 LillyBelle K. Deer
LillyBelle K. Deer Chen Su1
Chen Su1 Elysia Poggi Davis
Elysia Poggi Davis
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 08 May 2023
Sec. Experimental Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1111474
This article is part of the Research Topic The Fetal Origins of Metabolic Disorders, Volume II View all 6 articles
Cardiovascular disease (CVD) is a leading cause of death globally, with the prevalence projected to keep rising. Risk factors for adult CVD emerge at least as early as the prenatal period. Alterations in stress-responsive hormones in the prenatal period are hypothesized to contribute to CVD in adulthood, but little is known about relations between prenatal stress-responsive hormones and early precursors of CVD, such as cardiometabolic risk and health behaviors. The current review presents a theoretical model of the relation between prenatal stress-responsive hormones and adult CVD through cardiometabolic risk markers (e.g., rapid catch-up growth, high BMI/adiposity, high blood pressure, and altered blood glucose, lipids, and metabolic hormones) and health behaviors (e.g., substance use, poor sleep, poor diet and eating behaviors, and low physical activity levels). Emerging evidence in human and non-human animal literatures suggest that altered stress-responsive hormones during gestation predict higher cardiometabolic risk and poorer health behaviors in offspring. This review additionally highlights limitations of the current literature (e.g., lack of racial/ethnic diversity, lack of examination of sex differences), and discusses future directions for this promising area of research.
Cardiovascular disease (CVD) is a leading cause of death globally, killing 18.6 million people worldwide in 2019 (1). As a result, there is a strong public health imperative to identify the early factors that may predict the development of CVD or prevent its occurrence. Early risk factors for CVD, such as child obesity, atherosclerotic plaque formation, low physical activity, and poor diet can be detected early in life and contribute to poorer cardiovascular health (2–5). The Developmental Origins of Health and Disease (DOHaD) hypothesis posits that environmental exposures early in life, particularly during the prenatal period, can result in alterations in development that can have lasting health consequences for offspring (6, 7). A large epidemiological literature corroborates the DOHaD hypothesis showing that birth outcomes such as low birthweight and premature birth, are robustly associated with adult CVD (8–11). Small size at birth and premature birth do not cause CVD; rather, they are thought to reflect perturbations in the prenatal period that shape the development of physiological systems contributing to CVD later in life. Alterations in stress responsive hormones are one plausible mechanism by which exposures in the prenatal period affect offspring health, as stress-responsive hormones are sensitive to prenatal perturbations and also associated with poorer birth outcomes (12–20). However, less is known about relations between prenatal stress-responsive hormones and early precursors of CVD, including offspring cardiometabolic risk and health behaviors.
The current paper will review associations between prenatal stress-responsive hormones and early risk factors for later CVD, which include both cardiometabolic risk markers (e.g., rapid catch-up growth, high BMI/adiposity, high blood pressure, and altered blood glucose, lipids, and metabolic hormones) and health behaviors (e.g., substance use, poor sleep, poor diet and eating behaviors, and low physical activity levels). We begin by providing a brief overview of the dynamic changes in prenatal stress-responsive hormones focusing on the hypothalamic-pituitary-adrenal (HPA) and placental axis. We then provide a theoretical model of the relation between prenatal stress-responsive hormones and adult CVD. We present the state of the literature testing this model in both animal models, where experimental evidence is robust, and in human research. We conclude by discussing important gaps (e.g., lack of racial/ethnic diversity) and highlighting future directions for this promising area of research.
Prenatal stress-responsive hormones such as placental corticotrophin-releasing hormone (CRH) and cortisol have been posited as prenatal influences that contribute to the programming of adult CVD (21, 22). This section will provide a brief overview of the function of the HPA axis and how the HPA axis in the maternal-placental-fetal stress system changes over pregnancy (Figure 1). For a more comprehensive review of the maternal-placental-fetal stress system and the development of the fetal HPA axis, we refer readers to recent reviews (23, 24).
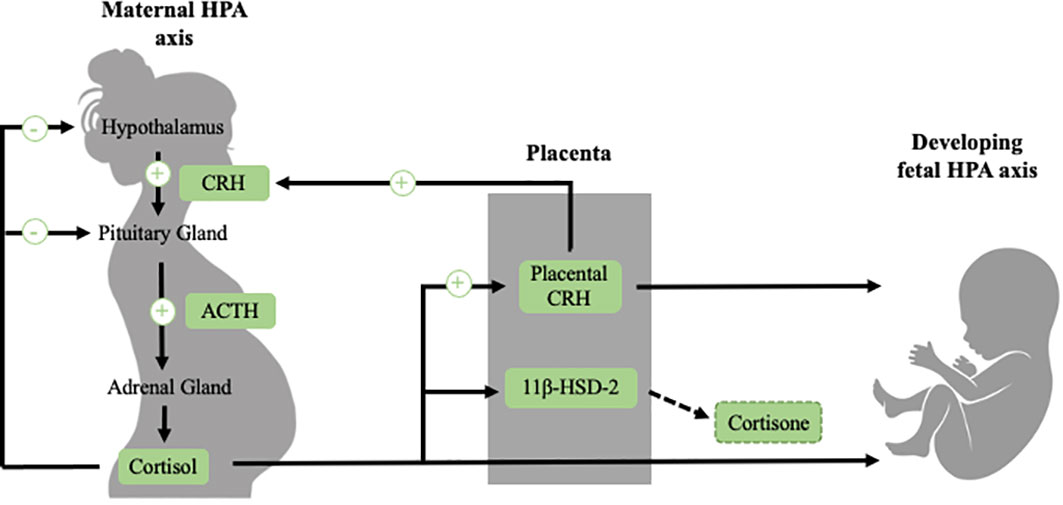
Figure 1 The regulation of the maternal HPA axis changes dramatically over the course of gestation, largely due to the development of the placenta, which is an organ of fetal origin. In non-pregnant individuals, exposure to a stressor activates the HPA axis, which involves the release of CRH, ACTH and cortisol. This stress system is regulated by a negative feedback loop (illustrated in black). During pregnancy (illustrated in green), CRH is released from the placenta into both the maternal and fetal compartments. In contrast to the inhibitory effect that cortisol has on hypothalamic CRH, maternal cortisol stimulates placental CRH production, producing a positive feedback loop. Placental CRH normatively increases exponentially over the course of gestation. In addition to placental CRH, maternal cortisol passes through the placenta to the fetus. However, transfer of maternal cortisol into the fetal compartment is somewhat blocked due to placental 11β-HSD-2, a placental enzyme which oxidizes cortisol into inactive cortisone. 11β-HSD-2 activity decreases late in pregnancy in order to allow maternal cortisol to reach the fetus to aid in maturation of vital organs such as the lungs. The fetal HPA axis begins to develop early in gestation and becomes increasingly active close to birth. See text for further description.
When an individual is confronted with a stressor, a cascade of physiological responses occurs to prepare the individual to cope with the stressor. When stressors are detected, neural signals are relayed to the paraventricular nucleus (PVN) in the hypothalamus, the amygdala, the hippocampus, and the locus coeruleus, which release the neuropeptides corticotropin-releasing hormone (CRH) and arginine-vasopressin (AVP) into the hypophyseal portal system (25–29). CRH and AVP stimulate the production of the prohormone proopiomelanocortin, which is cleaved by enzymes into adrenocorticotropic hormone (ACTH) and other peptides into the bloodstream (25, 30). ACTH binds to receptors in the cortices of the adrenal gland, which stimulate the production of glucocorticoid hormones from the zona fasciculata of the adrenal cortices (25, 26, 31). Cortisol, the primary glucocorticoid hormone in humans and non-human primates, binds to both mineralocorticoid receptors (MRs) and glucocorticoid receptors (GRs) throughout the body (26, 32). Corticosterone is the primary glucocorticoid in rodents and operates in a similar fashion. Under basal conditions, cortisol mainly binds to MRs due to its higher affinity, but under stress conditions it also binds to GRs. When cortisol binds to GRs, a negative feedback loop is triggered, such that in a healthy system the stress response is typically terminated (26, 32).
Over the course of gestation, many changes occur in the functioning of the maternal and the developing fetal HPA axes, largely due to the growth of a new organ, the placenta (33, 34). The placenta is a fetal organ that is responsible for changes in the maternal stress system and the development of the fetal stress system. A function of the placenta is the exchange of signals and information between the maternal and fetal stress systems. Additionally, the placenta produces a myriad of hormones into both the maternal and fetal systems, including a key stress responsive hormone CRH. Placental CRH is identical to hypothalamic CRH in its structure and bioactivity, and is an integrative stress signal that increases in response to many stressors from the maternal and fetal environment, such as nutrient restriction, infection, reduced intrauterine blood flow, and maternal depression, stress and anxiety (16, 35–38). Placental CRH normatively increases exponentially across gestation, approximately 40-fold from the end of the first trimester through term (12, 39). In contrast to the inhibitory effect that cortisol has on hypothalamic CRH, cortisol stimulates placental CRH production, producing a positive feedback loop whereby stressors in the maternal or fetal compartments that increase cortisol can stimulate the production of CRH from the placenta (40). Placental CRH plays a central role in both the regulation of fetal development and the timing of parturition (13, 33). While the normative increases in placental CRH are important for fetal maturation, accelerated production of CRH can alter fetal development. One of the most widely documented consequences of accelerated CRH production is shortened gestation and preterm birth (12, 13, 17–20, 41). Rapid increases in placental CRH may additionally alter the development of the fetal HPA axis, the brain, and have broad effects across the body (42–47).
Maternal cortisol levels also increase during pregnancy approximately three-to-five fold in comparison to pre-pregnancy levels (12, 48). Over most of gestation, transfer of maternal cortisol into the fetal compartment is partially blocked due to placental 11β-HSD-2, a placental enzyme which oxidizes cortisol into its inactive form, cortisone (49, 50). Later in gestation (around 34-35 weeks), 11β-HSD-2 activity decreases, facilitating the transfer of a greater proportion of maternal cortisol across the placenta, in order to support maturation of the fetus before birth (23, 51, 52). Placental 11β-HSD-2 additionally can be downregulated by a number of maternal stress signals such as proinflammatory cytokines, allowing a higher transfer of maternal cortisol to the fetus earlier in gestation (53, 54). Cortisol is important for fetal development, and it has been documented that cortisol levels that are too low over gestation are implicated in impaired lung (55, 56), as well as motor (57) and cognitive development (58, 59). However, levels of cortisol that are too high, especially experienced in early gestation, are also linked to altered offspring development (32, 60).
Accelerated increases in placental CRH and maternal cortisol have a role in the development of physiological systems linked to CVD. As a result, it is plausible that these prenatal hormones predict offspring cardiometabolic risk. Prenatal stress-responsive hormones are indeed implicated in fetal development and may have far-reaching influences. Prior work has documented that placental CRH may alter development of physiological systems that contribute to CVD. Placental CRH is associated with altered HPA axis activity postnatally (61, 62), as well as with physiological systems and processes involved in the development of CVD. CRH has been identified as an important regulator of adipocyte function (63), and therefore might be involved in fat storage. Additionally, placental CRH has been linked to altered brain development (44–47), which may be involved in the regulation of appetite and other health behaviors (64, 65).
Similarly, excessive exposure to glucocorticoids in utero has been linked to altered postnatal HPA axis activity in offspring (61, 66–68). The HPA axis is involved in metabolism and the regulation of appetite (69–71), which suggests a pathway through which glucocorticoids in utero may impact long-term CVD. In addition to programming of the postnatal HPA axis, exposure to glucocorticoids during gestation has been linked to multiple physiological systems and processes that are implicated in the development of CVD, including the development and accumulation of fat cells (72) and altered insulin production (73). Further, elevated glucocorticoid levels during pregnancy have been linked to altered brain and cognitive development (58, 74, 75), which may influence factors such as regulation of appetite and eating behaviors that are linked to later CVD (64, 65). The current literature provides evidence that prenatal stress-responsive hormones are involved in the development of CVD.
In the current paper, we will discuss associations between prenatal stress-responsive hormones with risk factors for adult CVD, such as cardiometabolic risk markers (e.g., rapid catch-up growth, high BMI/adiposity, high blood pressure, and altered blood glucose, lipids, and metabolic hormones) and health behaviors (e.g., substance use, poor sleep, poor diet and eating behaviors, and low physical activity levels). Figure 2 provides a hypothesized model for potential risk factors that may mediate the relation between prenatal stress-responsive hormones and adult CVD in offspring. The following sections will provide existing empirical evidence for each of these risk factors. In each section, research using non-human animal models, including experimental manipulation of the prenatal HPA axis, will be reviewed first. Then, observational human work will be described. Although the maternal-placental-fetal stress system is complex with many interactive factors, much of the current literature focuses on placental CRH and cortisol. As such, the current review of endogenous stress-responsive hormones will focus on placental CRH and cortisol. An important limitation to note in the human literature reviewed in this paper is that the majority of this work was conducted in largely White, WEIRD (Western, Educated, Industrialized, Rich, and Democratic) samples, limiting generalizability.
