 Elvina Almuradova
Elvina Almuradova Irfan Cicin
Irfan Cicin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 22 March 2023
Sec. Cancer Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1039490
This article is part of the Research Topic Cancer-related Hypercalcemia and Potential Treatments View all 7 articles
Cancer-related hypercalcemia is a common finding typically seen in patients with advanced cancer and occurs in about 20 to 30 percent of cases. The most common cause of hypercalcemia in hospitalized patients is hypercalcemia due to malignancy.This clinical problem is seen in patients with both solid tumors and patients with hematologic malignancies. Hypercalcemia is associated with a poor prognosis in oncology patients. This pathologic condition can occur due to many different mechanisms but is usually caused by abnormal calcium use resulting from bone resorption, intestinal absorption, or renal excretion. Hypercalcemia may present with a wide range of symptoms ranging from gastrointestinal system symptoms to neurologic symptoms. Timely diagnosis and initiation of treatment by the physician significantly reduce the risk of complications. Treatment aims to decrease serum calcium by increasing calciuresis, decreasing bone resorption, and decreasing intestinal calcium absorption. The mainstays of treatment are IV hydration, bisphosphonates and calcitonin, denosumab, and in some patients, prednisone, and cinacalcet. Patients with underlying advanced kidney disease and refractory severe hypercalcemia should be evaluated for hemodialysis. Every physician dealing with oncology patients should know the fastest and most effective management of hypercalcemia. We aimed to contribute in this sense.
Hypercalcemia of malignancy (HCM) is a condition in which the serum calcium level is above normal level (1–3). Symptoms resulting from hypercalcemia can range from mild to life-threatening. Malignancy is one of the most common causes of hypercalcemia, especially in patients with cancer associated with bone metastases (1). It is estimated that hipercalcemia affects aproximately 30% of patients with cancer (3). Common malignancies associated with HCM include multiple myeloma, breast, lung, squamous cell carcinomas, renal, ovarian cancer, and certain lymphomas. The severity of hypercalcemia is categorized according to the serum total calcium level (1). Hypercalcemia is a clinical problem that occurs as a result of abnormal bone formation and resorption process due to cancer. Although hypercalcemia due to malignancy has decreased with the introduction of new treatment agents, it is still a common clinical problem. The aim of this review is to contribute to the awareness of clinicians by summarizing the current literature on the mechanism, diagnosis and management of malignant hypercalcemia.
Calcium balance refers to the state of calcium stores in the body, especially in the bone. Bone calcium balance can be neutral, positive, or negative, depending on several factors, including growth, aging, and acquired or inherited disorders. Calcium homeostasis refers to the hormonal regulation of ionized serum calcium by parathyroid hormone, 1,25-dihydroxyvitamin D (calcitriol), and serum ionized calcium itself, and these factors together regulate calcium transport in the intestine, kidney, and bone (4–8) (Figure 1). Three main mechanisms cause hypercalcemia in malignancies. First, the most common (80%), is the secretion of PTHrP from tumors, which can cause hypercalcemia by acting similarly to parathyroid hormone (PTH) (9). The second is increased calcium absorption by the autonomous production of 1,25(OH)2D by 1α-hydroxylase in the tumor (10). The third mechanism is resorption, which occurs due to the increase in osteoclastic activity of tumor cells in the bone tissue (Figure 2) (11).
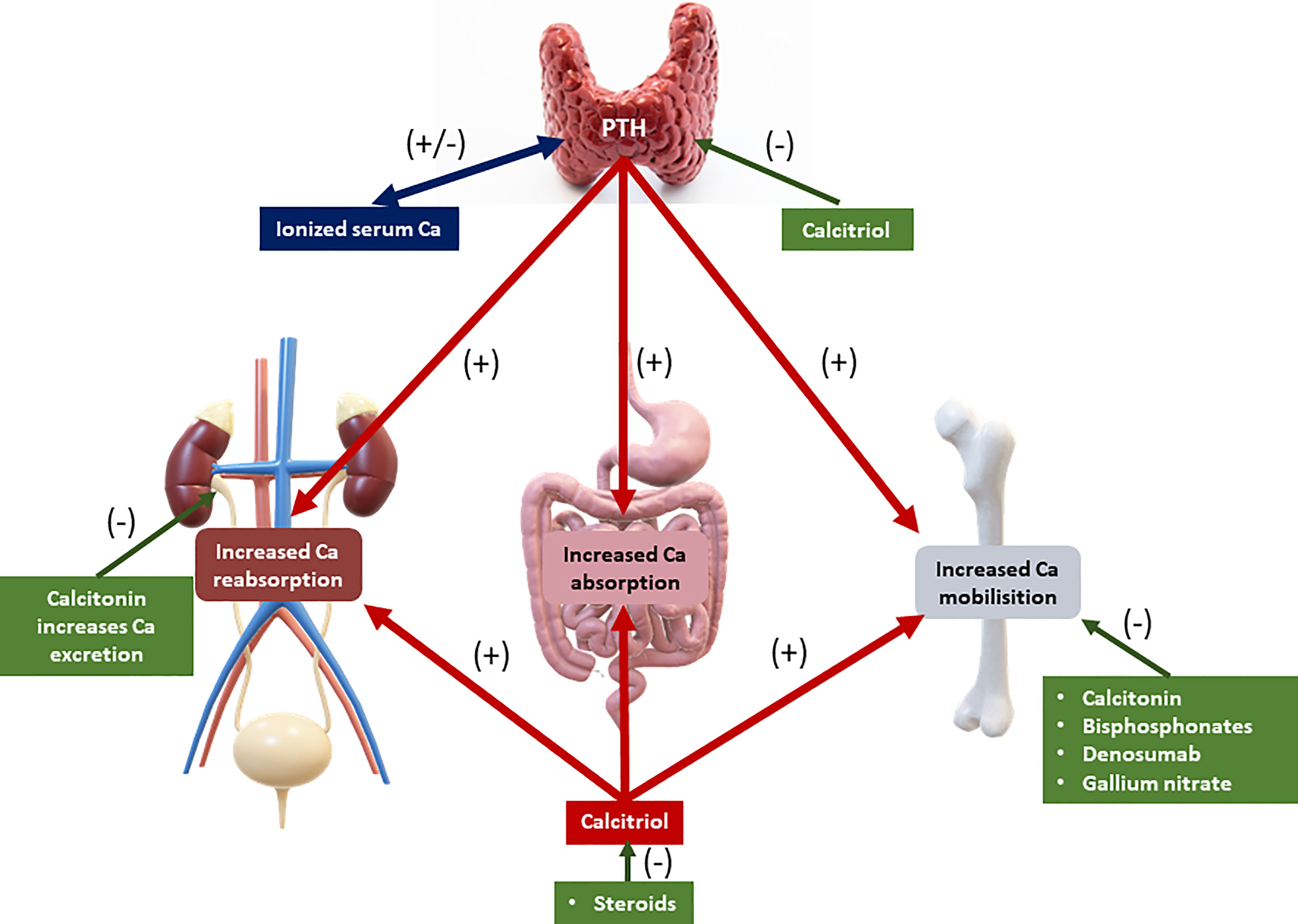
Figure 1 Calcium metabolism. PTH: parathyroid hormone. Green, decreasing effect on serum calcium level. Red: increasing effect on serum calcium level.
