 Nicole Bechmann
Nicole Bechmann Frank Westermann2,3
Frank Westermann2,3 Graeme Eisenhofer
Graeme Eisenhofer
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 08 June 2023
Sec. Pediatric Endocrinology
Volume 14 - 2023 | https://doi.org/10.3389/fendo.2023.1022192
This article is part of the Research TopicPediatric Adrenal NeoplasmsView all 12 articles
Pediatric neural crest-derived adrenal neoplasms include neuroblastoma and pheochromocytoma. Both entities are associated with a high degree of clinical heterogeneity, varying from spontaneous regression to malignant disease with poor outcome. Increased expression and stabilization of HIF2α appears to contribute to a more aggressive and undifferentiated phenotype in both adrenal neoplasms, whereas MYCN amplification is a valuable prognostic marker in neuroblastoma. The present review focuses on HIF- and MYC signaling in both neoplasms and discusses the interaction of associated pathways during neural crest and adrenal development as well as potential consequences on tumorigenesis. Emerging single-cell methods together with epigenetic and transcriptomic analyses provide further insights into the importance of a tight regulation of HIF and MYC signaling pathways during adrenal development and tumorigenesis. In this context, increased attention to HIF-MYC/MAX interactions may also provide new therapeutic options for these pediatric adrenal neoplasms.
The neural crest comprises a multipotent stem cell population that gives rise to a large variety of cell types, including sympathetic and parasympathetic ganglia and chromaffin cells. From these cell types neuroblastomas or pheochromocytomas/paragangliomas can originate. Both tumor entities can occur in the adrenal gland in some cases already soon after birth or in early childhood and are characterized by clinical and molecular heterogeneity. In both neural crest-derived neoplasms, it is discussed that increased expression and stabilization of HIF2α contributes to a more aggressive and undifferentiated phenotype (1–4). However, dependent on the context HIF2α may also promote tumor suppressive activities, which was reported for both neuroblastoma (5–7) and non-neural crest-derived neoplasms (8–10). The occurrence of MYCN amplification in approximately 20% of neuroblastomas, which correlates with high-risk disease and poor prognosis (11), further underscores the importance of MYC and HIF signaling pathways in these adrenal neoplasms derived from the neural crest.
The human adrenal gland composes two distinct tissues, the outer cortex and the inner medulla under a common capsule (Figure 1A). The steroid hormone producing adrenal cortex (mineralocorticoids, glucocorticoids and androgens) arises from the coelomic mesoderm of the urogenital ridge, while the adrenal medulla arises from the neural crest and secretes the catecholamines epinephrine and less prominently norepinephrine (13, 14). During neurulation, the neural folds arise at the boundary between non-neural and neural ectoderm and subsequently coalesce in the midline to form the neural tube. The pre-migrating neural crest cells localized at the neural fold are characterized by their epithelial phenotype associated with strong cell-cell junctions. To initiate migration, neural crest cells must undergo an epithelial-mesenchymal transition (EMT) to acquire a motile phenotype and lose their cell-cell junction (15).
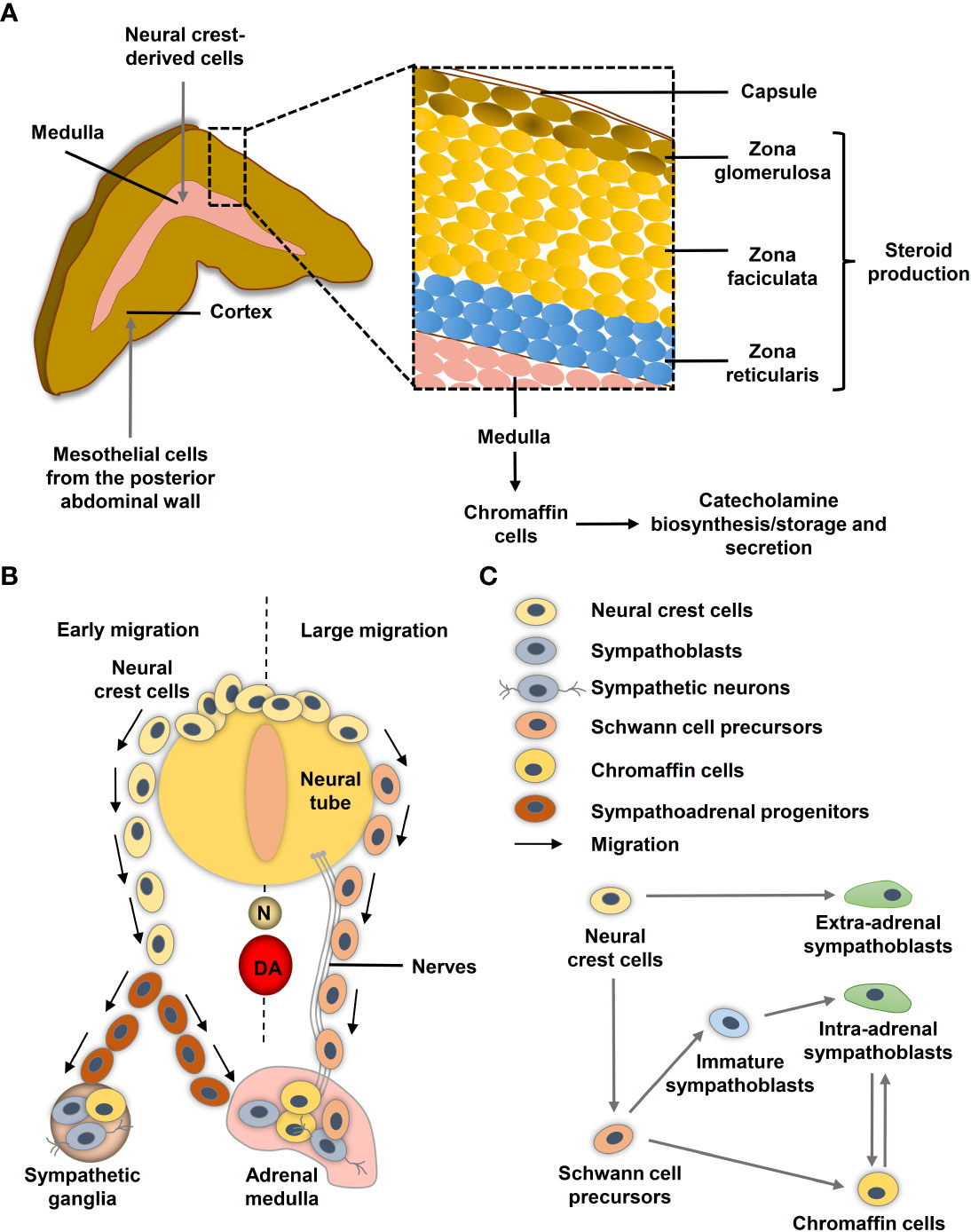
Figure 1 Neural crest and adrenal chromaffin cell development. (A) Human adult adrenal zonation. (B) Schematic illustration of human adrenal and sympathetic ganglia development from the neural crest. (C) Chromaffin cell and sympathoblast development from neural crest-derived cells in humans (adapted from (12)). DA, dorsal aorta; N, notochord.
During migration, neural crest cells proliferate extensively to generate enough precursor cells to colonize their targets. Ventrally migrating neural crest cells colonize the paravertebral sympathetic ganglia in the trunk and the chromaffin cells of the adrenal medulla and paraganglia (16) (Figure 1B). After reaching their targets, neural crest-derived cells differentiate into the appropriate cellular subtypes. Migration and differentiation of the distinct neural crest cell derivatives is orchestrated by expression of lineage specific markers that are influenced by signals of the microenvironment such as hypoxia.
Single-cell RNA sequencing (scRNA-seq) revealed differences in the transitions between sympathoadrenal fates in humans and mice (12). In human embryos, Schwann cell precursors (SCPs; SOX10+) derived from neural crest cells connect to STMN2+ISL1+PRPH− sympathoblasts and the CHGA+PENK+PNMT+ chromaffin cells through a ‘fork-like’ transition (Figure 1C) (12). Cells within this first transition show overlaps between both expression programs in form of SOX10+/ISL1+/HAND2+, SOX10+/ISL1+ and SOX10+/HAND2+ transitory cells (12). A second transition between sympathoblasts and chromaffin cells (markers: SOX4, BEX1, RAMP1, PENK) was identified (Figure 1C). In mice, chromaffin cells of the adrenal medulla arise from an intermediate bridge cell population (transition 1) and can transit to sympathoblasts (12, 17). While cellular properties, such as high proliferative capacity and motility, are crucial during embryogenesis, they may become problematic later in life when the same properties contribute to tumor aggressiveness and metastasis. Low-risk neuroblastomas share similarities to differentiated late sympathoblasts (also named neuroblasts in the context of neuroblastomas), whereas high-risk MYCN-amplified neuroblastomas resemble more undifferentiated sympathoblasts (18–20). Bedoya-Reina et al. further showed that tumor cells enriched in high-risk neuroblastomas resemble a subtype of TRKB+ cholinergic progenitor population that they previously identified in human postnatal adrenal glands (20). An undifferentiated phenotype is also associated with a higher risk of metastasis in pheochromocytomas and extra-adrenal paragangliomas (21, 22), although the exact cellular origin of the various subgroups is still unclear.
In the present review, we describe current knowledge about the interplay between MYC and HIF signaling during neural crest, adrenal development and the potential role of these signaling pathways in catecholamine-producing neural crest-derived neoplasms.
MYC proteins are key regulators of cell fate and part of a network of interacting transcription factors, that regulate expression of various genes involved in, for example, cell proliferation, differentiation and metabolism (23). In addition to c-MYC, the MYC protein family also includes MYCN and l-MYC. These helix–loop–helix leucine zipper transcription factors heterodimerize with MYC-associated protein X (MAX) (Figure 2). After heterodimerization, MYC/MAX binds to specific DNA sequence (CANNTG) called E-box to activate or repress transcription of more than 15% of all genes in cells (24–26). MAX itself is under tight control by a network of protein-protein interactions with MAX dimerization protein (MXD1, also known as MAD) and MAX interactor 1 (MXI1, also known as MAD2) (27). Other factors, such as the hypoxia inducible factor (HIF) 1α and 2α, further affect binding of MYC/MAX to the E-box.
