
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 26 October 2023
Sec. Clinical Diabetes
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1126661
This article is part of the Research Topic Diabetes and Obesity Effects on Lung Function - Volume II View all 9 articles
 Yimin Dai†
Yimin Dai† Shuang Zhou†
Shuang Zhou† Lin Qiao
Lin Qiao Zhao Peng
Zhao Peng Jiuliang Zhao
Jiuliang Zhao Dong XuChanyuan Wu*
Dong XuChanyuan Wu* Mengtao Li
Mengtao Li Xiaofeng Zeng
Xiaofeng Zeng Qian Wang*
Qian Wang*Diabetes mellitus (DM) is a chronic metabolic disorder that affects multiple organs and systems, including the pulmonary system. Pulmonary dysfunction in DM patients has been observed and studied for years, but the underlying mechanisms have not been fully understood. In addition to traditional mechanisms such as the production and accumulation of advanced glycation end products (AGEs), angiopathy, tissue glycation, oxidative stress, and systemic inflammation, recent studies have focused on programmed cell deaths (PCDs), especially the non-apoptotic ones, in diabetic pulmonary dysfunction. Non-apoptotic PCDs (NAPCDs) including autophagic cell death, necroptosis, pyroptosis, ferroptosis, and copper-induced cell death have been found to have certain correlations with diabetes and relevant complications. The AGE–AGE receptor (RAGE) axis not only plays an important role in the traditional pathogenesis of diabetes lung disease but also plays an important role in non-apoptotic cell death. In this review, we summarize novel studies about the roles of non-apoptotic PCDs in diabetic pulmonary dysfunction and focus on their interactions with the AGE–RAGE axis.
Diabetes mellitus (DM) has long been considered a systemic disease that is closely related to autoimmune disorders (1). Pulmonary involvement of DM patients was first mentioned in the 1970s (2, 3). From then on, multiple cases and retrospective studies linking pulmonary diseases such as chronic obstructive pulmonary disease (COPD) and lung fibrosis to DM have continuously been reported (4–10) with little clear and direct evidence obtained and contradictory conclusions appearing in different studies (11). Given that pulmonary function indexes are significant indicators of lung condition, clinical researchers have investigated its correlation with DM. In 1985, researchers summarized characteristics of the pulmonary function of DM patients documenting a significant decrease in forced vital capacity (FVC) and timed vital capacity (TVC), as well as a decrease in forced expiratory volume in 1 second (FEV1) and diffusion lung capacity for carbon monoxide (CO) (DLCO) in male DM patients (12). Subsequently, multiple studies have compared the pulmonary function of DM patients and healthy controls. Systemic reviews found bidirectional conclusions, but the decrease of FVC and FEV1 as well as the negative association between pulmonary function and severity of DM was frequently reported (13–15).
Although damage to pulmonary function in DM patients is frequently observed and analyzed, the possible underlying mechanisms are less reviewed in comparison (11). Previous perspectives on this matter have primarily focused on metabolic dysfunction, inflammation reaction, neurological deficit, and their combined effects (11). The impaired glucose metabolism in DM patients may lead to the deposition of glycolyzed serum proteins in the vascular system (16). Due to their rich capillary network, the lungs are vulnerable to diabetic microangiopathy, which can cause impaired pulmonary ventilation function (17). Additionally, the continuous pro-inflammatory response and damage to the autonomic nervous system of the lungs have also been mentioned (18, 19). While these traditional perspectives are convincing to some extent, more innovative mechanisms are expected to be uncovered.
In recent years, researchers have extensively studied programmed cell deaths (PCDs) in multiple systems and organs and have identified various pathogenic mechanisms of multiple diseases, among which the non-apoptotic ones have been newly identified (20–22). Although apoptosis has long been considered the most conservative form of PCD, non-apoptotic PCDs (NAPCDs) have gained increasing interest. These non-apoptotic PCDs possess unique characteristics that can compensate for failed apoptosis or independently contribute to disease development. Therefore, they have been considered potential new breakthroughs in the study of diseases (23). Non-apoptotic PCDs that have been commonly mentioned mainly include autophagic cell death, necroptosis, pyroptosis, ferroptosis, and the recently identified copper-induced cell death (21, 24–26).
Non-apoptotic PCDs have been found to have correlations with diabetes and its complications, primarily due to the activation of inflammatory reactions and the roles of inflammasomes (27). Non-apoptotic PCDs have been shown to have significant effects on diabetic cardiovascular disease and nephropathy (20, 28). However, considering the overlapping roles of non-apoptotic PCDs in pneumopathy and diabetes, it is necessary to investigate the possible effects of non-apoptotic PCDs on diabetic pulmonary dysfunction and its associations with traditional pathogenic mechanisms.
In this essay, we will review recent studies on the roles of non-apoptotic PCDs including autophagic cell death, necroptosis, pyroptosis, ferroptosis, and copper-induced cell death in the impairment of pulmonary function in DM and their interactions with traditional mechanisms especially with the advanced glycation end product (AGE)–RAGE axis. Previous studies have focused on mechanisms related to inflammation and metabolism dysfunction as well as their combined effects. By exploring the molecular level of pulmonary damage associated with DM, this review aims to provide novel perspectives and insights.
Abnormal glucose metabolism is closely related to DM, which involves complex organic reactions and the production of AGEs. These products have long been validated to be associated with various diabetic complications (29, 30). Specifically, the produced AGEs further accumulate and cause various lesions and tissue damage (29). The lung is a possible target organ of AGEs due to its rich constitution of capillaries, collagen, and elastin fibers that are prone to deposit AGEs (31, 32).
Non-enzymatic glycation reactions (Maillard reaction) occur in DM patients due to hyperglycemic conditions and largely result in the production of endogenous AGEs (33). Previous studies have already figured out the increase in the accumulation of AGEs in COPD patients (34–36). In addition, significantly increased levels of AGEs, as detected through skin autofluorescence (SAF), have also been observed in DM patients who exhibit only decreased FVC, FEV1, and DLCO yet no specific pulmonary diseases (37, 38), indicating that damage occurs before the onset of any diseases. The production of AGEs can result in a series of downstream reactions and can contribute to the development of various pathogenic states (39). One of the pulmonary reactions often associated with AGEs turns out to be the AGE receptor (RAGE) on alveolar epithelial cells (40, 41). An improvement in lung compliance and diminishing adherence of cells to matrix proteins were found after blocking the RAGE, which could partially explain the deficit in pulmonary function (42, 43).
The RAGE–AGE reaction has been identified to play a role in several classical signaling pathways and has been linked to lung homeostasis (44). Endogenous AGEs cross-link with extracellular collagen and hemoglobin extracellular (45), while intracellular responses mainly depend on pathways relevant to nuclear factor kappa-B (NF-κB) and Janus kinase (JAK)-signal transducer and the activator of the transcription (STAT) pathway reaction (44). Possible mechanisms behind this reaction may include increased vascular permeability, activation of inflammatory state, production of oxidative stress, angiogenesis, and dysfunction of the pulmonary epithelium (40, 44, 46).
The abundant vasculature in the lungs has long been considered a crucial target leading to diabetic lung injury, and pulmonary vascular disease (PVD) is a significant cause of damage to pulmonary function (47–49). In the early preclinical stages of diabetes, the subclinical vascular effects were found to be mainly manifested as dysfunction of the endothelium of conductance and resistance arteries, which is associated with a decrease in nitric oxide (NO) bioavailability and abnormal production of reactive oxygen species (ROS) in hyperglycemic states (50, 51). Additionally, the destruction of endothelial integrity due to pulmonary microvascular injury was found to mainly affect CO transfer capacity (52). This finding is consistent with the significant decrease in DLCO in DM patients observed in clinical studies (49).
The RAGE mentioned above is highly expressed in pulmonary small arteries and arteriolar capillaries (53, 54). Researchers have found that the RAGE–AGE reaction results in the upregulation of the vascular cell adhesion molecule-1 (VCAM-1) by increasing NF-κB. This can increase the permeability of inflammatory cells across vascular endothelium and accelerate pulmonary angiopathy (41, 55). Furthermore, NO is involved in the above pathway as well; the AGEs on endothelium may downregulate endothelial nitric oxide synthase (eNOS) and reduce the production of NO (56). This is possibly correlated with low shear stress resulting from atherosclerosis vessels, and the decreased NO further leads to activation of NF-κB (57). These signaling pathways intersect in a network and are involved in multiple other pathophysiological effects such as inflammation and oxidative stress.
In addition to several direct effects on vascular pathogenesis, AGEs have been found to stimulate the expression of transforming growth factor-β (TGF-β), which then facilitates the synthesis of collagen, laminin, and fibronectin in the extracellular matrix (ECM) (58). Hyperglycemia is known to significantly induce epithelial-to-mesenchymal transition (EMT) of alveolar epithelial cells by upregulating TGF-β, leading to lung fibrosis and decreased pulmonary function indexes such as FVC and total lung capacity (TLC) (59–61). The accumulation of extracellular collagen can deposit in the lung and chest wall, causing restrictive damage to pulmonary function (62). Moreover, AGEs can increase the stiffness of the vasculature by cross-linking elastin and collagen, enlarging the area of the ECM (63). AGEs can also bind to lipids, and the produced glycated low-density lipoprotein (LDL) has been shown to reduce the production of NO and mediate endothelial homeostasis (64). These extracellular changes in tissues result from glycation and AGE effects, forming the glycation network with the previously mentioned intracellular signaling, leading to pulmonary dysfunction (65).
Changes in the pulmonary interstitium, including the thickening of the alveolar epithelium and basal lamina of pulmonary capillaries, can significantly affect the viscoelasticity of lung tissue and cause alveolar collapse (66). These alterations also lead to decreased perfusion of pulmonary capillaries (67). Additionally, the apoptosis of alveolar epithelium cells as well as capillary endothelium cells can result in damage to the alveolar capillary network, destruction of pulmonary tissue, and the development of lesions such as emphysema (68). Possible relevant signaling pathways may involve c-Jun N-terminal kinase (JNK) and NF-κB (69). These injuries to alveoli and capillaries further result in a mismatch of ventilation perfusion and a reduction of diffusion capacity, that is, DLCO loss in pulmonary function (70). On a larger scale, the accumulation of collagen in the pulmonary interstitium and chest wall due to glycation in DM can rigidify the lung and rib cage, adding to the restrictive pathophysiology (62, 71).
Glycation and its products can stimulate tissues and organs in DM patients to respond abnormally, resulting in sustained oxidative stress due to the generation of ROS and subsequent inflammatory state with an attack of proinflammatory cytokines (46). This has long been considered a significant pathogenic mechanism for the development of DM complications in various organs and systems, including the respiratory system (30). The oxidative stress can exacerbate the inflammatory response, leading to tissue damage and further generation of more ROS and oxidative stress in turn. Such a vicious cycle also exists in the pathogenesis of pulmonary dysfunction (72).
Multiple studies have already identified the significant role of ROS in the development of pulmonary diseases and lesions, such as asthma, COPD, and lung fibrosis (73–76). The excess production of ROS disrupts the redox balance in lung tissues, leading to serious consequences such as telangiectasia and even pulmonary function failure (77). DM has been identified to be closely associated with endogenous ROS, largely resulting from AGE–RAGE reactions that activate the reduced form of nicotinamide adenine dinucleotide (NADH) oxidase to synthesize more ROS (78–80). The high production of ROS in turn facilitates more AGEs, leading to another vicious cycle (33, 46).
The destruction of pulmonary tissues by ROS is a serious issue that results in damage to multiple constituents, including DNA, lipids, and proteins (81–83). Moreover, ROS-induced mitochondrial injuries in the lungs have also been suspected to play roles in diabetic pulmonary dysfunction (80, 84). The decrease in the sirtuin 3 (SIRT3) protein in mitochondria under ROS stimulation from the diabetic lung has been found to support mitochondria injuries and possible SIRT3-dependent injury pathways as well (80, 85). However, as an important ROS producer, mitochondria generating excessive ROS under induction of hyperglycemia can also contribute to cellular injuries. An increased level of ROS has been identified to cause abnormal opening of the mitochondrial permeability transition pore (MPTP), followed by the release of cytochrome c, caspase activation, generation of more ROS, and finally cell apoptosis (86, 87). Furthermore, researchers also have figured out that the reaction between NO and ROS produces toxic reactive nitrogen species, which further results in vasoconstriction and endothelial injury (88). The involvement of oxidative stress in the formation of pulmonary angiopathy is also a significant constitution in the pathogenesis of pulmonary dysfunction.
The downstream inflammatory response resulting from ROS is also considered an important pathogenesis in pulmonary dysfunction. One of the central pathways is the NF-κB signaling pathway, which has been identified to regulate the expression of proinflammatory cytokines (89). Relevant cytokines include several interleukins (ILs; IL-1β, IL-4, IL-5, IL-6, IL-8, and IL-13) as well as tumor necrosis factor-α (TNF-α) and mucin. Together, these cytokines contribute to the formation of an inflammatory state and further lead to pulmonary dysfunction and even disease development (72, 90). Inflammatory responses modulated by NF-κB also involve inducible nitric oxide synthase (iNOS), resulting in excessive nitrosative stress, i.e., the formation of elevated levels of peroxynitrite (91–93). This has been linked to pulmonary vascular injuries (94). Moreover, followed by NF-κB activation, the upregulation of mitogen-activated protein kinases (MAPKs) is a result of the AGE–RAGE reaction (93). In addition, NF-κB also induces ROS production by activating NADPH oxidase through AGE–RAGE, which is independent of MAPK regulation (92). Research on diabetic rats has found significantly increased levels of NF-κB, along with upregulation of downstream iNOS in lung tissue (95). Conversely, inhibition of NF-κB can reduce the generation of proinflammatory cytokines IL-6 and TNF-α in respiratory tract epithelium, alleviating lung injury (95, 96). These findings provide strong evidence supporting the inflammatory response mediated by NF-κB in pulmonary dysfunction pathogenesis.
