30. DeBenedette M, Snow EC. Insulin modulates the interleukin 2 responsiveness of T lymphocytes. Reg Immunol (1990) 3:82–7.
31. Berman JS, Center DM. Chemotactic activity of porcine insulin for human T lymphocytes in vitro. J Immunol Baltim Md 1950 (1987) 138:2100–3. doi: 10.4049/jimmunol.138.7.2100
32. Fischer HJ, Sie C, Schumann E, Witte A-K, Dressel R, van den Brandt J, et al. The insulin receptor plays a critical role in T cell function and adaptive immunity. J Immunol Baltim Md 1950 (2017) 198:1910–20. doi: 10.4049/jimmunol.1601011
33. DiToro D, Harbour SN, Bando JK, Benavides G, Witte S, Laufer VA, et al. Insulin-like growth factors are key regulators of T helper 17 regulatory T cell balance in autoimmunity. Immunity (2020) 52:650–667.e10. doi: 10.1016/j.immuni.2020.03.013
34. Han JM, Patterson SJ, Speck M, Ehses JA, Levings MK. Insulin inhibits IL-10–mediated regulatory T cell function: Implications for obesity. J Immunol (2014) 192:623–9. doi: 10.4049/jimmunol.1302181
35. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, Castegna A. The metabolic signature of macrophage responses. Front Immunol (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
36. Jha AK, Huang SC-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
37. Wang Y, Li N, Zhang X, Horng T. Mitochondrial metabolism regulates macrophage biology. J Biol Chem (2021) 297:100904. doi: 10.1016/j.jbc.2021.100904
38. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab (2018) 28:463–475.e4. doi: 10.1016/j.cmet.2018.08.012
39. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
40. Jiang H, Westerterp M, Wang C, Zhu Y, Ai D. Macrophage mTORC1 disruption reduces inflammation and insulin resistance in obese mice. Diabetologia (2014) 57:2393–404. doi: 10.1007/s00125-014-3350-5
41. Bosc C, Broin N, Fanjul M, Saland E, Farge T, Courdy C, et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat Commun (2020) 11:4056. doi: 10.1038/s41467-020-17882-2
42. de-Brito NM, Duncan-Moretti J, da-Costa HC, Saldanha-Gama R, Paula-Neto HA, Dorighello GG, et al. Aerobic glycolysis is a metabolic requirement to maintain the M2-like polarization of tumor-associated macrophages. Biochim Biophys Acta BBA - Mol Cell Res (2020) 1867:118604. doi: 10.1016/j.bbamcr.2019.118604
43. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol (2008) 8:958–69. doi: 10.1038/nri2448
44. Hannigan MO, Huang CK, Wu DQ. Roles of PI3K in neutrophil function. Curr Top Microbiol Immunol (2004) 282:165–75. doi: 10.1007/978-3-642-18805-3_6
45. McInturff AM, Cody MJ, Elliott EA, Glenn JW, Rowley JW, Rondina MT, et al. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 α. Blood (2012) 120:3118–25. doi: 10.1182/blood-2012-01-405993
46. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
47. Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, et al. Oncogenic mTOR signaling recruits myeloid-derived suppressor cells to promote tumor initiation. Nat Cell Biol (2016) 18:632–44. doi: 10.1038/ncb3355
48. Salminen A, Kaarniranta K, Kauppinen A. Insulin/IGF-1 signaling promotes immunosuppression via the STAT3 pathway: Impact on the aging process and age-related diseases. Inflammation Res (2021) 70:1043–61. doi: 10.1007/s00011-021-01498-3
49. Alissafi T, Hatzioannou A, Mintzas K, Barouni RM, Banos A, Sormendi S, et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J Clin Invest (2018) 128:3840–52. doi: 10.1172/JCI120888
50. Weller PF, Spencer LA. Functions of tissue-resident eosinophils. Nat Rev Immunol (2017) 17:746–60. doi: 10.1038/nri.2017.95
51. Esnault S, Kelly EA, Schwantes EA, Liu LY, DeLain LP, Hauer JA, et al. Identification of genes expressed by human airway eosinophils after an in vivo allergen challenge. PloS One (2013) 8:e67560. doi: 10.1371/journal.pone.0067560
52. Porter L, Toepfner N, Bashant KR, Guck J, Ashcroft M, Farahi N, et al. Metabolic profiling of human eosinophils. Front Immunol (2018) 9:1404. doi: 10.3389/fimmu.2018.01404
53. Wu D, Molofsky AB, Liang H-E, Ricardo-Gonzalez RR, Jouihan HA, Bando JK, et al. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science (2011) 332:243–7. doi: 10.1126/science.1201475
54. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol (2014) 15:323–32. doi: 10.1038/ni.2833
55. Cao W, Manicassamy S, Tang H, Kasturi SP, Pirani A, Murthy N, et al. Toll-like receptor-mediated induction of type I interferon in plasmacytoid dendritic cells requires the rapamycin-sensitive PI(3)K-mTOR-p70S6K pathway. Nat Immunol (2008) 9:1157–64. doi: 10.1038/ni.1645
56. Sukhbaatar N, Hengstschläger M, Weichhart T. mTOR-mediated regulation of dendritic cell differentiation and function. Trends Immunol (2016) 37:778–89. doi: 10.1016/j.it.2016.08.009
57. Pelgrom LR, Patente TA, Otto F, Nouwen LV, Ozir-Fazalalikhan A, van der Ham AJ, et al. mTORC1 signaling in antigen-presenting cells of the skin restrains CD8+ T cell priming. Cell Rep (2022) 40:111032. doi: 10.1016/j.celrep.2022.111032
58. Almeida FF, Belz GT. Innate lymphoid cells: Models of plasticity for immune homeostasis and rapid responsiveness in protection. Mucosal Immunol (2016) 9:1103–12. doi: 10.1038/mi.2016.64