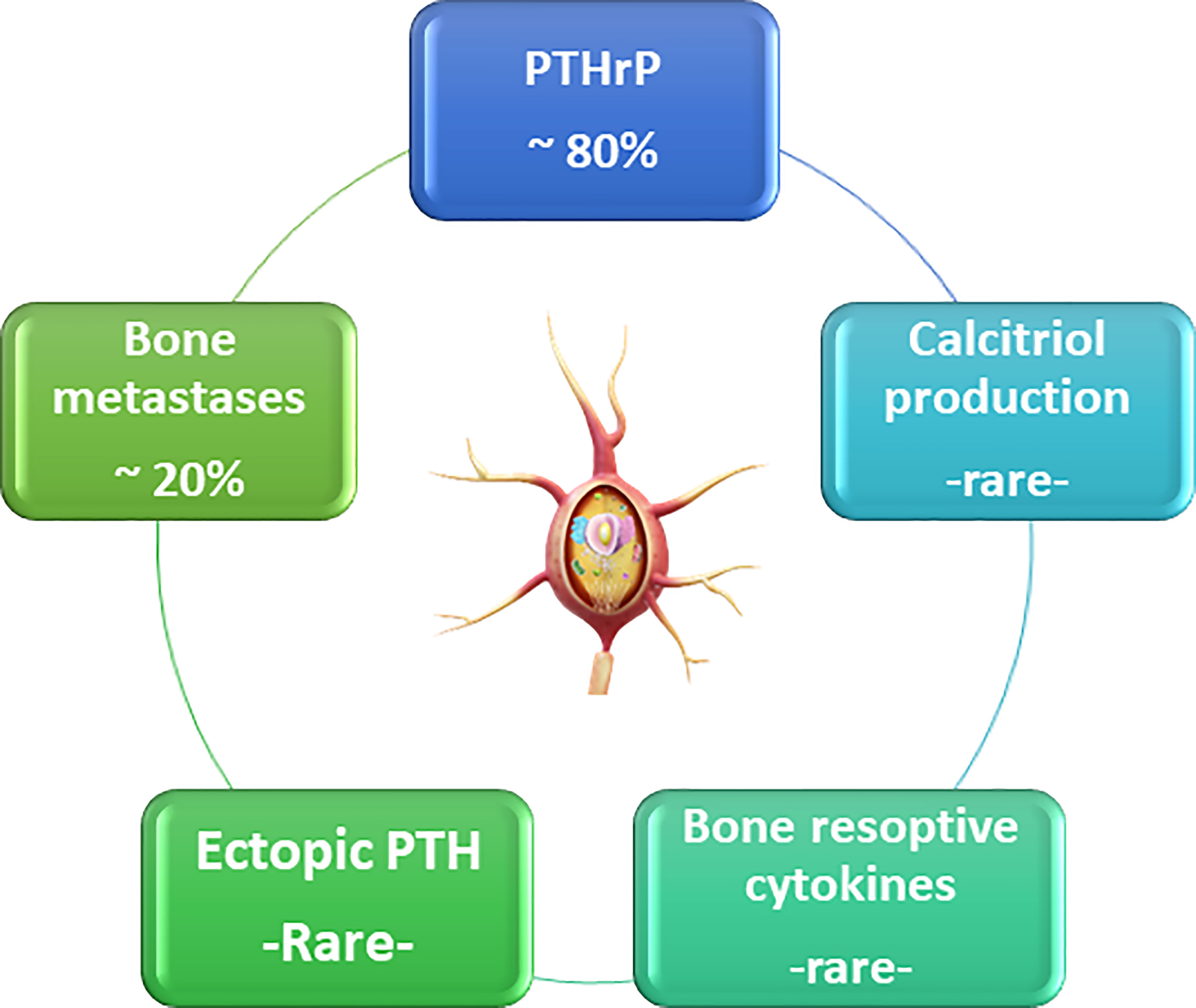
Figure 2 The reasons of malignant hypercalcemia.
The most common cause of hypercalcemia in patients with solid tumors is the secretion of PTHrP (12). This is also known as humoral hypercalcemia of malignancy. This can occur both in solid tumors such as lung, kidney, bladder, breast, head and neck cancers, and in diseases such as non-Hodgkin lymphoma, adult T-cell lymphoma, and chronic myeloid leukemia (13). Hypercalcemia due to PTHrP is frequently observed, especially in tumors with squamous histology (12–14). PTHrP contains homology with PTH, especially according to the sequence of the first 13 amino acids (15). As a result of this close similarity with PTH, PTHrP binds to the same PTH-1 receptor as PTH, thereby activating similar post-receptor pathways. The circulating PTHrP causes stimulation of the PTH receptor in the bone and kidney. This increases bone resorption and increases distal tubular calcium reabsorption, resulting in calcium release from the bone, as well as a decrease in the excretion ability of calcium (14, 15).
The structural difference after the first 13 amino acids of the molecule explains its immunologic difference from PTH. PTHrP is less likely than PTH to stimulate the production of 1,25-dihydroxyvitamin D, so the measurement of 1,25-dihydroxyvitamin D in patients with PTHrP-mediated hypercalcemia may be variable (4, 16). Typical laboratory findings in patients with humoral hypercalcemia are high serum PTHrP and very low or suppressed serum intact PTH and variable serum 1,25-dihydroxyvitamin D levels (16).
In normal individuals, 25-hydroxyvitamin D (calcidiol) is converted to 1,25-dihydroxyvitamin D (calcitriol, the most active form of vitamin D) through 1-hydroxylase in the renal tubules, under the influence of PTH (17). Fibroblast growth factor 23 (FGF-23) inhibits this transformation through hyperphosphatemia (17, 18). Hypercalcemia suppresses the release of PTH and thus the production of 1,25-dihydroxyvitamin D. However, in some tumor types, 25-dihydroxyvitamin D is produced extrarenally from 25-hydroxyvitamin D, independent of PTH control (19).
Uncontrolled production of 1,25-dihydroxyvitamin D (calcitriol) is the cause of almost all cases of hypercalcemia in Hodgkin lymphoma and about one-third of cases of non-Hodgkin lymphoma (20, 21). Hypercalcemia due to this mechanism has also been described in patients with ovarian dysgerminoma and lymphomatoid granulomatosis caused by 1,25-dihydroxyvitamin D (22, 23).
High serum 1,25-dihydroxyvitamin D concentrations may contribute to hypercalcemia by increasing both intestinal calcium absorption and bone resorption. Laboratory evaluation of these patients typically reveals low or suppressed serum PTH and elevated 1,25-dihydroxyvitamin D levels (16).
Hypercalcemia due to bone resorption occurs as a result of the release of mediators, which increase osteoclastic activity by tumor cells in the bone. The soluble protein RANKL, a member of the TNF family, is a central regulator of osteoclast formation, activity, and survival. This protein is synthesized by osteoblasts and T cells and directs the differentiation and activation of osteoclasts (24). Hematopoietic precursor cells exposed to macrophage colony-stimulating factor (M-CSF) express NFκB (RANK)-binding receptors. When RANK and RANKL combine, they induce differentiation into osteoclastic cells (25).
In addition, multiple RANKL-independent osteoclast (OCL) stimulatory factors, including macrophage-derived protein MIP-1a, ILs 3, -8, -6, -17, -18, and Activin A, are produced or induced by cancer cells (5). MIP-1α is a chemokine produced by multiple myeloma (MM) cells in 70% of patients and is a potent inducer of human OCL formation (26). MIP-1α gene expression is highly associated with bone resorption in MM, and high MIP-1α levels are associated with an extremely poor prognosis. MIP-1α acts as a chemotactic factor for OCL precursors and can induce differentiation of OCL progenitors, contributing to RANKL-independent OCL formation (27). In addition, MIP-1α potentiates both RANKL and interleukin (IL)-6-induced OCL formation (28). MIP-1a also increases the expression of β1 integrins on tumor cells, allowing them to settle in the bone marrow. This results in increased production of RANKL, IL-6, vascular endothelial growth factor (VEGF), and tumor necrosis factor-a (TNF-α) by bone marrow stromal cells, further enhancing tumor cell growth, angiogenesis, and bone resorption (29). IL-3 is another OCL-stimulating factor found in the bone marrow. IL-3 can also indirectly induce osteoclastogenesis by increasing the effects of RANKL on the development of OCLs (30). This factor also contributes to bone resorption by stimulating Activin A, which prevents osteoblast differentiation (31). TNF-α is a bifunctional cytokine that can induce OCL formation and suppress OB differentiation through its effects on Runx2 and Gfi-1 expression in bone marrow stromal cells (32). Transforming growth factor (TGF-β) is an upregulated factor in bone metastasis that has multiple effects on the tumor-bone microenvironment. TGF-β can increase the production of IL-6 and VEGF by tumor cells (33).
