
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 13 March 2023
Sec. Adrenal Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1095719
This article is part of the Research TopicRecent advances in diagnosis and treatment of congenital adrenal hyperplasia due to 21-hydroxylase deficiencyView all 12 articles
 Peng Tang1†
Peng Tang1† Jun Zhang1†
Jun Zhang1† Song Peng1
Song Peng1 Yapeng Wang1
Yapeng Wang1 Haoyang Li2
Haoyang Li2 Ze Wang1
Ze Wang1 Yao Zhang1
Yao Zhang1 Yiqiang Huang1
Yiqiang Huang1 Jing Xu1
Jing Xu1 Dianzheng Zhang3
Dianzheng Zhang3 Qiuli Liu1
Qiuli Liu1 Luofu Wang1
Luofu Wang1 Weihua Lan1*
Weihua Lan1* Jun Jiang1*
Jun Jiang1*Introduction: 21-hydroxylase deficiency (21OHD) is the most common cause of congenital adrenal hyperplasia (CAH). However, patients with 21OHD manifest various phenotypes due to a wide-spectrum residual enzyme activity of different CYP21A2 mutations.
Methods: A total of 15 individuals from three unrelated families were included in this study. Target Capture-Based Deep Sequencing and Restriction Fragment Length Polymorphism was conducted on peripheral blood DNA of the three probands to identify potential mutations/deletions in CYP21A2; Sanger sequencing was conducted with the DNA from the family members of the probands.
Results: Dramatically different phenotypes were seen in the three probands of CAH with different compound heterozygous mutations in CYP21A2. Proband 1 manifested simple virilizing with mutations of 30-kb deletion/c.[188A>T;518T>A], the latter is a novel double mutants classified as SV associated mutation. Although both probands carry the same compound mutations [293-13C>G]:[518T>A], gonadal dysfunction and giant bilateral adrenal myelolipoma were diagnosed for proband 2 and proband 3, respectively.
Conclusion: Both gender and mutations contribute to the phenotypes, and patients with the same compound mutations and gender could present with different phenotypes. Genetic analysis could help the etiologic diagnosis, especially for atypical 21OHD patients.
Most congenital adrenal hyperplasia (CAH) resulted from deficiencies of the enzymes, encoded by different cytochrome P450 (CYP) genes, in adrenal steroidogenesis (1). In humans, six CYPs including CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP19A1, and CYP21A2 are involved in the synthesis of steroid hormones and deficiencies in any of them can cause CAH. Of note, ~95% of CAH patients have a deficient 21-hydroxylase (21OH) due to the mutation of CYP21A2. Since adrenocorticotropic hormone (ACTH) is partially downregulated by glucocorticoid and mineralocorticoid through a feed-back mechanism, reduced levels of glucocorticoid and mineralocorticoid are accompanied by elevated level of ACTH, which in turn results in not only accumulation of precursors in adrenal androgenesis but also exacerbates CAH (2). However, a wide spectrum of clinical manifestations was observed in patients with different mutations of their CYPs.
Patients with 21OHD can be categorized into three subgroups based on their clinical phenotypes: classic salt‐wasting (SW), classic simple virilizing (SV), and the non-classic (NC) forms (3). SW is most severe and could be life‐threatening if occurring in early infancy. Classic SV is characterized by prenatal virilization in females and virilization in both sexes without salt wasting (4). NC usually shows mild symptoms of androgen excess and sometimes is even asymptomatic (5). The diverse phenotypes of 21OH-caused CAH are the results of varying degrees of enzymatic deficiency. For instance, Wilson RC et al. found that patients with mutations causing complete loss of enzymatic activity presented with more severe phenotype, whereas patients with partial loss of enzymatic activity exhibited non-classical CAH (6). Genotype-phenotype correlations in CAH have been reported previously (7, 8). In this study, we found that some phenotype is associated certain CYP21A2 mutation, and sometimes the same compound heterozygous mutation of CYP21A2, can lead to different phenotypes. These specific cases add to our knowledge of genotype-phenotype correlations of CYP21A2.
This study design was reviewed and approved by the ethics committee in the Daping Hospital of Army Medical University. A total of 13 individuals from three unrelated families with their medical records including computed tomography (CT) or magnetic resonance imaging (MRI) scan, and laboratory examinations as well as their family history were included in this study. All procedures were carried out in accordance with the ethical standards of the institutional research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Written informed consent for the use of medical records and related images was obtained from each patient.
Total DNA isolated from the peripheral blood leukocytes were obtained from the 15 individuals and used for sequencing to identify potential mutations and large deletions in CYP21A2 gene. To identify large gene deletions, primers CYP779f and Tena32F (available upon request) were used to amplify a fragment of 8515-bp followed the manufacturer’s recommendations. The 8.5-kb PCR product was digested with TaqI restriction endonuclease and the digested products were analyzed by electrophoresis in agarose gels.
Target Capture-Based Deep Sequencing (AmCare Genomic Laboratory, Guangzhou, Guangdong, China) was conducted with the PCR product amplified by CYP779f and Tena32F. The sequencing was carried out on NGS platform (HiSeq X system; Illumina) with PEx150 read length according to the manufactory’s instructions. Bioinformatics analyses were performed with in-house pipeline to identify rare or novel variants of CYP21A2 with Gnomad, HMGD, ClinVar, dbSNP for filtering and computational prediction algorithms. Software PolyPhen-2, SIFT, and MutationTaster were used for pathogenesis evaluation (Supplementary Table 1).