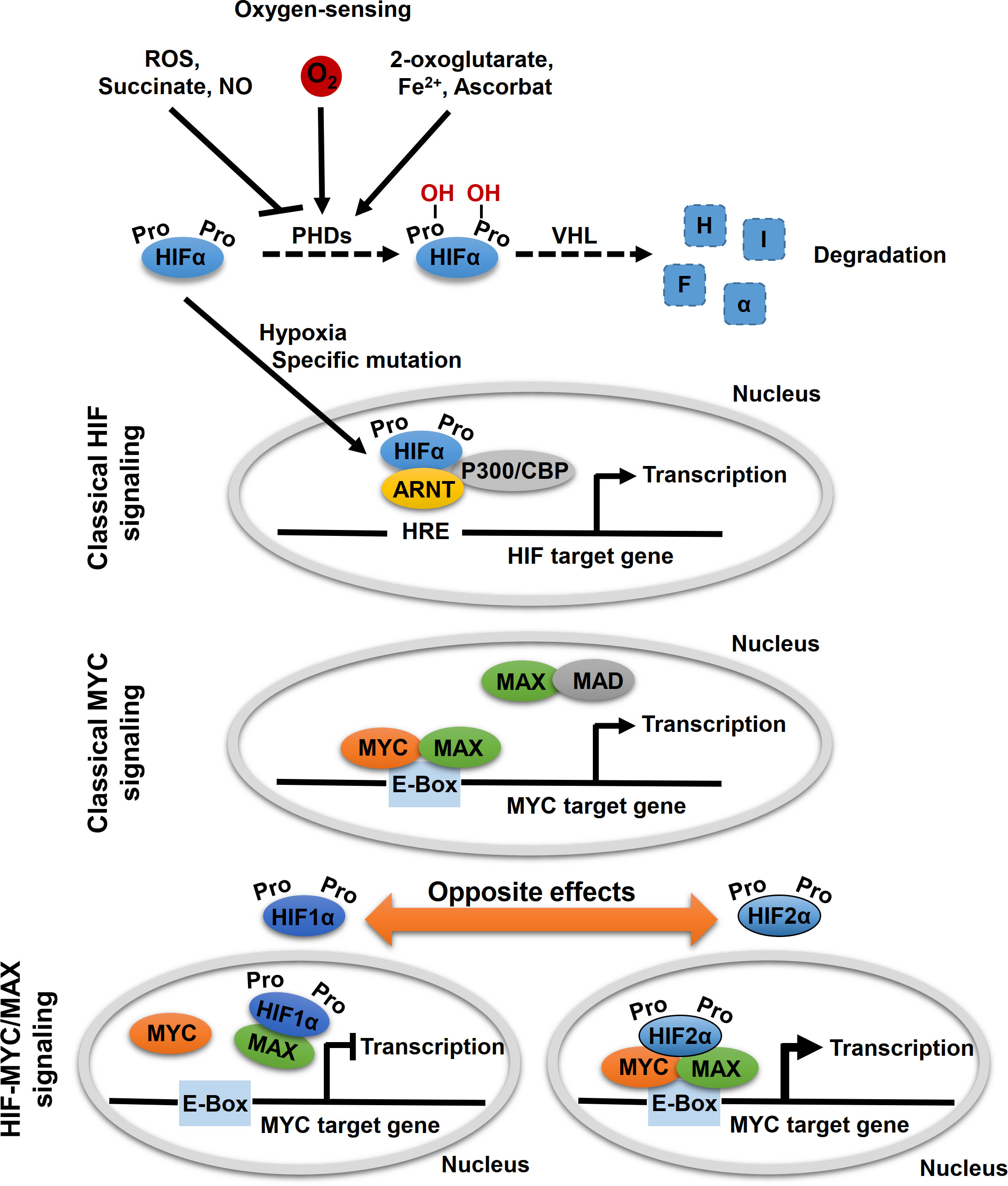
Figure 2 Oxygen-sensing and HIF-MYC signaling. Key molecule of oxygen-sensing system are hypoxia-inducible factors (HIFs), which regulate the transcription of a wide range of oxygen responsive genes. In presence of oxygen, HIF proline hydroxylases (PHDs) hydroxylate proline residues within HIFα subunits, which lead to proteasomal degradation of HIFα by the von Hippel-Lindau (VHL) tumor suppressor. Hypoxic conditions (absence of oxygen) or specific mutations, that affect HIFα degradation, lead to a stabilization of HIFα. Subsequently, HIFα subunits translocated to the nucleus where they form a complex with aryl hydrocarbon receptor nuclear translocator (ARNT, also known as HIFβ) and specific co-factors and bind to hypoxia-responsive elements (HREs) leading to transcription of HIF target genes (classical HIF signaling). MYC proteins encoded by MYC proto-oncogenes (c-MYC, MYCN, l-MYC) are localized in the nucleus and form heterodimers with myc-associated factor X (MAX), which enables recognition by the hexameric DNA sequence CACGTG (E-Box) and subsequent transcriptional activation of MYC target genes (classical MYC signaling). Moreover, other binding partners of MYC and MAX also regulate MYC target gene transcription, including MAX dimerization protein 1 (MAD) and HIFαs. Thereby, opposite effects on MYC target gene expression were described for HIF1α and HIF2α. While HIF1α dimerizes with MAX and thereby suppresses binding to the E-box, HIF2α leads to stabilization of the MYC/MAX complex and thus to activation of MYC target genes.
HIF proteins are the main regulators of oxygen sensing in cells (Figure 2). Under normoxic conditions (presence of oxygen), proline residues of the HIFα subunits are hydroxylated by oxygen- and α-ketoglutarate-dependent prolyl hydroxylases (PHDs). This allows for recognition of the HIFα subunits by the von Hippel–Lindau protein (VHL) and their subsequent degradation by proteasomes (Figure 2). Under hypoxic conditions (absence of oxygen), HIFα subunits are stabilized and form a complex with aryl hydrocarbon receptor nuclear translocator (ARNT, also known as HIFβ) and several co-factors, including CREB-binding protein and p300. The HIF complex binds to hypoxia-response element (HRE) of the gene promoter for transactivation, thereby regulating genes involved in angiogenesis, pH regulation, glycolysis, and glucose transport, which enable cellular adaptation to hypoxia.
The two main HIFα subunits, HIF1α and HIF2α, have mostly complementary functions, but their activity differs temporally: while HIF1α predominantly mediates the acute response to severe hypoxia, HIF2α modulates adaption to chronic or even mild hypoxia (4). In addition, HIF1α is ubiquitously expressed, while HIF2α has a more restricted expression profile (28). Both HIFαs differ in their targets; with HIF1α specifically activates genes involved in glycolysis and HIF2α preferentially activates VEGF, transforming growth factor-α (TGFα), lysyl oxidase (Lox), Oct4 and Cyclin D1 (29, 30). Evidence suggests that HIF2α preferentially promotes tumorigenesis and affects the differentiation status of different tumor entities (29). The effects on the tumor phenotype might be explained by the HIF2α-induced expression of the stem cell factor Oct4 and the transcriptional activation of c-Myc (29, 30). For example, in hepatocellular carcinoma (HCC) tumor tissue, c-Myc expression showed a positive correlation with HIF2α but not with HIF1α (31). Knockdown of HIF2α diminished expression of c-Myc in HCC cells, suppressed hypoxia-related proliferation, and induced apoptosis (31). On the other hand, Yang et al. revealed that HCC patients with high HIF2α protein levels had longer overall survival indicating a tumor suppressor function of HIF2α in these tumors (10). In colorectal cancer cell lines, HIF2α regulated expression of c-Myc under chronic hypoxia and thereby controls sensitivity to 5-flurouracil (32). HIF1α and HIF2α play distinct roles in colon cancer (9). It was also shown that EPAS1 was significantly reduced in primary adenocarcinoma samples of the colon compared to histopathologically non-neoplastic tissue, while no difference was found for HIF1α (33). Renal cell carcinoma cells expressing almost exclusively HIF2α exhibit lower genomic instability, which correlates with enhanced c-MYC-dependent expression of genes involved in DNA repair (34). Moreover, emerging evidence suggests that MYC regulates the levels and activity of HIF1α (35–38).
In addition, other HIF-dependent mechanisms modulating MYC/MAX complex formation and promoter occupancy have also been proposed, with HIF1α and HIF2α also exhibiting opposing effects (Figure 2) (26, 39, 40). HIF1α antagonized c-MYC activity by displacing c-MYC from transcription factor Sp1 binding that is required for MYC promotor activation, while phosphorylation of HIF2α prevents HIF2α from competing with MYC for Sp1, thereby enhancing Sp1-c-Myc activity (41–44). Under chronic hypoxia, HIF1α promotes c-MYC degradation and induces expression of MAX interactor 1 (MXI1) that furthermore inhibits MYC target gene expression (45, 46). HIF1α can further bind to MAX to thereby disrupt the formation of the MYC/MAX complex (39). On the other hand, HIF2α promotes MYC activity by enhancing Sp1-c-Myc activity as mentioned above. HIF2α enhances c-MYC activity by stabilizing the MYC/MAX complex (Figure 2) (45, 47). This effect appears to be much stronger than HIF1α-mediated degradation of c-MYC in tumor cells, leading to activation of MYC under hypoxic conditions (47). Moreover, HIF has been shown to promote proteosomal degradation of MYC under chronic hypoxic conditions in dependence of the used cell system (45, 48–50).
Most of the aforementioned studies addressing the interaction between HIF and MYC focused exclusively on the effect of c-Myc. Whether the mechanisms are also applicable to MYCN remains unclear. Due to the tissue- and cell-specific differences in c-MYC and MYCN expression and the resulting different roles (e.g. during neural crest development and tumorigenesis), it can be assumed that the interaction with HIF1α and HIF2α is also different.
The ability to sense oxygen (Figure 2) is crucial for the survival of water- and air-breathing organisms. Adrenomedullary chromaffin cells as well as glomus cells of the carotid body that both arise during neural crest development synthesize and release catecholamines in response to hypoxic stress (51, 52). Chemoreceptors in the carotid body are relatively non-sensitive to hypoxia in the neonatal period and achieve adult-like sensitivity by 2-3 weeks postnatally in rats. In contrast, rat adrenomedullary chromaffin cells are most sensitive to hypoxia in the perinatal period, and this sensitivity is gradually lost until it is largely gone by 2-3 weeks postnatally (53–55). The oxygen sensitivity of these neural crest-derived cells suggests involvement of the HIF signaling pathways already during embryogenic development.
Under physiological conditions, hypoxia occurs in amniotic embryos prior to the onset of a functional blood circulation (56). In rodents and chicken embryos, low oxygen concentrations (5%) have been shown to be required until the neural tube has closed and the cells of the cranial neural crest have emigrated (57, 58). Studies in Xenopus, chicken, and quail embryos indicate that HIF1α and ARNT are expressed together and ubiquitously in the developing embryo (up to HH14; HH stage in chicken embryo), whereas HIF2α has a more pronounced expression pattern that includes tissues that do not express HIF1α (58, 59). Trunk neural crest cells that give rise to the adrenal medulla and the sympathetic ganglia form mainly after vascularization and are not affected by high oxygen or deletion of HIF1α (57, 58, 60). However, stabilization of HIFα in neural crest cells seems to be controlled by oxygen-dependent (hypoxia) and oxygen-independent mechanisms (pseudohypoxia) (61), similar to what is already known for neuroblastoma, pheochromocytoma and paraganglioma (PPGL) (62, 63).