Apart from the classical NF-κB pathway, the JAK-STAT pathway has also been revealed to mediate inflammatory responses under the AGE–RAGE reaction, particularly the JAK2/STAT3 signaling pathway (97, 98). Animal studies have shown that negative regulation of JAK2/STAT3 can significantly relieve pulmonary epithelial injuries in response to hyperglycemia and pancreatic dysfunction while also reducing the levels of proinflammatory cytokines such as IL-6, IL-18, and TNF-α (98, 99). Activated by inflammatory cytokines such as IL-6, JAK2/STAT3 plays roles in inflammatory response (100). Furthermore, the upregulation of TGF-β via JAK2/STAT3 promotes profibrotic responses in lung tissue, leading to restrictive pulmonary dysfunction (101).
It is well established that poorly controlled diabetes in DM patients can increase the risk of infection in various tissues, organs, and systems, including the respiratory system (102, 103). This greatly increases hospitalization and mortality rates. Common pathogens that cause respiratory infection include bacteria such as Streptococcus pneumoniae, fungi such as Aspergillus fumigatus, tuberculosis, influenza virus, and certain viruses that cause specific epidemics, such as H1N1, MERS-CoV, and SARS-CoV-2 (104–106). The strike of respiratory infection can be devastating. According to clinical research conducted among hospitalized cystic fibrosis (CF) patients, those with A. fumigatus infection had significantly reduced FEV1 compared to those without infection. Moreover, persistent infection was identified as an independent risk factor for these patients (107).
Alterations of the immune system have been found in DM and are quite possibly associated with the increased risk of pulmonary infection (108). On the one hand, the killing effects of immune cells can be largely weakened in DM due to functional defects. For example, production of the bactericidal ROS, as well as the impaired migration function, can be reduced due to injured glucose metabolism (109, 110). In addition, the treatment of DM may also contribute to reduced generation of cytokines (111). On the other hand, the immune response can be excessively activated in DM under an infection state, resulting in damage to normal tissue (112). Such double effects in diabetic immune response toward invasion of pathogens may partially explain the mechanisms of lung injury caused by infection.
As types of regulated cell death differ from conservative apoptosis, non-apoptotic PCDs have become targets for intervention in diseases. Autophagic cell death, necroptosis, pyroptosis, ferroptosis, and even the newly reported copped cell death, e.g., cuproptosis, all play roles in certain diseases (23, 113). The regulation pathways of non-apoptotic PCDs were summarized in Table 1.
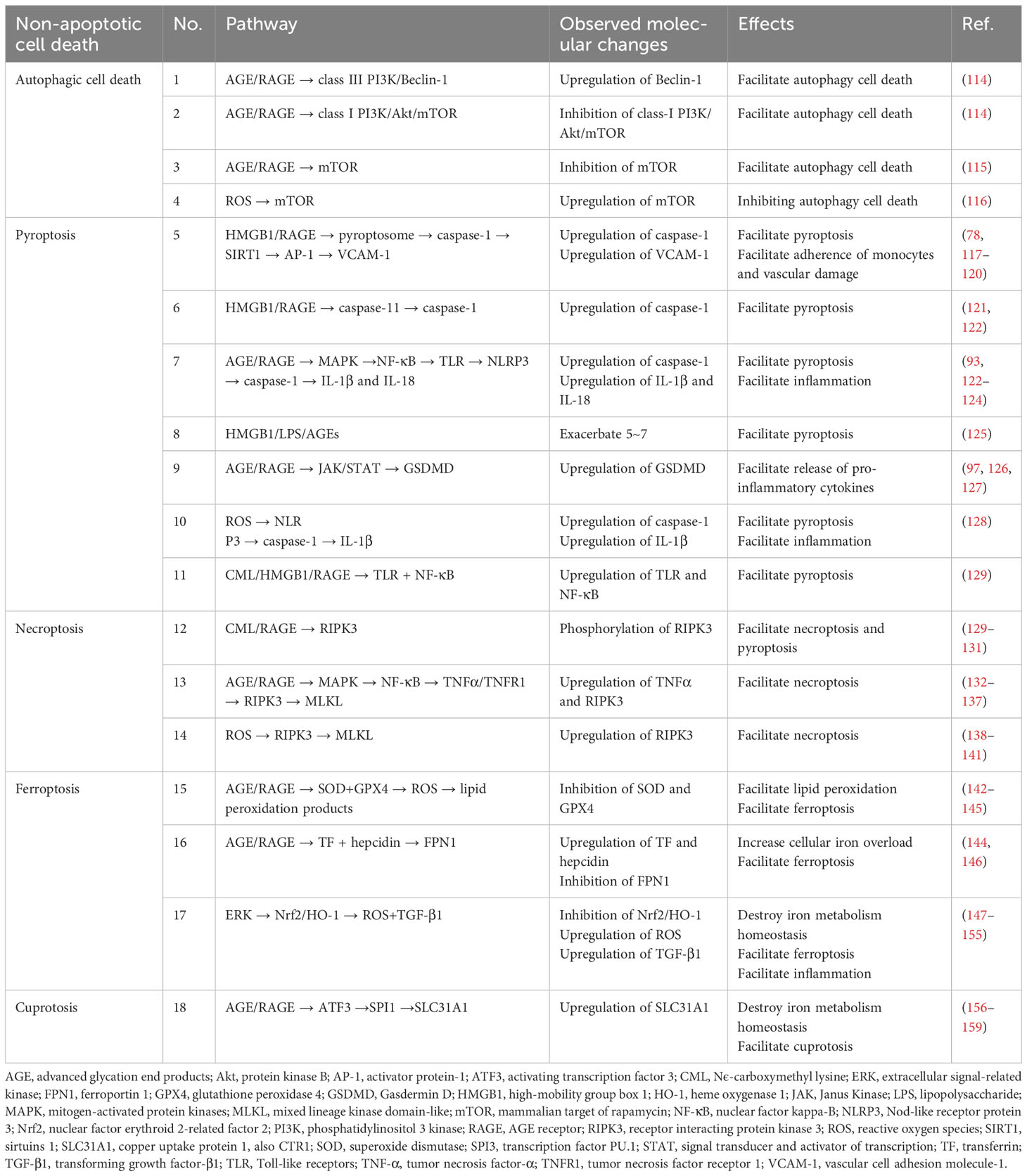
Table 1 The regulation pathways of different types of non-apoptotic programmed cell deaths.
Autophagic cell death is characterized by the formation of autophagosomes, followed by phagocytosis and lysosomal degradation (160). Vacuoles are produced due to the4 occurrence of isolated membranes in the cytoplasm of dying cells. These vacuoles accumulate in the cells and eventually mature into autophagosomes, which then fuse with lysosomes (160, 161). This fusion releases relevant enzymes that break down cellular materials, resulting in the autophagy of the cells (161). Essentially, autophagy is a process that recycles macromolecules within cells (162). It is common for cells to activate pro-survival mechanisms rather than trigger direct cell death in response to external stimuli such as starvation, oxygen deficit, and infection at the biological level (162). In such situations, any subsequent cell death that occurs after autophagy cannot be easily attributed to “autophagy-induced” cell death. This is because it is unclear whether the autophagosome itself is enough to rescue the cells from inevitable decompensation (163). In addition, several studies have found autophagic cell death in eukaryotes in vitro (164, 165). However, evidence of this type of PCD under in vivo conditions is still scarce, and its existence was once doubted (166). Inspiringly, follow-up studies gradually confirmed that cell death can result from autophagy per se, and the definition has been preserved (167). The autophagosome is considered a hallmark of autophagy, which is initiated by the effects of phosphatidylinositol 3 kinase (PI3K) and autophagy-related protein Beclin-1 (168). Additionally, autophagy is negatively regulated by the mammalian target of rapamycin (mTOR), which prevents excessive and uncontrolled autophagy (161). Pathological conditions of several diseases such as asthma and tumorigenesis have been identified to be correlated with autophagic cell death (169–172).
Cells undergoing necroptosis typically undergo dramatic morphological changes, such as rapid swelling and disruption of the plasma membrane. These changes are markedly different from the mild changes observed in apoptotic cells (173). Despite similar changes, necroptosis is still programmed to be regulated when compared to necrosis (174). Necroptosis also shares several characteristics with apoptosis. It is triggered by tumor necrosis factor receptor 1 (TNFR1) in response to certain stimuli, such as infection and sterile inflammation, with inhibition of caspase-8 of apoptosis (132). The receptor interacting protein kinases (RIPKs) (mainly RIPK1 and RIPK3) are at the center of necroptosis and mixed lineage kinase domain-like (MLKL) (133, 134). The ligation between TNF-α and TNFR1 could result in polyubiquitination of RIPK1, which further assembles the necrosome with RIPK3 and recruits MLKL to promote downstream cell death (135, 136).
The downstream mechanisms of necroptosis consist of the formation of plasma channels and rapid swelling of cells, which usually lead to rapid rupture of the plasma membrane and vigorous stimulation of innate immune responses (175). Necroptosis contributes to the pathogenesis of several diseases. For example, ischemic injury in several organisms, including the brain, myocardium, eyes, and kidneys, has been discovered (176–179). Additionally, the necroptosis of epithelial tissue has been linked to inflammation of the skin and even the pathogenesis of inflammatory bowel diseases (180, 181). Dysfunction of the barrier function is a consequence of necroptosis effects in the pathogenesis (182).
Pyroptosis is a type of PCD that is involved in the innate immune response and is mediated by pro-inflammatory mechanisms, including the effects of multiple inflammasomes (22). Although pyroptosis is dependent on the caspase family, it is typically considered a response to external stimulation such as bacterial infection, leading to the release of a series of pro-inflammatory cytokines and rapid cell death (183), unlike apoptosis, which occurs without inflammation. Caspase-1 is the major enzyme mediating pyroptosis but not apoptosis (184). Classical inflammasomes, such as Nod-like receptor protein 3 (NLRP3), help with the activation of pro-caspase-1, further promoting the perforation of the cell membrane by Gasdermin D (GSDMD), releasing pro-inflammatory cytokines such as IL-1β and IL-18, and finally causing disintegration of cells (26). The activation of NLRP3, mitochondria-producing ROS, and disruption of the lysosomal membrane are significant pathways (185, 186). Other caspases, including caspase-4/5/11, in addition to caspase-1 are also involved in non-classical inflammasome pathways (187). Recently, many studies have focused on the alteration of the above-mentioned key factors in diabetes and comorbidities (188). The pyroptosis of pulmonary epithelium under certain stimulation has also been observed (189, 190).
Ferroptosis is considered a novel type of non-apoptotic PCD. Particularly, it is mainly dependent on cellular iron rather than the key factors in apoptosis such as the caspases family (191). The two lethal incentives of ferroptosis were considered overloading of iron and excess accumulation of lipid peroxides (192).
As an essential nutritional element, iron is absorbed by the intestinal epithelial cell and stored in the form of ferric ion (Fe3+), combined with transferrin (TF), and involved in transmembrane transportation (193, 194). The pH value influences the release of Fe3+, which is reduced to ferrous iron (Fe2+) after release from TF under an acidic environment and further participates in downstream reactions (195). Under normal circumstances, the level of intracellular iron is maintained in homeostasis (196). While under certain pathogenic states, dysfunction of iron metabolism may produce overloaded iron and result in the production of hydroxyl radical and oxidative stress response due to a reaction between lipid peroxide and free Fe2+ (197). Fe2+ also promotes other metabolic processes, contributing to the production of more lipid ROS (195). The increased level of oxidative stress again enhanced the process of ferroptosis (195, 196). In a word, the metabolic dysfunction aggravates oxidative stress, which in turn results in ferroptosis and tissue damage. Such pathogenic states may further exacerbate under the hyperglycemic environment, which is characterized by metabolic dysfunction and imbalance of the oxidant-antioxidant system, leading to more generation of lipid peroxides (198). High blood sugar levels facilitate ferroptosis in diabetic complications such as myocardial ischemia and diabetic retinopathy (199, 200). Diabetic pneumopathy could also be predicted. In addition, the inhibition of lipid antioxidants such as glutathione peroxidase 4 (GPX4) would further decrease elimination of lipid peroxide, which also contributes to occurrence of ferroptosis (192).
Along with ferroptosis, other cell deaths dependent on heavy metals have been reported, including copper-induced cell death. This is a brand-new type of PCD, which differed from ferroptosis and necroptosis closely relating to oxidative stress, and has been named “cuprotosis” or “cuproptosis” (113). Though copper per se is an essential coenzyme factor, intracellular copper concentrations are still maintained at an extremely low level to prevent toxicities to cells (201). However, under certain pathological conditions, such as diabetes, copper metabolisms could be abnormal, leading to a higher concentration of circulation copper (156). This is possible due to the over-produced AGEs activating transcriptional factors, specifically transcription factor 3 (ATF3) and transcription factor PU.1 (SPI1), following the upregulated expression of the copper transmembrane transporter high-affinity copper uptake protein 1 (CTR1, also SLC31A1) and increased intracellular Cu concentration (157). The homeostasis of copper is then destroyed. Furthermore, ferredoxin 1 (FDX1), the mitochondrial reductase, was identified to reduce Cu2+ to Cu+ in the mitochondria, promoting more ROS to be released and ultimately cuprotosis of tissue cells (158). In fact, studies on copper-related cell death are only at an early stage (159). We are looking forward to more mechanisms of cuprotosis being explored.
When it comes to non-apoptotic PCDs, recent studies have shown that AGEs may directly induce ferroptosis of tissue cells and promote the release of iron, destroying iron homeostasis (202). Consequently, dysfunctional organs including the lung resulted, and downstream oxidative stress response was also activated (203, 204). Moreover, pyroptosis, necroptosis, and ferroptosis are involved to a certain extent.