After the identification of the variants in the probands, Sanger sequencing were conducted to determine family co-segregation. The corresponding primers were designed by Primer 3.0 and available upon request. The final pathogenicity of the variants was estimated using the American College of Medical Genetics and Genomics guidelines.
Proband 1 was an 8-year-old girl who was diagnosed as congenital adrenal hyperplasia when she was born with an atypical genitalia but no salt-wasting symptom in another hospital. She underwent clitoroplasty in that hospital at the age of 8 months and glucocorticoid and mineralocorticoid replacement therapy was prescribed. Laboratory investigations revealed elevated levels 17-hydroxyprogesterone (17-OHP, >75.75 nmol/L, normal 0.2–3.0 nmol/L) androgens, testosterone, and ACTH as shown in Table 1. Her karyotype is 46, XX.
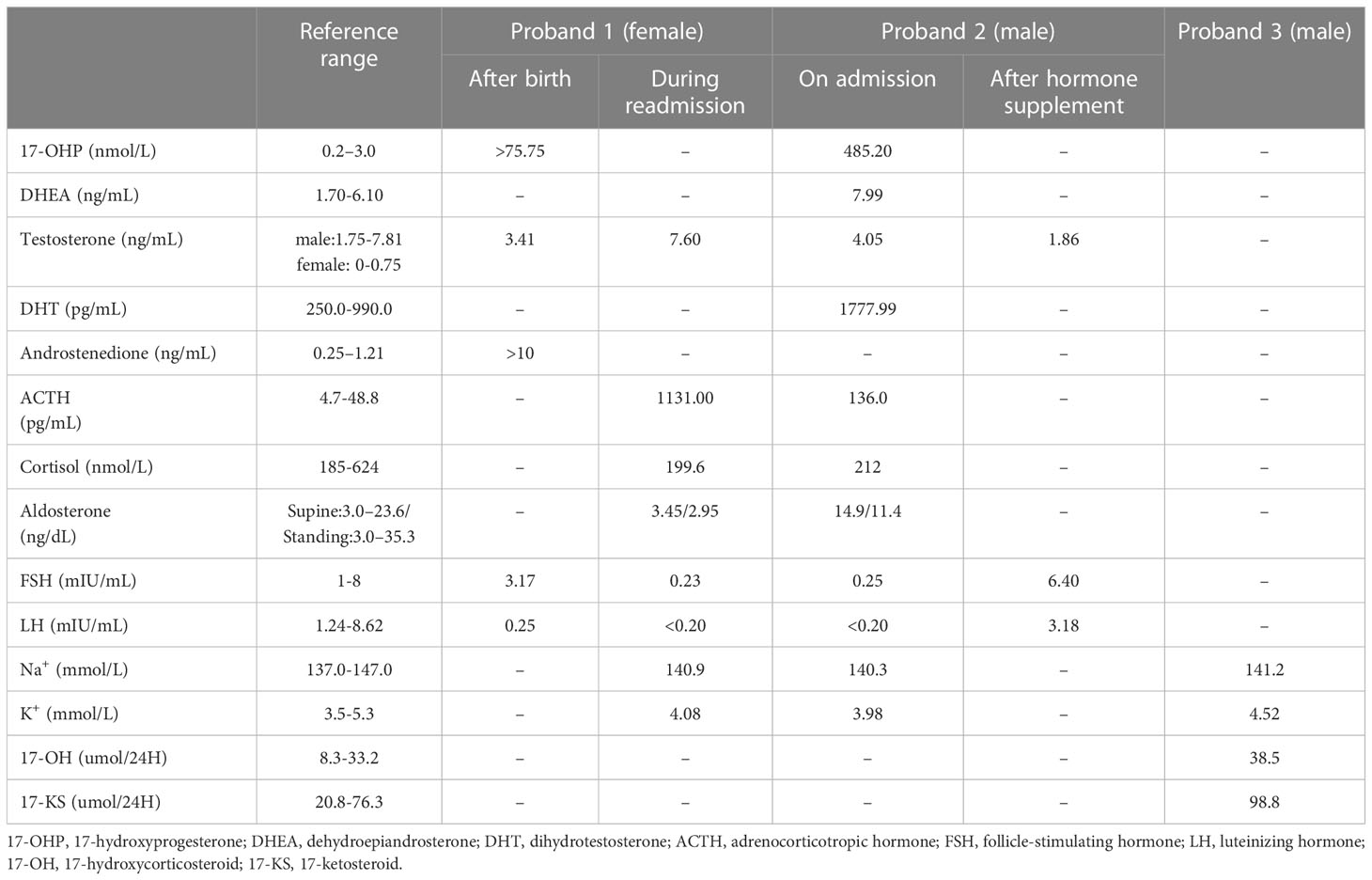
Table 1 Laboratory parameters of the probands.
She was referred to our hospital due to virilization and presented with high stature (140 cm tall, >97th percentile of girls of Han nationality in the same age group in China) and deep voice. Physical examination identified clitoral enlargement, partially fused labia majora, and a urogenital sinus in place of separate urethral and vaginal openings. She was taking hydrocortisone (20 mg per two days) before admission to our hospital. We noticed that based on her weight of 41 kg, she should be treated with 12.7-19.1 mg in a day according to the guideline (9). Laboratory examination revealed a markedly elevated testosterone level of 7.60 ng/mL and CT identified hyperplasia of bilateral adrenal gland, and the cavernous structure in the perineum as shown in Figure 1. She was advised to maintain her regular replacement therapy for now and will have genitoplastic surgery and vaginoplasty when she becomes an adult. None of her parents has any related symptom.