Apart from these three main mechanisms, there are other less common causes of hypercalcemia due to malignancy.
Tumors secreting ectopic PTH have been reported as case reports in the literature (34). Examples of these tumor types are ovarian carcinoma, small cell and squamous cell lung carcinomas, neuroectodermal tumors, thyroid papillary carcinoma, metastatic rhabdomyosarcoma, pancreatic malignancy, and gastric carcinoma (35). Laboratory analyses of these patients typically show elevated PTH, calcium, and low phosphorus levels (36).
Another rare cause of hypercalcemia is pseudohypercalcemia. This is due to measurement errors due to calcium binding to an abnormal immunoglobulin. It is possible to distinguish this using atomic absorption spectrophotometry (37).
The miRNAs act as subtle modulators in maintaining bone homeostasis. Basic evidence that miRNAs are essential for osteoclastogenesis is provided by genetic studies that delete DICER1, an enzyme essential for their biogenesis (38). DICER-deficient mice and osteoclast-specific DICER gene deficiency lead to impairment in both OC formation and activity (38). It is known that there are miRNAs that support and suppress the formation of OCs (39). Although the functions of a few of them are known, with increasing studies in the near future, miRNAs will also be used in the treatment of problems such as malignancy and hypercalcemia.
Symptoms of hypercalcemia depend on at least two factors: the degree of hypercalcemia and the rate of change in serum calcium. Degrees of hypercalcemia relative to serum total calcium level are as follows: mild hypercalcemia, 10.5 to 11.9 mg/dL; moderate hypercalcemia, 12 to 13.9 mg/dL; and severe hypercalcemia, 14 mg/dL or above (40). Mild hypercalcemia may be asymptomatic or associated with mild nonspecific symptoms such as numbness and pain (41). In contrast, severe, rapidly progressive hypercalcemia can be associated with a variety of life-threatening symptoms (41, 42).
It is important to note that at least two factors, such as the degree of hypercalcemia and the rate of its development, play a role in the development of clinically overt hypercalcemia. Patients with malignant hypercalcemia develop hypercalcemia at a very high level and in a short time, and therefore they are more symptomatic than patients with other causes of hypercalcemia (40). The major systems affected by hypercalcemia are neuropsychiatric, gastrointestinal, and renal systems (43–45). Almost all affected individuals have GI symptoms. Mild calcium elevation may manifest as anorexia and constipation. Nausea and vomiting may develop in patients with severe hypercalcemia, but these conditions can easily be confused with the adverse effects of tumor treatment or symptoms directly produced by the tumor itself (3, 45). Cramping abdominal pain, such as those seen in people with primary hyperparathyroidism, is rarely encountered, but severe outcomes such as peptic ulceration and pancreatitis are much less common in malignant hypercalcemia (46).
Hypercalcemia impairs the concentration ability of the kidney (47). Tubular damage causes acquired renal tubular acidosis, glycosuria, and aminoaciduria (47). Renal manifestations consist of nephrogenic diabetes insipidus resulting in polyuria. All patients with clinically overt hypercalcemia have volume depletion resulting in an increase in creatinine level and a decrease in glomerular filtration rate, explained by a decrease in oral intake due to polyuria and nausea and vomiting (47, 48). Nephrocalcinosis and nephrolithiasis require long-term hypercalcemia and therefore are not common in malignant hypercalcemia (49).
Neuropsychiatric symptoms such as apathy, mood changes, and fatigue are often seen as symptoms of hypercalcemia, which can be overlooked and can be attributed to the underlying neoplasm. In oncology patients, muscle strength defect leads to mobility restriction, which leads to more calcium resorption from the bone and increases hypercalcemia (50). As hypercalcemia continues to worsen, severe symptomatology may occur, including changes in mental status, confusion, and eventually coma (50, 51). Rarely, patients may even develop posterior reversible leukoencephalopathy syndrome (PRES), which is manifested by headaches, seizures, and imaging findings of subcortical edema (52).
Cardiovascular system findings are seen as a shortening of the QT interval on electrocardiograms (53). Malignant ventricular arrhythmias such as ventricular fibrillation may develop in patients with severe hypercalcemia (54).
Bone pain is a common symptom, both due to the malignancy itself and hypercalcemia. Bone pain may be associated with increased intramedullary pressure, ischemia, or the presence of metastases within the bone causing areas of microfracture, but the symptom is also present in the absence of demonstrable metastatic disease (55).
The first evaluation of patients with clinical signs of hypercalcemia starts with serum calcium measurement. Serum calcium is the sum of the physiologically inactive carrier-bound calcium and the active form of serum calcium, ionized calcium (56). Therefore, if the calcium level is found to be high in the analysis, it is necessary to know whether these tests measure ionized calcium or total calcium. Total calcium levels can be affected by many factors such as serum protein and pH. In such cases, the value of ionized calcium is more important (57). Serum albumin measurements are necessary for the interpretation of serum calcium levels because calcium homeostasis is greatly affected by albumin concentrations. If albumin is abnormal, serum calcium should be corrected using the following formula (58):
Corrected calcium = Total calcium + [0.8 × (4.0 – albumin)]
Protein levels and thus calcium levels may be elevated in patients with severe dehidratation (57). It is important to remember that the albumin-calcium system is highly sensitive to pH and changes in pH can alter the fraction of albumin-bound calcium ions (59). Therefore, it is always recommended to confirm high serum calcium with a repeat test (60).
The next step in the evaluation of a patient with malignancy-related hypercalcemia consists of measuring both PTH and PTHrP. PTH and PTHrP are similar molecules; therefore, both will not rise at the same time unless there is more than one cause. In most cases of malignancy, serum PTH levels appear to be suppressed or normal (10). High-normal PTH levels in the setting of hypercalcemia suggest the presence of PTH-mediated hypercalcemia or parathyroid carcinoma (61). Serum phosphorus and other electrolytes should be measured, as hypercalcemia due to PTHrP or PTH may cause hypophosphatemia, hyperchloremia, and mild metabolic alkalosis (62). If PTHrP levels are low, the next step should include measuring 1,25-dihydroxyvitamin D levels to screen for vitamin D-mediated hypercalcemia. In patients with low PTH, PTHrp, and 1,25-dihydroxyvitamin D, hypercalcemia due to osteolytic metastases may be considered the cause of malignancy-associated hypercalcemia (61).
Although rare, patients may have familial hypercalcemia symptoms together with malignancy. The 24-hour urinary calcium clearance-creatinine clearance ratio (FeCa) may be valuable for the assessment of familial hypocalciuric hypercalcemia (63). If FeCa is low (less than 0.01), familial hypocalciuric hypercalcemia should be suspected and definitive evaluation may include genetic testing for mutations in the CASR, AP2S1, or GNA11 genes (64). However, it is important to note that more than one malignancy-associated hypercalcemia mechanism can be seen in oncology patients.