Although divergences still exist, a consensus has been reached that autophagic cell death is a type of non-apoptotic PCD that relies on the formation of autophagosomes and the occurrence of autophagy (161). Previous studies have shown an increase in autophagy and activation of autophagic proteins in epithelial cells in pulmonary diseases such as COPD, and idiopathic pulmonary fibrosis was also observed (205, 206). Relevant signaling pathways mainly include class III PI3K/Beclin-1 and class I PI3K/Akt/mTOR, while recent studies have discovered the effects of AGEs acting on both of them (114). However, the class III PI3K/Beclin-1 can add a number of autophagic vacuoles and facilitate autophagy, promoting cell injuries under AGE administration, while class I PI3K/Akt/mTOR mainly manifested as a negative regulator of autophagy and can be inhibited by the AGE–RAGE interaction (114). The total effects turn out to be an exacerbation of autophagic cell death. AGE–RAGE interaction can also directly mediate the inactivation of mTOR, the final negative regulator of the classical pathway, which further facilitates the autophagy of tissue cells. Ma, J. et al. revealed that influenza virus infection, such as H5N1, could cause autophagic cell death by suppressing mTOR and do serious damage to pulmonary function, even acute respiratory distress syndrome (ARDS) (115). These studies have identified that the intensified AGE–RAGE interaction in diabetes can further intensify this process of autophagic cell death.
There seems to be some ambiguity regarding the regulation of mTOR by PI3K and protein kinase B (Akt). The classical PI3K/Akt/mTOR signaling pathway can inhibit autophagy, as mentioned above (207). However, another research study on rats has suggested that autophagy may be abnormally attenuated in diabetes, where a higher level of PI3K/Akt/mTOR and a low level of Beclin-1 are observed, and inhibiting the PI3K/Akt/mTOR pathway could help with promoting apoptosis and reducing tissue pathology (208). Nevertheless, the exact roles of autophagic cell death in diabetes complications remain unclear, especially with regard to pathogen clearance and the prevention of infection spread (116). The overproduced ROS could possibly express the relevance. Previous research has indicated that autophagy can reduce pathogen burden and facilitate adaptive immunity (209). However, increased ROS levels can facilitate the synthesis of inflammasome, followed by upregulating mTOR and leading to inhibition of normal autophagy (116). Given the overproduction of ROS in diabetes, normal autophagy could be impaired, further reducing the ability of organs to defend against infection, including the lungs (116). These findings highlight the complexity of the regulatory effects of PI3K, mTOR, and even autophagic cell death on pulmonary function in diabetes (Figure 1).
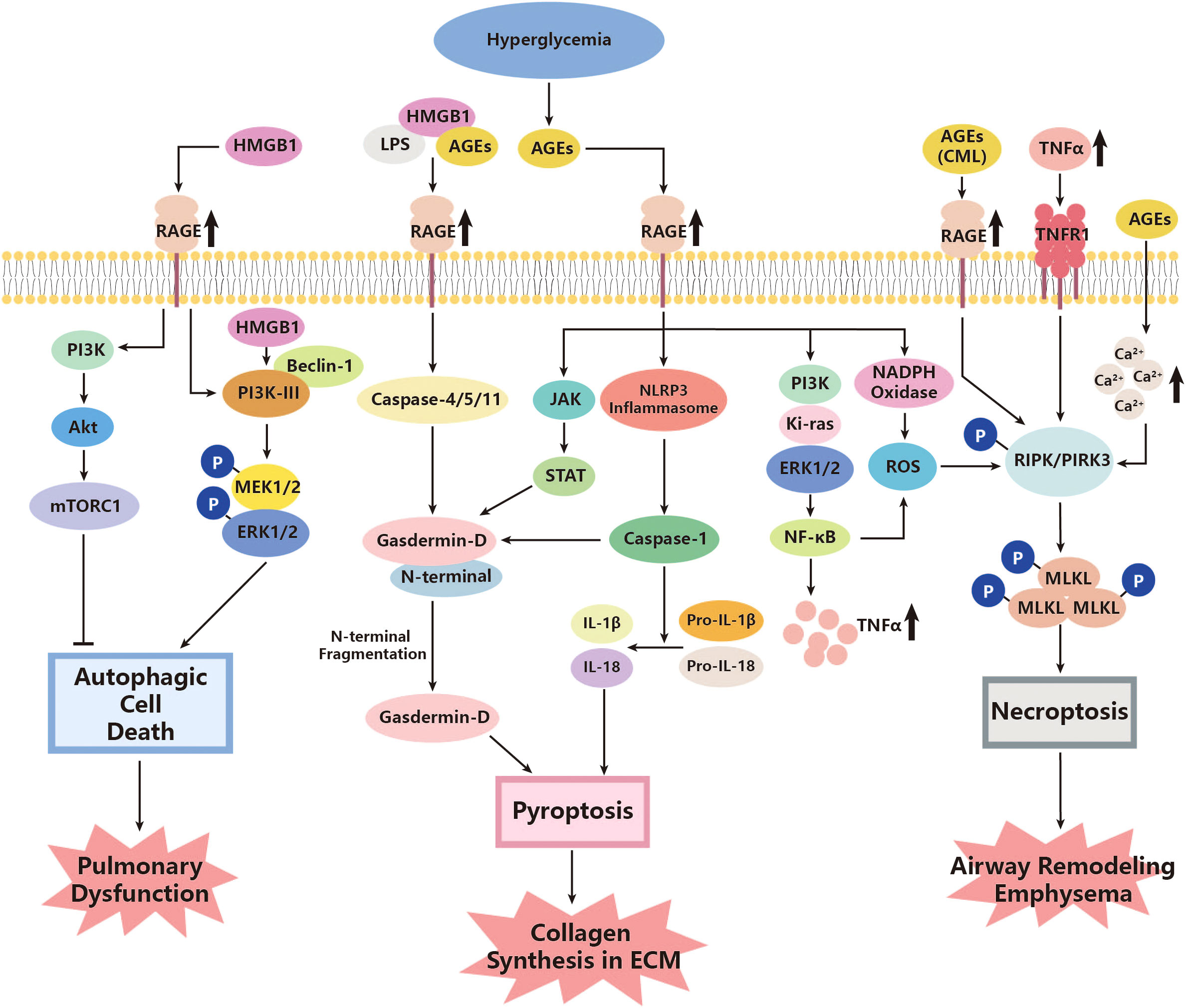
Figure 1 Roles of autophagic cell death, pyroptosis, and necroptosis in the pathogenesis of diabetic lung disease. AGEs, advanced glycation end products; Akt, protein kinase; CML, Nϵ-carboxymethyl lysine; ERK, extracellular signal-related kinase; HMGB1, high-mobility group box 1; JAK, Janus Kinase; Ki-ras, Kirsten rat sarcoma viral oncogene; LPS, lipopolysaccharide; MEK, mitogen-activated protein kinases (MAPK) kinases; MLKL, mixed lineage kinase domain-like; mTORC1, mammalian target of rapamycin C1; NADPH, Nicotinamide Adenine Dinucleotide Phosphate Oxidase; NF-κB, nuclear factor kappa-B; NLRP3, Nod-like receptor protein 3; PI3K, phosphoinositide 3 kinase; PIPK, receptor interacting protein kinases; RAGE, AGE receptor; ROS, reactive oxygen species; STAT, signal transducer and activator of transcription; TNF-α, tumor necrosis factor-α; TNFR1, TNFα activating TNF receptor 1.
On the whole, studies on autophagic cell death in relation to lung diseases and diabetes were relatively limited. Therefore, more attention needs to be given to this field in the future, especially the multifaceted effects of this unique form of non-apoptotic PCD.
Multiple roles of pyroptosis have been identified in pulmonary injury related to diabetes. One of the major pathways that show such associations turns out to be AGE–RAGE (117, 118). As a significant receptor in diabetes signaling, RAGE has also been found to be involved in pyroptosis and do harm to the lungs. According to a study by Yang J et al., under specific external stimuli such as hemorrhagic shock (HS), the release of high-mobility group box 1 (HMGB1) can be induced (117). It further interacts with RAGE to enter the pulmonary endothelial cells, triggering the formation of pyroptosome and activation of caspase-1 (117). Given the activation of RAGE and signaling of HMGB1 in diabetes, this could be a possible pathway for initiating pyroptosis of pulmonary endothelial cells and the pathogenesis of angiopathy in the lungs (119). Moreover, caspase-1, which is elevated in diabetes, cleaves SIRT1 to upregulate activator protein-1 (AP-1), ultimately leading to an increase in VCAM-1 of endothelial cells, promoting adherence of monocytes (120). Another crucial factor involved in pyroptosis is caspase-11, which has also been identified to play a role in this process (121). Researchers have found that HMGB1 and lipopolysaccharide, an endotoxin, can form a complex and internalize into the endo-lysosome of endothelial cells with the assistance of interaction with RAGE. This complex can further activate caspase-11 (121). The mature caspase-11 thus can directly activate caspase-1 without NLRP3 and promote pyroptosis (122). Regarding NF-κB, it has also been observed to increase its expression in diabetes, converting pro-caspase-1 to mature caspase-1 and facilitating pyroptosis of type I alveolar epithelial cells through Toll-like receptors (TLRs) and NLRP3 pathways (123). Furthermore, upregulation of both caspase-1 and caspase-11 was found to accelerate the cleavage of pro-IL-1β and pro-IL-18, promoting the release of mature IL-1β and IL-18, the inflammatory cytokines, which further participate in downstream pathogenesis (122, 124). In addition, a novel study has suggested that added AGEs could bind with HMGB1–lipopolysaccharide (HMGB1-LPS) complexes, forming a triplet and exacerbating the above-mentioned mediating RAGEs (84).
In diabetes, high glycemic levels can promote the production of ROS, which mainly originates from the endoplasmic reticulum (ER) stress, damaged mitochondria, and upregulated NADPH (210, 211). The increased oxidative stress, reflected in the growing production of ROS, may further exacerbate pyroptosis and relevant inflammation processes in the lung, especially endothelial dysfunction (210, 212). Heid ME et al. identified that ROS could activate NLRP3 inflammasome and increase the expression of caspase-1, hence facilitating proptosis and IL-1β release (128). The inflammatory cytokines were then released from the pyroptotic cells through the membrane pore made by the N-terminal domain (NTD) of GSDMD, which was cleaved by mature caspase-1 (212). Interestingly, the GSDMD protein was found to be regulated by the JAK/STAT pathway (152). Wenyi Z. et al. observed that the expression of GSDMD significantly decreased under treatment with the f JAK/STAT inhibitor, which was upregulated in response to AGE–RAGE action as mentioned above (97, 213). In addition, the largely released pro-inflammatory cytokine, IL-1β, could enhance the effects of ECM in human bronchial epithelial cells induced by TGF-β, thereby intensifying the pulmonary structural alterations and functional abnormalities mentioned above (126). In vitro experiments also support these effects. Yang F. et al. have recognized that downregulating caspase-1 in cells treated with high levels of glucose could strongly inhibit pyroptosis of the cells, decrease the release of IL-1β and TGF-β, and further relieve pathogenic processes such as collagen synthesis and ECM, which were due to the activation of TGF-β (127).
This evidence showed the possibility that pyroptosis could contribute to pulmonary dysfunction by means of cross-linking with the abnormal metabolic, oxidative stress, and inflammation pathways under the hyperglycemia condition of diabetes (Figure 1).
Several studies have identified that diabetes may be an important factor in inducing necroptosis in certain tissues through several classical pathways. Fiuza C. et al. found that AGE/RAGE could help with activating NF-κB and further facilitate TNF-α secretion in microvascular endothelial cells (137). The TNF-α/TNFR1 then stimulates downstream cell necroptosis through RIPK and MLKL, as described above (132–136). Yang et al. observed that Nϵ-carboxymethyl lysine (CML), a major member of advanced glycation end products, promotes the phosphorylation of RIPK3 and further facilitates necroptosis of tissue cells through interaction with RAGE (130). Although CML is expressed at a low level in normal physiological conditions, it can be upregulated under pathological conditions, such as diabetes. CML can bind with factors such as HMGB1, leading to the activation of TLRs and NF-κB, and even pyroptosis, as mentioned in the last section (129). HMGB1 is actually a type of damage-associated molecular pattern (DAMP) released from necroptotic cells such as airway epithelial cells (131). Necroptosis has also been found to positively interact with ROS, and such effects could intensify in diabetes (138). A high concentration of glucose has been observed to upregulate RIPK3 in tissue cells in vitro. Inhibiting the necroptosis of these cells decreased ROS production, while treating cells with ROS scavengers, in turn, decreased RIPK3 levels (138). These pieces of evidence indicate that the diabetic condition could not only directly promote necroptosis of tissue cells but also release pathological factors from necroptotic cells, further resulting in downstream reactions. What is worth mentioning is that necroptosis may co-exist with other types of CTDs, especially pyroptosis. In those with pulmonary dysfunction, including COPD or asthma, RIPK3 was shown to increase in the lung and airway tissues (139, 140), indicating the pathogenic effects of necroptosis in lung damage. Moreover, there is research also identifying attenuating RIP3K as well as the downstream MLKL, which can help with relieving airway remodeling and emphysema, which ameliorates pulmonary function to a certain extent (141) (Figure 1).
Previous studies showed that ferroptosis was related to ischemia-reperfusion injury (IRI)-relevant diseases (200). Under the high level of blood glucose in diabetes, vascular diseases were found in multiple systems and organs rich in blood supply, such as the cardiovascular system, optical system, and renal system (199, 214). As mentioned before, the lung has an abundant capillary network; thus, it might be inferred that the lung tends to be damaged in diabetes, including the pathways of ferroptosis. Moreover, the core mechanisms of ferroptosis, containing over accumulation of lipid peroxides as well as the overproduction of ROS, were found to play roles in the pathogenesis of several pulmonary diseases (146, 191). Combining the abnormal metabolic dysfunction and oxidative stress in diabetes, the pulmonary dysfunction due to ferroptosis under the corresponding circumstances could be significant to a certain extent.
