 Stéphane Dalle
Stéphane Dalle Amar Abderrahmani
Amar Abderrahmani Eric Renard1,3,4
Eric Renard1,3,4
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 15 March 2023
Sec. Cellular Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1076343
This article is part of the Research Topic Insights in Cellular Endocrinology: 2022 View all 9 articles
More than 500 million adults suffer from diabetes worldwide, and this number is constantly increasing. Diabetes causes 5 million deaths per year and huge healthcare costs per year. β-cell death is the major cause of type 1 diabetes. β-cell secretory dysfunction plays a key role in the development of type 2 diabetes. A loss of β-cell mass due to apoptotic death has also been proposed as critical for the pathogenesis of type 2 diabetes. Death of β-cells is caused by multiple factors including pro-inflammatory cytokines, chronic hyperglycemia (glucotoxicity), certain fatty acids at high concentrations (lipotoxicity), reactive oxygen species, endoplasmic reticulum stress, and islet amyloid deposits. Unfortunately, none of the currently available antidiabetic drugs favor the maintenance of endogenous β-cell functional mass, indicating an unmet medical need. Here, we comprehensively review over the last ten years the investigation and identification of molecules of pharmacological interest for protecting β-cells against dysfunction and apoptotic death which could pave the way for the development of innovative therapies for diabetes.
In type 1 diabetes (T1D), autoimmune destruction of pancreatic β-cells and reduction in β-cell mass are key characteristics of the disease (1–4). The destruction of β-cells is due to immune-mediated processes such as mononuclear infiltration into pancreatic islets leading to elevated intraislet concentrations of pro-inflammatory cytokines and chemokines (1–3). In type 2 diabetes (T2D), β-cell secretory dysfunction is a key event in the development of a clinically evident disease (5–8). A loss of β-cell mass by apoptotic death has also been proposed as critical for the pathogenesis of T2D (9, 10). Dysfunction and death by apoptosis of β-cells in T2D are caused by multiple factors, including chronic hyperglycemia (glucotoxicity), certain high-concentration fatty acids (lipotoxicity), reactive oxygen species (ROS), endoplasmic reticulum (ER) stress, and islet amyloid polypeptide deposits (11–15). Chronic, systemic and low grade inflammation was also proposed to lead to β-cell dysfunction and death, and ultimately to T2D (16, 17). Thus, in both T1D and T2D, a loss of β-cell survival and function contributes to the development of absolute or relative insulin insufficiency. Over the last years, several molecules and drugs have been developed and shown to inhibit β-cell apoptosis and dysfunction through different mechanisms of action (Figures 1, 2). In this review, we comprehensively highlight the development of these molecules and drugs over the last ten years that protect β-cells against apoptotic death and dysfunction which present key features for diabetes therapy (Table 1).
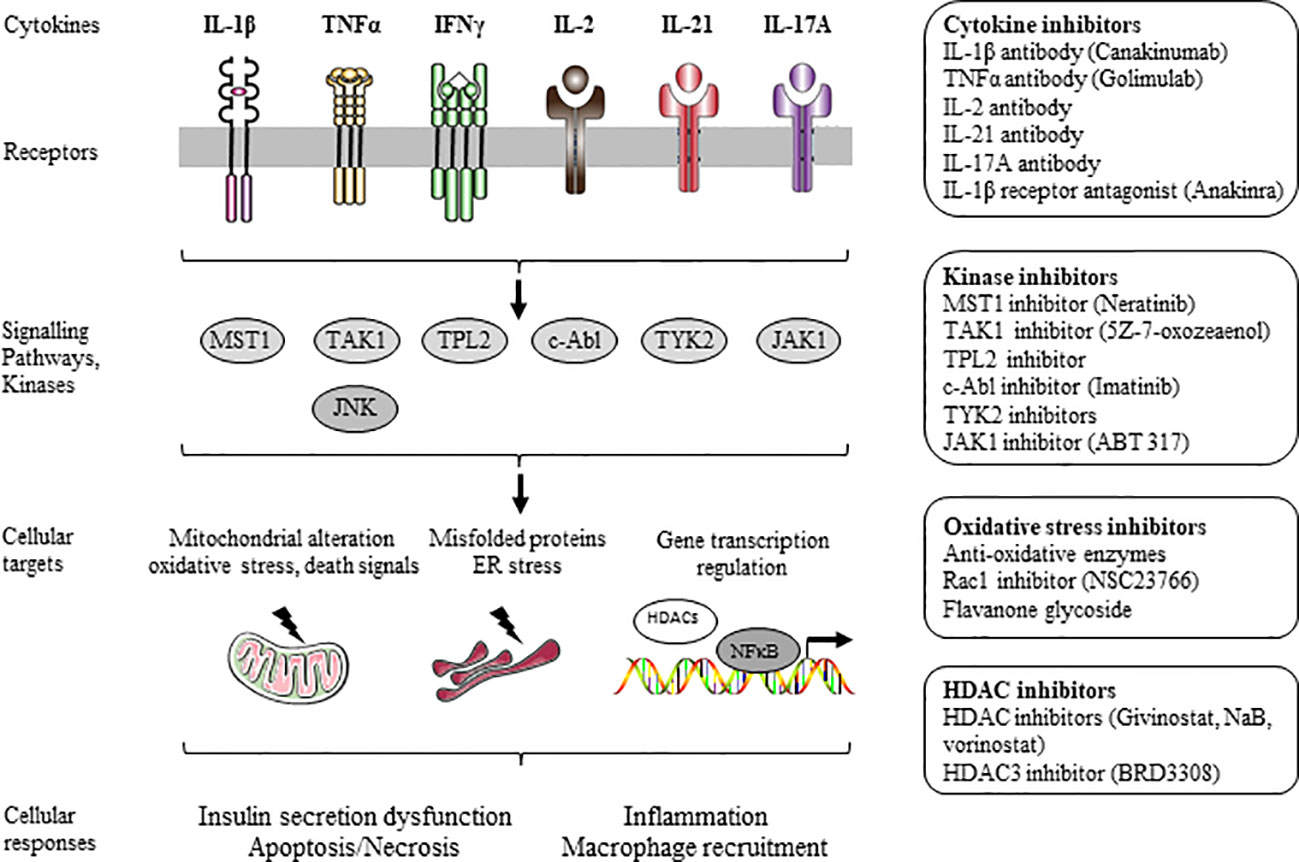
Figure 1 Inhibitors of β-cell dysfunction and death induced by inflammation.
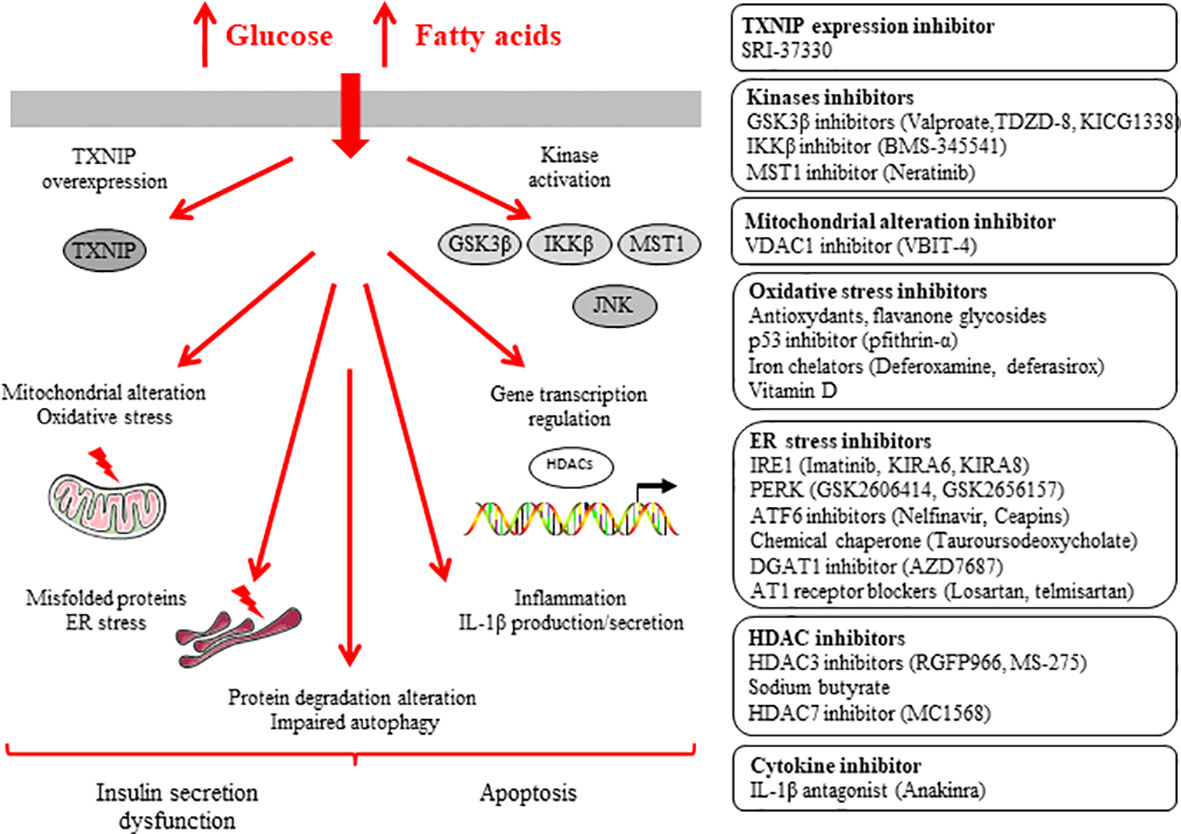
Figure 2 Inhibitors of β-cell dysfunction and death induced by glucotoxicity and glucolipotoxicity.
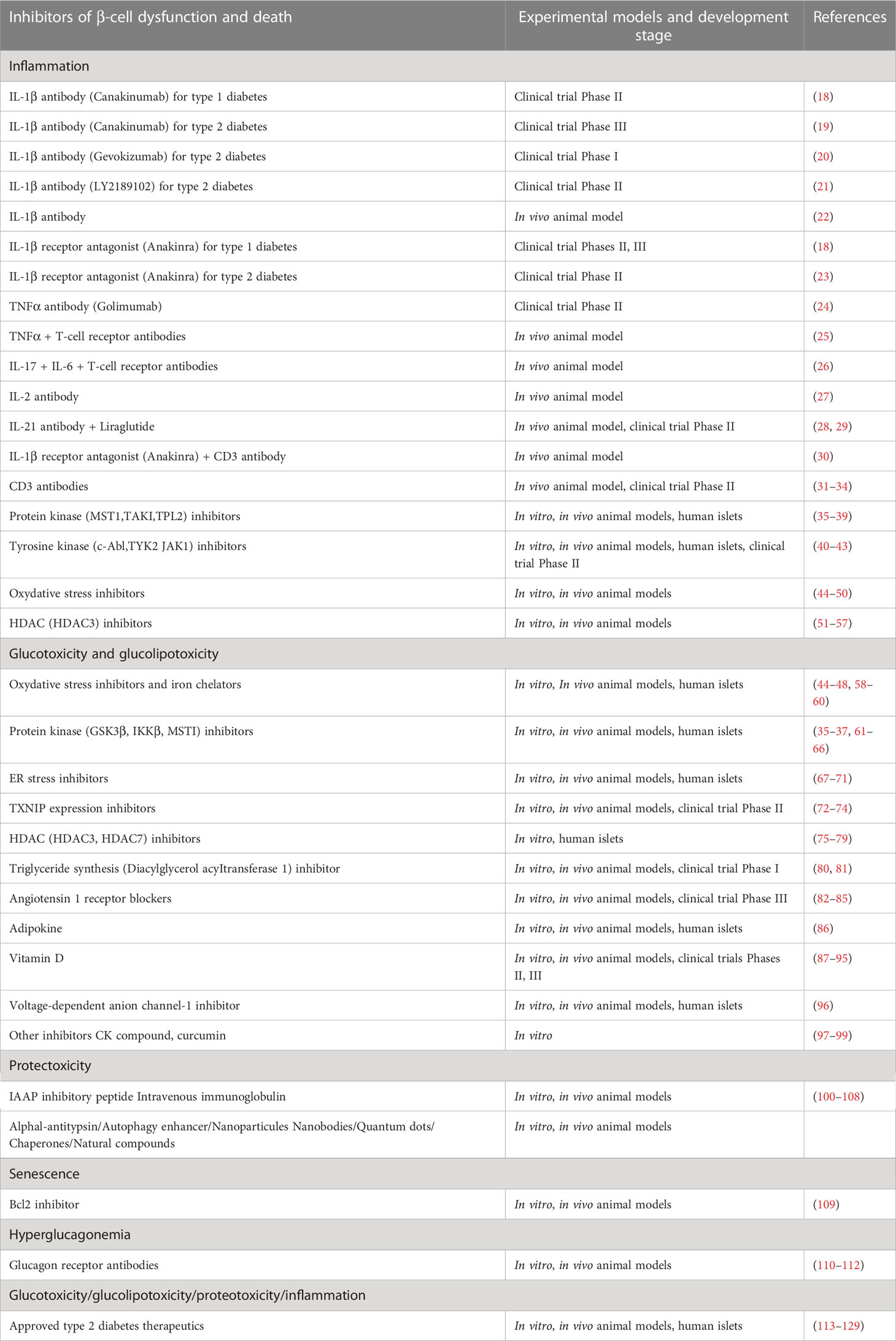
Table 1 Experimental models used to test and develop pharmacological inhibitors of β-cell dysfunction and death.