Inhibition of Hif1α in Xenopus laevis and zebrafish embryos results in complete blockade of neural crest migration by controlling chemotaxis and epithelial-mesenchymal transition (61). In mice selectively deficient in HIF1α in ISL+ cells, the development of sympathetic ganglia and chromaffin cells is impaired (64). Mice with deletion in Vhl restricted to tyrosine hydroxylase (Th)-positive cells exhibit atrophy of the carotid body, adrenal medulla and sympathetic ganglia; this was associated a striking intolerance to systemic hypoxia that could lead to death (65). HIF1α is diffusely expressed in all cell types of the developing human adrenal medulla with a stronger accumulation in the connecting progenitor cells and early chromaffin cells (Figures 3A, B), while expression of HIF2α (gene also known as EPAS1) appears to be restricted to specific cell populations (Figure 3C). snRNA-seq revealed a high expression of HIF2α in early chromaffin cells and neuroblasts as well as in the connecting progenitor cells, whereas SCPs and late neuroblasts as well as late chromaffin cells showed only limited expression of HIF2α in isolated cells (Figures 3C, D) (18).
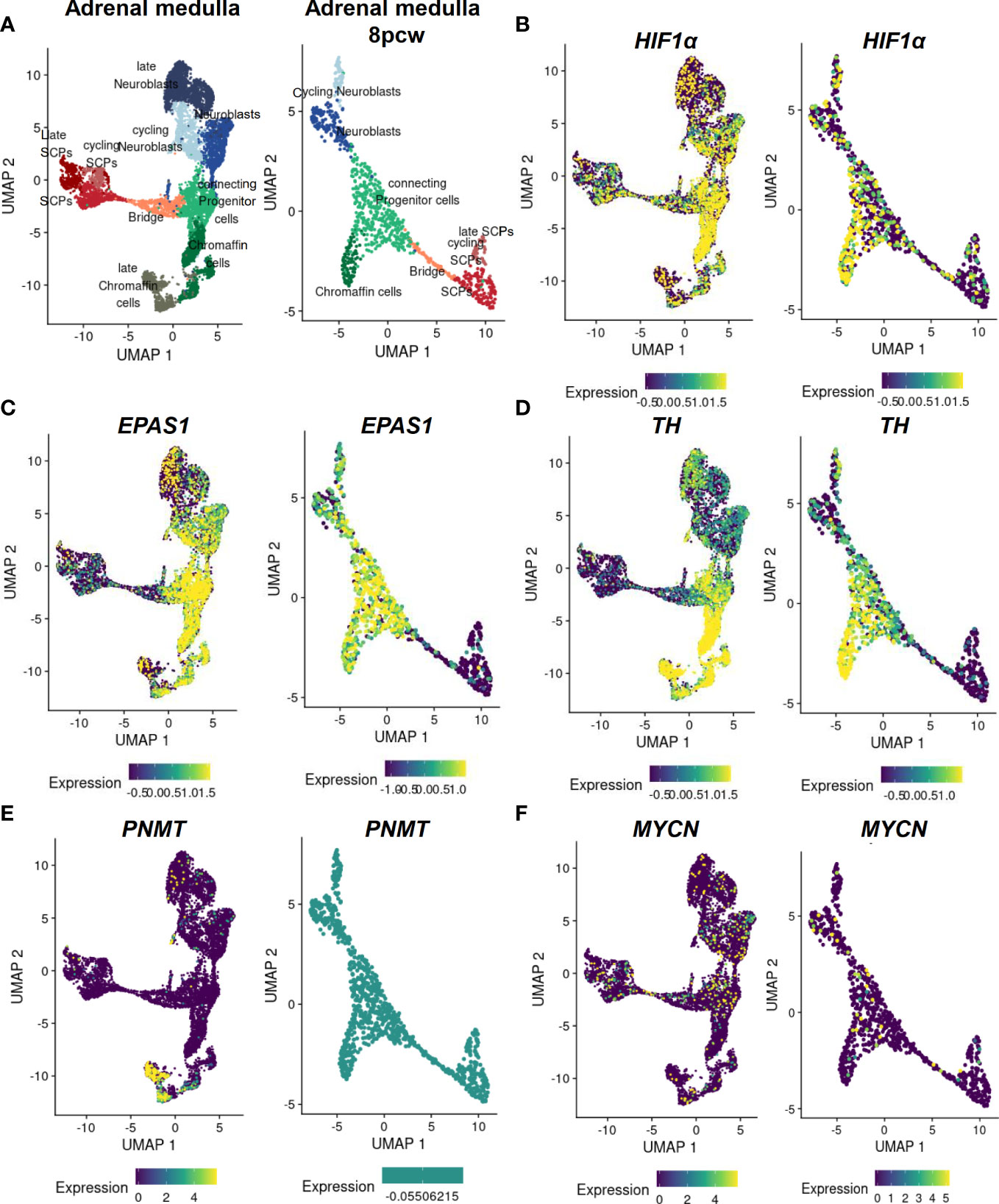
Figure 3 Expression of MYCN, HIFs and chromaffin cell markers in the developing human adrenal medulla. Expression patterns of genes of interest obtained by single nucleus RNA sequencing data (18) of the developing human adrenal medulla were visualized by https://adrenal.kitz-heidelberg.de/developmental_programs_NB_viz/ (last request: March 2022). (A) UMAP plot of adrenal medullary cells of developing adrenal and of the fetal adrenal eight weeks post conception (8pcw). Different colors highlight different cell populations. Expression pattern of (B) HIF1α, (C) EPAS1, (D) TH, (E) PNMT and (F) MYCN in the developing human adrenal and of the fetal adrenal eight weeks post conception. The color indicates the normalized gene expression (blue low expression; yellow high expression). Data on c-Myc and MAX expression were not available. UMAP: Uniform manifold approximation and projection; SCP, Schwann cell precursors.
During neural crest development, HIF2α is expressed in migrating trunk neural crest cells and sympathetic neuroblasts in human, murine, and avian embryos (66). In the sympathetic ganglia of human embryos, HIF2α is expressed at embryonic week 6.5, but expression is lost at later stages (67). HIF2α knockout mice exhibit severe sympathetic nervous system (SNS) abnormalities associated with mid-gestation lethality (68). Both overexpression and silencing of HIF2α in vivo delay neural crest development, induce proliferation and self-renewal capacity of neural crest cells, and reduce the proportion of neural crest cells that migrate ventrally to sympathoadrenal sites, suggesting that HIF2α needs to be tightly controlled during normal development of the trunk neural crest (66). These data underline the importance of HIF2α in chromaffin cell and sympathoblast differentiation, which may further provide a rational for the various differentiation states of adrenal neoplasms.
In mice, inactivation of Phd3 (responsible for the hydroxylation of the HIFαs that initiates their proteasomal degradation) promotes survival of sympathoadrenal neurons, which appear hypofunctional despite increased numbers of Th-positive cells in the adrenal medulla and carotid body, due at least in part to upregulation of Hif2α but not Hif1α (69). Moreover, hypoxia induced catecholamine release by chromaffin cells, and here in particular regulated by HIF2α (68, 70), is crucial to maintain physiological homeostasis of the fetus before sympathetic innervation is fully complete (71, 72). During birth, increased catecholamine release facilitates adequate hemodynamic adjustment and stimulates surfactant production by the lungs (73, 74). A deficiency of Phd2 in the adrenal medulla of mice results in a Hif2α-mediated reduction in phenylethanolamine N-methyltransferase (Pnmt) associated with a reduced epinephrine biosynthesis (75). This is also consistent with studies in Hif2α-/- mice, which showed reduced epinephrine levels in the adrenal gland (68, 76). In the developing human adrenal medulla, expression of PNMT is restricted to late chromaffin cells, which do not express HIF2α or express HIF2α in a restricted manner (Figure 3E) (18). Reduced oxygen promotes survival and catecholaminergic differentiation, characterized by the expression of tyrosine hydroxylase, which catalyzed conversion of tyrosine to L-DOPA (precursor catecholamine biosynthesis), in neural crest stem cells and central nervous system progenitors (77, 78). It is therefore possible that hypoxia mediates stem cell function by affecting Oct4, which is controlled by HIF2α as discussed in the previous section (30).
Mice studies revealed that c-Myc and Mycn, but not l-Myc, are fundamental for normal development since targeted deletions are lethal to the embryo at mid-gestation (79–81). Tissue- and cell-specific expression patterns of Mycn and c-Myc have been observed during embryogenesis. While Mycn expression is restricted to specific cell types of epithelial tissues, including those of the developing nervous system and organs characterized by epithelial-mesenchymal interaction, c-Myc expression is restricted to mesenchymal compartments (82). A time-dependent expression of c-Myc and Mycn in the developing neural crest and neural crest cells is observed in chicken embryos (83). In the neural plate during gastrulation throughout the anterior-to-posterior axis Mycn, but not c-Myc is expressed. Neither c-Myc nor Mycn is expressed in the neural plate boarder at stage HH4-7, which later form the neural crest. When the neural tube closure starts (HH8) expression of Mycn is restricted to the ventral parts, which form the central nervous system (CNS). The expression of c-Myc begins at later stage HH8 in the dorsal neural tube later forming the neural crest. In stage HH9, c-Myc expression remains restricted to the dorsal neural crest area, while Mycn is expressed in the remaining neural tube except in the dorsum (83).
In Xenopus, c-Myc is an essential early regulator of neural crest cell formation, with expression of c-Myc localized to the neural plate boundary prior to expression of early neural crest markers (84). It has been suggested that c-Myc prevents cell fate decisions during neural crest formation, possibly via the Myc target gene Id3 (85). Knockdown of Id3 leads to a lack of neural crest formation in Xenopus embryos, since Id3 maintains the precursor state of neural crest cells (85). Loss of c-Myc results in a drastic reduction in the number of emigrating cells of the neural crest, due to a reduced capacity for self-renewal, increased cell death, and a shorter duration of the emigration process in chicken embryos. In this regard, c-Myc appears to bind to Miz1 rather than to the E-box to activate the cell cycle (86).