Ferroptosis is a type of PCD that is dependent on ROS (215). In diabetes, there is an increase in ROS production in cells (142), which can lead to lipid peroxidation and further enhance ferroptosis (143). However, the effects of antioxidant agents are blocked under diabetic conditions. GPX4 is a powerful antioxidant enzyme that can inhibit lipid peroxidation and prevent ferroptosis. Previous studies have shown that GPX4 significantly decreases in certain types of pulmonary diseases such as lung fibrosis (147, 148) and tuberculosis (216). Recently, researchers have also found that diabetes can alter the levels of GPX4 (144). High blood glucose levels can increase the expression of RAGE and its ligands (145). According to Li YJ et al., overexpression of RAGE is associated with the suppression of superoxide dismutase (SOD), which inhibits the antioxidant pathway mainly mediated by GPX4. This leads to an increase in lipid peroxidation products and ferroptosis of tissue cells (144). Moreover, the same team has also revealed the possible effects of upregulated RAGE on iron uptake and storage. An increase in serum RAGE levels is significantly associated with a higher level of TF, which symbolizes more iron being transported into cells. It also decreases the export of iron through the elevation of hepcidin, which inhibits the iron exporter ferroportin1 (FPN1) (144). These all could contribute to cellular iron overload and exacerbate ferroptosis (146).
Apart from GPX4, the nuclear factor erythroid 2-related factor 2 (Nrf2) is another regulatory factor that functions as an antioxidant. Nrf2 defends against the damage caused by overloaded oxidative stress and inhibits ferroptosis (149). As a transcription factor for cellular antioxidants, Nrf2 is released in response to the production of ROS and promotes the production of more antioxidants (150). One significant downstream antioxidant is heme oxygenase 1 (HO-1), which promotes the catalyzation of heme, producing ferrous iron (151). The Nrf2/HO-1 pathway contributes to the homeostasis of iron metabolism and plays anti-inflammatory and antioxidant roles in cells (152). However, this defense system has been revealed to be destroyed in patients with pulmonary dysfunctional diseases such as lung fibrosis, COPD, and asthma (147, 153, 217). Interestingly, researchers have discovered the downregulation of Nrf2/HO-1 in DM patients (154), probably through the activation of extracellular signal-related kinase (ERK) (155). Therefore, it is reasonable to assume that diabetes could exacerbate ferroptosis in the lung and further contribute to the pathogenesis of pulmonary dysfunction. Additionally, downstream inflammation is also worthy of attention. Researchers found overexpression of TGF-β1 in lung fibrosis due to the inhibition of Nrf2 during ferroptosis. Therefore, ferroptosis can be a significant source of TGF-β1 under diabetic oxidative stress and do damage to the lung (148) (Figure 2).
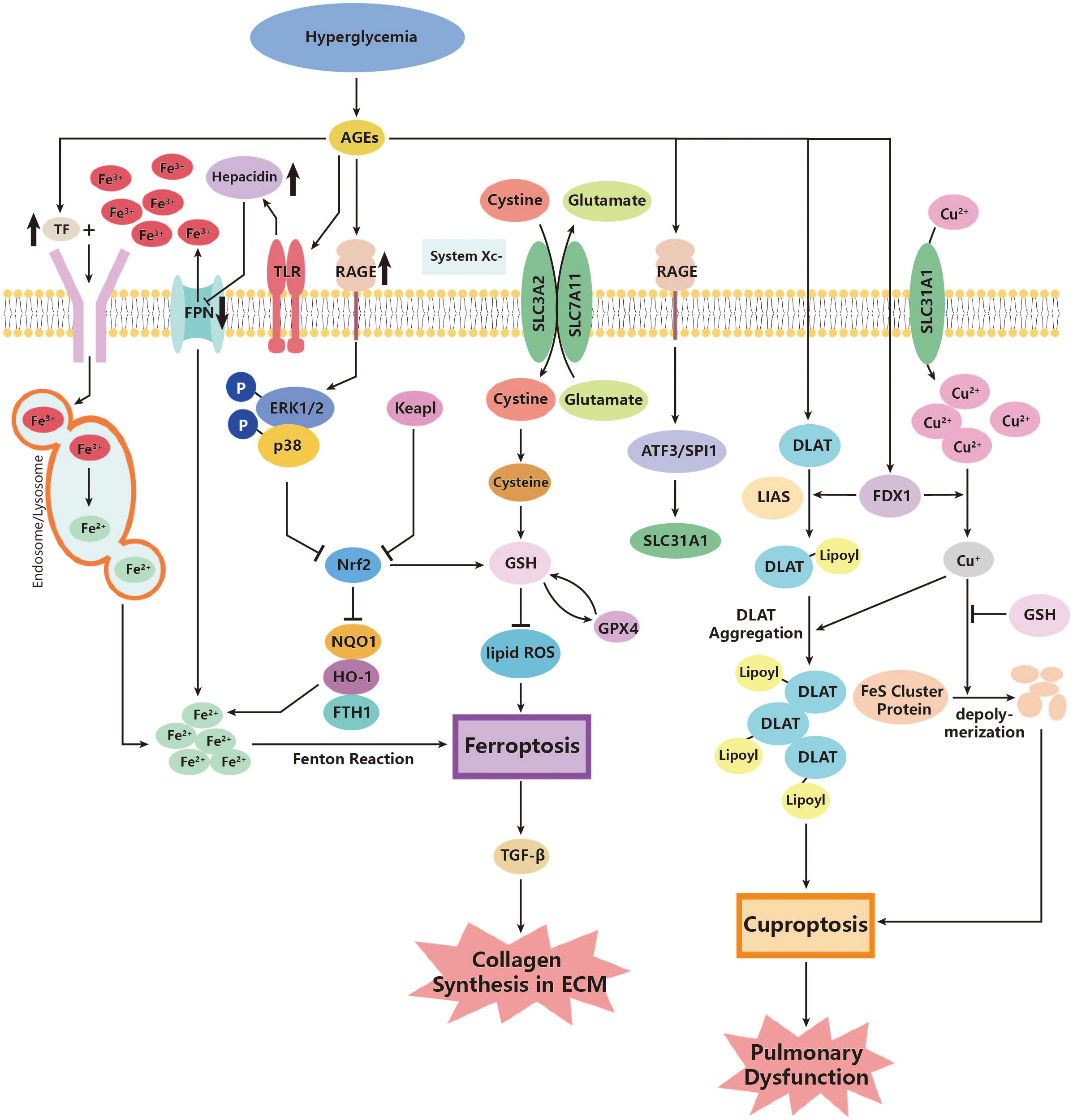
Figure 2 Roles of ferroptosis and cuproptosis in the pathogenesis of diabetic lung disease. AGEs, advanced glycation end products; ATF3, activating transcription factor 3; DLAT, dihydrolipoyl transacetylase; FDX1, ferredoxin 1; FPN, ferroportin; FTH1, ferritin heavy chain 1; GPX4, glutathione peroxidase 4; GSH, glutathione; HO-1, heme oxygenase 1; Keapl, Kelch-like ECH-associated protein 1; LIAS, lipoyl synthase; NOQ1, NADH dehydrogenase, quinone 1; Nrf2, nuclear factor erythroid 2-related factor 2; p38, p38 mitogen-activated protein kinases; RAGE, AGE receptor; ROS, reactive oxidative species; SLC3A2, Solute Carrier Family 3, Member 2; SLC7A11, Solute Carrier Family 7, Member 11; SLC31A1, high affinity copper uptake protein 1; SPI1, transcription factor PU; System Xc-, cysteine/glutamate transporter receptor; TF, transferrin; TGF-β, growth factor-β; TLR, Toll-like receptor.
Current studies on cuprotosis have focused on tumor cells and the life expectancy of relevant cancer diseases in various organs such as hepatocellular carcinoma, lung adenocarcinoma, clear cell renal carcinoma, and pancreatic cancer (218–221). Still, earlier studies that found abnormal copper metabolism in patients with diabetes compared to healthy controls demonstrate that cuprotosis may also play roles in several diabetic complications (222, 223). Few studies have mentioned the possible contribution of cuprotosis to diabetic pulmonary dysfunction; however, in view of cuprotosis often happening in cells with abundant mitochondria and energy consumption, the vascular endothelial cells of the lung, which are exposed directly to high concentrations of oxygen in gas exchange, may be potential targets of cuprotosis, especially in a pathological state like diabetes (224). In research using mouse experiments, researchers have found that an abnormally increased copper ion content can lead to higher production of ROS and decrease SOD expression in pulmonary tissue (225). Thus, it is reasonable to assume that pulmonary damage may be due to exacerbated cuprotosis in diabetic conditions. The above-mentioned signaling pathway, ATF3/SPI1/SLC31A1 (157), could be a possible pathway in diabetic lungs, though no direct studies have figured out the exact effect. Relevant studies can be a new way of studying non-apoptotic cell death effects in pulmonary dysfunction, not only cancer (Figure 2).
Though specific targeted drugs have not been largely studied or produced, some medicines are still found to contribute to influencing NAPCD-related pathways, leading to pulmonary dysfunction in DM. According to Huang et al., they have proved in mice that the drug, hispolon, can provide protection for pulmonary cells through inhibition of PI3K/Akt/mTOR, further resisting inflammation and oxidative stress under diabetic environments (226). Moreover, the chemical ingredients such as syringic and ascorbic acids, as well as nobiletin, have also been found to suppress autophagic cell death by downregulating the PI3K/Akt/mTOR pathway to prevent possible injuries to the lungs (227, 228). As for pyroptosis, several small molecules targeting the essential sites have already been developed to deal with conditions of multiple diseases. Possible molecules include MCC950, 3,4-methylenedioxy-β-nitrostyrene (MNS), and oridonin, which block NLRP3 and further play roles in DM and pulmonary injuries (229). Furthermore, necrosulfonamide and disulfiram have been identified to attenuate GSDMD to prevent LPS-induced lung fibrosis and pneumonia (229). Furthermore, there have been studies showing that blocking RIPK1, RIPK3, and MLKL can therefore improve the pulmonary function in diseases such as COPD and ARDS, as well as insulin resistance (230–232); the inhibitors include GW806742X and Nec‐1 (230, 232). Additionally, considering that ROS plays a significant role in the initiation of ferroptosis, vitamin E, which has weak chemical bonds, can help with trapping radicals and therefore protect the pulmonary from ferroptosis as well as oxidative damage under diabetic conditions (233, 234). Cuprotosis is a relatively novel concept. Though exact inhibitors have been rarely reported, they can be promising directions for future studies on diabetic pulmonary dysfunction.
SARS-Cov-2 has become a newly generated source of infectious pulmonary damage since the explosion of COVID-19. According to recent large-scale studies, diabetes is a significant risk factor for COVID-19, adding to the severity of the disease and mortality of the patients (235). During the process, non-apoptotic cell death has been proven to play a certain role. The most-mentioned one turned out to be ferroptosis. Bost, Pierre et al. found that the monocytes can promote the expression of cytokines and duplication of SARS-Cov-2, further ROS production in the mitochondria of endothelial cells under high-glucose level conditions in diabetics (236). Moreover, Jankauskas et al. revealed that the overproduced ROS exacerbates lipid oxidation and enhances ferroptosis, manifesting as increased levels of markers of ferroptosis, such as GPX4 (237). This can be a possible non-apoptotic cell death-mediated pathway for pulmonary damage in COVID-19 patients with diabetes. Furthermore, considering diabetes as a metabolic syndrome, the accompanying pro-inflammatory effects facilitate more production of inflammasome NLRP3, therefore adding to the pyroptosis of tissue cells in the lungs of COVID-19 patients (238, 239). Inhibition of NLRP3 by certain drugs can largely relieve the cytokine storm (240, 241).
This article provides a brief review of the traditional pathogenesis of diabetes pulmonary disease (DPD), various forms of NAPCD, and molecular regulatory pathways. Building on the foundation, it introduces the potential role of NAPCD in DPD.
Cell death plays a crucial role in the development and homeostasis of nearly all multicellular organisms. While apoptosis was once considered the primary pathway regulating cell death, recent years have witnessed the discovery of several forms of regulated cell death associated with inflammation, degenerative diseases, and cancer. This deepened understanding of regulated NAPCD has shed new light on the targeted treatment of these diseases. Inducing NAPCD has received increasing attention in the field of tumor therapy. For instance, in lung cancer studies, inhibitors of gamma-glutamyl cysteine ligase, glutathione, SLC7A11, and activators of iron have demonstrated anti-cancer effects by triggering ferroptosis. Some small molecules that target these pathways are now under consideration as candidate drugs for clinical trials. Despite diabetes having a lower mortality rate compared to tumors, diabetic complications impact vital organs such as the kidneys, eyes, and nervous system, significantly affecting patients’ quality of life. Presently, the role of NAPCD in the pathogenesis of diabetes is still in its infancy. For example, necroptosis can contribute to insulin resistance and damage to retinal, myocardial, and renal cells by mediating necrotizing inflammation, thereby influencing the onset and progression of diabetes and its associated chronic complications. Inhibitors targeting necroptosis are currently under development for various disease models, such as neurodegenerative diseases, acute heart failure, acute renal failure, systemic inflammation, and COPD. Consequently, the exploration of NAPCD in diabetic complications offers novel insights into the prevention and treatment of diabetes and its related health challenges.
All listed authors have made a substantial and direct contributions to this work. CW and QW conceptualized the review. YD and SZ are responsible for literature collection and article draft. LQ, ZP, and JZ are responsible for literature collection. DX, ML, XZ, and QW guided writing and revised the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by CAMS Innovation Fund for Medical Sciences (CIFMS) (2021-I2M-1-005 and 2019-I2M-2-008), the Beijing Municipal Science and Technology Commission (Z201100005520025), National High Level Hospital Clinical Research Funding (2022-PUMCH-A-107, 2022-PUMCH-C-020), the National Natural Science Foundation of China (81471615, 81601430), and Fundamental Research Funds for the Central Universities (3332021006).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Frazzei G, van Vollenhoven RF, de Jong BA, Siegelaar SE, van Schaardenburg D. Preclinical autoimmune disease: a comparison of rheumatoid arthritis, systemic lupus erythematosus, multiple sclerosis and type 1 diabetes. Front Immunol (2022) 13:899372. doi: 10.3389/fimmu.2022.899372
2. Schuyler MR, Niewoehner DE, Inkley SR, Kohn R. Abnormal lung elasticity in juvenile diabetes mellitus. Am Rev Respir Dis (1976) 113(1):37–41. doi: 10.1164/arrd.1976.113.1.37
3. Prugberger E, Kádas L. [Interstitial lung fibrosis and diabetes insipidus (author's transl)]. Z Erkr Atmungsorgane (1975) 143(3):289–93.