β-cell loss caused by immune-mediated processes is a key feature in the pathogenesis of T1D (1–4). Proinflammatory cytokines, including interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), and other mediators, are produced and released by immune cells invading the endocrine pancreas and promoting apoptotic death and dysfunction of β-cells (1, 2, 130). Pancreatic islet transplantation turns out to be a promising alternative therapy for some patients with T1D (131, 132). Nevertheless, clinical outcome is not always achieved because of significant loss of islet mass during and after transplantation (132, 133). Up to 80% of transplanted islets die during the post-transplantation period as a result of apoptotic death due to various mechanisms, such as the instant blood-mediated inflammatory response (IBMIR) and the release of various cytokines including IL-1β, TNF-α and IFN-γ (132–134). Chronic, systemic and low grade inflammation was also proposed to lead to β-cell dysfunction and death, and ultimately to T2D (16, 17).
β-cell dysfunction and death induced by cytokines, such as IL-1β, TNF-α, and IFN-γ, occur through stimulation of kinases, mitochondrial dysfunction and death signals, ER stress, and activation of gene transcription factors and regulators. Mitogen-activated protein kinases (MAPKs) (i.e. c-Jun N-terminal kinase (JNK) and p38 MAPK) activated by proinflammatory cytokines trigger the release of mitochondrial death signals and ER stress (1, 2, 135). The transcription factor NFκB is well described as a key mediator of cytokine-induced apoptosis (136, 137). The active transcriptional subunit p65 of NFκB is sequestered and inactivated in the cytoplasm by the inhibitor of NFκB (IκB) under resting conditions. Cytokines, such as IL-1β, induce phosphorylation-dependent degradation of IκB via activation of the IκB kinase (IKKβ), which favors the translocation of the active transcriptional subunit p65 to the nucleus and subsequent proinflammatory gene expression. Gene transcription is controlled by epigenetic mechanisms, including DNA methylation or demethylation, and/or histone acetylation or deacetylation. Enzymatic deacetylation of lysine residues within histone is regulated by histone deacetylase (HDAC) enzymes. HDACs were identified as key mediators of β-cell dysfunction and death induced by cytokines (138, 139) (Figure 1). Inhibition of these pathways using antibodies, antagonists, or small molecules inhibitors represents a key strategy to prevent cytokine-induced dysfunction and apoptotic death of β-cells for both type of diabetes. Hence, immune-modulatory strategies for T1D therapy, improvement of islet transplantation outcomes, and for T2D therapy have been extensively designed and developed (2, 16, 17, 134, 140) (Figure 1).
Over the last years, cytokine antibodies and combination immunotherapy approaches were designed and tested in their capacity to preserve the β-cell survival and function, and to reduce the progression of diabetes. The cytokine IL-1β was shown to induce β-cell death and dysfunction in both T1D and T2D, and blocking IL-1β deleterious actions using specific IL-1β antibodies was proposed. Randomised, controlled trials of blockade of IL-1β have been performed to investigate any improvement of β-cell function in recent-onset T1D using canakinumab, a human monoclonal anti-IL-1β antibody (18). Canakinumab was reported to be safe but was not effective as single immunomodulatory drug in recent-onset T1D. Authors proposed that IL-1β blockade might be more effective in combination with treatments that target adaptive immunity in organ-specific autoimmune disorders (18). Administration of a custom-made, rat-specific IL-1β monoclonal antibody to Cohen diabetes-sensitive rat, a genetic model of nutritionally induced diabetes when fed a high-sucrose/low-copper diet, counteracted β-cell dysfunction and glucose intolerance (22). Randomised, controlled trial of blockade of IL-1β using canakinumab has been also performed to investigate any improvement on cardiovascular events and T2D. Canakinumab treatment had similar effects on major cardiovascular events among patients with and without diabetes, however treatment did not reduce incident diabetes (19). Other IL-1β antibodies (i.e. gevokizumab, LY2189102) looked promising in several clinical trials for the treatment of T2D (20, 21). However, to date, no treatment for diabetes with anti-IL-1β antibodies has been approved. Despite data from preclinical and clinical studies, this reveals that an improvement in our knowledge is still necessary to develop this type of therapeutic strategy for diabetes.
TNF-α exerts toxic effects on β-cells (141). Patients with new-onset overt T1D generally have elevated serum TNF-α (142). Golimumab, a human monoclonal antibody specific for TNF-α, was tested to determine whether anti-TNF-α antibody could preserve β-cell function in youth with newly diagnosed overt T1D. In a phase 2, multicenter, placebo-controlled, double-blind, parallel-group clinical trial, golimumab treatment resulted in increased insulin production and less exogenous insulin use among children and young adults with newly diagnosed overt T1D (24). Therapy using anti-TNF-α antibody, alone or in combination with an anti-T-cell receptor (TCR) specific antibody was designed to prevent the emergence of T1D. Combination of these two antibodies increased β-cell proliferation, reduced apoptosis leading to a restoration of β-cell mass and function associated with decreased immune cell infiltration in the LEW.1AR1-iddm T1D rat model (25).
The cytokine IL-17A is well known as a key player in autoimmune processes, whereas the cytokine IL-6 is described for maintaining the inflammatory process (143, 144). Therapies with anti-IL-17A or anti-IL-6 antibody in combination with an anti-TCR antibody, or in a triple combination were tested for their capacity to reverse diabetes in the LEW.1AR1-iddm rat. The anti-TCR combination therapy with anti-IL-17 increased the β-cell proliferation and mass, while anti-IL-6 strongly reduced β-cell apoptosis and the islet immune cell infiltrate. The triple combination therapy achieved a complimentary anti-autoimmune and anti-inflammatory action resulting in sustained normoglycaemia (26).
Interleukin-2 was reported to suppress immune pathologies by preferentially expanding regulatory T cells (Tregs). Notably, administration of an human anti-IL-2 antibody (i.e. F5111.2), which promotes Treg expansion and function selectively, to non-obese diabetic mice (NOD), an animal model of T1D, reduced the progression of diabetes (27).
The cytokine IL-21 was proposed as a suitable target for an immuno-modulatory strategy, and glucagon-like peptide-1 receptor agonists were reported to be appropriate for β-cell protection in the context of T1D. Notably, an anti-IL-21 monoclonal antibody combined with a GLP-1 receptor agonist (i.e. liraglutide) reduced hyperglycemia in NOD mouse model of T1D (28). This combination of anti-IL-21 antibody and liraglutide preserved β-cell function in recently diagnosed T1D patients (29). Efficacy and safety of this combination deserve to be evaluated in a phase 3 trial programme (29).
The IL-1β receptor antagonist, anakinra, approved to treat rheumatoid arthritis, has been proposed to prevent autoimmune destruction of β-cells in patients with T1D. Randomised, controlled trials of blockade of IL-1β have been performed using anakinra (18). Anakinra was reported to be not effective as single immunomodulatory drug in recent-onset T1D (18). Administration of anakinra was found to exert antidiabetic effects in combination with anti-CD3 antibodies in NOD mice, suggesting that anakinra/anti-CD3 antibody combination therapy may be a potent treatment strategy in patients with T1D (30).
IL-1β receptor antagonist did show promise in clinical trials for the treatment of T2D (23). Treatment with anakinra led to improved glycemia by increasing β-cell function (23). Moreover, exposure to high glucose concentrations induces IL-1β production and secretion from β-cells leading to apoptosis which was prevented by IL-1β receptor antagonist. However, it should be noted that blocking IL-1β signalling by antagonist has shown modest improvement in β-cell function, although the long-term efficacy remains to be determined. It should also be noted that there are still no approved treatment for diabetes that targets innate immune mediators, despite large preclinical and clinical trial data and evidences demonstrating that targeting inflammatory pathways can prevent cardiovascular disease and complications in patients with diabetes (for review see 16, 17).
The signalling hexamer CD3 is a T-cell co-receptor which is described to be essential for the activity of the T-cell receptor. Anti-CD3 monoclonal antibodies bind to CD3 and induce tolerance of Tregs in autoimmune disease. The use of anti-CD3 monoclonal antibodies targeting the autoimmune destruction of β-cells in T1D was evaluated in clinical trials, and has been proposed as potential treatment providing key benefits for patients with T1D (for review see 31, 32). Oral anti CD3-specific monoclonal antibody treatment was reported to induce changes in effector and regulatory T cell compartments, and to reduce incidence of diabetes in NOD mice (33). Fingolimod (FTY720), a sphingosine-1-phosphate receptor modulator, is described to prevent islet damage and to preserve β-cell mass by inhibiting apoptosis and increasing β-cell survival. Combination therapy of an antibody directed against the T cell receptor of the TCR/CD3 complex with fingolimod increased the β-cell mass, blocked islet infiltration, and was effective to reverse T1D in the LEW.1AR1-iddm rat (34).
The serine/threonine kinase mammalian sterile 20-like kinase 1 (MST1) was found to mediate β-cell apoptotic death and dysfunction induced by cytokines (35, 36). MST1 is upregulated in β-cells exposed to cytokines, high glucose concentrations, activates downstream JNK kinases, apoptotic pathways, and impairs insulin secretion through proteasomal degradation of the β-cell transcription factor pancreatic and duodenal homeobox 1 (PDX1) which is critical for insulin production (35). MST1 deficiency restored normoglycemia and β-cell function and prevented the development of diabetes (35). Neratinib, an approved drug targeting HER2/EGFR dual kinases, was identified as a potent inhibitor of MST1, and shown to protect β-cells against the deleterious effects of autoimmune process in T1D (37).
The serine/threonine kinase transforming growth factor-β activated kinase-1 (TAK1, or MAP3kinase 7), a member of the mitogen-activated protein kinase kinase kinase (MAP3K) family, is well described to activate JNK and NFκB signalling pathways, and to be essential in innate and adaptive immune responses. Administration of 5Z-7-oxozeaenol, which inhibits both the kinase and ATPase activity of TAK1, decreased the incidence and delayed the onset of diabetes in NOD mice, reduced insulitis, preserved islet function, and inhibited downstream JNK and NF-κB signalling pathways in pancreatic tissues (38).
Multiple cytokines are likely present simultaneously within islets of T1D patients with powerful synergistic effects (1, 2, 130). IBMIR and the release of various cytokines including IL-1β, TNF-α and IFN-γ during the post-transplantation period destroy the transplanted islets (134). To protect β-cells in T1D and in pancreatic islet transplantation, the identification of a common regulator of β-cell apoptotic death induced by several pro-inflammatory would emerge as a key therapeutic target. Our group has discovered conditions in which targeting the tumor progression locus 2 kinase (TPL2, or MAP3kinase 8), produces potent anti-diabetic effects by preventing dysfunction and apoptosis of β-cells (39). TPL2 is a serine/threonine kinase member of the MAP3K family that stimulates downstream signalling kinases such as JNK and p38 MAPK, and is activated by IL-1β, TNF-α, several chemokines, and toll-like receptor (TLR) ligands. We reported that TPL2 expression was upregulated in β-cells following chronic exposure to cytokines. Pharmacological inactivation of TPL2 using a specific small molecule inhibitor prevented JNK and p38 kinase activation, apoptotic death, and dysfunction of β-cells and human islets induced by IL-1β and mixture of proinflammatory cytokines (IL-1β + TNFα + IFNγ) (39).
Tyrosine-kinase inhibitors have been proposed to prevent T1D progression (40–43). These tyrosine-kinase inhibitors include the approved small molecule kinase inhibitor imatinib which is used to treat chronic leukaemia. Although imatinib inhibits a number of tyrosine kinases, it is quite selective toward the tyrosine kinase c-Abl that is present in various cancers. Imatinib inhibits the c-Abl protein by binding to the ATP pocket in the active site, thus preventing downstream phosphorylation of target protein. c-Abl controls many downstream signalling pathways that are implicated in cellular proliferation, cellular motility, and apoptosis. Treatment of streptozotocin-injected mice with imatinib counteracted diabetes (40). Imatinib decreased the apoptotic death of human islets exposed to cytokines. NFκB signalling was proposed to mediate the antiapoptotic action of imatinib (40). Safety and efficacy of imatinib in preserving β-cell function in patients with recent-onset T1D were evaluated. Importantly, imatinib treatment preserved β-cell function in adults with recent-onset T1D (41). Members of the mammalian Janus kinases (JAK) family (i.e. JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2)) have been shown to mediate inflammation in β-cells. Pharmacological inhibitors of TYK2 were developed using structure-based drug design. Inhibitors were found to bind to the JH2 pseudokinase domain of TYK2 and to act as allosteric inhibitors of the kinase activity. The JH2-directed allosteric inhibition provides the high specificity for TYK2 over other JAK family members. These TYK2 inhibitors were reported to protect human β-cells against the deleterious effects of IFNγ (42). A JAK1-selective inhibitor, ABT 317, was found to reduce IL-21, IL-2, IL-15 and IL-7 signalling in T cells, and IFN-γ signalling in β cells. Notably, diabetes was reversed in mice treated with ABT 317 (43).