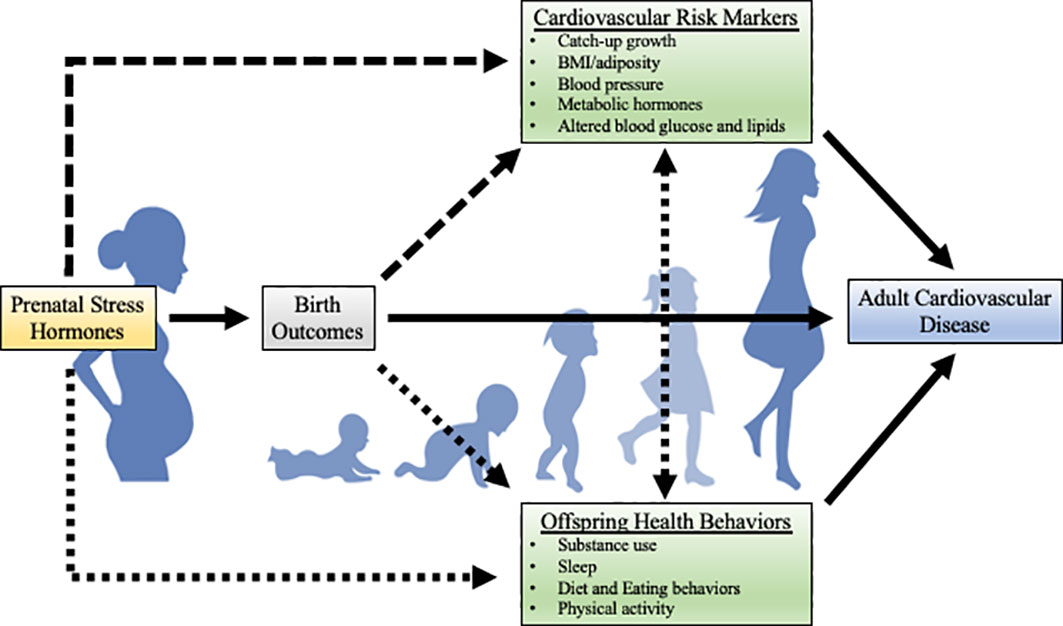
Figure 2 Theoretical model for potential risk factors for the development of adult CVD. The strength of the literature on each path is illustrated by the solidity of the line (solid lines indicate a larger body of research). While there are likely bidirectional relations between cardiometabolic risk markers and health behaviors, this literature is beyond the scope of the current review. Metabolic hormones include hormones such as leptin, ghrelin, and adiponectin.
Researchers have begun to examine associations between prenatal stress-responsive hormones and offspring postnatal cardiometabolic risk markers (e.g., catch-up growth, high BMI and adiposity, high blood pressure, and altered glucose, lipids, and metabolic hormones such as leptin and adiponectin; 42). These cardiometabolic risk markers are robust predictors of adult CVD risk (4, 76–78). A large body of extant work tests these associations in non-human animal models. In these non-human animal model studies, administration of glucocorticoids during pregnancy is used to test causal effects of glucocorticoids on offspring outcomes. Prenatal glucocorticoid administration is linked to lower fetal growth and lower birthweight in rodent models (79–82). As described in this section, a large literature has also examined long-term effects of prenatal glucocorticoid administration in rodent animal models. A recent meta-analysis of 114 studies of prenatal glucocorticoid administration in rodents was conducted to examine the effects of prenatal maternal glucocorticoid administration on offspring cardiometabolic risk markers in adulthood, such as body mass, adiposity, systolic blood pressure, and cardiometabolic hormones (82). Although most animal work in this area has focused on glucocorticoid administration in rodents, there is parallel, yet limited work with non-human primates. There is additionally a very small literature on the link between endogenous maternal cortisol and offspring cardiometabolic risk outcomes in non-human primates. These literatures will be reviewed in the following sections.
The relation between prenatal stress-responsive hormones and offspring cardiometabolic risk has also been examined in humans (see Table 1). Foundational evidence establishing prenatal stress-responsive hormones as a risk factor for CVD has been conducted to examine links between hormones such as placental CRH and glucocorticoids with birth outcomes. This research documents that placental CRH is robustly implicated in fetal development and gestational timing. CRH is in the causal pathway to delivery and rapid increases in placental CRH are associated with shortened gestation and preterm birth (12, 13, 16–20), which in turn are predictors of CVD risk. Meta-analytic work also links high maternal cortisol with low birthweight (15). Similarly, research has documented a pattern of high levels of maternal endogenous cortisol early in gestation predicting higher rates of preterm birth, though this is not always consistent (14, 93–98). In parallel to this work on maternal cortisol, a few studies have examined the relation between glucocorticoid administration during pregnancy and offspring cardiometabolic risk outcomes. Glucocorticoids are administered during pregnancy if there is a high risk for preterm birth in order to accelerate fetal lung maturation (99, 100), if the pregnant person has an autoimmune or inflammatory disease (101), or for other reasons. This work demonstrates that administration of glucocorticoids prenatally are linked to lower fetal growth and birthweight (102–105). As prenatal stress-responsive hormones are linked to poor birth outcomes, and poor birth outcomes are linked to CVD, this work indicates a role for prenatal stress-responsive hormones in the development of CVD in humans as well. As described in this section, the majority of the human work links endogenous maternal cortisol levels to cardiometabolic risk markers, but there are smaller literatures that examine the effects of placental CRH and glucocorticoid administration (Table 1).
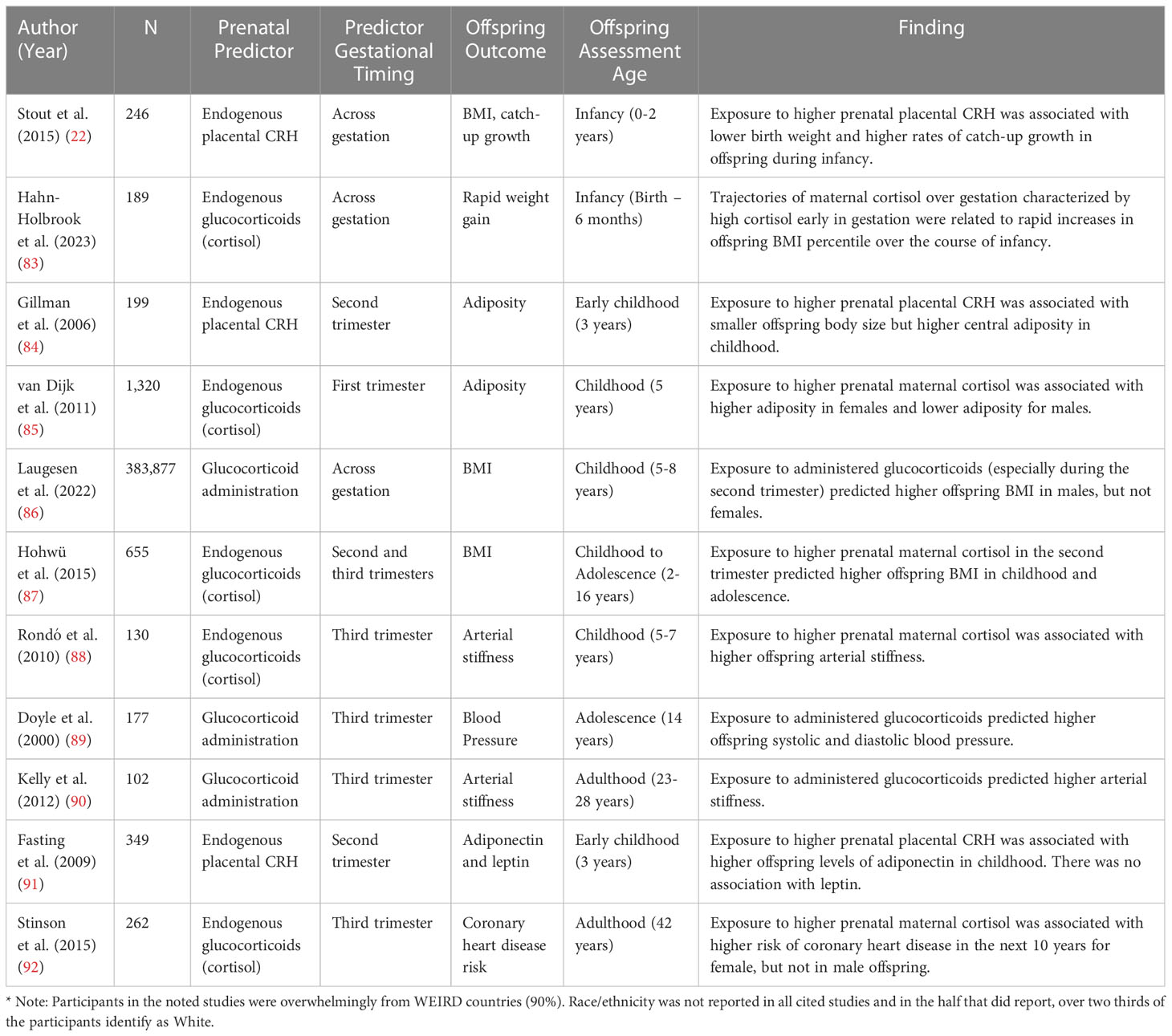
Table 1 Human studies linking prenatal stress-responsive hormones to offspring cardiometabolic risk.
In this section, we will first describe research conducted examining the relation between prenatal stress-responsive hormones and catch-up growth (section 4.1), BMI and adiposity (section 4.2), blood pressure (section 4.3), and other cardiometabolic risk markers such as altered glucose, lipids, and metabolic hormones such as leptin and adiponectin (section 4.4). Within each section, experimental non-human animal model research will be described first, followed by observational research in humans. A summary of the literature on prenatal stress-responsive hormones and cardiometabolic risk markers in humans is summarized in Table 1. As the literature on this relation in animal models is quite expansive and has been covered in meta-analytic work (82), we did not include a table overviewing links between prenatal stress responsive hormones and cardiometabolic risk in animal model (see 87 for review).
Catch-up growth, which is a pattern of growth characterized by small size at birth followed by rapid weight gain, is a strong predictor of later obesity and cardiometabolic disease (106, 107). One study conducted with marmosets examined the relation between endogenous maternal prenatal cortisol and offspring catch-up. This study demonstrated that elevated maternal cortisol, especially early in gestation, was linked to low offspring BMI change (compared to typical BMI increases) in the early postnatal period followed by a rapid catch-up growth period that lasted into adolescence (108).
One study has examined the link between placental CRH and catch-up growth in humans. Researchers in our group investigated the relation between exposure to placental CRH at five points during pregnancy (15, 19, 25, 30, and 37 gestational weeks) with offspring BMI trajectories over the first 24 postnatal months (22). Higher placental CRH at 30 gestational weeks predicted two patterns of accelerated BMI trajectories over the first 24 months of life. The first is a rapid-increase BMI trajectory, which was characterized by a low BMI percentile at birth and low birthweight followed by rapid increases in BMI percentile over the first year of life. The second is a delayed-increase BMI trajectory, which was characterized by a low birthweight, a subsequent reduction in BMI percentile over the first year, followed by a rapid increase in BMI percentile over the second year of life. Both of these growth profiles are indicative of catch-up growth, which is linked to an increased risk of obesity and metabolic diseases (22). The association between glucocorticoids and catch-up growth has not been examined in humans, but another study examined rapid postnatal weight gain over the course of infancy, another risk factor for CVD (109). This study found that offspring exposed to trajectories of high maternal cortisol levels early in gestation that later plateau, exhibit rapid increases in BMI percentile over the course of infancy (83).
The majority of rodent animal work has examined offspring body mass and adiposity, finding an overall pattern where dams who were administered prenatal glucocorticoids had adult offspring with lower total body mass, but higher adiposity (fat mass), suggesting a profile of lower-weight animals with higher body fat (82). Similarly, two studies of male baboon offspring found that offspring who were exposed to glucocorticoid administration in mid-pregnancy had higher adiposity compared to non-exposed offspring (110, 111).
In parallel with the non-human animal model work, the majority of human research assessing the relation between prenatal stress-responsive hormones and cardiometabolic risk has examined offspring body mass and adiposity. One study conducted by Gillman and colleagues, examined the relation between placental CRH at 27 gestational weeks and both BMI and central adiposity when offspring were three years of age (84). They found that although higher placental CRH at the end of the second trimester was related to lower offspring BMI at three years of age, higher placental CRH predicted higher central adiposity, which is a risk factor for later CVD (3, 112, 113).
A few studies have tested the relation between endogenous maternal cortisol and offspring body mass and adiposity. One study found that higher maternal cortisol at the end of the first trimester (13 gestational weeks) was linked to higher offspring fat mass in females, but lower fat mass in males at 5 years of age (85). Extending out further into childhood, Hohwü and colleagues found that higher maternal cortisol at the beginning of the second trimester (16 gestational weeks) was related to a higher likelihood of offspring being overweight between 2-6 and 12-16 years, but not at 7-11 years of age, suggesting potential timing effects (87).
Lastly, in parallel to the literature examining endogenous cortisol in humans, there is one study that assessed the link between glucocorticoid administration during pregnancy and offspring overweight status between 5-8 years of age using cohort data from Denmark (86). They found that, in boys, the prenatal administration of high-dose synthetic glucocorticoids (especially during the second trimester) predicted a higher likelihood of offspring being classified as overweight or obese between 5-8 years of age compared to unexposed offspring. Taken together, both non-human animal models and research conducted in humans indicate a pattern of higher body mass and potentially higher fat mass following exposure to heightened levels of prenatal stress-responsive hormones.
Fewer studies have examined effects of prenatal stress-responsive hormones on blood pressure in offspring. Studies reviewed in the meta-analysis of rodent model glucocorticoid administration revealed that prenatal maternal glucocorticoid administration led to higher systolic blood pressure in adult offspring (82). Research with blood pressure as an outcome in non-human primate work is less consistent. One study of adult marmosets found no effects of prenatal glucocorticoid administration in the last week of pregnancy on offspring blood pressure (114). In contrast, another study found that adult African vervet offspring who experienced glucocorticoid administration in mid-pregnancy had higher systolic and diastolic blood pressure in adulthood (115).