59. Watzl C, Long EO. Signal transduction during activation and inhibition of natural killer cells. Curr Protoc Immunol (2010) 90:11.9B.1–11.9B.17. doi: 10.1002/0471142735.im1109bs90
60. Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for interleukin-15 signaling during NK cell development and activation. Nat Immunol (2014) 15:749–57. doi: 10.1038/ni.2936
61. O’Sullivan TE, Geary CD, Weizman O-E, Geiger TL, Rapp M, Dorn GW, et al. Atg5 is essential for the development and survival of innate lymphocytes. Cell Rep (2016) 15:1910–9. doi: 10.1016/j.celrep.2016.04.082
62. Karagiannis F, Masouleh SK, Wunderling K, Surendar J, Schmitt V, Kazakov A, et al. Lipid-droplet formation drives pathogenic group 2 innate lymphoid cells in airway inflammation. Immunity (2020) 52:620–634.e6. doi: 10.1016/j.immuni.2020.03.003
63. Surace L, Doisne J-M, Croft CA, Thaller A, Escoll P, Marie S, et al. Dichotomous metabolic networks govern human ILC2 proliferation and function. Nat Immunol (2021) 22:1367–74. doi: 10.1038/s41590-021-01043-8
64. Di Luccia B, Gilfillan S, Cella M, Colonna M, Huang SC-C. ILC3s integrate glycolysis and mitochondrial production of reactive oxygen species to fulfill activation demands. J Exp Med (2019) 216:2231–41. doi: 10.1084/jem.20180549
65. Zhang Z, Amorosa LF, Coyle SM, Macor MA, Birnbaum MJ, Lee LY, et al. Insulin-dependent regulation of mTORC2-Akt-FoxO suppresses TLR4 signaling in human leukocytes: Relevance to type 2 diabetes. Diabetes (2016) 65:2224–34. doi: 10.2337/db16-0027
66. Ghanim H, Mohanty P, Deopurkar R, Ling Sia C, Korzeniewski K, Abuaysheh S, et al. Acute modulation of toll-like receptors by insulin. Diabetes Care (2008) 31:1827–31. doi: 10.2337/dc08-0561
67. Graves DT, Milovanova TN. Mucosal immunity and the FOXO1 transcription factors. Front Immunol (2019) 10:2530. doi: 10.3389/fimmu.2019.02530
68. Yan K, Da T-T, Bian Z-H, He Y, Liu M-C, Liu Q-Z, et al. Multi-omics analysis identifies FoxO1 as a regulator of macrophage function through metabolic reprogramming. Cell Death Dis (2020) 11:1–14. doi: 10.1038/s41419-020-02982-0
69. Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes (2011) 60(2):625–33. doi: 10.2337/db10-1164
70. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature (2018) 556:113–7. doi: 10.1038/nature25986
71. Ruiz HH, Nguyen A, Wang C, He L, Li H, Hallowell P, et al. AGE/RAGE/DIAPH1 axis is associated with immunometabolic markers and risk of insulin resistance in subcutaneous but not omental adipose tissue in human obesity. Int J Obes (2021) 45:2083–94. doi: 10.1038/s41366-021-00878-3
72. Helderman JH, Reynolds TC, Strom TB. The insulin receptor as a universal marker of activated lymphocytes. Eur J Immunol (1978) 8:589–95. doi: 10.1002/eji.1830080810
73. Viardot A, Heilbronn LK, Samocha-Bonet D, Mackay F, Campbell LV, Samaras K. Obesity is associated with activated and insulin resistant immune cells. Diabetes Metab Res Rev (2012) 28:447–54. doi: 10.1002/dmrr.2302
74. Baudler S, Baumgartl J, Hampel B, Buch T, Waisman A, Snapper CM, et al. Insulin-like growth factor-1 controls type 2 T cell-independent B cell response. J Immunol (2005) 174:5516–25. doi: 10.4049/jimmunol.174.9.5516
75. Šestan M, Marinović S, Kavazović I, Cekinović Đ, Wueest S, Turk Wensveen T, et al. Virus-induced interferon-γ causes insulin resistance in skeletal muscle and derails glycemic control in obesity. Immunity (2018) 49:164–177.e6. doi: 10.1016/j.immuni.2018.05.005
76. Wu D, Wong CK, Han JM, Orban PC, Huang Q, Gillies J, et al. T Reg-specific insulin receptor deletion prevents diet-induced and age-associated metabolic syndrome. J Exp Med (2020) 217:e20191542. doi: 10.1084/jem.20191542
77. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang L-S, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature (2009) 460:103–7. doi: 10.1038/nature08097
78. Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature (2009) 460:108–12. doi: 10.1038/nature08155
79. Kavazović I, Krapić M, Beumer-Chuwonpad A, Polić B, Turk Wensveen T, Lemmermann NA, et al. Hyperglycemia and not hyperinsulinemia mediates diabetes-induced memory CD8 T-cell dysfunction. Diabetes (2022) 71:706–21. doi: 10.2337/db21-0209
80. Mirdamadi Y, Bommhardt U, Goihl A, Guttek K, Zouboulis CC, Quist S, et al. Insulin and insulin-like growth factor-1 can activate the phosphoinositide-3-kinase /Akt/FoxO1 pathway in T cells in vitro. Dermatoendocrinol (2017) 9:e1356518. doi: 10.1080/19381980.2017.1356518
81. Bilbao D, Luciani L, Johannesson B, Piszczek A, Rosenthal N. Insulin-like growth factor-1 stimulates regulatory T cells and suppresses autoimmune disease. EMBO Mol Med (2014) 6:1423–35. doi: 10.15252/emmm.201303376
82. Miyagawa I, Nakayamada S, Nakano K, Yamagata K, Sakata K, Yamaoka K, et al. Induction of regulatory T cells and its regulation with insulin-like growth Factor/Insulin-like growth factor binding protein-4 by human mesenchymal stem cells. J Immunol Baltim Md 1950 (2017) 199:1616–25. doi: 10.4049/jimmunol.1600230