In chicken and mouse embryos, Mycn is expressed in early neural crest lineage, the central nervous system, a subset of mesoderm derivatives and endodermal epithelia, in particular (87). Deficiency of Mycn in mouse embryos leads to a reduction in mature neurons of the dorsal root ganglia and sympathetic ganglia, underscoring the importance of Mycn in the development of neurons from the neural crest (81). At early stages, Mycn appears to be involved in the proliferation of progenitor populations rather than in their differentiation per se (79). Later during neural development, Mycn has been linked to the maintenance of neural fate as it is expressed by slowly proliferating neural stem cells and is involved in the expansion and differentiation of neural progenitor cells in the CNS (81, 88, 89). In addition, Mycn promotes neural fate and differentiation in the peripheral nervous system (79, 87). After neural crest EMT, Mycn is only expressed at low levels in migrating neural crest cells followed by a further downregulation before the cells aggregate to form the ganglia (90). Mycn is expressed in regions of the neural plate destined to form the central nervous system, but not in the neighboring neural crest stem cell domain (83). Mycn expression in the neural crest domain biases cells toward a more CNS neural stem cell-like fate (expression SOX2), leading to improperly specified neural crest cells; this may play a role of priming in neuroblastoma development (83). However, overexpression of Mycn in mouse sympathoadrenal progenitors is insufficient for tumor formation in nude mice but leads to enhanced neural differentiation (91). Increased expression of Mycn in mouse neural crest cells results in neuroblastoma-like tumors, suggesting that premature exposure of neural crest cells to high Mycn levels may be important in the development of neuroblastoma (92). Our scRNA-seq data reveal an expression of MYCN only in single cells of the human developing adrenal medulla not restricted to a specific cell type (Figure 3F).
A possible time-dependent co-expression of HIFs and MYCs during neural crest and adrenal development may indicate a possible interaction of the two signaling pathways during these embryological processes, but precise data are unfortunately not available in this regard. The role of the cell-specific expression of the respective isoforms of HIF and MYC and how exactly they are related to each other remains unclear. Furthermore, most studies in this context rely on Xenopus, mouse and chicken embryo studies. Transferability to humans needs to be clarified. Nevertheless, data indicate that the embryogenic origin of the cells and the associated differentiation status influenced by HIF and MYC may contribute decisively to the phenotype of adrenal neoplasms.
Pediatric neuroblastomas and PPGLs are characterized by a high degree of heterogeneity in disease presentation associated with distinct differentiation stages, affected by HIF and MYC signaling pathways.
Neuroblastoma is the most common solid neoplasm in childhood and its clinical course varies from spontaneous tumor regression to fatal malignant progression (Figure 4); even differentiation into benign forms (ganglioneuroma or ganglionneuroblastoma) are described (93). Malignant expansion in neuroblastoma appears manly triggered by MYCN amplification or/and ALK and ATRX mutations (94). Additional factors that influence prognosis include the location of the primary tumor and the age of the patient. For example, adrenal neuroblastomas are more frequently MYCN-amplified and are often in tumor stage 4 (International Neuroblastoma Staging System Committee (INSS) system), in which the primary tumor has spread to distant organs, compared with extra-adrenal neuroblastomas (95). Children with stage 4 neuroblastoma who are younger than 1 year have a significantly better prognosis than older children at stage 4 (96).
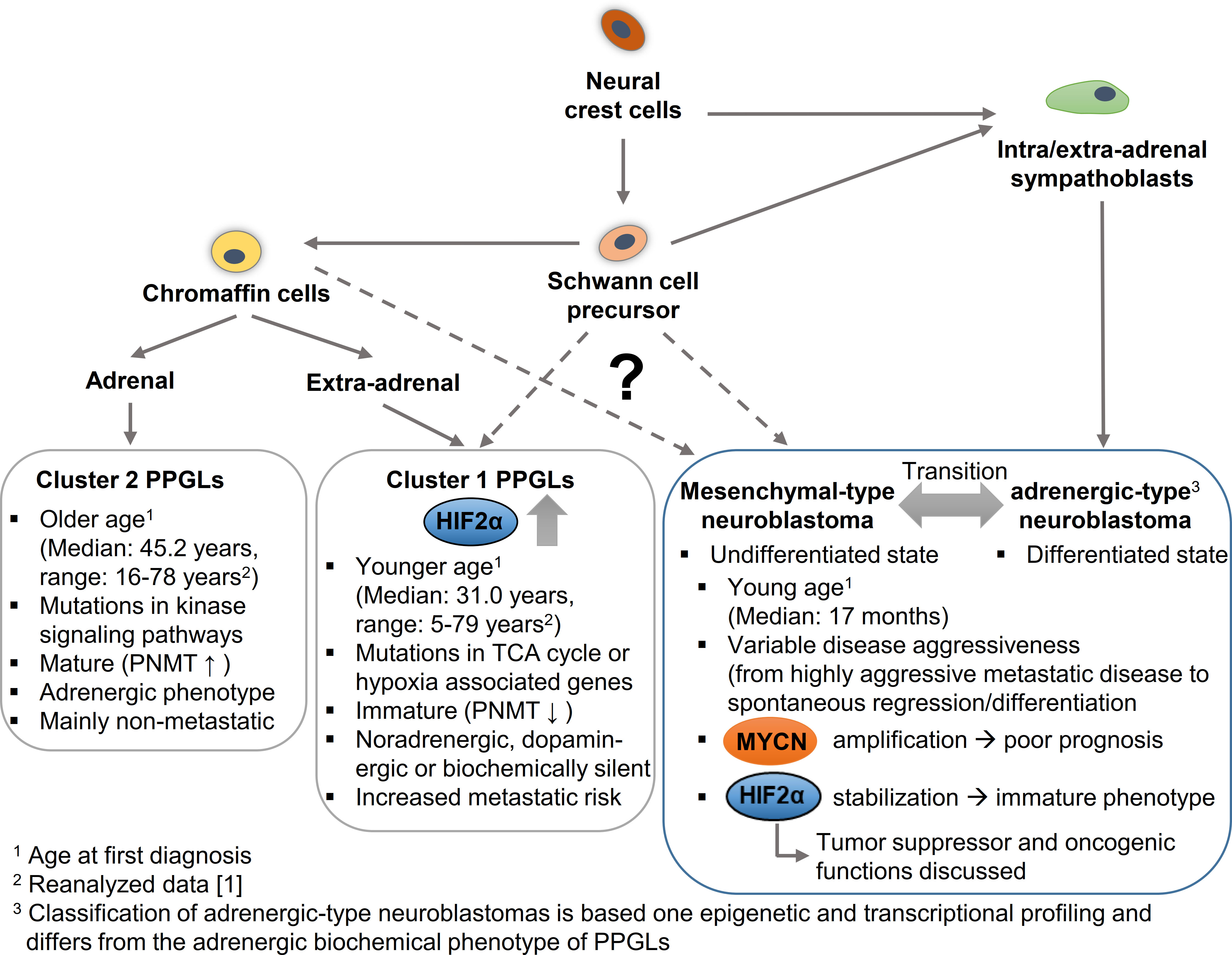
Figure 4 Hypothetical model to explain the cellular origin leading to different phenotypic features in pheochromocytomas/paragangliomas (PPGLs) and neuroblastomas in dependence of HIF2α and MYCN. PPGLs and neuroblastomas originate from neural crest cells; while mature neuroblastomas arise from a sympathoblast lineage, mature cluster 2 PPGLs originate form a chromaffin cell lineage. Both lineages arise via Schwann cell precursors that may give rise to more immature cluster 1 PPGLs and mesenchymal-type neuroblastomas. Increased expression and stabilization of HIF2α is associated with an immature phenotype and enhanced aggressiveness in PPGLs, while for HIF2α in neuroblastoma both an oncogenic and a tumor suppressor function are discussed. MYCN amplification correlates with a worse prognosis in neuroblastomas. PNMT, Phenylethanolamine N-methyltransferase; TCA cycle, Tricarboxylic acid cycle.
Epigenetic and transcriptional profiling in neuroblastoma cell lines identified two distinct neuroblastoma cell identities: the undifferentiated mesenchymal-type (MES) and adrenergic-type (ADRN; to be strictly distinguished from an adrenergic phenotype in PPGLs) that can interconvert and reflect cells from different stages of differentiation (Figure 4) (97). Patient samples allowed a further subclassification of the adrenergic group linked to distinct clinical outcomes into MYCN-amplified, MYCN non-amplified high-risk and MYCN non-amplified low-risk signatures (98). High MYCN/c-MYC target gene expression is a hallmark of malignant neuroblastoma progression (99). Elevated expression of a subset of MYCN/c-MYC target genes identifies a patient subtype with poor overall survival, independent of established risk markers such as MYCN amplification, disease stage, and age at diagnosis (99). Malignant progression, therapy resistance and disease relapse in neuroblastoma are associated with an undifferentiated phenotype, which is linked to the broad activation/repression of MYCN/c-MYC target genes (97, 98). Some results suggest that the phenotype and thus the clinical outcome strongly depend on the precise ratio of MAX to MYCN (100). In addition, hypoxia - in this case in particular the expression of HIF2α - is described to block differentiation and potentially also trigger dedifferentiation in neuroblastoma cells, which may contribute to a more aggressive tumor behavior (2, 101, 102). However, the role of this mechanism in MYCN-amplified neuroblastoma is not yet fully understood and there are also data attributing a tumor suppressor function to HIF2α in neuroblastoma (5–7). Westerlund and colleagues showed that EPAS1 expression correlates with features of low-risk neuroblastoma (6). For clear-cell carcinomas it has been shown that HIF2α sensitizes to ferroptosis, an iron-dependent form of cell death (103). MYCN-amplified neuroblastoma cells are highly sensitive to ferroptosis (104), which also suggests a rather tumor-suppressive role of HIF2α in neuroblastoma. Further studies are needed to finally clarify the role of HIF2 α in neuroblastoma. The complex regulation of both signaling pathways in neuroblastoma and the resulting consequences for differentiation and metastasis, suggests a close association of the MYC and HIF signaling pathways in neuroblastomas.
Neuroblastomas occur in many of the same regions as PPGLs. New insights into the putative cellular origins of both entities came from comparative snRNA-seq analyses of human embryonal/fetal adrenal glands and neuroblastoma/PPGLs (12, 18, 105). Whereas neuroblastomas showed prominent transcriptional similarity with early normal neuroblast populations, PPGLs were transcriptionally more similar to normal cells with chromaffin cell-like morphology (Figure 4) (18, 105). Regulatory super-enhancer elements involved in neuroblastoma-specific chromosomal rearrangements were predominantly active in early neuroblasts of the developing adrenal glands, indicating neuroblastoma-driving events may have occurred predominantly in early neuroblast populations and not in SCP or chromaffin cell-like populations (18). The different cellular origin may also explain why PPGLs are diagnosed at older ages (median: 41.8 years; range: 5.5-83.2 years (1)) and pediatric cases are comparatively rare. Nevertheless, taking into account the low growth rate of PPGLs (volume doubling time of 5-7 years (106, 107)), it seems reasonable that tumorigenesis may be initiated during embryogenesis in at least some of these tumors (discussed later in more detail). Increased expression and stabilization of HIF2α in a specific subset of PPGLs (pseudohypoxic PPGLs) is characterized by a more immature phenotype, enhanced disease aggressiveness and onset at younger age (1, 62).