While the link in humans between prenatal CRH and blood pressure has not yet been examined, there are studies testing the relations between both endogenous maternal cortisol and glucocorticoid administration with offspring blood pressure. Rondó and colleagues found that higher maternal cortisol in late pregnancy predicted lower arterial elasticity when offspring were 5-7 years of age (88), indicating a higher risk for hypertension. Doyle and colleagues tested the relation between prenatal glucocorticoid administration and offspring blood pressure at 14 years of age in preterm children (89). They found that children exposed to prenatal glucocorticoid administration had both higher systolic and diastolic blood pressure at 14 years of age. One other study that examined arterial stiffness in adult offspring who had received prenatal glucocorticoids documented that this treatment is related to higher arterial stiffness (90). While this body of literature is small, it indicates that there may be a relation between prenatal stress-responsive hormones and offspring blood pressure.
Other cardiometabolic risk markers, such as metabolic hormones and altered blood glucose and lipids have also been examined. Work reviewed in the rodent animal model meta-analysis documents that glucocorticoid administration caused higher levels of the metabolic hormone leptin, which is involved in the regulation of hunger (82). There were no consistent associations between prenatal glucocorticoid administration and offspring glucose metabolism, insulin, or triglycerides in rodent animal models reviewed in that meta-analysis and no literature on these associations in non-human primates.
In humans, one study has investigated the cardiometabolic hormones adiponectin and leptin in 3-year-old children in relation to placental CRH levels (91). They found that high placental CRH in the second trimester was related to higher level of offspring adiponectin, a cardiometabolic hormone involved in insulin sensitivity and metabolism (91). There was no relation between placental CRH and leptin (91). No work to our knowledge has examined links between either endogenous maternal cortisol or glucocorticoid administration with metabolic hormones or blood glucose in humans.
One study examined an alternative way of operationalizing cardiometabolic risk in humans. In this study, the researchers examined the relation between endogenous maternal cortisol levels in the third trimester of pregnancy and risk for developing coronary heart disease in the next 10 years (Framingham risk algorithm) when offspring were 42 years of age (92). They found that, in female offspring, higher maternal cortisol levels in the third trimester predicted a higher 10-year coronary heart disease risk (92).
The literature reviewed in this section indicates that prenatal stress-responsive hormones such as glucocorticoids and placental CRH are implicated in cardiometabolic risk (catch-up growth, high BMI and adiposity, high blood pressure, and altered blood glucose, lipids, and metabolic hormones). These signals indicate that offspring exposed to altered levels of prenatal stress-responsive hormones have higher cardiometabolic risk. The small human literature is bolstered by the robust animal model literature, where glucocorticoid levels can be manipulated through glucocorticoid administration and potential confounding variables can be tightly controlled. Further, prenatal stress-responsive hormones are strongly implicated in offspring birth outcomes, and the current literature points to a relation between offspring birth outcomes and cardiometabolic risk. As a result, this work lends further support to the hypothesis that prenatal stress-responsive hormones contribute to CVD later in life. Most of the studies described in this section examine outcomes in infancy and early childhood (22, 83–88, 91), but there are a few studies that examine adolescence (89, 116) and adulthood (90, 92). More research needs to be conducted, especially focused on endogenous maternal cortisol and placental CRH with offspring cardiometabolic risk in humans.
As described above, growing evidence from both research with humans and non-human animal models suggests there is an association between prenatal stress-responsive hormones and offspring cardiometabolic risk markers. However, much less is known about potential behavioral risk factors that may mediate the association between prenatal stress-responsive hormones and adult CVD. Health behaviors such as substance use, sleep, obesogenic eating behaviors, diet, and physical activity, are robust predictors of CVD (117–131). As described in the next section, there is a need for a new field of research on the role of prenatal stress-responsive hormones in the development of health behaviors relevant to CVD to fill this large gap. The following sections will describe empirical evidence for the relation between prenatal stress-responsive hormone exposure and substance use (section 5.1), sleep (section 5.2), obesogenic eating behaviors and diet (section 5.3), and physical activity (section 5.4). Within each section, experimental non-human animal model research will be described first, followed by observational research in humans. The current literature has examined both maternal endogenous cortisol and glucocorticoid administration in relation to these health behaviors but the literature on placental CRH in relation to health behaviors is sparse. A summary of the literature on prenatal stress-responsive hormones and health behaviors in non-human animal models is summarized in Tables 2, 3 summarizes this literature in humans.
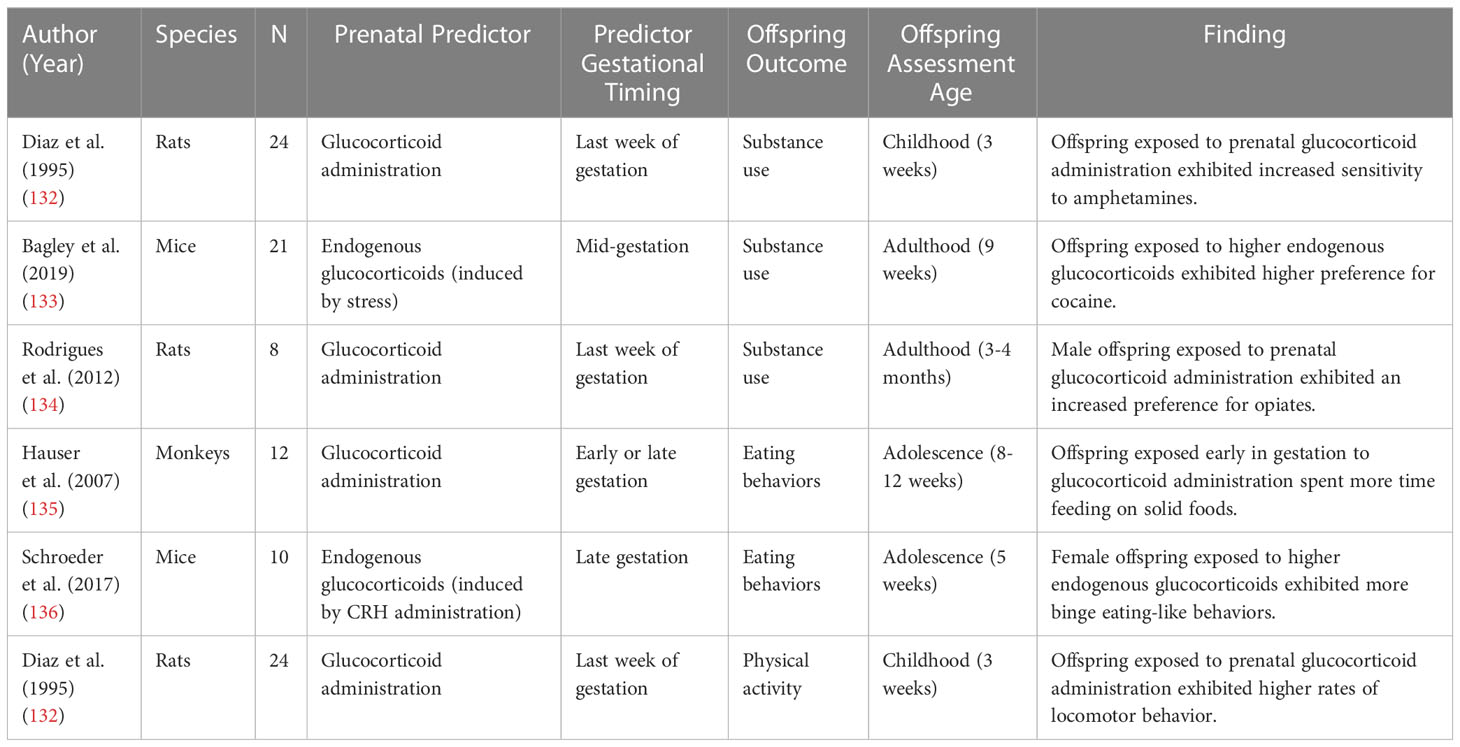
Table 2 Animal model studies examining effects of prenatal stress-responsive hormones on offspring health behaviors.
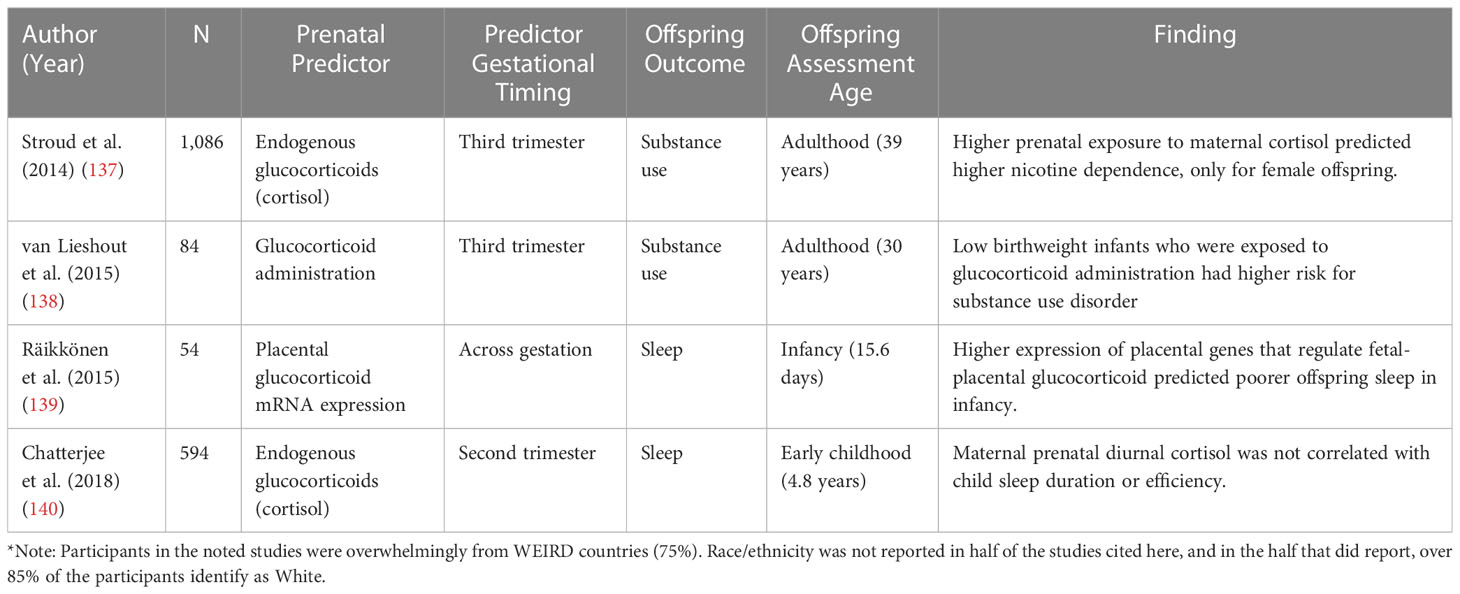
Table 3 Human studies linking prenatal stress-responsive hormones to offspring health behaviors.
There is evidence from non-human animal research that substance use might be a risk factor that mediates the relation between prenatal stress-responsive hormones and cardiovascular risk. Substance use behaviors such as cigarette smoking (118, 141, 142), excessive alcohol consumption (143, 144), and recreational drug use (117, 145), are well-documented risk factors for CVD (146). A few prior studies have examined the relation between prenatal stress-responsive hormones and substance use (132–134, 137, 138, 146). The majority of this work has been conducted in rodents, where glucocorticoids are manipulated during pregnancy (132–134). In two of these studies, pregnant rats were administered glucocorticoids during the last week of gestation. The offspring of rats that were administered glucocorticoids exhibited increased preference for amphetamines during childhood (132) and preference for opiates in adulthood (134). A third study found that adult mice who were exposed to stress-induced endogenous glucocorticoids exhibited a higher preference for cocaine (133). These findings serve as causal evidence that higher prenatal stress-responsive hormone levels increase proclivity towards substance use.
The literature testing the relation between stress-responsive hormones and substance use in humans is smaller but consistent with the experimental animal work (137, 138). In one prospective study, higher endogenous cortisol in the third trimester of pregnancy was related to higher nicotine dependence in adulthood for female offspring (137). Another study examining substance use disorder prevalence among adults with low birthweight, found that those who were exposed to prenatal glucocorticoid administration had a higher risk of substance use disorders in adulthood (138).
While the literature examining the relation between prenatal stress-responsive hormones and substance use is small, there is a larger number of studies aimed at establishing the association between prenatal exposures such as maternal stress, with offspring substance use in rodents (147–154). As prenatal maternal stress is a known activator of prenatal stress-responsive hormones such as placental CRH and cortisol (37, 155), these studies provide additional potential evidence that substance use could be a risk factor that mediates the relation between prenatal stress-responsive hormones and CVD. Studies in rodents that manipulate prenatal stress consistently find that offspring of prenatally stressed mothers exhibit increased preference to amphetamines (147), methamphetamine (149), ethanol (150), opioids (151, 152), and cocaine (153, 154). Taken together, the experimental animal and observational human research point to a relation between prenatal stress-responsive hormones and offspring substance use. As substance use is consistently predictive of CVD (117, 118, 141–146), this work positions substance use an important potential risk factor that mediates the relation between prenatal stress-responsive hormones and CVD.
Poorer sleep across the lifespan is related to higher CVD risk (120–123, 156). Stress-responsive hormones are involved in the regulation of circadian rhythm (157), and HPA axis dysregulation is often implicated in sleep disorders (158), as well as normative variability in sleep (159), pointing to the importance of stress-responsive hormones in regulating sleep. However, the relation between prenatal stress-responsive hormones and offspring sleep is less known. To date, only a few studies have assessed the link between prenatal stress-responsive hormones and sleep, and both of these studies are in humans (139, 140). One study examined the relation between mRNA expression of genes that regulate fetal-placental glucocorticoid exposure (HSD2B11 and NR3C1) and infant offspring sleep quality (139). This study found that higher mRNA expression of HSD2B11 and NR3C1, indicating greater likelihood of maternal glucocorticoid transfer to the fetus, was related to poorer offspring sleep in early infancy (139). This study indicates that higher glucocorticoid transfer and production in the fetal compartment are related to poorer offspring sleep. However, another study found no link between maternal cortisol in the second trimester and both child sleep duration and efficiency in early childhood (140). Taken together, these findings suggest that more research is needed to characterize the relation between prenatal stress-responsive hormones and offspring sleep, as the current small literature is mixed.