83. Johannesson B, Sattler S, Semenova E, Pastore S, Kennedy-Lydon TM, Sampson RD, et al. Insulin-like growth factor-1 induces regulatory T cell-mediated suppression of allergic contact dermatitis in mice. Dis Model Mech (2014) 7:977–85. doi: 10.1242/dmm.015362
84. Winer DA, Winer S, Chng MHY, Shen L, Engleman EG. B lymphocytes in obesity-related adipose tissue inflammation and insulin resistance. Cell Mol Life Sci CMLS (2014) 71:1033–43. doi: 10.1007/s00018-013-1486-y
85. Viardot A, Grey ST, Mackay F, Chisholm D. Potential antiinflammatory role of insulin via the preferential polarization of effector T cells toward a T helper 2 phenotype. Endocrinology (2007) 148:346–53. doi: 10.1210/en.2006-0686
86. Baracho GV, Cato MH, Zhu Z, Jaren OR, Hobeika E, Reth M, et al. PDK1 regulates B cell differentiation and homeostasis. Proc Natl Acad Sci U.S.A. (2014) 111:9573–8. doi: 10.1073/pnas.1314562111
87. Calamito M, Juntilla MM, Thomas M, Northrup DL, Rathmell J, Birnbaum MJ, et al. Akt1 and Akt2 promote peripheral B-cell maturation and survival. Blood (2010) 115:4043–50. doi: 10.1182/blood-2009-09-241638
88. Durand CA, Hartvigsen K, Fogelstrand L, Kim S, Iritani S, Vanhaesebroeck B, et al. Phosphoinositide 3-kinase p110 delta regulates natural antibody production, marginal zone and B-1 B cell function, and autoantibody responses. J Immunol Baltim Md 1950 (2009) 183:5673–84. doi: 10.4049/jimmunol.0900432
89. Omori SA, Cato MH, Anzelon-Mills A, Puri KD, Shapiro-Shelef M, Calame K, et al. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity (2006) 25:545–57. doi: 10.1016/j.immuni.2006.08.015
90. Suzuki A, Kaisho T, Ohishi M, Tsukio-Yamaguchi M, Tsubata T, Koni PA, et al. Critical roles of pten in B cell homeostasis and immunoglobulin class switch recombination. J Exp Med (2003) 197:657–67. doi: 10.1084/jem.20021101
91. Park S-G, Long M, Kang J-A, Kim W-S, Lee C-R, Im S-H, et al. The kinase PDK1 is essential for B-cell receptor mediated survival signaling. PloS One (2013) 8:e55378. doi: 10.1371/journal.pone.0055378
92. Baracho GV, Miletic AV, Omori SA, Cato MH, Rickert RC. Emergence of the PI3-kinase pathway as a central modulator of normal and aberrant B cell differentiation. Curr Opin Immunol (2011) 23:178–83. doi: 10.1016/j.coi.2011.01.001
93. Jellusova J, Miletic AV, Cato MH, Lin W-W, Hu Y, Bishop GA, et al. Context-specific BAFF-r signaling by the NF-κB and PI3K pathways. Cell Rep (2013) 5:1022–35. doi: 10.1016/j.celrep.2013.10.022
94. Kaileh M, Vazquez E, MacFarlane AW, Campbell K, Kurosaki T, Siebenlist U, et al. mTOR-dependent and independent survival signaling by PI3K in B lymphocytes. PloS One (2016) 11:e0146955. doi: 10.1371/journal.pone.0146955
95. Tamahara T, Ochiai K, Muto A, Kato Y, Sax N, Matsumoto M, et al. The mTOR-Bach2 cascade controls cell cycle and class switch recombination during B cell differentiation. Mol Cell Biol (2017) 37:e00418–17. doi: 10.1128/MCB.00418-17
96. Salih DAM, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol (2008) 20:126–36. doi: 10.1016/j.ceb.2008.02.005
97. Chen X, Ma J, Yao Y, Zhu J, Zhou Z, Zhao R, et al. Metformin prevents BAFF activation of Erk1/2 from B-cell proliferation and survival by impeding mTOR-PTEN/Akt signaling pathway. Int Immunopharmacol (2021) 96:107771. doi: 10.1016/j.intimp.2021.107771
98. Zhang T, Makondo KJ, Marshall AJ. p110δ phosphoinositide 3-kinase represses IgE switch by potentiating BCL6 expression. J Immunol Baltim Md 1950 (2012) 188:3700–8. doi: 10.4049/jimmunol.1103302
99. Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature (1995) 376:352–5. doi: 10.1038/376352a0
100. Jou S-T, Carpino N, Takahashi Y, Piekorz R, Chao J-R, Carpino N, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol (2002) 22:8580–91. doi: 10.1128/MCB.22.24.8580-8591.2002
101. Jellusova J, Rickert RC. The PI3K pathway in B cell metabolism. Crit Rev Biochem Mol Biol (2016) 51:359–78. doi: 10.1080/10409238.2016.1215288
102. Sciammas R, Shaffer AL, Schatz JH, Zhao H, Staudt LM, Singh H. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity (2006) 25:225–36. doi: 10.1016/j.immuni.2006.07.009
103. Minnich M, Tagoh H, Bönelt P, Axelsson E, Fischer M, Cebolla B, et al. Multifunctional role of the transcription factor Blimp-1 in coordinating plasma cell differentiation. Nat Immunol (2016) 17:331–43. doi: 10.1038/ni.3349
104. Pauls SD, Lafarge ST, Landego I, Zhang T, Marshall AJ. The phosphoinositide 3-kinase signaling pathway in normal and malignant B cells: Activation mechanisms, regulation and impact on cellular functions. Front Immunol (2012) 3:224. doi: 10.3389/fimmu.2012.00224
105. Sander S, Chu VT, Yasuda T, Franklin A, Graf R, Calado DP, et al. PI3 kinase and FOXO1 transcription factor activity differentially control B cells in the germinal center light and dark zones. Immunity (2015) 43:1075–86. doi: 10.1016/j.immuni.2015.10.021
106. Chen Z, Getahun A, Chen X, Dollin Y, Cambier JC, Wang JH. Imbalanced PTEN and PI3K signaling impairs class switch recombination. J Immunol Baltim Md 1950 (2015) 195:5461–71. doi: 10.4049/jimmunol.1501375