Proinflammatory cytokines induce the formation of ROS. β-cells are susceptible to formation of ROS because of low expression of anti-oxidative enzymes such as catalase, superoxide dismutase and glutathione peroxidase. Increased ROS formation induces β-cell damages (for review see 44–48). As example, excessive production of ROS by the phagocyte-like NADPH oxidase2 (Nox2) drives β-cells toward oxidative damages. Rac1, a small G-protein, is one of the members of Nox2 holoenzyme. NSC23766, a known inhibitor of Rac1, significantly attenuated cytokine-induced Nox2 activation and ROS formation in β-cells. Administration of NSC23766 to diabetic NOD mice suppressed Rac1 expression and activity, the endoplasmic reticulum stress, and prevented the development of diabetes (49).
The potential beneficial effects of hesperidin in T1D, a flavanone glycoside present in all citrus fruits, were investigated. Hesperidin exerted beneficial effects on β-cells reducing oxidative stress by increasing antioxidant SOD and GPx activities, decreasing nitrotyrosine and malondialdehyde levels, upregulating of anti-apoptotic Bcl-xL, downregulating of pro-apoptotic Bax and cleaved caspase-3. Treatment of streptozotocin diabetic rats with hesperidin reduced hyperglycemia, and increased serum and pancreatic insulin levels. In addition, concentrations of TNF-α and expressions of ER stress makers GRP78 and CHOP proteins were found diminished in the pancreas following the hesperidin treatment (50).
HDACs were proposed as key targets to develop therapeutics for T1D and T2D (for review see 138). Especially, HDACs 1 and 3 were identified as key mediators of β-cell cytotoxicity and death (138, 139). Specific knock-down of HDAC3 reduced binding of the NFκB subunit p65 to DNA and consequently reduced inflammatory gene transcription (139). HDAC inhibitors are natural products or synthetically produced, include pan-HDAC inhibitors and class-selective or isoform-selective inhibitors. The mechanisms of action of HDAC inhibitors are still unclear. However, these inhibitors are emerging therapeutic agents that have been clinically validated in cancer patients with hematologic malignancies.
Several HDAC inhibitors were reported to reduce β-cell apoptosis induced by proinflammatory cytokines (51–53). Vorinostat is the most advanced pan-HDAC inhibitor and was approved for the treatment of patients with hematologic malignancies including cutaneous T-cell lymphoma. Givinostat prevented β-cell apoptosis and expression of the β-cell toxic inflammatory molecule IL-1β (53). Sodium butyrate (NaB) is a short chain fatty acid having HDAC inhibition activity. NaB treatment decreased plasma glucose, improved plasma insulin level and glucose homeostasis through HDAC inhibition in streptozotocin injected-diabetic animal. NaB treatment was found to improve β-cell proliferation and function, and to reduce β-cell apoptosis by the modulation of p38/ERK MAPK and apoptotic pathway (54). Administration of BRD3308, an inhibitor of HDAC3, to NOD mice preserved β-cell function and survival, enhanced β-cell proliferation, decreased infiltration of mononuclear cells in islets, and reduced the emergence of diabetes (55).
Combination of the orally active form of givinostat with humanised CD3 antibodies (otelixizumab) synergised to prevent islet inflammation, to improve β-cell function and survival reducing diabetes in NOD mice (56). Combination of HDAC inhibitor vorinostat with the dipeptidyl peptidase-4 (DPP-4) inhibitor MK-626 increased β-cell mass without preventing diabetes. Authors proposed the combination of vorinostat and MK-626 as a beneficial adjunctive therapy in clinical trials for T1D prevention or remission (57).
T2D is characterized by a gradual loss of β-cell function and mass (5–10). It is now well described that nutrient-induced metabolic stresses, such as chronic hyperglycemia, are crucial factors underlying deterioration of β-cell function and mass. We and others have shown that chronic exposure of β-cells to hyperglycemia deteriorates insulin secretion and induces apoptosis (11–14, 145, 146). Studies in vitro provided evidences for synergistic adverse effects of elevated glucose and saturated fatty acid high concentrations on the function and survival of β-cells (for review see 14, 147, 148). High levels of glucose and fatty acids such as palmitate (alone or in combination) induce insulin secretion dysfunction and apoptosis in β-cells (i.e. glucotoxicity, lipotoxicity and glucolipotoxicity) through mitochondrial alteration, ROS formation, stimulation of kinases, ER stress, activation of HDACs, inflammation, alteration of protein degradation pathways and impaired autophagy (14, 15, 147–149) (Figure 2). Inhibitions of these pathways are key strategies to prevent β-cell dysfunction and apoptosis.
Chronic exposure of β-cells to high glucose leads to increased ROS formation (for review see 44–48). The β-oxidation of saturated non-esterified fatty acids in peroxisomes and mitochondria generate hydrogen peroxide. Several antioxidants were reported to protect β-cells against ROS-induced damages (44–48). As an example, a major flavanone glycoside in citrus species, naringin, was found to decrease ROS accumulation, and to exert protective effects on islet dysfunction and diabetes by amelioration of hyperglycemia (150).
The tumor suppressor p53 protein is activated by oxidative stress to induce mitochondrial dysfunction and has been proposed to play a role in T2D development. p53 deficiency protected against diabetes in streptozotocin-induced diabetes and db/db mouse. Moreover, treatment of db/db mouse with pfithrin-α, a reversible inhibitor of p53-mediated apoptosis and p53-dependent gene transcription, improved mitochondrial dysfunction and glucose intolerance (58).
Iron is a redox-reactive metal which can catalyse ROS formation. Increased expression of divalent metal transporter 1 correlates with increased β-cell iron content and ROS formation. Notably, knockout mice for the divalent metal transporter 1 were found to be protected against streptozotocin and high-fat diet-induced glucose intolerance (59). Iron chelators deferoxamine and deferasirox are approved drugs for the treatment of acute iron toxicity and transfusion-induced iron overload. Deferoxamine and deferasirox were shown to reduce ROS formation and β-cell apoptosis. Notably, increased levels of the iron-carrying plasma protein transferrin was reported to be associated with a risk of developing both type of diabetes (60).
Glycogen synthase kinase-3 (GSK3) is a ubiquitously expressed, highly conserved serine/threonine protein kinase. GSK3 is involved in various signalling pathways regulating glycogen metabolism, cell development, gene transcription, protein translation, cytoskeletal organization, cell cycle regulation, proliferation, and apoptosis. The role of GSK3β was particularly studied in pathways activated in β-cells and for the treatment of diabetes. Studies reported the mood stabilizer valproate (valproic acid) as an inhibitor of GSK-3β activity. In INS-1 β-cell line, valproate protected cells to palmitate-induced apoptosis (61). An inhibitor of GSK-3β, TDZD-8, was shown to exert same anti-apoptotic effect (61). Administration of KICG1338, another inhibitor of GSK-3β, preserved β-cell function in vivo (62). Using high-sensitivity mass spectrometry-based proteomics analysis, Sacco and colleagues confirmed that GSK3β is a key kinase involved in signalling network controlling the β-cell-specific transcription factor PDX1. GSK3β was found to mediate the insulin secretion dysfunction induced by chronic hyperglycemia. Pharmacological inhibition of GSK3β restored the ability of β-cells to secrete insuline in response to glucose (63). Treatment of Goto-Kakizaki rats, a spontaneous nonobese model that develops T2D, with infra-therapeutic doses of lithium, a widely used inhibitor of GSK3, reduced the expression of pro-inflammatory cytokines in the pancretic islets, partially restored the glucose-induced insulin secretion, and reduced the development of diabetes (64). GSK3β was further found to mediate the pro-apoptotic effects of glucocorticoids in β-cells (65).
A key role for IKKβ in β-cell dysfunction induced by fatty acids was demonstrated (66). An inhibitor of IKKβ, BMS-345541, and deletion of IKKβ prevented β-cell dysfunction in vitro and in vivo (66).
The serine/threonine kinase MST1 was found to be a key regulator of β-cell apoptotic death and dysfunction induced not only by cytokines but also by chronic hyperglycemia (35, 36). Administration of neratinib, a potent inhibitor of MST1, improved β-cell survival and preserved β-cell mass and function in db/db mouse model of T2D (37).
The ER stress has been reported to play an important role in β-cell dysfunction and death in the context of T2D (for review see 67–69). Restoring ER homeostasis, enhancing ER-associated degradation of misfolded protein, and boosting chaperoning activity are proposed as therapeutic approaches for the treatment of diabetes (67–70). Over the last years, reviews exhaustively listed inhibitor molecules existing against ER stress and tested to protect β-cells against diabetogenic conditions. Several inhibitors exist for each of the major molecules involved in ER stress such as IRE1, PERK and ATF6. As examples, imatinib, kinase-inhibiting RNase-attenuators 6 (KIRA6) and KIRA8 are three IRE1 inhibitors. GSK2606414 and GSK2656157 are two PERK inhibitors. Nelfinavir and ceapins are two ATF6 inhibitors which exert protective effects on β-cell function and mass against ER stress (for review see 67–70).
Tauroursodeoxycholic acid is a taurine conjugate of ursodeoxycholic acid, naturally occurring hydrophilic bile acid, and approved for primary biliary cholangitis treatment. Tauroursodeoxycholic acid was reported to exert therapeutic benefits in various models of diseases such as diabetes, obesity, and neurodegenerative diseases, due to its cytoprotective effect. The mechanisms underlying this cytoprotective effect have been attributed to reduction of ER stress and stabilization of the unfolded protein response, which contributed to naming tauroursodeoxycholic acid as a chemical chaperone. This chemical chaperone decreased palmitate-induced apoptosis in β-cells by reducing the expression of CHOP and ATF4 involved in ER stress (71).
Thioredoxin-interacting protein (TXNIP), a member of the alpha arrestin protein family, has emerged as a major factor regulating β-cell dysfunction and death in the pathogenesis of T2D and T1D (72–74, 151–154). Thioredoxin is a thiol-oxidoreductase and a major regulator of cellular redox signalling which protects cells from oxidative stress. TXNIP inhibits the antioxidative function of thioredoxin resulting in ROS accumulation. TXNIP also regulates the mitochondrial death pathway and ER stress. TXNIP expression was found to be up-regulated in diabetogenic conditions. Exposure to high glucose upregulates TXNIP expression in β-cells (152). TXNIP plays a key role in mediating the detrimental effects exerted by glucotoxicity. TXNIP promotes β-cell apoptosis, inhibits insulin production and β-cell function. Whole body TXNIP-deficient and β-cell-specific TXNIP knockout mice have decreased β-cell apoptosis, increased β-cell mass, elevated insulin levels, and are protected from diabetes (for review see 153). Exposure to inflammatory cytokines such as IFN-γ and cytokine mixture (i.e. IL-1β + TNF-α + IFN-γ) was also found to increase TXNIP expression in β-cells by distinct mechanisms (154).
Following a high-throughput screen, an orally bioavailable, non-toxic small molecule, SRI-37330, was found to effective in inhibiting TXNIP expression. In human islets, SRI-37330 inhibited TXNIP expression. SRI-37330 treatment rescued mice from obesity (db/db) - and streptozotocin-induced diabetes (72). In addition, SRI-37330 treatment inhibited glucagon secretion and function, reduced hepatic glucose production, and reversed hepatic steatosis (72).
The approved antihypertensive calcium-channel blocker, verapamil, was reported to promote β-cell survival by decreasing the expression of TXNIP. Verapamil is believed to decrease β-cell TXNIP expression by reducing cytosolic calcium, which in turn controls calcineurin/calcium-dependent protein phosphatase 2B signalling and leads to increased phosphorylation and nuclear exclusion of carbohydrate response element-binding protein (ChREBP) and results in decreased ChREBP mediated TXNIP transcription. Oral administration of verapamil reduced TXNIP expression and β-cell apoptosis, enhanced endogenous insulin levels, protected BTBR ob/ob mice as a model of T2D and mice from streptozotocin-induced diabetes (73). Retrospective studies suggest that verapamil use might be associated with a lower incidence of T2D in humans (for review see 153). A randomized double-blind placebo-controlled phase 2 clinical trial was further designed and conducted to assess the efficacy and safety of oral verapamil added to a standard insulin regimen in adult individuals with recent-onset T1D. Notably, addition of once-daily oral verapamil was shown to be a safe and effective strategy to promote β-cell function and to reduce insulin requirements in adult individuals with recent-onset T1D (74).