A large proportion of PPGLs are heritable due to germline pathogenic variants (PVs) in one of the described PPGL susceptibility genes (e.g. PVs in: FH, SDHA, SDHB, SDHC, SDHD, SDHAF2, MDH2, VHL, HIF2α, EGLN1/2, MAX, TMEM127, NF1, RET). Hereditary PPGLs are diagnosed at younger age and differ with respect to their plasma and urinary catecholamine/metanephrine profiles from sporadic PPGLs (108). Diagnosis of PPGLs largely depends on the biochemical assessment of catecholamine excess by measurement of plasma or urinary metanephrines. Due to slow growth, neglected consideration of PPGL in childhood and the immature or even non-functional catecholamine phenotype of pseudohypoxic PPGLs, the prevalence of PPGL in pediatrics may be underestimated. We recently reviewed the biochemical diagnosis of pediatric catecholamine-producing tumors and discuss in this context also the diagnosis of neuroblastoma (109).
Based on their transcriptional profile, PPGLs are divided into two main cluster groups (110, 111). Cluster 1 PPGLs bear PVs encoding two groups of genes, that either lead to a direct stabilization of HIF2α (cluster 1B, including mutations in VHL, HIF2α, EGLN1/2) or encode enzymes of the tricarboxylic acid (TCA) cycle (cluster 1A) that indirectly affect HIF2α stabilization (62). In contrast to the pseudohypoxic cluster 1 PPGLs, which are diagnosed earlier in life (median: 31.1 years; range: 5.5-79.3 years (1)), cluster 2 PPGLs (median: 45.2 years; range: 16.0-78-4 years) are characterized by an activation of kinase signaling pathways. The immature catecholamine phenotype of cluster 1 PPGLs is characterized by the absence of glucocorticoid-induced expression of PNMT, the rate-limiting enzyme that converts norepinephrine to epinephrine. In contrast to epinephrine-producing cluster 2 PPGLs that occur almost exclusively in the adrenal, cluster 1 PPGLs tend to metastasize more frequently and occur at intra- and extra-adrenal locations. In contrast to neuroblastomas, extra-adrenal tumor location in PPGL patients is associated with higher disease aggressiveness and shorter disease-specific survival (112). In addition, two other PPGL clusters were described based on mRNA expression analysis (111, 113). The WNT altered cluster is characterized by increased expression of genes in the WNT signaling pathway, while the cortical admixture cluster show expression of adrenal cortex markers (111).
Considering that 96% of pediatric PPGLs belong to the pseudohypoxic cluster (34% cluster 1A, 66% cluster 1B) (114), it becomes obvious that somatic mutations in cluster 1 genes likely occur during embryogenesis, while tumorigenesis of cluster 2 PPGLs may be initiated later in life. The HIF2α-mediated, pseudohypoxic PPGLs are often multifocal tumors and/or metastatic, which indicate somatic mutations even before the settlement of migratory neural crest progenitors at different locations (Figure 4). Moreover, paragangliomas with somatic gain-of-function mutations in HIF2α are associated with mosaicism and identical mutations in multiple tumors arising from postzygotic mutations at early stages of embryogenesis (115, 116). Remarkably, HIF2α mutations appear to be restricted to PPGLs, whereas they are mostly absent in tumor samples of other tumor entities included in The Cancer Genome Atlas (TCGA) and Genomics Evidence Neoplasia Information Exchange (GENIE) databases (117).
In PPGLs increased expression and stabilization of HIF2α is associated with a more immature/less differentiated phenotype that is accompanied by increased disease aggressiveness (1, 118). PPGLs with enhanced expression/stabilization of HIF2α do not express PNMT, whereas HIF2α expression in neuroblastomas marks a subpopulation of immature neural crest-like cells (2, 119). The induced expression of HIF2α target genes and the associated pseudohypoxic environment may provide cells of cluster 1 PPGLs with a selection advantage that favors tumorigenesis in these cells. This hypothesis is supported by patients with PPGL that occurred as a complication of cyanotic congenital heart disease (CCHD). These patients suffer from chronic hypoxia and frequently carry somatic PVs in HIF2α (120–122). PVs in HIF2α or already an increased HIF2α expression during neural crest may provide a survival benefit for neural crest derived cells. The organ of Zuckerkandl, the largest source of extra-adrenal chromaffin cells in mammals, disappears postnatally by a glucocorticoid-mediated mechanism of autophagy (123). An impairment of this mechanism during embryogenesis or immediately after birth may initiate premature paraganglioma formation. Unlike in neuroblastoma, where spontaneous but also drug-induced transitions from different stages of differentiation are observed (97), in the case of PPGLs this hypothesis would tend to suggest that the differentiation status is largely determined by the cellular origin and the underlying mutation (124). The slow growth and comparatively low aggressiveness of PPGLs, indicating a comparable low cellular flexibility, would potentially further support this hypothesis.
In hereditary PPGL patients, multifocal and bilateral tumors are associated with an earlier onset of the disease than in patients with solitary PPGLs (108). This might be explained by the initiation (second hit) of these tumors from a single tumor stem cell already during neural crest cell migration (Knudson two-hit hypothesis (125)). This would imply that in some patients with germline PVs the second chromosomal hit already occurs during embryogenesis, before the neural crest cells migrate to their paraganglial or adrenal localization.
Germline PVs in MAX are associated with bilateral and multifocal PPGLs (67%) (126, 127) and the occurrence of both neuroblastomas and PPGLs (128, 129), which highlights the importance of the MYC/MAX convergence point in these catecholamine-producing neoplasms. Furthermore, it suggests that the chromosomal second hit can occur early enough in embryogenesis to allow differentiation of the migrating neural crest cells into both, neuroblasts and chromaffin cells. Although tumors with MAX loss-of-function mutations classically belong to cluster 2 PPGLs, data on catecholamines suggest a biochemical phenotype intermediate between the established epinephrine-producing phenotype of cluster 2 PPGLs and a norepinephrine-producing phenotype of cluster 1 PPGL (22, 118, 127). PPGLs with MAX mutations belong to a specific molecular subgroup defined as cortical admixture subtype, which overexpress both PPGL markers and adrenal cortex markers (111). The intermediate phenotype of MAX-mutated PPGLs suggests that a fully functional MYC/MAX complex is required to facilitate differentiation. Whether c-Myc and MYCN assign different roles by the manifestation of the different phenotypic feature remain unclear. However, it would be reasonable to assume that they exert different functions, given their role in neural crest cell differentiation and migration.
A better understanding of the underlying cellular and molecular mechanisms also opens up for new therapeutic approaches to treat these catecholamine-producing neoplasms of the neural crest. In addition to the involvement of HIF2α in tumorigenesis of PPGLs, we and others also demonstrated the involvement of HIF2α in PPGL metastasis (1, 62, 130, 131), as reflected by the increased metastasis rate of pseudohypoxic cluster 1 PPGLs compared with cluster 2 PPGLs (1). Through the development of selective small-molecule inhibitors targeting HIF2α/ARNT dimerization, potentially suitable drugs are available, which are of particular interest for the more aggressive pseudohypoxic PPGLs and neuroblastomas. Nevertheless, some preclinical PPGL and neuroblastoma models show a lack of efficiency of these inhibitors (1, 63, 132), which is also in line with some clinical data from other HIF2α-dependent tumor entities that show resistance towards the available HIF2α inhibitors (133, 134). This partial lack of efficiency in HIF2α-dependent tumors may provide preliminary evidence that mechanisms independent of ARNT/HIF2α dimerization, for instance through interactions with the MYC/MAX complex (27, 34, 39), are involved in tumorigenesis and metastasis of these tumors. The ARNT-independent mechanisms of HIF2α may offer alternative therapeutic approaches for some patients who exhibit resistance to HIF2α inhibitors or may even be more efficient.
Direct therapeutic targeting of MYC/MYCN has been a challenge for decades considering its “undrugability” protein structure, but a least some small molecules are available that inhibit dimerization of MYC/MAX (135). However, this approach has shown only limited effectiveness (136). This may indicate other factors that influence the complex and thus MYC-target gene expression. For example, HIF2a expression in neuroblastomas is also associated with increased aggressiveness and a more undifferentiated phenotype. Similar to observations in PPGLs, ARNT-dependent inhibition of HIF2α by PT2385 (HIF2α inhibitor) is not sufficient to regulate HIF2α downstream target genes in neuroblastoma (63), which also indicates a ARNT-independent mechanisms of HIF2α in neuroblastoma (see discussion above). This further underlines the potential importance of the HIF-MYC/MAX interaction in these tumors.
Emerging single-cell methods have provide further insight in neural crest-derived cell linages, particularly in the adrenal gland, while epigenetic and transcriptomic data have increased our knowledge of tumorigenesis and progression of adrenal neoplasms. A tight regulation of HIF and MYC signaling pathways during neural crest development appears to be essential, among other things, for the complete and correct formation of the adrenal medulla and the sympathetic and parasympathetic ganglia. Dysregulation in either pathway may cause the emergence of neuroblastomas or pheochromocytomas, associated with an undifferentiated and more aggressive phenotype. Especially in pheochromocytomas/paragangliomas, increased expression and stabilization of HIF2α causes these tumors to appear at a younger age. An improved understanding of the cellular origin and pathogenesis of these pediatric adrenal neoplasms will contribute to a more efficient subtype classification and allow more precise and effective treatment of these young patients; in particular, the HIF-MYC/MAX interaction should be further considered.
NB carefully reviewed the literature and wrote the first version of the manuscript. FW and GE provided conceptual input, contributed to the editing, and revised the manuscript. All authors contributed to the article and approved the submitted version.