Although the prenatal stress-responsive hormone and sleep literature is small, there is a much larger literature that aims to determine the association between prenatal exposures such as maternal stress, depression, and anxiety with offspring sleep (160–165). Two of these studies were conducted in rodents, where prenatal stress was experimentally manipulated (160, 161). In both studies, offspring of prenatally stressed mothers exhibited disruptions in typical sleep-wake patterns. Correlational human literature is consistent with this experimental non-human animal work, finding that prenatal stress (164), as well as mood disorders such as depression and anxiety (162, 163, 165, 166), predict poorer offspring sleep in infancy and childhood. Because stress and mood disorders can disrupt regulation of stress-responsive hormones during pregnancy, there may be a relation between prenatal stress-responsive hormones and poor offspring sleep. As poor sleep has been repeatedly linked to greater cardiovascular risk, this work indicates that sleep may be a risk factor that mediates the relation between prenatal stress-responsive hormones and CVD. However, more work is needed directly testing sleep as a risk factor that mediates the relation between prenatal stress-responsive hormones and CVD.
Dietary quality and obesogenic eating behaviors are robust predictors of CVD (124–127), and there is strong evidence to suggest that stress influences diet, eating behaviors, and stress-responsive hormones (167–170). However, very little is known about the influence of prenatal stress-responsive hormones on offspring diet and eating behaviors. Currently, no research has examined the link between prenatal stress-responsive hormones and offspring dietary quality. However, two non-human animal studies provide evidence that prenatal stress-responsive hormones may influence offspring eating behaviors (135, 136). In one study, adolescent offspring of marmoset monkeys administered glucocorticoids early in gestation spent more time eating solid food in childhood (135). This increased time spent eating may indicate an increased appetite in the exposed offspring (135). In the second study, mice in late gestation were administered corticotrophin-releasing hormone in order to induce endogenous glucocorticoid production (136). Female offspring that experienced this increased prenatal glucocorticoid exposure exhibited more binge eating-like behaviors in adulthood (136). Additionally, low birthweight, which is indicative of perturbations in the prenatal period and has been linked to alterations in stress-responsive hormones (13), has been associated with increased intake of dietary fats and greater impulsive eating in early childhood (171). These studies provide preliminary evidence that prenatal stress-responsive hormones may influence offspring eating behaviors, though much more research is needed.
A related yet small human literature on other prenatal exposures related to alterations in prenatal stress-responsive hormones is consistent with this non-human animal work. Prenatal factors like maternal stress, depression, and anxiety activate prenatal stress-responsive hormones (16, 37, 38, 155, 172), which may then be associated with altered offspring diet and eating behaviors. Prenatal maternal stress has been examined in relation to offspring dietary quality and eating behaviors in humans (173, 174). Maternal stress across pregnancy is linked to lower preference for and consumption of healthy foods such as fruits, vegetables, and non-processed foods in childhood, indicating poor dietary quality (173). Similarly, maternal prenatal stress predicts greater offspring disordered eating behaviors in early adolescence (174). Given this preliminary evidence of associations between prenatal risk factors and diet/eating behaviors and robust work linking diet/eating behaviors to adult CVD, offspring diet and obesogenic eating behaviors may be promising risk factors to examine as mediators in the relation between prenatal stress-responsive hormones and adult CVD.
Low physical activity and greater sedentary behavior are well-established predictors of CVD (128–131). However, it is largely unknown whether prenatal stress-responsive hormones affect offspring physical activity levels, which could put them at risk for adult CVD. Just one study in rodents has examined physical activity levels following prenatal glucocorticoid administration (132). In this study, offspring whose mothers experienced glucocorticoid administration in the last week of gestation exhibited higher levels of locomotor behavior in childhood (132), suggesting that altered prenatal stress-responsive hormones could impact offspring motor development.
There is almost no research on the links between prenatal stress-responsive hormones and physical activity, but there are good reasons to believe that prenatal stress-responsive hormones may predict physical activity levels. High levels of stress and stress-responsive hormones are typically related to lower physical activity when they are measured concurrently (175–177). Additionally, the few studies that have examined the effects of experimentally manipulating prenatal stress in non-human animals suggest that prenatal stress is related to higher levels of offspring inactivity in both monkeys (178, 179) and rats (180). The current literature has not delved into mechanisms for this potential relation. A potential explanation is that the HPA axis is involved in the regulation of energy (181). It could be the case that individuals who have altered HPA axis activity may have less energy available for engagement in physical activity. Given this very small yet suggestive literature, it is important that future research further delve into the potential role of offspring physical activity as a potential risk factor that could mediate the association between prenatal stress-responsive hormones and offspring CVD.
The body of literature covered in this section illustrates that we know very little about the role of prenatal stress-responsive hormones in the development of each of these health behaviors (substance use, sleep, diet and obesogenic eating behaviors, and physical activity). What we do know indicates that offspring exposed to altered levels of prenatal stress-responsive hormones may be more likely to engage in substance use, poorer sleep, poorer diet, obesogenic eating behaviors, and lower physical activity. However, the very small number of studies in this area indicates a huge gap in the literature and opportunity for exciting further work to elucidate the developmental pathways to CVD. Due to suggestive research on prenatal hormones and other prenatal risk factors (stress, depression, anxiety) predicting each of these health behaviors, we argue that this area of research presents many opportunities to test the role of these behavioral risk factors in the development of adult CVD. Further, it is likely that these health behaviors are related. For example, there is robust evidence of a bidirectional association between sleep and physical activity (182). Therefore, future research should also examine potential interactions and temporal associations between these health behaviors.
As stated throughout this review, much more research is needed to fully understand the role of prenatal stress-responsive hormones in the development of CVD. While there is a more robust animal literature, there are a small number of studies on the relation between prenatal stress-responsive hormones and cardiometabolic risk in humans. The behavioral pathways to CVD have been overlooked, as there are even fewer studies examining health behaviors as potential mediators between prenatal stress-responsive hormones and offspring cardiometabolic risk. This area presents a rich potential for future work to fully characterize these possible developmental mechanisms.
There are many unanswered questions regarding the role of prenatal stress-responsive hormones in the development of CVD. We highlight here several needed directions for future research. Studies that examine trajectories of stress-responsive hormones across pregnancy rather than single timepoints are needed. Recent work documents that there are distinct trajectories of stress-responsive hormones and that these trajectories better predict offspring outcomes than single timepoint measures (83, 155). Additionally, most of the studies reviewed here assess outcomes at just one timepoint. A developmental approach is needed, as the current literature, in which outcomes are assessed at one timepoint, precludes identification of when alterations in risk markers and health behaviors begin. A related aspect of needed research are studies that consider the sensitive period of adolescence. In the non-human animal literature reviewed here, most studies examined outcomes when the offspring were adults. In the human literature, most studies evaluated offspring either early in childhood or in adulthood, with notable gaps in late childhood and adolescence. Recent work documents that adolescence may be a window of plasticity, as the HPA axis goes through a potential recalibration through puberty to match current environmental conditions following early stress (183, 184). Examining the pubertal period in relation to prenatal stress-responsive hormone regulation may be an important period for understanding the emergence of cardiometabolic risk and health behaviors. Future developmentally-informed work will provide opportunities to create interventions to improve health at multiple developmental periods.
There is a critical gap in our understanding of sex differences in the impact of prenatal experiences on development of CVD. There are known, dramatic sex differences in CVD risk and CVD-related mortality, such that while males and females develop CVD at the same rate, males are more likely to develop and die from CVD earlier in life (185, 186). Additionally, the presentation of CVD differs between males and females (185, 187–189). Importantly, there are also well-documented sex differences in fetal responses to adversity and stress-responsive hormones, as well as in fetal growth (190–195). Male fetuses are larger than females, and prior work documents that this is partially due to male fetuses prioritizing the energy demands of growth (192, 193). It is thought that this may make male fetuses less adaptable to prenatal adversity, potentially putting them at higher risk for adverse outcomes (190–195). Given these documented differences, it is surprising that very few studies in the literature reviewed here examine sex differences. Additionally, many studies with non-human animals reviewed here only included males. This lack of examination precludes a full understanding of the developmental pathways to CVD and potential targets for prevention efforts. In addition to the need to examine sex differences, the field would benefit from a standardization of covariates used in order to facilitate replicability and consistent findings. Covariates that are commonly used in the literature reviewed here and that we recommend utilizing include gestational age at the time of the prenatal stress-responsive hormone collection, child sex, and maternal factors (e.g., BMI, socioeconomic status, age, substance use during pregnancy).
Additionally, it is imperative that future work in humans is conducted in diverse samples. The overwhelming majority of the current human literature reviewed in this paper was conducted in WEIRD countries. When studies did report race/ethnicity, the participants were overwhelmingly White. Only a few of the studies reviewed included racially and ethnically diverse populations (22, 83, 84, 91). As there are known racial disparities in CVD risk (196, 197), it is imperative for research to include minoritized populations.
The literature could also benefit from a cross-species approach. Cross-species research allows for the parallel examination of experimental animal work and observational human work. This approach allows for the disentanglement of many factors that cannot be controlled in human research, such as shared genes, other prenatal influences than stress-responsive hormones, and postnatal influences such as parenting. This approach has been impactful in other areas (198–200) and would be useful here.
While the focus of the current paper was to review the potential influences of prenatal stress-responsive hormones in the development of offspring CVD, we recognize that these hormones do not exert their influence in isolation. It is likely that prenatal exposures such as environmental toxin exposure (201–203), maternal physical health (204–206), and maternal inflammation (207–209) impact the development of CVD. There is a need for future research examining the interaction of these factors. Further, environmental context, including stress, socioeconomic status, racism and discrimination, likely influence these stress-responsive hormones (16, 155, 210, 211). Finally, the continuity/discontinuity between the prenatal and postnatal environment may profoundly impact health (193, 212–214). Thus, there is a need for research evaluating the joint role of the pre- and postnatal environments to predict CVD.
Converging research across non-human animal and human research indicates that prenatal stress-responsive hormones have a role in the development of offspring CVD, potentially through intermediary cardiovascular risk and altered health behaviors. Altered levels of prenatal stress-responsive hormones are implicated in higher offspring cardiometabolic risk, and potentially, CVD-related health behaviors such as substance use, poor sleep, poor diet and eating behaviors, and physical activity. Continued research examining trajectories of prenatal hormones and offspring outcomes in diverse samples is an exciting opportunity for future research.
LKD, EPD, and JRD conceptualized the review. LKD, CS, NAT, EPD, and JRD drafted the manuscript. LKD and NAT drafted the figures and LKD and CS drafted the tables. All authors contributed to the article and approved the submitted version.
This manuscript was prepared with support from the National Heart, Lung, and Blood Institute grants F32HL165844 to LKD, K01HL143159 to JRD, and R01HL155744 to EPD and JRD as well as from the National Institute of Mental Health grant R01MH109662 to EPD.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Global Burden of Disease: GBD cause and risk summaries. Available at: https://www.thelancet.com/gbd/summaries.