The potential therapeutic effect of HDAC3 pharmacologic inhibition on T2D has been particularly investigated. RGFP966 (HDAC3 inhibitor) enhanced insulin secretion and synthesis in normal and diabetic mice islets and reduced partially palmitate-induced apoptosis in NIT-1 cells (75). We reported that MS-275 (HDAC3 inhibitor) prevented β-cell death induced by palmitate in human pancreatic islets (76). MS-275 was further found to potentiate insulin secretion (77). Other HDAC inhibitor such as sodium butyrate prevented β-cell dysfunction and death in diabetic rat and mice (78). Notably, the HDAC7 inhibitor MC1568 was reported to protect β-cells from dysfunction and death, suggesting that specific inhibitors for HDAC7 may be useful for T2D treatment (79).
Diacylglycerol acyltransferase 1 (DGAT1), using for the triglyceride synthesis, has been reported as a potential therapeutic target. DGAT1 inhibitor improved palmitate-induced apoptosis in β-cells and mice pancreatic islets. DGAT1 inhibitor was further found to protect β-cells against inflammation and endoplasmic reticulum stress. DGAT1 inhibitor treatment in db/db mice decreased hyperglycemia, triglyceride levels, and improved glucose tolerance (80).
Inhibition of DGAT1 was evaluated as a potential treatment modality for patients with obesity and T2D. A randomized, phase 1 study in overweight or obese men explored the effects and tolerability of AZD7687, a reversible and selective DGAT1 inhibitor. Dose-dependent reductions in postprandial serum triacylglycerol were demonstrated with AZD7687 treatment. However, gastrointestinal side effects were observed and several participants discontinued treatment due to diarrhea making the usefulness of DGAT1 inhibition as a novel treatment for diabetes and obesity questionable (81).
In a large clinical prospective trial, inhibition of the renin-angiotensin system was found to delay the onset of T2D in high-risk individuals (82). Losartan, a selective angiotensin 1 receptor (AT1R) blocker, was reported to protect human islets against glucotoxicity by inhibiting increased ER stress markers such as GRP78, sXBP1, ATF4 and Grp78. Losartan treatment improved insulin secretion (83). Telmisartan, another AT1R blocker, is a common drug used for hypertension treatment. Telmisartan was found to exert protective effects against high glucose/high lipid-induced β-cell apoptosis and to improve the insulin secretion by inhibiting oxidative and ER stress (84). Treatment of db/db mice with combination of telmisartan and the DPP-4 inhibitor linagliptin preserved islet cell function and morphology via reduction of oxidative stress (85).
The adipokine adipsin/complement factor D controls the alternative complement pathway and generation of complement component C3a, and was shown to increase insulin secretion. Higher concentrations of circulating adipsin were associated with a significantly lower risk of developing future diabetes among middle-aged adults. Administration of adipsin in db/db mice was found to improve hyperglycemia, to increase insulin levels, and to preserve β-cell function by blocking dedifferentiation and death. Adipsin/C3a was further found to decrease the phosphatase Dusp26 which negatively controls the expression of core β-cell identity genes and sensitizes cell to apoptotic death. Pharmacological inhibition of Dusp26 improved hyperglycemia in db/db mice and protected human islet cells from apoptotic death (86).
Impaired β-cell function has been reported with low blood 25-hydroxyvitamin D levels (87). Epidemiological and human genetic studies revealed a link between vitamin D and the vitamin D receptor (VDR) to both T1D and T2D (88). The VDR can bind to bromodomain-containing protein 9 (BRD9 protein). Wei Z and colleagues discovered that the pharmacological inhibition of BRD9 promotes the activation of a VDR-dependent transcriptional program that underlies β-cell survival. Activation of the VDR signalling by a synthetic ligand in combination with the BRD9 inhibitor partially restored β-cell function and glucose homeostasis in db/db mice and in mice treated with low doses of streptozotocin (89).
A low blood 25-hydroxyvitamin D level has emerged as a possible risk factor for T2D, and vitamin D supplementation has been proposed as a potential intervention to reduce diabetes risk (90, 91). In a multicenter, randomized, placebo-controlled trial involving persons at high risk for T2D not selected for vitamin D insufficiency, vitamin D3 supplementation did not result in a significantly lower risk of diabetes (92). In comparison, the Tromsø Vitamin D and T2DM Trial, which randomly assigned adults with prediabetes to vitamin D3 or placebo, the risk of diabetes was numerically lower in the vitamin D group than in the placebo group, but the difference was not significant (93). In other clinical trials which randomly assigned adults with prediabetes to an active form of vitamin D analogue (eldecalcitol) or placebo, the risk of diabetes was also lower in the vitamin D group than in the placebo group, but the difference was again not significant (94, 95).
The ATP-conducting mitochondrial outer membrane voltage-dependent anion channel-1 (VDAC1) was found to be upregulated in pancreatic islets of T2D organ donors. VDAC1 overexpression causes its mistargeting to the plasma membrane of the β-cells with loss of the metabolic coupling factor ATP. Through direct inhibition of VDAC1 conductance, metformin and specific VDAC1 inhibitors (VBIT-4) and antibodies were reported to restore the impaired generation of ATP and glucose-stimulated insulin secretion in T2D pancreatic islets. Treatment of db/db mice with VDAC1 inhibitor VBIT-4 prevented hyperglycemia, and maintained β-cell function and glucose tolerance (96).
Compound K (CK) has been described to possess anti-diabetic properties, but the mechanism of action is still not clear. Treatment of diabetic mice with CK decreased fasting plasma glucose, triacylglycerol, total cholesterol, elevated plasma insulin levels and improved glucose tolerance. CK treatment inhibited β-cell apoptosis and caspase-3 activity with a decrease in JNK activation (97).
The phytochemical curcumin decreased NAPDH oxidase and apoptotic factor expression induced by high glucose and palmitate involved in pancreatic islet death (98). Curcumin was further reported to protect RIN-m5F β-cells against apoptotic damages induced by oxidative stress (99). The mechanism of action of curcumin in β-cells is still not fully elucidated.
Amyloid deposition derived from amyloid polypeptide (IAPP), a protein which is synthesized and secreted with insulin by β-cells, are frequently found in pancreatic islets of T2D patients (155, 156). IAPP forms oligomers and subsequently insoluble fibrils in species at risk of developing diabetes (157–159). Supporting a role of IAPP in the development of T2D in humans, a rare missense mutation in the IAPP gene (S20G) that increases its amyloidogenicity has been reported to be associated with β-cell failure and the development of T2D (160, 161). In rodents, IAPP is well known to be nonamyloidogenic. Overexpression of human IAPP (hIAPP) in rodent models promotes amyloid deposits, β-cell dysfunction and apoptosis, leading to reduced β-cell functional mass and hyperglycemia (162, 163). IAPP induces β-cell dysfunction and apoptosis by promoting dysruption of mitochondrial network dynamics, oxidative stress, ER stress, defects in pathways regulating protein degradation, impairment of autophagy and inflammation (for review see 15, 155, 156, 164). The effects of drugs currently used for the treatment of T2D, hIAPP mimetics and peptides, small organic molecules, natural compounds, nanoparticles, nanobodies, quantum dots, metals and metal complexes, and chaperones found to potentially inhibit and/or reverse hIAPP aggregation have been extensively reviewed (100). As examples, we can cite an IAPP inhibitory peptide, an intravenous immunoglobulin, an alpha1-antitrypsin, and an autophagy enhancer.
Self-assembly into oligomers and fibrils during the process of aggregation by hIAPP can lead to β-cell failure. Some critical regions of hIAPP might contribute to the aggregation, and finding effective molecules, especially short-peptide inhibitors that bind to these regions and disrupt the aggregation of hIAPP are of interest. The IAPP inhibitory peptide, D-ANFLVH, was shown to prevent islet amyloid accumulation in cultured human islets. This D-ANFLVH peptide was administered into the hIAPP overexpressing transgenic mouse model, and found to be a potent inhibitor of islet amyloid deposition, resulting in decreased islet cell apoptosis and preservation of β-cell area leading to improved glucose tolerance (101).
Intravenous immunoglobulin (IVIg) is described to be an efficient anti-inflammatory and immunomodulatory agent for the treatment of several autoimmune or inflammatory neurological diseases. IVIg treatment in hIAPP transgenic mouse model significantly improved glucose control and insulin sensitivity, prevented β-cell apoptosis by lowering toxic IAPP oligomer levels, attenuating islet inflammation and activating autophagy (102).
Alpha1-antitrypsin (AAT) is a circulating protease inhibitor with anti-inflammatory properties. AAT treatment in mice overexpressing hIAPP in β-cells improved glucose tolerance and restored the insulin secretory response to glucose. AAT prevented the formation of amyloid deposits and apoptosis induced by high glucose concentrations. Notably, AAT protected β-cells against the cytotoxic effects of conditioned medium from hIAPP-treated macrophages. AAT also prevented the cytotoxic effects of proinflammatory cytokines on β-cells, and protected β-cells from the cytotoxic actions of hIAPP mediated by macrophages (103).
Autophagy is crucial for clearance of hIAPP oligomer, suggesting that an autophagy enhancer could be a therapeutic modality against human diabetes with amyloid accumulation. A recent identified autophagy enhancer (MSL-7) was found to reduce hIAPP oligomer accumulation in human induced pluripotent stem cell-derived β-cells and oligomer-mediated apoptosis of β-cells. MSL-7 administration improved glucose tolerance and β-cell function of hIAPP transgenic mice on high-fat diet, associated with reduced hIAPP oligomer/amyloid accumulation and β-cell apoptosis (104).
Studies have showed that selenium-containing phycocyanin inhibited the fibrillation of hIAPP to form nanoscale particles (105, 106). Anti-amyloidogenic potential of azadirachtin, a metabolite isolated of medicinal plant, acts on hIAPP in β-cells. Azadirachtin reduced oxidative and ER stresses with an improvement of glucose-stimulated insulin secretion (107). Chitosan oligosaccharides (COS) have been reported to exhibit a potential antidiabetic effect. COS have the capacity to diassembled preformed hIAPP and inhibited its aggregation. COS reduced IAPP-induced apoptosis in β-cells (108).
A subset population of β-cells was observed and reported to acquire a senescence-associated secretory phenotype during T1D development in diabetic NOD mice and humans. Senescent β-cells displayed upregulation of the pro-survival mediator Bcl-2. Notably, treatment of NOD mice with a Bcl-2 inhibitor selectively eliminated these cells without altering the abundance of the immune cell types involved in the disease. Elimination of senescent β-cells prevented immune-mediated β-cell destruction and diabetes, suggesting that clearance of senescent β-cells could be an innovative therapeutic approach for T1D (109).
Glucagon is a peptide hormone secreted from the α-cells of the islets of Langerhans in response to hypoglycemia. Glucagon exerts its intracellular effects through binding to its specific receptor (Glucagon receptor, GCGR) that spans the plasma membrane (for review see 165, 166). The GCGR belongs to the class II (or B) secretin/glucagon/vasoactive intestinal peptide superfamily of 7-transmembrane receptor guanine nucleotide-binding protein (G-protein) coupled receptors (GPCRs) (165, 166). The GCGR is expressed in liver, adipose tissues, kidney, gastrointestinal tract, brain, heart, and β-cells (165, 166). Glucagon increases glucose production and lipid oxidation in the liver, energy expenditure, glomerular filtration in the kidney, reduces motility in the gastrointestinal tract and food intake (165). Hyperglucagonemia is present in all forms of diabetes. Despite controversy regarding the role of glucagon in metabolic disorders associated with diabetes, the recognized importance of hyperglucagonemia in the pathophysiology of T1D and T2D fostered the development of therapeutic strategies aimed at glucagon action reduction (165, 167). Increasing studies propose that blocking glucagon and/or GCGR can reduce hyperglycemia (for review see 165, 167).
Anti-glucagon receptor antibody treatment has been proposed and tested to reverse T1D. Blockade glucagon action using a monoclonal antibody of the GCGR (Ab-4) was found to improve glycemia (110). Notably, treatment with this GCGR monoclonal antibody promoted β-cell survival, and enhanced formation of functional β-cell mass and production of insulin-positive cells from α-cell precursors in NOD mice (110). An antidiabetic effect and a β-cell regenerative action using GCGR antibody were recently confirmed (111). When combined with the immune modulator anti-CD3 teplizumab, GCGR antibody efficiently increased β-cell mass and reversed diabetes in NOD mice (111). GCGR antagonism ameliorated hyperglycemia and promoted β-cell regeneration in db/db mice and high-fat diet and streptozotocin-induced mice with T2D (112). The liver-derived fibroblast growth factor 21 (FGF21) was shown to be involved in the GCGR antagonism-induced β-cell regeneration (112).