107. Rolf J, Bell SE, Kovesdi D, Janas ML, Soond DR, Webb LMC, et al. Phosphoinositide 3-kinase activity in T cells regulates the magnitude of the germinal center reaction. J Immunol Baltim Md 1950 (2010) 185:4042–52. doi: 10.4049/jimmunol.1001730
108. Zhang T-T, Okkenhaug K, Nashed BF, Puri KD, Knight ZA, Shokat KM, et al. Genetic or pharmaceutical blockade of p110delta phosphoinositide 3-kinase enhances IgE production. J Allergy Clin Immunol (2008) 122:811–819.e2. doi: 10.1016/j.jaci.2008.08.008
109. Williams JW, Tjota MY, Sperling AI. The contribution of allergen-specific IgG to the development of Th2-mediated airway inflammation. J Allergy (2012) 2012:236075. doi: 10.1155/2012/236075
110. Jacob A, Cooney D, Pradhan M, Coggeshall KM. Convergence of signaling pathways on the activation of ERK in B cells. J Biol Chem (2002) 277:23420–6. doi: 10.1074/jbc.M202485200
111. Greaves SA, Peterson JN, Torres RM, Pelanda R. Activation of the MEK-ERK pathway is necessary but not sufficient for breaking central B cell tolerance. Front Immunol (2018) 9:707. doi: 10.3389/fimmu.2018.00707
112. Rowland SL, DePersis CL, Torres RM, Pelanda R. Ras activation of Erk restores impaired tonic BCR signaling and rescues immature B cell differentiation. J Exp Med (2010) 207:607–21. doi: 10.1084/jem.20091673
113. Sanjo H, Hikida M, Aiba Y, Mori Y, Hatano N, Ogata M, et al. Extracellular signal-regulated protein kinase 2 is required for efficient generation of B cells bearing antigen-specific immunoglobulin G. Mol Cell Biol (2007) 27:1236–46. doi: 10.1128/MCB.01530-06
114. Yasuda T, Kometani K, Takahashi N, Imai Y, Aiba Y, Kurosaki T. ERKs induce expression of the transcriptional repressor Blimp-1 and subsequent plasma cell differentiation. Sci Signal (2011) 4:ra25. doi: 10.1126/scisignal.2001592
115. Niu H, Ye BH, Dalla-Favera R. Antigen receptor signaling induces MAP kinase-mediated phosphorylation and degradation of the BCL-6 transcription factor. Genes Dev (1998) 12:1953–61. doi: 10.1101/gad.12.13.1953
116. Douglas RS, Naik V, Hwang CJ, Afifiyan NF, Gianoukakis AG, Sand D, et al. B cells from patients with graves’ disease aberrantly express the IGF-1 receptor: Implications for disease pathogenesis. J Immunol Baltim Md 1950 (2008) 181:5768–74. doi: 10.4049/jimmunol.181.8.5768
117. Kimata H, Fujimoto M. Growth hormone and insulin-like growth factor I induce immunoglobulin (Ig)E and IgG4 production by human B cells. J Exp Med (1994) 180:727–32. doi: 10.1084/jem.180.2.727
118. Kimata H, Yoshida A. Differential effect of growth hormone and insulin-like growth factor-I, insulin-like growth factor-II, and insulin on Ig production and growth in human plasma cells. Blood (1994) 83:1569–74. doi: 10.1182/blood.V83.6.1569.1569
119. Geng X-R, Yang G, Li M, Song J-P, Liu Z-Q, Qiu S, et al. Insulin-like growth factor-2 enhances functions of antigen (Ag)-specific regulatory B cells. J Biol Chem (2014) 289:17941–50. doi: 10.1074/jbc.M113.515262
120. Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, et al. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol Baltim Md 1950 (2014) 192:3626–36. doi: 10.4049/jimmunol.1302062
121. Doughty CA, Bleiman BF, Wagner DJ, Dufort FJ, Mataraza JM, Roberts MF, et al. Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: Role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood (2006) 107:4458–65. doi: 10.1182/blood-2005-12-4788
122. Donnelly RP, Finlay DK. Glucose, glycolysis and lymphocyte responses. Mol Immunol (2015) 68:513–9. doi: 10.1016/j.molimm.2015.07.034
123. Dufort FJ, Gumina MR, Ta NL, Tao Y, Heyse SA, Scott DA, et al. Glucose-dependent de novo lipogenesis in B lymphocytes: A requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. J Biol Chem (2014) 289:7011–24. doi: 10.1074/jbc.M114.551051
124. Lam WY, Becker AM, Kennerly KM, Wong R, Curtis JD, Llufrio EM, et al. Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity (2016) 45:60–73. doi: 10.1016/j.immuni.2016.06.011
125. Waters LR, Ahsan FM, Wolf DM, Shirihai O, Teitell MA. Initial B cell activation induces metabolic reprogramming and mitochondrial remodeling. iScience (2018) 5:99–109. doi: 10.1016/j.isci.2018.07.005
126. Khan S, Chan YT, Revelo XS, Winer DA. The immune landscape of visceral adipose tissue during obesity and aging. Front Endocrinol (2020) 11:267. doi: 10.3389/fendo.2020.00267
127. Pedersen DJ, Guilherme A, Danai LV, Heyda L, Matevossian A, Cohen J, et al. A major role of insulin in promoting obesity-associated adipose tissue inflammation. Mol Metab (2015) 4:507–18. doi: 10.1016/j.molmet.2015.04.003
128. O’Sullivan TE, Rapp M, Fan X, Weizman O-E, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
129. Ghazarian M, Luck H, Revelo XS, Winer S, Winer DA. Immunopathology of adipose tissue during metabolic syndrome. Turk Patoloji Derg (2015) 31(Suppl 1):172–80. doi: 10.5146/tjpath.2015.01323
130. Ghazarian M, Revelo XS, Nøhr MK, Luck H, Zeng K, Lei H, et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci Immunol (2017) 2:eaai7616. doi: 10.1126/sciimmunol.aai7616
131. Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo XS, et al. Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab (2015) 21:527–42. doi: 10.1016/j.cmet.2015.03.001
132. Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, et al. Gut-associated IgA+ immune cells regulate obesity-related insulin resistance. Nat Commun (2019) 10:3650. doi: 10.1038/s41467-019-11370-y
133. Winer DA, Winer S, Shen L, Wadia PP, Yantha J, Paltser G, et al. B lymphocytes promote insulin resistance through modulation of T lymphocytes and production of pathogenic IgG antibody. Nat Med (2011) 17:610–7. doi: 10.1038/nm.2353
134. Paich HA, Sheridan PA, Handy J, Karlsson EA, Schultz-Cherry S, Hudgens MG, et al. Overweight and obese adult humans have a defective cellular immune response to pandemic H1N1 influenza A virus. Obes Silver Spring Md (2013) 21:2377–86. doi: 10.1002/oby.20383
135. Sheridan PA, Paich HA, Handy J, Karlsson EA, Hudgens MG, Sammon AB, et al. Obesity is associated with impaired immune response to influenza vaccination in humans. Int J Obes 2005 (2012) 36:1072–7. doi: 10.1038/ijo.2011.208
136. Hu J, Zhao M, Zhao Y, Dong W, Huang X, Zhang S. Increased body mass index linked to decreased neutralizing antibody titers of inactivated SARS-CoV-2 vaccine in healthcare workers. Obes Sci Pract (2022) 9(1):23–29. doi: 10.1002/osp4.626
137. Milner JJ, Rebeles J, Dhungana S, Stewart DA, Sumner SCJ, Meyers MH, et al. Obesity increases mortality and modulates the lung metabolome during pandemic H1N1 influenza virus infection in mice. J Immunol Baltim Md 1950 (2015) 194:4846–59. doi: 10.4049/jimmunol.1402295
138. Ieronymaki E, Theodorakis EM, Lyroni K, Vergadi E, Lagoudaki E, Al-Qahtani A, et al. Insulin resistance in macrophages alters their metabolism and promotes an M2-like phenotype. J Immunol (2019) 202(6):1786–97. doi: 10.4049/jimmunol.1800065
139. Cai Q, Chen F, Wang T, Luo F, Liu X, Wu Q, et al. Obesity and COVID-19 severity in a designated hospital in shenzhen, China. Diabetes Care (2020) 43:1392–8. doi: 10.2337/dc20-0576
140. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 183:996-1012.e19. doi: 10.1016/j.cell.2020.09.038
141. Klonoff DC, Messler JC, Umpierrez GE, Peng L, Booth R, Crowe J, et al. Association between achieving inpatient glycemic control and clinical outcomes in hospitalized patients with COVID-19: A multicenter, retrospective hospital-based analysis. Diabetes Care (2021) 44:578–85. doi: 10.2337/dc20-1857
142. Desai N, Neyaz A, Szabolcs A, Shih AR, Chen JH, Thapar V, et al. Temporal and spatial heterogeneity of host response to SARS-CoV-2 pulmonary infection. Nat Commun (2020) 11:6319. doi: 10.1038/s41467-020-20139-7
143. Wu Y-Y, Wang S-H, Wu C-H, Yen L-C, Lai H-F, Ho C-L, et al. In silico immune infiltration profiling combined with functional enrichment analysis reveals a potential role for naïve B cells as a trigger for severe immune responses in the lungs of COVID-19 patients. PloS One (2020) 15:e0242900. doi: 10.1371/journal.pone.0242900
144. Sethi JK, Hotamisligil GS. Metabolic messengers: Tumour necrosis factor. Nat Metab (2021) 3:1302–12. doi: 10.1038/s42255-021-00470-z
145. Kruszynska YT, Worrall DS, Ofrecio J, Frias JP, Macaraeg G, Olefsky JM. Fatty acid-induced insulin resistance: Decreased muscle PI3K activation but unchanged akt phosphorylation. J Clin Endocrinol Metab (2002) 87:226–34. doi: 10.1210/jcem.87.1.8187
146. Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol Baltim Md 1950 (2017) 198:1006–14. doi: 10.4049/jimmunol.1601515
147. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci (2012) 109:9517–22. doi: 10.1073/pnas.1119038109
148. Chen J, Tang H, Hay N, Xu J, Ye RD. Akt isoforms differentially regulate neutrophil functions. Blood (2010) 115:4237–46. doi: 10.1182/blood-2009-11-255323
149. Abdullah L, Hills LB, Winter EB, Huang YH. Diverse roles of Akt in T cells. Immunometabolism (2021) 3:e210007. doi: 10.20900/immunometab20210007
150. Liang C-P, Han S, Okamoto H, Carnemolla R, Tabas I, Accili D, et al. Increased CD36 protein as a response to defective insulin signaling in macrophages. J Clin Invest (2004) 113:764–73. doi: 10.1172/JCI19528
151. Khadke S, Ahmed N, Ahmed N, Ratts R, Raju S, Gallogly M, et al. Harnessing the immune system to overcome cytokine storm and reduce viral load in COVID-19: A review of the phases of illness and therapeutic agents. Virol J (2020) 17:154. doi: 10.1186/s12985-020-01415-w
152. Yu B, Li C, Sun Y, Wang DW. Insulin treatment is associated with increased mortality in patients with COVID-19 and type 2 diabetes. Cell Metab (2021) 33:65–77.e2. doi: 10.1016/j.cmet.2020.11.014
153. Donohoe CL, Lysaght J, O’Sullivan J, Reynolds JV. Emerging concepts linking obesity with the hallmarks of cancer. Trends Endocrinol Metab (2017) 28:46–62. doi: 10.1016/j.tem.2016.08.004
154. Zhang AMY, Chu KH, Daly BF, Ruiter T, Dou Y, Yang JCC, et al. Effects of hyperinsulinemia on pancreatic cancer development and the immune microenvironment revealed through single-cell transcriptomics. Cancer Metab (2022) 10:5. doi: 10.1186/s40170-022-00282-z