2. Berenson GS, Srinivasan SR, Bao W, Newman WP, Tracy RE, Wattigney WA. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. New Engl J Med (1998) 338(23):1650–6. doi: 10.1056/NEJM199806043382302
3. Freedman DS, Khan LK, Dietz WH, Srinivasan SR, Berenson GS. Relationship of childhood obesity to coronary heart disease risk factors in adulthood: the bogalusa heart study. Pediatrics (2001) 108(3):712–8. doi: 10.1542/peds.108.3.712
4. Jacobs DR, Woo JG, Sinaiko AR, Daniels SR, Ikonen J, Juonala M, et al. Childhood cardiovascular risk factors and adult cardiovascular events. New Engl J Med (2022) 386:1877–88. doi: 10.1056/NEJMoa2109191
5. Kaikkonen JE, Mikkilä V, Raitakari OT. Role of childhood food patterns on adult cardiovascular disease risk. Curr Atheroscler Rep (2014) 16(10):443. doi: 10.1007/s11883-014-0443-z
6. Barker DJ. In utero Programming of chronic disease. Clin Sci (Lond) (1998) 95(2):115–28. doi: 10.1042/CS19980019
7. Barker DJP, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol (2002) 31(6):1235–9. doi: 10.1093/ije/31.6.1235
8. Crump C, Howell EA, Stroustrup A, McLaughlin MA, Sundquist J, Sundquist K. Association of preterm birth with risk of ischemic heart disease in adulthood. JAMA Pediatrics (2019) 173(8):736–43. doi: 10.1001/jamapediatrics.2019.1327
9. de Jong F, Monuteaux MC, van Elburg RM, Gillman MW, Belfort MB. Systematic review and meta-analysis of preterm birth and later systolic blood pressure. Hypertension (2012) 59(2):226–34. doi: 10.1161/HYPERTENSIONAHA.111.181784
10. Hofman PL, Regan F, Jackson WE, Jefferies C, Knight DB, Robinson EM, et al. Premature birth and later insulin resistance. N Engl J Med (2004) 351(21):2179–86. doi: 10.1056/NEJMoa042275
11. Visentin S, Grumolato F, Nardelli GB, Di Camillo B, Grisan E, Cosmi E. Early origins of adult disease: low birth weight and vascular remodeling. Atherosclerosis (2014) 237(2):391–9. doi: 10.1016/j.atherosclerosis.2014.09.027
12. Sandman CA, Glynn L, Schetter CD, Wadhwa P, Garite T, Chicz-DeMet A, et al. Elevated maternal cortisol early in pregnancy predicts third trimester levels of placental corticotropin releasing hormone (CRH): priming the placental clock. Peptides (2006) 27(6):1457–63. doi: 10.1016/j.peptides.2005.10.002
13. Wadhwa PD, Garite TJ, Porto M, Glynn L, Chicz-DeMet A, Dunkel-Schetter C, et al. Placental corticotropin-releasing hormone (CRH), spontaneous preterm birth, and fetal growth restriction: a prospective investigation. Am J Obstetrics Gynecol (2004) 191(4):1063–9. doi: 10.1016/j.ajog.2004.06.070
14. Giurgescu C. Are maternal cortisol levels related to preterm birth? J Obstetric Gynecol Neonatal Nurs (2009) 38(4):377–90. doi: 10.1111/j.1552-6909.2009.01034.x
15. Cherak SJ, Giesbrecht GF, Metcalfe A, Ronksley PE, Malebranche ME. The effect of gestational period on the association between maternal prenatal salivary cortisol and birth weight: a systematic review and meta-analysis. Psychoneuroendocrinology (2018) 94:49–62. doi: 10.1016/j.psyneuen.2018.04.023
16. Hobel CJ, Arora CP, Korst LM. Corticotrophin-releasing hormone and CRH-binding protein. differences between patients at risk for preterm birth and hypertension. Ann N Y Acad Sci (1999) 897:54–65. doi: 10.1111/j.1749-6632.1999.tb07878.x
17. Holzman C, Jetton J, Siler-Khodr T, Fisher R, Rip T. Second trimester corticotropin-releasing hormone levels in relation to preterm delivery and ethnicity. Obstetrics Gynecol (2001) 97(5, Part 1):657–63. doi: 10.1016/s0029-7844(00)01209-6
18. McLean M, Bisits A, Davies J, Woods R, Lowry P, Smith R. A placental clock controlling the length of human pregnancy. Nat Med (1995) 1(5):460–3. doi: 10.1038/nm0595-460
19. Ruiz RJ, Gennaro S, O’Connor C, Dwivedi A, Gibeau A, Keshinover T, et al. CRH as a predictor of preterm birth in minority women. Biol Res Nurs (2016) 18(3):316–21. doi: 10.1177/1099800415611248
20. Sandman CA, Wadhwa P, Glynn L, Chicz-Demet A, Porto M, Garite TJ. Corticotrophin-releasing hormone and fetal responses in human pregnancy. Ann New York Acad Sci (1999) 897(1):66–75. doi: 10.1111/j.1749-6632.1999.tb07879.x
21. Entringer S. Impact of stress and stress physiology during pregnancy on child metabolic function and obesity risk. Curr Opin Clin Nutr Metab Care (2013) 16(3):320–7. doi: 10.1097/MCO.0b013e32835e8d80
22. Stout SA, Espel EV, Sandman CA, Glynn LM, Davis EP. Fetal programming of children’s obesity risk. Psychoneuroendocrinology (2015) 53:29–39. doi: 10.1016/j.psyneuen.2014.12.009
23. Howland MA, Sandman CA, Glynn LM. Developmental origins of the human hypothalamic-pituitary-adrenal axis. Expert Rev Endocrinol Metab (2017) 12(5):321–39. doi: 10.1080/17446651.2017.1356222
24. Noroña AN, Doom JR, Davis EP, Gunnar MR. The effects of stress on early brain and behavioral development. In: Rubenstein J, Rakic P, Chen B, Kwan KY, editors. Neural circuit and cognitive development (Second edition). Academic Press (2020). p. 561–84. Available at: https://www.sciencedirect.com/science/article/pii/B9780128144114000263.
25. Levy BH, Tasker JG. Synaptic regulation of the hypothalamic–pituitary–adrenal axis and its modulation by glucocorticoids and stress. Front Cell Neurosci (2012) 6:24. doi: 10.3389/fncel.2012.00024
26. Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci (2009) 10(6):459–66. doi: 10.1038/nrn2632
27. Koob GF. A role for brain stress systems in addiction. Neuron (2008) 59(1):11–34. doi: 10.1016/j.neuron.2008.06.012
28. Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci (2001) 21(18):7171–81. doi: 10.1523/JNEUROSCI.21-18-07171.2001
29. Valentino RJ, Van Bockstaele E. Convergent regulation of locus coeruleus activity as an adaptive response to stress. Eur J Pharmacol (2008) 583(2–3):194–203. doi: 10.1016/j.ejphar.2007.11.062
30. Harbuz MS, Rees RG, Eckland D, Jessop DS, Brewerton D, Lightman SL. Paradoxical responses of hypothalamic corticotropin-releasing factor (CRF) messenger ribonucleic acid (mRNA) and CRF-41 peptide and adenohypophysial proopiomelanocortin mRNA during chronic inflammatory stress. Endocrinology (1992) 130(3):1394–400. doi: 10.1210/endo.130.3.1537299
31. Gunnar MR, Doom JR, Esposito EA. Psychoneuroendocrinology of stress: normative development and individual differences. In: Hoboken NJ, editor. Handbook of child psychology and developmental science: socioemotional processes, vol 3, 7th ed. US: John Wiley & Sons, Inc (2015). p. 106–51.
32. De Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr Rev (1998) 19(3):269–301. doi: 10.1210/edrv.19.3.0331
33. Sandman CA. Fetal exposure to placental corticotropin-releasing hormone (pCRH) programs developmental trajectories. Peptides (2015) 72:145–53. doi: 10.1016/j.peptides.2015.03.020
34. Reis FM, Petraglia F. The placenta as a neuroendocrine organ. Front Horm Res (2001) 27:216–28. doi: 10.1159/000061028
35. Petraglia F, Garuti GC, De Ramundo B, Angioni S, Genazzani AR, Bilezikjian LM. Mechanism of action of interleukin-1 beta in increasing corticotropin-releasing factor and adrenocorticotropin hormone release from cultured human placental cells. Am J Obstet Gynecol (1990) 163(4 Pt 1):1307–12. doi: 10.1016/0002-9378(90)90711-F
36. Herrmann TS, Siega-Riz AM, Hobel CJ, Aurora C, Dunkel-Schetter C. Prolonged periods without food intake during pregnancy increase risk for elevated maternal corticotropin-releasing hormone concentrations. Am J Obstet Gynecol (2001) 185(2):403–12. doi: 10.1067/mob.2001.115863
37. Mancuso RA, Schetter CD, Rini CM, Roesch SC, Hobel CJ. Maternal prenatal anxiety and corticotropin-releasing hormone associated with timing of delivery. Psychosom Med (2004) 66(5):762–9. doi: 10.1097/01.psy.0000138284.70670.d5
38. Rich-Edwards JW, Mohllajee AP, Kleinman K, Hacker MR, Majzoub J, Wright RJ, et al. Elevated midpregnancy corticotropin-releasing hormone is associated with prenatal, but not postpartum, maternal depression. J Clin Endocrinol Metab (2008) 93(5):1946–51. doi: 10.1210/jc.2007-2535
39. Frim DM, Emanuel RL, Robinson BG, Smas CM, Adler GK, Majzoub JA. Characterization and gestational regulation of corticotropin-releasing hormone messenger RNA in human placenta. J Clin Invest (1988) 82(1):287–92. doi: 10.1172/JCI113585
40. Robinson BG, Emanuel RL, Frim DM, Majzoub JA. Glucocorticoid stimulates expression of corticotropin-releasing hormone gene in human placenta. Proc Natl Acad Sci USA (1988) 85(14):5244–8. doi: 10.1073/pnas.85.14.5244
41. Hobel CJ, Dunkel-Schetter C, Roesch SC, Castro LC, Arora CP. Maternal plasma corticotropin-releasing hormone associated with stress at 20 weeks’ gestation in pregnancies ending in preterm delivery. Am J Obstetrics Gynecol (1999) 180(1, Supplement 2):S257–63. doi: 10.1016/S0002-9378(99)70712-X
42. Charil A, Laplante DP, Vaillancourt C, King S. Prenatal stress and brain development. Brain Res Rev (2010) 65(1):56–79. doi: 10.1016/j.brainresrev.2010.06.002
43. Avishai-Eliner S, Brunson K, Sandman CA, Baram TZ. Stressed-out, or in (utero)? Trends Neurosci (2002) 25(10):518–24. doi: 10.1016/S0166-2236(02)02241-5
44. Curran MM, Sandman CA, Davis EP, Glynn LM, Baram TZ. Abnormal dendritic maturation of developing cortical neurons exposed to corticotropin releasing hormone (CRH): insights into effects of prenatal adversity? PloS One (2017) 12(6):e0180311. doi: 10.1371/journal.pone.0180311
45. Chen Y, Bender RA, Brunson KL, Pomper JK, Grigoriadis DE, Wurst W, et al. Modulation of dendritic differentiation by corticotropin-releasing factor in the developing hippocampus. Proc Natl Acad Sci (2004) 101(44):15782–7. doi: 10.1073/pnas.0403975101
46. Sandman CA, Curran MM, Davis EP, Glynn LM, Head K, Baram TZ. Cortical thinning and neuropsychiatric outcomes in children exposed to prenatal adversity: a role for placental CRH? AJP (2018) 175(5):471–9. doi: 10.1176/appi.ajp.2017.16121433
47. Howland MA, Sandman CA, Glynn LM, Crippen C, Davis EP. Fetal exposure to placental corticotropin-releasing hormone is associated with child self-reported internalizing symptoms. Psychoneuroendocrinology (2016) 67:10–7. doi: 10.1016/j.psyneuen.2016.01.023
48. Mastorakos G, Ilias I. Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann N Y Acad Sci (2003) 997:136–49. doi: 10.1196/annals.1290.016
49. Kajantie E, Raivio T, Jänne OA, Hovi P, Dunkel L, Andersson S. Circulating glucocorticoid bioactivity in the preterm newborn after antenatal betamethasone treatment. J Clin Endocrinol Metab (2004) 89(8):3999–4003. doi: 10.1210/jc.2004-0013
50. Salvante KG, Milano K, Kliman HJ, Nepomnaschy PA. Placental 11 β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) expression very early during human pregnancy. J Dev Orig Health Dis (2017) 8(2):149–54. doi: 10.1017/S2040174416000611
51. Shams M, Kilby MD, Somerset DA, Howie AJ, Gupta A, Wood PJ, et al. 11Beta-hydroxysteroid dehydrogenase type 2 in human pregnancy and reduced expression in intrauterine growth restriction. Hum Reprod (1998) 13(4):799–804. doi: 10.1093/humrep/13.4.799
52. Schoof E, Girstl M, Frobenius W, Kirschbaum M, Repp R, Knerr I, et al. Course of placental 11beta-hydroxysteroid dehydrogenase type 2 and 15-hydroxyprostaglandin dehydrogenase mRNA expression during human gestation. Eur J Endocrinol (2001) 145(2):187–92. doi: 10.1530/eje.0.1450187
53. Johnstone JF, Bocking AD, Unlugedik E, Challis JRG. The effects of chorioamnionitis and betamethasone on 11beta hydroxysteroid dehydrogenase types 1 and 2 and the glucocorticoid receptor in preterm human placenta. J Soc Gynecol Investig (2005) 12(4):238–45. doi: 10.1016/j.jsgi.2005.01.029
54. Kossintseva I, Wong S, Johnstone E, Guilbert L, Olson DM, Mitchell BF. Proinflammatory cytokines inhibit human placental 11beta-hydroxysteroid dehydrogenase type 2 activity through Ca2+ and cAMP pathways. Am J Physiol Endocrinol Metab (2006) 290(2):E282–288. doi: 10.1152/ajpendo.00328.2005
55. Austin MP, Leader L. Maternal stress and obstetric and infant outcomes: epidemiological findings and neuroendocrine mechanisms. Aust N Z J Obstet Gynaecol (2000) 40(3):331–7. doi: 10.1111/j.1479-828X.2000.tb03344.x
56. Hacking D, Watkins A, Fraser S, Wolfe R, Nolan T. Respiratory distress syndrome and antenatal corticosteroid treatment in premature twins. Arch Dis Child Fetal Neonatal Ed (2001) 85(1):F77–78. doi: 10.1136/fn.85.1.F75g
57. Huizink AC, Robles de Medina PG, Mulder EJH, Visser GHA, Buitelaar JK. Stress during pregnancy is associated with developmental outcome in infancy. J Child Psychol Psychiatry (2003) 44(6):810–8. doi: 10.1111/1469-7610.00166
58. Davis EP, Sandman CA. The timing of prenatal exposure to maternal cortisol and psychosocial stress is associated with human infant cognitive development. Child Dev (2010) 81(1):131–48. doi: 10.1111/j.1467-8624.2009.01385.x
59. Ram S, Howland MA, Sandman CA, Davis EP, Glynn LM. Prenatal risk for ASD: fetal cortisol exposure predicts child autism-spectrum disorder symptoms. Clin Psychol Sci (2019) 7(2):349–61. doi: 10.1177/2167702618811079
60. Seckl JR, Meaney MJ. Glucocorticoid “programming” and PTSD risk. Ann N Y Acad Sci (2006) 1071:351–78. doi: 10.1196/annals.1364.027
61. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 1: outcomes. Nat Rev Endocrinol (2014) 10(7):391–402. doi: 10.1038/nrendo.2014.73
62. Rinne GR, Somers JA, Ramos IF, Ross KM, Coussons-Read M, Schetter CD. Increases in maternal depressive symptoms during pregnancy and infant cortisol reactivity: mediation by placental corticotropin-releasing hormone. Dev Psychopathol (2022) 1–14. doi: 10.1017/S0954579422000621
63. Grammatopoulos D. The family of corticotropin-releasing hormone (CRH) peptides: important regulators of adipocyte function. Endocrine Abstracts (2008), 16. https://www.endocrine-abstracts.org/ea/0016/ea0016s19.1.