4. Ehrlich SF, Quesenberry CP Jr., Van Den Eeden SK, Shan J, Ferrara A. Patients diagnosed with diabetes are at increased risk for asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, and pneumonia but not lung cancer. Diabetes Care (2010) 33(1):55–60. doi: 10.2337/dc09-0880
5. Parappil A, Depczynski B, Collett P, Marks GB. Effect of comorbid diabetes on length of stay and risk of death in patients admitted with acute exacerbations of COPD. Respirology (2010) 15(6):918–22. doi: 10.1111/j.1440-1843.2010.01781.x
6. Carlsson AC, Wändell P, Ösby U, Zarrinkoub R, Wettermark B, Ljunggren G. High prevalence of diagnosis of diabetes, depression, anxiety, hypertension, asthma and COPD in the total population of stockholm, sweden - a challenge for public health. BMC Public Health (2013) 13:670. doi: 10.1186/1471-2458-13-670
7. Caughey GE, Preiss AK, Vitry AI, Gilbert AL, Roughead EE. Comorbid diabetes and COPD: impact of corticosteroid use on diabetes complications. Diabetes Care (2013) 36(10):3009–14. doi: 10.2337/dc12-2197
8. Bai L, Zhang L, Pan T, Wang W, Wang D, Turner C, et al. Idiopathic pulmonary fibrosis and diabetes mellitus: a meta-analysis and systematic review. Respir Res (2021) 22(1):175. doi: 10.1186/s12931-021-01760-6
9. Kumar V, Nawroth PP. Is the association between diabetes mellitus and pulmonary fibrosis real? Nat Rev Endocrinol (2021) 17(12):703–4. doi: 10.1038/s41574-021-00577-z
10. Teague TT, Payne SR, Kelly BT, Dempsey TM, McCoy RG, Sangaralingham LR, et al. Evaluation for clinical benefit of metformin in patients with idiopathic pulmonary fibrosis and type 2 diabetes mellitus: a national claims-based cohort analysis. Respir Res (2022) 23(1):91. doi: 10.1186/s12931-022-02001-0
11. Kopf S, Kumar V, Kender Z, Han Z, Fleming T, Herzig S, et al. Diabetic pneumopathy-a new diabetes-associated complication: Mechanisms, consequences and treatment considerations. Front Endocrinol (Lausanne) (2021) 12:765201. doi: 10.3389/fendo.2021.765201
12. Asanuma Y, Fujiya S, Ide H, Agishi Y. Characteristics of pulmonary function in patients with diabetes mellitus. Diabetes Res Clin Pract (1985) 1(2):95–101. doi: 10.1016/s0168-8227(85)80034-6
13. Klein OL, Krishnan JA, Glick S, Smith LJ. Systematic review of the association between lung function and type 2 diabetes mellitus. Diabetes Med (2010) 27(9):977–87. doi: 10.1111/j.1464-5491.2010.03073.x
14. Zhou Y, Meng F, Wang M, Li L, Yu P, Jiang Y. Reduced lung function predicts risk of incident type 2 diabetes: insights from a meta-analysis of prospective studies. Endocr J (2022) 69(3):299–305. doi: 10.1507/endocrj.EJ21-0403
15. Zhang RH, Cai YH, Shu LP, Yang J, Qi L, Han M, et al. Bidirectional relationship between diabetes and pulmonary function: a systematic review and meta-analysis. Diabetes Metab (2021) 47(5):101186. doi: 10.1016/j.diabet.2020.08.003
16. Orasanu G, Plutzky J. The pathologic continuum of diabetic vascular disease. J Am Coll Cardiol (2009) 53(5 Suppl):S35–42. doi: 10.1016/j.jacc.2008.09.055
17. Hsia CC, Raskin P. Lung involvement in diabetes: does it matter? Diabetes Care (2008) 31(4):828–9. doi: 10.2337/dc08-0103
18. Antonelli Incalzi R, Fuso L, Giordano A, Pitocco D, Maiolo C, Calcagni ML, et al. Neuroadrenergic denervation of the lung in type i diabetes mellitus complicated by autonomic neuropathy. Chest (2002) 121(2):443–51. doi: 10.1378/chest.121.2.443
19. Antonelli Incalzi R, Fuso L, Pitocco D, Basso S, Trové A, Longobardi A, et al. Decline of neuroadrenergic bronchial innervation and respiratory function in type 1 diabetes mellitus: a longitudinal study. Diabetes Metab Res Rev (2007) 23(4):311–6. doi: 10.1002/dmrr.688
20. Dorn GW 2nd. Mechanisms of non-apoptotic programmed cell death in diabetes and heart failure. Cell Cycle (2010) 9(17):3442–8. doi: 10.4161/cc.9.17.12944
21. Sauler M, Bazan IS, Lee PJ. Cell death in the lung: The apoptosis-necroptosis axis. Annu Rev Physiol (2019) 81:375–402. doi: 10.1146/annurev-physiol-020518-114320
22. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol (2021) 18(9):2114–27. doi: 10.1038/s41423-021-00740-6
23. Tait SW, Ichim G, Green DR. Die another way–non-apoptotic mechanisms of cell death. J Cell Sci (2014) 127(Pt 10):2135–44. doi: 10.1242/jcs.093575
24. Kahlson MA, Dixon SJ. Copper-induced cell death. Science (2022) 375(6586):1231–2. doi: 10.1126/science.abo3959
25. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid peroxidation-dependent cell death regulated by GPx4 and ferroptosis. Curr Top Microbiol Immunol (2017) 403:143–70. doi: 10.1007/82_2016_508
26. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol (2009) 7(2):99–109. doi: 10.1038/nrmicro2070
27. Ding S, Xu S, Ma Y, Liu G, Jang H, Fang J. Modulatory mechanisms of the NLRP3 inflammasomes in diabetes. Biomolecules (2019) 9(12). doi: 10.3390/biom9120850
28. Al Mamun A, Ara Mimi A, Wu Y, Zaeem M, Abdul Aziz M, Aktar Suchi S, et al. Pyroptosis in diabetic nephropathy. Clin Chim Acta (2021) 523:131–43. doi: 10.1016/j.cca.2021.09.003
29. Zhao XW, Yue WX, Zhang SW, Chen Q. Correlation between the accumulation of skin glycosylation end products and the development of type 2 diabetic peripheral neuropathy. BMC Endocr Disord (2022) 22(1):106. doi: 10.1186/s12902-022-00997-6
30. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature (2001) 414(6865):813–20. doi: 10.1038/414813a
31. Hsia CC, Raskin P. Lung function changes related to diabetes mellitus. Diabetes Technol Ther (2007) 9(Suppl 1):S73–82. doi: 10.1089/dia.2007.0227
32. Lecube A, Simó R, Pallayova M, Punjabi NM, López-Cano C, Turino C, et al. Pulmonary function and sleep breathing: Two new targets for type 2 diabetes care. Endocr Rev (2017) 38(6):550–73. doi: 10.1210/er.2017-00173
33. Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, et al. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab (2018) 28(3):337–52. doi: 10.1016/j.cmet.2018.08.014
34. Hoonhorst SJ, Lo Tam Loi AT, Hartman JE, Telenga ED, van den Berge M, Koenderman L, et al. Advanced glycation end products in the skin are enhanced in COPD. Metabolism (2014) 63(9):1149–56. doi: 10.1016/j.metabol.2014.06.006
35. Hoonhorst SJ, Lo Tam Loi AT, Pouwels SD, Faiz A, Telenga ED, van den Berge M, et al. Advanced glycation endproducts and their receptor in different body compartments in COPD. Respir Res (2016) 17:46. doi: 10.1186/s12931-016-0363-2
36. Gopal P, Reynaert NL, Scheijen JL, Engelen L, Schalkwijk CG, Franssen FM, et al. Plasma advanced glycation end-products and skin autofluorescence are increased in COPD. Eur Respir J (2014) 43(2):430–8. doi: 10.1183/09031936.00135312
37. Sánchez E, Lecube A, Betriu À, Hernández C, López-Cano C, Gutiérrez-Carrasquilla L, et al. Subcutaneous advanced glycation end-products and lung function according to glucose abnormalities: The ILERVAS project. Diabetes Metab (2019) 45(6):595–8. doi: 10.1016/j.diabet.2018.04.002
38. Zaigham S, Persson M, Jujic A, Frantz S, Borné Y, Malinovschi A, et al. Measures of lung function and their relationship with advanced glycation end-products. ERJ Open Res (2020) 6(2). doi: 10.1183/23120541.00356-2019
39. Reynaert NL, Gopal P, Rutten EPA, Wouters EFM, Schalkwijk CG. Advanced glycation end products and their receptor in age-related, non-communicable chronic inflammatory diseases; overview of clinical evidence and potential contributions to disease. Int J Biochem Cell Biol (2016) 81(Pt B):403–18. doi: 10.1016/j.biocel.2016.06.016
40. Ota C, Ishizawa K, Yamada M, Tando Y, He M, Takahashi T, et al. Receptor for advanced glycation end products expressed on alveolar epithelial cells is the main target for hyperoxia-induced lung injury. Respir Investig (2016) 54(2):98–108. doi: 10.1016/j.resinv.2015.08.009
41. Mangalmurti NS, Friedman JL, Wang LC, Stolz D, Muthukumaran G, Siegel DL, et al. The receptor for advanced glycation end products mediates lung endothelial activation by RBCs. Am J Physiol Lung Cell Mol Physiol (2013) 304(4):L250–63. doi: 10.1152/ajplung.00278.2012
42. Al-Robaiy S, Weber B, Simm A, Diez C, Rolewska P, Silber RE, et al. The receptor for advanced glycation end-products supports lung tissue biomechanics. Am J Physiol Lung Cell Mol Physiol (2013) 305(7):L491–500. doi: 10.1152/ajplung.00090.2013
43. Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type i cells. Cell Tissue Res (2006) 323(3):475–88. doi: 10.1007/s00441-005-0069-0
44. Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A. Role of advanced glycation end products in cellular signaling. Redox Biol (2014) 2:411–29. doi: 10.1016/j.redox.2013.12.016
45. Kasper M, Funk RH. Age-related changes in cells and tissues due to advanced glycation end products (AGEs). Arch Gerontol Geriatr (2001) 32(3):233–43. doi: 10.1016/s0167-4943(01)00103-0
46. Nowotny K, Jung T, Höhn A, Weber D, Grune T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules (2015) 5(1):194–222. doi: 10.3390/biom5010194
47. Roberts TJ, Burns AT, MacIsaac RJ, MacIsaac AI, Prior DL, La Gerche A. Diagnosis and significance of pulmonary microvascular disease in diabetes. Diabetes Care (2018) 41(4):854–61. doi: 10.2337/dc17-1904
48. Kuziemski K, Pieńkowska J, Słomiński W, Jassem and M. Studniarek E. Pulmonary capillary permeability and pulmonary microangiopathy in diabetes mellitus. Diabetes Res Clin Pract (2015) 108(3):e56–9. doi: 10.1016/j.diabres.2015.02.033
49. Tai H, Jiang XL, Yao SC, Liu Y, Wei H, Li LB, et al. Vascular endothelial function as a valid predictor of variations in pulmonary function in T2DM patients without related complications. Front Endocrinol (Lausanne) (2021) 12:622768. doi: 10.3389/fendo.2021.622768
50. Williams SB, Cusco JA, Roddy MA, Johnstone and M.A. Creager MT. Impaired nitric oxide-mediated vasodilation in patients with non-insulin-dependent diabetes mellitus. J Am Coll Cardiol (1996) 27(3):567–74. doi: 10.1016/0735-1097(95)00522-6
51. Senoner T, Dichtl W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients (2019) 11(9). doi: 10.3390/nu11092090
52. Barnes PJ. The role of inflammation and anti-inflammatory medication in asthma. Respir Med (2002) 96(Suppl A):S9–15.
53. Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem (1992) 267(21):14998–5004.
54. Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol (1993) 143(6):1699–712.
55. Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, et al. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. a potential mechanism for the accelerated vasculopathy of diabetes. J Clin Invest (1995) 96(3):1395–403. doi: 10.1172/jci118175
56. Vlassara H, Fuh H, Makita Z, Krungkrai S, Cerami A, Bucala R. Exogenous advanced glycosylation end products induce complex vascular dysfunction in normal animals: a model for diabetic and aging complications. Proc Natl Acad Sci USA (1992) 89(24):12043–7. doi: 10.1073/pnas.89.24.12043
57. Mohan S, Hamuro M, Sorescu GP, Koyoma K, Sprague EA, Jo H, et al. IkappaBalpha-dependent regulation of low-shear flow-induced NF-kappa b activity: role of nitric oxide. Am J Physiol Cell Physiol (2003) 284(4):C1039–47. doi: 10.1152/ajpcell.00464.2001
58. Throckmorton DC, Brogden AP, Min B, Rasmussen and M. Kashgarian H. PDGF and TGF-beta mediate collagen production by mesangial cells exposed to advanced glycosylation end products. Kidney Int (1995) 48(1):111–7. doi: 10.1038/ki.1995.274
59. Amal Abd El-Azeem A, Hamdy G, Amin M, Rashad A. Pulmonary function changes in diabetic lung. Egyptian J Chest Dis Tuberculosis (2013) 62(3):513–7. doi: 10.1016/j.ejcdt.2013.07.006
60. Pitocco D, Fuso L, Conte EG, Zaccardi F, Condoluci C, Scavone G, et al. The diabetic lung–a new target organ? Rev Diabetes Stud (2012) 9(1):23–35. doi: 10.1900/rds.2012.9.23
61. Talakatta G, Sarikhani M, Muhamed J, Dhanya K, Somashekar BS, Mahesh PA, et al. Diabetes induces fibrotic changes in the lung through the activation of TGF-β signaling pathways. Sci Rep (2018) 8(1):11920. doi: 10.1038/s41598-018-30449-y
62. Yeh HC, Punjabi NM, Wang NY, Pankow JS, Duncan BB, Cox CE, et al. Cross-sectional and prospective study of lung function in adults with type 2 diabetes: the atherosclerosis risk in communities (ARIC) study. Diabetes Care (2008) 31(4):741–6. doi: 10.2337/dc07-1464
63. Kass DA, Shapiro EP, Kawaguchi M, Capriotti AR, Scuteri A, deGroof RC, et al. Improved arterial compliance by a novel advanced glycation end-product crosslink breaker. Circulation (2001) 104(13):1464–70. doi: 10.1161/hc3801.097806
64. Posch K, Simecek S, Wascher TC, Jürgens G, Baumgartner-Parzer S, Kostner GM, et al. Glycated low-density lipoprotein attenuates shear stress-induced nitric oxide synthesis by inhibition of shear stress-activated l-arginine uptake in endothelial cells. Diabetes (1999) 48(6):1331–7. doi: 10.2337/diabetes.48.6.1331
65. Goldin A, Beckman JA, Schmidt and M.A. Creager AM. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation (2006) 114(6):597–605. doi: 10.1161/circulationaha.106.621854
66. Südy R, Schranc Á, Fodor GH, Tolnai J, Babik B, Peták F. Lung volume dependence of respiratory function in rodent models of diabetes mellitus. Respir Res (2020) 21(1):82. doi: 10.1186/s12931-020-01334-y
67. Ozşahin K, Tuğrul A, Mert S, Yüksel M, Tuğrul G. Evaluation of pulmonary alveolo-capillary permeability in type 2 diabetes mellitus: using technetium 99mTc-DTPA aerosol scintigraphy and carbon monoxide diffusion capacity. J Diabetes Complications (2006) 20(4):205–9. doi: 10.1016/j.jdiacomp.2005.07.003
69. Alikhani M, Maclellan CM, Raptis M, Vora S, Trackman PC, Graves DT. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am J Physiol Cell Physiol (2007) 292(2):C850–6. doi: 10.1152/ajpcell.00356.2006
70. Rajasurya V, Gunasekaran K, Surani S. Interstitial lung disease and diabetes. World J Diabetes (2020) 11(8):351–7. doi: 10.4239/wjd.v11.i8.351
71. Sternberg M, Cohen-Forterre L, Peyroux J. Connective tissue in diabetes mellitus: biochemical alterations of the intercellular matrix with special reference to proteoglycans, collagens and basement membranes. Diabete Metab (1985) 11(1):27–50.
72. Zheng H, Wu J, Jin and L.J. Yan Z. Potential biochemical mechanisms of lung injury in diabetes. Aging Dis (2017) 8(1):7–16. doi: 10.14336/ad.2016.0627
73. Fu L, Zhao H, Xiang Y, Xiang HX, Hu B, Tan ZX, et al. Reactive oxygen species-evoked endoplasmic reticulum stress mediates 1-nitropyrene-induced epithelial-mesenchymal transition and pulmonary fibrosis. Environ pollut (2021) 283:117134. doi: 10.1016/j.envpol.2021.117134
74. Barnes PJ. Cellular and molecular mechanisms of asthma and COPD. Clin Sci (Lond) (2017) 131(13):1541–58. doi: 10.1042/cs20160487
75. Kirkham PA, Barnes PJ. Oxidative stress in COPD. Chest (2013) 144(1):266–73. doi: 10.1378/chest.12-2664
76. Brown RK, Wyatt H, Price JF, Kelly FJ. Pulmonary dysfunction in cystic fibrosis is associated with oxidative stress. Eur Respir J (1996) 9(2):334–9. doi: 10.1183/09031936.96.09020334
77. Duecker R, Baer P, Eickmeier O, Strecker M, Kurz J, Schaible A, et al. Oxidative stress-driven pulmonary inflammation and fibrosis in a mouse model of human ataxia-telangiectasia. Redox Biol (2018) 14:645–55. doi: 10.1016/j.redox.2017.11.006
78. Guimarães EL, Empsen C, Geerts and L.A. van Grunsven A. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol (2010) 52(3):389–97. doi: 10.1016/j.jhep.2009.12.007
79. Zhang M, Kho AL, Anilkumar N, Chibber R, Pagano PJ, Shah AM, et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)-containing NADPH oxidase. Circulation (2006) 113(9):1235–43. doi: 10.1161/circulationaha.105.581397
80. Wu J, Jin Z, Yan LJ. Redox imbalance and mitochondrial abnormalities in the diabetic lung. Redox Biol (2017) 11:51–9. doi: 10.1016/j.redox.2016.11.003
81. Yan LJ. Protein redox modification as a cellular defense mechanism against tissue ischemic injury. Oxid Med Cell Longev (2014) 2014:343154. doi: 10.1155/2014/343154
82. Valencia JV, Weldon SC, Quinn D, Kiers GH, DeGroot J, TeKoppele JM, et al. Advanced glycation end product ligands for the receptor for advanced glycation end products: biochemical characterization and formation kinetics. Anal Biochem (2004) 324(1):68–78. doi: 10.1016/j.ab.2003.09.013
83. Redmann M, Darley-Usmar V, Zhang J. The role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and proteotoxic damage: Implications for neurodegenerative diseases. Aging Dis (2016) 7(2):150–62. doi: 10.14336/ad.2015.0820
84. Koliaki C, Roden M. Alterations of mitochondrial function and insulin sensitivity in human obesity and diabetes mellitus. Annu Rev Nutr (2016) 36:337–67. doi: 10.1146/annurev-nutr-071715-050656
85. Chen Y, Zhang F, Wang D, Li L, Si H, Wang C, et al. Mesenchymal stem cells attenuate diabetic lung fibrosis via adjusting Sirt3-mediated stress responses in rats. Oxid Med Cell Longev (2020) 2020:8076105. doi: 10.1155/2020/8076105
86. Skulachev VP. Cytochrome c in the apoptotic and antioxidant cascades. FEBS Lett (1998) 423(3):275–80. doi: 10.1016/s0014-5793(98)00061-1
87. Yu T, Sheu SS, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc Res (2008) 79(2):341–51. doi: 10.1093/cvr/cvn104
88. Tousoulis D, Kampoli AM, Tentolouris C, Papageorgiou N, Stefanadis C. The role of nitric oxide on endothelial function. Curr Vasc Pharmacol (2012) 10(1):4–18. doi: 10.2174/157016112798829760
89. Yagi O, Aoshiba K, Nagai A. Activation of nuclear factor-kappaB in airway epithelial cells in patients with chronic obstructive pulmonary disease. Respiration (2006) 73(5):610–6. doi: 10.1159/000090050
90. Kelly C, Shields MD, Elborn JS, Schock BC. A20 regulation of nuclear factor-κB: perspectives for inflammatory lung disease. Am J Respir Cell Mol Biol (2011) 44(6):743–8. doi: 10.1165/rcmb.2010-0339TR
91. Wong A, Dukic-Stefanovic S, Gasic-Milenkovic J, Schinzel R, Wiesinger H, Riederer P, et al. Anti-inflammatory antioxidants attenuate the expression of inducible nitric oxide synthase mediated by advanced glycation endproducts in murine microglia. Eur J Neurosci (2001) 14(12):1961–7. doi: 10.1046/j.0953-816x.2001.01820.x
92. San Martin A, Foncea R, Laurindo FR, Ebensperger R, Griendling KK, Leighton F. Nox1-based NADPH oxidase-derived superoxide is required for VSMC activation by advanced glycation end-products. Free Radic Biol Med (2007) 42(11):1671–9. doi: 10.1016/j.freeradbiomed.2007.02.002
93. Nah SS, Choi IY, Lee CK, Oh JS, Kim YG, Moon HB, et al. Effects of advanced glycation end products on the expression of COX-2, PGE2 and NO in human osteoarthritic chondrocytes. Rheumatol (Oxford) (2008) 47(4):425–31. doi: 10.1093/rheumatology/kem376
94. Farmer DG, Kennedy S. RAGE, vascular tone and vascular disease. Pharmacol Ther (2009) 124(2):185–94. doi: 10.1016/j.pharmthera.2009.06.013
95. Zhang F, Yang F, Zhao H, An Y. Curcumin alleviates lung injury in diabetic rats by inhibiting nuclear factor-κB pathway. Clin Exp Pharmacol Physiol (2015) 42(9):956–63. doi: 10.1111/1440-1681.12438
96. Sun J, Huang N, Ma W, Zhou H, Lai K. Protective effects of metformin on lipopolysaccharide−induced airway epithelial cell injury via NF−κB signaling inhibition. Mol Med Rep (2019) 19(3):1817–23. doi: 10.3892/mmr.2019.9807
97. Oh H, Park SH, Kang MK, Kim YH, Lee EJ, Kim DY, et al. Asaronic acid attenuates macrophage activation toward M1 phenotype through inhibition of NF-κB pathway and JAK-STAT signaling in glucose-loaded murine macrophages. J Agric Food Chem (2019) 67(36):10069–78. doi: 10.1021/acs.jafc.9b03926
98. Duan WN, Xia ZY, Liu M, Sun Q, Lei SQ, Wu XJ, et al. Protective effects of SOCS3 overexpression in high glucose−induced lung epithelial cell injury through the JAK2/STAT3 pathway. Mol Med Rep (2017) 16(3):2668–74. doi: 10.3892/mmr.2017.6941
99. Qin MZ, Qin MB, Liang ZH, Tang GD. Effect of SOCS3 on lung injury in rats with severe acute pancreatitis through regulating JAK2/STAT3 signaling pathway. Eur Rev Med Pharmacol Sci (2019) 23(22):10123–31. doi: 10.26355/eurrev_201911_19582
100. Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood (2007) 109(1):353–8. doi: 10.1182/blood-2006-07-033969
101. Zhang Y, Dees C, Beyer C, Lin NY, Distler A, Zerr P, et al. Inhibition of casein kinase II reduces TGFβ induced fibroblast activation and ameliorates experimental fibrosis. Ann Rheum Dis (2015) 74(5):936–43. doi: 10.1136/annrheumdis-2013-204256
102. Critchley JA, Carey IM, Harris T, DeWilde S, Hosking FJ, Cook DG. Glycemic control and risk of infections among people with type 1 or type 2 diabetes in a large primary care cohort study. Diabetes Care (2018) 41(10):2127–35. doi: 10.2337/dc18-0287
103. Hine JL, de Lusignan S, Burleigh D, Pathirannehelage S, McGovern A, Gatenby P, et al. Association between glycaemic control and common infections in people with type 2 diabetes: a cohort study. Diabetes Med (2017) 34(4):551–7. doi: 10.1111/dme.13205
104. Erener S. Diabetes, infection risk and COVID-19. Mol Metab (2020) 39:101044. doi: 10.1016/j.molmet.2020.101044
105. Kulcsar KA, Coleman CM, Beck and M.B. Frieman SE. Comorbid diabetes results in immune dysregulation and enhanced disease severity following MERS-CoV infection. JCI Insight (2019) 4(20). doi: 10.1172/jci.insight.131774
106. Ye Y, Chen Y, Sun J, Zhang H, Li W, Wang W, et al. Repressed hypoxia inducible factor-1 in diabetes aggravates pulmonary aspergillus fumigatus infection through modulation of inflammatory responses. Clin Transl Med (2021) 11(1):e273. doi: 10.1002/ctm2.273
107. Amin R, Dupuis A, Aaron SD, Ratjen F. The effect of chronic infection with aspergillus fumigatus on lung function and hospitalization in patients with cystic fibrosis. Chest (2010) 137(1):171–6. doi: 10.1378/chest.09-1103
108. Hodgson K, Morris J, Bridson T, Govan B, Rush C, Ketheesan N. Immunological mechanisms contributing to the double burden of diabetes and intracellular bacterial infections. Immunology (2015) 144(2):171–85. doi: 10.1111/imm.12394
109. Chanchamroen S, Kewcharoenwong C, Susaengrat W, Ato M, Lertmemongkolchai G. Human polymorphonuclear neutrophil responses to burkholderia pseudomallei in healthy and diabetic subjects. Infect Immun (2009) 77(1):456–63. doi: 10.1128/iai.00503-08
110. Alba-Loureiro TC, Hirabara SM, Mendonça JR, Curi R, Pithon-Curi TC. Diabetes causes marked changes in function and metabolism of rat neutrophils. J Endocrinol (2006) 188(2):295–303. doi: 10.1677/joe.1.06438
111. Kewcharoenwong C, Rinchai D, Utispan K, Suwannasaen D, Bancroft GJ, Ato M, et al. Glibenclamide reduces pro-inflammatory cytokine production by neutrophils of diabetes patients in response to bacterial infection. Sci Rep (2013) 3:3363. doi: 10.1038/srep03363
112. Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell (2013) 153(3):521–34. doi: 10.1016/j.cell.2013.03.022
113. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science (2022) 375(6586):1254–61. doi: 10.1126/science.abf0529
114. Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y, et al. Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc Diabetol (2014) 13:78. doi: 10.1186/1475-2840-13-78