The main goal in the treatment of hypercalcemia is to find the underlying cause and initiate treatment (Table 1). Although all attention can be given to this goal, especially in mild hypercalcemia, moderate-to-severe hypercalcemia also requires concomitant symptomatic treatment. Patients with a calcium value of >14 mg/dL (>3.5 mmol/L) require more aggressive treatment. In addition, patients with neurologic symptoms (eg, lethargy, stupor), regardless of serum calcium levels, require urgent aggressive treatment (65, 66).
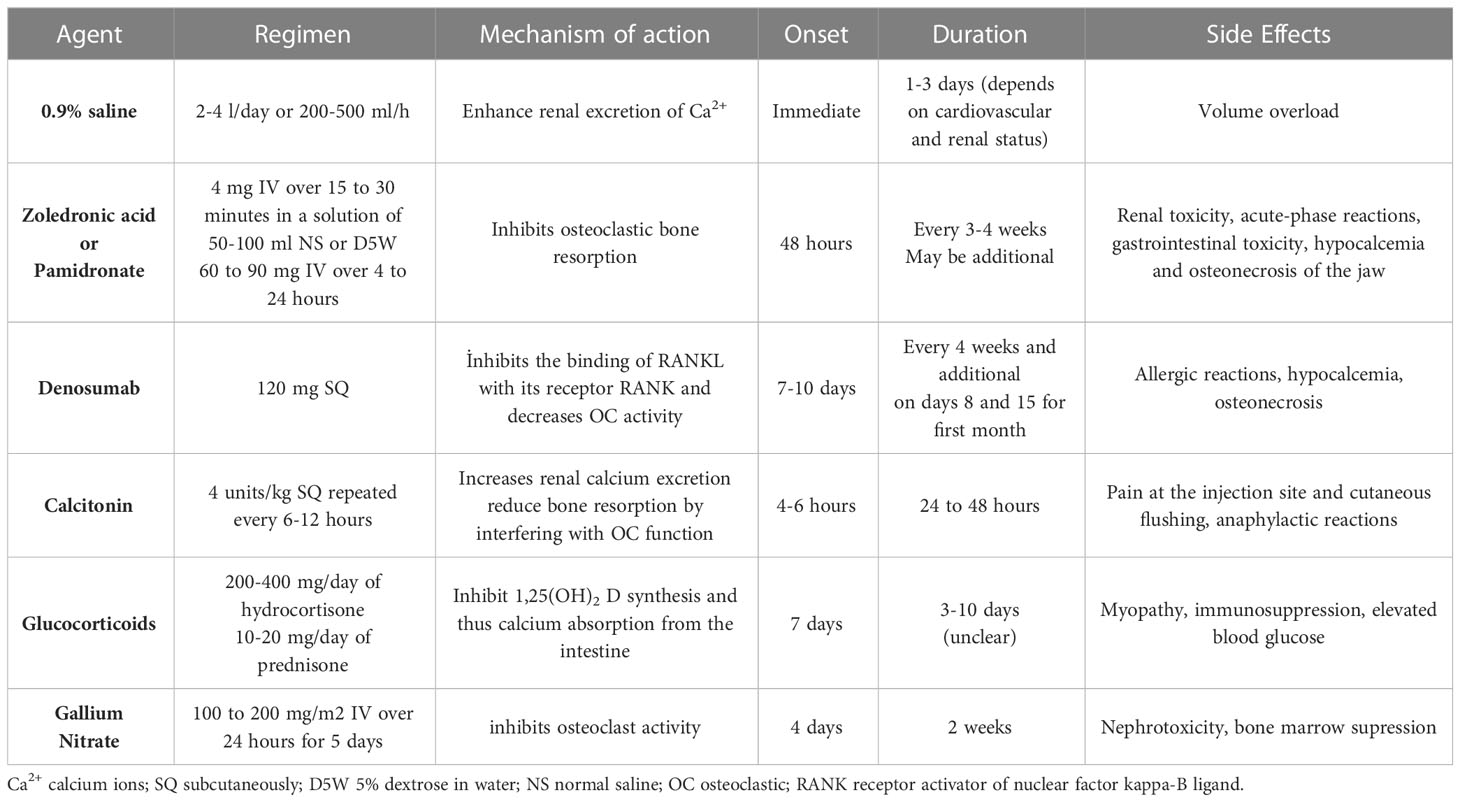
Table 1 Treatment options for hypercalcemia of malignancy.
As mentioned before, patients with hypercalcemia present with nausea, vomiting, inadequate hydration due to altered mental status, and a dehydrated state due to nephrogenic diabetes insipidus caused by hypercalcemia (47, 67). In addition, the volume reduction itself also reduces the renal clearance of calcium due to increased calcium reabsorption due to hypovolemia in the kidneys, resulting in a vicious circle. Therefore, hydration is one of the cornerstones of treatment in hypercalcemia. Isotonic crystalloid solutions (e.g. normal saline) should be used for IV hydration. Typically, patients with advanced hypercalcemia should start at a rate of approximately 200-300 mL/hour (68). Patients should be evaluated periodically for signs of fluid overload (e.g.shortness of breath, edema). IV hydration rate should be reduced in patients with underlying heart and kidney disease to minimize the risk of symptomatic fluid overload. It is important to note that the routine use of loop diuretics such as furosemide is not recommended due to volume depletion and the development of electrolyte abnormalities. The use of furosemide should be reserved for patients who develop signs of fluid overload while receiving IV hydration (69).
Calcitonin is a potent hypocalcemic hormone produced by the C-cells of the thyroid (70). Pharmacologic doses of calcitonin reduce serum calcium concentration by increasing renal calcium excretion and, more importantly, by reducing bone resorption by interfering with OC function (71). In addition, calcitonin inhibits the osteoclastogenic effects of the NF-kB ligand (RANKL) receptor activator. It should be administered intramuscularly or subcutaneously (72). Calcitonin is safe and adverse effects are not expected except for a hypersensitivity reaction with mild nausea. The starting dose is 4 units/kg (73). Serum calcium is repeated after 4 to 6 hours. If a hypocalcemic response is noted, the patient is calcitonin sensitive and calcitonin may be repeated every 12 hours for a total of 24 to 48 hours. If the response is unsatisfactory, the dose may be increased to 8 units/kg every 6 to 12 hours (total treatment duration 24 to 48 hours) (74). Although a relatively weak agent, it works rapidly and lowers serum calcium concentrations by a maximum of 1 to 2 mg/dL (0.3 to 0.5 mmol/L) starting within 4 to 6 hours. The efficacy of calcitonin is limited to the first 48 hours, even with repeated doses; this is probably due to the development of tachyphylaxis due to receptor downregulation (75). Due to its limited duration of action, calcitonin is more beneficial in symptomatic patients with calcium >14 mg/dL (3.5 mmol/L) when combined with hydration and bisphosphonates (or denosumab in bisphosphonate-resistant patients).
Bisphosphonates inhibit osteoclastic bone resorption by binding to hydroxyapatite binding sites on bone surfaces (76). When OCs begin to resorb bisphosphonate-impregnated bone, the bisphosphonate released during resorption impairs the OCs’ ability to form folded edges, adhere to the bone surface, and produce protons necessary for sustained bone resorption. Bisphosphonates also reduce their activity by reducing OC progenitor development and recruitment and promoting OC apoptosis (77, 78).
In addition to their inhibitory effects on OCs, bisphosphonates appear to have a beneficial effect on osteoblasts. The mechanism of this effect has been attributed to connexin 43, a gap junction protein that facilitates the activation of protein kinases. However, this anti-apoptotic effect probably does not contribute significantly to the anti-osteoporotic efficacy of bisphosphonates above their potent antiresorptive effects (79).