64. Russell CG, Russell A. “Food” and “non-food” self-regulation in childhood: a review and reciprocal analysis. Int J Behav Nutr Phys Act (2020) 17(1):33. doi: 10.1186/s12966-020-00928-5
65. Broberger C. Brain regulation of food intake and appetite: molecules and networks. J Internal Med (2005) 258(4):301–27. doi: 10.1111/j.1365-2796.2005.01553.x
66. Davis EP, Waffarn F, Sandman CA. Prenatal treatment with glucocorticoids sensitizes the hpa axis response to stress among full-term infants. Dev Psychobiol (2011) 53(2):175–83. doi: 10.1002/dev.20510
67. Irwin MR. Human psychoneuroimmunology: 20 years of discovery. Brain Behavior Immunity. (2008) 22(2):129–39. doi: 10.1016/j.bbi.2007.07.013
68. Gutteling BM, de Weerth C, Buitelaar JK. Prenatal stress and children’s cortisol reaction to the first day of school. Psychoneuroendocrinology (2005) 30(6):541–9. doi: 10.1016/j.psyneuen.2005.01.002
69. Newcomer JW, Selke G, Melson AK, Gross J, Vogler GP, Dagogo-Jack S. Dose-dependent cortisol-induced increases in plasma leptin concentration in healthy humans. Arch Gen Psychiatry (1998) 55(11):995–1000. doi: 10.1001/archpsyc.55.11.995
70. Leal-Cerro A, Soto A, Martínez MA, Dieguez C, Casanueva FF. Influence of cortisol status on leptin secretion. Pituitary (2001) 4(1–2):111–6. doi: 10.1023/A:1012903330944
71. Bouret SG. Early life origins of obesity: role of hypothalamic programming. J Pediatr Gastroenterol Nutr (2009) 48:S31. doi: 10.1097/MPG.0b013e3181977375
72. Campbell JE, Peckett AJ, D’souza AM, Hawke TJ, Riddell MC. Adipogenic and lipolytic effects of chronic glucocorticoid exposure. Am J Physiol Cell Physiol (2011) 300(1):C198–209. doi: 10.1152/ajpcell.00045.2010
73. Schwitzgebel VM, Somm E, Klee P. Modeling intrauterine growth retardation in rodents: impact on pancreas development and glucose homeostasis. Mol Cell Endocrinol (2009) 304(1–2):78–83. doi: 10.1016/j.mce.2009.02.019
74. Bergman K, Sarkar P, Glover V, O’Connor TG. Maternal prenatal cortisol and infant cognitive development: moderation by infant–mother attachment. Biol Psychiatry (2010) 67(11):1026–32. doi: 10.1016/j.biopsych.2010.01.002
75. Glynn LM, Sandman CA. Sex moderates associations between prenatal glucocorticoid exposure and human fetal neurological development. Dev Sci (2012) 15(5):601–10. doi: 10.1111/j.1467-7687.2012.01159.x
76. Morrison JA, Glueck CJ, Horn PS, Yeramaneni S, Wang P. PEDIATRIC TRIGLYCERIDES PREDICT CARDIOVASCULAR DISEASE EVENTS IN THE 4TH-5TH DECADE OF LIFE. Metabolism (2009) 58(9):1277–84. doi: 10.1016/j.metabol.2009.04.009
77. Yang L, Magnussen CG, Yang L, Bovet P, Xi B. Elevated blood pressure in childhood or adolescence and cardiovascular outcomes in adulthood. Hypertension (2020) 75(4):948–55. doi: 10.1161/HYPERTENSIONAHA.119.14168
78. Umer A, Kelley GA, Cottrell LE, Giacobbi P, Innes KE, Lilly CL. Childhood obesity and adult cardiovascular disease risk factors: a systematic review with meta-analysis. BMC Public Health (2017) 17:683. doi: 10.1186/s12889-017-4691-z
79. Newnham JP. Is prenatal glucocorticoid administration another origin of adult disease? Clin Exp Pharmacol Physiol (2001) 28(11):957–61. doi: 10.1046/j.1440-1681.2001.03559.x
80. Newnham JP, Evans SF, Godfrey M, Huang W, Ikegami M, Jobe A. Maternal, but not fetal, administration of corticosteroids restricts fetal growth. J Maternal-Fetal Med (1999) 8(3):81–7. doi: 10.1002/(SICI)1520-6661(199905/06)8:3<81::AID-MFM3>3.0.CO;2-N
81. Takahashi T, Fee EL, Takahashi Y, Saito M, Yaegashi N, Usuda H, et al. Betamethasone phosphate reduces the efficacy of antenatal steroid therapy and is associated with lower birthweights when administered to pregnant sheep in combination with betamethasone acetate. Am J Obstetrics Gynecol (2022) 226(4):564.e1–564.e14. doi: 10.1016/j.ajog.2021.10.001
82. Burgueño AL, Juárez YR, Genaro AM, Tellechea ML. Prenatal stress and later metabolic consequences: systematic review and meta-analysis in rodents. Psychoneuroendocrinology (2020) 113:104560. doi: 10.1016/j.psyneuen.2019.104560
83. Hahn-Holbrook J, Davis EP, Sandman CA, Glynn LM. Maternal prenatal cortisol trajectories predict accelerated growth in infancy. Psychoneuroendocrinology (2023) 147:105957. doi: 10.1016/j.psyneuen.2022.105957
84. Gillman MW, Rich-Edwards JW, Huh S, Majzoub JA, Oken E, Taveras EM, et al. Maternal corticotropin-releasing hormone levels during pregnancy and offspring adiposity. Obes (Silver Spring) (2006) 14(9):1647–53. doi: 10.1038/oby.2006.189
85. Van Dijk AE, Van Eijsden M, Stronks K, Gemke RJBJ, Vrijkotte TGM. The relation of maternal job strain and cortisol levels during early pregnancy with body composition later in the 5-year-old child: the ABCD study. Early Hum Dev (2012) 88(6):351–6. doi: 10.1016/j.earlhumdev.2011.09.009
86. Laugesen K, Sørensen HT, Jorgensen JOL, Petersen I. Prenatal exposure to glucocorticoids and the prevalence of overweight or obesity in childhood. Eur J Endocrinol (2022) 186(4):429–40. doi: 10.1530/EJE-21-0846
87. Hohwü L, Henriksen TB, Grønborg TK, Hedegaard M, Sørensen TIA, Obel C. Maternal salivary cortisol levels during pregnancy are positively associated with overweight children. Psychoneuroendocrinology (2015) 52:143–52. doi: 10.1016/j.psyneuen.2014.11.006
88. Rondó PHC, Lemos JO, Pereira JA, Souza JMP. The relationship between cortisol concentrations in pregnancy and systemic vascular resistance in childhood. Early Hum Dev (2010) 86(2):127–31. doi: 10.1016/j.earlhumdev.2010.02.002
89. Doyle LW, Ford GW, Davis NM, Callanan C. Antenatal corticosteroid therapy and blood pressure at 14 years of age in preterm children. Clin Sci (2000) 98(2):137–42. doi: 10.1042/CS19990211
90. Kelly BA, Lewandowski AJ, Worton SA, Davis EF, Lazdam M, Francis J, et al. Antenatal glucocorticoid exposure and long-term alterations in aortic function and glucose metabolism. Pediatrics (2012) 129(5):e1282–1290. doi: 10.1542/peds.2011-3175
91. Fasting MH, Oken E, Mantzoros CS, Rich-Edwards JW, Majzoub JA, Kleinman K, et al. Maternal levels of corticotropin-releasing hormone during pregnancy in relation to adiponectin and leptin in early childhood. J Clin Endocrinol Metab (2009) 94(4):1409–15. doi: 10.1210/jc.2008-1424
92. Stinson LJ, Stroud LR, Buka SL, Eaton CB, Lu B, Niaura R, et al. Prospective evaluation of associations between prenatal cortisol and adulthood coronary heart disease risk: the new England family study. Psychosomatic Med (2015) 77(3):237–45. doi: 10.1097/PSY.0000000000000164
93. Stewart CP, Oaks BM, Laugero KD, Ashorn U, Harjunmaa U, Kumwenda C, et al. Maternal cortisol and stress are associated with birth outcomes, but are not affected by lipid-based nutrient supplements during pregnancy: an analysis of data from a randomized controlled trial in rural Malawi. BMC Pregnancy Childbirth (2015) 15:346. doi: 10.1186/s12884-015-0793-8
94. Bandoli G, Jelliffe-Pawlowski LL, Feuer SK, Liang L, Oltman SP, Paynter R, et al. Second trimester serum cortisol and preterm birth: an analysis by timing and subtype. J Perinatol (2018) 38(8):973–81. doi: 10.1038/s41372-018-0128-5
95. Entringer S, Buss C, Andersen J, Chicz-DeMet A, Wadhwa PD. Ecological momentary assessment of maternal cortisol profiles over a multiple-day period predicts the length of human gestation. Psychosom Med (2011) 73(6):469–74. doi: 10.1097/PSY.0b013e31821fbf9a
96. Hoffman MC, Mazzoni SE, Wagner BD, Laudenslager ML, Ross RG. Measures of maternal stress and mood in relation to preterm birth. Obstet Gynecol (2016) 127(3):545–52. doi: 10.1097/AOG.0000000000001287
97. Ruiz RJ, Fullerton J, Brown CE, Schoolfield J. Relationships of cortisol, perceived stress, genitourinary infections, and fetal fibronectin to gestational age at birth. Biol Res Nurs (2001) 3(1):39–48. doi: 10.1177/109980040100300106
98. Yoon BH, Romero R, Jun JK, Maymon E, Gomez R, Mazor M, et al. An increase in fetal plasma cortisol but not dehydroepiandrosterone sulfate is followed by the onset of preterm labor in patients with preterm premature rupture of the membranes. Am J Obstet Gynecol (1998) 179(5):1107–14. doi: 10.1016/S0002-9378(98)70114-0
99. Bolt RJ, van Weissenbruch MM, Lafeber HN, Delemarre-van de Waal HA. Glucocorticoids and lung development in the fetus and preterm infant. Pediatr Pulmonol (2001) 32(1):76–91. doi: 10.1002/ppul.1092
100. Agnew EJ, Ivy JR, Stock SJ, Chapman KE. Glucocorticoids, antenatal corticosteroid therapy and fetal heart maturation. J Mol Endocrinol (2018) 61(1):R61–73. doi: 10.1530/JME-18-0077
101. Bandoli G, Palmsten K, Forbess Smith CJ, Chambers CD. A review of systemic corticosteroid use in pregnancy and the risk of select pregnancy and birth outcomes. Rheum Dis Clin North Am (2017) 43(3):489–502. doi: 10.1016/j.rdc.2017.04.013
102. Braun T, Sloboda DM, Tutschek B, Harder T, Challis JRG, Dudenhausen JW, et al. Fetal and neonatal outcomes after term and preterm delivery following betamethasone administration. Int J Gynaecol Obstet (2015) 130(1):64–9. doi: 10.1016/j.ijgo.2015.01.013
103. Bloom SL, Sheffield JS, McIntire DD, Leveno KJ. Antenatal dexamethasone and decreased birth weight. Obstetrics Gynecol (2001) 97(4):485–90. doi: 10.1016/s0029-7844(00)01206-0
104. Rizzo G, Mappa I, Bitsadze V, Khizroeva J, Makatsariya A, D’Antonio F. Administration of antenatal corticosteroid is associated with reduced fetal growth velocity: a longitudinal study. J Maternal-Fetal Neonatal Med (2022) 35(14):2775–80. doi: 10.1080/14767058.2020.1800634
105. Thorp JA, Jones PG, Knox E, Clark RH. Does antenatal corticosteroid therapy affect birth weight and head circumference? Obstetrics Gynecol (2002) 99(1):101–8. doi: 10.1097/00006250-200201000-00020
106. Nobili V, Alisi A, Panera N, Agostoni C. Low birth weight and catch-up-growth associated with metabolic syndrome: a ten year systematic review. Pediatr Endocrinol Rev (2008) 6(2):241–7.