115. Ma J, Sun Q, Mi R, Zhang H. Avian influenza a virus H5N1 causes autophagy-mediated cell death through suppression of mTOR signaling. J Genet Genomics (2011) 38(11):533–7. doi: 10.1016/j.jgg.2011.10.002
116. Krakauer T. Inflammasome, mTORC1 activation, and metabolic derangement contribute to the susceptibility of diabetics to infections. Med Hypotheses (2015) 85(6):997–1001. doi: 10.1016/j.mehy.2015.08.019
117. Yang J, Zhao Y, Zhang P, Li Y, Yang Y, Yang Y, et al. Hemorrhagic shock primes for lung vascular endothelial cell pyroptosis: role in pulmonary inflammation following LPS. Cell Death Dis (2016) 7(9):e2363. doi: 10.1038/cddis.2016.274
118. Cao Z, Huang D, Tang C, Lu Y, Huang S, Peng C, et al. Pyroptosis in diabetes and diabetic nephropathy. Clin Chim Acta (2022) 531:188–96. doi: 10.1016/j.cca.2022.04.011
119. Behl T, Sharma E, Sehgal A, Kaur I, Kumar A, Arora R, et al. Expatiating the molecular approaches of HMGB1 in diabetes mellitus: Highlighting signalling pathways via RAGE and TLRs. Mol Biol Rep (2021) 48(2):1869–81. doi: 10.1007/s11033-020-06130-x
120. Yin Y, Li X, Sha X, Xi H, Li YF, Shao Y, et al. Early hyperlipidemia promotes endothelial activation via a caspase-1-sirtuin 1 pathway. Arterioscler Thromb Vasc Biol (2015) 35(4):804–16. doi: 10.1161/atvbaha.115.305282
121. Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, et al. The endotoxin delivery protein HMGB1 mediates caspase-11-Dependent lethality in sepsis. Immunity (2018) 49(4):740–753.e7. doi: 10.1016/j.immuni.2018.08.016
122. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479(7371):117–21. doi: 10.1038/nature10558
123. Tian L, Yan J, Li K, Zhang W, Lin B, Lai W, et al. Ozone exposure promotes pyroptosis in rat lungs via the TLR2/4-NF-κB-NLRP3 signaling pathway. Toxicology (2021) 450:152668. doi: 10.1016/j.tox.2020.152668
124. Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger? Science (2010) 327(5963):296–300. doi: 10.1126/science.1184003
125. Watanabe M, Toyomura T, Tomiyama M, Wake H, Liu K, Teshigawara K, et al. Advanced glycation end products (AGEs) synergistically potentiated the proinflammatory action of lipopolysaccharide (LPS) and high mobility group box-1 (HMGB1) through their direct interactions. Mol Biol Rep (2020) 47(9):7153–9. doi: 10.1007/s11033-020-05783-y
126. Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res (2009) 10(1):100. doi: 10.1186/1465-9921-10-100
127. Yang F, Qin Y, Lv J, Wang Y, Che H, Chen X, et al. Silencing long non-coding RNA Kcnq1ot1 alleviates pyroptosis and fibrosis in diabetic cardiomyopathy. Cell Death Dis (2018) 9(10):1000. doi: 10.1038/s41419-018-1029-4
128. Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial reactive oxygen species induces NLRP3-dependent lysosomal damage and inflammasome activation. J Immunol (2013) 191(10):5230–8. doi: 10.4049/jimmunol.1301490
129. Waghela BN, Vaidya FU, Ranjan K, Chhipa AS, Tiwari BS, Pathak C. AGE-RAGE synergy influences programmed cell death signaling to promote cancer. Mol Cell Biochem (2021) 476(2):585–98. doi: 10.1007/s11010-020-03928-y
130. Yang J, Zhang F, Shi H, Gao Y, Dong Z, Ma L, et al. Neutrophil-derived advanced glycation end products-nϵ-(carboxymethyl) lysine promotes RIP3-mediated myocardial necroptosis via RAGE and exacerbates myocardial ischemia/reperfusion injury. FASEB J (2019) 33(12):14410–22. doi: 10.1096/fj.201900115RR
131. Simpson J, Loh Z, Ullah MA, Lynch JP, Werder RB, Collinson N, et al. Respiratory syncytial virus infection promotes necroptosis and HMGB1 release by airway epithelial cells. Am J Respir Crit Care Med (2020) 201(11):1358–71. doi: 10.1164/rccm.201906-1149OC
132. Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G, Declercq W, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med (1998) 187(9):1477–85. doi: 10.1084/jem.187.9.1477
133. Berger SB, Bertin J, Gough PJ. Life after death: RIP1 and RIP3 move beyond necroptosis. Cell Death Discovery (2016) 2:16056. doi: 10.1038/cddiscovery.2016.56
134. Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity (2013) 38(1):27–40. doi: 10.1016/j.immuni.2012.09.015
135. Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y, et al. Mlkl knockout mice demonstrate the indispensable role of mlkl in necroptosis. Cell Res (2013) 23(8):994–1006. doi: 10.1038/cr.2013.91
136. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148(1-2):213–27. doi: 10.1016/j.cell.2011.11.031
137. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood (2003) 101(7):2652–60. doi: 10.1182/blood-2002-05-1300
138. Liang W, Chen M, Zheng D, He J, Song M, Mo L, et al. A novel damage mechanism: Contribution of the interaction between necroptosis and ROS to high glucose-induced injury and inflammation in H9c2 cardiac cells. Int J Mol Med (2017) 40(1):201–8. doi: 10.3892/ijmm.2017.3006
139. Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest (2014) 124(9):3987–4003. doi: 10.1172/jci74985
140. Oikonomou N, Schuijs MJ, Chatzigiagkos A, Androulidaki A, Aidinis V, Hammad H, et al. Airway epithelial cell necroptosis contributes to asthma exacerbation in a mouse model of house dust mite-induced allergic inflammation. Mucosal Immunol (2021) 14(5):1160–71. doi: 10.1038/s41385-021-00415-5
141. Lu Z, Van Eeckhoutte HP, Liu G, Nair PM, Jones B, Gillis CM, et al. Necroptosis signaling promotes inflammation, airway remodeling, and emphysema in chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2021) 204(6):667–81. doi: 10.1164/rccm.202009-3442OC
142. Yan LJ. Pathogenesis of chronic hyperglycemia: from reductive stress to oxidative stress. J Diabetes Res (2014) 2014:137919. doi: 10.1155/2014/137919
143. Yang L, Cao LM, Zhang XJ, Chu B. Targeting ferroptosis as a vulnerability in pulmonary diseases. Cell Death Dis (2022) 13(7):649. doi: 10.1038/s41419-022-05070-7
144. Li Y, Qin M, Zhong W, Liu C, Deng G, Yang M, et al. RAGE promotes dysregulation of iron and lipid metabolism in alcoholic liver disease. Redox Biol (2022). doi: 10.1016/j.redox.2022.102559:102559.https://doi.org/https://doi.org/10.1016/j.redox.2022.102559
145. Yao D, Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes (2010) 59(1):249–55. doi: 10.2337/db09-0801
146. Tao N, Li K, Liu J. Molecular mechanisms of ferroptosis and its role in pulmonary disease. Oxid Med Cell Longev (2020) 2020:9547127. doi: 10.1155/2020/9547127
147. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun (2019) 519(2):240–5. doi: 10.1016/j.bbrc.2019.08.165
148. Li X, Duan L, Yuan S, Zhuang X, Qiao T, He J. Ferroptosis inhibitor alleviates radiation-induced lung fibrosis (RILF) via down-regulation of TGF-β1. . J Inflammation (Lond) (2019) 16:11. doi: 10.1186/s12950-019-0216-0
149. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
150. Sun X, Niu X, Chen R, He W, Chen D, Kang R, et al. Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology (2016) 64(2):488–500. doi: 10.1002/hep.28574
151. Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol (2019) 23:101107. doi: 10.1016/j.redox.2019.101107
152. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
153. Lim JO, Song KH, Lee IS, Lee SJ, Kim WI, Pak SW, et al. Cimicifugae rhizoma extract attenuates oxidative stress and airway inflammation via the upregulation of Nrf2/HO-1/NQO1 and downregulation of NF-κB phosphorylation in ovalbumin-induced asthma. Antioxidants (Basel) (2021) 10(10). doi: 10.3390/antiox10101626
154. Siewert S, Gonz¨¢lez I, Santill¨¢n L, Lucero R, Ojeda MS, Gimenez MAS. Downregulation of Nrf2 and HO-1 expression contributes to oxidative stress in type 2 diabetes mellitus: A study in juana koslay city, san luis, argentina. J Diabetes Mellitus (2013) 03(02):8. doi: 10.4236/jdm.2013.32011
155. Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes (2011) 60(2):625–33. doi: 10.2337/db10-1164
156. Masad A, Hayes L, Tabner BJ, Turnbull S, Cooper LJ, Fullwood NJ, et al. Copper-mediated formation of hydrogen peroxide from the amylin peptide: a novel mechanism for degeneration of islet cells in type-2 diabetes mellitus? FEBS Lett (2007) 581(18):3489–93. doi: 10.1016/j.febslet.2007.06.061
157. Huo S, Wang Q, Shi W, Peng L, Jiang Y, Zhu M, et al. ATF3/SPI1/SLC31A1 signaling promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int J Mol Sci (2023) 24(2). doi: 10.3390/ijms24021667
158. Soma S, Latimer AJ, Chun H, Vicary AC, Timbalia SA, Boulet A, et al. Elesclomol restores mitochondrial function in genetic models of copper deficiency. Proc Natl Acad Sci USA (2018) 115(32):8161–6. doi: 10.1073/pnas.1806296115
159. Tang D, Chen X, Kroemer G. Cuproptosis: a copper-triggered modality of mitochondrial cell death. Cell Res (2022) 32(5):417–8. doi: 10.1038/s41422-022-00653-7
160. Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol (2011) 76:397–402. doi: 10.1101/sqb.2011.76.011023
161. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol (2010) 221(1):3–12. doi: 10.1002/path.2697
162. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med (2013) 368(7):651–62. doi: 10.1056/NEJMra1205406
163. Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest (2005) 115(10):2679–88. doi: 10.1172/jci26390
164. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, et al. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell (2005) 122(6):927–39. doi: 10.1016/j.cell.2005.07.002
165. Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol (2004) 6(12):1221–8. doi: 10.1038/ncb1192
166. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol (2008) 9(12):1004–10. doi: 10.1038/nrm2529
167. Clarke PG, Puyal J. Autophagic cell death exists. Autophagy (2012) 8(6):867–9. doi: 10.4161/auto.20380
168. He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature (2012) 481(7382):511–5. doi: 10.1038/nature10758
169. Curtiss ML, Deshane JS. "Stick a fork in me; i'm done": Epithelial cell expression of ORMDL sphingolipid biosynthesis regulator 3 mediates autophagic cell death. Am J Respir Cell Mol Biol (2022) 66(6):593–5. doi: 10.1165/rcmb.2022-0023ED
170. Song CF, Hu YH, Mang ZG, Ye Z, Chen HD, Jing DS, et al. Hernandezine induces autophagic cell death in human pancreatic cancer cells via activation of the ROS/AMPK signaling pathway. Acta Pharmacol Sin (2022). doi: 10.1038/s41401-022-01006-1
171. Yu JE, Yeo IJ, Lee DW, Chang JY, Son DJ, Yun J, et al. Snake venom induces an autophagic cell death via activation of the JNK pathway in colorectal cancer cells. J Cancer (2022) 13(12):3333–41. doi: 10.7150/jca.75791
172. Zhang L, Xu S, Cheng X, Wu J, Wu L, Wang Y, et al. Curcumin induces autophagic cell death in human thyroid cancer cells. Toxicol In Vitro (2022) 78:105254. doi: 10.1016/j.tiv.2021.105254
173. de Almagro MC, Vucic D. Necroptosis: Pathway diversity and characteristics. Semin Cell Dev Biol (2015) 39:56–62. doi: 10.1016/j.semcdb.2015.02.002
174. Bertheloot D, Latz E, Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol (2021) 18(5):1106–21. doi: 10.1038/s41423-020-00630-3
175. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity (2013) 38(2):209–23. doi: 10.1016/j.immuni.2013.02.003
176. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol (2005) 1(2):112–9. doi: 10.1038/nchembio711
177. Oerlemans MI, Liu J, Arslan F, den Ouden K, van Middelaar BJ, Doevendans PA, et al. Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia-reperfusion in vivo. Basic Res Cardiol (2012) 107(4):270. doi: 10.1007/s00395-012-0270-8
178. Lee YS, Dayma Y, Park MY, Kim KI, Yoo SE, Kim E. Daxx is a key downstream component of receptor interacting protein kinase 3 mediating retinal ischemic cell death. FEBS Lett (2013) 587(3):266–71. doi: 10.1016/j.febslet.2012.12.004
179. Linkermann A, Bräsen JH, Himmerkus N, Liu S, Huber TB, Kunzendorf U, et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int (2012) 81(8):751–61. doi: 10.1038/ki.2011.450
180. Bonnet MC, Preukschat D, Welz PS, van Loo G, Ermolaeva MA, Bloch W, et al. The adaptor protein FADD protects epidermal keratinocytes from necroptosis in vivo and prevents skin inflammation. Immunity (2011) 35(4):572–82. doi: 10.1016/j.immuni.2011.08.014
181. Günther C, Martini E, Wittkopf N, Amann K, Weigmann B, Neumann H, et al. Caspase-8 regulates TNF-α-induced epithelial necroptosis and terminal ileitis. Nature (2011) 477(7364):335–9. doi: 10.1038/nature10400
182. Linkermann A, Green DR. Necroptosis. N Engl J Med (2014) 370(5):455–65. doi: 10.1056/NEJMra1310050
183. Deng W, Bai Y, Deng F, Pan Y, Mei S, Zheng Z, et al. Streptococcal pyrogenic exotoxin b cleaves GSDMA and triggers pyroptosis. Nature (2022) 602(7897):496–502. doi: 10.1038/s41586-021-04384-4
184. Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, et al. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science (1995) 267(5206):2000–3. doi: 10.1126/science.7535475
185. Zhou Y, Tong Z, Jiang S, Zheng W, Zhao J, Zhou X. The roles of endoplasmic reticulum in NLRP3 inflammasome activation. Cells (2020) 9(5). doi: 10.3390/cells9051219
186. Rajamäki K, Lappalainen J, Oörni K, Välimäki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PloS One (2010) 5(7):e11765. doi: 10.1371/journal.pone.0011765
187. Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology (2017) 152(2):207–17. doi: 10.1111/imm.12787
188. Mamun AA, Wu Y, Nasrin F, Akter A, Taniya MA, Munir F, et al. Role of pyroptosis in diabetes and its therapeutic implications. J Inflammation Res (2021) 14:2187–206. doi: 10.2147/jir.S291453
189. Zhang MY, Jiang YX, Yang YC, Liu JY, Huo C, Ji XL, et al. Cigarette smoke extract induces pyroptosis in human bronchial epithelial cells through the ROS/NLRP3/caspase-1 pathway. Life Sci (2021) 269:119090. doi: 10.1016/j.lfs.2021.119090
190. Wan X, Li J, Wang Y, Yu X, He X, Shi J, et al. H7N9 virus infection triggers lethal cytokine storm by activating gasdermin e-mediated pyroptosis of lung alveolar epithelial cells. Natl Sci Rev (2022) 9(1):nwab137. doi: 10.1093/nsr/nwab137
191. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
192. Lei P, Bai T, Sun Y. Mechanisms of ferroptosis and relations with regulated cell death: A review. Front Physiol (2019) 10:139. doi: 10.3389/fphys.2019.00139
193. Das NK, Schwartz AJ, Barthel G, Inohara N, Liu Q, Sankar A, et al. Microbial metabolite signaling is required for systemic iron homeostasis. Cell Metab (2020) 31(1):115–30. doi: 10.1016/j.cmet.2019.10.005
194. Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell (2004) 116(4):565–76. doi: 10.1016/s0092-8674(04)00130-8
195. Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer (2013) 13(5):342–55. doi: 10.1038/nrc3495
196. Ganz T. Systemic iron homeostasis. Physiol Rev (2013) 93(4):1721–41. doi: 10.1152/physrev.00008.2013
197. Shen Z, Liu T, Li Y, Lau J, Yang Z, Fan W, et al. Fenton-Reaction-Acceleratable magnetic nanoparticles for ferroptosis therapy of orthotopic brain tumors. ACS Nano (2018) 12(11):11355–65. doi: 10.1021/acsnano.8b06201
198. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res (2021) 2021:9999612. doi: 10.1155/2021/9999612
199. Lascar N, Brown J, Pattison H, Barnett AH, Bailey CJ, Bellary S. Type 2 diabetes in adolescents and young adults. Lancet Diabetes Endocrinol (2018) 6(1):69–80. doi: 10.1016/s2213-8587(17)30186-9
200. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial Ischemia/Reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol (2020) 39(2):210–25. doi: 10.1089/dna.2019.5097
201. Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol (2008) 4(3):176–85. doi: 10.1038/nchembio.72
202. Ge W, Jie J, Yao J, Li W, Cheng Y, Lu W. Advanced glycation end products promote osteoporosis by inducing ferroptosis in osteoblasts. Mol Med Rep (2022) 25(4). doi: 10.3892/mmr.2022.12656
203. Ghio AJ, Hilborn ED, Stonehuerner JG, Dailey LA, Carter JD, Richards JH, et al. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am J Respir Crit Care Med (2008) 178(11):1130–8. doi: 10.1164/rccm.200802-334OC
204. Yoshida M, Minagawa S, Araya J, Sakamoto T, Hara H, Tsubouchi K, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun (2019) 10(1):3145. doi: 10.1038/s41467-019-10991-7
205. Ryter SW, Lee SJ, Choi AM. Autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. Expert Rev Respir Med (2010) 4(5):573–84. doi: 10.1586/ers.10.61
206. Kuwano K, Araya J, Hara H, Minagawa S, Takasaka N, Ito S, et al. Cellular senescence and autophagy in the pathogenesis of chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). Respir Investig (2016) 54(6):397–406. doi: 10.1016/j.resinv.2016.03.010
207. Liu MW, Su MX, Tang DY, Hao L, Xun XH, Huang YQ. Ligustrazin increases lung cell autophagy and ameliorates paraquat-induced pulmonary fibrosis by inhibiting PI3K/Akt/mTOR and hedgehog signalling via increasing miR-193a expression. BMC Pulm Med (2019) 19(1):35. doi: 10.1186/s12890-019-0799-5
208. Liu K, Yang Y, Zhou F, Xiao and L. Shi Y. Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy and relieves hyperalgesia in diabetic rats. Neuroreport (2020) 31(9):644–9. doi: 10.1097/wnr.0000000000001461
209. Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol (2007) 7(10):767–77. doi: 10.1038/nri2161
210. Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis (2020) 11(9):776. doi: 10.1038/s41419-020-02985-x
211. Iannantuoni F, Diaz-Morales N, Escribano-Lopez I, Sola E, Roldan-Torres I, Apostolova N, et al. Does glycemic control modulate the impairment of NLRP3 inflammasome activation in type 2 diabetes? Antioxid Redox Signal (2019) 30(2):232–40. doi: 10.1089/ars.2018.7582
212. Feng Y, Li M, Yangzhong X, Zhang X, Zu A, Hou Y, et al. Pyroptosis in inflammation-related respiratory disease. J Physiol Biochem (2022) 78(4):721–37. doi: 10.1007/s13105-022-00909-1
213. Zhao W, Yang H, Lyu L, Zhang J, Xu Q, Jiang N, et al. GSDMD, an executor of pyroptosis, is involved in IL-1β secretion in aspergillus fumigatus keratitis. Exp Eye Res (2021) 202:108375. doi: 10.1016/j.exer.2020.108375
214. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic Biol Med (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
215. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res (2021) 31(2):107–25. doi: 10.1038/s41422-020-00441-1
216. Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, et al. A major role for ferroptosis in mycobacterium tuberculosis-induced cell death and tissue necrosis. J Exp Med (2019) 216(3):556–70. doi: 10.1084/jem.20181776
217. Li J, Baker J, Higham A, Shah R, Montero-Fernandez A, Murray C, et al. COPD lung studies of Nrf2 expression and the effects of Nrf2 activators. Inflammopharmacology (2022) 30(4):1431–43. doi: 10.1007/s10787-022-00967-3
218. Liu T, Cai L, Hua H, Jiang X, Xu X, Zhang T, et al. Cuprotosis patterns are associated with tumor mutation burden and immune landscape in lung adenocarcinoma. J Oncol (2022) 2022:9772208. doi: 10.1155/2022/9772208
219. Xiao J, Liu Z, Wang J, Zhang S, Zhang Y. Identification of cuprotosis-mediated subtypes, the development of a prognosis model, and influence immune microenvironment in hepatocellular carcinoma. Front Oncol (2022) 12:941211. doi: 10.3389/fonc.2022.941211
220. Zhang WT, Gong YM, Zhang CY, Pan JS, Huang T, Li YX. A novel cuprotosis-related gene FDX1 signature for overall survival prediction in clear cell renal cell carcinoma patients. BioMed Res Int (2022) 2022:9196540. doi: 10.1155/2022/9196540
221. Xu Y, Li H, Lan A, Wu Q, Tang Z, Shu D, et al. Cuprotosis-related genes: Predicting prognosis and immunotherapy sensitivity in pancreatic cancer patients. J Oncol (2022) 2022:2363043. doi: 10.1155/2022/2363043
222. Cooper GJ, Chan YK, Dissanayake AM, Leahy FE, Keogh GF, Frampton CM, et al. Demonstration of a hyperglycemia-driven pathogenic abnormality of copper homeostasis in diabetes and its reversibility by selective chelation: quantitative comparisons between the biology of copper and eight other nutritionally essential elements in normal and diabetic individuals. Diabetes (2005) 54(5):1468–76. doi: 10.2337/diabetes.54.5.1468
223. Ito S, Fujita H, Narita T, Yaginuma T, Kawarada Y, Kawagoe M, et al. Urinary copper excretion in type 2 diabetic patients with nephropathy. Nephron (2001) 88(4):307–12. doi: 10.1159/000046013
224. Cui X, Wang Y, Liu H, Shi M, Wang J, Wang Y. The molecular mechanisms of defective copper metabolism in diabetic cardiomyopathy. Oxid Med Cell Longev (2022) 2022:5418376. doi: 10.1155/2022/5418376
225. Jian Z, Guo H, Liu H, Cui H, Fang J, Zuo Z, et al. Oxidative stress, apoptosis and inflammatory responses involved in copper-induced pulmonary toxicity in mice. Aging (Albany NY) (2020) 12(17):16867–86. doi: 10.18632/aging.103585
226. Huang CY, Deng JS, Huang WC, Jiang WP, Huang GJ. Attenuation of lipopolysaccharide-induced acute lung injury by hispolon in mice, through regulating the TLR4/PI3K/Akt/mTOR and Keap1/Nrf2/HO-1 pathways, and suppressing oxidative stress-mediated ER stress-induced apoptosis and autophagy. Nutrients (2020) 12(6). doi: 10.3390/nu12061742
227. Somade OT, Adeyi OE, Ajayi BO, Asunde OO, Iloh PD, Adesanya AA, et al. Syringic and ascorbic acids prevent NDMA-induced pulmonary fibrogenesis, inflammation, apoptosis, and oxidative stress through the regulation of PI3K-Akt/PKB-mTOR-PTEN signaling pathway. Metabol Open (2022) 14:100179. doi: 10.1016/j.metop.2022.100179
228. El Tabaa MM, El Tabaa MM, Elgharabawy RM, Abdelhamid WG. Suppressing NLRP3 activation and PI3K/AKT/mTOR signaling ameliorates amiodarone-induced pulmonary fibrosis in rats: a possible protective role of nobiletin. Inflammopharmacology (2023) 31(3):1373–86. doi: 10.1007/s10787-023-01168-2
229. Coll RC, Schroder K, Pelegrín P. NLRP3 and pyroptosis blockers for treating inflammatory diseases. Trends Pharmacol Sci (2022) 43(8):653–68. doi: 10.1016/j.tips.2022.04.003
230. Pan L, Yao DC, Yu YZ, Li SJ, Chen BJ, Hu GH, et al. Necrostatin-1 protects against oleic acid-induced acute respiratory distress syndrome in rats. Biochem Biophys Res Commun (2016) 478(4):1602–8. doi: 10.1016/j.bbrc.2016.08.163
231. Xu H, Du X, Liu G, Huang S, Du W, Zou S, et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab (2019) 23:14–23. doi: 10.1016/j.molmet.2019.02.003
232. Shlomovitz I, Erlich Z, Speir M, Zargarian S, Baram N, Engler M, et al. Necroptosis directly induces the release of full-length biologically active IL-33 in vitro and in an inflammatory disease model. FEBS J (2019) 286(3):507–22. doi: 10.1111/febs.14738
233. Angeli JPF, Shah R, Pratt and M. Conrad DA. Ferroptosis inhibition: Mechanisms and opportunities. Trends Pharmacol Sci (2017) 38(5):489–98. doi: 10.1016/j.tips.2017.02.005
234. Agler AH, Kurth T, Gaziano JM, Buring JE, Cassano PA. Randomised vitamin e supplementation and risk of chronic lung disease in the women's health study. Thorax (2011) 66(4):320–5. doi: 10.1136/thx.2010.155028
235. Abdi A, Jalilian M, Sarbarzeh PA, Vlaisavljevic Z. Diabetes and COVID-19: A systematic review on the current evidences. Diabetes Res Clin Pract (2020) 166:108347. doi: 10.1016/j.diabres.2020.108347
236. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-viral infection maps reveal signatures of severe COVID-19 patients. Cell (2020) 181(7):1475–1488.e12. doi: 10.1016/j.cell.2020.05.006
237. Jankauskas SS, Kansakar U, Sardu C, Varzideh F, Avvisato R, Wang X, et al. COVID-19 causes ferroptosis and oxidative stress in human endothelial cells. Antioxidants (Basel) (2023). doi: 10.3390/antiox12020326
238. Wood LG, Li Q, Scott HA, Rutting S, Berthon BS, Gibson PG, et al. Saturated fatty acids, obesity, and the nucleotide oligomerization domain-like receptor protein 3 (NLRP3) inflammasome in asthmatic patients. J Allergy Clin Immunol (2019) 143(1):305–15. doi: 10.1016/j.jaci.2018.04.037
239. Korbecki J, Bajdak-Rusinek K. The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflammation Res (2019) 68(11):915–32. doi: 10.1007/s00011-019-01273-5
240. Demidowich AP, Davis AI, Dedhia N, Yanovski JA. Colchicine to decrease NLRP3-activated inflammation and improve obesity-related metabolic dysregulation. Med Hypotheses (2016) 92:67–73. doi: 10.1016/j.mehy.2016.04.039
241. Scarsi M, Piantoni S, Colombo E, Airó P, Richini D, Miclini M, et al. Association between treatment with colchicine and improved survival in a single-centre cohort of adult hospitalised patients with COVID-19 pneumonia and acute respiratory distress syndrome. Ann Rheum Dis (2020) 79(10):1286–9. doi: 10.1136/annrheumdis-2020-217712
Keywords: diabetic pulmonary dysfunction, advanced glycosylation end products, non-apoptotic programmed cell deaths, inflammation, oxidative stress
Citation: Dai Y, Zhou S, Qiao L, Peng Z, Zhao J, Xu D, Wu C, Li M, Zeng X and Wang Q (2023) Non-apoptotic programmed cell deaths in diabetic pulmonary dysfunction: the new side of advanced glycation end products. Front. Endocrinol. 14:1126661. doi: 10.3389/fendo.2023.1126661
Received: 18 December 2022; Accepted: 26 September 2023;
Published: 26 October 2023.
Edited by:
Xiaoqiang Tang, Sichuan University, ChinaReviewed by:
Sichong Ren, Chengdu Medical College, ChinaCopyright © 2023 Dai, Zhou, Qiao, Peng, Zhao, Xu, Wu, Li, Zeng and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chanyuan Wu, d2NoeXB1bWNoQGhvdG1haWwuY29t; Qian Wang, d2FuZ3FpYW5fcHVtY2hAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.