155. Trovato R, Fiore A, Sartori S, Canè S, Giugno R, Cascione L, et al. Immunosuppression by monocytic myeloid-derived suppressor cells in patients with pancreatic ductal carcinoma is orchestrated by STAT3. J Immunother Cancer (2019) 7:255. doi: 10.1186/s40425-019-0734-6
156. Germic N, Frangez Z, Yousefi S, Simon H-U. Regulation of the innate immune system by autophagy: Monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differ (2019) 26:715–27. doi: 10.1038/s41418-019-0297-6
157. Dyck L, Prendeville H, Raverdeau M, Wilk MM, Loftus RM, Douglas A, et al. Suppressive effects of the obese tumor microenvironment on CD8 T cell infiltration and effector function. J Exp Med (2022) 219:e20210042. doi: 10.1084/jem.20210042
158. Ringel AE, Drijvers JM, Baker GJ, Catozzi A, García-Cañaveras JC, Gassaway BM, et al. Obesity shapes metabolism in the tumor microenvironment to suppress anti-tumor immunity. Cell (2020) 183:1848–1866.e26. doi: 10.1016/j.cell.2020.11.009
159. Chen W, Zhang Y, Yue C, Ye Y, Chen P, Peng W, et al. Accumulation of advanced glycation end products involved in inflammation and contributing to severe preeclampsia, in maternal blood, umbilical blood and placental tissues. Gynecol Obstet Invest (2017) 82:388–97. doi: 10.1159/000448141
160. van Niekerk G, Christowitz C, Engelbrecht A-M. Insulin-mediated immune dysfunction in the development of preeclampsia. J Mol Med (2021) 99:889–97. doi: 10.1007/s00109-021-02068-0
161. Eghbal-Fard S, Yousefi M, Heydarlou H, Ahmadi M, Taghavi S, Movasaghpour A, et al. The imbalance of Th17/Treg axis involved in the pathogenesis of preeclampsia. J Cell Physiol (2019) 234:5106–16. doi: 10.1002/jcp.27315
162. Chávez-Reyes J, Escárcega-González CE, Chavira-Suárez E, León-Buitimea A, Vázquez-León P, Morones-Ramírez JR, et al. Susceptibility for some infectious diseases in patients with diabetes: The key role of glycemia. Front Public Health (2021) 9:559595. doi: 10.3389/fpubh.2021.559595
163. Alba-Loureiro TC, Hirabara SM, Mendonca JR, Curi R, Pithon-Curi TC. Diabetes causes marked changes in function and metabolism of rat neutrophils. J Endocrinol (2006) 188(2):295–303. doi: 10.1677/joe.1.06438
164. Wensveen FM, Šestan M, Turk Wensveen T, Polić B. Blood glucose regulation in context of infection. Vitam Horm (2021) 117:253–318. doi: 10.1016/bs.vh.2021.06.009
165. Liu Y, Liu F-J, Guan Z-C, Dong F-T, Cheng J-H, Gao Y-P, et al. The extracellular domain of staphylococcus aureus LtaS binds insulin and induces insulin resistance during infection. Nat Microbiol (2018) 3:622–31. doi: 10.1038/s41564-018-0146-2
166. Finucane FM, Davenport C. Coronavirus and obesity: Could insulin resistance mediate the severity of COVID-19 infection? Front Public Health (2020) 8:184. doi: 10.3389/fpubh.2020.00184
167. Langouche L, Van den Berghe G, Gunst J. Hyperglycemia and insulin resistance in COVID-19 versus non-COVID critical illness: Are they really different? Crit Care (2021) 25:437. doi: 10.1186/s13054-021-03861-6
168. He X, Liu C, Peng J, Li Z, Li F, Wang J, et al. COVID-19 induces new-onset insulin resistance and lipid metabolic dysregulation via regulation of secreted metabolic factors. Signal Transduct Target Ther (2021) 6:1–12. doi: 10.1038/s41392-021-00822-x
169. Chen M, Zhu B, Chen D, Hu X, Xu X, Shen W-J, et al. COVID-19 may increase the risk of insulin resistance in adult patients without diabetes: A 6-month prospective study. Endocr Pract Off J Am Coll Endocrinol Am Assoc Clin Endocrinol (2021) 27:834–41. doi: 10.1016/j.eprac.2021.04.004
170. Montefusco L, Ben Nasr M, D’Addio F, Loretelli C, Rossi A, Pastore I, et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat Metab (2021) 3:774–85. doi: 10.1038/s42255-021-00407-6
171. Wu C-T, Lidsky PV, Xiao Y, Lee IT, Cheng R, Nakayama T, et al. SARS-CoV-2 infects human pancreatic β cells and elicits β cell impairment. Cell Metab (2021) 33:1565–1576.e5. doi: 10.1016/j.cmet.2021.05.013
172. Reiterer M, Rajan M, Gómez-Banoy N, Lau JD, Gomez-Escobar LG, Ma L, et al. Hyperglycemia in acute COVID-19 is characterized by insulin resistance and adipose tissue infectivity by SARS-CoV-2. Cell Metab (2021) 33:2174–2188.e5. doi: 10.1016/j.cmet.2021.09.009
173. Samuel SM, Varghese E, Büsselberg D. Therapeutic potential of metformin in COVID-19: Reasoning for its protective role. Trends Microbiol (2021) 29:894–907. doi: 10.1016/j.tim.2021.03.004
174. Mirabella S, Gomez-Paz S, Lam E, Gonzalez-Mosquera L, Fogel J, Rubinstein S. Glucose dysregulation and its association with COVID-19 mortality and hospital length of stay. Diabetes Metab Syndr (2022) 16:102439. doi: 10.1016/j.dsx.2022.102439
175. Cameron AR, Morrison VL, Levin D, Mohan M, Forteath C, Beall C, et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res (2016) 119:652–65. doi: 10.1161/CIRCRESAHA.116.308445
176. Mastrototaro L, Roden M. Insulin resistance and insulin sensitizing agents. Metabolism (2021) 125:154892. doi: 10.1016/j.metabol.2021.154892
177. Bailey CJ, Gwilt M. Diabetes, metformin and the clinical course of COVID-19: Outcomes, mechanisms and suggestions on the therapeutic use of metformin. Front Pharmacol (2022) 13:784459. doi: 10.3389/fphar.2022.784459