Several drugs currently used for the treatment of T2D have been reported to protect β-cell function and survival against glucotoxicity, lipotoxicity, proteotoxicity, and inflammation in preclinical studies (113–129).
Metformin has been shown to protect rat and human islets from oxidative and ER stress, metabolic dysfunction, apoptosis induced by glucotoxicity and lipotoxicity (113–116). Metformin was further reported to promote INS-1 β-cell proliferation and survival by activating AMPK/SIRT1/PGC-1α signalling pathway and inducing autophagy in high-glucose environment (117). Metformin was found to restore the impaired generation of ATP, through inhibition of VDAC1 conductance, and glucose-stimulated insulin secretion in T2D pancreatic islets (96).
Thiazolidinediones are small molecules that activate the nuclear receptor PPARγ to increase insulin sensitivity. The potential direct effects of thiazolidinediones on β-cells have been controversial. However, rosiglitazone or pioglitazone were shown to protect β-cells against proinflammatory cytokines (118). Pioglitazone treatment in db/db mice protected β-cell function (119). A new thiazolidinedione, lobeglitazone, was found to exert beneficial effects on β-cell function and survival in db/db mice (120). Thiazolidinedione treatment may be a promising approach to preserve β-cell mass and function by inhibiting islet amyloid formation and decreasing ER stress induced by hIAPP. Rosiglitazone was recently reported to improve the survival of INS-1E cells exposed to hIAPP. Rosiglitazone inhibited hIAPP fibrillation and decreased hIAPP-induced expression of C/EBP homologous protein (CHOP) (121).
GLP-1 and GIP are the two primary incretin hormones secreted from the intestine on ingestion of glucose or nutrients to stimulate insulin secretion from β-cells. GLP-1 and GIP exert their actions by binding to the GLP-1 receptor (GLP-1R) and GIP receptor (GIPR), respectively, two receptors that belong to the GPCR family (for review see 122–124). GLP-1 receptor agonists are successfully used for the treatment of T2D (122–124). Using β-cell lines, mouse, rat, human islets, and animal models of T2D, GLP-1 receptor agonists were widely reported to improve β-cell function, to protect β-cells against glucotoxicity, lipotoxicity, and inflammation (for review see 15, 122–124).
DPP-4 inhibitors work by blocking the action of DPP-4, an enzyme that destroys the incretins GLP-1 and GIP. Studies on β-cell lines and human islets have shown that DPP-4 inhibitors protected β-cells against cytokine-induced apoptosis and increased glucose-stimulating insulin secretion (125).
SGLT2 are a class of prescribed medicines that are approved for use with diet and exercise to lower blood sugar in adults with T2D. SGLT2 inhibitors lower blood sugar by causing the kidneys to remove sugar from the body through the urine. Dapagliflozin treatment was shown to reduce hyperglycemia in db/db mice associated with an increase of β-cell mass (126). Dapagliflozin was also found to be effective to protect β-cell survival in db/db mice (127). Empagliflozin has been studied in Zucker diabetic fatty rat model and was found to preserve β-cell function and mass (128, 129).
The European Medicine Agency and the Food Drug Agency recall that there is an unmet and urgent medical need for therapies that act on the pathogenesis of the disease, rather than on the symptoms, and prevent dysfunction and death of β-cells. Treatment strategies focus on insulin replacement therapy (T1D) (4), reducing insulin resistance and preserving insulin secretion (T2D) (7, 8). None of the approved anti-diabetic treatments are effective in protecting functional β-cell mass against diabetogenic stressors. The research has made interesting progress in identifying multiple types of immune cells, soluble factors, and signalling pathways that destroy insulin-producing β-cells. This knowledge produced in recent years on the pathogenesis of the disease has made it possible to propose the development of innovative therapies to prevent and modify T1D and T2D.
As described in this review, a large number of antibodies, receptor antagonists, natural compounds or small molecules inhibitors with diverse chemical structures have been validated both in vitro and in vivo experimental models to potentially treat T1D and T2D by inhibiting β-cell dysfunction and death. Nevertheless, these antibodies, antagonists, natural compounds or small molecules inhibitors are need to be evaluated for pharmacokinetic properties, toxicity, and efficacy. Clinical trials in humans will be also required to assess the relevance of some of these pharmacological agents as a therapeutic strategy for assessing the safety, dose and/or durability of treatment. A major concern in therapeutic strategies based on the use of pharmacological inhibitors is the high risk of serious side effects, by inhibiting ubiquitously expressed targets and targets with pleiotropic properties. The specificity and selectivity of pharmacological inhibitors in a physiological system have to be studied and represent fundamental informations. However, it should be noted that the use of tyrosine kinase inhibitors and HDAC inhibitors in some cancer treatment is a feasible and promising approach. Another important aspect to consider is the translation of pre-clinical data into clinical trials, which may lead to negative results, as shown by the clinical trials using an IL-1β blockade strategy.
A key question that remains to be solved Is a specific therapeutic strategy to preserve the function and survival of β cells sufficient to prevent diabetes? A functional β-cell mass is essential in preventing the development of T1D and T2D. Theoretically, a therapeutic strategy targeting β-cells seems appropriate, but targeting multiple pathways in combination therapies. The release of a new antidiabetic medicine targeting both GLP-1R and GIPR (168) supports the idea that the next antidiabetic therapy for the future management of diabetes could involve the combination of pharmacological agents with complementary mechanisms of action.
SD, ER, AA wrote the manuscript. All authors contributed to the article and approved the submitted version.
The authors declare that the writing of this review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cnop M, Welsh N, Jonas JC, Jörns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: Many differences, few similarities. Diabetes (2005) 54 Suppl 2:S97–107. doi: 10.2337/diabetes.54.suppl_2.s97
2. Mandrup-Poulsen T. Interleukin-1 antagonists and other cytokine blockade strategies for type 1 diabetes. Rev Diabetes Stud (2012) 9:338–47. doi: 10.1900/RDS.2012.9.338
3. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
4. Holt RIG, DeVries JH, Hess-Fischl A, Hirsch IB, Kirkman MS, Klupa T, et al. The management of type 1 diabetes in adults. A consensus report by the American diabetes association (ADA) and the European association for the study of diabetes (EASD). Diabetologia (2021) 64:2609–52. doi: 10.1007/s00125-021-05568-3
5. Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia (2009) 52:1003–12. doi: 10.1007/s00125-009-1321-z
6. Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, et al. β-cell failure in type 2 diabetes: Postulated mechanisms and prospects for prevention and treatment. Diabetes Care (2014) 37:1751–58. doi: 10.2337/dc14-0396
7. DeFronzo RA, Ferrannini E, Groop L, Henry RR, Herman WH, Holst JJ, et al. Type 2 diabetes mellitus. Nat Rev Dis Primers (2015) 1:15019. doi: 10.1038/nrdp.2015.19
8. Holman RR, Clark A, Rorsman P. β-cell secretory dysfunction: A key cause of type 2 diabetes. Lancet Diabetes Endocrinol (2020) 8:370. doi: 10.1016/S2213-8587(20)30119-4
9. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
10. Weir GC, Gaglia J, Bonner-Weir S. Inadequate β-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol (2020) 8:249–56. doi: 10.1016/S2213-8587(20)30022-X
11. Bensellam M, Laybutt DR, Jonas JC. The molecular mechanisms of pancreatic β-cell glucotoxicity: Recent findings and future research directions. Mol Cell Endocrinol (2012) 364:1–27. doi: 10.1016/j.mce.2012.08.003
12. Ashcroft FM, Rohm M, Clark A, Brereton MF. Is type 2 diabetes a glycogen storage disease of pancreatic β-cells. Cell Metab (2017) 26:17–23. doi: 10.1016/j.cmet.2017.05.014
13. Weir GC. Glucolipotoxicity, β-cells, and diabetes: The emperor has no clothes. Diabetes (2020) 69:273–78. doi: 10.2337/db19-0138
14. Prentki M, Peyot ML, Masiello P, Madiraju SRM. Nutrient-induced metabolic stress, adaptation, detoxification, and toxicity in the pancreatic β-cell. Diabetes (2020) 69:279–90. doi: 10.2337/dbi19-0014
15. Costes S, Bertrand G, Ravier MA. Mechanisms of beta-cell apoptosis in type 2 diabetes-prone situations and potential protection by GLP-1-based therapies. Int J Mol Sci (2021) 22:5303. doi: 10.3390/ijms22105303
16. Rohm TV, Meier DT, Olefsky JM, Donath MY. Inflammation in obesity, diabetes, and related disorders. Immunity (2022) 55:31–55. doi: 10.1016/j.immuni.2021.12.013
17. Donath MY, Dinarello CA, Mandrup-Poulsen T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat Rev Immunol (2019) 19:734–46. doi: 10.1038/s41577-019-0213-9
18. Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: Two multicentre, randomised, double-blind, placebo-controlled trials. Lancet (2013) 381:1905–15. doi: 10.1016/S0140-6736(13)60023-9
19. Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, et al. Anti-inflammatory therapy with canakinumab for the prevention and management of diabetes. J Am Coll Cardiol (2018) 71:2392–401. doi: 10.1016/j.jacc.2018.03.002
20. Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, et al. Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care (2012) 35:1654–62. doi: 10.2337/dc11-2219
21. Sloan-Lancaster J, Abu-Raddad E, Polzer J, et al. Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1β antibody, in patients with type 2 diabetes. Diabetes Care (2013) 36:2239–46. doi: 10.2337/dc12-1835
22. Aharon-Hananel G, Jörns A, Lenzen S, Raz I, Weksler-Zangen S. Antidiabetic effect of interleukin-1β antibody therapy through β-cell protection in the Cohen diabetes-sensitive rat. Diabetes (2015) 64:1780–85. doi: 10.2337/db14-1018
23. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med (2007) 356:1517–26. doi: 10.1056/NEJMoa065213
24. Quattrin T, Haller MJ, Steck AK, Felner EI, Li Y, Xia Y, et al. Golimumab and beta-cell function in youth with new-onset type 1 diabetes. N Engl J Med (2020) 383:2007–17. doi: 10.1056/NEJMoa2006136
25. Jörns A, Ertekin ÜG, Arndt T, Terbish T, Wedekind D, Lenzen S. TNF-α antibody therapy in combination with the T-cell-specific antibody anti-TCR reverses the diabetic metabolic state in the LEW. 1AR1-iddm Rat. Diabetes (2015) 64:2880–91. doi: 10.2337/db14-1866
26. Jörns A, Ishikawa D, Teraoku H, Yoshimoto T, Wedekind D, Lenzen S. Remission of autoimmune diabetes by anti-TCR combination therapies with anti-IL-17A or/and anti-IL-6 in the IDDM rat model of type 1 diabetes. BMC Med (2020) 18:33. doi: 10.1186/s12916-020-1503-6