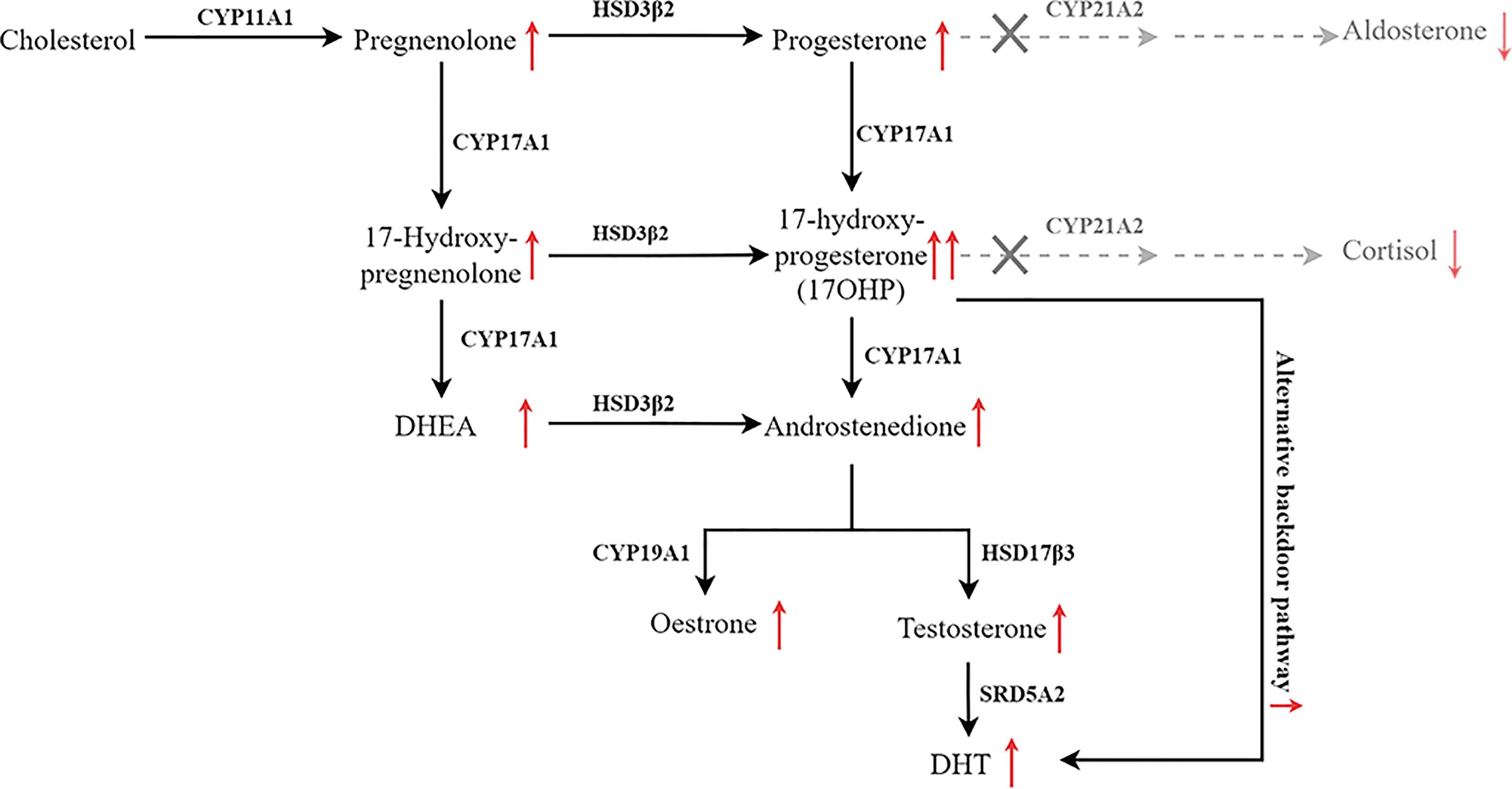
Figure 1 Pathway of steroid biosynthesis in the adrenal cortex. 21-hydroxylase deficiency results in decreased cortisol and aldosterone, and elevated cortisol precursors and adrenal androgens. The grey denotes deficient pathways.
Proband 2 was a 37-year-old male referred to our hospital due to the azoospermia, without other symptoms. He had been diagnosed as idiopathic hypogonadotropic hypogonadism (IHH) based on small testes and significantly decreased serum levels of FSH (0.07 mIU/mL) and LH (0.00 mIU/mL, Table 1) but without salt wasting. The patient had sex premature with masculinization and precocious pubarche when he was only 7 years old. He had a short stature (149 cm in height), small testis (left: 11 mL; right: 12 mL), and mild hypospadias. CT scan revealed left adrenal hyperplasia and right adrenal adenomatous hyperplasia (4.3*3.7 cm, Figure 2). Scrotal ultrasound detected an 8 mm*6mm mass in his left testis, likely to be testicular adrenal rest tumors (TARTs). Laboratory tests showed elevated serum 17OHP (485.20 nmol/L, normal 0.2–3.0 nmol/L), dehydroepiandrosterone (DHEA) (7.99ng/mL, normal 1.70-6.10), and dihydrotestosterone (DHT) (1777.99 pg/mL, normal 250.0-990.0, Table 1). Gonadotropin-releasing hormone (GnRH) stimulation test was negative (Supplementary Table 2). We concluded that he has congenital adrenal hyperplasia and secondary hypogonadotropic hypogonadism. After eight months of replacement therapy, his semen count was relatively normal (Supplementary Table 3). His parents were normal.