178. Luo P, Qiu L, Liu Y, Liu X-L, Zheng J-L, Xue H-Y, et al. Metformin treatment was associated with decreased mortality in COVID-19 patients with diabetes in a retrospective analysis. Am J Trop Med Hyg (2020) 103:69–72. doi: 10.4269/ajtmh.20-0375
179. Li W, Li J, Wei Q, McCowen K, Xiong W, Liu J, et al. Inpatient use of metformin and acarbose is associated with reduced mortality of COVID-19 patients with type 2 diabetes mellitus. Endocrinol Diab Metab (2022) 5:e301. doi: 10.21203/rs.3.rs-287308/v1
180. Lalau J-D, Al-Salameh A, Hadjadj S, Goronflot T, Wiernsperger N, Pichelin M, et al. Metformin use is associated with a reduced risk of mortality in patients with diabetes hospitalised for COVID-19. Diabetes Metab (2021) 47:101216. doi: 10.1016/j.diabet.2020.101216
181. Lim S, Bae JH, Kwon H-S, Nauck MA. COVID-19 and diabetes mellitus: From pathophysiology to clinical management. Nat Rev Endocrinol (2021) 17:11–30. doi: 10.1038/s41574-020-00435-4
182. Apicella M, Campopiano MC, Mantuano M, Mazoni L, Coppelli A, Del Prato S. COVID-19 in people with diabetes: Understanding the reasons for worse outcomes. Lancet Diabetes Endocrinol (2020) 8:782–92. doi: 10.1016/S2213-8587(20)30238-2
183. Wan L, Gao Q, Deng Y, Ke Y, Ma E, Yang H, et al. GP73 is a glucogenic hormone contributing to SARS-CoV-2-induced hyperglycemia. Nat Metab (2022) 4:29–43. doi: 10.1038/s42255-021-00508-2
184. Ieronymaki E, Daskalaki MG, Lyroni K, Tsatsanis C. Insulin signaling and insulin resistance facilitate trained immunity in macrophages through metabolic and epigenetic changes. Front Immunol (2019) 10:1330. doi: 10.3389/fimmu.2019.01330
185. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020) 20:375–88. doi: 10.1038/s41577-020-0285-6
186. Divangahi M, Aaby P, Khader SA, Barreiro LB, Bekkering S, Chavakis T, et al. Trained immunity, tolerance, priming and differentiation: Distinct immunological processes. Nat Immunol (2021) 22:2–6. doi: 10.1038/s41590-020-00845-6
187. Benn CS, Netea MG, Selin LK, Aaby P. A small jab - a big effect: Nonspecific immunomodulation by vaccines. Trends Immunol (2013) 34:431–9. doi: 10.1016/j.it.2013.04.004
188. Lumeng CN, Deyoung SM, Bodzin JL, Saltiel AR. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes (2007) 56:16–23. doi: 10.2337/db06-1076
189. Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345:1250684. doi: 10.1126/science.1250684
190. Arts RJW, Novakovic B, Ter Horst R, Carvalho A, Bekkering S, Lachmandas E, et al. Glutaminolysis and fumarate accumulation integrate immunometabolic and epigenetic programs in trained immunity. Cell Metab (2016) 24:807–19. doi: 10.1016/j.cmet.2016.10.008
191. Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes (2009) 58:1229–36. doi: 10.2337/db08-1666
192. Bekkering S, Quintin J, Joosten LAB, van der Meer JWM, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
193. de Laval B, Maurizio J, Kandalla PK, Brisou G, Simonnet L, Huber C, et al. C/EBPβ-dependent epigenetic memory induces trained immunity in hematopoietic stem cells. Cell Stem Cell (2020) 26:657–674.e8. doi: 10.1016/j.stem.2020.01.017
194. Thomas AL, Alarcon PC, Divanovic S, Chougnet CA, Hildeman DA, Moreno-Fernandez ME. Implications of inflammatory states on dysfunctional immune responses in aging and obesity. Front Aging (2021) 2:732414. doi: 10.3389/fragi.2021.732414
195. Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. Daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditiis elegans. Science (1997) 277:942–6. doi: 10.1126/science.277.5328.942
196. Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science (2001) 292:107–10. doi: 10.1126/science.1057987
197. Yan H, Opachaloemphan C, Carmona-Aldana F, Mancini G, Mlejnek J, Descostes N, et al. Insulin signaling in the long-lived reproductive caste of ants. Science (2022) 377:1092–9. doi: 10.1126/science.abm8767
198. Bartke A, Wright JC, Mattison JA, Ingram DK, Miller RA, Roth GS. Extending the lifespan of long-lived mice. Nature (2001) 414:412. doi: 10.1038/35106646
199. Hsieh C-C, DeFord JH, Flurkey K, Harrison DE, Papaconstantinou J. Implications for the insulin signaling pathway in Snell dwarf mouse longevity: A similarity with the C. elegans longevity paradigm. Mech Ageing Dev (2002) 123:1229–44. doi: 10.1016/s0047-6374(02)00036-2
200. Marmentini C, Soares GM, Bronczek GA, Piovan S, Mareze-Costa CE, Carneiro EM, et al. Aging reduces insulin clearance in mice. Front Endocrinol (2021) 12:679492. doi: 10.3389/fendo.2021.679492
201. Janssen JAMJL. Hyperinsulinemia and its pivotal role in aging, obesity, type 2 diabetes, cardiovascular disease and cancer. Int J Mol Sci (2021) 22:7797. doi: 10.3390/ijms22157797
202. Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, et al. Comprehensive profiling of an aging immune system reveals clonal GZMK+ CD8+ T cells as conserved hallmark of inflammaging. Immunity (2021) 54:99–115.e12. doi: 10.1016/j.immuni.2020.11.005
203. Ouyang W, Beckett O, Ma Q, Paik J, DePinho RA, Li MO. Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol (2010) 11:618–27. doi: 10.1038/ni.1884