The work has been supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the CRC/Transregio 205, Project No. 314061271 – TRR ´The Adrenal: Central Relay in Health and Disease`.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Bechmann N, Moskopp ML, Ullrich M, Calsina B, Wallace PW, Richter S, et al. HIF2α supports pro-metastatic behavior in pheochromocytomas/paragangliomas. Endocrine-Related Cancer (2020) 27:625–40. doi: 10.1530/ERC-20-0205
2. Jögi A, Øra I, Nilsson H, Lindeheim A, Makino Y, Poellinger L, et al. Hypoxia alters gene expression in human neuroblastoma cells toward an immature and neural crest-like phenotype. Proc Natl Acad Sci (2002) 99:7021–6. doi: 10.1073/pnas.102660199
3. Pietras A, Hansford LM, Johnsson AS, Bridges E, Sjölund J, Gisselsson D, et al. HIF-2α maintains an undifferentiated state in neural crest-like human neuroblastoma tumor-initiating cells. Proc Natl Acad Sci (2009) 106:16805–10. doi: 10.1073/pnas.0904606106
4. Holmquist-Mengelbier L, Fredlund E, Löfstedt T, Noguera R, Navarro S, Nilsson H, et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell (2006) 10:413–23. doi: 10.1016/j.ccr.2006.08.026
5. Westerlund I, Shi Y, Toskas K, Fell SM, Li SSurova O, et al. Combined epigenetic and differentiation-based treatment inhibits neuroblastoma tumor growth and links HIF2α to tumor suppression. Proc Natl Acad Sci (2017) 114:E6137–46. doi: 10.1073/pnas.1700655114
6. Westerlund I, Shi Y, Holmberg J. EPAS1/HIF2α correlates with features of low-risk neuroblastoma and with adrenal chromaffin cell differentiation during sympathoadrenal development. Biochem Biophys Res Commun (2019) 508:1233–9. doi: 10.1016/j.bbrc.2018.12.076
7. Westerlund I, Shi Y, Toskas K, Fell SM, Li S, Södersten E, et al. High levels of EPAS1 are closely associated with key features of low-risk neuroblastoma. Proc Natl Acad Sci (2017) 114:E10859–60. doi: 10.1073/pnas.1718429115
8. Sun HX, Xu Y, Yang XR, Wang WM, Bai H, Shi RY, et al. Hypoxia inducible factor 2 alpha inhibits hepatocellular carcinoma growth through the transcription factor dimerization partner 3/E2F transcription factor 1–dependent apoptotic pathway. Hepatology (2013) 57:1088–97. doi: 10.1002/hep.26188
9. Imamura T, Kikuchi H, Herraiz MT, Park DY, Mizukami Y, Mino-Kenduson M, et al. HIF-1α and HIF-2α have divergent roles in colon cancer. Int J Cancer (2009) 124:763–71. doi: 10.1002/ijc.24032
10. Yang S-l, Liu L-p, Niu L, Sun YF, Yang XR, Fan J, et al. Downregulation and pro-apoptotic effect of hypoxia-inducible factor 2 alpha in hepatocellular carcinoma. Oncotarget (2016) 7:34571. doi: 10.18632/oncotarget.8952
11. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harbor Perspect Med (2013) 3:a014415. doi: 10.1101/cshperspect.a014415
12. Kameneva P, Artemov AV, Kastriti ME, Faure L, Olsen TK, Otte J, et al. Single-cell transcriptomics of human embryos identifies multiple sympathoblast lineages with potential implications for neuroblastoma origin. Nat Genet (2021) 53:694-706. doi: 10.1038/s41588-021-00818-x
13. Goldstein DS. Adrenal responses to stress. Cell Mol Neurobiol (2010) 53:694–706. doi: 10.1007/s10571-010-9606-9
14. Bechmann N, Berger I, Bornstein SR, Steenblock C. Adrenal medulla development and medullary-cortical interactions. Mol Cell Endocrinol (2021) 528:111258. doi: 10.1016/j.mce.2021.111258
15. Maguire LH, Thomas AR, Goldstein AM. Tumors of the neural crest: common themes in development and cancer. Dev Dynam (2015) 244:311–22. doi: 10.1002/dvdy.24226
16. Anderson DJ, Carnahan JF, Michelsohn A, Patterson PH. Antibody markers identify a common progenitor to sympathetic neurons and chromaffin cells in vivo and reveal the timing of commitment to neuronal differentiation in the sympathoadrenal lineage. J Neurosci (1991) 11:3507–19. doi: 10.1523/JNEUROSCI.11-11-03507.1991
17. Furlan A, Dyachuk V, Kastriti ME, Calvo-Enrique L, Abdo H, Hadjab S, et al. Multipotent peripheral glial cells generate neuroendocrine cells of the adrenal medulla. Science (2017) 357:eaal3753. doi: 10.1126/science.aal3753
18. Jansky S, Sharma AK, Körber V, Quintero A, Toprak UH, Wecht EM, et al. Single-cell transcriptomic analyses provide insights into the developmental origins of neuroblastoma. Nat Genet (2021) 53:683–93. doi: 10.1038/s41588-021-00806-1
19. Stainczyk SA, Westermann F. Neuroblastoma–telomere maintenance, deregulated signaling transduction and beyond. Int J Cancer (2021) 150:903–15. doi: 10.1002/ijc.33839
20. Bedoya-Reina O, Li W, Arceo M, Plescher M, Bullova P, Pui H, et al. Single-nuclei transcriptomes from human adrenal gland reveal distinct cellular identities of low and high-risk neuroblastoma tumors. Nat Commun (2021) 12:1–15. doi: 10.1038/s41467-021-24870-7
21. Crona J, Taïeb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocrine Rev (2017) 38:489–515. doi: 10.1210/er.2017-00062
22. Eisenhofer G, Pacak K, Huynh T-T, Qin N, Bratslavsky G, Linehan WM, et al. Catecholamine metabolomic and secretory phenotypes in phaeochromocytoma. Endocrine-related Cancer (2011) 18:97–111. doi: 10.1677/ERC-10-0211
24. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer (2008) 8:976–90. doi: 10.1038/nrc2231
25. Otte J, Dyberg C, Pepich A, Johnsen JI. MYCN function in neuroblastoma development. Front Oncol (2021) 3210. doi: 10.3389/fonc.2020.624079
26. Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer (2008) 8:51–6. doi: 10.1038/nrc2274
27. Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol (2000) 16:653–99. doi: 10.1146/annurev.cellbio.16.1.653
28. Wiesener MS, Jürgensen JS, Rosenberger C, Scholze C, Hörstrup JH, Warnecke C, et al. Widespread, hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J (2003) 17:271–3. doi: 10.1096/fj.02-0445fje
29. Gordan JD, Simon MC. Hypoxia-inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev (2007) 17:71–7. doi: 10.1016/j.gde.2006.12.006
30. Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, et al. HIF-2α regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev (2006) 20:557–70. doi: 10.1101/gad.1399906
31. Mu H, Yu G, Li H, Wang M, Cui Y, Zhang T, et al. Mild chronic hypoxia-induced HIF-2α interacts with c-MYC through competition with HIF-1α to induce hepatocellular carcinoma cell proliferation. Cell Oncol (2021) 44:1151–66. doi: 10.1007/s13402-021-00625-w
32. Wang L, Xue M, Chung DC. C-myc is regulated by HIF-2α in chronic hypoxia and influences sensitivity to 5-FU in colon cancer. Oncotarget (2016) 7:78910. doi: 10.18632/oncotarget.12911
33. Rawłuszko-Wieczorek AA, Horbacka K, Krokowicz P, Misztal M, Jagodziński P. Prognostic potential of dna methylation and transcript levels of HIF1A and EPAS1 in colorectal cancer. Mol Cancer Res (2014) 12:1112–27. doi: 10.1158/1541-7786.MCR-14-0054
34. Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIF-α effects on c-myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell (2008) 14:435–46. doi: 10.1016/j.ccr.2008.10.016
35. Zhang J, Sattler M, Tonon G, Grabher C, Lababidi S, Zimmerhackl A, et al. Targeting angiogenesis via a c-myc/hypoxia-inducible factor-1α–dependent pathway in multiple myeloma. Cancer Res (2009) 69:5082–90. doi: 10.1158/0008-5472.CAN-08-4603
36. Chen C, Cai S, Wang G, Cao X, Yang X, Luo X, et al. C-myc enhances colon cancer cell-mediated angiogenesis through the regulation of HIF-1α. Biochem Biophys Res Commun (2013) 430:505–11. doi: 10.1016/j.bbrc.2012.12.006
37. Weili Z, Zhikun L, Jianmin W, Qingbao T. Knockdown of USP28 enhances the radiosensitivity of esophageal cancer cells via the c-myc/hypoxia-inducible factor-1 alpha pathway. J Cell Biochem (2019) 120:201–12. doi: 10.1002/jcb.27305
38. Doe MR, Ascano JM, Kaur M, Cole MD. Myc posttranscriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res (2012) 72:949–57. doi: 10.1158/0008-5472.CAN-11-2371
39. Gordan JD, Bertout JA, Hu C-J, Diehl JA, Simon MC. HIF-2α promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell (2007) 11:335–47. doi: 10.1016/j.ccr.2007.02.006
40. Gordan JD, Thompson CB, Simon MC. HIF and c-myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell (2007) 12:108–13. doi: 10.1016/j.ccr.2007.07.006
41. Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1α induces cell cycle arrest by functionally counteracting myc. EMBO J (2004) 23:1949–56. doi: 10.1038/sj.emboj.7600196
42. Koshiji M, To KK-W, Hammer S, Kumamoto K, Harris AL, Modrich P, et al. HIF-1α induces genetic instability by transcriptionally downregulating MutSα expression. Mol Cell (2005) 17:793–803. doi: 10.1016/j.molcel.2005.02.015
43. To KKW, Sedelnikova OA, Samons M, Bonner WM, Huang LE. The phosphorylation status of PAS-b distinguishes HIF-1α from HIF-2α in NBS1 repression. EMBO J (2006) 25:4784–94. doi: 10.1038/sj.emboj.7601369
44. Florczyk U, Czauderna S, Stachurska A, Tertil M, Nowak W, Kozakowska M, et al. Opposite effects of HIF-1α and HIF-2α on the regulation of IL-8 expression in endothelial cells. Free Radical Biol Med (2011) 51:1882–92. doi: 10.1016/j.freeradbiomed.2011.08.023
45. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of c-MYC activity. Cancer Cell (2007) 11:407–20. doi: 10.1016/j.ccr.2007.04.001
46. Corn PG, Ricci MS, Scata KA, Arsham AM, Simon MC, Dicker DT, et al. Mxi1 is induced by hypoxia in a HIF-1–dependent manner and protects cells from c-myc-induced apoptosis. Cancer Biol Ther (2005) 4:1285–94. doi: 10.4161/cbt.4.11.2299
47. Xue G, Yan H, Zhang Y, Zhu XT, Mei Q, Sun SH. C-myc-mediated repression of miR-15-16 in hypoxia is induced by increased HIF-2α and promotes tumor angiogenesis and metastasis by upregulating FGF2. Oncogene (2015) 34:1393–406. doi: 10.1038/onc.2014.82
48. Wong WJ, Qiu B, Nakazawa MS, Qing G, Simon MC. MYC degradation under low O2 tension promotes survival by evading hypoxia-induced cell death. Mol Cell Biol (2013) 33:3494–504. doi: 10.1128/MCB.00853-12
49. Zarrabi AJ, Kao D, Nguyen DT, Loscalzo J, Handy D. Hypoxia-induced suppression of c-myc by HIF-2α in human pulmonary endothelial cells attenuates TFAM expression. Cell signalling (2017) 38:230–7. doi: 10.1016/j.cellsig.2017.07.008
50. Li Q, Kluz T, Sun H, Costa M. Mechanisms of c-myc degradation by nickel compounds and hypoxia. PloS One (2009) 4:e8531. doi: 10.1371/journal.pone.0008531
51. Kokovay E, Temple S. Taking neural crest stem cells to new heights. Cell (2007) 131:234–6. doi: 10.1016/j.cell.2007.10.006
52. Hockman D, Adameyko I, Kaucka M, Barraud P, Otani T, Hunt A, et al. Striking parallels between carotid body glomus cell and adrenal chromaffin cell development. Dev Biol (2018) 444:S308–24. doi: 10.1016/j.ydbio.2018.05.016
53. Seidler F, Slotkin TA. Adrenomedullary function in the neonatal rat: responses to acute hypoxia. J Physiol (1985) 358:1–16. doi: 10.1113/jphysiol.1985.sp015536
54. Slotkin T, Seidler F. Adrenomedullary catecholamine release in the fetus and newborn: secretory mechanisms and their role in stress and survival. J Dev Physiol (1988) 10:1–16.