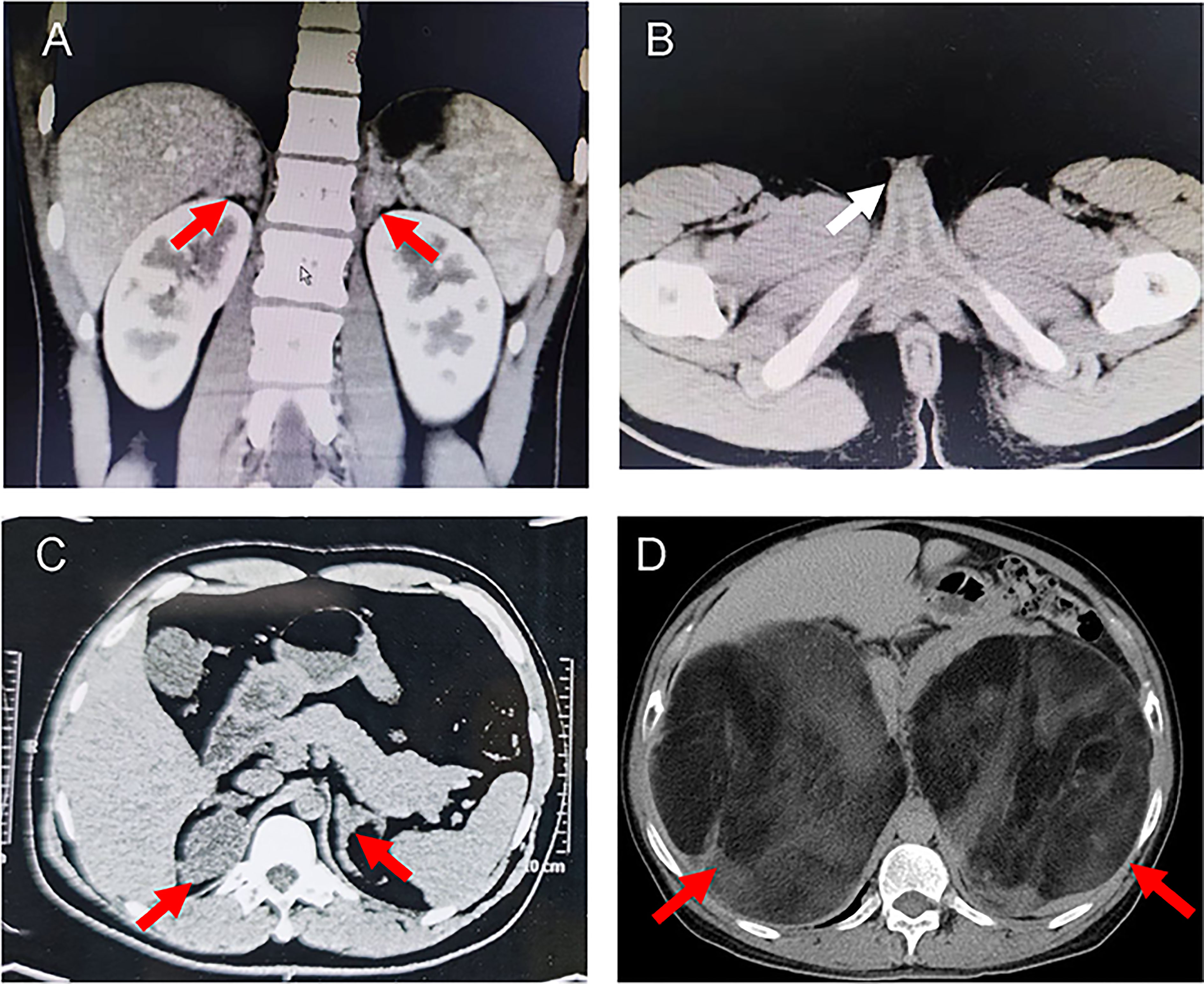
Figure 2 Representative computed tomography (CT) scans of the probands. (A) CT images showing bilateral adrenal hyperplasia in the proband 1. (B) Pelvic CT image of proband 1 indicated the cavernous structure in the perineum (white arrow). (C) CT images showing left adrenal hyperplasia and right adrenal adenomatous hyperplasia (4.3*3.7 cm) in the proband 2. (D) CT image showing giant bilateral adrenal masses (left: 20*25 cm; right: 30*40 cm) in the proband 3. The red arrows indicate the adrenal lesions.
Proband 3 was a 59-year-old male admitted to our hospital with marked abdominal distension. CT scan revealed giant bilateral myelolipoma (left: 20*25 cm; right: 30*40 cm; Figure 2). His 24-hour urine 17-hydroxycorticosteroid (17-OH) and 17-ketosteroid (17-KS) were a bit higher (Table 1). The proband underwent resection of his myelolipoma accompanied with glucocorticoid and mineralocorticoid replacement therapy. Patient history indicates that he had a short stature at birth but no salt wasting. Both his parents died with unknown causes. His brother also underwent resection of bilateral giant myelolipomas at age of 50. But his three sisters and three daughters appear to be normal.