27. Trotta E, Bessette PH, Silveria SL, Ely LK, Jude KM, Le DT, et al. A human anti-IL-2 antibody that potentiates regulatory T cells by a structure-based mechanism. Nat Med (2018) 24:1005–14. doi: 10.1038/s41591-018-0070-2
28. Rydén AK, Perdue NR, Pagni PP, Gibson CB, Ratliff SS, Kirk RK, et al. Anti-IL-21 monoclonal antibody combined with liraglutide effectively reverses established hyperglycemia in mouse models of type 1 diabetes. J Autoimmun (2017) 84:65–74. doi: 10.1016/j.jaut.2017.07.006
29. von Herrath M, Bain SC, Bode B, Clausen JO, Coppieters K, Gaysina L, et al. Anti-IL-21-liraglutide study group investigators and contributors. anti-interleukin-21 antibody and liraglutide for the preservation of β-cell function in adults with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol (2021) 9:212–24. doi: 10.1016/S2213-8587(21)00019-X
30. Ablamunits V, Henegariu O, Hansen JB, Opare-Addo L, Preston-Hurlburt P, Santamaria P, et al. Synergistic reversal of type 1 diabetes in NOD mice with anti-CD3 and interleukin-1 blockade: Evidence of improved immune regulation. Diabetes (2012) 61:145–54. doi: 10.2337/db11-1033
31. Mignogna C, Maddaloni E, D’Onofrio L, Buzzetti R. Investigational therapies targeting CD3 for prevention and treatment of type 1 diabetes. Expert Opin Investig Drugs (2021) 30:1209–19. doi: 10.1080/13543784.2022.2022119
32. LeFevre JD, Cyriac SL, Tokmic A, Pitlick JM. Anti-CD3 monoclonal antibodies for the prevention and treatment of type 1 diabetes: A literature review. Am J Health Syst Pharm (2022) 79:2099–2117. zxac244. doi: 10.1093/ajhp/zxac244
33. Kuhn C, Rezende RM, da Cunha AP, Valette F, Quintana FJ, Chatenoud L, et al. Mucosal administration of CD3-specific monoclonal antibody inhibits diabetes in NOD mice and in a preclinical mouse model transgenic for the CD3 epsilon chain. J Autoimmun (2017) 76:115–22. doi: 10.1016/j.jaut.2016.10.001
34. Jörns A, Akin M, Arndt T, Terbish T, Zu Vilsendorf AM, Wedekind D, et al. Anti-TCR therapy combined with fingolimod for reversal of diabetic hyperglycemia by β cell regeneration in the LEW.1AR1-iddm rat model of type 1 diabetes. J Mol Med (Berl) (2014) 92:743–55. doi: 10.1007/s00109-014-1137-2
35. Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med (2014) 20:385–97. doi: 10.1038/nm.3482
36. Ardestani A, Maedler K. MST1: A promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia (2016) 59:1843–49. doi: 10.1007/s00125-016-3892-9
37. Ardestani A, Li S, Annamalai K, Lupse B, Geravandi S, Dobrowolski A, et al. Neratinib protects pancreatic beta cells in diabetes. Nat Commun (2019) 10:5015. doi: 10.1038/s41467-019-12880-5
38. Cao H, Lu J, Du J, Xia F, Wei S, Liu X, et al. TAK1 inhibition prevents the development of autoimmune diabetes in NOD mice. Sci Rep (2015) 13:14593. doi: 10.1038/srep14593
39. Varin EM, Wojtusciszyn A, Broca C, Muller D, Ravier MA, Ceppo F, et al. Inhibition of the MAP3 kinase Tpl2 protects rodent and human β-cells from apoptosis and dysfunction induced by cytokines and enhances anti-inflammatory actions of exendin-4. Cell Death Dis (2016) 7:e2065. doi: 10.1038/cddis.2015.399
40. Hägerkvist R, Sandler S, Mokhtari D, Welsh N. Amelioration of diabetes by imatinib mesylate (Gleevec): Role of beta-cell NF-kappaB activation and anti-apoptotic preconditioning. FASEB J (2007) 21:618–28. doi: 10.1096/fj.06-6910com
41. Gitelman SE, Bundy BN, Ferrannini E, Lim N, Blanchfield JL, DiMeglio LA, et al. Imatinib therapy for patients with recent-onset type 1 diabetes: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Diabetes Endocrinol (2021) 9:502–14. doi: 10.1016/S2213-8587(21)00139-X
42. Coomans de Brachène A, Castela A, Op de Beeck A, Mirmira RG, Marselli L, Marchetti P, et al. Preclinical evaluation of tyrosine kinase 2 inhibitors for human beta-cell protection in type 1 diabetes. Diabetes Obes Metab (2020) 22:1827–36. doi: 10.1111/dom.14104
43. Ge T, Jhala G, Fynch S, Akazawa S, Litwak S, Pappas EG, et al. The JAK1 selective inhibitor ABT 317 blocks signaling through interferon-γ and common γ chain cytokine receptors to reverse autoimmune diabetes in NOD mice. Front Immunol (2020) 11:588543. doi: 10.3389/fimmu.2020.588543
44. Newsholme P, Cruzat VF, Keane KN, Carlessi R, de Bittencourt PI Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem J (2016) 473:4527–50. doi: 10.1042/BCJ20160503C
45. Gerber PA, Rutter GA. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid Redox Signal (2017) 26:501–18. doi: 10.1089/ars.2016.6755
46. Roma LP, Jonas JC. Nutrient metabolism, subcellular redox state, and oxidative stress in pancreatic islets and β-cells. J Mol Biol (2020) 432:1461–93. doi: 10.1016/j.jmb.2019.10.012
47. Eguchi N, Vaziri ND, Dafoe DC, Ichii H. The role of oxidative stress in pancreatic β cell dysfunction in diabetes. Int J Mol Sci (2021) 22:1509. doi: 10.3390/ijms22041509
48. Mukai E, Fujimoto S, Inagaki N. Role of reactive oxygen species in glucose metabolism disorder in diabetic pancreatic β-cells. Biomolecules (2022) 12:1228. doi: 10.3390/biom12091228
49. Veluthakal R, Sidarala V, Kowluru A. NSC23766, a known inhibitor of Tiam1-Rac1 signaling module, prevents the onset of type 1 diabetes in the NOD mouse model. Cell Physiol Biochem (2016) 39:760–67. doi: 10.1159/000445666
50. Hanchang W, Khamchan A, Wongmanee N, Seedadee C. Hesperidin ameliorates pancreatic β-cell dysfunction and apoptosis in streptozotocin-induced diabetic rat model. Life Sci (2019) 235:116858. doi: 10.1016/j.lfs.2019.116858
51. Larsen L, Tonnesen M, Ronn SG, Størling J, Jørgensen S, Mascagni P, et al. Inhibition of histone deacetylases prevents cytokine-induced toxicity in beta cells. Diabetologia (2007) 50:779–89. doi: 10.1007/s00125-006-0562-3
52. Patel T, Patel V, Singh R, Jayaraman S. Chromatin remodeling resets the immune system to protect against autoimmune diabetes in mice. Immunol Cell Biol (2011) 89:640–49. doi: 10.1038/icb.2010.144
53. Dahllöf MS, Christensen DP, Lundh M, Dinarello CA, Mascagni P, Grunnet LG, et al. The lysine deacetylase inhibitor givinostat inhibits β-cell IL-1β induced IL-1β transcription and processing. Islets (2012) 4:417–22. doi: 10.4161/isl.23541
54. Khan S, Jena GB. Protective role of sodium butyrate, a HDAC inhibitor on beta-cell proliferation, function and glucose homeostasis through modulation of p38/ERK MAPK and apoptotic pathways: Study in juvenile diabetic rat. Chem Biol Interact (2014) 213:1–12. doi: 10.1016/j.cbi.2014.02.001
55. Dirice E, Ng RWS, Martinez R, Hu J, Wagner FF, Holson EB, et al. Isoform-selective inhibitor of histone deacetylase 3 (HDAC3) limits pancreatic islet infiltration and protects female nonobese diabetic mice from diabetes. J Biol Chem (2017) 292:17598–08. doi: 10.1074/jbc.M117.804328
56. Besançon A, Goncalves T, Valette F, Dahllöf MS, Mandrup-Poulsen T, Chatenoud L, et al. Oral histone deacetylase inhibitor synergises with T cell targeted immunotherapy to preserve beta cell metabolic function and induce stable remission of new-onset autoimmune diabetes in NOD mice. Diabetologia (2018) 61:389–98. doi: 10.1007/s00125-017-4459-0
57. Cabrera SM, Colvin SC, Tersey SA, Maier B, Nadler JL, Mirmira RG. Effects of combination therapy with dipeptidyl peptidase-IV and histone deacetylase inhibitors in the non-obese diabetic mouse model of type 1 diabetes. Clin Exp Immunol (2013) 172:375–82. doi: 10.1111/cei.12068
58. Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, et al. Inhibition of p53 preserves parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc Natl Acad Sci USA (2014) 111:3116–21. doi: 10.1073/pnas.1318951111
59. Hansen JB, Tonnesen MF, Madsen AN, Hagedorn PH, Friberg J, Grunnet LG, et al. Divalent metal transporter 1 regulates iron-mediated ROS and pancreatic β cell fate in response to cytokines. Cell Metab (2012) 16:449–61. doi: 10.1016/j.cmet.2012.09.001
60. Ellervik C, Mandrup-Poulsen T, Andersen HU, Tybjærg-Hansen A, Frandsen M, Birgens H, et al. Elevated transferrin saturation and risk of diabetes: Three population-based studies. Diabetes Care (2011) 34:2256–58. doi: 10.2337/dc11-0416
61. Huang S, Zhu M, Wu W, Rashid A, Liang Y, Hou L, et al. Valproate pretreatment protects pancreatic β-cells from palmitate-induced ER stress and apoptosis by inhibiting glycogen synthase kinase-3β. J BioMed Sci (2014) 21:38. doi: 10.1186/1423-0127-21-38
62. Kim KM, Lee KS, Lee GY, Jin H, Durrance ES, Park HS, et al. Anti-diabetic efficacy of KICG1338, a novel glycogen synthase kinase-3β inhibitor, and its molecular characterization in animal models of type 2 diabetes and insulin resistance. Mol Cell Endocrinol (2015) 409:1–10. doi: 10.1016/j.mce.2015.03.011
63. Sacco F, Seelig A, Humphrey SJ, Krahmer N, Volta F, Reggio A, et al. Phosphoproteomics reveals the GSK3-PDX1 axis as a key pathogenic signaling node in diabetic islets. Cell Metab (2019) 29:1422–32. doi: 10.1016/j.cmet.2019.02.012
64. Pitasi CL, Liu J, Gausserès B, Pommier G, Delangre E, Armanet M, et al. Implication of glycogen synthase kinase 3 in diabetes-associated islet inflammation. J Endocrinol (2020) 244:133–48. doi: 10.1530/JOE-19-0239
65. Delangre E, Liu J, Tolu S, Maouche K, Armanet M, Cattan P, et al. Underlying mechanisms of glucocorticoid-induced β-cell death and dysfunction: A new role for glycogen synthase kinase 3. Cell Death Dis (2021) 12:1136. doi: 10.1038/s41419-021-04419-8
66. Ivovic A, Oprescu AI, Koulajian K, Mori Y, Eversley JA, Zhang L, et al. IKKβ inhibition prevents fat-induced beta cell dysfunction in vitro and in vivo in rodents. Diabetologia (2017) 60:2021–32. doi: 10.1007/s00125-017-4345-9
67. Cnop M, Toivonen S, Igoillo-Esteve M, Salpea P. Endoplasmic reticulum stress and eIF2α phosphorylation: The Achilles heel of pancreatic β cells. Mol Metab (2017) 6:1024–39. doi: 10.1016/j.molmet.2017.06.001
68. Shrestha N, De Franco E, Arvan P, Cnop M. Pathological β-cell endoplasmic reticulum stress in type 2 diabetes: Current evidence. Front Endocrinol (Lausanne) (2021) 12:650158. doi: 10.3389/fendo.2021.650158
69. Bilekova S, Sachs S, Lickert H. Pharmacological targeting of endoplasmic reticulum stress in pancreatic beta cells. Trends Pharmacol Sci (2021) 42:85–95. doi: 10.1016/j.tips.2020.11.011
70. Yong J, Johnson JD, Arvan P, Han J, Kaufman RJ. Therapeutic opportunities for pancreatic β-cell ER stress in diabetes mellitus. Nat Rev Endocrinol (2021) 17:455–67. doi: 10.1038/s41574-021-00510-4
71. Zhu Q, Zhong JJ, Jin JF, Yin XM, Miao H. Tauroursodeoxycholate, a chemical chaperone, prevents palmitate-induced apoptosis in pancreatic β-cells by reducing ER stress. Exp Clin Endocrinol Diabetes (2013) 121:43–7. doi: 10.1055/s-0032-1321787
72. Thielen LA, Chen J, Jing G, Moukha-Chafiq O, Xu G, Jo S, et al. Identification of an anti-diabetic, orally available small molecule that regulates TXNIP expression and glucagon action. Cell Metab (2020) 32:353–65. doi: 10.1016/j.cmet.2020.07.002
73. Xu G, Chen J, Jing G, Shalev A. Preventing β-cell loss and diabetes with calcium channel blockers. Diabetes (2012) 61:848–56. doi: 10.2337/db11-0955