204. Rao RR, Li Q, Gubbels Bupp MR, Shrikant PA. Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity (2012) 36:374–87. doi: 10.1016/j.immuni.2012.01.015
205. Hess Michelini R, Doedens AL, Goldrath AW, Hedrick SM. Differentiation of CD8 memory T cells depends on Foxo1. J Exp Med (2013) 210:1189–200. doi: 10.1084/jem.20130392
206. Delpoux A, Marcel N, Hess Michelini R, Katayama CD, Allison KA, Glass CK, et al. FOXO1 constrains activation and regulates senescence in CD8 T cells. Cell Rep (2021) 34:108674. doi: 10.1016/j.celrep.2020.108674
207. Raynor J, Lages CS, Shehata H, Hildeman DA, Chougnet CA. Homeostasis and function of regulatory T cells in aging. Curr Opin Immunol (2012) 24:482–7. doi: 10.1016/j.coi.2012.04.005
208. Raynor J, Karns R, Almanan M, Li K-P, Divanovic S, Chougnet CA, et al. IL-6 and ICOS antagonize bim and promote regulatory T cell accrual with age. J Immunol Baltim Md 1950 (2015) 195:944–52. doi: 10.4049/jimmunol.1500443
209. Elyahu Y, Hekselman I, Eizenberg-Magar I, Berner O, Strominger I, Schiller M, et al. Aging promotes reorganization of the CD4 T cell landscape toward extreme regulatory and effector phenotypes. Sci Adv (2019) 5:eaaw8330. doi: 10.1126/sciadv.aaw8330
210. Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, et al. Depletion of fat-resident T reg cells prevents age-associated insulin resistance. Nature (2015) 528:137–41. doi: 10.1038/nature16151
211. Juricic P, Lu Y-X, Leech T, Drews LF, Paulitz J, Lu J, et al. Long-lasting geroprotection from brief rapamycin treatment in early adulthood by persistently increased intestinal autophagy. Nat Aging (2022) 2:824–36. doi: 10.1038/s43587-022-00278-w
212. Mannick JB, Morris M, Hockey H-UP, Roma G, Beibel M, Kulmatycki K, et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med (2018) 10:eaaq1564. doi: 10.1126/scitranslmed.aaq1564
213. Halloran J, Hussong SA, Burbank R, Podlutskaya N, Fischer KE, Sloane LB, et al. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non-cognitive components of behavior throughout lifespan in mice. Neuroscience (2012) 223:102–13. doi: 10.1016/j.neuroscience.2012.06.054
214. Dai D-F, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, et al. Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell (2014) 13:529–39. doi: 10.1111/acel.12203
215. Chen C, Liu Y, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal (2009) 2:ra75. doi: 10.1126/scisignal.2000559
216. den Hartigh LJ, Goodspeed L, Wang SA, Kenerson HL, Omer M, O’Brien KD, et al. Chronic oral rapamycin decreases adiposity, hepatic triglycerides and insulin resistance in male mice fed a diet high in sucrose and saturated fat. Exp Physiol (2018) 103:1469–80. doi: 10.1113/EP087207
217. Bartleson JM, Radenkovic D, Covarrubias AJ, Furman D, Winer DA, Verdin E. SARS-CoV-2, COVID-19 and the ageing immune system. Nat Aging (2021) 1:769–82. doi: 10.1038/s43587-021-00114-7
218. Kulkarni AS, Gubbi S, Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab (2020) 32:15–30. doi: 10.1016/j.cmet.2020.04.001
219. Saeedi-Boroujeni A, Mahmoudian-Sani M-R. Anti-inflammatory potential of quercetin in COVID-19 treatment. J Inflammation Lond Engl (2021) 18:3. doi: 10.1186/s12950-021-00268-6
220. Gu Y-Y, Zhang M, Cen H, Wu Y-F, Lu Z, Lu F, et al. Quercetin as a potential treatment for COVID-19-induced acute kidney injury: Based on network pharmacology and molecular docking study. PloS One (2021) 16:e0245209. doi: 10.1371/journal.pone.0245209
221. Shisheva A, Shechter Y. Quercetin selectively inhibits insulin receptor function in vitro and the bioresponses of insulin and insulinomimetic agents in rat adipocytes. Biochemistry (1992) 31:8059–63. doi: 10.1021/bi00149a041
222. Cox LS, Bellantuono I, Lord JM, Sapey E, Mannick JB, Partridge L, et al. Tackling immunosenescence to improve COVID-19 outcomes and vaccine response in older adults. Lancet Healthy Longev (2020) 1:e55–7. doi: 10.1016/S2666-7568(20)30011-8
Keywords: insulin resistance, immunometabolism, obesity, cancer, infection, inflammation, aging, pre-eclampsia
Citation: Makhijani P, Basso PJ, Chan YT, Chen N, Baechle J, Khan S, Furman D, Tsai S and Winer DA (2023) Regulation of the immune system by the insulin receptor in health and disease. Front. Endocrinol. 14:1128622. doi: 10.3389/fendo.2023.1128622
Received: 20 December 2022; Accepted: 08 February 2023;
Published: 13 March 2023.
Edited by:
Rajakumar Anbazhagan, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NIH), United StatesReviewed by:
Jennifer Lee, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesCopyright © 2023 Makhijani, Basso, Chan, Chen, Baechle, Khan, Furman, Tsai and Winer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sue Tsai, c3RzYWlAdWFsYmVydGEuY2E=; Daniel A. Winer, RFdpbmVyQGJ1Y2tpbnN0aXR1dGUub3Jn
†These authors jointly supervised this work
‡ORCID: Priya Makhijani, orcid.org/0000-0002-1775-3303
Paulo José Basso, orcid.org/0000-0003-4856-9207
David Furman, orcid.org/0000-0002-3654-9519
Sue Tsai, orcid.org/0000-0002-6261-5233
Daniel A. Winer, orcid.org/0000-0002-8702-0957
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.