55. Thompson RJ, Farragher SM, Cutz E, Nurse CA. Developmental regulation of O2 sensing in neonatal adrenal chromaffin cells from wild-type and NADPH-oxidase-deficient mice. Pflügers Archiv (2002) 444:539–48. doi: 10.1007/s00424-002-0853-6
56. Dunwoodie SL. The role of hypoxia in development of the mammalian embryo. Dev Cell (2009) 17:755–73. doi: 10.1016/j.devcel.2009.11.008
57. Morriss GM, New D. Effect of oxygen concentration on morphogenesis of cranial neural folds and neural crest in cultured rat embryos.J Embryol Exp Morphol (1979) 54:17–35. doi: 10.1242/dev.54.1.17
58. Scully D, Keane E, Batt E, Karunakaran P, Higgins DF, Itasaki N, et al. Hypoxia promotes production of neural crest cells in the embryonic head. Development (2016) 143:1742–52. doi: 10.1242/dev.131912
59. Naňka O, Valášek P, Dvořáková M, Grim M. Experimental hypoxia and embryonic angiogenesis. Dev dynamics (2006) 235:723–33. doi: 10.1002/dvdy.20689
60. Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev (1998) 12:149–62. doi: 10.1101/gad.12.2.149
61. Barriga EH, Maxwell PH, Reyes AE, Mayor R. The hypoxia factor hif-1α controls neural crest chemotaxis and epithelial to mesenchymal transition. J Cell Biol (2013) 201:759–76. doi: 10.1083/jcb.201212100
62. Bechmann N, Eisenhofer G. Hypoxia-inducible factor 2α: a key player in tumorigenesis and metastasis of pheochromocytoma and paraganglioma? Exp Clin Endocrinol Diabetes (2021) 130:282–9. doi: 10.1055/a-1526-5263
63. Persson CU, von Stedingk K, Fredlund E, Bexell D, Påhlman S, Wigerup C, et al. ARNT-dependent HIF-2 transcriptional activity is not sufficient to regulate downstream target genes in neuroblastoma. Exp Cell Res (2020) 388:111845. doi: 10.1016/j.yexcr.2020.111845
64. Bohuslavova R, Cerychova R, Papousek F, Olejnickova V, Bartos M, Görlach A, et al. HIF-1α is required for development of the sympathetic nervous system. Proc Natl Acad Sci (2019) 116:13414–23. doi: 10.1073/pnas.1903510116
65. Macías D, Fernández-Agüera MC, Bonilla-Henao V, Bonilla‐Henao V, López‐Barneo J. Deletion of the von hippel–lindau gene causes sympathoadrenal cell death and impairs chemoreceptor-mediated adaptation to hypoxia. EMBO Mol Med (2014) 6:1577–92. doi: 10.15252/emmm.201404153
66. Niklasson CU, Fredlund E, Monni E, Lindvall JM, Kokaia Z, Hammarlund EU, et al. Hypoxia inducible factor-2α importance for migration, proliferation, and self-renewal of trunk neural crest cells. Dev Dynam (2021) 250:191–236. doi: 10.1002/dvdy.253
67. Mohlin S, Hamidian A, Påhlman S. HIF2A and IGF2 expression correlates in human neuroblastoma cells and normal immature sympathetic neuroblasts. Neoplasia (2013) 15:328–IN338. doi: 10.1593/neo.121706
68. Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev (1998) 12:3320–4. doi: 10.1101/gad.12.21.3320
69. Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, et al. Abnormal sympathoadrenal development and systemic hypotension in PHD3–/– mice. Mol Cell Biol (2008) 28:3386–400. doi: 10.1128/MCB.02041-07
70. Bechmann N, Poser I, Seifert V, Greunke C, Ullrich M, Qin N, et al. Impact of extrinsic and intrinsic hypoxia on catecholamine biosynthesis in absence or presence of HIF2α in pheochromocytoma cells. Cancers (2019) 11:594. doi: 10.3390/cancers11050594
71. Phillippe M. Fetal catecholamines. Am J obstetrics gynecol (1983) 146:840–55. doi: 10.1016/0002-9378(83)91088-8
72. Ream MA, Chandra R, Peavey M, Ray AM, Roffler-Tarlov S, Kim HG, et al. High oxygen prevents fetal lethality due to lack of catecholamines. Am J Physiology-Regulatory Integr Comp Physiol (2008) 295:R942–53. doi: 10.1152/ajpregu.00860.2007
73. Padbury JF. Functional maturation of the adrenal medulla and peripheral sympathetic nervous system. Bailliere's Clin Endocrinol Metab (1989) 3:689–705. doi: 10.1016/S0950-351X(89)80049-7
74. Paulick R, Kastendieck E, Wernze H. Catecholamines in arterial and venous umbilical blood: placental extraction, correlation with fetal hypoxia, and transcutaneous partial oxygen tension. J Perinat Med (1985) 13:31–42. doi: 10.1515/jpme.1985.13.1.31
75. Watts D, Bechmann N, Meneses A, Poutakidou IK, Kaden D, Conrad C, et al. HIF2α regulates the synthesis and release of epinephrine in the adrenal medulla. J Mol Med (2021) 99:1655–66. doi: 10.1007/s00109-021-02121-y
76. Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan LJ, et al. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1–/– mice. Nat Genet (2003) 35:331–40. doi: 10.1038/ng1266
77. Studer L, Csete M, Lee S-H, Kabbani N, Walikonis J, Wold B, et al. Enhanced proliferation, survival, and dopaminergic differentiation of CNS precursors in lowered oxygen. J Neurosci (2000) 20:7377–83. doi: 10.1523/JNEUROSCI.20-19-07377.2000
78. Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ, et al. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci (2000) 20:7370–6. doi: 10.1523/JNEUROSCI.20-19-07370.2000
79. Charron J, Malynn BA, Fisher P, Stewart V, Jeannotte L, Goff SP, et al. Embryonic lethality in mice homozygous for a targeted disruption of the n-myc gene. Genes Dev (1992) 6:2248–57. doi: 10.1101/gad.6.12a.2248
80. Davis AC, Wims M, Spotts GD, Hann SR, Bradley A, et al. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev (1993) 7:671–82. doi: 10.1101/gad.7.4.671
81. Sawai S, Shimono A, Wakamatsu Y, Palmes C, Hanaoka K, Kondoh H, et al. Defects of embryonic organogenesis resulting from targeted disruption of the n-myc gene in the mouse. Development (1993) 117:1445–55. doi: 10.1242/dev.117.4.1445
82. Stanton BR, Perkins AS, Tessarollo L, Sassoon DA, Parada LF. Loss of n-myc function results in embryonic lethality and failure of the epithelial component of the embryo to develop. Genes Dev (1992) 6:2235–47. doi: 10.1101/gad.6.12a.2235
83. Kerosuo L, Neppala P, Hsin J, Mohlin S, Vieceli FM, Török Z, et al. Enhanced expression of MycN/CIP2A drives neural crest toward a neural stem cell-like fate: implications for priming of neuroblastoma. Proc Natl Acad Sci (2018) 115:E7351–60. doi: 10.1073/pnas.1800039115
84. Bellmeyer A, Krase J, Lindgren J, LaBonne C. The protooncogene c-myc is an essential regulator of neural crest formation in xenopus. Dev Cell (2003) 4:827–39. doi: 10.1016/S1534-5807(03)00160-6
85. Light W, Vernon AE, Lasorella A, LaBonne C. Xenopus Id3 is required downstream of myc for the formation of multipotent neural crest progenitor cells. Development (2005) 132:1831–41. doi: 10.1242/dev.01734
86. Kerosuo L, Bronner ME. cMyc regulates the size of the premigratory neural crest stem cell pool. Cell Rep (2016) 17:2648–59. doi: 10.1016/j.celrep.2016.11.025
87. Wakamatsu Y, Watanabe Y, Nakamura H, Kondoh H. Regulation of the neural crest cell fate by n-myc: promotion of ventral migration and neuronal differentiation. Development (1997) 124:1953–62. doi: 10.1242/dev.124.10.1953
88. Knoepfler PS, Cheng PF, Eisenman RN. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev (2002) 16:2699–712. doi: 10.1101/gad.1021202
89. Zinin N, Adameyko I, Wilhelm M, Fritz N, Uhlén P, Ernfors P, et al. MYC proteins promote neuronal differentiation by controlling the mode of progenitor cell division. EMBO Rep (2014) 15:383–91. doi: 10.1002/embr.201337424
90. Khudyakov J, Bronner-Fraser M. Comprehensive spatiotemporal analysis of early chick neural crest network genes. Dev dynamics (2009) 238:716–23. doi: 10.1002/dvdy.21881
91. Mobley BC, Kwon M, Kraemer BR, Hickman FE, Qiao J, Chung DH, et al. Expression of MYCN in multipotent sympathoadrenal progenitors induces proliferation and neural differentiation, but is not sufficient for tumorigenesis. PloS One (2015) 10:e0133897. doi: 10.1371/journal.pone.0133897
92. Olsen R, Otero J, Garcia-Lopez J, Wallace K, Finkelstein D, Rehg J, et al. MYCN induces neuroblastoma in primary neural crest cells. Oncogene (2017) 36:5075–82. doi: 10.1038/onc.2017.128
93. Ackermann S, Cartolano M, Hero B, Welte A, Kahlert Y, Roderwieser A, et al. A mechanistic classification of clinical phenotypes in neuroblastoma. Science (2018) 362:1165–70. doi: 10.1126/science.aat6768
94. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45:279–84. doi: 10.1038/ng.2529
95. Vo KT, Matthay KK, Neuhaus J, London WB, Hero B, Ambros PF, et al. Clinical, biologic, and prognostic differences on the basis of primary tumor site in neuroblastoma: a report from the international neuroblastoma risk group project. J Clin Oncol (2014) 32:3169. doi: 10.1200/JCO.2014.56.1621
96. Jereb B, Bretsky SS, Vogel R, Helson L. Age and prognosis in neuroblastoma. review of 112 patients younger than 2 years. Am J Pediatr hematology/oncol (1984) 6:233–43. doi: 10.1097/00043426-198423000-00002
97. Van Groningen T, Koster J, Valentijn LJ, Zwijnenburg DA, Akogul N, Hasselt NE, et al. Neuroblastoma is composed of two super-enhancer-associated differentiation states. Nat Genet (2017) 49:1261–6. doi: 10.1038/ng.3899