107. Calkins K, Devaskar SU. Fetal origins of adult disease. Curr Probl Pediatr Adolesc Health Care (2011) 41(6):158–76. doi: 10.1016/j.cppeds.2011.01.001
108. Mustoe AC, Birnie AK, Korgan AC, Santo JB, French JA. Natural variation in gestational cortisol is associated with patterns of growth in marmoset monkeys (Callithrix geoffroyi). Gen Comp Endocrinol (2012) 175(3):519–26. doi: 10.1016/j.ygcen.2011.12.020
109. Hulst AV, Barnett TA, Paradis G, Roy-Gagnon M, Gomez-Lopez L, Henderson M. Birth weight, postnatal weight gain, and childhood adiposity in relation to lipid profile and blood pressure during early adolescence. J Am Heart Assoc (2017) 6(8):e006302. doi: 10.1161/JAHA.117.006302
110. Kuo AH, Li J, Li C, Huber HF, Schwab M, Nathanielsz PW, et al. Prenatal steroid administration leads to adult pericardial and hepatic steatosis in Male baboons. Int J Obes (Lond) (2017) 41(8):1299–302. doi: 10.1038/ijo.2017.82
111. Huber HF, Kuo AH, Li C, Jenkins SL, Gerow KG, Clarke GD, et al. Antenatal synthetic glucocorticoid exposure at human therapeutic equivalent doses predisposes middle-age Male offspring baboons to an obese phenotype that emerges with aging. Reprod Sci (2019) 26(5):591–9. doi: 10.1177/1933719118778794
112. Savva SC, Tornaritis M, Savva ME, Kourides Y, Panagi A, Silikiotou N, et al. Waist circumference and waist-to-height ratio are better predictors of cardiovascular disease risk factors in children than body mass index. Int J Obes Relat Metab Disord (2000) 24(11):1453–8. doi: 10.1038/sj.ijo.0801401
113. Yang H, Huynh QL, Venn AJ, Dwyer T, Marwick TH. Associations of childhood and adult obesity with left ventricular structure and function. Int J Obes (2017) 41(4):560–8. doi: 10.1038/ijo.2016.234
114. Bramlage CP, Schlumbohm C, Pryce CR, Mirza S, Schnell C, Amann K, et al. Prenatal dexamethasone exposure does not alter blood pressure and nephron number in the young adult marmoset monkey. Hypertension (2009) 54(5):1115–22. doi: 10.1161/HYPERTENSIONAHA.109.136580
115. de Vries A, Holmes MC, Heijnis A, Seier JV, Heerden J, Louw J, et al. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J Clin Invest (2007) 117(4):1058–67. doi: 10.1172/JCI30982
116. Hohwü L, Zhu JL, Graversen L, Li J, Sørensen TIA, Obel C, et al. Prenatal parental separation and body weight, including development of overweight and obesity later in childhood. PloS One (2015) 10(3):e0119138. doi: 10.1371/journal.pone.0119138
117. Gan WQ, Buxton JA, Scheuermeyer FX, Palis H, Zhao B, Desai R, et al. Risk of cardiovascular diseases in relation to substance use disorders. Drug Alcohol Dependence (2021) 229:109132. doi: 10.1016/j.drugalcdep.2021.109132
118. Schulte MT, Hser YI. Substance use and associated health conditions throughout the lifespan. Public Health Rev (2013) 35(2):1–27. doi: 10.1007/BF03391702
119. Nagata JM, Ganson KT, Cunningham ML, Mitchison D, Lavender JM, Blashill AJ, et al. Associations between legal performance-enhancing substance use and future cardiovascular disease risk factors in young adults: a prospective cohort study. PloS One (2020) 15(12):e0244018. doi: 10.1371/journal.pone.0244018
120. Cappuccio FP, Taggart FM, Kandala NB, Currie A, Peile E, Stranges S, et al. Meta-analysis of short sleep duration and obesity in children and adults. Sleep (2008) 31(5):619–26. doi: 10.1093/sleep/31.5.619
121. Taveras EM, Rifas-Shiman SL, Oken E, Gunderson EP, Gillman MW. Short sleep duration in infancy and risk of childhood overweight. Arch Pediatr Adolesc Med (2008) 162(4):305–11. doi: 10.1001/archpedi.162.4.305
122. Wolk R, Gami AS, Garcia-Touchard A, Somers VK. Sleep and cardiovascular disease. Curr Problems Cardiol (2005) 30(12):625–62. doi: 10.1016/j.cpcardiol.2005.07.002
123. Fobian AD, Elliott L, Louie T. A systematic review of sleep, hypertension, and cardiovascular risk in children and adolescents. Curr Hypertens Rep (2018) 20(5):42. doi: 10.1007/s11906-018-0841-7
124. Mozaffarian D. Dietary and policy priorities for cardiovascular disease, diabetes, and obesity. Circulation (2016) 133(2):187–225. doi: 10.1161/CIRCULATIONAHA.115.018585
126. Scarborough P, Nnoaham KE, Clarke D, Capewell S, Rayner M. Modelling the impact of a healthy diet on cardiovascular disease and cancer mortality. J Epidemiol Community Health (2012) 66(5):420–6. doi: 10.1136/jech.2010.114520
127. Tanofsky-Kraff M, Shomaker LB, Stern EA, Miller R, Sebring N, Dellavalle D, et al. Children’s binge eating and development of metabolic syndrome. Int J Obes (Lond) (2012) 36(7):956–62. doi: 10.1038/ijo.2011.259
128. Ahmed HM, Blaha MJ, Nasir K, Rivera JJ, Blumenthal RS. Effects of physical activity on cardiovascular disease. Am J Cardiol (2012) 109(2):288–95. doi: 10.1016/j.amjcard.2011.08.042
129. Kohl HW. Physical activity and cardiovascular disease: evidence for a dose response. Med Sci Sports Exerc (2001) 33(6 Suppl):S472–83. doi: 10.1097/00005768-200106001-00017
130. Anderson E, Durstine JL. Physical activity, exercise, and chronic diseases: a brief review. Sports Med Health Sci (2019) 1(1):3–10. doi: 10.1016/j.smhs.2019.08.006
131. Carter S, Hartman Y, Holder S, Thijssen DH, Hopkins ND. Sedentary behavior and cardiovascular disease risk: mediating mechanisms. Exercise Sport Sci Rev (2017) 45(2):80–6. doi: 10.1249/JES.0000000000000106
132. Diaz R, Ögren SO, Blum M, Fuxe K. Prenatal corticosterone increases spontaneous and d-amphetamine induced locomotor activity and brain dopamine metabolism in prepubertal male and female rats. Neuroscience (1995) 66(2):467–73. doi: 10.1016/0306-4522(94)00605-5
133. Bagley JR, Adams J, Bozadjian RV, Bubalo L, Kippin TE. Strain differences in maternal neuroendocrine and behavioral responses to stress and the relation to offspring cocaine responsiveness. Int J Dev Neurosci (2019) 78:130–8. doi: 10.1016/j.ijdevneu.2019.06.009
134. Rodrigues AJ, Leão P, Pêgo JM, Cardona D, Carvalho MM, Oliveira M, et al. Mechanisms of initiation and reversal of drug-seeking behavior induced by prenatal exposure to glucocorticoids. Mol Psychiatry (2012) 17(12):1295–305. doi: 10.1038/mp.2011.126
135. Hauser J, Dettling-Artho A, Pilloud S, Maier C, Knapman A, Feldon J, et al. Effects of prenatal dexamethasone treatment on postnatal physical, endocrine, and social development in the common marmoset monkey. Endocrinology (2007) 148(4):1813–22. doi: 10.1210/en.2006-1306
136. Schroeder M, Jakovcevski M, Polacheck T, Lebow M, Drori Y, Engel M, et al. A methyl-balanced diet prevents CRF-induced prenatal stress-triggered predisposition to binge eating-like phenotype. Cell Metab (2017) 25(6):1269–81. doi: 10.1016/j.cmet.2017.05.001
137. Stroud LR, Papandonatos GD, Shenassa E, Rodriguez D, Niaura R, LeWinn KZ, et al. Prenatal glucocorticoids and maternal smoking during pregnancy independently program adult nicotine dependence in daughters: a 40-year prospective study. Biol Psychiatry (2014) 75(1):47–55. doi: 10.1016/j.biopsych.2013.07.024
138. Van Lieshout RJ, Boyle MH, Saigal S, Morrison K, Schmidt LA. Mental health of extremely low birth weight survivors in their 30s. Pediatrics (2015) 135(3):452–9. doi: 10.1542/peds.2014-3143
139. Räikkönen K, Pesonen AK, O’Reilly JR, Tuovinen S, Lahti M, Kajantie E, et al. Maternal depressive symptoms during pregnancy, placental expression of genes regulating glucocorticoid and serotonin function and infant regulatory behaviors. Psychol Med (2015) 45(15):3217–26. doi: 10.1017/S003329171500121X
140. Chatterjee A, Thompson JW, Svensson K, Tamayo Y Ortiz M, Wright R, Wright R, et al. Maternal antenatal stress has little impact on child sleep: results from a prebirth cohort in Mexico city. Sleep Health (2018) 4(5):397–404. doi: 10.1016/j.sleh.2018.07.013
141. Khoramdad M, Vahedian-azimi A, Karimi L, Rahimi-Bashar F, Amini H, Sahebkar A. Association between passive smoking and cardiovascular disease: a systematic review and meta-analysis. IUBMB Life (2020) 72(4):677–86. doi: 10.1002/iub.2207
142. Messner B, Bernhard D. Smoking and cardiovascular disease. Arteriosclerosis Thrombosis Vasc Biol (2014) 34(3):509–15. doi: 10.1161/ATVBAHA.113.300156
143. Corrao G, Rubbiati L, Bagnardi V, Zambon A, Poikolainen K. Alcohol and coronary heart disease: a meta-analysis. Addiction (2000) 95(10):1505–23. doi: 10.1046/j.1360-0443.2000.951015056.x
144. Ronksley PE, Brien SE, Turner BJ, Mukamal KJ, Ghali WA. Association of alcohol consumption with selected cardiovascular disease outcomes: a systematic review and meta-analysis. BMJ (2011) 342:d671. doi: 10.1136/bmj.d671
145. Arenas DJ, Beltran S, Zhou S, Goldberg LR. Cocaine, cardiomyopathy, and heart failure: a systematic review and meta-analysis. Sci Rep (2020) 10(1):19795. doi: 10.1038/s41598-020-76273-1
146. Horn SR, Roos LE, Berkman ET, Fisher PA. Neuroendocrine and immune pathways from pre- and perinatal stress to substance abuse. Neurobiol Stress (2018) 9:140–50. doi: 10.1016/j.ynstr.2018.09.004
147. Deminière JM, Piazza PV, Guegan G, Abrous N, Maccari S, Moal ML, et al. Increased locomotor response to novelty and propensity to intravenous amphetamine self-administration in adult offspring of stressed mothers. Brain Res (1992) 586(1):135–9. doi: 10.1016/0006-8993(92)91383-P
148. Henry C, Guegant G, Cador M, Arnauld E, Arsaut J, Le Moal M, et al. Prenatal stress in rats facilitates amphetamine-induced sensitization and induces long-lasting changes in dopamine receptors in the nucleus accumbens. Brain Res (1995) 685(1–2):179–86. doi: 10.1016/0006-8993(95)00430-X
149. Morley-Fletcher S, Puopolo M, Gentili S, Gerra G, Macchia T, Laviola G. Prenatal stress affects 3,4-methylenedioxymethamphetamine pharmacokinetics and drug-induced motor alterations in adolescent female rats. Eur J Pharmacol (2004) 489:89–92. doi: 10.1016/j.ejphar.2004.02.028
150. Biggio F, Talani G, Locci V, Pisu MG, Boero G, Ciarlo B, et al. Low doses of prenatal ethanol exposure and maternal separation alter HPA axis function and ethanol consumption in adult male rats. Neuropharmacology (2018) 131:271–81. doi: 10.1016/j.neuropharm.2017.12.005
151. Deroche V, Piazza PV, Casolini P, Maccari S, Le Moal M, Simon H. Stress-induced sensitization to amphetamine and morphine psychomotor effects depend on stress-induced corticosterone secretion. Brain Res (1992) 598(1):343–8. doi: 10.1016/0006-8993(92)90205-N
152. Deroche V, Marinelli M, Maccari S, Moal ML, Simon H, Piazza PV. Stress-induced sensitization and glucocorticoids. i. sensitization of dopamine-dependent locomotor effects of amphetamine and morphine depends on stress-induced corticosterone secretion. J Neurosci (1995) 15(11):7181–8. doi: 10.1016/0006-8993(92)90205-n
153. Kippin TE, Szumlinski KK, Kapasova Z, Rezner B, See RE. Prenatal stress enhances responsiveness to cocaine. Neuropsychopharmacol (2008) 33(4):769–82. doi: 10.1038/sj.npp.1301447
154. Pastor V, Pallarés ME, Antonelli MC. Prenatal stress increases adult vulnerability to cocaine reward without affecting pubertal anxiety or novelty response. Behav Brain Res (2018) 339:186–94. doi: 10.1016/j.bbr.2017.11.035
155. Peterson GF, Espel EV, Davis EP, Sandman CA, Glynn LM. Characterizing prenatal maternal distress with unique prenatal cortisol trajectories. Health Psychol (2020) 39(11):1013–9. doi: 10.1037/hea0001018
156. Nevarez-Brewster M, Demers CH, Mejia A, Haase MH, Bagonis MM, Kim SH, et al. Longitudinal and prospective assessment of prenatal maternal sleep quality and associations with newborn hippocampal and amygdala volume. Dev Cogn Neurosci (2022) 58:101174. doi: 10.1016/j.dcn.2022.101174
157. Nicolaides NC, Charmandari E, Chrousos GP, Kino T. Circadian endocrine rhythms: the hypothalamic–pituitary–adrenal axis and its actions. Ann N Y Acad Sci (2014) 1318:71–80. doi: 10.1111/nyas.12464
158. Hirotsu C, Tufik S, Andersen ML. Interactions between sleep, stress, and metabolism: from physiological to pathological conditions. Sleep Sci (2015) 8(3):143–52. doi: 10.1016/j.slsci.2015.09.002
159. Tuladhar CT, Schwartz S, St. John AM, Meyer JS, Tarullo AR. Infant diurnal cortisol predicts sleep. J Sleep Res (2021) 30(6):e13357. doi: 10.1111/jsr.13357
160. Dugovic C, Maccari S, Weibel L, Turek FW, Reeth and OV. High corticosterone levels in prenatally stressed rats predict persistent paradoxical sleep alterations. J Neurosci (1999) 19(19):8656–64. doi: 10.1523/JNEUROSCI.19-19-08656.1999
161. Rao U, McGinty DJ, Shinde A, McCracken JT, Poland RE. Prenatal stress is associated with depression-related electroencephalographic sleep changes in adult male rats: a preliminary report. Prog Neuropsychopharmacol Biol Psychiatry (1999) 23(5):929–39. doi: 10.1016/S0278-5846(99)00036-6