Bisphosphonates should be given within 48 hours of diagnosis at the latest because they take approximately 2 to 4 days to take effect. Pamidronate is given as 60 to 90 mg IV over 4 to 24 hours (80). Zoledronic acid is given as 4 mg IV over 15 to 30 minutes (81). One of the serious adverse effects of bisphosphonates is nephrotoxicity (82). In patients presenting with abnormal renal function due to underlying renal disease or hypercalcemia, the benefit of treatment should be reviewed and the dose should be reduced if necessary. In addition to bisphosphonate therapy, adequate hydration can help maintain kidney function. For refractory hypercalcemia, retreatment with zoledronic acid may be considered, but a second dose may be administered as soon as 7 days after the first treatment (83). Renal function should be carefully monitored with serum creatinine level before additional doses of zoledronic acid are given. Recommended dose reduction based on creatinine clearance is follows: GFR >60 mL/min, 4 mg; GFR 50 to 60 mL/min, 3.5 mg; GFR 40 to 49 mL/min, 3.3 mg; and GFR 30 to 39 mL/min, 3.0 mg (61).
The most common adverse effect of bisphosphonates is related to impaired renal function. We have already mentioned the adjustment required for this. Other common adverse effects include bone pain and flu symptoms during the first 1 to 2 days after the infusion. Osteonecrosis of the jaw, which is very rare but very important for the patient’s quality of life, can be seen in patients who receive high-dose and long-term treatment, those who have invasive dental procedures during treatment, and patients with poor oral care (84, 85).
Glucocorticoids help lower serum calcium levels by several mechanisms. Glucocorticoids can suppress the synthesis of extrarenal calcitriol through activated mononuclear cells in the lung and lymph nodes. Namely, by inhibiting 1-alpha hydroxylase, they inhibit 1,25(OH)2 D synthesis and thus calcium absorption from the intestine (86). In addition, they also have inhibitory properties on cytokines released directly from tumor cells, which inhibit osteoclastic bone resorption that will occur with these cytokines (87). Glucocorticoids are usually given as 200 to 400 mg/day of hydrocortisone for 3 to 4 days followed by 10 to 20 mg/day of prednisone for 7 days. Treatment should be continued for a maximum of 10 days and should not be continued if hypercalcemia does not respond (30).
Denosumab is a humanized monoclonal antibody that inhibits the binding of RANKL with its receptor RANK (88). It is the agent used in the second-line treatment of patients with bisphosphonate-resistant malignancy hypercalcemia. In a study in which 120 mg subcutaneous denosumab every 4 weeks was compared with 4 mg IV zoledronic acid intravenously every 4 weeks, it was reported that denosumab was more effective in preventing malignant hypercalcemia in patients with metastatic bone disease (89). This study showed that denosumab delayed the first episode of malignancy-associated hypercalcemia (hazard ratio [HR] 0.63)and also reduced the risk of developing recurrent hypercalcemia by 52%. Compared with 40% of the zoledronic acid group, only 31% of those receiving denosumab developed hypercalcemia (90).
The adverse-effect profile of denosumab is also different from that of bisphosphonates. Denosumab, unlike bisphosphonates, is not excreted by the kidney and, as a result, there is no restriction on its use in patients with chronic kidney disease or a decrease in GFR for any reason, for whom bisphosphonates are used with caution or contraindicated (91). Given that pharmacokinetics and pharmacodynamics are not affected by renal status, renal adjustment has not been reported to be necessary. However, because it is a more potent agent, the risk of hypocalcemia is higher in bisphosphonates (92). Careful monitoring of serum calcium levels is required because this risk is seen to be higher in patients with renal failure. Denosumab has also been reported to be effective in cases of parathyroid carcinoma resistant to cinacalcet and IV bisphosphonates (93, 94). Denosumab cesassion may lead to a rapid increase in the concentrations of bone turnover markers. Usually, this markers elevated to above pre-treatment levels (95). This fenomenon also associated with a decline in bone mineral density at skeletal sites and it described as “rebound phenomenon” (96). The underlying mechanism of this phenomenon is thought to be increased RANKL due to discontinuation of antiresorptive agent. Abnormally increased RANKL expression is lead to a mass increase in osteoclastogenesis by premature osteoclasts accumulated during RANKL inhibition (95). For prevention fracture risk and hypercalcemia, bifosfonates could be preferred as subsequent treatment in patients discontinuing denosumab (97).
Cinacalcet directly decreases PTH levels by increasing the sensitivity of the calcium-sensing receptor to extracellular calcium, thus leading to a decrease in serum calcium levels (95). The drug is approved for use in tertiary and secondary hyperparathyroidism and refractory parathyroid carcinoma. Parathyroid carcinoma is the only malignancyfor which cinacalcet is approved (96).
Gallium nitrate is thought to exert its hypocalcemic effect by inhibiting calcium absorption from bone (97). Gallium nitrate is localized where bone remodeling occurs and inhibits osteoclast activity. Compared with IV pamidronate, gallium appeared to have generally similar calcium-lowering effects, although it was more successful than pamidronate in epidermoid tumors (98). Gallium nitrate was found to be more potent in a study compared with calcitonin (99). It has also been shown that gallium is effective in tamoxifen-induced hypercalcemia and provides normocalcemia while patients continue tamoxifen treatment (100). The recommended dose is 100 to 200 mg/m2 IV over 24 hours for 5 days (101). It is well tolerated and does not show significant nephrotoxicity, but is not currently FDA approved. Gallium nitrate was removed from the US market in 2012
Dialysis is the method used for the treatment of patients in whom optimal hydration cannot be achieved safely due to heart or kidney failure or who have hypercalcemia unresponsive to other treatments (6). It can also be performed as an emergency treatment in patients who develop arrhythmia due to severe hypercalcemia (102). The dialysis solution used for this purpose consists of a calcium-free acetate solution or a dialysate with a very low calcium level (103).
Hypercalcemia due to malignancy is a very important issue in terms of both being the most common cause of hypercalcemia and affecting the prognosis of patients with cancer. Hypercalcemia can present with a wide range of findings from mild symptoms to life-threatening symptoms. Hematology,oncology, internal medicine,and palliative care specialists should have sufficient knowledge about the diagnosis and management of hypercalcemia.