Proband 1 inherited a 30kb deletion (a classic category of chimeric CYP21A1P/CYP21A2 genes: CH-2, which carries common mutations including: c.92C>T, p.P31L; c.188A>T, p.H63L; c.293-13C>G; c.332_339del, p.G111Vfs*21; c.518T>A, p.I173N) (10) and a double mutants in cis (c.[188A>T;518T>A], p.[H63L;(I173N)]) (Figure 3). Both proband 2 and 3 have a compound heterozygous mutation of CYP21A2 (c.293-13C>G and c.518T>A, p.I173N, Figure 3), the detailed pedigree for family of proband 3 has been previously reported (11). We found that proband 2 inherited c.293-13C>G from his mother and c.518T>A, p.I173N from his father. However, we cannot determine his genetic heritage since both his parents are dead. His brother harbored the same compound mutations. Each of the other family members of the three probands harbored a monoallelic CYP21A2 variant. The genotypes and phenotypes of the individuals were summarized in Table 2 and Supplementary Table 4.
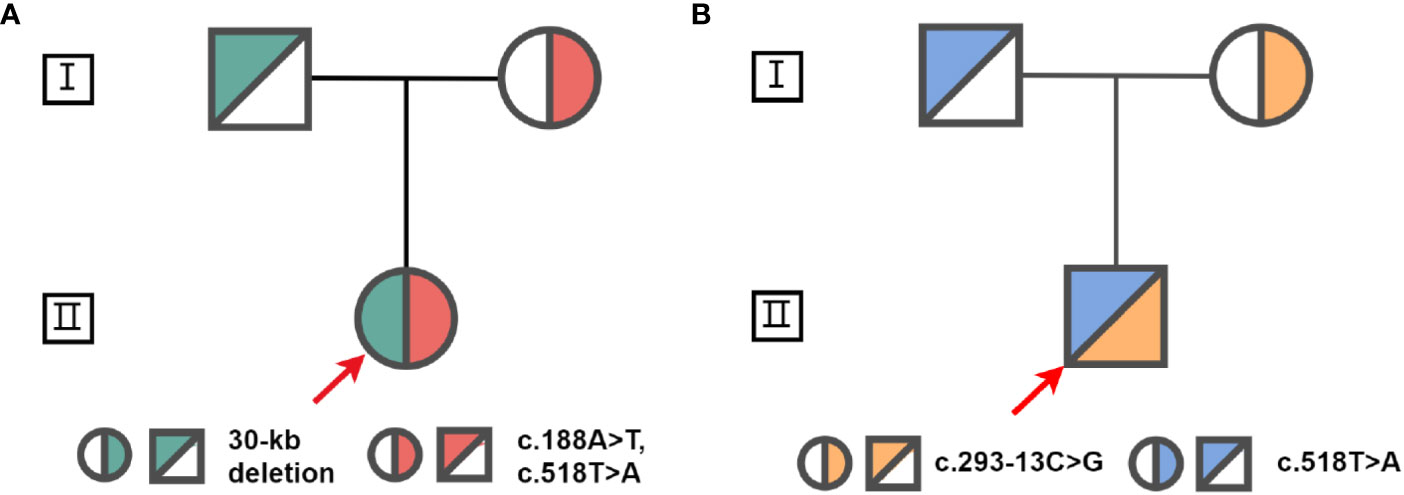
Figure 3 Pedigrees for families of proband 1 (A) and s proband 2 (B). The red arrow indicates the probands.
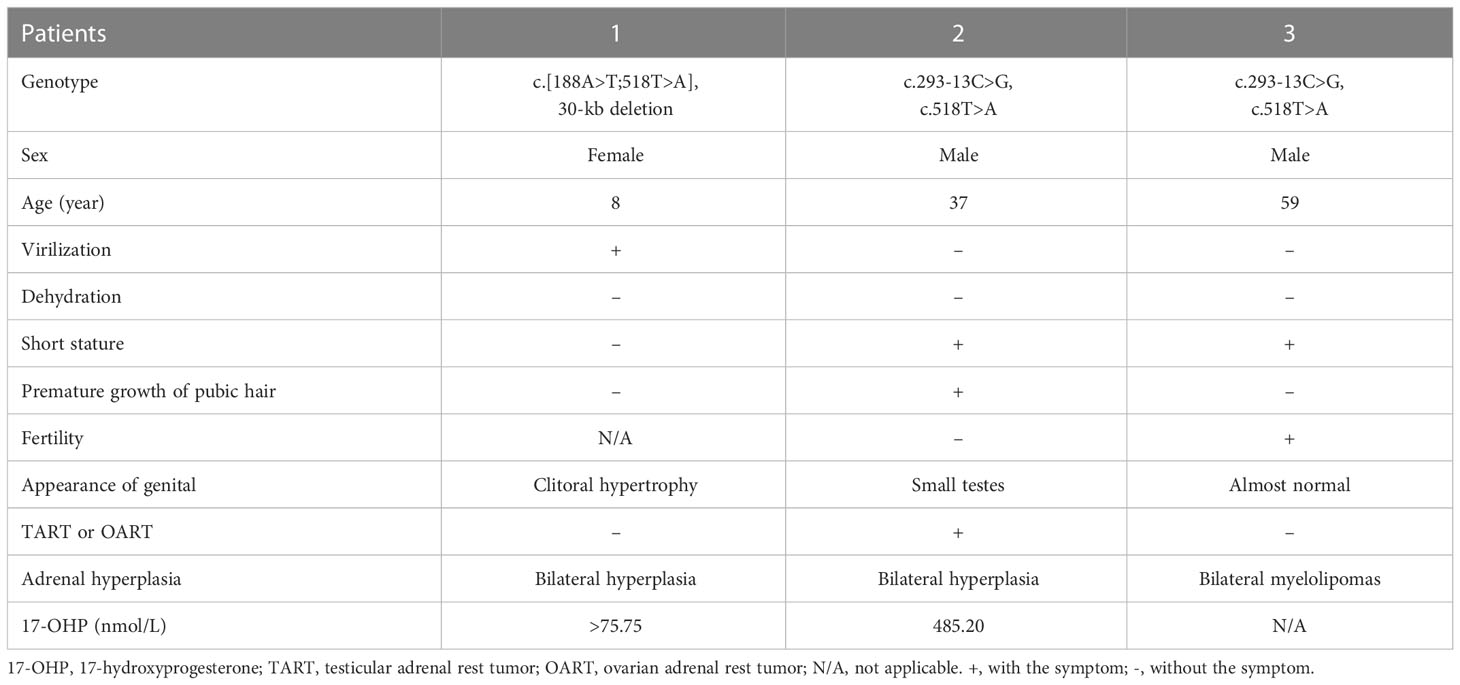
Table 2 The genotype-phenotype of the probands.