162. O’Connor TG, Caprariello P, Blackmore ER, Gregory AM, Glover V, Fleming P. Prenatal mood disturbance predicts sleep problems in infancy and toddlerhood. Early Hum Dev (2007) 83(7):451–8. doi: 10.1016/j.earlhumdev.2006.08.006
163. Ma S, Yin X, Tao R, Jiang X, Xie J, Li P, et al. Association of maternal prenatal depression and anxiety with toddler sleep: the China-anhui birth cohort study. Arch Womens Ment Health (2022) 25(2):431–9. doi: 10.1007/s00737-021-01200-w
164. van den Heuvel MI, Hect JL, Smarr BL, Qawasmeh T, Kriegsfeld LJ, Barcelona J, et al. Maternal stress during pregnancy alters fetal cortico-cerebellar connectivity in utero and increases child sleep problems after birth. Sci Rep (2021) 11(1):2228. doi: 10.1038/s41598-021-81681-y
165. Toffol E, Lahti-Pulkkinen M, Lahti J, Lipsanen J, Heinonen K, Pesonen AK, et al. Maternal depressive symptoms during and after pregnancy are associated with poorer sleep quantity and quality and sleep disorders in 3.5-year-old offspring. Sleep Med (2019) 56:201–10. doi: 10.1016/j.sleep.2018.10.042
166. Field T, Diego M, Hernandez-Reif M, Figueiredo B, Schanberg S, Kuhn C. Sleep disturbances in depressed pregnant women and their newborns. Infant Behav Dev (2007) 30(1):127–33. doi: 10.1016/j.infbeh.2006.08.002
167. Hill DC, Moss RH, Sykes-Muskett B, Conner M, O’Connor DB. Stress and eating behaviors in children and adolescents: systematic review and meta-analysis. Appetite (2018) 123:14–22. doi: 10.1016/j.appet.2017.11.109
169. Khaled K, Tsofliou F, Hundley V, Helmreich R, Almilaji O. Perceived stress and diet quality in women of reproductive age: a systematic review and meta-analysis. Nutr J (2020) 19(1):92. doi: 10.1186/s12937-020-00609-w
170. Adam TC, Epel ES. Stress, eating and the reward system. Physiol Behavior (2007) 91(4):449–58. doi: 10.1016/j.physbeh.2007.04.011
171. Silveira PP, Agranonik M, Faras H, Portella AK, Meaney MJ, Levitan RD, et al. Preliminary evidence for an impulsivity-based thrifty eating phenotype. Pediatr Res (2012) 71(3):293–8. doi: 10.1038/pr.2011.39
172. Kassotaki I, Valsamakis G, Mastorakos G, Grammatopoulos DK. Placental CRH as a signal of pregnancy adversity and impact on fetal neurodevelopment. Front Endocrinol (Lausanne) (2021) 12:714214. doi: 10.3389/fendo.2021.714214
173. Vitt N, Vecchi M, James J, Belot M. Maternal stress during pregnancy and children’s diet: evidence from a population of low socioeconomic status. Nutrition (2022) 93:111423. doi: 10.1016/j.nut.2021.111423
174. St-Hilaire A, Steiger H, Liu A, Laplante DP, Thaler L, Magill T, et al. A prospective study of effects of prenatal maternal stress on later eating-disorder manifestations in affected offspring: preliminary indications based on the project ice storm cohort. Int J Eat Disord (2015) 48(5):512–6. doi: 10.1002/eat.22391
175. Lines RLJ, Ducker KJ, Ntoumanis N, Thøgersen-Ntoumani C, Fletcher D, Gucciardi DF. Stress, physical activity, sedentary behavior, and resilience–the effects of naturalistic periods of elevated stress: a measurement-burst study. Psychophysiology (2021) 58(8):e13846. doi: 10.1111/psyp.13846
176. Burg MM, Schwartz JE, Kronish IM, Diaz KM, Alcantara C, Duer-Hefele J, et al. Does stress result in you exercising less? or does exercising result in you being less stressed? or is it both? testing the bi-directional stress-exercise association at the group and person (N of 1) level. Ann Behav Med (2017) 51(6):799–809. doi: 10.1007/s12160-017-9902-4
177. Stults-Kolehmainen MA, Sinha R. The effects of stress on physical activity and exercise. Sports Med (2014) 44(1):81–121. doi: 10.1007/s40279-013-0090-5
178. Clarke AS, Soto A, Bergholz T, Schneider ML. Maternal gestational stress alters adaptive and social behavior in adolescent rhesus monkey offspring. Infant Behav Dev (1996) 19(4):451–61. doi: 10.1016/S0163-6383(96)90006-5
179. Schneider ML. Prenatal stress exposure alters postnatal behavioral expression under conditions of novelty challenge in rhesus monkey infants. Dev Psychobiol (1992) 25(7):529–40. doi: 10.1002/dev.420250706
180. Takahashi LK, Haglin C, Kalin NH. Prenatal stress potentiates stress-induced behavior and reduces the propensity to play in juvenile rats. Physiol Behavior (1992) 51(2):319–23. doi: 10.1016/0031-9384(92)90147-T
181. McEWEN BS, Seeman T. Protective and damaging effects of mediators of stress: elaborating and testing the concepts of allostasis and allostatic load. Ann New York Acad Sci (1999) 896(1):30–47. doi: 10.1111/j.1749-6632.1999.tb08103.x
182. Kline CE. The bidirectional relationship between exercise and sleep: implications for exercise adherence and sleep improvement. Am J Lifestyle Med (2014) 8(6):375–9. doi: 10.1177/1559827614544437
183. Gunnar MR, DePasquale CE, Reid BM, Donzella B, Miller BS. Pubertal stress recalibration reverses the effects of early life stress in postinstitutionalized children. Proc Natl Acad Sci (2019) 116(48):23984–8. doi: 10.1073/pnas.1909699116
184. Guyer AE, Pérez-Edgar K, Crone EA. Opportunities for neurodevelopmental plasticity from infancy through early adulthood. Child Dev (2018) 89(3):687–97. doi: 10.1111/cdev.13073
185. Parvar SL, Thiyagarajah A, Nerlekar N, King P, Nicholls SJ. A systematic review and meta-analysis of gender differences in long-term mortality and cardiovascular events in peripheral artery disease. J Vasc Surg (2021) 73(4):1456–65. doi: 10.1016/j.jvs.2020.09.039
186. Bots SH, Peters SAE, Woodward M. Sex differences in coronary heart disease and stroke mortality: a global assessment of the effect of ageing between 1980 and 2010. BMJ Global Health (2017) 2(2):e000298. doi: 10.1136/bmjgh-2017-000298
187. Wei YC, George NI, Chang CW, Hicks KA. Assessing sex differences in the risk of cardiovascular disease and mortality per increment in systolic blood pressure: a systematic review and meta-analysis of follow-up studies in the united states. PloS One (2017) 12(1):e0170218. doi: 10.1371/journal.pone.0170218
188. Appelman Y, van Rijn BB, ten Haaf ME, Boersma E, Peters SAE. Sex differences in cardiovascular risk factors and disease prevention. Atherosclerosis (2015) 241(1):211–8. doi: 10.1016/j.atherosclerosis.2015.01.027
189. Mosca L, Barrett-Connor E, Wenger NK. Sex/Gender differences in cardiovascular disease prevention what a difference a decade makes. Circulation (2011) 124(19):2145–54. doi: 10.1161/CIRCULATIONAHA.110.968792
190. Clifton VL. Review: sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta (2010) 31:S33–9. doi: 10.1016/j.placenta.2009.11.010
191. Cuffe JSM, Saif Z, Perkins AV, Moritz KM, Clifton VL. Dexamethasone and sex regulate placental glucocorticoid receptor isoforms in mice. J Endocrinol (2017) 234(2):89–100. doi: 10.1530/JOE-17-0171
192. Meakin AS, Cuffe JSM, Darby JRT, Morrison JL, Clifton VL. Let’s talk about placental sex, baby: understanding mechanisms that drive female- and Male-specific fetal growth and developmental outcomes. Int J Mol Sci (2021) 22(12):6386. doi: 10.3390/ijms22126386
193. Sandman CA, Glynn LM, Davis EP. Is there a viability-vulnerability tradeoff? sex differences in fetal programming. J Psychosom Res (2013) 75(4):327–35. doi: 10.1016/j.jpsychores.2013.07.009
194. Davis EP, Pfaff D. Sexually dimorphic responses to early adversity: implications for affective problems and autism spectrum disorder. Psychoneuroendocrinology (2014) 49:11–25. doi: 10.1016/j.psyneuen.2014.06.014
195. Kim DJ, Davis EP, Sandman CA, Sporns O, O’Donnell BF, Buss C, et al. Prenatal maternal cortisol has sex-specific associations with child brain network properties. Cereb Cortex (2017) 27(11):5230–41. doi: 10.1093/cercor/bhw303
196. Hackler E, Lew J, Gore MO, Ayers CR, Atzler D, Khera A, et al. Racial differences in cardiovascular biomarkers in the general population. J Am Heart Assoc (2019) 8(18):e012729. doi: 10.1161/JAHA.119.012729
197. He J, Zhu Z, Bundy JD, Dorans KS, Chen J, Hamm LL. Trends in cardiovascular risk factors in US adults by race and ethnicity and socioeconomic status, 1999-2018. JAMA (2021) 326(13):1286–98. doi: 10.1001/jama.2021.15187
198. Davis EP, Stout SA, Molet J, Vegetabile B, Glynn LM, Sandman CA, et al. Exposure to unpredictable maternal sensory signals influences cognitive development across species. Proc Natl Acad Sci (2017) 114(39):10390–5. doi: 10.1073/pnas.1703444114
199. Devinsky O, Boesch JM, Cerda-Gonzalez S, Coffey B, Davis K, Friedman D, et al. A cross-species approach to disorders affecting brain and behaviour. Nat Rev Neurol (2018) 14(11):677–86. doi: 10.1038/s41582-018-0074-z
200. Mishra J, Gazzaley A. Cross-species approaches to cognitive neuroplasticity research. Neuroimage (2016) 131:4–12. doi: 10.1016/j.neuroimage.2015.09.002
201. Peyvandi S, Baer RJ, Chambers CD, Norton ME, Rajagopal S, Ryckman KK, et al. Environmental and socioeconomic factors influence the live-born incidence of congenital heart disease: a population-based study in California. J Am Heart Assoc (2020) 9(8):e015255. doi: 10.1161/JAHA.119.015255
202. Sørensen N, Murata K, Budtz-Jørgensen E, Weihe P, Grandjean P. Prenatal methylmercury exposure as a cardiovascular risk factor at seven years of age. Epidemiology (1999) 10(4):370.
203. Perera F, Herbstman J. Prenatal environmental exposures, epigenetics, and disease. Reprod Toxicol (2011) 31(3):363–73. doi: 10.1016/j.reprotox.2010.12.055
204. Nahum Sacks K, Friger M, Shoham-Vardi I, Spiegel E, Sergienko R, Landau D, et al. Prenatal exposure to preeclampsia as an independent risk factor for long-term cardiovascular morbidity of the offspring. Pregnancy Hypertension (2018) 13:181–6. doi: 10.1016/j.preghy.2018.06.013
205. Taittonen L, Nuutinen M, Turtinen J, Uhari M. Prenatal and postnatal factors in predicting later blood pressure among children: cardiovascular risk in young finns. Pediatr Res (1996) 40(4):627–32. doi: 10.1203/00006450-199610000-00019
206. Reynolds RM, Allan KM, Raja EA, Bhattacharya S, McNeill G, Hannaford PC, et al. Maternal obesity during pregnancy and premature mortality from cardiovascular event in adult offspring: follow-up of 1 323 275 person years. BMJ (2013) 347:f4539. doi: 10.1136/bmj.f4539
207. Radaelli T, Uvena-Celebrezze J, Minium J, Huston-Presley L, Catalano P, Hauguel-de Mouzon S. Maternal interleukin-6: marker of fetal growth and adiposity. J Soc Gynecol Investig (2006) 13(1):53–7. doi: 10.1016/j.jsgi.2005.10.003
208. Rogers LK, Velten M. Maternal inflammation, growth retardation, and preterm birth: insights into adult cardiovascular disease. Life Sci (2011) 89(13):417–21. doi: 10.1016/j.lfs.2011.07.017
209. Nguyen MU, Wallace MJ, Pepe S, Menheniott TR, Moss TJ, Burgner D. Perinatal inflammation: a common factor in the early origins of cardiovascular disease? Clin Sci (2015) 129(8):769–84. doi: 10.1042/CS20150045
210. Busse D, Yim IS, Campos B, Marshburn CK. Discrimination and the HPA axis: current evidence and future directions. J Behav Med (2017) 40(4):539–52. doi: 10.1007/s10865-017-9830-6
211. Mustonen P, Karlsson L, Kataja EL, Scheinin NM, Kortesluoma S, Coimbra B, et al. Maternal prenatal hair cortisol is associated with prenatal depressive symptom trajectories. Psychoneuroendocrinology (2019) 109:104383. doi: 10.1016/j.psyneuen.2019.104383
212. Gluckman PD, Hanson MA. Developmental origins of disease paradigm: a mechanistic and evolutionary perspective. Pediatr Res (2004) 56(3):311–7. doi: 10.1203/01.PDR.0000135998.08025.FB
213. Painter R, Osmond C, Gluckman P, Hanson M, Phillips D, Roseboom T. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG: Int J Obstetrics Gynaecol (2008) 115(10):1243–9. doi: 10.1111/j.1471-0528.2008.01822.x
Keywords: cardiovascular disease (CVD), cortisol, placental CRH, health behaviors, cardiometabolic risk
Citation: Deer LK, Su C, Thwaites NA, Davis EP and Doom JR (2023) A framework for testing pathways from prenatal stress-responsive hormones to cardiovascular disease risk. Front. Endocrinol. 14:1111474. doi: 10.3389/fendo.2023.1111474
Received: 29 November 2022; Accepted: 10 April 2023;
Published: 08 May 2023.
Edited by:
Takahiro Nemoto, Nippon Medical School, JapanReviewed by:
Kristina Laugesen, Aarhus University, DenmarkCopyright © 2023 Deer, Su, Thwaites, Davis and Doom. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: LillyBelle K. Deer, bGlsbHliZWxsZS5kZWVyQGR1LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.