74. Ovalle F, Grimes T, Xu G, Patel AJ, Grayson TB, Thielen LA, et al. Verapamil and beta cell function in adults with recent-onset type 1 diabetes. Nat Med (2018) 24:1108–12. doi: 10.1038/s41591-018-0089-4
75. Lei L, Bai G, Wang X, Liu S, Xia J, Wu S, et al. Histone deacetylase 3-selective inhibitor RGFP966 ameliorates impaired glucose tolerance through β-cell protection. Toxicol Appl Pharmacol (2020) 406:115189. doi: 10.1016/j.taap.2020.115189
76. Plaisance V, Rolland L, Gmyr V, Annicotte JS, Kerr-Conte J, Pattou F, et al. The class I histone deacetylase inhibitor MS-275 prevents pancreatic beta cell death induced by palmitate. J Diabetes Res (2014) 2014:195739. doi: 10.1155/2014/195739
77. Zhang Y, Li M, Wang Y, Liu X, Zhou L, Zhang C, et al. Histone deacetylase inhibition by MS-275 potentiates glucose-stimulated insulin secretion without affecting glucose oxidation. Life Sci (2020) 257:118073. doi: 10.1016/j.lfs.2020.118073
78. Hu Y, Liu J, Yuan Y, Chen J, Cheng S, Wang H, et al. Sodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ Toxicol Pharmacol (2018) 64:112–21. doi: 10.1016/j.etap.2018.09.002
79. Daneshpajooh M, Eliasson L, Bacos K, Ling C. MC1568 improves insulin secretion in islets from type 2 diabetes patients and rescues β-cell dysfunction caused by Hdac7 upregulation. Acta Diabetol (2018) 55:1231–35. doi: 10.1007/s00592-018-1201-4
80. Huang JS, Guo BB, Wang GH, Zeng LM, Hu YH, Wang T, et al. DGAT1 inhibitors protect pancreatic β-cells from palmitic acid-induced apoptosis. Acta Pharmacol Sin (2021) 42:264–71. doi: 10.1038/s41401-020-0482-7
81. Denison H, Nilsson C, Löfgren L, Himmelmann A, Mårtensson G, Knutsson M, et al. Diacylglycerol acyltransferase 1 inhibition with AZD7687 alters lipid handling and hormone secretion in the gut with intolerable side effects: A randomized clinical trial. Diabetes Obes Metab (2014) 16:334–43. doi: 10.1111/dom.12221
82. NAVIGATOR Study Group, McMurray JJ, Holman RR, Haffner SM, Bethel MA, Holzhauer B, et al. Effect of valsartan on the incidence of diabetes and cardiovascular events. N Engl J Med (2010) 362:1477–90. doi: 10.1056/NEJMoa1001121
83. Madec AM, Cassel R, Dubois S, Ducreux S, Vial G, Chauvin MA, et al. Losartan, an angiotensin II type 1 receptor blocker, protects human islets from glucotoxicity through the phospholipase c pathway. FASEB J (2013) 27:5122–30. doi: 10.1096/fj.13-234104
84. Wang Y, Xue J, Li Y, Zhou X, Qiao S, Han D. Telmisartan protects against high glucose/high lipid-induced apoptosis and insulin secretion by reducing the oxidative and ER stress. Cell Biochem Funct (2019) 37:161–68. doi: 10.1002/cbf.3383
85. Zhao S, Chan LK, Chen L, Cheng TW, Klein T, Leung PS. Combination of telmisartan and linagliptin preserves pancreatic islet cell function and morphology in db/db mice. Pancreas (2016) 45:584–92. doi: 10.1097/MPA.0000000000000505
86. Gómez-Banoy N, Guseh JS, Li G, Rubio-Navarro A, Chen T, Poirier B, et al. Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat Med (2019) 25:1739–47. doi: 10.1038/s41591-019-0610-4
87. Kayaniyil S, Vieth R, Retnakaran R, Knight JA, Qi Y, Gerstein HC, et al. Association of vitamin d with insulin resistance and beta cell dysfunction in subjects at risk with type 2 diabetes. Diabetes Care (2010) 33:1379–81. doi: 10.2337/dc09-2321
88. Takiishi T, Gysemans C, Bouillon R, Mathieu C. Vitamin d and diabetes. Endocrinol Metab Clin North Am (2010) 39:419–46. doi: 10.1016/j.ecl.2010.02.013
89. Wei Z, Yoshihara E, He N, Hah N, Fan W, Pinto AFM, et al. Vitamin d switches BAF complexes to protect β cells. Cell (2018) 173:1135–49. doi: 10.1016/j.cell.2018.04.013
90. Pittas AG, Lau J, Hu FB, Dawson-Hughes B. The role of vitamin d and calcium in type 2 diabetes. A systematic review and meta-analysis. J Clin Endocrinol Metab (2007) 92:2017–29. doi: 10.1210/jc.2007-0298
91. Lu L, Bennett DA, Millwood IY, Parish S, McCarthy MI, Mahajan A, et al. Association of vitamin d with risk of type 2 diabetes: A mendelian ransdomisation study in european and chineses adults. PloS Med (2018) 15(5):e1002566. doi: 10.1371/journal.pmed.1002566
92. Pittas AG, Dawson-Hughes B, Sheehan P, Ware JH, Knowler WC, Aroda VR, et al. Vitamin d supplementation and prevention of type 2 diabetes. N Engl J Med (2019) 381:520–30. doi: 10.1056/NEJMoa1900906
93. Jorde R, Sollid ST, Svartberg J, Schirmer H, Joakimsen RM, Njølstad I, et al. Vitamin d 20,000 IU per week for five years does not prevent progression from prediabetes to diabetes. J Clin Endocrinol Metab (2016) 101:1647–55. doi: 10.1210/jc.2015-4013
94. Kawahara T, Suzuki G, Inazu T, Mizuno S, Kasagi F, Okada Y, et al. Rationale and design of diabetes prevention with active vitamin d (DPVD): A randomised, double-blind, placebo-controlled study. BMJ Open (2016) 6(7):e011183. doi: 10.1136/bmjopen-2016-011183
95. Kawahara T, Suzuki G, Mizuno S, Inazu T, Kasagi F, Kawahara C, et al. Effect of active vitamin d treatment on development of type 2 diabetes: DPVD randomised controlled trial in Japanese population. BMJ (2022) 377:e066222. doi: 10.1136/bmj-2021-066222
96. Zhang E, Mohammed Al-Amily I, Mohammed S, Luan C, Asplund O, Ahmed M, et al. Preserving insulin secretion in diabetes by inhibiting VDAC1 overexpression and surface translocation in β cells. Cell Metab (2019) 29:64–77. doi: 10.1016/j.cmet.2018.09.008
97. Guan FY, Gu J, Li W, Zhang M, Ji Y, Li J, et al. Compound K protects pancreatic islet cells against apoptosis through inhibition of the AMPK/JNK pathway in type 2 diabetic mice and in MIN6 β-cells. Life Sci (2014) 107:42–9. doi: 10.1016/j.lfs.2014.04.034
98. Li J, Wu N, Chen X, Chen H, Yang X, Liu C. Curcumin protects islet cells from glucolipotoxicity by inhibiting oxidative stress and NADPH oxidase activity both in vitro and in vivo. Islets (2019) 11:152–64. doi: 10.1080/19382014.2019.1690944
99. Xia ZH, Jiang X, Li K, Li LX, Chen WB, Wang YX, et al. Curcumin inhibits alloxan-induced pancreatic islet cell damage via antioxidation and antiapoptosis. J Biochem Mol Toxicol (2020) 34:e22499. doi: 10.1002/jbt.22499
100. Roham PH, Save SN, Sharma S. Human islet amyloid polypeptide: A therapeutic target for the management of type 2 diabetes mellitus. J Pharm Anal (2022) 12:556–69. doi: 10.1016/j.jpha.2022.04.001
101. Wijesekara N, Ahrens R, Wu L, Ha K, Liu Y, Wheeler MB, et al. Islet amyloid inhibitors improve glucose homeostasis in a transgenic mouse model of type 2 diabetes. Diabetes Obes Metab (2015) 17:1003–06. doi: 10.1111/dom.12529
102. Zhang Y, Yu XL, Zhu J, Liu SY, Liu XM, Dong QX, et al. Intravenous immunoglobulin improves glucose control and β-cell function in human IAPP transgenic mice by attenuating islet inflammation and reducing IAPP oligomers. Int Immunopharmacol (2018) 54:145–52. doi: 10.1016/j.intimp.2017.11.012
103. Rodríguez-Comas J, Moreno-Vedia J, Obach M, Castaño C, de Pablo S, Alcarraz-Vizán G, et al. Alpha1-antitrypsin ameliorates islet amyloid-induced glucose intolerance and β-cell dysfunction. Mol Metab (2020) 37:100984. doi: 10.1016/j.molmet.2020.100984
104. Kim J, Park K, Kim MJ, Lim H, Kim KH, Kim SW, et al. An autophagy enhancer ameliorates diabetes of human IAPP-transgenic mice through clearance of amyloidogenic oligomer. Nat Commun (2021) 12:183. doi: 10.1038/s41467-020-20454-z
105. Li X, Ma L, Zheng W, Chen T. Inhibition of islet amyloid polypeptide fibril formation by selenium-containing phycocyanin and prevention of beta cell apoptosis. Biomaterials (2014) 35:8596–04. doi: 10.1016/j.biomaterials.2014.06.056
106. Li XL, Wong YS, Xu G, Chan JC. Selenium-enriched spirulina protects INS-1E pancreatic beta cells from human islet amyloid polypeptide-induced apoptosis through suppression of ROS-mediated mitochondrial dysfunction and PI3/AKT pathway. Eur J Nutr (2015) 54:509–22. doi: 10.1007/s00394-014-0732-x
107. Dubey R, Patil K, Dantu SC, Sardesai DM, Bhatia P, Malik N, et al. Azadirachtin inhibits amyloid formation, disaggregates pre-formed fibrils and protects pancreatic β-cells from human islet amyloid polypeptide/amylin-induced cytotoxicity. Biochem J (2019) 476:889–07. doi: 10.1042/BCJ20180820
108. Meng QY, Wang H, Cui ZB, Yu WG, Lu XZ. Chitosan oligosaccharides attenuate amyloid formation of hIAPP and protect pancreatic β-cells from cytotoxicity. Molecules (2020) 25:1314. doi: 10.3390/molecules25061314
109. Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, Bhushan A. Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab (2019) 29:1045–60. doi: 10.1016/j.cmet.2019.01.021
110. Wang MY, Dean ED, Quittner-Strom E, Zhu Y, Chowdhury KH, Zhang Z, et al. Glucagon blockade restores functional β-cell mass in type 1 diabetic mice and enhances function of human islets. Proc Natl Acad Sci USA (2021) 118:e2022142118. doi: 10.1073/pnas.2022142118
111. Xi Y, Song B, Ngan I, Solloway MJ, Humphrey M, Wang Y, et al. Glucagon-receptor-antagonism-mediated β-cell regeneration as an effective anti-diabetic therapy. Cell Rep (2022) 39:110872. doi: 10.1016/j.celrep.2022.110872
112. Cui X, Feng J, Wei T, Zhang L, Lang S, Yang K, et al. Pancreatic alpha cell glucagon-liver FGF21 axis regulates beta cell regeneration in a mouse model of type 2 diabetes. Diabetologia (2022) 66:535–550. doi: 10.1007/s00125-022-05822-2
113. Simon-Szabó L, Kokas M, Mandl J, Kéri G, Csala M. Metformin attenuates palmitate-induced endoplasmic reticulum stress, serine phosphorylation of IRS-1 and apoptosis in rat insulinoma cells. PloS One (2014) 9:e97868. doi: 10.1371/journal.pone.0097868
114. Moon JS, Karunakaran U, Elumalai S, Lee IK, Lee HW, Kim YW, et al. Metformin prevents glucotoxicity by alleviating oxidative and ER stress-induced CD36 expression in pancreatic beta cells. J Diabetes Complications (2017) 31:21–30. doi: 10.1016/j.jdiacomp.2016.09.001
115. Baidwan S, Chekuri A, Hynds DL, Kowluru A. Glucotoxicity promotes aberrant activation and mislocalization of ras-related C3 botulinum toxin substrate 1 [Rac1] and metabolic dysfunction in pancreatic islet β-cells: Reversal of such metabolic defects by metformin. Apoptosis (2017) 22:1380–93. doi: 10.1007/s10495-017-1409-8
116. Cen J, Sargsyan E, Forslund A, Bergsten P. Mechanisms of beneficial effects of metformin on fatty acid-treated human islets. J Mol Endocrinol (2018) 61:91–9. doi: 10.1530/JME-17-0304
117. Li Q, Jia S, Xu L, Li B, Chen N. Metformin-induced autophagy and irisin improves INS-1 cell function and survival in high-glucose environment via AMPK/SIRT1/PGC-1α signal pathway. Food Sci Nutr (2019) 7:1695–03. doi: 10.1002/fsn3.1006
118. Wang AP, Li X, Zheng Y, Liu BL, Huang G, Yan X, et al. Thiazolidinediones protect mouse pancreatic β-cells directly from cytokine-induced cytotoxicity through PPARγ-dependent mechanisms. Acta Diabetol (2013) 50:163–73. doi: 10.1007/s00592-010-0239-8
119. Kimura T, Kaneto H, Shimoda M, Hirukawa H, Okauchi S, Kohara K, et al. Protective effects of pioglitazone and/or liraglutide on pancreatic β-cells in db/db mice: Comparison of their effects between in an early and advanced stage of diabetes. Mol Cell Endocrinol (2015) 400:78–89. doi: 10.1016/j.mce.2014.11.018