CAH is an autosomal recessive disease caused by deficient enzymes including 21OH, 11β-hydroxylase, 17α-hydroxylase, 3β-hydroxysteroid dehydrogenase type 2, steroidogenic acute regulatory protein, and P450 oxidoreductase (12). 21OHD-caused CAH is found 1 in every 10 000 to 20 000 new born (13) with diverse phenotype. Complete inactive CYP21A2 results in deficiencies of both glucocorticoid and mineralocorticoid, and severe adrenal-derived androgen excess. In newborn females, excess androgen is clinically evident due to virilization of their external genitalia. However, males with excess androgen appear to be normal at birth but with premature growth of pubic and axillary hair, oily skin, rapid somatic and skeletal maturation. Proband 1 described in this study is a girl and diagnosed after birth, who carried compound mutations with the 30-kb deletion inherited from her father and c.[188A>T;518T>A], a novel double mutants in cis (14), from her mother. The c.188A>T mutation has a mild effect on the enzyme; whilst the c.518T>A affects the enzymatic activity more dramatically (15, 16). The double mutants in the young girl (proband 1) appears to be associated with simple virilization according to her clinical presentations. On the other hand, both probands 2 and 3 in this study are male and their azoospermia and giant bilateral adrenal myelolipomas were not recognized until much later. Thus, the gender could be one of the main factor that contributes to the diverse phenotype (1, 17).
Findings from previous studies indicate that phenotypes of 21OHD-caused CAH maybe mutation site-specific. When the genotype-phenotype correlation was examined with more than 230 mutations (18), 70–75% of the CAH cases were caused by the ten most common variants (7, 19, 20). The p.V282L, one of the ten most common variants, accounts for 0.9% cases in Chinese people, compared with 26.2% and 23.9% in Argentinian and other heterogeneous western population respectively (21). In this study, we found that proband 2 and 3 have the same compound mutations [293-13C>G]:[518T>A] but totally different clinical manifestations. Previous study reported patients with this compound heterozygous mutation shows phenotypic variability (NC : SV:SW = 1:36:13) (7). In our report, proband 2 presented with azoospermia mainly due to secondary gonadal dysfunction. The proband has been initially misdiagnosed as IHH, a rare type of congenital disease characterized with dysfunction in the secretion of hypothalamic GnRH and reduced serum levels of sex steroids (22). In fact, the lower normal range of testosterone with suppressed gonadotropins is also the typical profile of secondary hypogonadotropic hypogonadism (23). Previous studies have reported that the accumulation of excessive progesterone, 17OHP, estrogen, and androgens jointly contribute to hypothalamic-pituitary -gonadal (HPG) axis suppression and the secondary gonadal failure (23–25). Based on the elevated levels of 17OHP, DHT, and DHEA, proband 2 is diagnosed as CAH and nearly full recover of semen count after glucocorticoid supplementary therapy further confirmed his gonadal dysfunction is secondary to hypogonadotropic hypogonadism. In addition, the testicular mass might be TART, since it is well known that upregulated ACTH secretion causes TARTs (25). Previous study has shown that hypogonadism and azoospermia could be associated with bilateral TARTs (26). Noteworthy, proband 2 had relatively high levels of DHT, which might be due to the activation of alternative backdoor pathway as described previously (27). Although carrying the same compound mutations, proband 3 presented with giant bilateral adrenal myelolipomas, which are caused by chronic ACTH hyperstimulation (28). One of the potential explanations is that different c.293-13C>G were transcribed in these patients (29).
Glucocorticoid replacement is a life-long treatment and the options for glucocorticoid are hydrocortisone or long-acting synthetic glucocorticoid (2). To avoid suppressive effect of growth and chronic cushingoid complication, hydrocortisone is usually prescribed for children (30). The long-acting synthetic glucocorticoid is generally recommended for adolescents and adults (31). Although hydrocortisone was prescribed for proband 1, virilization has developed progressively. This could be the result of insufficient dosage and/or unresponsive surveillance (9). Intensified glucocorticoid treatment is the first choice for gonadal dysfunction (24). For proband 2, normal level of FSH after one month dexamethasone supplement suggests the rehabilitation of HPG axis. After eight months of treatment his sperm production became relatively normal. In addition, differences of sex development (DSD) often occur in females with classic 21OHD. Decision for sex mainly depends on karyotype and the degree of virilization. Timely diagnosis is the key for early treatment of CAH and the ratio between the intermediates and the hormones is the mainstay for the diagnosis. Since elevated 17OHP is a typical indicator of 21OHD-associated CAH (32), the levels of 17OHP should be monitored closely for individuals showing evidence of androgen excess.