98. Gartlgruber M, Sharma AK, Quintero A, Dreidax D, Jansky S, Park YG, et al. Super enhancers define regulatory subtypes and cell identity in neuroblastoma. Nat Cancer (2021) 2:114–28. doi: 10.1038/s43018-020-00145-w
99. Westermann F, Muth D, Benner A, Bauer T, Henrich KO, Oberthuer A, et al. Distinct transcriptional MYCN/c-MYC activities are associated with spontaneous regression or malignant progression in neuroblastomas. Genome Biol (2008) 9:1–14. doi: 10.1186/gb-2008-9-10-r150
100. Ferrucci F, Ciaccio R, Monticelli S, Pigini P, di Giacomo S, Purgato S, et al. MAX to MYCN intracellular ratio drives the aggressive phenotype and clinical outcome of high risk neuroblastoma. Biochim Biophys Acta (BBA)-Gene Regul Mech (2018) 1861:235–45. doi: 10.1016/j.bbagrm.2018.01.007
101. Jögi A, Øra I, Nilsson H, Poellinger L, Axelson H, Påhlman S. Hypoxia-induced dedifferentiation in neuroblastoma cells. Cancer Lett (2003) 197:145–50. doi: 10.1016/S0304-3835(03)00095-8
102. Mohlin S, Wigerup C, Jögi A, Påhlman S. Hypoxia, pseudohypoxia and cellular differentiation. Exp Cell Res (2017) 356:192–6. doi: 10.1016/j.yexcr.2017.03.007
103. Zou Y, Palte MJ, Deik AA, Li H, Eaton JK, Wang W, et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun (2019) 10:1617. doi: 10.1038/s41467-019-09277-9
104. Alborzinia H, Flórez AF, Kreth S, Brückner LM, Yildiz U, Gartlgruber M, et al. MYCN mediates cysteine addiction and sensitizes neuroblastoma to ferroptosis. Nat Cancer (2022) 3:471–85. doi: 10.1038/s43018-022-00355-4
105. Zethoven M, Martelotto L, Pattison A, Bowen B, Balachander S, Flynn A, et al. Single-nuclei and bulk-tissue gene-expression analysis of pheochromocytoma and paraganglioma links disease subtypes with tumor microenvironment. Nat Commun (2022) 13:6262. doi: 10.1038/s41467-022-34011-3
106. Michałowska I, Ćwikła J, Michalski W, Wyrwicz LS, Prejbisz A, Szperl M, et al. Growth rate of paragangliomas related to germline mutations of the SDHX genes. Endocrine Pract (2017) 23:342–52. doi: 10.4158/EP161377.OR
107. Powers JF, Pacak K, Tischler AS. Pathology of human pheochromocytoma and paraganglioma xenografts in NSG mice. Endocrine Pathol (2017) 28:2–6. doi: 10.1007/s12022-016-9452-5
108. Eisenhofer G, Timmers HJ, Lenders JW, Bornstein SR, Tiebel O, Mannelli M, et al. Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab (2011) 96:375–84. doi: 10.1210/jc.2010-1588
109. Eisenhofer G, Peitzsch M, Bechmann N, Huebner A. Biochemical diagnosis of catecholamine-producing tumors of childhood: neuroblastoma, pheochromocytoma and paraganglioma. Front Endocrinol (2022) 1446. doi: 10.3389/fendo.2022.901760
110. Eisenhofer G, Huynh T, Pacak K, Brouwers FM, Walther MM, Linehan WM, et al. Distinct gene expression profiles in norepinephrine-and epinephrine-producing hereditary and sporadic pheochromocytomas: activation of hypoxia-driven angiogenic pathways in von hippel–lindau syndrome. Endocrine-Related Cancer (2004) 11:897–911. doi: 10.1677/erc.1.00838
111. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell (2017) 31:181–93. doi: 10.1016/j.ccell.2017.01.001
112. Pamporaki C, Prodanov T, Meuter L, Berends AM, Bechmann N, Constantinescu G, et al. Determinants of disease-specific survival in patients with and without metastatic pheochromocytoma and paraganglioma. Eur J Cancer (2022) 169:32–41. doi: 10.1016/j.ejca.2022.03.032
113. Juhlin CC. Challenges in paragangliomas and pheochromocytomas: from histology to molecular immunohistochemistry. Endocrine Pathol (2021) 32:228–44. doi: 10.1007/s12022-021-09675-0
114. Redlich A, Pamporaki C, Lessel L, Frühwald MC, Vorwerk P, Kuhlen M. Pseudohypoxic pheochromocytomas and paragangliomas dominate in children. Pediatr Blood Cancer (2021) 68:e28981. doi: 10.1002/pbc.28981
115. Buffet A, Smati S, Mansuy L, Ménara M, Lebras M, Heymann MF, et al. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab (2014) 99:E369–73. doi: 10.1210/jc.2013-2600
116. Yang C, Hong CS, Prchal JT, Balint MT, Pacak K, Zhuang Z. Somatic mosaicism of EPAS1 mutations in the syndrome of paraganglioma and somatostatinoma associated with polycythemia. Hum Genome Variation (2015) 2:1–2. doi: 10.1038/hgv.2015.53
117. Dahia PL, Toledo RA. Recognizing hypoxia in phaeochromocytomas and paragangliomas. Nat Rev Endocrinol (2020) 16:191–2. doi: 10.1038/s41574-020-0324-1
118. Qin N, De Cubas AA, Garcia-Martin R, Richter S, Peitzsch M, Menschikowski M, et al. Opposing effects of HIF1α and HIF2α on chromaffin cell phenotypic features and tumor cell proliferation: insights from MYC-associated factor X. Int J Cancer (2014) 135:2054–64. doi: 10.1002/ijc.28868
119. Påhlman S, Mohlin S. Hypoxia and hypoxia-inducible factors in neuroblastoma. Cell Tissue Res (2018) 372:269–75. doi: 10.1007/s00441-017-2701-1
120. Vaidya A, Flores SK, Cheng Z-M, Nicolas M, Deng Y, Opotowsky AR, et al. EPAS1 mutations and paragangliomas in cyanotic congenital heart disease. New Engl J Med (2018) 378:1259–61. doi: 10.1056/NEJMc1716652
121. Agarwal S, Jindal I, Balazs A, Paul D. Catecholamine-secreting tumors in pediatric patients with cyanotic congenital heart disease. J Endocrine Soc (2019) 3:2135–50. doi: 10.1210/js.2019-00226
122. Ogasawara T, Fujii Y, Kakiuchi N, Shiozawa Y, Sakamoto R, Ogawa Y, et al. Genetic analysis of pheochromocytoma and paraganglioma complicating cyanotic congenital heart disease. J Clin Endocrinol Metab (2022) 107:2545–55. doi: 10.1210/clinem/dgac362
123. Schober A, Parlato R, Huber K, Kinscherf R, Hartleben B, Huber TB, et al. Cell loss and autophagy in the extra-adrenal chromaffin organ of zuckerkandl are regulated by glucocorticoid signalling. J Neuroendocrinol (2013) 25:34–47. doi: 10.1111/j.1365-2826.2012.02367.x
124. Jiang J, Zhang J, Pang Y, Bechmann N, Li M, Monteagudo M, et al. Sino-European differences in the genetic landscape and clinical presentation of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab (2020) 105:3295–307. doi: 10.1210/clinem/dgaa502
125. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci (1971) 68:820–3. doi: 10.1073/pnas.68.4.820
126. Nölting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized management of pheochromocytoma and paraganglioma. Endocrine Rev (2021) 43:199–239. doi: 10.1210/endrev/bnab019
127. Burnichon N, Cascón A, Schiavi F, Morales NP, Comino-Méndez I, Abermil N, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res (2012) 18:2828–37. doi: 10.1158/1078-0432.Ccr-12-0160
128. Duarte DB, Ferreira L, Santos AP, Costa C, Lima J, Santos C, et al. Case report: pheochromocytoma and synchronous neuroblastoma in a family with hereditary pheochromocytoma associated with a MAX deleterious variant. Front Endocrinol (2021) 12:255. doi: 10.3389/fendo.2021.609263
129. Seabrook AJ, Harris JE, Velosa SB, Kim E, McInerney-Leo AM, Dwight T, et al. Multiple endocrine tumors associated with germline MAX mutations: multiple endocrine neoplasia type 5? J Clin Endocrinol Metab (2021) 106:1163–82. doi: 10.1210/clinem/dgaa957
130. Morin A, Goncalves J, Moog S, Castro-Vega LJ, Job S, Buffet A, et al. TET-mediated hypermethylation primes SDH-deficient cells for HIF2α-driven mesenchymal transition. Cell Rep (2020) 30:4551–4566. e4557. doi: 10.1016/j.celrep.2020.03.022
131. Helm J, Drukewitz S, Poser I, Richter S, Friedemann M, William D, et al. Treatment of pheochromocytoma cells with recurrent cycles of hypoxia: a new pseudohypoxic In vitro model. Cells (2022) 11:560. doi: 10.3390/cells11030560
132. Moog S, Salgues B, Braik-Djellas Y, Viel T, Balvay D, Autret G, et al. Preclinical evaluation of targeted therapies in sdhb-mutated tumors. Endocrine-Related Cancer (2022) 29:375–88 . doi: 10.1530/ERC-22-0030
133. Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature (2016) 539:112–7. doi: 10.1038/nature19796
134. Courtney KD, Ma Y, de Leon AD, Christie A, Xie Z, Woolford L, et al. HIF-2 complex dissociation, target inhibition, and acquired resistance with PT2385, a first-in-class HIF-2 inhibitor, in patients with clear cell renal cell carcinoma. Clin Cancer Res (2020) 26:793–803. doi: 10.1158/1078-0432.CCR-19-1459
135. Chen H, Liu H, Qing G. Targeting oncogenic myc as a strategy for cancer treatment. Signal transduction targeted Ther (2018) 3:1–7. doi: 10.1038/s41392-018-0008-7
Keywords: pheochromocytoma, neuroblastoma, catecholamines, sympathoadrenal cell lineage, hypoxia, MYC, neural crest, paraganglioma
Citation: Bechmann N, Westermann F and Eisenhofer G (2023) HIF and MYC signaling in adrenal neoplasms of the neural crest: implications for pediatrics. Front. Endocrinol. 14:1022192. doi: 10.3389/fendo.2023.1022192
Received: 18 August 2022; Accepted: 09 May 2023;
Published: 08 June 2023.
Edited by:
Antje Redlich, University Hospital Magdeburg, GermanyReviewed by:
C. Christofer Juhlin, Karolinska Institutet (KI), SwedenCopyright © 2023 Bechmann, Westermann and Eisenhofer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicole Bechmann, Tmljb2xlLmJlY2htYW5uQHVuaWtsaW5pa3VtLWRyZXNkZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.