All authors meet the ICMJE authorship criteria and were involved in the whole writing process.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jick S, Li L, Gastanaga VM, Liede A. Prevalence of hypercalcemia of malignancy among cancer patients in the UK: analysis of the clinical practice research datalink database. Cancer Epidemiol (2015) 39(6):901–7. doi: 10.1016/j.canep.2015.10.012
2. Burt ME, Brennan MF. Incidence of hypercalcemia and malignant neoplasm. Arch Surg (1980) 115:704–7. doi: 10.1001/archsurg.1980.01380060012004
3. Endres DB. Investigation of hypercalcemia. Clin Biochem (2012) 45:954–63. doi: 10.1016/j.clinbiochem.2012.04.025
4. Mirrakhimov AE. Hypercalcemia of malignancy: An update on pathogenesis and management. N Am J Med Sci (2015) 7(11):483–93. doi: 10.4103/1947-2714.170600
5. Sternlicht H, Glezerman IG. Hypercalcemia of malignancy and new treatment options. Ther Clin Risk Manag (2015) 11:1779–88. doi: 10.2147/TCRM.S83681
6. Koo WS, Jeon DS, Ahn SJ, Kim YS, Yoon YS, Bang BK. Calcium-free hemodialysis for the management of hypercalcemia. Nephron (1996) 72:424. doi: 10.1159/000188907
7. Lambers TT, Bindels RJ, Hoenderop JG. Coordinated control of renal Ca2+ handling. Kidney Int (2006) 69:650. doi: 10.1038/sj.ki.5000169
8. Bronner F. Mechanisms of intestinal calcium absorption. J Cell Biochem (2003) 88:387–93. doi: 10.1002/jcb.10330
9. Wysolmerski JJ. Parathyroid hormone-related protein: An update. J Clin Endocrinol Metab (2012) 97:2947–56. doi: 10.1210/jc.2012-2142
11. Clines GA, Guise TA. Hypercalcaemia of malignancy and basic research on mechanisms responsible for osteolytic and osteoblastic metastasis to bone. EndocrRelat Cancer (2005) 12(3):549–83. doi: 10.1677/erc.1.00543
12. Burtis WJ, Brady TG, Orloff JJ, Ersbak JB, Warrell RP Jr, Olson BR, et al. Immunochemical characterization of circulating parathyroid hormone-related protein in patients with humoral hypercalcemia of cancer. New Engl J Med (1990) 322(16):1106–12. doi: 10.1056/NEJM199004193221603
13. Orloff JJ, Wu TL, Stewart AF. Parathyroid hormone-like proteins: biochemical responses and receptor interactions. Endocr Rev (1989) 10(4):476–95. doi: 10.1210/edrv-10-4-476
14. Budayr AA, Nissenson RA, Klein RF, Pun KK, Clark OH, Diep D, et al. Increased serum levels of a parathyroid hormone-like protein in malignancy-associated hypercalcemia. Ann Intern Med (1989) 111(10):807–12. doi: 10.7326/0003-4819-111-10-807
15. Johnson RW, Nguyen MP, Padalecki SS, Grubbs BG, Merkel AR, Oyajobi BO, et al. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical hedgehog signaling. Cancer Res (2011) 71:822–31. doi: 10.1158/0008-5472
16. Lafferty FW. Differential diagnosis of hypercalcemia. J Bone Miner Res (1991) 6 Suppl 2:S51–9. doi: 10.1002/jbmr.5650061413
19. Roodman GD. Mechanisms of bone lesions in multiple myeloma and lymphoma. Cancer (1997) 80:1557. doi: 10.1002/(SICI)1097-0142(19971015)80:8+<1557::AID-CNCR5>3.0.CO;2-H
20. Seymour JF, Grill V, Martin TJ, Lee N, Firkin F. Hypercalcemia in the blastic phase of chronic myeloid leukemia associated with elevated parathyroid hormone-related protein. Leukemia (1993) 7:1672.
21. Shivnani SB, Shelton JM, Richardson JA, Maalouf NM. Hypercalcemia of malignancy with simultaneous elevation in serum parathyroid hormone–related peptide and 1,25-dihydroxyvitamin d in a patient with metastatic renal cell carcinoma. Endocr Pract (2009) 15:234. doi: 10.4158/EP.15.3.234
22. Lim D, Oliva E. Gynecological neoplasms associated with paraneoplastic hypercalcemia. Semin Diagn Pathol (2019) 36(4):246–59. doi: 10.1053/j.semdp.2019.01.003
23. Hosseini B, Leibl M, Stoffman J, Morris A. Two cases of hypercalcemia in pediatric ovarian dysgerminoma. J Obstet Gynaecol Can (2019) 41(5):660–5. doi: 10.1016/j.jogc.2018.05.004
24. Roodman GD. Mechanisms of bone metastasis. New Engl J Med (2004) 350(16):1655–64. doi: 10.1056/NEJMra030831
25. Horwitz M, Hodak S, Stewart A. Favus M, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism. Washington, DC: American Society of Bone and Mineral Research (2008). p. 307–12.
26. Choi SJ, Cruz JC, Craig F, Chung H, Devlin RD, Roodman GD, et al. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood (2000) 96(2):671–5. doi: 10.1182/blood.V96.2.671
27. Yee J. Hypercalcemia. xPharm: Compr Pharmacol Reference (2007), 1–6. doi: 10.1016/B978-008055232-3.60633-6
28. Politou M, Terpos E, Anagnostopoulos A, Szydlo R, Laffan M, Layton M, et al. Role of receptor activator of nuclear factor-kappa b ligand (RANKL), osteoprotegerin and macrophage protein 1-alpha (MIP-1a) in monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol (2004) 126:686–9. doi: 10.1111/j.1365-2141.2004.05092.x
29. Terpos E, Christoulas D, Gavriatopoulou M. Biology and treatment of myeloma related bone disease. Metabolism (2018) 80:80–90. doi: 10.1016/j.metabol.2017.11.012
30. Goldner W. Cancer-related hypercalcemia. J Oncol Pract (2016) 12(5):426–32. doi: 10.1200/JOP.2016.011155
31. Sugatani T, Alvarez UM, Hruska KA. Activin a stimulates IkappaB-alpha/NFkappaB and RANKexpression for osteoclast differentiation, but not AKT survival pathway in osteoclast precursors. J Cell Biochem (2003) 90:59–67. doi: 10.1002/jcb.10613
32. Abildgaard N, Glerup H, Rungby J, Bendix-Hansen K, Kassem M, Brixen K, et al. Biochemical markers of bone metabolism reflect osteoclastic and osteoblastic activity in multiple myeloma. Eur J Haematol (2000) 64:121–9. doi: 10.1034/j.1600-0609.2000.90074.x
33. Elliott P, Kostenuik P, Chen C, Kelley M, Hawkins N, Housman J, et al. Denosumab is a selective inhibitor of human receptor activator of NF-KB ligand that blocks osteoclast formation. Vitro vivo. Eur J Cancer Suppl (2006) 4:62. doi: 10.1016/S1359-6349(06)70202-6
34. Yoshimoto K, Yamasaki R, Sakai H, Tezuka U, Takahashi M, Iizuka M, et al. Ectopic production of parathyroid hormone by small cell lung cancer in a patient with hypercalcemia. J Clin Endocrinol Metab (1989) 68:976. doi: 10.1210/jcem-68-5-976
35. VanHouten JN, Yu N, Rimm D, Dotto J, Arnold A, Wysolmerski JJ, et al. Hypercalcemia of malignancy due to ectopic transactivation of the parathyroid hormone gene. J Clin Endocrinol Metab (2006) 91:580. doi: 10.1210/jc.2005-2095
36. Nakajima K, Tamai M, Okaniwa S, Nakamura Y, Kobayashi M, Niwa T, et al. Humoral hypercalcemia associated with gastric carcinoma secreting parathyroid hormone: a case report and review of the literature. Endocr J (2013) 60:557. doi: 10.1507/endocrj.ej12-0406
37. Schwab JD, Strack MA, Hughes LD, Shaker JL. Pseudohypercalcemia in an elderly patient with multiple myeloma: report of a case and review of literature. Endocr Pract (1995) 1:390–2. doi: 10.4158/EP.1.6.390
38. Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE. Mello CC.Potent and specific genetic interference by double-stranded RNAin caenorhabditis elegans. Nature (1998) 391:806–11. doi: 10.1038/35888
39. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol (2014) 15:509–24. doi: 10.1038/nrm3838
40. Stewart AF. Clinical practice. hypercalcemia associated with cancer. N Engl J Med (2005) 352(4):373–9. doi: 10.1056/NEJMcp042806
41. Wright JD, Tergas AI, Ananth CV, Burke WM, Hou JY, Chen L, et al. Quality and outcomes of treatment of hypercalcemia of malignancy. Cancer Invest (2015) 33(8):331–9. doi: 10.3109/07357907.2015.1047506
42. Hamilton F, Carroll R, Hamilton W, Salisbury C. The risk of cancer in primary care patients with hypercalcaemia: A cohort study using electronic records. Br J Cancer (2014) 111:1410–2. doi: 10.1038/bjc.2014.433
43. Levi M, Ellis MA, Berl T. Control of renal hemodynamics and glomerular filtration rate in chronic hypercalcemia. role of prostaglandins, renin-angiotensin system, and calcium. J Clin Invest (1983) 71(6):1624–32. doi: 10.1172/JCI110918
44. Inzucchi SE. Understanding hypercalcemia. Its Metab basis signs symptoms. Postgrad Med (2004) 115(4):69–70. doi: 10.3810/pgm.2004.04.1486
45. Carnaille B, Oudar C, Pattou F, Combemale F, Rocha J, Proye C. Pancreatitis and primary hyperparathyroidism: forty cases. Aust N Z J Surg (1998) 68(2):117–9. doi: 10.1111/j.1445-2197.1998.tb04719.x
46. Gardner EC Jr, Hersh T. Primary hyperparathyroidism and the gastrointestinal tract. South Med J (1981) 74:197–9. doi: 10.1097/00007611-198102000-00019
47. Lins LE. Reversible renal failure caused by hypercalcemia. A retrospective study. Acta Med Scand (1978) 203(4):309–14. doi: 10.1111/j.0954-6820.1978.tb14879.x
48. Caruana RJ, Buckalew VM Jr. The syndrome of distal renal tubular acidosis. Clin Lab findings 58 cases. Med Baltimore (1988) 67:84–99. doi: 10.1097/00005792-198803000-00002
49. Parfitt AM. Hypercalcaemic nephropathy. Med J Aust (1964) 2:127–34. doi: 10.5694/j.1326-5377.1964.tb115035.x
50. Nakajima N, Ueda M, Nagayama H, Yamazaki M, Katayama Y. Posterior reversible encephalopathy syndrome due to hypercalcemia associated with parathyroid hormone-related peptide: a case report and review of the literature. Intern Med (2013) 52(21):2465–8. doi: 10.2169/internalmedicine.52.0444
51. Camara-Lemarroy CR, Gonzalez-Moreno EI, Ortiz-Corona JJ, Yeverino-Castro SG, Sanchez-Cardenas M, Nuñez-Aguirre S, et al. Posterior reversible encephalopathy syndrome due to malignant hypercalcemia: physiopathological considerations. J Clin Endocrinol Metab (2014) 99(4):1112–6. doi: 10.1210/jc.2013-3487
52. Shane E, Irani D. Favus MJ, editor. Primer on the metabolic bone diseases and disorders of mineral metabolism, 6. Washington, DC: American Society for Bone and Mineral Research (2006).
53. Diercks DB, Shumaik GM, Harrigan RA, Brady WJ, Chan TC. Electrocardiographic manifestations: electrolyte abnormalities. J Emerg Med (2004) 27(2):153–60. doi: 10.1016/j.jemermed.2004.04.006
54. Kiewiet RM, Ponssen HH, Janssens ENW, Fels W. Ventricular fibrillation in hypercalcaemic crisis due to primary hyperparathyroidism. Neth J Med (2004) 62(3):94–6.
55. Zagzag J, Hu MI, Fisher SB, Perrier ND. Hypercalcemia and cancer: Differential diagnosis and treatment. CA Cancer J Clin (2018) 68(5):377–86. doi: 10.3322/caac.21489
56. Baird GS. Ionized calcium. Clin Chim Acta (2011) 412(9-10):696–701. doi: 10.1016/j.cca.2011.01.004
57. Meng QH, Wagar EA. Laboratory approaches for the diagnosis and assessment of hypercalcemia. Crit Rev Clin Lab Sci (2015) 52(3):107–19. doi: 10.3109/10408363.2014.970266
58. Ladenson JH, Lewis JW, McDonald JM, Slatopolsky E, Boyd JC. Relationship of free and total calcium in hypercalcemic conditions. J Clin Endocrinol Metab (1979) 48:393–7. doi: 10.1210/jcem-48-3-393
59. Sargent JT, Smith OP. Haematological emergencies managing hypercalcaemia in adults and children with haematological disorders. Br J Haematol (2010) 149(4):465–77. doi: 10.1111/j.1365-2141.2010.08173.x
60. Morton R, Lipton A. Abeloff's clinical oncology, 4th edition part II – problems common to cancer and its therapy section d – metabolic and paraneoplastic syndromes. Chapter 48 – Hypercalcemia (2020).
61. Schilling T, Pecherstorfer M, Blind E, Leidig G, Ziegler R, Raue F. Parathyroid hormone-related protein (PTHrP) does not regulate 1,25-dihydroxyvitamin d serum levels in hypercalcemia of malignancy. J Clin Endocrinol Metab (1993) 76:801. doi: 10.1210/jcem.76.3.8445039
62. Kremer R, Shustik C, Tabak T, Papavasiliou V, Goltzman D. Parathyroid-hormone-related peptide in hematologic malignancies. Am J Med (1996) 100:406. doi: 10.1016/S0002-9343(97)89515-0
63. Afzal M, Kathuria P. Familial hypocalciuric hypercalcemia. [Updated 2022 jul 19]. In: StatPearls. Treasure Island (FL: StatPearls Publishing (2022).
64. Ruggeri RM, Campennì A, Cannavo S. Familial hypocalciuric hypercalcemia: grey zones of the differential diagnosis from primary hyperparathyroidism: a case report. APMB (2021) 109(2):11. doi: 10.13129/1828-6550/APMB.109.2.2021.CCS3
65. Bilezikian JP. Clinical review 51: Management of hypercalcemia. J Clin Endocrinol Metab (1993) 77:1445.
66. Bilezikian JP. Management of acute hypercalcemia. N Engl J Med (1992) 326:1196. doi: 10.1056/NEJM199204303261806
67. Hosking DJ, Cowley A, Bucknall CA. Rehydration in the treatment of severe hypercalcaemia. Q J Med (1981) 50:473.
69. Makras P, Papapoulos SE. Medical treatment of hypercalcaemia. Hormones (Athens) (2009) 8:83. doi: 10.14310/horm.2002.1225
70. Wimalawansa SJ. Chapter 53 - calcitonin: History, physiology, pathophysiology and therapeutic applications. Osteoporosis Men (Second Edition) Effects Gender Skeletal Health (2010), 653–66. doi: 10.1016/B978-0-12-374602-3.00053-5
71. Tobeiha M, Moghadasian MH, Amin N, Jafarnejad S. RANKL/RANK/OPG pathway: A mechanism involved in exercise-induced bone remodeling. BioMed Res Int (2020) 6910312. doi: 10.1155/2020/6910312
72. Deftos LJ, First BP. Calcitonin as a drug. Ann Intern Med (1981) 95:192. doi: 10.7326/0003-4819-95-2-192
73. Chevallier B, Peyron R, Basuyau JP, Bastit P, Comoz M. Human calcitonin in neoplastic hypercalcemia. Results prospective randomized trial. Presse Med (1988) 17:2375.
74. Austin LA, Heath H. Calcitonin: physiology and pathophysiology. N Engl J Med (1981) 304:269. doi: 10.1056/NEJM198101293040505
75. Wisneski LA. Salmon calcitonin in the acute management of hypercalcemia. Calcif Tissue Int (1990) 46 Suppl:S26. doi: 10.1007/BF02553290
76. Fleisch H. Bisphosphonates in bone disease. In: From the laboratory to the patient, 4th edn. New York: Academic Press (2000).
77. Ebetino FH, Dansereau SM. Bisphosphonate antiresorptive structure–activity relationships. In: Bijvoet OLM, Fleisch HA, Canfield RE, editors. Russell RGG (eds) bisphosphonate on bones. Elsevier, Amsterdam (1995). p. pp139–153.