In conclusion, we described three probands with diverse phenotypes of CAH carrying different compound heterozygous mutations of CYP21A2. The novel double mutants (c.[188A>T;518T>A]) is a SV subtype. In addition to be consistent with that genders and different mutations contribute to various phenotypes, we found that diverse phenotypes could be presented in patients with identical compound mutations in the same gender. Moreover, genetic analysis could help the etiologic diagnosis, especially for atypical 21OHD patients. These specific cases add to our knowledge of genotype-phenotype correlations of CYP21A2.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author/s.
The studies involving human participants were reviewed and approved by the ethics committee in the Daping Hospital. The patients/participants provided their written informed consent to participate in this study.
Study design were conducted by JJ, WL, and JZ. Samples and clinical data were collected by YH, LW, YZ, JX, and YW. Analysis of data were performed by PT, SP, ZW, YZ, and HL. DZ, QL, JZ and PT wrote the manuscript which was revised and approved by all authors.
This work was supported by University Research Project of Army Medical University (2018XLC3073 for JZ; 2018XLC1014 and 2019CXLCB006 for JJ).
The authors thank the patients and their family members in this study for their collaboration and the AmCare-Guangzhou for conducting the sequencing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2023.1095719/full#supplementary-material
CAH, congenital adrenal hyperplasia; CYP, cytochrome P450; 21OH, 21-hydroxylase; SW, salt‐wasting; SV simple virilizing; NC, non-classic; DHEA, dehydroepiandrosterone; IHH, idiopathic hypogonadotropic hypogonadism; 17-OHP, 17-hydroxyprogesterone; GnRH, Gonadotropin-releasing hormone; DHT, dihydrotestosterone; 17-OH, 17-hydroxycorticosteroid; 17-KS, 17-ketosteroid; HPG, hypothalamic pituitary gonadal; TART, testicular adrenal rest tumor; DSD, differences of sex development.
1. El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplasia. Lancet (2017) 390(10108):2194–210. doi: 10.1016/S0140-6736(17)31431-9
2. Merke DP, Auchus RJ. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. N Engl J Med (2020) 383(13):1248–61. doi: 10.1056/NEJMra1909786
3. Carvalho B, Marques CJ, Santos-Silva R, Fontoura M, Carvalho D, Carvalho F. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: An update on genetic analysis of Cyp21a2 gene. Exp Clin Endocrinol Diabetes (2021) 129(7):477–81. doi: 10.1055/a-1108-1419
4. Forest MG. Recent advances in the diagnosis and management of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Reprod Update (2004) 10(6):469–85. doi: 10.1093/humupd/dmh047
5. Hayashi GY, Carvalho DF, de Miranda MC, Faure C, Vallejos C, Brito VN, et al. Neonatal 17-hydroxyprogesterone levels adjusted according to age at sample collection and birthweight improve the efficacy of congenital adrenal hyperplasia newborn screening. Clin Endocrinol (Oxf) (2017) 86(4):480–7. doi: 10.1111/cen.13292
6. Wilson RC, Nimkarn S, Dumic M, Obeid J, Azar MR, Najmabadi H, et al. Ethnic-specific distribution of mutations in 716 patients with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Mol Genet Metab (2007) 90(4):414–21. doi: 10.1016/j.ymgme.2006.12.005
7. New MI, Abraham M, Gonzalez B, Dumic M, Razzaghy-Azar M, Chitayat D, et al. Genotype-phenotype correlation in 1,507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U.S.A. (2013) 110(7):2611–6. doi: 10.1073/pnas.1300057110
8. Xu C, Jia W, Cheng X, Ying H, Chen J, Xu J, et al. Genotype-phenotype correlation study and mutational and hormonal analysis in a Chinese cohort with 21-hydroxylase deficiency. Mol Genet Genom Med (2019) 7(6):e671. doi: 10.1002/mgg3.671
9. Speiser PW, Arlt W, Auchus RJ, Baskin LS, Conway GS, Merke DP, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J Clin Endocrinol Metab (2018) 103(11):4043–88. doi: 10.1210/jc.2018-01865
10. Chen W, Xu Z, Sullivan A, Finkielstain GP, Van Ryzin C, Merke DP, et al. Junction site analysis of chimeric Cyp21a1p/Cyp21a2 genes in 21-hydroxylase deficiency. Clin Chem (2012) 58(2):421–30. doi: 10.1373/clinchem.2011.174037
11. Liu Q, Wang LA, Su J, Tong D, Lan W, Wang L, et al. Giant bilateral adrenal myelolipomas in two Chinese families with congenital adrenal hyperplasia. Endocr Connect (2018) 7(11):1136–41. doi: 10.1530/EC-18-0273
12. Krone N, Dhir V, Ivison HE, Arlt W. Congenital adrenal hyperplasia and P450 oxidoreductase deficiency. Clin Endocrinol (Oxf) (2007) 66(2):162–72. doi: 10.1111/j.1365-2265.2006.02740.x
13. Nordenstrom A, Lajic S, Falhammar H. Clinical outcomes in 21-hydroxylase deficiency. Curr Opin Endocrinol Diabetes Obes (2021) 28(3):318–24. doi: 10.1097/MED.0000000000000625