120. Kwon MJ, Lee YJ, Jung HS, Shin HM, Kim TN, Lee SH, et al. The direct effect of lobeglitazone, a new thiazolidinedione, on pancreatic beta cells: A comparison with other thiazolidinediones. Diabetes Res Clin Pract (2019) 151:209–23. doi: 10.1016/j.diabres.2019.04.006
121. Marmentini C, Guimarães DSPSF, de Lima TI, Teófilo FBS, da Silva NS, Soares GM, et al. Rosiglitazone protects INS-1E cells from human islet amyloid polypeptide toxicity. Eur J Pharmacol (2022) 928:175122. doi: 10.1016/j.ejphar.2022.175122
122. Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab (2018) 27:740–56. doi: 10.1016/j.cmet.2018.03.001
123. Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
124. Nauck MA, Quast DR, Wefers J, Meier JJ. GLP-1 receptor agonists in the treatment of type 2 diabetes. state-of-the-art. Mol Metab (2021) 46:101102. doi: 10.1016/j.molmet.2020.101102
125. Bugliani M, Syed F, Paula FMM, Omar BA, Suleiman M, Mossuto S, et al. DPP-4 is expressed in human pancreatic beta cells and its direct inhibition improves beta cell function and survival in type 2 diabetes. Mol Cell Endocrinol (2018) 473:186–93. doi: 10.1016/j.mce.2018.01.019
126. Wei R, Cui X, Feng J, Gu L, Lang S, Wei T, et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism (2020) 111:154324. doi: 10.1016/j.metabol.2020.154324
127. Kanno A, Asahara SI, Kawamura M, Furubayashi A, Tsuchiya S, Suzuki E, et al. Early administration of dapagliflozin preserves pancreatic β-cell mass through a legacy effect in a mouse model of type 2 diabetes. J Diabetes Investig (2019) 10:577–90. doi: 10.1111/jdi.12945
128. Hansen HH, Jelsing J, Hansen CF, Hansen G, Vrang N, Mark M, et al. The sodium glucose cotransporter type 2 inhibitor empagliflozin preserves β-cell mass and restores glucose homeostasis in the male zucker diabetic fatty rat. J Pharmacol Exp Ther (2014) 350:657–64. doi: 10.1124/jpet.114.213454
129. Steven S, Oelze M, Hanf A, Kröller-Schön S, Kashani F, Roohani S, et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol (2017) 13:370–85. doi: 10.1016/j.redox.2017.06.009
130. Jörns A, Arndt T, Meyer zu Vilsendorf A, Klempnauer J, Wedekind D, Hedrich HJ, et al. Islet infiltration, cytokine expression and beta cell death in the NOD mouse, BB rat, komeda rat, LEW.1AR1-iddm rat and humans with type 1 diabetes. Diabetologia (2014) 57:512–21. doi: 10.1007/s00125-013-3125-4
131. Barton FB, Rickels MR, Alejandro R, Hering BJ, Wease S, Naziruddin B, et al. Improvement in outcomes of clinical islet transplantation: 1999-2010. Diabetes Care (2012) 35:1436–45. doi: 10.2337/dc12-0063
132. Marfil-Garza BA, Imes S, Verhoeff K, Hefler J, Lam A, Dajani K, et al. Pancreatic islet transplantation in type 1 diabetes: 20-year experience from a single-centre cohort in Canada. Lancet Diabetes Endocrinol (2022) 10:519–32. doi: 10.1016/S2213-8587(22)00114-0
133. Lablanche S, Borot S, Wojtusciszyn A, Bayle F, Tétaz R, Badet L, et al. Five-year metabolic, functional, and safety results of patients with type 1 diabetes transplanted with allogenic islets within the swiss-french GRAGIL network. Diabetes Care (2015) 38:1714–22. doi: 10.2337/dc15-0094
134. Barshes NR, Wyllie S, Goss JA. Inflammation-mediated dysfunction and apoptosis in pancreatic islet transplantation: Implications for intrahepatic grafts. J Leukoc Biol (2005) 77:587–97. doi: 10.1189/jlb.1104649
135. Clark AL, Urano F. Endoplasmic reticulum stress in beta cells and autoimmune diabetes. Curr Opin Immunol (2016) 43:60–6. doi: 10.1016/j.coi.2016.09.006
136. Cardozo AK, Heimberg H, Heremans Y, Leeman R, Kutlu B, Kruhøffer M, et al. A comprehensive analysis of cytokine-induced and nuclear factor-kappa b-dependent genes in primary rat pancreatic beta-cells. J Biol Chem (2001) 276:48879–86. doi: 10.1074/jbc.M108658200
137. Heimberg H, Heremans Y, Jobin C, Leemans R, Cardozo AK, Darville M, et al. Inhibition of cytokine-induced NF-kappaB activation by adenovirus-mediated expression of a NF-kappaB super-repressor prevents beta-cell apoptosis. Diabetes (2001) 50:2219–24. doi: 10.2337/diabetes.50.10.2219
138. Meier BC, Wagner BK. Inhibition of HDAC3 as a strategy for developing novel diabetes therapeutics. Epigenomics (2014) 6:209–14. doi: 10.2217/epi.14.11
139. Lundh M, Christensen DP, Damgaard Nielsen M, Richardson SJ, Dahllöf MS, Skovgaard T, et al. Histone deacetylases 1 and 3 but not 2 mediate cytokine-induced beta cell apoptosis in INS-1 cells and dispersed primary islets from rats and are differentially regulated in the islets of type 1 diabetic children. Diabetologia (2012) 55:2421–31. doi: 10.1007/s00125-012-2615-0
140. Bluestone JA, Buckner JH, Herold KC. Immunotherapy: Building a bridge to a cure for type 1 diabetes. Science (2021) 373:510–16. doi: 10.1126/science.abh1654
141. Koulmanda M, Bhasin M, Awdeh Z, Qipo A, Fan Z, Hanidziar D, et al. The role of TNF-α in mice with type 1. and 2. diabetes. PloS One (2012) 7:e33254. doi: 10.1371/journal.pone.0033254
142. Cavallo MG, Pozzilli P, Bird C, Wadhwa M, Meager A, Visalli N, et al. Cytokines in sera from insulin-dependent diabetic patients at diagnosis. Clin Exp Immunol (1991) 86:256–59. doi: 10.1111/j.1365-2249.1991.tb05806.x
143. Kim BS, Park YJ, Chung Y. Targeting IL-17 in autoimmunity and inflammation. Arch Pharm Res (2016) 39:1537–47. doi: 10.1007/s12272-016-0823-8
144. Allocca M, Jovani M, Fiorino G, Schreiber S, Danese S. Anti-IL-6 treatment for inflammatory bowel diseases: next cytokine, next target. Curr Drug Targets (2013) 14:1508–21. doi: 10.2174/13894501113146660224
145. Costes S, Vandewalle B, Tourrel-Cuzin C, Broca C, Linck N, Bertrand G, et al. Degradation of cAMP-responsive element-binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in beta-cells and human pancreatic islets. Diabetes (2009) 58:1105–15. doi: 10.2337/db08-0926
146. Ruiz L, Gurlo T, Ravier MA, Wojtusciszyn A, Mathieu J, Brown MR, et al. Proteasomal degradation of the histone acetyl transferase p300 contributes to beta-cell injury in a diabetes environment. Cell Death Dis (2018) 9:600. doi: 10.1038/s41419-018-0603-0
147. Lytrivi M, Castell AL, Poitout V, Cnop M. Recent insights into mechanisms of β-cell lipo. and glucolipotoxicity in type 2 diabetes. J Mol Biol (2020) 432:1514–34. doi: 10.1016/j.jmb.2019.09.016
148. Lytrivi M, Ghaddar K, Lopes M, Rosengren V, Piron A, Yi X, et al. Combined transcriptome and proteome profiling of the pancreatic β-cell response to palmitate unveils key pathways of β-cell lipotoxicity. BMC Genomics (2020) 21:590. doi: 10.1186/s12864-020-07003-0
149. You S, Zheng J, Chen Y, Huang H. Research progress on the mechanism of beta-cell apoptosis in type 2 diabetes mellitus. Front Endocrinol (Lausanne) (2022) 13:976465. doi: 10.3389/fendo.2022.976465
150. Lim YJ, Kim JH, Pan JH, Kim JK, Park TS, Kim YJ, et al. Naringin protects pancreatic β-cells against oxidative stress-induced apoptosis by inhibiting both intrinsic and extrinsic pathways in insulin-deficient diabetic mice. Mol Nutr Food Res (2018) 62. doi: 10.1002/mnfr.201700810
151. Oslowski CM, Hara T, O’Sullivan-Murphy B, Kanekura K, Lu S, Hara M, et al. Thioredoxin-interacting protein mediates ER stress-induced β cell death through initiation of the inflammasome. Cell Metab (2012) 16:265–73. doi: 10.1016/j.cmet.2012.07.005
152. Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A. Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes (2008) 57:938–44. doi: 10.2337/db07-0715
153. Thielen L, Shalev A. Diabetes pathogenic mechanisms and potential new therapies based upon a novel target called TXNIP. Curr Opin Endocrinol Diabetes Obes (2018) 25:75–80. doi: 10.1097/MED.0000000000000391
154. Hong K, Xu G, Grayson TB, Shalev A. Cytokines regulate β-cell thioredoxin interacting protein (TXNIP) via distinct mechanisms and pathways. J Biol Chem (2016) 291:8428–39. doi: 10.1074/jbc.M115.698365
155. Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased a-cells, reduced b-cells and exocrine fibrosis: Quantitative changes in the pancreas in type 2 diabetes. Diabetes Res (1988) 9:151–59.
156. Ueberberg S, Nauck MA, Uhl W, Montemurro C, Tannapfel A, Clark A, et al. Islet amyloid in patients with diabetes due to exocrine pancreatic disorders, type 2 diabetes, and nondiabetic patients. J Clin Endocrinol Metab (2020) 105:dgaa176. doi: 10.1210/clinem/dgaa176
157. Raleigh D, Zhang X, Hastoy B, Clark A. The β-cell assassin: IAPP cytotoxicity. J Mol Endocrinol (2017) 59:R121–40. doi: 10.1530/JME-17-0105
158. Bishoyi AK, Roham PH, Rachineni K, Save S, Hazari MA, Sharma S, et al. Human islet amyloid polypeptide (hIAPP). a curse in type II diabetes mellitus: Insights from structure and toxicity studies. Biol Chem (2020) 402:133–53. doi: 10.1515/hsz-2020-0174
159. Sevcuka A, White K, Terry C. Factors that contribute to hIAPP amyloidosis in type 2 diabetes mellitus. Life (Basel) (2022) 12:583. doi: 10.3390/life12040583
160. Sakagashira S, Sanke T, Hanabusa T, Shimomura H, Ohagi S, Kumagaye KY, et al. Missense mutation of amylin gene (S20G) in japanese NIDDM patients. Diabetes (1996) 45:1279–81. doi: 10.2337/diab.45.9.1279
161. Meier DT, Entrup L, Templin AT, Hogan MF, Mellati M, Zraika S, et al. The S20G substitution in hIAPP is more amyloidogenic and cytotoxic than wild-type hIAPP in mouse islets. Diabetologia (2016) 59:2166–71. doi: 10.1007/s00125-016-4045-x
162. Matveyenko AV, Butler PC. Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J (2006) 47:225–33. doi: 10.1093/ilar.47.3.225
163. Haataja L, Gurlo T, Huang CJ, Butler PC. Islet amyloid in type 2 diabetes, and the toxic oligomer hypothesis. Endocr Rev (2008) 29:303–16. doi: 10.1210/er.2007-0037
164. Guillemain G, Lacapere JJ, Khemtemourian L. Targeting hIAPP fibrillation: A new paradigm to prevent β-cell death? Biochim Biophys Acta Biomembr (2022) 1864:184002. doi: 10.1016/j.bbamem.2022.184002
165. Campbell JE, Drucker DJ. Islet a cells and glucagon. critical regulators of energy homeostasis. Nat Rev Endocrinol (2015) 372:329–38. doi: 10.1038/nrendo.2015.51
166. Mayo KE, Miller LJ, Bataille D, Dalle S, Göke B, Thorens B, et al. International union of pharmacology. XXXV. the glucagon receptor family. Pharmacol Rev (2003) 55:167–94. doi: 10.1124/pr.55.1.6
167. Jia Y, Liu Y, Feng L, Sun S, Sun G. Role of glucagon and its receptor in the pathogenesis of diabetes. Front Endocrinol (Lausanne) (2022) 13:928016. doi: 10.3389/fendo.2022.928016
168. Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet (2018) 392:2180–93. doi: 10.1016/S0140-6736(18)32260-8
Keywords: diabetes, pancreatic β-cell, insulin secretion dysfunction, apoptosis, pharmacological inhibitors, therapeutic strategies
Citation: Dalle S, Abderrahmani A and Renard E (2023) Pharmacological inhibitors of β-cell dysfunction and death as therapeutics for diabetes. Front. Endocrinol. 14:1076343. doi: 10.3389/fendo.2023.1076343
Received: 21 October 2022; Accepted: 20 February 2023;
Published: 15 March 2023.
Edited by:
Ralf Jockers, Université Paris Cité, FranceReviewed by:
Yumi Imai, The University of Iowa, United StatesCopyright © 2023 Dalle, Abderrahmani and Renard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stéphane Dalle, c3RlcGhhbmUuZGFsbGVAaWdmLmNucnMuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.