78. Luckman SP, Coxon FP, Ebetino FH Russell RG, Rogers MJ. Heterocycle-containing bisphosphonates cause apoptosis and inhibit boneresorption by preventing protein prenylation: evidence fromstructure-activity relationships in J774 macrophages. J Bone Miner Res (1998) 13(11):1668–78. doi: 10.1359/jbmr.1998.13.11.1668
79. Kavanagh KL, Guo K, Dunford JE, Wu X, Knapp S, Ebetino FH, et al. The molecularmechanism of nitrogen-containing bisphosphonates as antiosteoporosis drugs. Proc Natl Acad Sci USA (2006) 103(20):7829–34. doi: 10.1073/pnas.0601643103
80. Major P, Lortholary A, Hon J, Abdi E, Mills G, Menssen HD, et al. Zoledronic acid is superior to pamidronate in the treatment of hypercalcemia of malignancy: a pooled analysis of two randomized, controlled clinical trials. J Clin Oncol (2001) 19:558. doi: 10.1200/JCO.2001.19.2.558
81. Ralston SH, Gallacher SJ, Patel U, Dryburgh FJ, Fraser WD, Cowan RA, et al. Comparison of three intravenous bisphosphonates in cancer-associated hypercalcaemia. Lancet (1989) 2:1180. doi: 10.1016/s0140-6736(89)91791-1
82. Major PP, Coleman RE. Zoledronic acid in the treatment of hypercalcemia of malignancy: Results of the international clinical development program. Semin Oncol (2001) 28(6):7–24. doi: 10.1016/S0093-7754(01)90261-1
83. Terpos E, Zamagni E, Lentzsch S, Drake MT, García-Sanz R, Abildgaard N. Treatment of multiple myeloma-related bone disease: recommendations from the bone working group of the international myeloma working group. Lancet Oncol (2021) 22(3):e119–30. doi: 10.1016/S1470-2045(20)30559-3
84. Henrich D, Hoffmann M, Uppenkamp M, Bergner R. Ibandronate for the treatment of hypercalcemia or nephrocalcinosis in patients with multiple myeloma and acute renal failure: Case reports. Acta Haematol (2006) 116:165. doi: 10.1159/000094676
85. Tanvetyanon T, Stiff PJ. Management of the adverse effects associated with intravenous bisphosphonates. Ann Oncol (2006) 17:897. doi: 10.1093/annonc/mdj105
86. Adams JS. Vitamin d metabolite-mediated hypercalcemia. Endocrinol Metab Clin North Am (1989) 18(3):765–78. doi: 10.1016/S0889-8529(18)30365-7
87. Fardet L, Flahault A, Kettaneh A, Tiev KP, Généreau T, Tolédano C, et al. Corticosteroid-induced clinical adverse events: Frequency, risk factors and patient’s opinion. Br J Dermatol (2007) 157:142–8. doi: 10.1111/j.1365-2133.2007.07950.x
88. Hanley DA, Adachi JD, Bell A, Brown V. Denosumab: mechanism of action and clinical outcomes. Int J Clin Pract (2012) 66(12):1139–46. doi: 10.1111/ijcp.12022
89. Henry DH, Costa L, Goldwasser F, Hirsh V, Hungria V, Prausova J, et al. Randomized, double-blind study of denosumab versus zoledronic acid in the treatment of bone metastases in patients with advanced cancer (excluding breast and prostate cancer) or multiple myeloma. J Clin Oncol (2011) 29(9):1125–32. doi: 10.1200/JCO.2010.31.3304
90. Fizazi K, Carducci M, Smith M, Damião R, Brown J, Karsh L, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet (2011) 377(9768):813–22. doi: 10.1016/S0140-6736(10)62344-6
91. Hu MI, Glezerman IG, Leboulleux S, Insogna K, Gucalp R, Misiorowski W, et al. Denosumab for treatment of hypercalcemia of malignancy. J Clin Endocrinol Metab (2014) 99:3144. doi: 10.1210/jc.2014-1001
92. Pittman K, Antill YC, Goldrick A, Goh J, Boer RH. Denosumab: Prevention and management of hypocalcemia, osteonecrosis of the jaw and atypical fractures. Asia Pac J Clin Oncol (2017) 13(4):266–76. doi: 10.1111/ajco.12517
93. Vellanki P, Lange K, Elaraj D, Kopp PA, El Muayed M. Denosumab for management of parathyroid carcinoma-mediated hypercalcemia. J Clin Endocrinol Metab (2014) 99(2):387–90. doi: 10.1210/jc.2013-3031
94. Roukain A, Alwan H, Bongiovanni M, Sykiotis GP, Kopp PA. Denosumab for the treatment of hypercalcemia in a patient with parathyroid carcinoma: A case report. Front Endocrinol (Lausanne) (2022) 12:794988. doi: 10.3389/fendo.2021.794988
95. O'Callaghan S, Yau H. Treatment of malignancy-associated hypercalcemia with cinacalcet: a paradigm shift. Endocr Connect (2021) 10(1):R13–24. doi: 10.1530/EC-20-0487
96. Storvall S, Ryhänen E, Bensch FV, Heiskanen I, Kytölä S, Ebeling T, et al. Recurrent metastasized parathyroid carcinoma-Long-Term remission after combined treatments with surgery, radiotherapy, cinacalcet, zoledronic acid, and temozolomide. JBMR Plus (2018) 3(4):e10114. doi: 10.1002/jbm4.10114
97. Warrell RP Jr, Bockman RS, Coonley CJ, Isaacs M, Staszewski H. Gallium nitrate inhibits calcium resorption from bone and is effective treatment for cancer-related hypercalcemia. J Clin Invest (1984) 73:1487–90. doi: 10.1172/JCI111353
98. Cvitkovic F, Armand JP, Tubiana-Hulin M, Rossi JF, Warrell RP Jr. Randomized, double-blind, phase II trial of gallium nitrate compared with pamidronate for acute control of cancer-related hypercalcemia. Cancer J (2006) 12:47–53. doi: 10.1097/00130404-200601000-00009
99. Warrell RP Jr, Israel R, Frisone M, Snyder T, Gaynor JJ, Bockman RS. Gallium nitrate for acute treatment of cancer-related hypercalcemia. a randomized, double-blind comparison to calcitonin. Ann Int Med (1988) 108:669–74. doi: 10.7326/0003-4819-108-5-669
100. Arumugam GP, Sundravel S, Shanthi P, Sachdanandam P. Tamoxifen flare hypercalcemia: an additional support for gallium nitrate usage. J Bone Miner Metab (2006) 24(3):243–7. doi: 10.1007/s00774-005-0678-4
101. Hall TJ, Chambers TJ. Gallium inhibits bone resorption by a direct effect on osteoclasts. Bone miner (1990) 8:211–6. doi: 10.1016/0169-6009(90)90106-P
102. Pittaway JF, Srirangalingam U, Hanson PL, Jones P, Drake WM. Renal replacement therapy as a treatment for severe refractory hypercalcemia. Minerva Endocrinol (2014) 39(3):231–3.
Keywords: cancer, hypercalcaemia, treatment, maligancy, therapy
Citation: Almuradova E and Cicin I (2023) Cancer-related hypercalcemia and potential treatments. Front. Endocrinol. 14:1039490. doi: 10.3389/fendo.2023.1039490
Received: 08 September 2022; Accepted: 13 March 2023;
Published: 22 March 2023.
Edited by:
Lorenzo Scappaticcio, University Hospital “Luigi Vanvitelli”, ItalyReviewed by:
Matthew T. Drake, Mayo Clinic, United StatesCopyright © 2023 Almuradova and Cicin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irfan Cicin, aXJmYW5jaWNpbmxAdHJha3lhLmVkdS50cg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.