14. Simonetti L, Bruque CD, Fernandez CS, Benavides-Mori B, Delea M, Kolomenski JE, et al. Cyp21a2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum Mutat (2018) 39(1):5–22. doi: 10.1002/humu.23351
15. Soardi FC, Barbaro M, Lau IF, Lemos-Marini SH, Baptista MT, Guerra-Junior G, et al. Inhibition of Cyp21a2 enzyme activity caused by novel missense mutations identified in Brazilian and Scandinavian patients. J Clin Endocrinol Metab (2008) 93(6):2416–20. doi: 10.1210/jc.2007-2594
16. Menassa R, Tardy V, Despert F, Bouvattier-Morel C, Brossier JP, Cartigny M, et al. P.H62l, a rare mutation of the Cyp21 gene identified in two forms of 21-hydroxylase deficiency. J Clin Endocrinol Metab (2008) 93(5):1901–8. doi: 10.1210/jc.2007-2701
17. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med (2003) 349(8):776–88. doi: 10.1056/NEJMra021561
18. Concolino P, Costella A. Congenital adrenal hyperplasia (Cah) due to 21-hydroxylase deficiency: A comprehensive focus on 233 pathogenic variants of Cyp21a2 gene. Mol Diagn Ther (2018) 22(3):261–80. doi: 10.1007/s40291-018-0319-y
19. Hannah-Shmouni F, Chen W, Merke DP. Genetics of congenital adrenal hyperplasia. Endocrinol Metab Clin North Am (2017) 46(2):435–58. doi: 10.1016/j.ecl.2017.01.008
20. Marques CJ, Pignatelli D, Carvalho B, Barcelo J, Almeida AC, Fernandes S, et al. Mutational characterization of steroid 21-hydroxylase gene in Portuguese patients with congenital adrenal hyperplasia. Exp Clin Endocrinol Diabetes (2010) 118(8):505–12. doi: 10.1055/s-0029-1237363
21. Xia Y, Shi P, Gao S, Liu N, Zhang H, Kong X. Genetic analysis and novel variation identification in Chinese patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Steroid Biochem Mol Biol (2022) 222:106156. doi: 10.1016/j.jsbmb.2022.106156
22. Liu Q, Yin X, Li P. Clinical, hormonal, and genetic characteristics of 25 Chinese patients with idiopathic hypogonadotropic hypogonadism. BMC Endocr Disord (2022) 22(1):30. doi: 10.1186/s12902-022-00940-9
23. Engels M, Gehrmann K, Falhammar H, Webb EA, Nordenstrom A, Sweep FC, et al. Gonadal function in adult Male patients with congenital adrenal hyperplasia. Eur J Endocrinol (2018) 178(3):285–94. doi: 10.1530/EJE-17-0862
24. Claahsen-van der Grinten HL, Stikkelbroeck N, Falhammar H, Reisch N. Management of endocrine disease: Gonadal dysfunction in congenital adrenal hyperplasia. Eur J Endocrinol (2021) 184(3):R85–97. doi: 10.1530/EJE-20-1093
25. Pitteloud N, Dwyer AA, DeCruz S, Lee H, Boepple PA, Crowley WF Jr., et al. Inhibition of luteinizing hormone secretion by testosterone in men requires aromatization for its pituitary but not its hypothalamic effects: Evidence from the tandem study of normal and gonadotropin-releasing hormone-deficient men. J Clin Endocrinol Metab (2008) 93(3):784–91. doi: 10.1210/jc.2007-2156
26. Claahsen-van der Grinten HL, Otten BJ, Stikkelbroeck MM, Sweep FC, Hermus AR. Testicular adrenal rest tumours in congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab (2009) 23(2):209–20. doi: 10.1016/j.beem.2008.09.007
27. Suminska M, Bogusz-Gorna K, Wegner D, Fichna M. Non-classic disorder of adrenal steroidogenesis and clinical dilemmas in 21-hydroxylase deficiency combined with backdoor androgen pathway. mini-review and case report. Int J Mol Sci (2020) 21(13):4622. doi: 10.3390/ijms21134622
28. Hagiwara H, Usui T, Kimura T, Tagami T, Naruse M, Minamiguchi S, et al. Lack of acth and androgen receptor expression in a giant adrenal myelolipoma associated with 21-hydroxylase deficiency. Endocr Pathol (2008) 19(2):122–7. doi: 10.1007/s12022-008-9034-2
29. Kocova M, Concolino P, Falhammar H. Characteristics of In2g variant in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Front Endocrinol (Lausanne) (2021) 12:788812. doi: 10.3389/fendo.2021.788812
30. Auchus RJ, Arlt W. Approach to the patient: The adult with congenital adrenal hyperplasia. J Clin Endocrinol Metab (2013) 98(7):2645–55. doi: 10.1210/jc.2013-1440
31. Auchus RJ. Management considerations for the adult with congenital adrenal hyperplasia. Mol Cell Endocrinol (2015) 408:190–7. doi: 10.1016/j.mce.2015.01.039
Keywords: congenital adrenal hyperplasia, 21OHD, genotype, phenotype, CYP21A2
Citation: Tang P, Zhang J, Peng S, Wang Y, Li H, Wang Z, Zhang Y, Huang Y, Xu J, Zhang D, Liu Q, Wang L, Lan W and Jiang J (2023) Genotype-phenotype correlation in patients with 21-hydroxylase deficiency. Front. Endocrinol. 14:1095719. doi: 10.3389/fendo.2023.1095719
Received: 11 November 2022; Accepted: 26 January 2023;
Published: 13 March 2023.
Edited by:
Semra Çaglar Çetinkaya, University of Health Sciences, TürkiyeReviewed by:
Sifeng Qu, Shandong University, ChinaCopyright © 2023 Tang, Zhang, Peng, Wang, Li, Wang, Zhang, Huang, Xu, Zhang, Liu, Wang, Lan and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Jiang, amlhbmdqdW5fNjRAMTYzLmNvbQ==; Weihua Lan, ZG9jbGFuQHllYWgubmV0
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.